

Abstract
We search for ischemic stroke treatment knowing we have failed—intensely and often—to translate mechanistic knowledge into treatments that alleviate our patients’ functional impairments. Lessons can be derived from our shared failures that may point to new directions and new strategies. First, the principle criticisms of both preclinical and clinical assessments are summarized. Next, previous efforts to develop single-mechanism treatments are reviewed. Finally, new definitions, novel approaches, and different directions are presented. In previous development efforts, the basic science and preclinical assessment of candidate treatments often lacked rigor and sufficiency; the clinical trials may have lacked power, rigor, or rectitude; or most likely both preclinical and clinical investigations were flawed. Single-target agents directed against specific molecular mechanisms proved unsuccessful. The term neuroprotection should be replaced as it has become ambiguous: protection of the entire neurovascular unit may be called cerebral cytoprotection or cerebroprotection. Success in developing cerebroprotection—either as an adjunct to recanalization or as stand-alone treatment—will require new definitions that recognize the importance of differential vulnerability in the neurovascular unit. Recent focus on pleiotropic multi-target agents that act via multiple mechanisms of action to interrupt ischemia at multiple steps may be more fruitful. Examples of pleiotropic treatments include therapeutic hypothermia and 3K3A-APC. Alternatively, the single-target drug NA-1 triggers multiple downstream signaling events. Renewed commitment to scientific rigor is essential, and funding agencies and journals may enforce quality principles of rigor in preclinical science. Appropriate animal models should be selected that are suited to the purpose of the investigation. Prior to clinical trials, preclinical assessment could include subjects that are aged, of both sexes, and harbor co-morbid conditions such as diabetes or hypertension. With these new definitions, novel approaches, and renewed attention to rigor, the prospect for successful cerebroprotective therapy should improve.
The search for stroke treatments began with the earliest physicians, seeking to alleviate functional impairments in patients with ‘apoplexy’1. Remedies described in Greek, Roman, Persian, and medieval texts included manipulations of diet, herbs, and some surgeries2. These treatments—which may seem naïve or whimsical to us now—were devised to address the known mechanisms of apoplexy: a lack of balance among the four humors. For example, bloodletting and purging gained popularity along with cranial cauterization to allow release of the bad humor. The search for stroke treatment as we know it began in earnest after two critical mechanistic discoveries: the ischemic penumbra and the excitotoxic hypothesis3, 4. Then, with the advent of recanalization therapies—thrombolysis and thrombectomy—the search for stroke treatment shifted from neuroprotection to treating reperfusion injury, including mechanisms related to free radical generation5.
Yet the real story of neuroprotection lies not in the successful elucidation of the molecular mechanisms underlying ischemia/reperfusion, leading to the rational design of effective treatments. Rather, we now search for ischemic stroke treatment knowing we have failed—intensely and often—to translate mechanistic knowledge into treatments that alleviate our patients’ functional impairments. No doubt, two millennia from now physicians will look back on our notions of ischemia and treatment with the same awe and bemusement we hold for Galen, Aristotle, and Avicenna.
What then are we to do next? Given our extraordinary track record (of failure), how do we re-focus and re-organize our search for ischemic stroke treatment? Sifting through the flotsam and jetsam of innumerable—really: too many to count—failed trials, can we learn anything that might guide future research? Perhaps lessons can be derived from our shared failures that may point to new directions and new strategies.
PRECLINICAL ℃ CLINICAL FAILURE
Several prior authors thoroughly documented the magnitude of our collective failure to find effective treatment besides recanalization for acute ischemic stroke6–12. A plethora of putative protective treatments emerged from laboratories; many qualified in Phase 1 and Phase 2 trials; and some proceeded to Phase 3 definitive trials where all disappointed. A cottage industry emerged to explain all these failures and the literature here is vast. To simplify: either the basic science and preclinical assessment of the candidate treatment lacked rigor and sufficiency, or the clinical trial design lacked power, rigor, or rectitude. Likely both are true: we probably neglected to properly assess candidate treatments at the preclinical stage, and we probably lacked the optimal approach to definitive clinical trials.
The principle criticisms of both preclinical and clinical assessments can be summarized (Table). Difficulties in selecting an appropriate animal model for qualifying a candidate treatment of stroke are reviewed elsewhere6, 13–15. Some limiting features of animal models are well known: studies typically include only young male rodents free of any co-morbid diseases while stroke patients tend to be older, with co-existing diseases that moderate stroke outcome, e.g., diabetes and hypertension. Although changing, there has been a tendency among preclinical investigators to test their candidate drugs early after stroke onset, while in clinical practice, patients present hours or many hours after stroke onset. Pressure to publish causes a positive publication bias, which has been well described and quantified16. Recanalization with thrombectomy can be easily modeled with transient occlusion of the middle cerebral artery (MCAo)17.
Table. Summary of Issues in Preclinical and Clinical Assessment of Neuroprotectants.
The author’s personal appraisal of the differences between typical preclinical and clinical investigations. These differences may partly help explain the failure to translate candidate cerebroprotectants from preclinical assessment to pivotal clinical trials.
| Issue | Preclinical | Clinical |
|---|---|---|
| Time Window | Usually short | Usually long |
| Age | Usually young | Usually old |
| Sex | Usually male | Always both |
| Dose | Optimized | Limited by side effects |
| Validity | Publication bias towards positive results | Pre-specified clinical trial protocol |
| Reperfusion | Transient MCAo models | Endovascular therapy |
| Outcomes | Often lesion volume | Always behavior (modified Rankin) |
| Subjects | Usually homogeneous | Heterogeneous |
Considerable confusion remains associated with the choice of endpoints, both in clinical and preclinical assessments (Table). Regulatory agencies require a demonstration of “substantial evidence to support claims of effectiveness for new drugs” (21CFR314.126 (a)). It is understood that effectiveness must be shown in terms of something the patient understands, e.g., survival or improvement in functional capacity. Usually, clinical trial protocols use the modified Rankin score as their primary endpoint because the mRS describes the patient’s ability to care for themselves and accomplish activities of daily living18. The Rankin is widely accepted and understood, is easy to administer, can be administered by telephone, and performs well in clinimetric analyses18–20. Other outcome scales, however, could serve as well if they were widely accepted in the stroke community and quantified how the patient feels, functions, or survives21, 22. Functional rating instruments, such as the Rankin score, are mis-understood, and frequently criticized for being insensitive21. In fact, the modified Rankin holds great power to detect meaningful differences between treated groups in a clinical trial. On the other hand, volume of infarction—or its inverse, the volume of remaining intact brain—is intuitively and obviously important to the subject, and easier to measure quantitatively. In preclinical assessment, infarct volume is usually the primary outcome measure, although increasingly investigators are including behavioral endpoints in preclinical assessment studies23. To date, infarct volume has not been used as a primary outcome measure in Phase 3 clinical trials intended for regulatory licensure.
For the purpose of looking ahead to the next generation of stroke treatments, endpoints chosen for preclinical assessment must come into concordance with human clinical trial design. Almost certainly, the optimal approach will include both behavioral and histomorphometric measurements.
SINGLE-AGENT SINGLE-TARGET
In 1983 the term “Ischemic Cascade” appeared in print for the first time (that I could find) in the Neurologic Clinics, Volume 1, Number 1, although in that same year the word ‘cascade’ appeared in numerous symposium reports and review papers addressing cerebral ischemia and reperfusion injury24. For the next 20 years, countless drawings of the ischemic cascade appeared in print, always drawn with reverent arrows connecting disparate observations as if to imply a causal, orderly sequence. One such example appears in Figure 1, although in this version there was no attempt to communicate a causal sequence. Students and investigators came to believe in a cascade that had a beginning, middle, and an end. The search for stroke treatment turned entirely toward finding the ‘master switch’: the single molecular step that would control the ischemic cascade.
Figure 1. Excitatory and inhibitory influences on post-synaptic neurons.
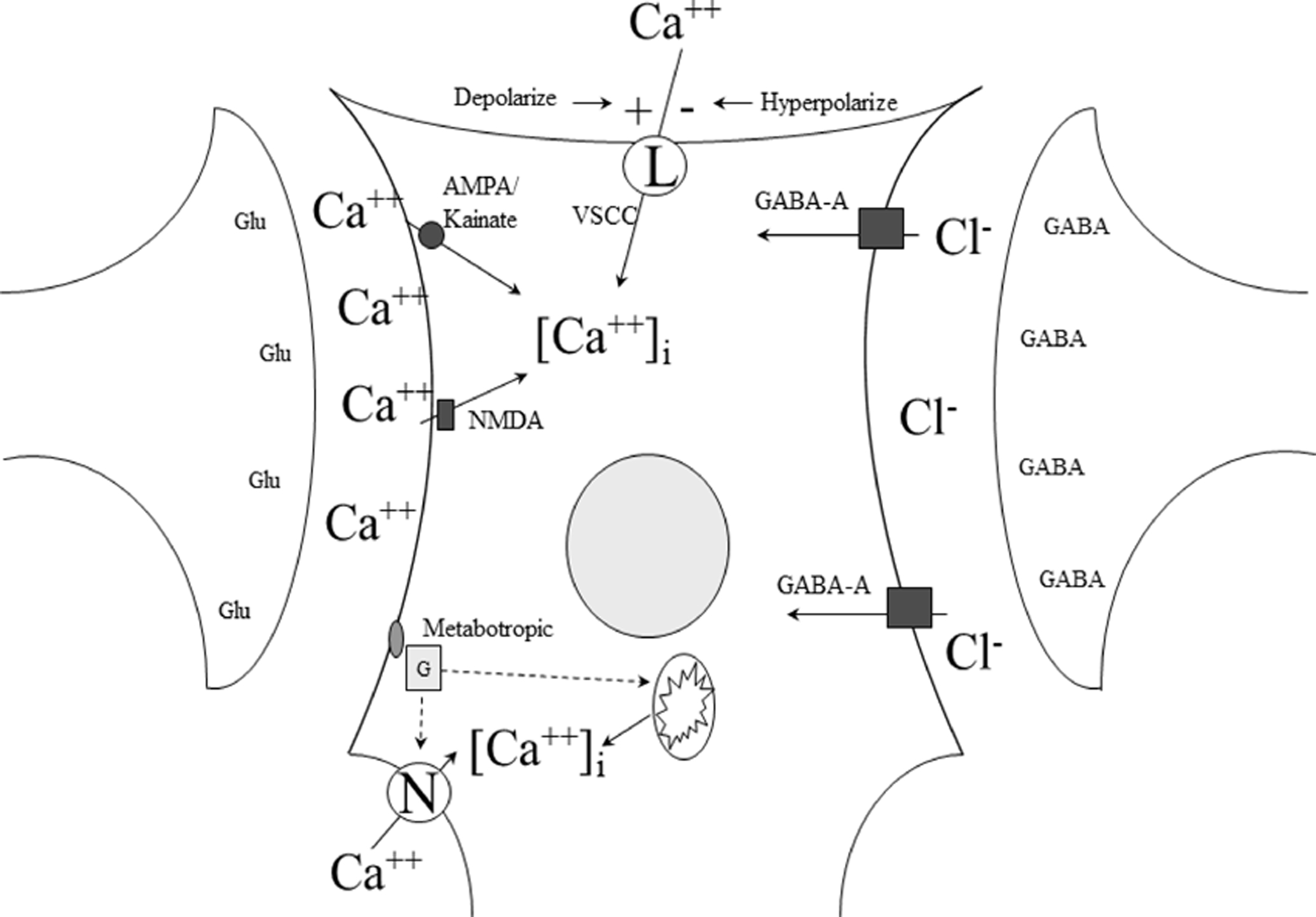
In the development of neuronoprotective treatment, antagonists of the glutamate receptors succeeded in preclinical models. Antagonists targeting the NMDA, AMPA/kainite, and metabotropic receptors all failed in clinical trials. Agents acting on the voltage gated calcium, or L-type, channel are used in treating post-hemorrhage vasospasm, but did not succeed as cerebroprotectants. Agonists of the GABA-A receptor, though promising in preclinical assessment, failed in large, pivotal clinical trials. Figure from the author. Abbreviations: NMDA N-methyl-d-aspartate; AMPA α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid; GABA γ-aminobutyric acid
Study sections and journal reviewers insisted that putative stroke treatments should have a known mechanism of action, which should involve only one step or action. Drugs that harbored multiple mechanisms were derided as ‘dirty drugs’.
The prototypical single-mechanism target may be the glutamate receptor, as in Figure 1. Recognizing that ischemia produced a flood of pre-synaptic glutamate release, it was determined that excess glutamate stimulation and neuronal depolarization led to elevated intra-cellular calcium that activated a variety of calcium dependent toxic enzymes4, 25. Receptor specific glutamate antagonists were found, or re-purposed, and rushed into clinical trials that failed26–28. Knowing that activation at the GABA receptor caused hyperpolarization that blocked depolarization-gated calcium influx, we showed that GABA agonists were as effective in animals models as glutamate antagonists29, 30. Again, however, clinical trials in patients failed31–33.
Many investigators began to question the single-target, single-agent approach to stroke therapy. In an early stab at pleiotropic neuroprotection, we showed the combination of glutamate antagonists and GABA agonists could be used synergistically, although demonstrating true synergism required an attentive experimental design34–36. Others assembled ‘cocktails’ or combinatorial therapies, most of which looked promising in experimental models37–39. Acceptance of multi-targeted approaches lagged, however, at funding and regulatory agencies.
As the current investigative focus shifts from single-target to pleiotropic stroke treatments, a few obvious conclusions emerge from nearly 3 decades of preclinical and clinical stroke drug development. Firstly, the role that dogma played in limiting investigation must be admitted. Dramatic, beneficial results arising from combinatorial approaches were ignored or belittled in peer review and labeled as violations of agreed-upon convention: scientists must be driven by an understanding of the molecular and cellular mechanism of any candidate treatment. In contrast, many have pointed out that the most effective neuroprotectant in animal stroke models, therapeutic hypothermia, works by many mechansims40–43.
The second important lesson from the single-target drug development years emphasizes scientific rigor. Most studies failed to consider key issues: randomization, blinding, sample size, and using appropriate statistics. Stroke treatment development has suffered greatly due to the failure to adhere to principles of rigorous design13, 16, 44–46. Important new initiatives are underway to correct this failure, and to require standards of rigor at journals and at grant review47–49.
NEW DIRECTIONS
The graphic artist M. C. Escher specialized in complex drawings that appear banal until suddenly the viewer solves the optical illusion, and the drawing inverts into a completely distinct perspective. Similarly, we who search for stroke treatment require a novel perspective, a new viewpoint that will allow us to find a way past previous translational failure. We have learned much in our failures and we have progressed significantly. Although much of what we propose today will tomorrow go the way of bloodletting and purging bad humors, yet we can formulate some new directions and ideas, at least enough to get us started.
Nothing speaks to new perspectives so well as re-defining terms. In a separate publication, a STAIR (Stroke Treatment Academic Industry Roundtable) workshop on neuroprotection has proposed new terminology (Lyden et al, in press). The term neuroprotection, having outlived its usefulness, is proposed to be replaced with terms more specific and more appropriate. At the earlier STAIR X, workshop participants proposed to rename the process of protecting the entire brain during stroke “cerebral cytoprotection”50. The term “neurovascular unit” was proposed to indicate the brain consists of several different cell types, each playing a unique role51, 52. In the NVU, consisting of neurons, astrocytes, endothelial cells, pericytes and other glia subtypes, each element plays a different role and there is considerable cross-cell communication53, 54. At STAIR XI, participants proposed to define cerebral cytoprotection in terms of the NVU. Preclinical and clinical research targeting neurons would be called “Neuronoprotection”; that targeting astrocytes “glioprotection”, and that targeting the blood brain barrier (BBB), “vasculoprotection”. Cerebral cytoprotection or just cerebroprotection connotes treatment designed to benefit the entire brain and presumably neurological function. But what good are new definitions if they do not influence or assist preclinical investigation? New investigative directions have opened, and novel insights gained, in response to our new understanding of the NVU. Three such insights include: our understanding of reperfusion injury, our understanding of NVU response to injury, and ‘help-me’ signaling in the NVU.
The first insight arising from a new appreciation of the NVU concerns the effect of reperfusion after a prolonged period of ischemia. Reperfusion affects all elements of the NVU and causes a new set of pathological mechanisms not found during ischemia without reperfusion (Figure 2)5. If the ischemia lasts long enough, then with reperfusion several deleterious effects result: astrocyte swelling, pericyte contraction, and platelet accumulation on the abluminal wall of the dysfunctional endothelial cell. Eventually, leukocytes adhere, clotting factors activate, and micro-clotting begins. Post-ischemic microcirculatory failure was identified in the 1960’s as the “no-reflow” phenomenon55, 56, but in contemporary parlance we call it reperfusion failure. Using the Thrombolysis in Cerebral Infarction (TICI) scale, the extent of recanalization can be described57. After successful recanalization, any score worse than TICI-3 may include areas of poor capillary perfusion, which is an imaging approximation of no-reflow, i.e., failure to reperfuse the microcirculation downstream of the recanalized vessel. It is crucial in discussing these issues, by the way, to clearly define recanalization as the opening of a large, feeding artery; we define reperfusion as the opening of the microcirculation allowing blood to reach the tissue supplied by that feeding artery. This distinction is often confused in practice because microcirculatory reperfusion depends mainly on successful upstream recanalization. Augmentation of collateral flow is another way to improve perfusion, and does not require recanalization, so therapies targeting recanalization should remain distinct from those targeting reperfusion.
Figure 2. Reperfusion injury in the neurovascular unit.
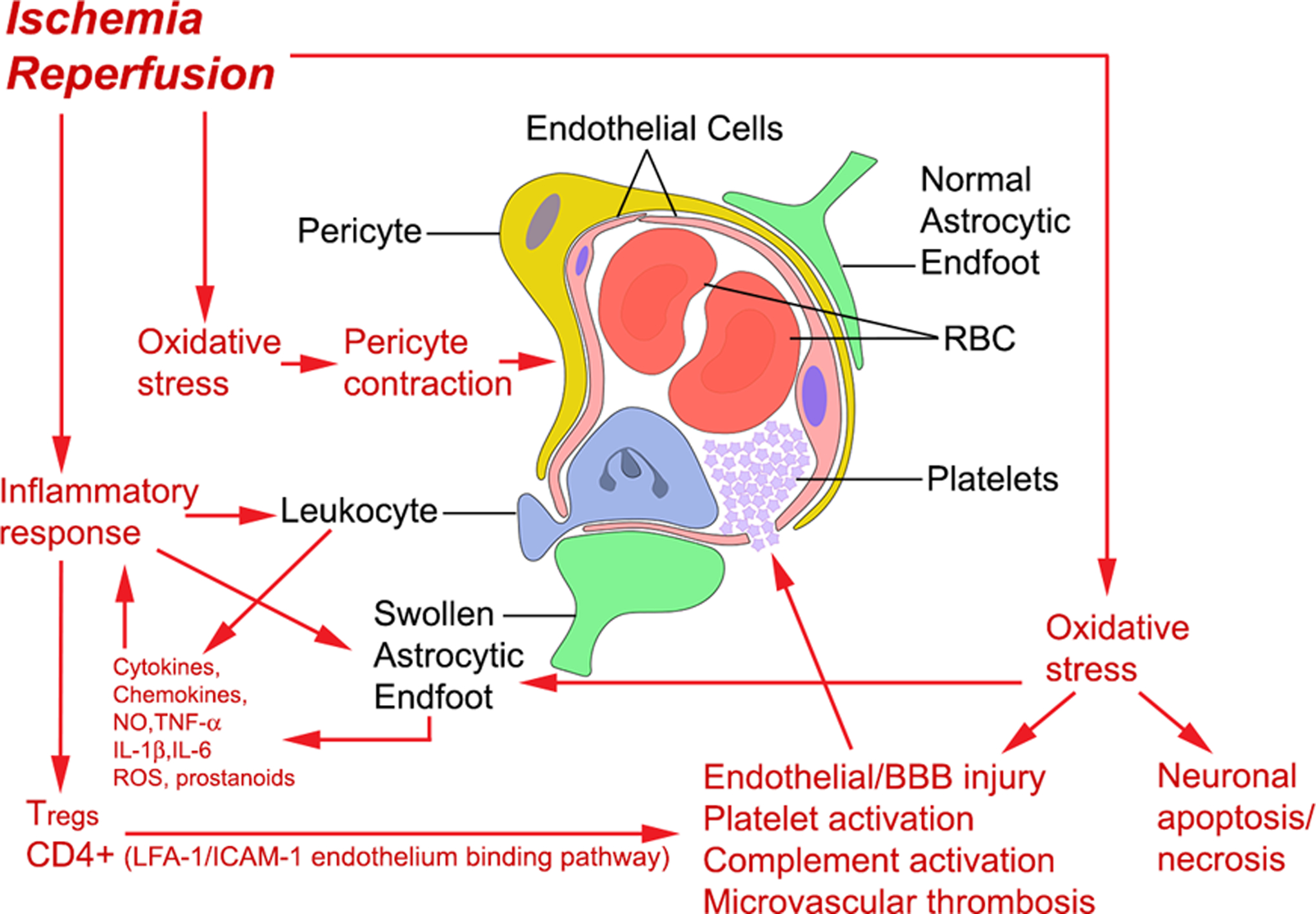
The NVU includes neurons, astrocytes, endothelial cells, pericytes, among other cell types. During reperfusion injury, several processes occur to impede microvascular reflow as well as open the blood brain barrier. During reperfusion, impaired mitochondria generate oxygen and nitrogen free radicals that mediate cell injury pathways throughout the NVU. Injury to endothelial cells triggers platelet aggregation and microthrombosis that can exacerbate perfusion failure. Figure reprinted5 with permission of Sage Publications.
The availability of mechanical thrombectomy in clinical practice has not only saved thousands of patients but allowed investigators to define and understand the role played by reperfusion in mediating brain injury. Prior to the clinical deployment of thrombectomy, cerebroprotective therapies probably failed to enter the ischemic brain in large quantities. Collateral flow might carry some amount of the test agent into ischemic brain, but only recanalization allows proper delivery of the test agent in enough amounts to influence outcome. The first clinical trial of stroke treatment to enroll patients after mechanical thrombectomy was actually ongoing when thrombectomy received regulatory approval – the study was amended part way through58. Subsequently, contemporary clinical trial design allows—if not requires—enrollment of patients after documented recanalization.
An argument can be made that some cerebral cytoprotectants, by virtue of their mechanism of action, could preserve brain, pending recanalization. Therapeutic hypothermia, or specifically head cooling with local cooling devices59, 60, lowers cerebral metabolic demand, and allows brain to survive pending recanalization or augmentation of collateral flow. So far therapeutic hypothermia has not succeeded in clinical trials61, 62. Transcranial near-infrared light therapy was another treatment proposed for ischemic stroke patients without recanalization, by providing light energy directly to mitochondria, thus preserving metabolic function until recanalization or augmentation of collateral flow63. Despite early promise, this treatment failed in a large, pivotal trial64. Undoubtedly, future, novel cerebral cytoprotectants will emerge, and some may likely prove useful, but for the foreseeable future, it seems prudent to require documented recanalization in the context of a clinical trial of a putative cerebral cytoprotectant.
A second insight following the definition of the NVU arose out of an attempt to understand the differences among the elements comprising the NVU. The different elements—neurons, astrocytes, pericytes, endothelial cells—differ markedly in their tolerance for ischemia65. The notion that some areas of brain, and some cell types, are selectively vulnerable dates back a few decades66–68. Direct comparisons of the various NVU cell types was accomplished only recently, and the mechanism for this differential vulnerability remains unclear65. The mechanism of regional selective vulnerability relates, perhaps, to differences in the ratio of excitotoxic versus inhibitory transmitter efflux during ischemia, called the excitotoxic index66. Regional selective vulnerability would depend on intrinsic differences in the tolerance to ischemia of each NVU cell type; on the regional variation in cerebral blood flow; and on the variation in the distribution of glutamatergic versus GABAergic receptors subtypes. In contrast, differential vulnerability depends solely on the innate resistance to ischemia in each cell type65.
Regional selective vulnerability in the brain and differential vulnerability among elements of the NVU together imply many cautions while developing treatments for acute ischemic stroke. There is no doubt, for example, that some treatments targeting the BBB, i.e., vasculoprotectants, may impact neurons very differently69. It would be predicted that treatment dose and duration of treatment would differ70. Further, the time window in which treatment might be predicted to remain effective should differ considerably among different NVU elements.
Knowledge of the differential vulnerability among elements in the NVU yields a powerful tool that can be used to target therapies at different NVU elements. For example, if one were targeting neurons with a novel compound that was solely neuronoprotective, then using a lower dose very early after ischemia onset would preferentially benefit neurons. In contrast, vasculoprotective therapy might be given hours or days later, when post-ischemic BBB damage was evolving. Such targeting could avoid side effects that result from excessive dosing or excessive treatment duration65.
A third insight to emerge from our new understanding of the NVU is the ‘help-me’ signaling concept. It is now very apparent that cell-cell communication occurs among the different elements in the NVU. Some of this communication proceeds via canonical synaptic release of neurotransmitters. In the previous decade, non-canonical calcium flux among astrocytes was discovered and related to regional control of cerebral blood flow71, 72. Most recently, however, non-cell autonomous or paracrine communication among NVU elements has been demonstrated. In one direction, neurons seek assistance during ischemia or other insult, and in another direction, glia appear to secrete protective factors (Fig. 3).
Figure 3. Help-me signaling in the neurovascular unit.
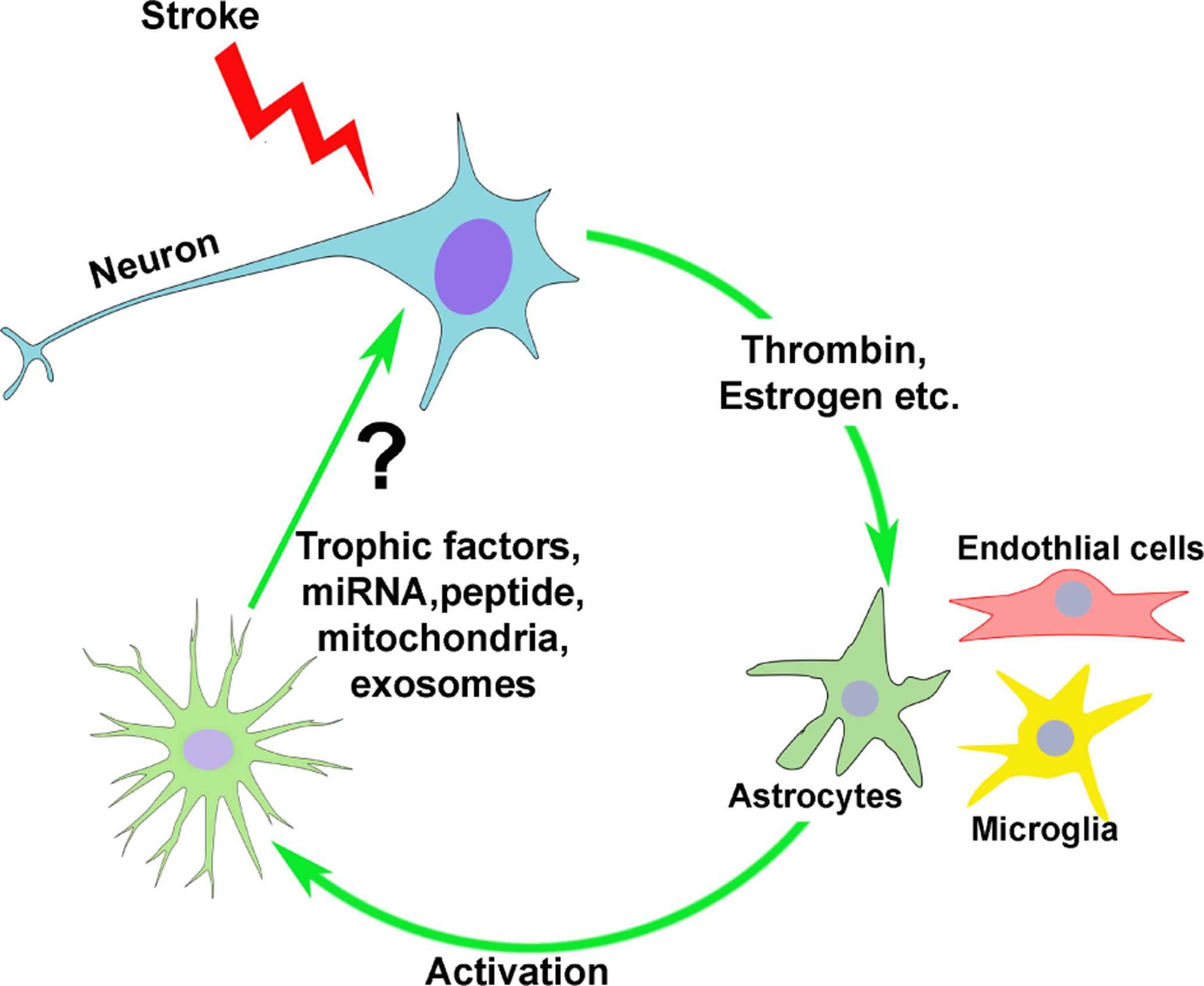
In response to injury, neurons generate paracrine signals that reach adjacent astrocytes and microglia, causing activation. Glial activation is pleiotropic, with some protective and some toxic responses. After activation, astrocytes generate paracrine factors that protect neurons from further injury, and promote regeneration. Figure from the author and Dr. Padmesh Rajput, PhD.
Neurons seek assistance during ischemia by secreting activating substances that act on adjacent astrocytes (Fig. 3)73. The serine protease prothrombin is released from ischemic neurons and promotes astrocyte activation74. The resulting astrocyte activation stimulates gene expression changes consistent with the so-called protective astroglial phenotype, but also some genes associated with the toxic astrocyte phenotype. Activated astrocytes then secrete protective factors—that remain to be delineated—that protect neurons from ischemia74. Another help-me signal recently identified is estrogen75. Like prothrombin, under conditions of ischemia, neurons appear to secrete estrogen that activate adjacent astrocytes in a paracrine manner. Finally, the peptide lipocalin-2 was shown to activate neurons and microglia in a paracrine fashion76. Undoubtably other signaling molecules exist and future effort will be required to determine which are fully functional and relevant in human stroke or cardiac arrest patients suffering brain ischemia.
In response to neuronal ‘help-me’ signals, astrocytes respond with a protective response. Likely other elements of the NVU—notably microglia—also participate. The key components of the astrocyte protective response remain undefined and provide a rich opportunity for future pharmacological development. In a highly novel experiment, Lo and colleagues demonstrated transfer of mitochondria from astrocytes to injured neurons, with resultant salvage77. This truly remarkable observation will require further delineation, but potentially opens a considerable therapeutic opportunity. In response to ischemia, astrocytes also secrete a variety of peptides that are known to function as cytoprotectants, including growth factors. Likely, protection from injury utilizes the same functions that astrocytes serve during normal neuronal growth, survival, and synaptogenesis. Neurotoxic astroglial responses do occur as well, however, and astrocytes may contribute to the death of adjacent neurons78. In another recent, stunning observation, astrocytes were documented to phagocytose neurons in the adult hippocampus as part of activity-related pruning79.
Clearly the role of astrocytes in protecting neurons requires considerable further exploration and definition. One immediate implication of these recent developments is that cerebroprotectant development must proceed cautiously, because treatments designed to benefit one element of the NVU may alter or even impede protective responses from other NVU elements. To demonstrate this pitfall, therapeutic hypothermia was tested for effects on help-me signaling and the astrocyte protective response65. Hypothermia is one of the most effective cerebroprotectant treatments ever studied in preclinical models of stroke and cardiac arrest40, 80, yet clinical benefit in patients has been difficult to prove. Hypothermia significantly impaired the astrocyte protective response after neuronal help-me signaling65. This finding demonstrates the principle that neuronoprotective and glioprotective treatments may clash—thoughtful approaches are required to dosing and timing of new, candidate cerebroprotectants.
PRECLINICAL ASSESSMENT OF STROKE TREATMENTS
The search for effective cerebroprotectants requires an efficient and valid experimental paradigm81. Candidate treatments may emerge from an understanding of mechanisms—e.g., understanding the molecular and cellular pathophysiology of ischemia—or from agnostic pharmacological screening. By whatever route, investigators must show evidence that the candidate treatment shows efficacy. Much has been written about preclinical disease models, and stroke models in particular6, 49, 82, 83. Mostly, examining the models leads to more questions than answers, but a few key hypotheses are available for testing.
The first and most relevant question to ask concerns the purpose of the planned investigation. If an investigator wishes to test a hypothesis about the molecular mechanism underlying an aspect of ischemia, then models using OGD in cell culture or with brain slices are appropriate. Each element of the NVU can be studied in monocellular cultures, or together in co-cultures or transwells. The OGD model mimics ischemia sufficiently well that candidate treatments can be screened in these in vitro models. A key misinterpretation of this approach is that treatment of monocellular culture cells predicts the results of the assembled NVU in the whole brain. Rather, studies involving OGD reveal the isolated behavior of each cell type of the NVU. Ultimately, in vivo studies must be done to complement and confirm such in vitro work. Another key limitation of OGD in monocellular cultures is that the conditions resemble those of the ischemic core and not penumbra; in the penumbra residual blood flow may support cell survival and this aspect is difficult to model in vitro.
Despite the limitations of OGD models, they are simple (relative to an in vivo model) and rapid. Thus, OGD models allow high throughput screening of candidate treatments. A candidate treatment that benefits neurons during OGD would emerge as a neuronoprotectant, one that benefits astrocytes in monocellular culture a glioprotectant, and so on. Such studies allow for the determination of cell-type differences in dose, duration and timing of administration.
Whole animal stroke models employing young, disease-free animals can provide appropriate, efficient, and valid test environments, if the molecular process under study is known to act similarly with aging, sex differences, or in the face of co-morbidities. Here the investigator should choose thoughtfully. A variety of animal stroke models have been proposed over the years to simulate focal or global ischemia82. There are two main approaches: occluding the middle cerebral artery (MCAo) using a mechanical approach or using a thrombo-embolic approach. While a thrombo-embolic model may seem more ‘natural’ or in some way replicate human stroke more faithfully, in practice such models are highly variable and difficult. For studies of thrombolytic drugs, a thromboembolic model may be ideal84. On the other hand, if the investigative purpose is to demonstrate benefit of a candidate cerebroprotective therapy using as few subjects as possible, then a mechanical MCAo model is more appropriate; it has been suggested that the nylon filament MCAo model85, 86 faithfully replicates the sudden, total recanalization seen during mechanical thrombectomy in stroke patients5.
If the purpose of the planned investigation is to demonstrate efficacy, and perhaps safety, of a treatment candidate prior to a clinical trial, then additional considerations enter into choosing an animal model. In the ideal scenario, we would like a preclinical assessment that can reliably predict the outcome of human clinical trials. If we screen dozens of candidate treatments in a preclinical assessment, we would like to know which ones are most likely to succeed in a clinical development program that includes Phase 2 dose-finding and definitive Phase 3 trials in humans. This ideal scenario may be asking too much of preclinical modeling; we may find that preclinical assessment tools can establish efficacy, and perhaps safety, of a candidate treatment, but only the actual human clinical trials can establish benefit in human patients. At the very least, we would like a preclinical assessment that biases our selection of candidate treatments towards success in human clinical stroke trials, a process sometimes called de-risking.
To strengthen the preclinical assessment of candidate cerebroprotectants, many authors recommend essential changes to the traditional paradigm13, 16, 44, 81, 87. Age must be accounted for at some point in the development process, as there is no proof that cerebroprotectants that function well in young animals will also function well in older humans. Sex has proven to be a problem as well. For example, the cerebroprotectant drug tirilazad—after a long and expensive preclinical assessment—was found in human clinical trials to require different dosing in females88. Other drugs likewise may require different dosing in males compared to females89, 90.
Some attention must also be given to assessing candidate cerebroprotectants in the setting of comorbid conditions such as diabetes or chronic hypertension. As yet, there is no consensus on the optimal approach to modeling the role of age, sex, and co-morbid conditions on the preclinical assessment, but the hypothesis is that such enhanced models will provide a superior approach. This hypothesis remains to be tested.
The National Institute of Neurological Disorders and Stroke (NINDS) issued a call (RFA-NS-18–033 and RFA-NS-18-034) for investigators to propose a multi-site network, called the Stroke Preclinical Assessment Network, or SPAN. The call followed an NINDS sponsored symposium that gathered a large number of experienced investigators who surveyed past failures and recommended future directions48, 49. The bulk of the innovations to be included in SPAN concern rigor and the reduction of bias. For example, SPAN will include centralized subject randomization, masking of the test compounds when they are administered, and blinded outcome measurement. Most importantly, SPAN includes 6 study sites, all doing the same stroke models and studying the same candidate cerebroprotectants. This network approach will avoid several sources of bias and harness the power of heterogeneity across sites to identify effective treatments87. It remains to be determined whether such rigor and heterogeneity proves effective in selecting candidate treatments for eventual success in clinical trials, i.e., de-risking.
PLEIOTROPIC AGENTS
Currently, a small number of candidate cerebroprotectants are under study in human trials91. Interestingly, in a recent review the authors included non-pharmacological treatments such as remote ischemic conditioning and transcranial electrical stimulation, again testifying to the interest in pleiotropic agents that act via multiple—or even unknown—mechanisms91. The efficacy of conditioning for acute ischemic stroke has been reviewed92. While no evidence has yet emerged that ischemic conditioning benefits patients, significant gaps in our knowledge base prevent firm conclusions; further clinical trials are underway.
The ultimate example of a pleiotropic candidate cerebroprotectant is therapeutic hypothermia, which acts to interrupt a large number of death pathways in ischemia41. Although hypothermia has not succeeded in planned, clinical trials61, 62, this was due to failure to recruit enough subjects and fear that prolonged, whole-body hypothermia might prove deleterious. Currently, focal cooling via the embolectomy catheter is under study93. Certainly more work will be needed to optimize the delivery of therapeutic hypothermia to stroke patients65.
An example of a pleiotropic effect based on a single molecule concerns the effect of thrombin, a naturally occurring, blood circulating serine protease also called activated Factor II in the coagulation cascade. In addition to cleaving fibrinogen to fibrin, thrombin acts on the G protein-coupled receptor PAR (protease activated receptor), of which there are four main subtypes94, 95. PARs are found on neurons, astrocytes, and endothelial cells, although the effects on each cell type differ69. Thrombin activation of PAR1 leads to cytotoxic effects96, 97. In preclinical assessment, it was shown that the direct thrombin inhibitor, argatroban, powerfully ameliorated infarction and behavioral deficits after MCAo in animals98. A clinical trial of argatroban (NCT03735979) is underway in the StrokeNet. In a striking example of biased agonism, PAR1 activation by other serine proteases, e.g., activated protein C (APC) results in cytoprotective rather than cytotoxic effects99. Several laboratories have demonstrated significant and powerful benefit after MCAo using APC analogues, and a large, Phase III clinical trial testing the drug 3K3A-APC, which acts protectively on PAR1 has been proposed95, 100–102.
In contrast to agents with pleiotropic effects, a single molecular target with multiple downstream effects is the post-synaptic density protein PSD-95. Specific agents that decouple this protein from its effector molecules were shown in preclinical assessments to be neuronoprotective103. Preclinical assessment of an agent targeting PSD-95 included a study in a gyrencephalic animal model104. Although a pivotal clinical trial that was properly powered failed to show benefit of the PSD-95 targeting agent nerinitide, follow up studies are planned that will target a potentially more appropriate subgroup of patients105.
Interventions targeting neuroinflammation tend to mimic a pleiotropic agent due to the multiple feedback, feed forward, and cross talk loops in the neuroinflammatory response to ischemia106. Early efforts such as corticosteroids appeared to fail, although it must be said that steroids were tested prior to the advent of modern clinical trial design107–109. A biological agent targeting the ICAM-1 receptor should have prevented neutrophil entry into brain and reduce stroke related injury, but instead, patients appeared to worsen after anti-ICAM-1 treatment110. Other targets in the neuroinflammatory response to ischemia include IL-1, the IL-1 receptor, and IL-6111, 112. Inasmuch as a large number of receptors in neuroinflammatory pathways are tyrosine kinases, another example of a single-molecule target with pleiotropic effects are the tyrosine kinase inhibitors113. Several inhibitors of tyrosine kinase are in clinical use for cancer treatment and many are being explored as possible cerebroprotectants. Cell based therapies, using a variety of engineered progenitor-like cells, or exosomes derived from such cells, illustrate another approach that may act on many targets114–116.
Early studies that are pursuing mitochondrial transfer are based on the extraordinary finding that astrocytes and neurons exchange mitochondria77. The initial step in cell death during ischemia is the deprivation of glucose and oxygen, resulting in mitochondrial failure to generate energy117. Thus, it would make sense to target energy failure in stroke treatment, although energy failure occurs so early after blood flow interruption it may prove an unwieldy target. An initial preclinical study showed extraordinary success in salvaging neurons with astrocyte mitochondria77. A treatment using laser light to attempt to deliver energy (photons) to impaired mitochondria failed64.
Pleiotropic biological therapies for stroke include the use of exosomes and microRNAs. Exosomes are an example of an extracellular vesical produced by exocytosis to transfer material between cells. Traditionally defined as 40–100 nm in diameter, exosomes generated from brain cells may contain peptides, lipids, RNA, or other undefined material. Although neuronoprotective and cerebroprotective effects of exosomes can be demonstrated, this treatment modality will require further development114. A huge number of microRNAs have been tested in stroke models, with wildly mixed results118.
CONCLUSIONS
A generation of stroke researchers has grown up watching large clinical trials of cerebroprotective treatments fail, while simultaneously celebrating the success of recanalization therapies, thrombolysis and thrombectomy. Looking ahead, success in developing cerebroprotection—either as an adjunct to recanalization or as stand-alone treatment—will require new definitions that recognize the importance of differential vulnerability in the NVU. Success will require new focus on pleiotropic agents that act via multiple mechanisms of action. Renewed commitment to scientific rigor is essential to success, as embodied in the new SPAN effort as well as resolve among grant agencies and journals to enforce principles of quality in preclinical science. With these new definitions, novel approaches, and renewed attention to rigor, the prospect for successful cerebroprotective should improve.
Acknowledgements.
The author is grateful to Dr. Padmesh Rajput, PhD, for creating Figure 3.
Sources of Funding
Dr. Lyden is supported by the National Institute of Neurological Disorders and Stroke grants U24 NS113452 and R01NS075930
Non-standard abbreviations and acronyms
- APC
Activated protein C
- BBB
Blood brain barrier
- GABA
gamma amino butyric acid
- ICAM
Intercellular adhesion molecule
- LD50
50% lethal dose
- MCAo
Middle cerebral artery occlusion
- NVU
Neurovascular Unit
- NINDS
National Institute of Neurological Disorders and Stroke
- OGD
Oxygen glucose deprivation
- PAR
Protease activated receptor
- PSD-95
Post synaptic density protein 95
- STAIR
Stroke Treatment Academic Industry Roundtable
- TICI
Thrombolysis in Cerebral Infarction
Footnotes
Disclosures
Dr. Lyden is the Principal Investigator of the NIH sponsored Stroke Preclinical Assessment Network; a DSMB member for the Basilar Artery International Cooperation Study (unpaid) and the “PORTICO™ Re-sheathable Transcatheter Aortic Valve System US IDE Trial (PORTICO)” (Baim Institute); received royalties for “Thrombolytic Therapy for Acute Stroke”, 3rd Ed., Springer Press; and serves as a consultant to various plaintiff and defense legal firms and Apex Innovations.
References
- 1.Hort I, Karenberg A. Medieval descriptions and doctrines of stroke: Preliminary analysis of select sources. Part i: The struggle for terms and theories - late antiquity and early middle ages (300–800). Journal of the History of the Neurosciences. 1998;7:162–173 [DOI] [PubMed] [Google Scholar]
- 2.Karenberg A, Hort I. Medieval descriptions and doctrines of stroke: Preliminary analysis of select sources. Part ii: Between galenism and aristotelism – islamic theories of apoplexy (800–1200). Journal of the History of the Neurosciences. 1998;7:174–185 [DOI] [PubMed] [Google Scholar]
- 3.Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia: The ischemic penumbra. Stroke. 1981;12:723. [DOI] [PubMed] [Google Scholar]
- 4.Rothman SM, Olney JW. Excitotoxicity and the nmda receptor. Trends in Neuroscience. 1987;10:299. [DOI] [PubMed] [Google Scholar]
- 5.Bai J, Lyden PD. Revisiting cerebral postischemic reperfusion injury: New insights in understanding reperfusion failure, hemorrhage, and edema. Int J Stroke. 2015;10:143–152 [DOI] [PubMed] [Google Scholar]
- 6.O’Collins VE, Macleod MR, Cox SF, Van Raay L, Aleksoska E, Donnan GA, Howells DW. Preclinical drug evaluation for combination therapy in acute stroke using systematic review, meta-analysis, and subsequent experimental testing. J Cereb Blood Flow Metab. 2011;31:962–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tymianski M Can molecular and cellular neuroprotection be translated into therapies for patients?: Yes, but not the way we tried it before. Stroke. 2010;41:S87–90 [DOI] [PubMed] [Google Scholar]
- 8.Auriel E, Bornstein NM. Neuroprotection in acute ischemic stroke--current status. J Cell Mol Med. 2010;14:2200–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantz J, Degos V, Laigle C. Recent advances in pharmacologic neuroprotection. Eur J Anaesthesiol. 2010;27:6–10 [DOI] [PubMed] [Google Scholar]
- 10.Lapchak PA, Araujo DM. Advances in ischemic stroke treatment: Neuroprotective and combination therapies. Expert Opin Emerg Drugs. 2007;12:97–112 [DOI] [PubMed] [Google Scholar]
- 11.Ginsberg MD. Life after cerovive: A personal perspective on ischemic neuroprotection in the post-nxy-059 era. Stroke. 2007;38:1967–1972 [DOI] [PubMed] [Google Scholar]
- 12.Savitz SI. A critical appraisal of the nxy-059 neuroprotection studies for acute stroke: A need for more rigorous testing of neuroprotective agents in animal models of stroke. Exp Neurol. 2007;205:20–25 [DOI] [PubMed] [Google Scholar]
- 13.Macleod MR, Fisher M, O’Collins V, Sena ES, Dirnagl U, Bath PM, Buchan A, van der Worp HB, Traystman R, Minematsu K, et al. Good laboratory practice: Preventing introduction of bias at the bench. Stroke. 2009;40:e50–52 [DOI] [PubMed] [Google Scholar]
- 14.DeGraba TJ, Pettigrew LC. Why do neuroprotective drugs work in animals but not humans? Neurol Clin. 2000;18:475. [DOI] [PubMed] [Google Scholar]
- 15.Grotta J Why do all drugs work in animals but none in stroke patients? 2. Neuroprotective therapy. J Intern Med. 1995;237:89–94 [DOI] [PubMed] [Google Scholar]
- 16.Sena ES, van der Worp HB, Bath PM, Howells DW, Macleod MR. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. PLoS Biol. 2010;8:e1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linfante I, Cipolla MJ. Improving reperfusion therapies in the era of mechanical thrombectomy. Transl Stroke Res. 2016;7:294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broderick JP, Adeoye O, Elm J. Evolution of the modified rankin scale and its use in future stroke trials. Stroke. 2017;48:2007–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Candelise L, Pinardi G, Aritzu E, Musicco M. Telephone interview for stroke outcome assessment. Cerebrovascular Diseases. 1994;4:341 [Google Scholar]
- 20.Lees KR, Bath PM, Schellinger PD, Kerr DM, Fulton R, Hacke W, Matchar D, Sehra R, Toni D. Contemporary outcome measures in acute stroke research: Choice of primary outcome measure. Stroke. 2012;43:1163–1170 [DOI] [PubMed] [Google Scholar]
- 21.Quinn TJ, Singh S, Lees KR, Bath PM, Myint PK. Validating and comparing stroke prognosis scales. Neurology. 2017;89:997–1002 [DOI] [PubMed] [Google Scholar]
- 22.Bath PM, Lees KR, Schellinger PD, Altman H, Bland M, Hogg C, Howard G, Saver JL. Statistical analysis of the primary outcome in acute stroke trials. Stroke. 2012;43:1171–1178 [DOI] [PubMed] [Google Scholar]
- 23.Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med. 2010;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hass WK. The cerebral ischemic cascade. Neurologic clinics. 1983;1:345–353 [PubMed] [Google Scholar]
- 25.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999;22:391–397 [DOI] [PubMed] [Google Scholar]
- 26.Albers GW, Saenz RE, Moses JA Jr., Choi DW. Safety and tolerance of oral dextromethorphan in patients at risk for brain ischemia. Stroke. 1991;22:1075. [DOI] [PubMed] [Google Scholar]
- 27.Muir KW, Lees KR. Excitatory amino acid antagonists for acute stroke. Cochrane Database Syst Rev. 2003:CD001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grotta J, Clark W, Coull B, Pettigrew LC, Mackay B, Goldstein LB, Meissner I, Murphy D, LaRue L. Safety and tolerability of the glutamate antagonist cgs 19755 (selfotel) in patients with acute ischemic stroke. Results of a phase iia randomized trial. Stroke. 1995;26:602. [DOI] [PubMed] [Google Scholar]
- 29.Lyden PD. Gaba and neuroprotection. Int Rev Neurobiol. 1997;40:233–258 [PubMed] [Google Scholar]
- 30.Lyden PD, Hedges B. Protective effect of synaptic inhibition during cerebral ischemia. Stroke. 1992;23:1463. [DOI] [PubMed] [Google Scholar]
- 31.Lyden P, Jacoby M, Schim J, Albers GW, Mazzeo P, Ashwood T, Nordlund A, Odergren T. The clomethiazole acute stroke study in tissue-type plasminogen activator-treated stroke (class-t): Final results. Neurology. 2001;57:1199. [DOI] [PubMed] [Google Scholar]
- 32.Lyden P, Shuaib A, Ng K, Atkinson RP, Ashwood T, Nordlund A, Odergren T. The clomethiazole acute stroke study in hemorrhagic stroke (class-h): Final results. J Stroke Cerebr Dis. 2000;9:8 [Google Scholar]
- 33.Lyden P, Shuaib A, Ng K, Levin K, Atkinson RP, Rajput A, Wechsler L, Ashwood T, Claesson L, Odergren T, et al. Clomethiazole acute stroke study in ischemic stroke (class-i): Final results. Stroke. 2002;33:122. [DOI] [PubMed] [Google Scholar]
- 34.Lyden P, Jackson-Friedman C, Shin C, Hassid S. Synergistic combinatorial stroke therapy: A quantal bioassay of a gaba agonist and a glutamate antagonist. Experimental Neurology. 2000;163:477. [DOI] [PubMed] [Google Scholar]
- 35.Lyden PD, Lonzo L, Nunez S. Combination chemotherapy extends the therapeutic window to 60 minutes after stroke. Journal of Neurotrauma. 1995;12:223. [DOI] [PubMed] [Google Scholar]
- 36.Lyden PD, Lonzo L. Combination therapy protects ischemic brain in rats: A glutamate antagonist plus a gamma-aminobutyric acid agonist. Stroke. 1994;25:189. [DOI] [PubMed] [Google Scholar]
- 37.Weng YC, Kriz J. Differential neuroprotective effects of a minocycline-based drug cocktail in transient and permanent focal cerebral ischemia. Experimental neurology. 2007;204:433–442 [DOI] [PubMed] [Google Scholar]
- 38.Aronowski J, Strong R, Shirzadi A, Grotta JC. Ethanol plus caffeine (caffeinol) for treatment of ischemic stroke: Preclinical experience. Stroke. 2003;34:1246–1251 [DOI] [PubMed] [Google Scholar]
- 39.Auer RN. Combination therapy with u74006f (tirilazad mesylate), mk-801, insulin and diazepam in transient forebrain ischaemia. Neurol Res. 1995;17:132. [DOI] [PubMed] [Google Scholar]
- 40.Dumitrascu OM, Lamb J, Lyden PD. Still cooling after all these years: Meta-analysis of pre-clinical trials of therapeutic hypothermia for acute ischemic stroke. J Cereb Blood Flow Metab. 2016: 10.1177/0271678X16645112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu TC, Grotta JC. Hypothermia for acute ischaemic stroke. Lancet Neurol. 2013;12:275–284 [DOI] [PubMed] [Google Scholar]
- 42.Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186–202 [DOI] [PubMed] [Google Scholar]
- 43.Maher J, Hachinski V. Hypothermia as a potential treatment for cerebral ischemia. Cerebrovasc Brain Metab Rev. 1993;5:277–300 [PubMed] [Google Scholar]
- 44.van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O’Collins V, Macleod MR. Can animal models of disease reliably inform human studies? PLoS Med. 2010;7:e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crossley NA, Sena E, Goehler J, Horn J, van der Worp B, Bath PM, Macleod M, Dirnagl U. Empirical evidence of bias in the design of experimental stroke studies: A metaepidemiologic approach. Stroke. 2008;39:929–934 [DOI] [PubMed] [Google Scholar]
- 46.Sena E, van der Worp HB, Howells D, Macleod M. How can we improve the pre-clinical development of drugs for stroke? Trends Neurosci. 2007;30:433–439 [DOI] [PubMed] [Google Scholar]
- 47.Lapchak PA, Zhang JH, Noble-Haeusslein LJ. Rigor guidelines: Escalating stair and steps for effective translational research. Transl Stroke Res. 2013;4:279–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bix GJ, Fraser JF, Mack WJ, Carmichael ST, Perez-Pinzon M, Offner H, Sansing L, Bosetti F, Ayata C, Pennypacker KR. Uncovering the rosetta stone: Report from the first annual conference on key elements in translating stroke therapeutics from pre-clinical to clinical. Transl Stroke Res. 2018;9:258–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bosetti F, Koenig JI, Ayata C, Back SA, Becker K, Broderick JP, Carmichael ST, Cho S, Cipolla MJ, Corbett D, et al. Translational stroke research: Vision and opportunities. Stroke. 2017;48:2632–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savitz SI, Baron JC, Fisher M, Consortium SX. Stroke treatment academic industry roundtable x: Brain cytoprotection therapies in the reperfusion era. Stroke. 2019;50:1026–1031 [DOI] [PubMed] [Google Scholar]
- 51.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185 [DOI] [PubMed] [Google Scholar]
- 52.del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267:156–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jha MK, Seo M, Kim JH, Kim BG, Cho JY, Suk K. The secretome signature of reactive glial cells and its pathological implications. Biochim Biophys Acta. 2013;1834:2418–2428 [DOI] [PubMed] [Google Scholar]
- 54.Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031 [DOI] [PubMed] [Google Scholar]
- 55.Schmid-Sch”nbein GW. Capillary plugging by granulocytes and the no-reflow phenomenon in the microcirculation. ProcFedAmerSocExpBiol. 1987;46:2397. [PubMed] [Google Scholar]
- 56.Ames A, Wright RL, Kowada M, Thurston JM, Majno G. Cerebral ischemia 2. No-reflow phenomenon. Am J Path. 1968;52:437. [PMC free article] [PubMed] [Google Scholar]
- 57.Higashida RT, Furlan AJ, Roberts H, Tomsick T, Connors B, Barr J, Dillon W, Warach S, Broderick J, Tilley B, et al. Trial design and reporting standards for intra-arterial cerebral thrombolysis for acute ischemic stroke. Stroke. 2003;34:e109–137 [DOI] [PubMed] [Google Scholar]
- 58.Lyden P, Weymer S, Coffey C, Cudkowicz M, Berg S, O’Brien S, Fisher M, Haley EC, Khatri P, Saver J, et al. Selecting patients for intra-arterial therapy in the context of a clinical trial for neuroprotection. Stroke. 2016;47:2979–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poli S, Purrucker J, Priglinger M, Diedler J, Sykora M, Popp E, Steiner T, Veltkamp R, Bosel J, Rupp A, et al. Induction of cooling with a passive head and neck cooling device: Effects on brain temperature after stroke. Stroke. 2013;44:708–713 [DOI] [PubMed] [Google Scholar]
- 60.Wang H, Olivero W, Lanzino G, Elkins W, Rose J, Honings D, Rodde M, Burnham J, Wang D. Rapid and selective cerebral hypothermia achieved using a cooling helmet. J Neurosurg. 2004;100:272–277 [DOI] [PubMed] [Google Scholar]
- 61.van der Worp HB, Macleod MR, Bath PM, Bathula R, Christensen H, Colam B, Cordonnier C, Demotes-Mainard J, Durand-Zaleski I, Gluud C, et al. Therapeutic hypothermia for acute ischaemic stroke. Results of a european multicentre, randomised, phase iii clinical trial. Eur Stroke J. 2019;4:254–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lyden P, Hemmen T, Grotta J, Rapp K, Ernstrom K, Rzesiewicz T, Parker S, Concha M, Hussain S, Agarwal S, et al. Results of the ictus 2 trial (intravascular cooling in the treatment of stroke 2). Stroke. 2016;47:2888–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zivin JA, Albers GW, Bornstein N, Chippendale T, Dahlof B, Devlin T, Fisher M, Hacke W, Holt W, Ilic S, et al. Effectiveness and safety of transcranial laser therapy for acute ischemic stroke. Stroke. 2009;40:1359–1364 [DOI] [PubMed] [Google Scholar]
- 64.Hacke W, Schellinger PD, Albers GW, Bornstein NM, Dahlof BL, Fulton R, Kasner SE, Shuaib A, Richieri SP, Dilly SG, et al. Transcranial laser therapy in acute stroke treatment: Results of neurothera effectiveness and safety trial 3, a phase iii clinical end point device trial. Stroke. 2014;45:3187–3193 [DOI] [PubMed] [Google Scholar]
- 65.Lyden PD, Lamb J, Kothari S, Toossi S, Boitano P, Rajput PS. Differential effects of hypothermia on neurovascular unit determine protective or toxic results: Toward optimized therapeutic hypothermia. J Cereb Blood Flow Metab. 2018:271678X18814614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mordecai Y, Globus T, Busto R, Martinez E, Valdes I, Ginsberg MD. Excitotoxic index - a biochemical marker of selective vulnerability. Stroke. 1991;22(1):128. [DOI] [PubMed] [Google Scholar]
- 67.Collins RC, Dobkin BH, Choi DW. Selective vulnerability of the brain: New insights into the pathophysiology of stroke. Annals of Internal Medicine. 1989;110:992. [DOI] [PubMed] [Google Scholar]
- 68.Kirino T, Sano K. Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neurolpathol. 1984;62:201. [DOI] [PubMed] [Google Scholar]
- 69.Rajput PS, Lamb JA, Fernández JÁ, Bai J, Pereira BR, Lei IF, Leung J, Griffin JH, Lyden PD. Neuroprotection and vasculoprotection using genetically targeted protease-ligands. Brain Research. 2019;1715:13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Colbourne F, Li H, Buchan AM. Continuing postischemic neuronal death in ca1. Influence of ischemia duration and cytoprotective doses of nbqx and snx-111 in rats. Stroke. 1999;30:662. [DOI] [PubMed] [Google Scholar]
- 71.Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci. 2003;23:9254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530 [DOI] [PubMed] [Google Scholar]
- 73.Xing C, Lo EH. Help-me signaling: Non-cell autonomous mechanisms of neuroprotection and neurorecovery. Progress in Neurobiology. 2017;152:181–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rajput PS, Lamb J, Kothari S, Pereira B, Soetkamp D, Wang Y, Tang J, Van Eyk JE, Mullins ES, Lyden PD. Neuron-generated thrombin induces a protective astrocyte response via protease activated receptors. Glia. 2020;68:246–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lu Y, Sareddy GR, Wang J, Zhang Q, Tang F-L, Pratap UP, Tekmal RR, Vadlamudi RK, Brann DW. Neuron-derived estrogen is critical for astrocyte activation and neuroprotection of the ischemic brain. The Journal of Neuroscience. 2020;40:7355–7374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xing C, Wang X, Cheng C, Montaner J, Mandeville E, Leung W, van Leyen K, Lok J, Wang X, Lo EH. Neuronal production of lipocalin-2 as a help-me signal for glial activation. Stroke. 2014;45:2085–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee J-H, Kim J-y, Noh S, Lee H, Lee SY, Mun JY, Park H, Chung W-S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature. 2020 [DOI] [PubMed] [Google Scholar]
- 80.van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR. Hypothermia in animal models of acute ischaemic stroke: A systematic review and meta-analysis. Brain. 2007;130:3063–3074 [DOI] [PubMed] [Google Scholar]
- 81.Dirnagl U, Endres M. Found in translation: Preclinical stroke research predicts human pathophysiology, clinical phenotypes, and therapeutic outcomes. Stroke. 2014;45:1510–1518 [DOI] [PubMed] [Google Scholar]
- 82.Kumar A, Aakriti, Gupta V. A review on animal models of stroke: An update. Brain Res Bull. 2016;122:35–44 [DOI] [PubMed] [Google Scholar]
- 83.Sena ES, Currie GL, McCann SK, Macleod MR, Howells DW. Systematic reviews and meta-analysis of preclinical studies: Why perform them and how to appraise them critically. J Cereb Blood Flow Metab. 2014;34:737–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zivin JA, Fisher M, DeGirolami U, Hemenway CC, Stashak KA. Tissue plasminogen activator reduces neurological damage after cerebral embolism. Science. 1985;230:1289. [DOI] [PubMed] [Google Scholar]
- 85.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral-artery occlusion - evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472. [DOI] [PubMed] [Google Scholar]
- 86.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84. [DOI] [PubMed] [Google Scholar]
- 87.Voelkl B, Vogt L, Sena ES, Wurbel H. Reproducibility of preclinical animal research improves with heterogeneity of study samples. PLoS Biol. 2018;16:e2003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fleishaker JC, Hulst-Pearson LK, Peters GR. Effect of gender and menopausal status on the pharmacokinetics of tirilazad mesylate in healthy subjects. Am J Ther. 1995;2:553–560 [DOI] [PubMed] [Google Scholar]
- 89.Meyer DM, Eastwood JA, Compton MP, Gylys K, Zivin JA, Ovbiagele B. Sex differences in antiplatelet response in ischemic stroke. Womens Health (Lond Engl). 2011;7:465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen CH, Toung TJ, Hurn PD, Koehler RC, Bhardwaj A. Ischemic neuroprotection with selective kappa-opioid receptor agonist is gender specific. Stroke. 2005;36:1557–1561 [DOI] [PubMed] [Google Scholar]
- 91.Chamorro Á, Lo EH, Renú A, van Leyden K, Lyden PD. The future of neuroprotection in stroke. Journal of Neurology, Neurosurgery & Psychiatry. 2021;92:129–135 [DOI] [PubMed] [Google Scholar]
- 92.Landman TR, Schoon Y, Warlé MC, de Leeuw F-E, Thijssen DH. Remote ischemic conditioning as an additional treatment for acute ischemic stroke: The preclinical and clinical evidence. Stroke. 2019;50:1934–1939 [DOI] [PubMed] [Google Scholar]
- 93.Wu C, Zhao W, An H, Wu L, Chen J, Hussain M, Ding Y, Li C, Wei W, Duan J, et al. Safety, feasibility, and potential efficacy of intraarterial selective cooling infusion for stroke patients treated with mechanical thrombectomy. J Cereb Blood Flow Metab. 2018;38:2251–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Luca C, Virtuoso A, Maggio N, Papa M. Neuro-coagulopathy: Blood coagulation factors in central nervous system diseases. Int J Mol Sci. 2017;18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Griffin JH, Fernandez JA, Lyden PD, Zlokovic BV. Activated protein c promotes neuroprotection: Mechanisms and translation to the clinic. Thromb Res. 2016;141Suppl 2:S62–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen B, Friedman B, Whitney MA, Winkle JA, Lei IF, Olson ES, Cheng Q, Pereira B, Zhao L, Tsien RY, et al. Thrombin activity associated with neuronal damage during acute focal ischemia. J Neurosci. 2012;32:7622–7631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rajput PS, Lyden PD, Chen B, Lamb JA, Pereira B, Lamb A, Zhao L, Lei IF, Bai J. Protease activated receptor-1 mediates cytotoxicity during ischemia using in vivo and in vitro models. Neuroscience. 2014;281C:229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lyden P, Pereira B, Chen B, Zhao L, Lamb J, Lei IF, Rajput P. Direct thrombin inhibitor argatroban reduces stroke damage in 2 different models. Stroke. 2014;45:896–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein c caused by noncanonical cleavage at arg46. Blood. 2012;120:5237–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lyden P, Pryor KE, Coffey CS, Cudkowicz M, Conwit R, Jadhav A, Sawyer RN Jr., Claassen J, Adeoye O, Song S, et al. Final results of the rhapsody trial: A multi-center, phase 2 trial using a continual reassessment method to determine the safety and tolerability of 3k3a-apc, a recombinant variant of human activated protein c, in combination with tissue plasminogen activator, mechanical thrombectomy or both in moderate to severe acute ischemic stroke. Ann Neurol. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sinha RK, Wang Y, Zhao Z, Xu X, Burnier L, Gupta N, Fernandez JA, Martin G, Kupriyanov S, Mosnier LO, et al. Par1 biased signaling is required for activated protein c in vivo benefits in sepsis and stroke. Blood. 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Griffin JH, Mosnier LO, Fernandez JA, Zlokovic BV. 2016 scientific sessions sol sherry distinguished lecturer in thrombosis: Thrombotic stroke: Neuroprotective therapy by recombinant-activated protein c. Arterioscler Thromb Vasc Biol. 2016;36:2143–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cui H, Hayashi A, Sun H-S, Belmares MP, Cobey C, Phan T, Schweizer J, Salter MW, Wang YT, Tasker RA, et al. Pdz protein interactions underlying nmda receptor-mediated excitotoxicity and neuroprotection by psd-95 inhibitors. The Journal of Neuroscience. 2007;27:9901–9915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cook DJ, Teves L, Tymianski M. Treatment of stroke with a psd-95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483:213–217 [DOI] [PubMed] [Google Scholar]
- 105.Hill MD, Goyal M, Menon BK, Nogueira RG, McTaggart RA, Demchuk AM, Poppe AY, Buck BH, Field TS, Dowlatshahi D, et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (escape-na1): A multicentre, double-blind, randomised controlled trial. Lancet. 2020;395:878–887 [DOI] [PubMed] [Google Scholar]
- 106.De Luca C, Colangelo AM, Alberghina L, Papa M. Neuro-immune hemostasis: Homeostasis and diseases in the central nervous system. Frontiers in Cellular Neuroscience. 2018;12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Koide T, Wieloch TW, Siesj” BK. Chronic dexamethasone pretreatment aggravates ischemic neuronal necrosis. JCerebBlood Flow and Metab. 1986;6:395. [DOI] [PubMed] [Google Scholar]
- 108.Norris JW, Hachinski VC. High dose steroid treatment in cerebral infarction. Br Med J (Clin Res Ed). 1986;292:21–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mulley G, Wilcox RG, Mitchell JR. Dexamethasone in acute stroke. Br Med J. 1978;2:994–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Use of anti-icam-1 therapy in ischemic stroke: Results of the enlimomab acute stroke trial. Neurology. 2001;57:1428–1434 [DOI] [PubMed] [Google Scholar]
- 111.Saito K, Suyama K, Nishida K, Sei Y, Basile AS. Early increases in tnf-alpha, il-6 and il-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett. 1996;206:149. [DOI] [PubMed] [Google Scholar]
- 112.McCann SK, Cramond F, Macleod MR, Sena ES. Systematic review and meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke: An update. Translational Stroke Research. 2016;7:395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gagalo I, Rusiecka I, Kocic I. Tyrosine kinase inhibitor as a new therapy for ischemic stroke and other neurologic diseases: Is there any hope for a better outcome? Curr Neuropharmacol. 2015;13:836–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang ZG, Chopp M. Exosomes in stroke pathogenesis and therapy. The Journal of Clinical Investigation. 2016;126:1190–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Uccelli A, Benvenuto F, Laroni A, Giunti D. Neuroprotective features of mesenchymal stem cells. Best Pract Res Clin Haematol. 2011;24:59–64 [DOI] [PubMed] [Google Scholar]
- 116.Kassis I, Vaknin-Dembinsky A, Karussis D. Bone marrow mesenchymal stem cells: Agents of immunomodulation and neuroprotection. Curr Stem Cell Res Ther. 2011;6:63–68 [DOI] [PubMed] [Google Scholar]
- 117.Russo E, Nguyen H, Lippert T, Tuazon J, Borlongan CV, Napoli E. Mitochondrial targeting as a novel therapy for stroke. Brain Circ. 2018;4:84–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sun P, Liu DZ, Jickling GC, Sharp FR, Yin K-J. Microrna-based therapeutics in central nervous system injuries. Journal of Cerebral Blood Flow & Metabolism. 2018;38:1125–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]