

Abstract
A protein performs its task by binding a variety of ligands in its local region that is also known as the ligand-binding-site (LBS). Therefore, accurate prediction, characterization, and refinement of LBS can facilitate protein functional annotations and structure-based drug design. In this work, we present CHARMM-GUI LBS Finder & Refiner (https://www.charmm-gui.org/input/lbsfinder) that predicts potential LBS, offers interactive features for local LBS structure analysis, and prepares various molecular dynamics (MD) systems and inputs by setting up distance restraint potentials for LBS structure refinement. LBS Finder & Refiner supports 5 different commonly used simulation programs, such as NAMD, AMBER, GROMACS, GENESIS, and OpenMM, for LBS structure refinement together with hydrogen mass repartitioning. The capability of LBS Finder & Refiner is illustrated through LBS structure predictions and refinements of 48 modeled and 20 apo benchmark target proteins. Overall, successful LBS structure predictions and refinements are seen in our benchmark tests. We hope that LBS Finder & Refiner is useful to predict, characterize, and refine potential LBS on any given protein of interest.
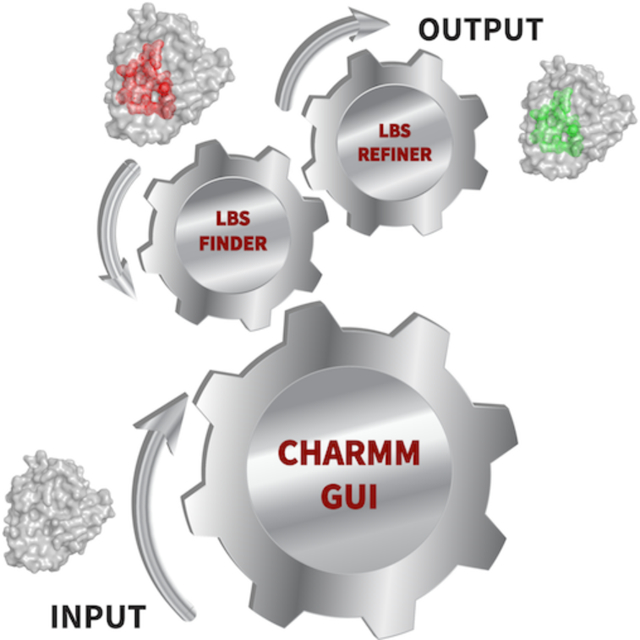
Graphical Abstract
INTRODUCTION
Proteins perform various essential cellular functions. A remarkable feature of proteins is their ability to recognize small molecules with specificity and reversibly bind them. Ligands bind to the local regions of the protein surface, commonly known as the ligand-binding-site (LBS).1 A protein LBS is a key location that should be identified and characterized properly for a successful structure-based drug design campaign.2 In addition, locating LBS on a protein can facilitate functional annotation through their LBS structural similarity comparisons.3–5 This is particularly useful for the characterization of a large number of protein structures solved via high-throughput methods from structural genomics.6
Over the years, many computational tools have been developed to predict protein LBS. They can be classified into three broad categories, Three-dimensional (3D) structure-based, template similarity-based, and machine learning-based.7 Two main 3D structure-based approaches utilize spatial geometry and energy features. A spatial geometry approach finds cavities on the protein surface, while an energy-based method works with probes to estimate interaction energy with the protein. Some of the available tools include Surfnet,8 POCKET,9 LigSite,10 CASTp,11 Fpocket,12 QSiteFinder,13 CurPocket,14 FTSite,15 SiteComp,16 CAVITY,17 GalaxySite,18 and AutoSite.19 In template similarity-based approaches, known protein templates are used to predict LBS on a query protein. Some examples include G-LoSA,1 FINDSITE,4 3DLigandSite,20 S-SITE,21 bSiteFinder,22 and LIBRA.23 Recently, machine learning-based prediction methods have become more popular, ranging from traditional Random Forest algorithms to the more sophisticated deep learning methods. They include LigandDSES,24 COACH-D,25 DEEPSite,26 DeepDrug3D,27 ISMBLab-LIG,28 and P2Rank.29
One of the main goals of finding potential LBS is to help in virtual screening (VS) for drug discovery. However, LBS prediction on a protein surface is commonly performed on apo protein (without bound ligand) structures and computationally predicted structures, that are not suitable for VS, as the LBS region tends to undergo conformational changes upon ligand binding.30, 31 Therefore, additional optimization of the LBS structure is important to prime the binding pocket of apo/homology models for VS. As we and others have shown that VS performances of apo LBS are significantly inferior to holo (ligand bound) LBS structures.32–34 Apo LBS conformations tend to have incorrect sidechain orientations or loops structures that occlude ligand docking.
Implementation of freely available and user-friendly computational tools to predict, analyze, and refine LBS is an important step to facilitate drug design research. CHARMM-GUI (https://www.charmm-gui.org) is a well-established and widely used web-based tool to prepare complex molecular simulation systems and inputs in various enviroments.35–47 In this work, we introduce LBS Finder & Refiner that runs G-LoSA (Graph-based Local Structure Alignment) to predict protein LBS and prepare simulation inputs for molecular dynamics (MD)-based LBS structure refinement.1, 48–51 LBS Finder & Refiner is different from the aforementioned LBS prediction methods in that it provides interactive tools with 3D structure visualization of multiple aligned templates to facilitate LBS structural similarity comparisons. In addition, our efficient workflow allows users to select any LBS template for LBS refinement of their query protein. We have recently shown that this MD-based refinement protocol can successfully refine the local LBS structures of homology models and apo crystal structures to generate holo-like conformations.34, 51 System preparations, including setting up distance restraint potentials and protein solvation processes are integrated seamlessly in our workflow. For the different simulation program community, we also provide input files with the LBS refinement restraints for the commonly used MD software packages, such as NAMD,52 AMBER,53 GROMACS,54 OpenMM,55 and GENESIS.56 In order to guide users for successful practical applications of LBS Finder & Refiner, we have made a video demonstration available in the CHARMM-GUI website (https://www.charmm-gui.org/demo/lbs).
METHODS
Workflow of LBS Finder & Refiner
G-LoSA is our robust computational tool for local structure alignment that aligns protein LBS structures based on their geometry and physicochemical features in a sequence order independent manner.1 A comparative performance evaluation study by Govindaraj et al. showed that G-LoSA was an efficient local structure alignment tool to calculate similarities in LBS structures.57 G-LoSA showed better overall performance than other widely used tools, including APoc58 and SiteEngine.59 As shown in Figure 1, LBS Finder & Refiner starts by reading a protein (apo / holo / model) structure file. PDB Reader & Manipulator is the next step that provides a variety of options to manipulate a structure, including adding missing residues, disulfide bonds, protonation, mutation, phosphorylation and glycosylation sites.60 Note that no ligand should be selected during PDB reading process of LBS Finder & Refiner, as only protein structures should be submitted for G-LoSA LBS search. G-LoSA LBS search entails extensive alignment of all LBS structures in our PDB library (41,443 templates, as of March 2021) on a query structure. We built our LBS library using G-LoSA Toolkit by collecting experimentally solved PDB structures containing at least one ligand and one protein with crystal resolutions < 3.5 Å.48 G-LoSA LBS search calculation is submitted to our computer server. Users have an option to be notified by email when the calculation is completed. In addition, a unique job ID number is created for each job that can be used to retrieve and check the job at any time through Job Retriever. Figure S1 shows the estimated running time for G-LoSA LBS search that depends on the size of the query protein. For a protein with ~200 residues, it takes about 71 minutes for a G-LoSA LBS search job to be completed. In the following sections, important features of LBS Finder & Refiner result sections are discussed in detail.
Figure 1.

Schematic workflow of LBS Finder & Refiner.
Locating and Selecting LBS Templates
As shown in Figure 2, the LBS Finder & Refiner result page provides 3D structure visualization using NGL viewer.61 Next to the protein structure is a table that contains top 10 ranked LBS templates based on their G-LoSA alignment score (GA-score). The GA-score is a normalized similarity scoring function between two LBS local structures, which is calculated based on their aligned distance and physicochemical features. The GA-score ranges from 0 to 1 with 1 being a perfect alignment.48 In addition, global structure and sequence similarity information are also provided using TM-score and sequence identity between each LBS template and the query protein.62, 63 Since all our LBS templates are holo structures with bound ligands, the RCSB ligand IDs are also included in the table.64 The user can find more information regarding the bound ligand, such as molecular weight, chemical formula, and 2D structure, by hovering the mouse on the ligand ID. Upon selecting one of the LBS templates in the table, aligned LBS structure (and its ligand) is displayed on the query structure in grey sticks representations (Figure 2). Noncovalent protein-ligand interactions, including hydrogen bonds, salt bridge, and hydrophobic interactions are also displayed. By clicking the LBS template PDB ID, one can see a list of LBS residues along with their equivalent aligned residues on the query protein below the table. Equivalent aligned residues are identified using the shortest augmenting path algorithm to solve the linear sum assignment problem.65 When the distance between the Cα atoms of aligned residues is less than 5 Å, they are assigned as equivalent aligned residues. From the top 10 ranked templates, multiple possible LBS can be identified on the query protein. For the example case in Figure 2, protein dihydrofolate reductase (PDB ID: 1PDB) binds DHF (rank 1) and a cofactor NDP (rank 10). Such interactive features of LBS Finder & Refiner is expected to guide users in locating potential LBS with a thorough local structure analysis by comparing many aligned LBS structures (and their diverse ligands) on their query protein. All top 10 aligned template structures can be downloaded at the end of MD system preparation step. All aligned templates are located in the downloadable CHARMM-GUI subdirectory called “Top 10”. In addition, the equivalent residues between the query and templates structures are also provided in the same directory in text file formats.
Figure 2.

LBS Finder & Refiner result page allows for interactive LBS structure analysis and template selection. Top 10 aligned templates from G-LoSA search are displayed with their PDB ID, ligand ID, sequence identity, TM-score, and GA-score. Template number 1 & 10 are selected and bound ligands, DHF and NDP, are displayed along with their binding site residues aligned onto a query protein. Equivalent LBS residues between the query (s1) and template (s2) are shown below the table.
Clustering of Binding Site Templates
In order to eliminate redundancy of binding site templates, we provide the top 10 clusters of binding sites in the second tab in the table (Figure 2). In our example case, templates 1, 3, 4, 5, 6, 7, 8 all reside in the same LBS vicinity with different bound ligands. LBS templates are grouped together using single hierarchical clustering.66 The distance between LBS templates are defined by the difference of the distances between all pairs of the equivalent LBS residues from G-LoSA alignment. The cutoff value was empirically determined at 2 Å to cluster similar templates together. The clusters are ranked by the number of templates in each cluster, and the template with the best GA-score within each cluster becomes the cluster’s representative. When multiple diverse LBS templates (and their ligands) aligned to the same local structure, it increases the chance that the predicted LBS is a real binding pocket. In addition, top 10 clusters allow for display of different aligned LBS on a query protein by clustering similar ones together.
Setting up Distance Restraint Potentials
From the user-selected template(s) for LBS structure refinement, we provide two options to derive distance restraint potentials. Method 1 includes both Cα and sidechain center-of-mass (SC-COM) distance restraints and method 2 is Cα-only distance restraints. We have recently shown that adding SC-COM distance restraints improved side-chain orientations of the LBS residues.34 To derive the restraint potentials, we calculate a distance matrix (M) between Cα atoms of the selected LBS template and their equivalent atoms on the query protein using Equation 1. For SC-COM restraint potentials, we calculate the SC-COM of each residue and obtain a distance matrix (M) between SC-COM points to derive a similar harmonic restraint potential using Equation 1.
| (1) |
where k is the force constant, r0,ij is the distance between ith and jth atoms in the LBS template, and rij is the distance between the equivalent atoms in the query protein. The force constant value has been optimized to be 1.5 kcal/(mol·Å2) in our previous work.51 We also add weak positional restraints with a force constant of 0.5 kcal/(mol·Å2) to Cα atoms of each residue that are not part of the LBS in the query protein. This approach has been shown to be effective in keeping the overall protein fold from drifting away from their native structure during MD simulations.51
Generating Systems and Inputs for Various MD Simulation Programs
After selecting the LBS template, we use Solution Builder to prepare the protein system in water solution.45, 67 Users can define the system box size (using TIP3P water model), as well as the types of ions (NaCl, KCl), and ion placing method (distance and Monte Carlo) to solvate their system.68 Following this, periodic boundary conditions are applied to the system. Then, users can generate simulation input files together with the LBS (Cα + SC-COM or Cα-only) refinement distance restraints for 5 different MD simulations programs (NAMD,52 AMBER,53 GROMACS,54 OpenMM,55 and GENESIS).56 In addition, we support hydrogen mass repartitioning (HMR) as the default option to accelerate MD simulations with 4 fs time steps.69, 70
APPLICATIONS
For our benchmark tests, we selected datasets from the directory of useful decoys-enhanced (DUD-E) and Astex.71, 72 For computationally predicted structures benchmark test, we obtained protein sequences from DUD-E that have amino acid residues < 300. We used I-TASSER suite to predict their 3D protein structures with a benchmark option to exclude templates with > 30% sequence identity when building the homology model.73 I-TASSER results are listed in Table S1 with their confidence scores (C-scores) that range from −5 to 2, where a higher score means a better-quality structure. For apo crystal structures, we obtained Astex holo structure dataset that had apo counterpart in the Protein Data Bank (PDB) with 100% sequence identity.64
Aligned LBS Structure Analysis
For our benchmark test, we excluded all homologous proteins in our library that have > 30% sequence identity to the target protein. In total, there are 68 structures in our benchmark test: 48 from DUD-E set (for model structure testing) and 20 from Astex set (for apo structure testing). The list of our target proteins along with their best LBS template, GA-score, TM-score, and sequence identity are shown in Table S2 (DUD-E dataset) and Table S3 (Astex dataset). The average sequence identity between the target and template proteins is 24% (DUD-E) and 25% (Astex). Their TM-scores that measure global structure similarities between the template and target proteins have an average of 0.55 (DUD-E) and 0.50 (Astex). In total 29/68 (43%) of target proteins have TM-scores < 0.5, indicating that their global folds are not similar.74 We and others have shown previously that many nonhomologous proteins with dissimilar global folds can have similar local LBS structures.1, 49, 75
LBS Finder & Refiner does a good job in predicting the location of LBS on the query protein from our benchmark dataset. For 48 DUD-E modeled structures, the predicted templates contain on average 70% of the LBS residues from the crystal structures (Table S2). Out of 48 structures, 44 of them have greater than 50% coverage. For 20 Astex apo structures, the predicted templates cover on average 79% of the LBS residues (Table S3). And all of them have greater than 50% coverage. Additional data on LBS prediction by G-LoSA can be found in our previous study.42
Significant LBS Structure Refinements Are Achieved in Both Modeled and Apo Benchmark Targets
Our benchmark structures are categorized into three groups based on their LBS all-heavy-atom RMSD relative to their holo crystal structures: group 1 (<2 Å), group 2 (2–3 Å), and group 3 (>3 Å). In benchmark 1 (modeled structures from 48 DUD-E targets), we aim to do structure refinement of their LBS toward the quality of experimentally solved structures. Whereas, in benchmark 2 (apo crystal structures from 20 Astex targets), we aim to refine their LBS structures by generating holo structure conformations. For this, we used the default options for the system setup in LBS Finder & Refiner and the LBS refinement simulations were performed using OpenMM with 3 replicas for each target. The average structures over 3 replicas were compared with the holo crystal structures to assess the LBS refinement quality.
For 48 DUD-E modeled structures (Figure 3), an average improvement is 0.96 Å (initial 2.94 Å, refined 1.98 Å) using method 1 (Cα and SC-COM restraints). A slightly smaller average improvement of 0.64 Å is obtained using method 2 (Cα-only restraints) (Table S4). There are 42/48 (88%) structures that undergo improvements with lower LBS all-heavy-atom RMSD compared to their holo crystal structures after refinement. In group 1, the average improvement is 29% (0.52 Å). Larger improvement is seen in group 2 with 38% (0.96 Å). Group 3 shows an average improvement of 30% (1.19 Å). There are 6 structures that show no improvement after refinement, 1 from group 1, 2 from group 2 and 3 from group 3. These unsuccessful cases result from unsuitable LBS templates from G-LoSA search. For example, number 40, LFA-1 (leukocyte function-associated antigen-1) protein has a closed conformation in its modeled structure, but the best G-LoSA LBS template is a nucleotide-binding UspA (universal stress protein) that does not have the correct holo conformation of LFA-1 protein (Table S2).
Figure 3.

Ligand-binding-site all-heavy-atom RMSD values of the initial model structures and MD-refined structures against their crystal structures for 48 targets in the DUD-E dataset. The average improvement is 0.96 Å. The structures are categorized into three groups based on their initial binding site RMSD relative to their crystal structure: group 1 (<2 Å, red), group 2 (2–3 Å, green), and group 3 (>3 Å, blue).
Using method 1, apo structures refinement to generate holo conformations from 20 Astex crystal structures undergo an average improvement of 0.30 Å (initial 1.07 Å, refined 0.77 Å) (Figure S2). Using method 2 there is only an average improvement of 0.09 Å (Table S5). Clearly, more significant improvements are seen in modeled structures refinement (DUD-E) than apo crystal structures (Astex), simply because LBS of modeled structures are poorly predicted and apo crystal structures tend to have only slight (sidechain) differences between their apo-holo pairs. For example, 90% (18/20) structures in the apo dataset fall in group 1 with an average LBS RMSD of 0.85 Å relative to their holo counterparts. There are 14/20 (70%) cases where the refined structures show better LBS RMSD than the initial apo structures. The other 6 structures that do not improve after refinement are all coming from group 1, and their apo and refined LBS structures are already very similar to their holo crystal structure (i.e., initial and refined RMSD are 0.47 Å and 0.65 Å, respectively).
In total, there are 82% (56/68) success rate in LBS structure refinement using our benchmark dataset. Additionally, we show that successful refinements originate from the distance restraints because the same simulations without distance restraints in the control group do not show improvement (Table S4, S5)
Consistent Refinement Results Are Seen Across Different Simulation Programs
We tested and validated the performance of 5 simulation input files from LBS Finder & Refiner, including NAMD,52 AMBER,53 GROMACS,54 OpenMM,55 and GENESIS.56 For each program, we used the default CHARMM-GUI settings (see SI). Our results show that their LBS all-heavy-atom RMSD improvements are consistent across all programs (Table 1). In addition to a clear validation of our implementation of LBS refinement distance restraints in different programs, this result is also significant in that automated setup of distance restraints in MD simulations is not trivial and requires many different steps, including defining atom groups, force constant, and types of restraints. Furthermore, different functionalities are used to setup restraints in different MD simulation programs. For example, in OpenMM, the custom bond force is called to setup Cα restraints and the custom centroid bond force is called to setup SC-COM restraints.55 Whereas, in NAMD, colvars (collective variables) are used to setup distance restraints.52 Handling all the relevant information to properly prepare simulation systems with specific restraints can be challenging and time consuming. Therefore, an automated workflow that derives restraint potentials for any given templates can be very useful and time-saving for both experts and beginners in the field.
Table 1.
Results of 5 targets from DUD-E dataset comparing all-heavy-atom RMSD of the ligand-binding-site (LBS) residues across 5 different simulation programs.a
| PDB | Program | LBS all-heavy-atom RMSD | |||
|---|---|---|---|---|---|
| Initial | Method 1b | Method 2c | Controld | ||
| 2nnq | NAMD | 1.99 | 1.10 | 1.48 | 2.10 |
| AMBER | 1.99 | 1.18 | 1.46 | 2.04 | |
| GROMACS | 1.99 | 1.04 | 1.48 | 2.55 | |
| OpenMM | 1.99 | 1.03 | 1.32 | 2.17 | |
| GENESIS | 1.99 | 1.00 | 1.22 | 2.01 | |
| 1h00 | NAMD | 2.13 | 1.06 | 1.28 | 2.07 |
| AMBER | 2.13 | 1.05 | 1.15 | 2.91 | |
| GROMACS | 2.13 | 1.05 | 1.19 | 2.25 | |
| OpenMM | 2.13 | 1.12 | 1.44 | 2.04 | |
| GENESIS | 2.13 | 1.15 | 1.37 | 2.61 | |
| 2fsz | NAMD | 2.15 | 1.32 | 1.68 | 2.37 |
| AMBER | 2.15 | 1.44 | 1.77 | 2.49 | |
| GROMACS | 2.15 | 1.32 | 1.52 | 1.97 | |
| OpenMM | 2.15 | 1.35 | 1.65 | 2.04 | |
| GENESIS | 2.15 | 1.45 | 1.76 | 2.10 | |
| 3m2w | NAMD | 2.35 | 1.28 | 1.75 | 2.36 |
| AMBER | 2.35 | 1.34 | 1.82 | 2.48 | |
| GROMACS | 2.35 | 1.28 | 1.68 | 1.96 | |
| OpenMM | 2.35 | 1.31 | 1.77 | 2.18 | |
| GENESIS | 2.35 | 1.24 | 1.56 | 2.35 | |
| 3bwm | NAMD | 3.43 | 1.75 | 2.60 | 3.81 |
| AMBER | 3.43 | 1.82 | 2.54 | 3.67 | |
| GROMACS | 3.43 | 1.65 | 2.22 | 4.21 | |
| OpenMM | 3.43 | 1.68 | 2.55 | 3.74 | |
| GENESIS | 3.43 | 1.77 | 2.29 | 3.44 | |
Improved cases with lower RMSD are highlighted in bold.
Method 1 using both Cα and SC-COM distance restraints.
Method 2 using Cα-only distance restraints.
Control simulations without distance restraints.
CONCLUSIONS
An accurate identification, characterization, and refinement of a protein LBS can significantly facilitate structure-based drug design.2 In this work, we introduce LBS Finder & Refiner, a new functional module in CHARMM-GUI to help users to predict, characterize, and refine LBS on a protein structure. We expect that our module can properly prepare ligand-binding-site on homology models and apo proteins for virtual screening whenever their holo structures are not available. Homology models in particular are becoming more attractive due to recent advances in computationally predicted protein structures with good structural qualities as shown in the latest CASP14 (critical assessment of structure prediction) contest.76, 77 For structure refinement with distance restraints from LBS templates, MD simulation inputs can be generated for 5 different programs, including NAMD,52 AMBER,53 GROMACS,54 OpenMM55 and GENESIS.56 We have shown previously that structure refinement of protein LBS could improve virtual screening results and ligand binding modes.34, 51 Therefore, we expect that refined structures from this module will be useful for virtual screening.
A distinctive feature of LBS Finder & Refiner is its rich and interactive result page that includes multiple aligned templates on a query protein and their detail structural and ligand information. We expect that this feature can help users characterize potential LBS on their proteins. Our benchmark tests on two different datasets, include modeled DUD-E targets and apo Astex targets, achieve significant LBS structure refinements. In addition, we show that their results are consistent across all 5 different simulation programs that our module generates.
For structure refinement, our application results have the limitation of running refinement based on a single holo structure template for each target. As a result, a refined structure does not account for all possible induced fit effects that can occur from binding of different ligands. To improve this limitation, our result page offers top 10 templates and top 10 clusters of templates, from which users can judiciously choose one or multiple appropriate best template(s) (possibly based on the types of bound ligands) to be used for LBS refinement.
Since LBS Finder & Refiner works as an automated system to prepare binding pockets for docking, going forward we plan to build a new docking workflow that can accommodate induced-fit ligand docking. We and others have observed that cross-docking tends to have very low success rate due to the induced-fit problem, where changes are observed in the protein structure during ligand binding.78, 79 Using LBS Finder & Refiner, we will prepare an ensemble of binding pocket conformations using different LBS templates. Successively, rigid receptor docking will be performed on the various LBS conformations and a high-throughput short MD simulations will be used to evaluate the docking results.78 We expect to provide a combined scoring function that can properly solve the induced-fit problem in docking.
Supplementary Material
ACKNOWLEDGEMENT
This work has been supported by NIH GM126140 and GM138472.
Footnotes
Supporting Information
The Supporting Information is available free of charge.
MD simulation methods; Table S1, List of 48 computationally predicted structures with their C-scores; Table S2, DUD-E targets showing their G-LoSA template, GA-score, sequence identity, and TM-score; Table S3, Astex targets showing their G-LoSA template, GA-score, sequence identity, and TM-score; Table S4, LBS refinement results from 48 modeled DUD-E dataset; Table S5, LBS refinement results from 20 apo crystal structure from Astex dataset; Figure S1, Estimated running time for G-LoSA search; Figure S2, LBS RMSD values of the initial apo structures and MD-refined against their crystal structures from 20 Astex dataset.
Data and software availability
All the protein structures and molecular dynamics data are available upon request. LBS Finder & Refiner module can be accessed through the following link: https://www.charmm-gui.org/input/lbsfinder.
CONFLICT OF INTEREST
No conflict of interest.
References:
- 1.Lee HS; Im W, G-LoSA: An Efficient Computational Tool for Local Structure-Centric Biological Studies and Drug Design. Protein Sci 2016, 25, 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lionta E; Spyrou G; Vassilatis DK; Cournia Z, Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr Top Med Chem 2014, 14, 1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell SJ; Gold ND; Jackson RM; Westhead DR, Ligand Binding: Functional Site Location, Similarity and Docking. Curr Opin Struct Biol 2003, 13, 389–395. [DOI] [PubMed] [Google Scholar]
- 4.Brylinski M; Skolnick J, A Threading-Based Method (FINDSITE) for Ligand-Binding Site Prediction and Functional Annotation. Proc Natl Acad Sci U S A 2008, 105, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mills CL; Garg R; Lee JS; Tian L; Suciu A; Cooperman GD; Beuning PJ; Ondrechen MJ, Functional Classification of Protein Structures by Local Structure Matching in Graph Representation. Protein Sci 2018, 27, 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grabowski M; Niedzialkowska E; Zimmerman MD; Minor W, The Impact of Structural Genomics: The First Quindecennial. J Struct Funct Genomics 2016, 17, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao J; Cao Y; Zhang L, Exploring the Computational Methods for Protein-Ligand Binding Site Prediction. Comput Struct Biotechnol J 2020, 18, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laskowski RA, SURFNET: A Program for Visualizing Molecular Surfaces, Cavities, and Intermolecular Interactions. J Mol Graph 1995, 13, 323–330. [DOI] [PubMed] [Google Scholar]
- 9.Levitt DG; Banaszak LJ, POCKET: A Computer Graphics Method for Identifying and Displaying Protein Cavities and Their Surrounding Amino Acids. J Mol Graph 1992, 10, 229–234. [DOI] [PubMed] [Google Scholar]
- 10.Hendlich M; Rippmann F; Barnickel G, LIGSITE: Automatic and Efficient Detection of Potential Small Molecule-Binding Sites in Proteins. J Mol Graph Model 1997, 15, 359–363. [DOI] [PubMed] [Google Scholar]
- 11.Dundas J; Ouyang Z; Tseng J; Binkowski A; Turpaz Y; Liang J, CASTp: Computed Atlas of Surface Topography of Proteins with Structural and Topographical Mapping of Functionally Annotated Residues. Nucleic Acids Res 2006, 34, W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Le Guilloux V; Schmidtke P; Tuffery P, Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinformatics 2009, 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laurie AT; Jackson RM, Q-SiteFinder: An Energy-Based Method for the Prediction of Protein-Ligand Binding Sites. Bioinformatics 2005, 21, 1908–1916. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y; Grimm M; Dai WT; Hou MC; Xiao ZX; Cao Y, CB-Dock: A Web Server for Cavity Detection-Guided Protein-Ligand Blind Docking. Acta Pharmacol Sin 2020, 41, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ngan CH; Hall DR; Zerbe B; Grove LE; Kozakov D; Vajda S, FTSite: High Accuracy Detection of Ligand Binding Sites on Unbound Protein Structures. Bioinformatics 2012, 28, 286–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Y; Yoo S; Sanchez R, SiteComp: A Server for Ligand Binding Site Analysis in Protein Structures. Bioinformatics 2012, 28, 1172–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan Y; Pei J; Lai L, Binding Site Detection and Druggability Prediction of Protein Targets for Structure-Based Drug Design. Curr Pharm Des 2013, 19, 2326–2333. [DOI] [PubMed] [Google Scholar]
- 18.Heo L; Shin WH; Lee MS; Seok C, GalaxySite: Ligand-Binding-Site Prediction by using Molecular Docking. Nucleic Acids Res 2014, 42, W210–W214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ravindranath PA; Sanner MF, AutoSite: An Automated Approach for Pseudo-Ligands Prediction-from Ligand-Binding Sites Identification to Predicting Key Ligand Atoms. Bioinformatics 2016, 32, 3142–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wass MN; Kelley LA; Sternberg MJ, 3DLigandSite: Predicting Ligand-Binding Sites Using Similar Structures. Nucleic Acids Res 2010, 38, W469–W473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J; Roy A; Zhang Y, Protein-Ligand Binding Site Recognition Using Complementary Binding-Specific Substructure Comparison and Sequence Profile Alignment. Bioinformatics 2013, 29, 2588–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao J; Zhang Q; Liu M; Zhu L; Wu D; Cao Z; Zhu R, bSiteFinder, an Improved Protein-Binding Sites Prediction Server Based on Structural Alignment: More Accurate and Less Time-Consuming. J Cheminform 2016, 8, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toti D; Viet Hung L; Tortosa V; Brandi V; Polticelli F, LIBRA-WA: A Web Application for Ligand Binding Site Detection and Protein Function Recognition. Bioinformatics 2018, 34, 878–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen P; Hu S; Zhang J; Gao X; Li J; Xia J; Wang B, A Sequence-Based Dynamic Ensemble Learning System for Protein Ligand-Binding Site Prediction. IEEE/ACM Trans Comput Biol Bioinform 2016, 13, 901–912. [DOI] [PubMed] [Google Scholar]
- 25.Wu Q; Peng Z; Zhang Y; Yang J, COACH-D: Improved Protein-Ligand Binding Sites Prediction with Refined Ligand-Binding Poses Through Molecular Docking. Nucleic Acids Res 2018, 46, W438–W442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jimenez J; Doerr S; Martinez-Rosell G; Rose AS; De Fabritiis G, DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 2017, 33, 3036–3042. [DOI] [PubMed] [Google Scholar]
- 27.Pu L; Govindaraj RG; Lemoine JM; Wu HC; Brylinski M, DeepDrug3D: Classification of Ligand-Binding Pockets in Proteins with a Convolutional Neural Network. PLoS Comput Biol 2019, 15, e1006718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jian JW; Elumalai P; Pitti T; Wu CY; Tsai KC; Chang JY; Peng HP; Yang AS, Predicting Ligand Binding Sites on Protein Surfaces by 3-Dimensional Probability Density Distributions of Interacting Atoms. PLoS One 2016, 11, e0160315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krivak R; Hoksza D, P2Rank: Machine Learning Based Tool for Rapid and Accurate Prediction of Ligand Binding Sites from Protein Structure. J Cheminform 2018, 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaudreault F; Chartier M; Najmanovich R, Side-Chain Rotamer Changes Upon Ligand Binding: Common, Crucial, Correlate with Entropy and Rearrange Hydrogen Bonding. Bioinformatics 2012, 28, i423–i430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark JJ; Benson ML; Smith RD; Carlson HA, Inherent Versus Induced Protein Flexibility: Comparisons Within and Between Apo and Holo Structures. PLoS Comput Biol 2019, 15, e1006705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGovern SL; Shoichet BK, Information Decay in Molecular Docking Screens Against Holo, Apo, and Modeled Conformations of Enzymes. J Med Chem 2003, 46, 2895–2907. [DOI] [PubMed] [Google Scholar]
- 33.Lee HS; Lee CS; Kim JS; Kim DH; Choe H, Improving Virtual Screening Performance Against Conformational Variations of Receptors by Shape Matching with Ligand Binding Pocket. J Chem Inf Model 2009, 49, 2419–2428. [DOI] [PubMed] [Google Scholar]
- 34.Guterres H; Park SJ; Jiang W; Im W, Ligand-Binding-Site Refinement to Generate Reliable Holo Protein Structure Conformations from Apo Structures. J Chem Inf Model 2021, 61, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jo S; Kim T; Iyer VG; Im W, CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J Comput Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 36.Park SJ; Lee J; Qi Y; Kern NR; Lee HS; Jo S; Joung I; Joo K; Lee J; Im W, CHARMM-GUI Glycan Modeler for Modeling and Simulation of Carbohydrates and Glycoconjugates. Glycobiology 2019, 29, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qi Y; Lee J; Klauda JB; Im W, CHARMM-GUI Nanodisc Builder for Modeling and Simulation of Various Nanodisc Systems. J Comput Chem 2019, 40, 893–899. [DOI] [PubMed] [Google Scholar]
- 38.Lee J; Patel DS; Stahle J; Park SJ; Kern NR; Kim S; Lee J; Cheng X; Valvano MA; Holst O; Knirel YA; Qi Y; Jo S; Klauda JB; Widmalm G; Im W, CHARMM-GUI Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J Chem Theory Comput 2019, 15, 775–786. [DOI] [PubMed] [Google Scholar]
- 39.Kim S; Lee J; Jo S; Brooks CL 3rd; Lee HS; Im W, CHARMM-GUI Ligand Reader and Modeler for CHARMM Force Field Generation of Small Molecules. J Comput Chem 2017, 38, 1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S; Oshima H; Zhang H; Kern NR; Re S; Lee J; Roux B; Sugita Y; Jiang W; Im W, CHARMM-GUI Free Energy Calculator for Absolute and Relative Ligand Solvation and Binding Free Energy Simulations. J Chem Theory Comput 2020, 16, 7207–7218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu EL; Cheng X; Jo S; Rui H; Song KC; Davila-Contreras EM; Qi Y; Lee J; Monje-Galvan V; Venable RM; Klauda JB; Im W, CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J Comput Chem 2014, 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng X; Jo S; Lee HS; Klauda JB; Im W, CHARMM-GUI Micelle Builder for Pure/Mixed Micelle and Protein/Micelle Complex Systems. J Chem Inf Model 2013, 53, 2171–2180. [DOI] [PubMed] [Google Scholar]
- 43.Jo S; Lim JB; Klauda JB; Im W, CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys J 2009, 97, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park SJ; Lee J; Patel DS; Ma H; Lee HS; Jo S; Im W, Glycan Reader is Improved to Recognize Most Sugar Types and Chemical Modifications in the Protein Data Bank. Bioinformatics 2017, 33, 3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y; Jo S; Pande VS; Case DA; Brooks CL 3rd; MacKerell AD Jr.; Klauda JB; Im W, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput 2016, 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J; Hitzenberger M; Rieger M; Kern NR; Zacharias M; Im W, CHARMM-GUI Supports the Amber Force Fields. J Chem Phys 2020, 153, 035103. [DOI] [PubMed] [Google Scholar]
- 47.Jo S; Jiang W; Lee HS; Roux B; Im W, CHARMM-GUI Ligand Binder for Absolute Binding Free Energy Calculations and Its Application. J Chem Inf Model 2013, 53, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee HS; Im W, G-LoSA for Prediction of Protein-Ligand Binding Sites and Structures. Methods Mol Biol 2017, 1611, 97–108. [DOI] [PubMed] [Google Scholar]
- 49.Lee HS; Im W, Ligand Binding Site Detection by Local Structure Alignment and Its Performance Complementarity. J Chem Inf Model 2013, 53, 2462–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HS; Im W, Identification of Ligand Templates using Local Structure Alignment for Structure-Based Drug Design. J Chem Inf Model 2012, 52, 2784–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guterres H; Lee HS; Im W, Ligand-Binding-Site Structure Refinement Using Molecular Dynamics with Restraints Derived from Predicted Binding Site Templates. J Chem Theory Comput 2019, 15, 6524–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phillips JC; Hardy DJ; Maia JDC; Stone JE; Ribeiro JV; Bernardi RC; Buch R; Fiorin G; Henin J; Jiang W; McGreevy R; Melo MCR; Radak BK; Skeel RD; Singharoy A; Wang Y; Roux B; Aksimentiev A; Luthey-Schulten Z; Kale LV; Schulten K; Chipot C; Tajkhorshid E, Scalable Molecular Dynamics on CPU and GPU architectures with NAMD. J Chem Phys 2020, 153, 044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr.; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber Biomolecular Simulation Programs. J Comput Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hess B; Kutzner C; van der Spoel D; Lindahl E, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- 55.Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS, OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput Biol 2017, 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jung J; Mori T; Kobayashi C; Matsunaga Y; Yoda T; Feig M; Sugita Y, GENESIS: A Hybrid-Parallel and Multi-Scale Molecular Dynamics Simulator with Enhanced Sampling Algorithms for Biomolecular and Cellular Simulations. Wiley Interdiscip Rev Comput Mol Sci 2015, 5, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Govindaraj RG; Brylinski M, Comparative Assessment of Strategies to Identify Similar Ligand-Binding Pockets in Proteins. BMC Bioinformatics 2018, 19, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao M; Skolnick J, APoc: Large-Scale Identification of Similar Protein Pockets. Bioinformatics 2013, 29, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shulman-Peleg A; Nussinov R; Wolfson HJ, SiteEngines: Recognition and Comparison of Binding Sites and Protein-Protein Interfaces. Nucleic Acids Res 2005, 33, W337–W341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jo S; Cheng X; Islam SM; Huang L; Rui H; Zhu A; Lee HS; Qi Y; Han W; Vanommeslaeghe K; MacKerell AD Jr.; Roux B; Im W, CHARMM-GUI PDB Manipulator for Advanced Modeling and Simulations of Proteins Containing Nonstandard Residues. Adv Protein Chem Struct Biol 2014, 96, 235–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rose AS; Hildebrand PW, NGL Viewer: A Web Application for Molecular Visualization. Nucleic Acids Res 2015, 43, W576–W579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y; Skolnick J, Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins 2004, 57, 702–710. [DOI] [PubMed] [Google Scholar]
- 63.Johnson M; Zaretskaya I; Raytselis Y; Merezhuk Y; McGinnis S; Madden TL, NCBI BLAST: A Better Web Interface. Nucleic Acids Res 2008, 36, W5–W9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE, The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Derigs U, The Shortest Augmenting Path Method for Solving Assignment Problems - Motivation and Computational Experience. Annals of Operations Research 1985, 4, 57–102. [Google Scholar]
- 66.Jain AKDRC, Algorithms for Clustering Data. Prentice Hall: Englewood Cliffs, 1998. [Google Scholar]
- 67.Brooks BR; Brooks CL 3rd; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M, CHARMM: The Biomolecular Simulation Program. J Comput Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983, 79, 926–935. [Google Scholar]
- 69.Gao Y; Lee J; Smith IPS; Lee H; Kim S; Qi Y; Klauda JB; Widmalm G; Khalid S; Im W, CHARMM-GUI Supports Hydrogen Mass Repartitioning and Different Protonation States of Phosphates in Lipopolysaccharides. J Chem Inf Model 2021, 61, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hopkins CW; Le Grand S; Walker RC; Roitberg AE, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J Chem Theory Comput 2015, 11, 1864–1874. [DOI] [PubMed] [Google Scholar]
- 71.Mysinger MM; Carchia M; Irwin JJ; Shoichet BK, Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J Med Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hartshorn MJ; Verdonk ML; Chessari G; Brewerton SC; Mooij WT; Mortenson PN; Murray CW, Diverse, High-Quality Test Set for the Validation of Protein-Ligand Docking Performance. J Med Chem 2007, 50, 726–741. [DOI] [PubMed] [Google Scholar]
- 73.Roy A; Kucukural A; Zhang Y, I-TASSER: A Unified Platform for Automated Protein Structure and Function Prediction. Nat Protoc 2010, 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu J; Zhang Y, How Significant is a Protein Structure Similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gherardini PF; Wass MN; Helmer-Citterich M; Sternberg MJ, Convergent Evolution of Enzyme Active Sites is Not a Rare Phenomenon. J Mol Biol 2007, 372, 817–845. [DOI] [PubMed] [Google Scholar]
- 76.Kryshtafovych A; Schwede T; Topf M; Fidelis K; Moult J, Critical Assessment of Methods of Protein Structure Prediction (CASP)-Round XIII. Proteins 2019, 87, 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heo L; Arbour CF; Janson G; Feig M, Improved Sampling Strategies for Protein Model Refinement Based on Molecular Dynamics Simulation. J Chem Theory Comput 2021, 17, 1931–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guterres H; Im W, Improving Protein-Ligand Docking Results with High-Throughput Molecular Dynamics Simulations. J Chem Inf Model 2020, 60, 2189–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clark AJ; Tiwary P; Borrelli K; Feng S; Miller EB; Abel R; Friesner RA; Berne BJ, Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. J Chem Theory Comput 2016, 12, 2990–2998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.