

Abstract
Responsible for interpreting histone post-translational modifications (PTMs), epigenetic reader proteins have emerged as novel therapeutic targets for a wide range of diseases. Chemical probes have been critical in enabling target validation studies and have led to translational advances in cancer and inflammation-related pathologies. Here, we present the most recently reported probes of reader proteins that recognize acylated and methylated lysine. We will discuss challenges associated with achieving potent antagonism of reader domains and review ongoing efforts to overcome these hurdles, focusing on targeting strategies including the use of peptidomimetic ligands, allosteric modulators, and protein degraders.
Introduction
Proper regulation of gene transcription by chromatin regulatory factors is essential to nearly every aspect of normal biology in multicellular organisms. Consequently, growing research has demonstrated the importance of the dysregulation of this network of transcription factors, chromatin remodelers, and histone post-translational modifications (PTMs) in diseases ranging from cancer to autoimmune and neurological disorders [1–3]. However, complex genetic association data and uncertain disease etiology has created a need for chemical tools to both investigate and pharmacologically perturb the function of these proteins. Potent, selective, and cell-permeable chemical probes have numerous advantageous over genetic methods of protein-target manipulation as they provide temporal resolution, domain-specific antagonism, and, most excitingly, the potential for therapeutic development [4]. In this review we discuss recent advancements in chemical probes targeting ‘readers’ of histone PTMs, which are responsible for binding to a specific PTM to elicit a downstream biological output. Due to their central and highly regulated role in gene transcription, the development of validated probes for these targets has been of great interest in the last 20 years and led to the progression of small molecules to the clinic (Table 1, ClinicalTrials.gov).
Table 1.
Antagonists of epigenetic reader domains in clinical trials
| Antagonist | Antagonist Class | Target | Disease State | Phase(s) | NCT Number(s) |
|---|---|---|---|---|---|
| ABBV-075 | Kac Reader | BET | Advanced Cancers | I | NCT02391480 |
| Myelofibrosis | I | NCT04480086 | |||
| ABBV-744 | Kac Reader | BET (BD2) | AML | I | NCT03360006 |
| Myelofibrosis | I | NCT04454658 | |||
| BMS-986158 | Kac Reader | BET | Pediatric Tumors Lymphomas | I | NCT03936465 |
| Advanced Cancers | I-II | NCT02419417 | |||
| FT-1101 | Kac Reader | BET | Hematologic Malignancies | I | NCT02543879 |
| GS-5829 | Kac Reader | BET | Solid Tumors Lymphomas | I | NCT02392611 |
| Breast Cancer | I-II | NCT02983604 | |||
| Castration-Resistant Prostate Cancer | I-II | NCT02607228 | |||
| GSK2820151 | Kac Reader | BET | Solid Tumors | I | NCT02630251 |
| GSK4027 | Kac Reader | PCAF/GCN5 | COPD Emphysema Chronic Bronchitis | IV | NCT00175565 |
| I-BET762 (GSK525762) | Kac Reader | BET | Hematologic Malignancies | I II |
NCT03925428
NCT01943851 |
| Castration-Resistant Prostate Cancer | I | NCT03150056 | |||
| Solid Tumors | I I II |
NCT02259114
NCT03925428 NCT03266159 |
|||
| Breast Cancer | II | NCT02964507 | |||
| Carcinoma | I I-II |
NCT01587703
NCT04116359 |
|||
| INCB054329 | Kac Reader | BET | Hematologic Malignancies Solid Tumors | I-II | NCT02431260 |
| INCB-057643 | Kac Reader | BET | Myelofibrosis | I | NCT04279847 |
| Solid Tumors | I-II | NCT02959437 | |||
| MAK683 | Kme Reader | EED | DLBCL | I-II | NCT02900651 |
| MK-8628 (OTX015) | Kac Reader | BET | Hematologic Malignancies | I | NCT01713582 |
| Advanced Cancers | I | NCT02259114 | |||
| GBM | II | NCT02296476 | |||
| PLX51107 | Kac Reader | BRD4 | AML Myelodysplastic Syndromes | I | NCT04022785 |
| Hematologic Malignancies Solid Tumors | I | NCT02683395 | |||
| RO6870810 | Kac Reader | BET | AML Myelodysplastic Syndromes | I | NCT02308761 |
| Multiple Myeloma | I | NCT03068351 | |||
| Solid Tumors | I | NCT01987362 | |||
| Ovarian Cancer TNBC | I | NCT03292172 | |||
| B-cell Lymphomas | I | NCT03255096 | |||
| RVX-208 | Kac Reader | BET | Diabetes | II III |
NCT01728467
NCT02586155 |
| Fabry Disease | I-II | NCT03228940 | |||
| Kidney Failure | I-II | NCT03160430 | |||
| Cardiovascular Diseases | II I-II II III II |
NCT01058018
NCT00768274 NCT01423188 NCT02586155 NCT01067820 |
|||
| SYHA1801 | Kac Reader | BRD4 | Solid Tumors | I | NCT04309968 |
| ZEN003694 | Kac Reader | BET | Castration-Resistant Prostate Cancer | I I-II I-II II |
NCT02705469
NCT04145375 NCT02711956 NCT04471974 |
| TNBC | II | NCT03901469 |
Despite growing evidence validating the importance and druggability of some readers, the development of antagonists toward these proteins has overall been slow, in part due to the nature of their endogenous binding modes. Typically, reader domains interact with their histone binding partners through extended protein-protein interactions (PPIs) that can be difficult to replicate with drug-like small molecules [5]. Additionally, while some readers show high affinity and specificity for their histone targets, others do not, binding with affinities in the mM range in in vitro assays [6]. Finally, it has been shown that some readers bind their substrates through an induced-fit binding mode, where binding to the PTM induces a substantial conformational change in the reader domain to engender high-affinity binding [5,7]. This can make structure-based and rational design of antagonists challenging. Recent developments in the field have therefore focused on various targeting strategies in addition to traditional small molecule antagonism. Here, we discuss the use of peptides and peptidomimetic ligands, allosteric modulators, and targeted chemical degraders as alternative approaches to block the function of reader domains.
Acyl-lysine readers
Acetyl-lysine (Kac) is a common histone PTM typically associated with active transcription [5]. By neutralizing the positive charge of lysine on histone tails, Kac weakens the interaction between histones and the negatively charged DNA and thereby promotes a more open and accessible chromatin state. Furthermore, readers of Kac often act as transcriptional coactivators that recruit transcription factors and other effectors such as RNA polymerase II to these relaxed chromatin sites to initiate and maintain active transcription [8]. Accordingly, dysregulation of these proteins is implicated in a wide array of diseases including cancer [8,9].
Bromodomains.
Bromodomains (BRDs) are structurally conserved Kac reading modules found in 46 human proteins, separated into eight subfamilies according to sequence similarity (Figure 1A) [10]. In these proteins, Kac is recognized by a hydrophobic cavity formed from a left-handed bundle of four α helices (αZ, αA, αB, and αC) connected through interhelical ZA and BC loops. Binding to Kac is typically stabilized through a direct hydrogen bond with a conserved asparagine and a water-mediated hydrogen bond with a conserved tyrosine residue [8]. Despite the modest affinity of BRDs to isolated acetylated peptides in vitro, numerous highly potent nanomolar antagonists and chemical probes have been developed for this class, making BRDs an exceptionally well-studied and successfully targeted class of reader proteins. The profound biological effects observed with (+)-JQ1 and I-BET762, the first-in-class chemical probes targeting the bromodomain and extra-terminal (BET) subfamily of BRDs, established the tractability of targeting reader modules and fueled the rapid development of new antagonists to investigate the therapeutic relevance of the BET proteins [5,11,12]. Since the discovery of (+)-JQ1 and I-BET762 ten years ago, >30 chemical probes for the BET family and >20 chemical probes for non-BET BRD proteins (e.g. KAT2A/B, CERC2, CREBBP/EP300, ATAD2A/B, BRD1, BRPF1/3, BRD7/9, TAF1/1L, PBRM1, SMARCA2/4, and BAZ2A/B ) have been developed as discussed in previous reviews [9,13,14].
Figure 1.
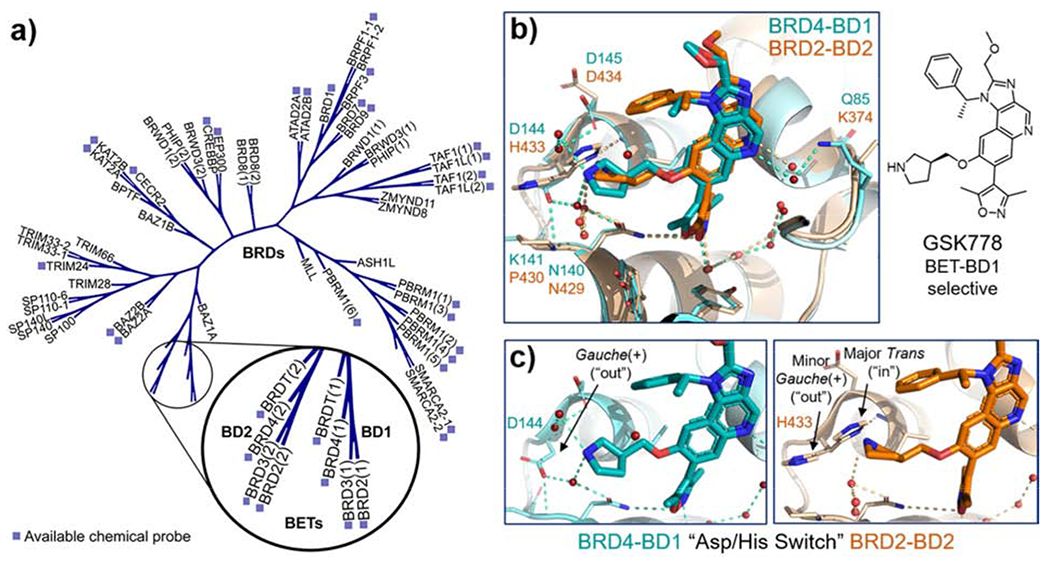
Developing selective chemical probes for the BET subfamily. (a) Phylogenetic tree of bromodomains, with available chemical probes noted; the BET subfamily and the divergence of its first and second bromodomains, BD1 and BD2, are highlighted (adapted from chromohub.thesgc.org); (b) BET BD1-selective GSK778 bound to BRD4-BD1 (in cyan, PDB ID: 6SWN [27]) overlayed with GSK778 bound to BRD2-BD2 (in orange, PDB ID: 6SWO [27]); (c) (Left) Isolated view of GSK778 bound to BRD4-BD1 highlighting the gauche(+), or “out”, conformation of BD1-specific residue Asp144; (right) isolated view of GSK778 bound to BRD2-BD2 highlighting the major trans, or “in,” conformation of BD2-specific residue His433.
Domain selectivity within the BET family.
In recent years, in addition to the ongoing chemical probe efforts in this area, there has been a renewed interest in developing BET antagonists with selectivity for only one of the two tandem BRDs, BD1 or BD2, that are present in BET family members BRD2, −3, −4, and −T. In vitro evidence supporting unique roles for BD1 and BD2 highlights the need for selective BD1 and BD2 chemical probes to deconvolute the associated biological activity of these domains [15]. Although pan-BET antagonists that target BD1 and BD2 have been efficacious in oncology and inflammation-related clinical trials, several on-target and dose-limiting adverse events have been observed, with thrombocytopenia and gastrointestinal toxicity now associated as severe safety signals of pan-BET antagonism [9]. Intriguingly, the early BD2-biased RVX-208 is the only BET antagonist to have advanced to phase III clinical trials, in this case for type II diabetes mellitus, coronary artery disease, and cardiovascular disease (Table 1), suggesting that selective antagonists may increase the tolerability profile of these compounds [16,17].
Using a “bump-and-hole” chemical genetics approach, pioneered by the Shokat lab, in which subtle, structurally nondisruptive mutations of individual BET BRDs were paired with orthogonal “bumped” analogs of pan-BET antagonist I-BET762, the BD1 domain of BRD4 was shown to be required for chromatin binding, while BD2 appeared less essential [18–20]. More recent cellular studies using a “clickable” BD2-biased molecular probe and chromatin immunoprecipitation confirm that BRD4 is bound to chromatin through its BD1 domain [21]. Meanwhile, the interactions of BRD4-BD2 with transcription factors including P-TEFb and TWIST support its unique role in recruiting non-histone proteins to target genes [22,23]. In contrast, BD1 of BRD3 has been shown to bind the hematopoietic transcription factor GATA1, further emphasizing the complex biological roles of these domains and the need for selective antagonists to better understand their function [24,25]. With >90% sequence identity at the Kac binding pocket of BET BRDs, developing isoform-selective antagonists is a formidable task [10]. Due to the lower conservation between BD1 and BD2 across isoforms, selective antagonism of these domains can more readily be achieved. By taking advantage of specific residue differences and alternate dynamics of the ZA and BC loops of BD1 and BD2, several domain-selective pan-BD1 (binding BD1 of BRD2, −3, and −4) and −BD2 (binding BD2 of BRD2, −3, and −4) chemical probes have been developed within the last year (Figure 1A) [15].
The first reported pan-BD1 antagonist, LT052, is a nanomolar binder of BRD3, 4, and T-BD1 (AlphaScreen IC50 = 88-357 nM) that is 138-fold selective for BRD4-BD1 over BRD4-BD2 and 202-fold selective for BRD3-BD1 over BRD3-BD2 [26]. An unexpected loss in BRD2-BD1 binding and selectivity was observed with an IC50 of 9 μM and only 2-fold BD1/2 selectivity. In a dual luciferase reporter gene assay, LT052 was more potent in downregulating NF-κB activity compared to BD2-biased RVX-208 eliciting an 11- versus 2-fold change relative to the model. LT052 was also shown to be efficacious in a rat model of acute gout arthritis. Together, these results suggest that BD1 antagonism has superior anti-inflammatory activity than BD2 antagonism. However, separate studies using superior pan-BD2 antagonists report contrasting roles of BD2 in models of inflammatory and autoimmune disease (vide infra), underlining the need for follow-up studies.
Developed from the pan-antagonist I-BET151, GSK778 (iBET-BD1) exhibits potent pan-BD1 activity (TR-FRET IC50 = 40–158 nM) and 100 to 158-fold selectivity for BRDT- and BRD4-BD1 over their respective BD2 domains [27,28]. Lower selectivity was observed for the BD1 domains of BRD2 and −3 over their corresponding BD2 domains (40- and 25-fold, respectively). X-ray co-crystal structures of GSK778 bound to BRD4-BD1 and BRD2-BD2 attribute its BD1 selectivity to the “Asp/His switch,” which refers to a key residue difference between the BD1 and BD2 domains (PDB IDs: 6SWN and 6SWO; Figure 1B–C) [27]. When bound to BRD4-BD1, the 3-methylene pyrrolidine substituent of GSK778 extends toward the BD1-specific Asp144 and another nearby aspartic acid, Asp145, rotates to engage the pyrrolidine nitrogen through a water-mediated hydrogen bond. In BRD2-BD2, the corresponding BD2-specific residue, His433, adopts an “in” conformation that restricts the rotation of the analogous nearby Asp434, thus accounting for the loss in affinity. Similar rationale has been used to explain the affinity differences of other domain-selective antagonists, with the Asp/His switch, as well as Lys/Pro and gatekeeper residues Ile/Val BD1/BD2 structural differences shown to be responsible[16,17,26,29–33]. Encouragingly, GSK778 was efficacious in vivo in oncology and inflammation models [27,28].
During the development of ATAD2 chemical probe GSK8814, several compounds with submicromolar potency and >60-fold selectivity for BRD4-BD1 over BRD4-BD2 were identified and progressed to pan-BD1 antagonist, GSK789 [29]. With >1000-fold selectivity over all BET BD2 domains and >500-fold selectivity over other non-BET BRDs, GSK789 is the most selective pan-BD1 antagonist developed to date. However, GSK789 was not progressed to an in vivo probe due to suboptimal pharmacokinetics [29].
The first reported pan-BD2 chemical probe with superior selectivity compared to BD2-biased RVX-208 is ABBV-744, currently in early-stage clinical trials for acute myeloid leukemia (AML) and myelofibrosis [30,31]. Encouraging preclinical in vivo efficacy and reduced toxicity compared to pan-BET antagonist ABBV-075 suggests that ABBV-744 may also have enhanced tolerability in these trials [30]. With low nanomolar BET-BD2 potencies (TR-FRET IC50 = 1–5 nM) and >252-fold selectivity for the BD2 domains of BRD2, −3, and −4 over their respective BD1 domains [31], ABBV-744 has achieved a tremendous leap in selectivity compared to the first BD2-biased antagonist RVX-208, which exhibited a modest 11- to 45-fold BD1 selectivity [16,17].
Recently, pan-BD2 and -BD1 selective antagonists were characterized alongside one another in a series of in vitro and in vivo phenotypic assays [28]. Specifically, GSK046 (iBET-BD2) was utilized as a pan-BD2 ligand which exhibits nanomolar potency for its respective BD2 targets and an impressive selectivity over BD1 domains (>1200-fold for BRD4 and ~40–398 for BRD2, −3, and −T) and other non-BET BRDs [32]. GSK046 was shown to be ineffective at displacing chromatin-bound BET proteins, while pan-BD1 antagonist GSK778 showed similar efficiency as pan-BET antagonist I-BET151, supporting previous reports that BD1 plays a more significant role in localizing BET proteins to chromatin [28]. Additionally, while GSK778 phenocopied I-BET151 in terms of anti-proliferative effects on a range of human cancer cells, GSK046 was less effective. Instead, a unique effect of BD2-selective antagonism was revealed with GSK046 affecting the induction of gene expression more so than the expression of steady-state genes, in contrast to GSK778 [28].
Although effective in in vivo inflammation studies, GSK046 was further optimized, leading to the development of an improved pan-BD2 antagonist, GSK620, with retained potency and high selectivity (80 to 316-fold for BD2/1) along with improved pharmacokinetic properties [33]. GSK620 was highly effective in a collagen-induced arthritis model in rats, outperforming the pan-BET antagonist I-BET151 in terms of reduced joint swelling and overall disease score, again supporting the advantage of domain-selective BET antagonism. Chemically related pan-BD2 antagonist GSK549 has slightly improved potency (TR-FRET IC50 ≈ 16-63 nM) and superior selectivity (~501 to 1260-fold for BD2/1), although its pharmacokinetic properties are subpar [33].
GSK973 retains the excellent BD2 selectivity of GSK549 (>1000-fold selective against all BD1 domains) and is also suitable in vivo, with the added benefit of a unique chemical composition relative to other pan-BD2 antagonist and a negative control compound for further studies [34].
In summary, these potent pan-domain-selective chemical probes have only recently been disclosed and future efforts, as well as clinical studies, will enable further investigations into the individual roles of BET BD1 and BD2 domains, as well as additional insight regarding the efficacy and tolerability profiles of BET domain-selective antagonism.
Proteolysis-targeting chimeras.
In addition to traditional small molecules, alternate modalities for achieving potent and selective antagonism, including bivalent antagonists and proteolysis targeting chimeras (PROTACs), have hugely impacted the development of BRD antagonists [35–38]. PROTACs are bivalent molecules that link a ligand for a protein-of-interest (POI) to an E3 ligase-targeting ligand in order to recruit ubiquitin-proteasome machinery to a POI and induce targeted degradation of the protein. Extensive reviews have summarized the advantages of degraders over their more conventional small-molecule counterparts, which include (1) prolonged efficacy due to target removal rather than occupancy-driven target antagonism, (2) improved selectivity due to the formation of cooperative ternary complexes (POI:Degrader:E3), and (3) reduced off-target effects due to the catalytic mechanism of these compounds and the lower doses that are required for efficacy [39–41]. Chemical degradation is an especially important strategy for targeting reader domain containing proteins/protein complexes because the reader domain itself may not be the critical functional module in terms of disease phenotype.
Despite the ongoing success in selectively targeting BD1 and BD2 domains within BRDs outlined above, limited progress has been made in selectively targeting individual BET family members with conventional small molecule antagonists. Due to the potential functional redundancy of the BET proteins, there is much interest in developing chemical tools to better decipher the roles of these related proteins. FL-411, ZL0420, and ZL0454 are the most successful compounds reported thus far, with submicromolar to low nanomolar potency for BRD4 and 50 to 200-fold selectivity against other BET BRDs [42,43]. PROTACs offer another way to engineer potent and selective—even isoform-specific (e.g. BRD4 alone)—BET antagonists. For example, the JQ1-based PROTAC MZ1 demonstrates selective degradation of BRD4 over BRD2 and BRD3 despite equipotent binding of JQ1 to the three proteins [44]. This selectivity profile was rationalized by the extensive protein-protein interactions observed in the X-ray co-crystal structure of the BRD4:MZ1:VHL ternary complex [45]. Since this initial report, inducing shape complementarity at the interface between a POI and E3 ligase to achieve potency and selectivity has become a common design strategy of PROTACs [46–48], with similar successes achieved by AT1 [45], as well as macroPROTAC-1, the first cyclic PROTAC targeting BRD4 [49].
Beyond the BET family, several PROTACs targeting BRDs including BRD7/9, TRIM24, PCAF/GCN5, and SMARCA2/4 have also been developed [50–54]. Interestingly, the TRIM24 degrader, dTRIM24, demonstrates unique potency on acute leukemia cells compared to conventional small molecule antagonists that had no functional output [51]. The success of dTRIM24 demonstrates that chemical degradation is especially applicable for targeting chromatin regulatory complexes containing reader domains when ligands for these domains do not lead to similar phenotypic activity as protein knockout experiments ([51]). Chemical probes and degraders for additional BRDs will undoubtedly progress understanding of biological mechanisms and therapeutic opportunities within this target class.
YEATs domain.
Lysine crotonylation is a more elusive PTM than acetyl-lysine, with only four crotonyl-lysine (Kcr) readers currently known to be encoded by the human genome: YEATS proteins ENL (MLLT1), AF9 (MLLT3), YEATS2, and GAS41 [55]. Similar to BRDs, dysregulation of these reader domains impacts transcriptional programs affecting oncogenic gene expression in aggressive cancer types [56,57]. Unlike BRDs, the open-ended and tunnel-like binding cavity of YEATS domains enables the recognition of Kac as well as larger acyl-marks such as Kcr [58]. However, the unique π-π-π stacking interaction between two conserved aromatic residues and the conjugated crotonyl-amide group of Kcr enhances its binding affinity over Kac by 2- to 5-fold [55,58]. SGC-iMMLT is the first small molecule chemical probe of a YEATs domain that is equipotent and selective for ENL (ITC Kd = 129 nM) and AF9 (ITC Kd = 77 nM). In MV4;11 AML cells, SGC-iMLLT downregulated pro-oncogene c-Myc and altered the expression of several other target genes, supporting previous ENL knockdown/out studies [59]. The submicromolar tripeptide antagonist, XL-13m, had a slightly improved 5-fold selectivity for ENL over AF9 and, likewise, antagonized the leukemia gene signature in MOLM-13, MV4;11, and HEL cell lines [60]. Effective targeting of the YEATs domain is an emerging area for probe discovery and future progress in probe potency and selectivity, as well as degrader development, may reveal new therapeutic opportunities.
Methyl-lysine readers
Methyl-lysine (Kme) is the most versatile histone PTM. Lysine can be mono-, di-, or trimethylated (Kme1/2/3) across a range of sites along the flexible histone tails. Unlike acetylation, lysine methylation does not alter the charge of the amino group, but rather, its effect on hydrophobicity and size is enough to potently and specifically recruit Kme reader proteins to the PTM [7]. Through their own catalytic functions or the subsequent recruitment of effector proteins, Kme readers translate their recognition of the methylation signal into a diverse set of downstream biological outcomes [5].
Kme readers recognize their histone targets through a cage comprised of 2–4 aromatic residues that form cation-π interactions with the lysine methylammonium group. The size and composition of the aromatic cage plays a key role in dictating selectivity for its substrate [5,7]. Typically, readers of lower methylation states (Kme1/2) have smaller cages with one of the aromatic cage residues replaced by a negatively charged aspartic or glutamic acid. This charged residue forms a hydrogen bond with the N+-H donor present in these methylation states while making binding of Kme3, which lacks this hydrogen bond donating capability, energetically unfavorable with Kme1/2 readers [61]. In addition, this larger PTM is often sterically occluded from these smaller binding pockets. Further specificity for a particular methylated lysine is imparted by some Kme readers through interaction with the surrounding residues. This is typically found in readers that utilize a surface-groove binding mode, where the aromatic cage is accessible from the surface of the protein, allowing the flanking histone residues to make important contacts outside of the Kme binding pocket [62]. Surface-groove Kme readers have been difficult to target using traditional small molecules due to the extensive protein-protein interaction (PPI) surface created by the histone peptide with its reader. On the other hand, Kme readers that utilize a cavity-insertion binding mode often have deep binding pockets that retain localized interactions between the aromatic cage and the lysine methylammonium group, making them more amenable for small molecule binding.
Small-molecule antagonists.
Using targeted screening and ligand-based design, our lab developed UNC1215, the first chemical probe for a Kme reader protein, targeting the MBT domains of L3MBTL3 [63]. UNC1215 binds L3MBTL3 with a Kd of 120 nM and results in a cellular EC50 of 50–100 nM. UNC1215 has continued to demonstrate its utility as a chemical probe and has been widely used to investigate the biology of L3MBTL3 [64,65], but its use in disease-related biology has been limited. Despite the apparent druggability of cavity-insertion reader domains and this initial success, the development of small-molecule antagonists for Kme reader proteins has been slow relative to those for Kac readers. Recent advances in small molecule discovery have been concentrated largely on the WD40 domain of EED, resulting in the potent antagonists EED226 [66] and A-395 [67] from Novartis and AbbVie/SGC, respectively. Interestingly, co-crystallization of EED226 with EED suggests that the high binding affinity of this compound is dependent on the rearrangement of an aromatic cage tryptophan residue upon antagonist binding, a phenomenon that could not have been rationally predicted (Figure 2A) [66]. It is therefore unsurprising that many small molecule successes for Kme reader targets have been discovered by unbiased, industry-scale high-throughput screening.
Figure 2.
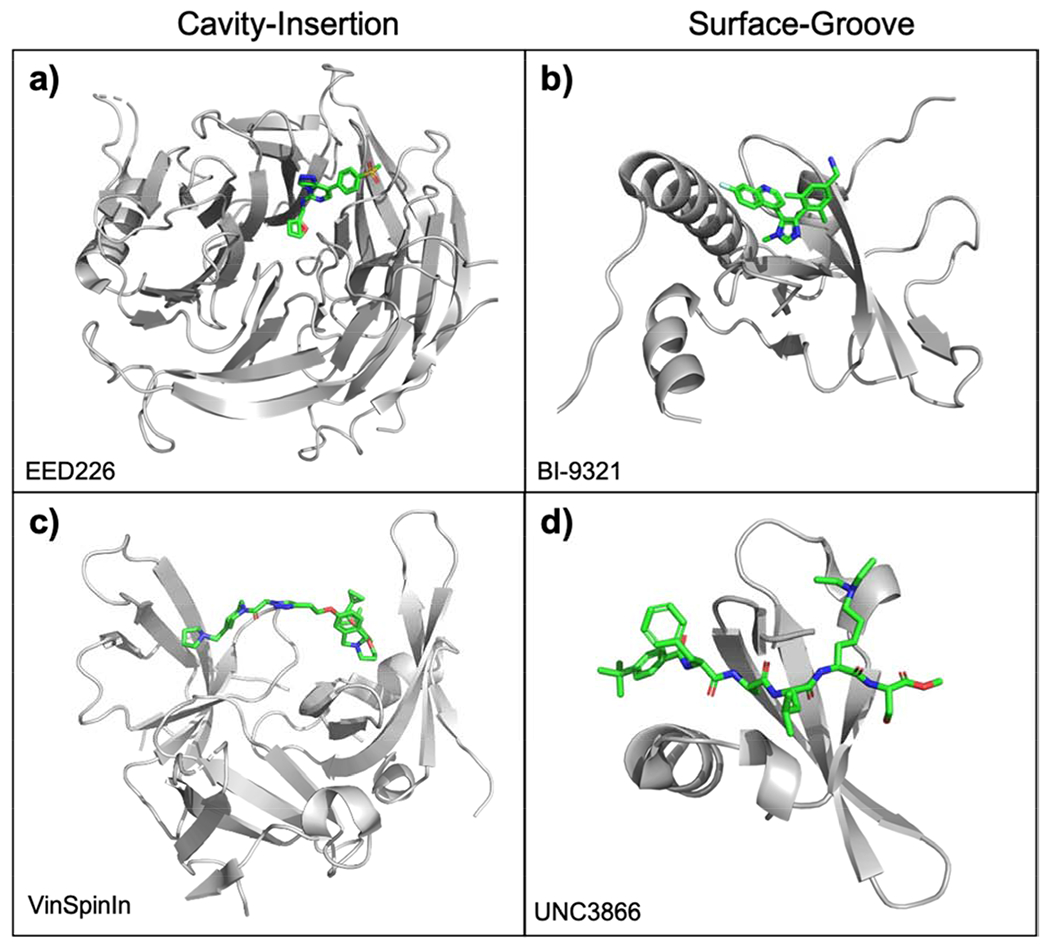
Representative co-crystal structures of Kme reader antagonist binding. (a) EED226 inserts into EED (PDB ID: 5WUK); (b) BI-9321 utilizes fragment linking to engage in multiple contacts throughout NSD3PWWP1 (PDB ID: 6G2O); (c) VinSpinIn binds Tudor domains 1 and 2 of SPIN1 through a cavity-insertion binding mode (PDB ID: 6I8B); (d) UNC3866 creates an extended PPI with CBX7 chromodomain (PDB ID: 5EPJ).
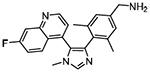
The application of fragment-based drug discovery (FBDD) to the development of Kme reader antagonists has created new opportunities for small molecule discovery. FBDD uses sensitive biophysical techniques such as NMR or surface plasmon resonance (SPR) to detect weak binding of small fragment molecules (<20 non-hydrogen atoms) to the protein of interest [68]. From there, these fragments can be grown through rational design or linked together to create larger, more potent molecules. FBDD is particularly amenable to Kme reader antagonism for two key reasons: (1) the small fragments can bind the shallow binding pockets that occlude binding of larger molecules and (2) linking and growing these fragments creates peptide-like interactions with the protein of interest that can mimic the endogenous PPIs while maintaining drug-like properties. FBDD efforts toward Kme reader domains have led to the development of high-quality chemical probes for previously undruggable families including the PWWP and Tudor domains. BI-9321, which targets PWWP1 of NSD3, was developed through FBDD collaborations with the Structural Genomics Consortium (SGC) (Figure 2B). [69]. Using a small fragment library of less than 2,000 fragments, Böttcher et al. discovered weak binders of NSD3PWWPI through NMR and differential scanning fluorimetry (DSF). Fragment hits that showed binding to the histone binding pocket by two-dimensional 1H/15N-transverse relaxation optimized spectroscopy (TROSY) NMR and NMR Kds <2 mM were progressed to structural analysis by co-crystallization. Subsequent optimization using a combination of virtual screening based on the validated fragment hits and ligand-based rational design resulted in the highly potent and selective chemical probe, BI-9321. With an ITC Kd of 445 nM and cellular EC50 of 5.5 μM, BI-9321 had a selective effect on proliferation of cancer cell lines sensitive to NSD3 loss-of-function mutations (MOLM-13 and RN2), likely through an observed downregulation of MYC. BI-9321 has demonstrated the therapeutic potential of single PWWP domain antagonism, and has already paved the way for other antagonists of this family (See: https://www.thesgc.org/chemical-probes/UNC6934). In a similar method, the first-in-class Tudor domain antagonist for Spindlin1, VinSpinIn, linked previously discovered SPIN1 binders, A-366 and EML631, to create a bidentate, or dual-domain, ligand that interacts with Tudor domains 1 and 2 of the protein (Figure 2C) [70]. While A-366 demonstrates high in vitro potency toward SPIN1 (IC50 = 186 nM), the fragment was originally developed for the methyltransferase G9a (IC50 = 3 nM) and would therefore have poor SPIN1 selectivity [71]. Using structure-based design, A-366 was modified for potent and selective binding of SPIN1 Tudor 2. However, the optimized compound showed high toxicity in cells at concentrations >3 μM, leading Fagan et al. to develop a new series of bidentate ligands. Bidentate antagonists have the potential not only for increased selectivity through elongated interactions, but also for increased functional efficacy in cells by targeting multiple functional domains within the protein. Fagan et al. again utilized rational design to combine and optimize the previously discovered EML631, which targets SPIN1 Tudor 1 [72], with the optimized Tudor 2 antagonist to create VinSpinIn. VinSpinIn demonstrated high in vitro and cellular potency (IC50 = 33 nM, EC50 = 270 nM), excellent selectivity (no significant off-target binding to 16 related Kme/Rme reader domains and 33 methyltransferases including G9a), and, importantly, no observed toxicity.
While VinSpinIn demonstrated only moderate effects on cell proliferation, the well-characterized probe will be exceptionally useful for investigation of SPIN family protein biology in normal and disease-relevant contexts.
Peptides and peptidomimetics.
While small molecules are often seen as the preferred mode of antagonism due to their drug-like properties, peptides and peptidomimetic ligands can be especially useful tool compounds due to their high affinity and selectivity combined with their generally low off-target effects and toxicity [73]. Peptide and peptidomimetic ligands are of particular interest for Kme reader antagonists as the natural histone binding partners provide useful starting points for ligand development. In particular, peptide-based ligands have the potential to replicate and improve upon the surface-groove binding modes of Kme readers that can be difficult to target by small molecules.
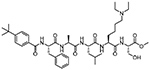
To date, the most successful examples of Kme reader peptidomimetics have targeted the CBX family of chromodomains within the Polycomb Repressive Complex 1 (PRC1) which bind H3K27me3 in a surface-groove binding mode. Initial efforts toward development of small molecule antagonists of CBX proteins led to MS452 [74] and MS351 [75], which demonstrated moderate binding to CBX7 in vitro (MS452 Kd = 29 μM; MS351 Kd ~500 μM) but lacked cellular efficacy likely due to limited solubility at the required concentrations [76]. Development of UNC4976 and its predecessor, UNC3866, therefore exemplify the utility of peptidic ligands for these readers by demonstrating high potency, selectivity, and cellular activity against CBX7 (UNC4976 Kd = 59 nM; UNC3866 Kd = 97 nM) (Figure 2D) [76,77]. UNC3866 and UNC4976 make use of a few key design strategies that have been essential for the development of potent, cell permeable, and proteolytically stable peptidic ligands. First, both peptides contain a hydrophobic lysine mimic at the Kme3 site which both increases engagement with the aromatic cage through hydrophobic interactions and allows for removal of the quaternary amine that can be detrimental to cell permeability. Additionally, the peptides are ‘capped’ with a methyl ester on the C-terminus and a non-aminoacid, aromatic N-terminus. While the C-terminal cap provides added permeability relative to an amide and the potential for intracellular trapping by esterase conversion to the corresponding acid, the N-terminal cap has been optimized to improve potency of the peptide [78]. It is worth noting that both UNC3866 and UNC4976 show acceptable permeabilities in the chloroalkane penetration assay (CAPA) with CP50 values in the low micromolar range [76]. In related studies, through a DNA encoded library approach the Dykhuizen and Krusemark labs discovered SW2_110A, which binds the chromodomain of the closely-related CBX8 with a Kd of 800 nM and shows moderate anti-proliferative effects in MLL-AF9 transformed leukemia [79]. Similar to UNC3866 and UNC4976, SW2_110A swaps the Kme3 for a more hydrophobic tertiary amine (diethyl-lysine) and includes a heterocyclic aromatic N-terminal cap. With these modifications, SW2_110A demonstrated modest permeability by CAPA with a CP50 of 26 μM. Continued work on CBX antagonists is ongoing in our lab and others through a variety of approaches [80].
Allosteric modulation within a multicomponent complex.
Kme readers play key scaffolding and modulating roles in larger complexes binding to nucleic acids and/or regulating catalytic domains responsible for propagating these marks. Recent discoveries have highlighted the importance of exploiting allosteric interactions to achieve cellular potency and efficacy with Kme probes [81].
The aforementioned UNC4976 is a good case study for allosteric modulation. While UNC3866 and UNC4976 exhibit similar Kd values in vitro, UNC4976 demonstrated a 14-fold increase in cellular potency (EC50 = 3.2 μM) from UNC3866 (EC50 = 42 μM). Lamb etal. proposed that this discrepancy arises from the unique ability of UNC4976 but not UNC3866 to increase the affinity of CBX7 for DNA while simultaneously antagonizing H3K27me3 recognition, thereby further diminishing the specific binding of CBX7 to H3K27me3 sites. In this way, UNC4976 acts both as a competitive antagonist of H3K27me3 and as a positive allosteric modulator (PAM) of CBX7 binding to DNA, increasing its overall efficacy in cells. This PAM mechanism was first proposed by the Zhou lab in the characterization of MS351 [75].
One of the most prolific uses of allosteric regulation has been through the use of EED antagonists as allosteric modulators of EZH2. EED functions as a reader of H3K27me3 in the Polycomb Repressive Complex 2 (PRC2), which also propagates the repressive mark through the methyltransferase EZH2. Binding of H3K27me3 to EED induces a conformational change in EZH2 that allosterically activates its catalytic activity. Accordingly, positive and negative modulators of EED have been discovered that allosterically regulate EZH2 activity. As previously mentioned, EED226 is a potent antagonist of EED, which recently led to the development of MAK683, the first small molecule targeting a Kme reader that has progressed into clinical trials (Table 1). Like EED226, binding of MAK683 in the H3K27me3-binding pocket is a very high affinity interaction (AlphaScreen IC50 = 26 nM) which prevents a conformational change in EZH2 required to activate PRC2 catalytic activity. Importantly, these EED antagonists have been shown to be effective against EZH2 mutants that are resistant to EZH2 inhibitors as a first line treatment [66]. Allosteric binders have also been used by our lab to develop selective allosteric agonists of the mutant EED-I363M, which results in PRC2 loss of function and plays a driving role in myeloid dysplasia. Excitingly, rational design led to UNC5635 and UNC5636, which are capable of compensating for the loss of EZH2 activation in this mutant. In a catalytic assay, UNC5635 and UNC5636 showed agonism of PRC2-EED-I363M but not PRC2-EED-WT activity, demonstrating their selective activity toward the disease-relevant mutant [82].
PROTACs revisited.
Finally, additional methods to target EED and the PRC2 complex have recently been achieved through the development of PRC2-targeted PROTACs. Namely, UNC6852 [83] and PROTAC1/PROTAC2 [84] potently target EED but have also been shown to degrade all three core components of the PRC2 complex: EED, EZH2, and SUZ12. This large-scale degradation is highly effective in antagonizing PRC2 activity, as re-establishment of PRC2 function requires re-synthesis of all three components. Both groups showed significant anti-proliferative effects toward PRC2-dependent cancer cell lines.
Conclusions
Chromatin regulation is a complex network consisting, in part, of histone PTMs and the readers that translate them. The development of chemical probes for readers offers tools to map these networks and has enriched our understanding of normal and disease biology. As shown with (+)-JQ1, these efforts have led to translational discoveries targeting the BET family, with Kme reader EED antagonists also moving into the clinic. Future success regarding BET antagonism will rely on selective modulation of individual BRD domains, BD1 and BD2. As epigenetic research has grown outside the BET and Kme reader family, so has the number of chemical probes targeting non-BET BRDs and YEATS domains. Among Kme readers, antagonists with new binding modalities and mechanisms-of-action are being actively pursued to overcome inherent challenges of targeting this class. In particular, this review highlights the lack of chemical probes targeting the large subset of Kme readers making up the PHD family, as well as the smaller BAH, ADD, Ankyrin, and chromo-barrel families. Probes for these families have remained elusive due to their shallow binding pockets and limited chemical space explored for hit discovery. New techniques such as DNA-encoded libraries applied to reader domains will hopefully provide new opportunities for ligand discovery for these challenging targets. Overall, chemical probe development will continue to play a leading role in the investigation of the therapeutic potential and biological role of epigenetic readers.
Table 2.
Representative chemical probes of Kme readers with unique binding modes and mechanisms
| Kme Reader | Chemical Probe |
||
|---|---|---|---|
| Name | Modality | Structure | |
| NSD3 (PWWP) | BI-9321 | Small Molecule Cavity Binder |
|
| SPIN1 (Tudor) | VinSpinIn | Bidentate Small Molecule |
|
| CBX7 (CBX) | UNC3866 | Peptidic Surface-Groove Binder |
|
| EED (WD40) | MAK683 | Allosteric Modulator |
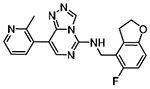
|
| EED (WD40) | UNC6852 | PROTAC Degrader |
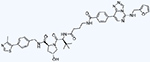
|
Acknowledgements
Our research described herein was supported by the National Cancer Institute NIH (Grant R01CA218392) and the National Institute of General Medical Sciences, U.S. National Institutes of Health (NIH) (Grant R01GM100919) to S.V.F; and the National Institute on Drug Abuse NIH (Grant R61DA047023), the National Cancer Institute, NIH (Grant R01CA242305), and the University Cancer Research Fund, University of North Carolina at Chapel Hill to L.I.J. The authors also wish to recognize the collaborative contributions of all co-authors in our cited references, especially the Structural Genomics Consortium.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Baylin SB, Jones PA: Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zaware N, Zhou M-M: Chemical modulators for epigenome reader domains as emerging epigenetic therapies for cancer and inflammation. Curr Opin Chem Biol 2017, 39:116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jakovcevski M, Akbarian S: Epigenetic mechanisms in neurological disease. Nat Med 2012, 18:1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huston A, Arrowsmith CH, Knapp S, Schapira M: Probing the epigenome. Nat Chem Biol 2015, 11:542–545. [DOI] [PubMed] [Google Scholar]
- 5.Musselman CA, Lalonde ME, Côté J, Kutateladze TG: Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol 2012, 19:1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu H, Galka M, Iberg A, Wang Z, Li L, Voss C, Jiang X, Lajoie G, Huang Z, Bedford MT, et al. : Systematic identification of methyllysine-driven interactions for histone and nonhistone targets. J Proteome Res 2010, 9:5827–5836. [DOI] [PubMed] [Google Scholar]
- 7.Yun M, Wu J, Workman JL, Li B: Readers of histone modifications. Cell Res 2011, 21:564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaware N, Zhou M-M: Bromodomain biology and drug discovery. Nat Struct Mol Biol 2019, 26:870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cochran AG, Conery AR, Sims RJ: Bromodomains: a new target class for drug development. Nat Rev Drug Discov 2019, 18:609–628. [DOI] [PubMed] [Google Scholar]
- 10.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al. : Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. : Selective inhibition of BET bromodomains. Nature 2010, 468:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. : Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468:1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galdeano C, Ciulli A: Selectivity on-target of bromodomain chemical probes by structure-guided medicinal chemistry and chemical biology. Future Med Chem 2016, 8:1655–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moustakim M, Clark PGK, Hay DA, Dixon DJ, Brennan PE: Chemical probes and inhibitors of bromodomains outside the BET family. Medchemcomm 2016, 7:2246–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petretich M, Demont EH, Grandi P: Domain-selective targeting of BET proteins in cancer and immunological diseases. Curr Opin Chem Biol 2020, 57:184–193. [DOI] [PubMed] [Google Scholar]; • This article reviews evidence supporting the unique roles of the BET BD1 and BD2 domains, as well as recently disclosed pan-BD1 and -BD2 chemical probes of the BET proteins.
- 16.Mclure KG, Gesner EM, Tsujikawa L, Kharenko OA, Attwell S: RVX-208, an Inducer of ApoA-I in Humans, Is a BET Bromodomain Antagonist. PLoS One 2013, 8:e83190–e83190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, et al. : RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. PNAS 2013, 110:19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Shah K, Yang F, Witucki L, Shokat KM: Engineering Src family protein kinases with unnatural nucleotide specificity. Chem Biol 1998, 5:91–101. [DOI] [PubMed] [Google Scholar]
- 19.Baud MGJ, Lin-Shiao E, Cardote T, Tallant C, Pschibul A, Chan K-H, Zengerle M, Garcia JR, Kwan TT-L, Ferguson FM, et al. : A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 2014, 346:638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Runcie AC, Zengerle M, Chan KH, Testa A, van Beurden L, Baud MGJ, Epemolu O, Ellis LCJ, Read KD, Coulthard V, et al. : Optimization of a “bump-and-hole” approach to allele-selective BET bromodomain inhibition. Chem Sci 2018, 9:2452–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyler DS, Vappiani J, Cañeque T, Lam EYN, Ward A, Gilan O, Chan Y-C, Hienzsch A, Rutkowska A, Werner T, et al. : Click chemistry enables preclinical evaluation of targeted epigenetic therapies. Science 2017, 356:1397–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This article describes the use of BET chemical probes and click-chemistry followed by chromatin immunoprecipitation to investigate the roles of the BD1 and BD2 domains on recruitment of BRD4 to chromatin.
- 22.Bisgrove DA, Mahmoudi T, Henklein P, Verdin E: Conserved P-TEFb-Interacting Domain of BRD4 Inhibits HIV Transcription. PNAS 2007, 104:13690–13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, et al. : Disrupting the Interaction of BRD4 with Diacetylated Twist Suppresses Tumorigenesis in Basal-like Breast Cancer. Cancer Cell 2014, 25:210–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamonica JM, Deng W, Kadauke S, Campbell AE, Gamsjaeger R, Wang H, Cheng Y, Billin AN, Hardison RC, Mackay JP, et al. : Bromodomain protein BRD3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. PNAS 2011, 108:8927–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamsjaeger R, Webb SR, Lamonica JM, Billin A, Blobel GA, Mackay JP: Structural Basis and Specificity of Acetylated Transcription Factor GATA1 Recognition by BET Family Bromodomain Protein BRD3. Mol Cell Biol 2011, 31:2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang F, Hu Q, Zhang Z, Li H, Li H, Zhang D, Li H, Ma Y, Xu J, Chen H, et al. : Discovery of Benzo[cd]indol-2(1H)-ones and Pyrrolo[4,3,2-de]quinolin-2(1H)-ones as Bromodomain and Extra-Terminal Domain (BET) Inhibitors with Selectivity for the First Bromodomain with Potential High Efficiency against Acute Gouty Arthritis. J Med Chem 2019, 62:11080–11107. [DOI] [PubMed] [Google Scholar]; •• This article discloses the first pan-BD1 BET antagonist to be classified as a chemical probe.
- 27.Wellaway CR, Bamborough P, Bernard SG, Chung CW, Craggs PD, Cutler L, Demont EH, Evans JP, Gordon L, Karamshi B, et al. : Structure-Based Design of a Bromodomain and Extraterminal Domain (BET) Inhibitor Selective for the N-Terminal Bromodomains That Retains an Anti-inflammatory and Antiproliferative Phenotype. J Med Chem 2020, 63:9020–9044. [DOI] [PubMed] [Google Scholar]
- 28.Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, Lam EYN, Chung C-W, Bamborough P, Petretich M, et al. : Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This article presents the first side-by-side comparison of pan-BD1 and -BD2 BET chemical probes in in vitro and in vivo phenotypic assays. Previous studies relied on antagonists with biased affinity for BD1 or BD2, such as BD2-biased RVX-208, which are not classified as chemical probes due to insufficient in vitro potency and/or selectivity.
- 29.Watson RJ, Bamborough P, Barnett H, Chung C-W, Davis R, Gordon L, Grandi P, Petretich M, Phillipou A, Prinjha RK, et al. : GSK789: A Selective Inhibitor of the First Bromodomains (BD1) of the Bromo and Extra Terminal Domain (BET) Proteins. J Med Chem 2020, 63:22. [DOI] [PubMed] [Google Scholar]
- 30.Faivre EJ, Mcdaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, Zhang L, Bui MH, Sheppard GS, Wang L, et al. : Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578:306–310. [DOI] [PubMed] [Google Scholar]; •• This article discloses the first pan-BD2 antagonist to be classified as a chemical probe.
- 31.Sheppard GS, Wang L, Fidanze SD, Hasvold LA, Liu D, Pratt JK, Park CH, Longenecker K, Qiu W, Torrent M, et al. : Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J Med Chem 2020, 63:5585–5623. [DOI] [PubMed] [Google Scholar]; •• This article discloses the first pan-BD2 BET antagonist to be classified as a chemical probe.
- 32.Preston A, Atkinson S, Bamborough P, Chung C-W, Craggs PD, Gordon L, Grandi P, Gray JRJ, Jones EJ, Lindon M, et al. : Design and Synthesis of a Highly Selective and In Vivo-Capable Inhibitor of the Second Bromodomain of the Bromodomain and Extra Terminal Domain Family of Proteins. J Med Chem 2020, 63:45. [DOI] [PubMed] [Google Scholar]
- 33.Seal JT, Atkinson SJ, Aylott H, Bamborough P, Chung C-W, Copley RCB, Gordon L, Grandi P, Gray JRJ, Harrison LA, et al. : The Optimization of a Novel, Weak Bromo and Extra Terminal Domain (BET) Bromodomain Fragment Ligand to a Potent and Selective Second Bromodomain (BD2) Inhibitor. J Med Chem 2020, 63:9093–9126. [DOI] [PubMed] [Google Scholar]
- 34.Preston A, Atkinson SJ, Bamborough P, Chung C-W, Gordon LJ, Grandi P, Gray JRJ, Harrison LA, Lewis AJ, Lugo D, et al. : GSK973 Is an Inhibitor of the Second Bromodomains (BD2s) of the Bromodomain and Extra-Terminal (BET) Family. ACS Med Chem Lett 2020, 11:1581–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waring MJ, Chen H, Rabow AA, Walker G, Bobby R, Boiko S, Bradbury RH, Callis R, Clark E, Dale I, et al. : Potent and selective bivalent inhibitors of BET bromodomains. Nat Chem Biol 2016, 12:1097–1104. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka M, Roberts JM, Seo HS, Souza A, Paulk J, Scott TG, Deangelo SL, Dhe-Paganon S, Bradner JE: Design and characterization of bivalent BET inhibitors. Nat Chem Biol 2016, 12:1089–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren C, Zhang G, Han F, Fu S, Cao Y, Zhang F, Zhang Q, Meslamani J, Xu Y, Ji D, et al. : Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. PNAS 2018, 115:7949–7954.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang CY, Qin C, Bai L, Wang S: Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins — A review. Drug Discov Today Technol 2019, 31:43–51. [DOI] [PubMed] [Google Scholar]
- 39.Pettersson M, Crews CM: PROteolysis TArgeting Chimeras (PROTACs) — Past, present and future. Drug Discov Today Technol 2019, 31:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This article reviews the development of chemical degraders known as PROTACs, providing an excellent overview of their unique mechanism-of-action, as well as their advantages over conventional small-molecule antagonists.
- 40.Maniaci C, Ciulli A: Bifunctional chemical probes inducing protein–protein interactions. Curr Opin Chem Biol 2019, 52:145–156. [DOI] [PubMed] [Google Scholar]
- 41.Cromm PM, Crews CM: Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem Biol 2017, 24:1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouyang L, Zhang L, Liu J, Fu L, Yao D, Zhao Y, Zhang S, Wang G, He G, Liu B: Discovery of a Small-Molecule Bromodomain-Containing Protein 4 (BRD4) Inhibitor That Induces AMP-Activated Protein Kinase-Modulated Autophagy-Associated Cell Death in Breast Cancer. J Med Chem 2017, 60:9990–10012. [DOI] [PubMed] [Google Scholar]
- 43.Liu Z, Tian B, Chen H, Wang P, Brasier AR, Zhou J: Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur J Med Chem 2018, 151:450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zengerle M, Chan K-H, Ciulli A: Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 2015, 10:1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gadd MS, Testa A, Lucas X, Chan K-H, Chen W, Lamont DJ, Zengerle M, Ciulli A: Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol 2017, 13:514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article presents the co-crystal structure of the BRD4:MZ1:VHL ternary complex as rationale for the selectivity of MZ1 towards BRD4 alone. Using this structural information, an improved BRD4-selective PROTAC, AT1, is developed.
- 46.Chan K-H, Zengerle M, Testa A, Ciulli A: Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J Med Chem 2018, 61:504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, Crews CM: Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol 2018, 25:78–87.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, et al. : Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol 2018, 14:706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Testa A, Hughes SJ, Lucas X, Wright JE, Ciulli A: Structured-Based Design of a Macrocyclic PROTAC. Angew Chemie Int Ed 2020, 59:1727–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bassi ZI, Fillmore MC, Miah AH, Chapman TD, Maller C, Roberts EJ, Davis LC, Lewis DE, Galwey NW, Waddington KE, et al. : Modulating PCAF/GCN5 Immune Cell Function through a PROTAC Approach. ACS Chem Biol 2018, 13:2862–2867. [DOI] [PubMed] [Google Scholar]
- 51.Gechijian LN, Buckley DL, Lawlor MA, Reyes JM, Paulk J, Ott CJ, Winter GE, Erb MA, Scott TG, Xu M, et al. : Functional TRIM24 degrader via conjugation of ineffectual bromodomain and VHL ligands. Nat Chem Biol 2018, 14:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This article describes a case in which conventional antagonism of the TRIM24 reader domain had no functional output, whereas PROTAC-mediated degradation of the protein demonstrated potent cellular activity.
- 52.Remillard D, Buckley DL, Paulk J, Brien GL, Sonnett M, Seo H-S, Dastjerdi S, Wühr M, Dhe-Paganon S, Armstrong SA, et al. : Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chemie Int Ed 2017, 56:5738–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zoppi V, Hughes SJ, Maniaci C, Testa A, Gmaschitz T, Wieshofer C, Koegl M, Riching KM, Daniels DL, Spallarossa A, et al. : Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel-Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J Med Chem 2019, 62:699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farnaby W, Koegl M, Roy MJ, Whitworth C, Diers E, Trainor N, Zollman D, Steurer S, Karolyi-Oezguer J, Riedmueller C, et al. : BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat Chem Biol 2019, 15:672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y, Zhao D, Chen Z, Li H: YEATS domain: Linking histone crotonylation to gene regulation. Transcription 2017, 8:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YHE, Erb MA, et al. : ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. : Transcription control by the ENL YEATS domain in acute leukaemia. Nature 2017, 543:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klein BJ, Vann KR, Andrews FH, Wang WW, Zhang J, Zhang Y, Beloglazkina AA, Mi W, Li Y, Li H, et al. : Structural insights into the π-π-π stacking mechanism and DNA-binding activity of the YEATS domain. Nat Commun 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moustakim M, Christott T, ctovia Monteiro OP, ames Bennett J, Giroud C, Ward J, Rogers CM, aul Smith P, Panagakou I, Díaz-Sμez L, et al. : Discovery of an MLLT1/3 YEATS Domain Chemical Probe. Angew Chem Int Ed 2018, 57:16302–16307. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article discloses the first-in-class antagonist of a Kcr reader domain to be classified as a chemical probe.
- 60.Li X, Li XM, Jiang Y, Liu Z, Cui Y, Fung KY, van der Beelen SHE, Tian G, Wan L, Shi X. et al. : Structure-guided development of YEATS domain inhibitors by targeting π-π-π stacking. Nat Chem Biol 2018, 14:1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamps JJAG, Huang J, Poater J, Xu C, Pieters BJGE, Dong A, Min J, Sherman W, Beuming T, Matthias Bickelhaupt F, et al. : Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat Commun 2015, 6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ: How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat Struct Mol Biol 2007, 14:1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, MacNevin CJ, Norris JL, Sagum CA, Tempel W, et al. : Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol 2013, 9:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang C, Leng F, Saxena L, Hoang N, Yu J, Alejo S, Lee L, Qi D, Lu F, Sun XH, et al. : Proteolysis of methylated SOX2 protein is regulated by L3MBTL3 and CRL4DCAF5 ubiquitin ligase. J Biol Chem 2019, 294:476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh AN, Sharma N: Epigenetic Modulators as Potential Multi-targeted Drugs Against Hedgehog Pathway for Treatment of Cancer. Protein J 2019, 38:537–550. [DOI] [PubMed] [Google Scholar]
- 66.Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, et al. : An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol 2017, 13:381–388. [DOI] [PubMed] [Google Scholar]
- 67.He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, Shaw B, The J, Lima-Fernandes E, Szewczyk MM, et al. : The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol 2017, 13:389–395. [DOI] [PubMed] [Google Scholar]
- 68.Erlanson DA, McDowell RS, O’Brien T: Fragment-based drug discovery. J Med Chem 2004, 47:3463–3482. [DOI] [PubMed] [Google Scholar]
- 69.Böttcher J, Dilworth D, Reiser U, Neumüller RA, Schleicher M, Petronczki M, Zeeb M, Mischerikow N, Allali-Hassani A, Szewczyk MM, et al. : Fragment-based discovery of a chemical probe for the PWWP1 domain of NSD3. Nat Chem Biol 2019, 15:822–829. [DOI] [PubMed] [Google Scholar]; •• This article describes the discovery of the first-in-class antagonist of a PWWP Kme reader domain, BI-9321.
- 70.Fagan V, Johansson C, Gileadi C, Monteiro O, Dunford JE, Nibhani R, Philpott M, Malzahn J, Wells G, Faram R, et al. : A Chemical Probe for Tudor Domain Protein Spindlin1 to Investigate Chromatin Function. J Med Chem 2019, 62:9008–9025. [DOI] [PubMed] [Google Scholar]
- 71.Wagner T, Greschik H, Burgahn T, Schmidtkunz K, Schott AK, McMillan J, Baranauskiene L, Xiong Y, Fedorov O, Jin J, et al. : Identification of a small-molecule ligand of the epigenetic reader protein Spindlin1 via a versatile screening platform. Nucleic Acids Res 2016, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article describes the discovery of the first-in-class antagonist of a Tudor Kme reader domain, VinSpinIn.
- 72.Bae N, Viviano M, Su X, Lv J, Cheng D, Sagum C, Castellano S, Bai X, Johnson C, Khalil MI, et al. : Developing Spindlin1 small-molecule inhibitors by using protein microarrays. Nat Chem Biol 2017, 13:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fosgerau K, Hoffmann T: Peptide therapeutics: current status and future directions. Drug Discov Today 2015, 20:122–128. [DOI] [PubMed] [Google Scholar]
- 74.Ren C, Morohashi K, Plotnikov AN, Jakoncic J, Smith SG, Li J, Zeng L, Rodriguez Y, Stojanoff V, Walsh M, et al. : Small-molecule modulators of methyl-lysine binding for the CBX7 chromodomain. Chem Biol 2015, 22:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ren C, Smith SG, Yap K, Li S, Li J, Mezei M, Rodriguez Y, Vincek A, Aguilo F, Walsh MJ, et al. : Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7. ACS Med Chem Lett 2016, 7:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lamb KN, Bsteh D, Dishman SN, Moussa HF, Fan H, Stuckey JI, Norris JL, Cholensky SH, Li D, Wang J, et al. : Discovery and Characterization of a Cellular Potent Positive Allosteric Modulator of the Polycomb Repressive Complex 1 Chromodomain, CBX7. Cell Chem Biol 2019, 26:1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This article describes the discovery and characterization of UNC4976, an in vivo chemical probe for chromodomain Kme reader, CBX7. In particular, this article describes the mechanism of a positive allosteric modulation (PAM) by nucleic acid binding.
- 77.Stuckey JI, Dickson BM, Cheng N, Liu Y, Norris JL, Cholensky SH, Tempel W, Qin S, Huber KG, Sagum C, et al. : A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat Chem Biol 2016, 12:180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stuckey JI, Simpson C, Norris-Drouin JL, Cholensky SH, Lee J, Pasca R, Cheng N, Dickson BM, Pearce KH, Frye SV, et al. : Structure-Activity Relationships and Kinetic Studies of Peptidic Antagonists of CBX Chromodomains. J Med Chem 2016, 59:8913–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang S, Denton KE, Hobbs KF, Weaver T, McFarlane JMB, Connelly KE, Gignac MC, Milosevich N, Hof F, Paci I, et al. : Optimization of Ligands Using Focused DNA-Encoded Libraries to Develop a Selective, Cell-Permeable CBX8 Chromodomain Inhibitor. ACS Chem Biol 2020, 15:112–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barnash KD, Lamb KN, Stuckey JI, Norris JL, Cholensky SH, Kireev DB, Frye SV, James LI: Chromodomain Ligand Optimization via Target-Class Directed Combinatorial Repurposing. ACS Chem Biol 2016, 11:2475–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weaver TM, Morrison EA, Musselman CA: Reading more than Histones: The prevalence of nucleic acid binding among reader domains. Molecules 2018, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article gives a detailed description on the importance of nucleic acid binding by reader domains, which is becoming an important topic of investigation. Nucleic acid binding must be taken into account both to understand the role of reader domain biology and develop cellularly active antagonists.
- 82.Suh JL, Barnash KD, Abramyan TM, Li F, The J, Engelberg IA, Vedadi M, Brown PJ, Kireev DB, Arrowsmith CH, et al. : Discovery of selective activators of PRC2 mutant EED-I363M. Sci Rep 2019, 9:6524. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Novel proof-of-concept study for selective activation of a loss-of-function Kme reader mutant.
- 83.Potjewyd F, Turner AMW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, Margolis DM, Pearce KH, Herring LE, James LI: Degradation of Polycomb Repressive Complex 2 with an EED-Targeted Bivalent Chemical Degrader. Cell Chem Biol 2020, 27:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hsu JHR, Rasmusson T, Robinson J, Pachl F, Read J, Kawatkar S, O’ Donovan DH, Bagal S, Code E, Rawlins P, et al. : EED-Targeted PROTACs Degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem Biol 2020, 27:41–46. [DOI] [PubMed] [Google Scholar]