

Abstract
Background:
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder, and the most common type of dementia. A growing body of evidence has implicated neuroinflammation as an essential player in the etiology of AD. Inflammasomes are intracellular multiprotein complexes and essential components of innate immunity in response to pathogen- and danger-associated molecular patterns. Among the known inflammasomes, the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome plays a critical role in the pathogenesis of AD.
Objective:
We recently developed a novel class of small molecule inhibitors that selectively target the NLRP3 inflammasome. One of the lead compounds, JC124, has shown therapeutic efficacy in a transgenic animal model of AD. In this study we will test the preventative efficacy of JC124 in another strain of transgenic AD mice.
Methods:
In this study, 5-month-old female APP/PS1 and matched wild type mice were treated orally with JC124 for 3 months. After completion of treatment, cognitive functions and AD pathologies, as well as protein expression levels of synaptic proteins, were assessed.
Results:
We found that inhibition of NLRP3 inflammasome with JC124 significantly decreased multiple AD pathologies in APP/PS1 mice, including amyloid-β (Aβ) load, neuroinflammation, and neuronal cell cycle re-entry, accompanied by preserved synaptic plasticity with higher expression of pre- and post-synaptic proteins, increased hippocampal neurogenesis, and improved cognitive functions.
Conclusion:
Our study demonstrates the importance of the NLRP3 inflammasome in AD pathological development, and pharmacological inhibition of NLRP3 inflammasome with small molecule inhibitors represents a potential therapy for AD.
Keywords: Alzheimer’s disease, cell cycle re-entry, cognitive function, neuroinflammation, NLRP3 inflammasome
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common type of dementia. Although significant advances have been made in understanding the pathophysiology of AD, there is still no effective treatment to cure or slow the progression of this disease. A growing body of evidence has implicated neuroinflammation as an essential player in the etiology of AD [1, 2]. Epidemiological studies have provided evidence that the incidence of systemic infection correlates with the severity of dementia [3, 4]. Preclinical studies in AD animal models also suggest that non-steroidal anti-inflammatory drug (NSAID) treatment could be beneficial [5–7]. Furthermore, genome-wide association studies have identified risk genes associated with innate immunity for late-onset AD [8, 9]. Studies also point to a causative role of inflammation upstream of amyloid-β (Aβ) and tau pathology [10, 11]. To support this notion, immunosuppression suppressed tau pathology and increased lifespan in a P301S tauopathy mouse model [12].
Inflammasomes are large multimeric protein complexes that detect pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) to initiate innate immune responses [13]. Structurally, the formation of inflammasomes requires a cytosolic pattern-recognition receptor as the sensor, an adaptor protein (apoptosis-associated speck-like protein, or ASC), and the cysteine protease, caspase-1, as an effector [13, 14]. Upon sensing a wide range of stimuli, self-assembly and activation lead to the cleavage of pro-caspase-1 and production of the pro-inflammatory cytokines, IL-1β, and IL-18 [15, 16]. The nucleotide-binding domain leucine-rich repeats family protein 3 (NLRP3) inflammasome, the best characterized of all known inflammasomes, can respond to a diverse set of stimuli including reactive oxygen species, mitochondrial damage, ATP and ion flux from injured cells following tissue damage [17, 18]. Dysregulation of the NLRP3 inflammasome pathway plays important pathophysiological roles in AD. In both AD animal models and AD patients, the protein expression levels of NLRP3, ASC, caspase-1, and IL-1β are upregulated [8, 19, 20]. Aβ can trigger the NLRP3 inflammasome to recruit and activate microglia [19, 21]. Notably, APP/PS1 mice with NLRP3 or Caspase-1 gene knock out have reduced Aβ plaques and improved spatial memory functions [21]. Studies employing 5xFAD mice carrying the ASC+/− genotype also supported this notion [22]. Conversely, NLRP3 over-activation could promote differentiation of T cells into pro-inflammatory Th1 and Th17 effector phenotypes contributing to sustained inflammation [23]. Collectively, these observations strongly implicate the importance of NLPR3 inflammasome in mediating inflammatory process and pathogenesis of AD, and suggest it as a viable target to develop effective treatment for AD.
In this study, we explored the effects of a novel NLRP3 inflammasome inhibitor as a potential AD therapy. Recently, our group developed a class of sulfonamide-based NLRP3 inhibitors, and one of the lead compounds, JC124, exhibited selective inhibition of NLRP3 inflammasome formation and the activation of caspase-1, and reduced the production of IL-1β both in vitro and in vivo [24]. In a mouse acute myocardial infarction model, short term JC124 treatment blocked inflammasome formation and significantly reduced myocardial infarct size while exhibiting no hypoglycemic effects, suggesting its target engagement and confirming in vivo activities [25, 26]. In a rat traumatic brain injury model, JC124 treatment at the acute stage following injury significantly inhibited injury-induced NLRP3 activation, reduced the expression of its downstream effector proteins and provided neuroprotective effect [27]. Furthermore, a therapeutic regime in which JC124 treatment started at the age when AD pathology was already apparent, yielded reduced inflammatory responses and Aβ load in TgCRND8 mice [28], and improved cognitive function in APP/PS1 mice [25]. Here, we report the preventive efficacy of JC124 in AD with the treatment starting before the onset of AD pathology using APP/PS1 mice.
MATERIALS AND METHODS
Animals
Female APP/PS1 mice (B6C3-Tg (APPSWE, PSEN1dE9)85Dbo/Mmjax) and matching wild type female littermates were purchased from the Jackson Laboratory (Bar Harbor, ME). According to the vendor information, APP/PS1 mice develop Aβ plaques by 6 to 7 months old, and female mice have 5–10-fold more pronounced Aβ deposits than males. A total of 28 APP/PS1 mice and 16 wild type littermate controls were included in this study. Animals were housed in the Virginia Commonwealth University (VCU) animal facility with a 12-h light/dark cycle, and water and food were provided ad libitum. All procedures were approved by the VCU Institutional Animal Care and Use Committee.
Treatment procedures
Treatment commenced when the animals were 5 months old, before the onset of AD pathology, to test the preventative efficacy of JC124 for AD. Animals were randomized into the following groups: Wild type (WT): vehicle group (n = 5), JC124–50 mg/kg (n = 5), JC124–100 mg/kg group (n = 6). APP/PS1 transgenic mice (Tg): vehicle group (n = 8), JC124–50 mg/kg (n = 10), JC124–100 mg/kg group (n = 10). The choice of dosing was based on our previous studies showing the efficacy of JC124 in inhibition of NLRP3 inflammasome activation in the TBI model and the TgCRND8 mice [27, 28]. JC124 was dissolved in 2% DMSO in corn oil at the concentration of 30 mg/mL, and was administrated through oral gavage with single daily treatments from Monday through Friday, followed by two days without treatment. Animals received JC124 or vehicle treatment continually by this protocol for 3 months. Control animals were treated with an equal volume of vehicle solution (2% DMSO in corn oil). To assess if JC124 treatment had effect on neurogenesis in AD mice, APP/PS1 mice received BrdU (50 mg/kg) or vehicle administration i.p. beginning at the 5th week of JC124 treatment; a single BrdU dose was then administered daily for the next 5 days. After completing JC124 treatment, a battery of cognitive functional assessments was performed.
Novel object recognition (NOR) test
Animals were first assessed by NOR, a “pure” working-memory, non-spatial test completely free of reference memory components. NOR tests were performed after completion of drug treatment. Briefly, each animal was first acquainted with the testing chamber (22.5 × 22.5 cm) for 2 × 5 min intervals. 24 h later, the animal completed a 5 min sample phase in the chamber to explore two identical objects. At the testing phase to assess short-term memory after a 2 h delay, the animal was returned to the chamber with one of the familiar objects replaced with a novel object. To assess long-term memory, the animals were tested again after a 24 h delay with a different novel object. The time the animals spent exploring each object during sample and testing phases was video recorded and scored by Any-Maze software (Stoelting). Results of the testing phase were expressed as a discrimination index (DI = time spent with novel object/[time spent with familiar object + time spent with novel object] × 100). A DI score greater than 50 indicates preference for the novel object and an intact memory capacity, whereas an DI score of less than 50 suggests poor object recognition and a memory deficit.
Fear conditioning (FC) test
This test was performed after the NOR test. The fear conditioning apparatus consists of a box (30.5 × 24.1 × 21 cm, Coulbourn Instruments, Lehigh Valley, PA) with an electric grid floor. For the cued and contextual fear-conditioning task, each animal was first exposed to the box for 10 min for habituation. The next day, for training the animal was placed in the chamber for 120 s, then a 30 s tone (85 dB, 3 kHz) was sounded and during the last 1 s of the tone, a 1.0 mA footshock was delivered through the grid floor. The box was cleaned with 70% ethanol between animals. At 24 h after training, contextual fear memory was assessed by returning the mouse to the original training box for 5 min, after which freezing behavior was measured. At 1 h after contextual fear testing, cued fear memory was assessed by placing the animals into a novel context (for general environmental changes a wall was covered with a dark sheet, the ambient light was altered, and the box was cleaned with vanilla as a novel odorant). The animals freezing behavior was measured for 3 min, during which the tone (85 dB, 3 kHz) was delivered for the last 1 min. Freezing behavior was quantified using a video-based analysis (FreezeFrame, Coulburn Instruments).
Tissue preparation
Animals sacrificed after completing FC were deeply anesthetized with an overdose of isoflurane inhalation and perfused with 150 ml ice-cold phosphate-buffer saline (PBS). Each brain was quickly dissected on ice and bisected through the midline, after which one hemisphere was fixed by immersion in 4% paraformaldehyde for pathology and the other hemisphere was used for protein extraction for western blotting. For protein extraction, cerebral cortex and hippocampus were dissected separately and homogenized with RIPA buffer (Stock 10X RIPA, EMD Millipore, MA) supplemented to contain 10% TritonX-100, 10% SDS, a protease inhibitor cocktail and 0.5 M EDTA. Homogenates were centrifuged at 14,000 rpm for 25 min, and the supernatants were collected and stored at −80°C until use. The total protein concentration was determined by the BCA method (Pierce, Rockford, IL). For histology, the brains were post-fixed in 4% paraformaldehyde for 48 h at 4°C and then cut coronally at 50 μm thickness with a vibratome throughout the rostro-caudal extent of the brain. Sections were collected in 48-well plates filled with PBS plus 0.05% sodium azide and stored at 4°C until use.
Western blotting
Tissue lysates of the cerebral cortex were processed for western blotting. For each sample, 20 μg of protein was loaded in each well of 4–12% SDS-PAGE Criterion Gel TGX stain free gels (Bio-Rad, Hercules, CA, USA). The Gels were activated by the UV light on a Chemidoc MP imaging system (Bio-Rad, USA) for 45 s before blotting, and then were blotted to PVDF membranes using a Trans-Turbo Blot transfer system (Bio-Rad, USA). The transferred membranes were then blocked for 1 h in 5% powdered milk made in Tris buffered saline with 1% Tween 20 (TBST) at room temperature, and incubated with primary antibodies. The following primary antibodies were used: high-mobility group box protein-1 (HMGB1, 1:1000, rabbit polyclonal, cat# NB100-2322, Novus Biologicals), GFAP (1:1000, rabbit polyclonal, Dake, cat# Z0334), pre-synaptic markers including synapsin1 (1:1000, rabbit polyclonal, Abcam, cat# ab64581), synaptophysin (1:10000, rabbit polyclonal, Abcam, cat#: ab32127), post-synaptic marker PSD95 (1:500, rabbit polyclonal, Millipore, cat#: AB9708), cell cycle marker cyclin D1 (1:10000, rabbit polyclonal, Abcam, cat# ab134175). After the primary antibody incubation, membranes were thoroughly washed 5 times for 5 min each with 5% milk in TBST. The membranes were then incubated with the appropriate secondary antibodies for 1 h at room temperature. The secondary antibodies used were horse radish peroxidase-conjugated anti-rabbit, anti-mouse or anti-goat IgG (1:5000; Cell Signaling, MA, USA). The membranes were then washed 5 times and developed with the Clarity Western ECL ChemiDoc MP imaging system (Bio-Rad, USA). The analysis of the images was done using the Image Lab 6.0 software (Bio-Rad, USA). The stain-free images of blots were used for the total protein normalization against the chemiluminescence images. The normalized volume intensity was plotted as densitometric values in the form of histograms as published before [29].
Immunohistochemistry
To assess AD pathology and inflammatory cell response, we used antibodies against Aβ, the microglial marker, Iba1, and the astrocyte marker, GFAP. For each brain, every 8th section at the level of hippocampus from 2.56 mm to 5 mm of the bregma was processed for Aβ, Iba1, or GFAP immunostaining following our previously published protocol [30]. Briefly, the sections were washed with PBS and endogenous peroxidase was blocked using 3% H2O2. Following an overnight serum blocking with 5% normal horse serum in PBS, sections were incubated with mouse anti-Aβ 6E10 (1:2000, Biolegend, Sig-39300), goat anti-Iba1 (1:1000, Wako), rabbit anti-GFAP (1:1000, Dako) in PBST (PBS with 0.4% Triton) plus 5% normal horse serum at 4°C for 48 h with agitation on shaker. After rinsing with phosphate buffered saline with 1% Tween 20 (PBST), sections were incubated with biotin-conjugated anti goat, or rabbit-IgG (1:200, Jackson Laboratory) overnight at 4°C and then incubated with ABC complex for 2 h at room temperature before visualization with 5.5 diaminobenzidine (DAB). Sections were mounted on glass slides, lightly counterstained with 0.1% cresyl violet and covered with a coverslip. We also used immunofluorescent double labeling to assess the effect of JC124 treatment on hippocampal neurogenesis and cell cycle re-entry. Four coronal sequential sections were processed for BrdU and NeuN, or NeuN and CyclinD1 for double labeling. For BrdU/NeuN double-labeling, the sections first underwent DNA denaturation with 50% formamide for 60 min at 65°C followed by a rinse in 2 × saline-sodium citrate buffer (SSC) and then incubated with 2N HCl for 30 min at 37°C as we previously published [30]. Sections for NeuN/Cyclin D1 were not denatured. After endogenous peroxidase blocking and serum blocking, sections were incubated with the primary antibodies for BrdU (1:200, rat monoclonal, Immunologicals Direct, UK) and NeuN (1:1000, rabbit polyclonal, Abcam, cat#ab104225), or NeuN (1:500, mouse monoclonal, Millipore, cat#: MAB 377) and cyclin D1 (1:200, rabbit polyclonal, Abcam, cat# ab16663). Secondary antibodies were Alexa Fluor anti-rat or mouse 488 and anti-mouse 568 or anti-rabbit 594 (1:200, Molecular Probes). After secondary antibody incubation, sections were washed, mounted, and covered with a coverslipped sealed by Vectashield (Vector Lab). To verify that immunofluorescent staining does not result from non-specific labeling or tissue auto-fluorescence, control sections were included with primary antibodies omitted.
Immunostaining quantification
For Aβ staining, the expression level of plaques in cerebral cortex and the hippocampus was quantified from each section. Sections were examined with an Olympus microscope using a x20 objective lens and images were captured. Aβ in the cortex was examined according to sub-regions, including frontal, parietal, piriform, and entorhinal. For Aβ staining, the percentage of total tissue area with positive staining was measured by a blinded observer using the ImageJ version 1.52 automated counting program. The average of the percentage from four sequential sections was used for each brain. Microglial activation state was assessed with Iba1 labeling according to the size of cell body, and the shape and branches of the processes, as we published previously [31]. Briefly, in Iba1 stained sections, four random regions in the cortex were imaged and captured with a x20 objective lens. The number of cells with each of the three different morphologies were counted separately by a blinded observer with the ImageJ automated counting program. The number of counted cells from 4 sections per brain was averaged and expressed as number of cells per mm2.
For quantification of BrdU/NeuN double-labeling, BrdU-labeled newly generated cells and BrdU + cells that were double-labeled with NeuN in the dentate gyrus (DG) of the hippocampus were detected by confocal microscopy (Zeiss LCS710) by a blinded observer. The entire granule cell layer of the DG was assessed, and every BrdU-labeled cell was examined to assess the co-labeling. A minimum of 100 BrdU + cells from at least three sections per brain were examined. Each BrdU + cell was manually examined in its full “z” dimension. The percentage of double-labeled cells was calculated as the number of cells which were stained with both BrdU and NeuN against the total number of BrdU + cells on the same section.
Statistical analysis
The data generated were analyzed using the Graph-Pad Prism 7.0 software. Behavior data were analyzed by two-way ANOVA with repeated measures and post-hoc adjustments using Tukey’s test. Western blotting and immunostaining data were analyzed by one-way ANOVA followed by Tukey’s post hoc test for the multiple comparison or the Student t-test. Data were presented as mean ± SEM in all figures, with p value less than 0.05 taken as statistically significant.
RESULTS
JC124 improves cognitive function and reduces Aβ plaques
In AD, cognitive impairment is the dominant clinic symptom. To assess the effect of inhibition of the NLRP3 inflammasome on cognitive functions, we assessed the APP/PS1 mice after completion of the JC124 treatment with NOR and FC tests. The NOR test was performed at 2 and 24 h delay after the familiar phase to assess both short- and long-term non-spatial, hippocampal-mediated recognition memory. At both time points, the discrimination index in all groups was above 50% regardless of genotype or treatment, and no statistical significance was found between any categories within the group (Fig. 1A, B). In the FC test, contextual fear conditioning is a hippocampal-dependent task whereas the cued fear is a hippocampal-amygdala-dependent task. Significant differences were found in the freezing time in both contextual and cued fear tests between Tg mice treated with vehicle in comparison to the matched wild type control, and between Tg mice treated with either 50 or 100 mg/kg JC124. No difference was observed between wild type mice and Tg mice receiving the same dose of JC124 (Fig. 1C, D). Collectively, the behavior results suggested that Tg mice showed significant hippocampal-dependent and hippocampal-amygdala-dependent fear memory deficits compared to wild type mice and JC124 treatment exhibited protective effects. The results also suggested that FC is a more sensitive measure than the NOR under our current experimental conditions.
Fig. 1.
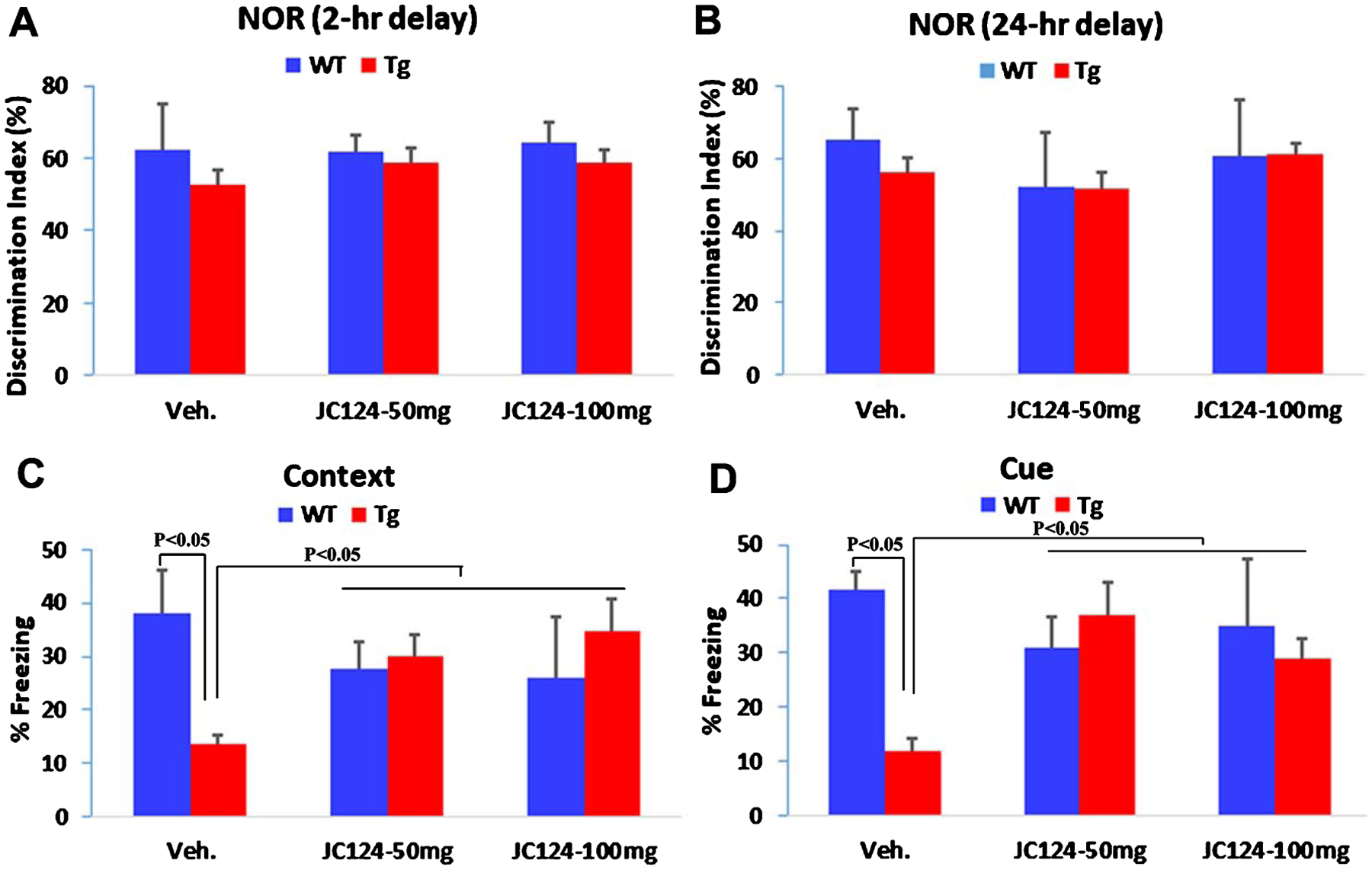
NLRP3 inflammasome inhibition by JC 124 improves functional recovery. The behavior changes of animals after treatment were assessed with novel object recognition (NOR) and fear conditioning (FC) tests. A, B) In the NOR test, the choice phase at 2 h (A) and 24 h (B) delay after the familiar phase, the discrimination index in all groups was above 50% regardless of genotype or treatment, and no statistical significance was found between groups. C). In the Contextual FC test, Tg animals with vehicle treatment showed significantly shorter freezing time compared to WT controls or Tg mice treated with either 50 mg/kg or 100 mg/kg JC124. D). In the Cued FC test, a significantly shorter freezing time was also found in the vehicle treated Tg mice in comparison to WT controls or JC124-treated Tg mice. No difference was found between WT controls and Tg-JC124-treated groups in both Contextual and Cued FC tests.
To assess the AD pathological changes, we first examined the brain tissues for Aβ plaques with immunostaining using the 6E10 antibody against Aβ. In the vehicle-treated Tg animals, widespread Aβ-positive plaques were observed in both the cerebral cortex and the hippocampus. Compared to vehicle-treated Tg animals, JC124-treated Tg animals exhibited reduced number of Aβ plaques (Fig. 2A). Using ImageJ, we quantified the Aβ staining percentage in sub-regions of the cerebral cortical regions and the hippocampus. As shown in Fig. 2B, compared to vehicle-treated Tg animals, the Tg mice treated with JC124 dosed at 50 mg/kg showed significant reduction of Aβ staining in the frontal cortex and the piriform cortex, whereas with the 100 mg/kg dose, reduction was observed in all regions assessed, including hippocampus, parietal cortex, piriform cortex, frontal cortex, and entorhinal cortex (Fig. 2B).
Fig. 2.

NLRP3 inflammasome inhibition by JC 124 reduces Aβ plaques in APP/PS1 mice. A) Representative images of the cerebral cortex and DG taken from Aβ stained coronal sections from Tg mice following vehicle or JC124 treatment. B). Quantification analysis showed in all regions assessed, including hippocampus, parietal cortex (Par), Piriform cortex (Pir), frontal cortex (Fr), and entorinal cortex (Ent), significantly less Aβ plaque staining was found in JC124-treated groups, particularly at 100 mg/kg dosing, as compared to the vehicle treated group.
JC124 treatment reduces microglia activation and astrogliosis
To assess changes of inflammatory responses following JC124 treatment, we examined the morphological changes of microglia in the cerebral cortex with immunostaining using an antibody to the microglia specific marker, Iba1. Microglia labeled with anti-Iba1 in three different phenotypes/morphologies (Type 1-resting/ramified, Type 2-activated, Type 3-activated/amoeboid) were quantified as we described before [28]. Using western blotting, we also measured the protein expression levels of Iba1 and HMGB1 in cerebral cortex. HMGB1 is a chromatin-binding factor that is localized to the nucleus and released by microglia under pathological conditions [32, 33], and has been implicated as a key factor mediating neuroinflammatory responses [34, 35].
The results of Iba1 staining revealed that in Tg mice many microglia were in an activated state characterized by an enlarged cell body and bushy, stubby processes, and and that such microglia accumulated prominently at the plaque areas (Fig. 3). Quantification revealed that the Type-2 microglia were the major sub-type, and that JC124 treatment at 100 mg/kg significantly reduced the percentage of Type-2 microglia in the cerebral cortex (Fig. 3).
Fig. 3.
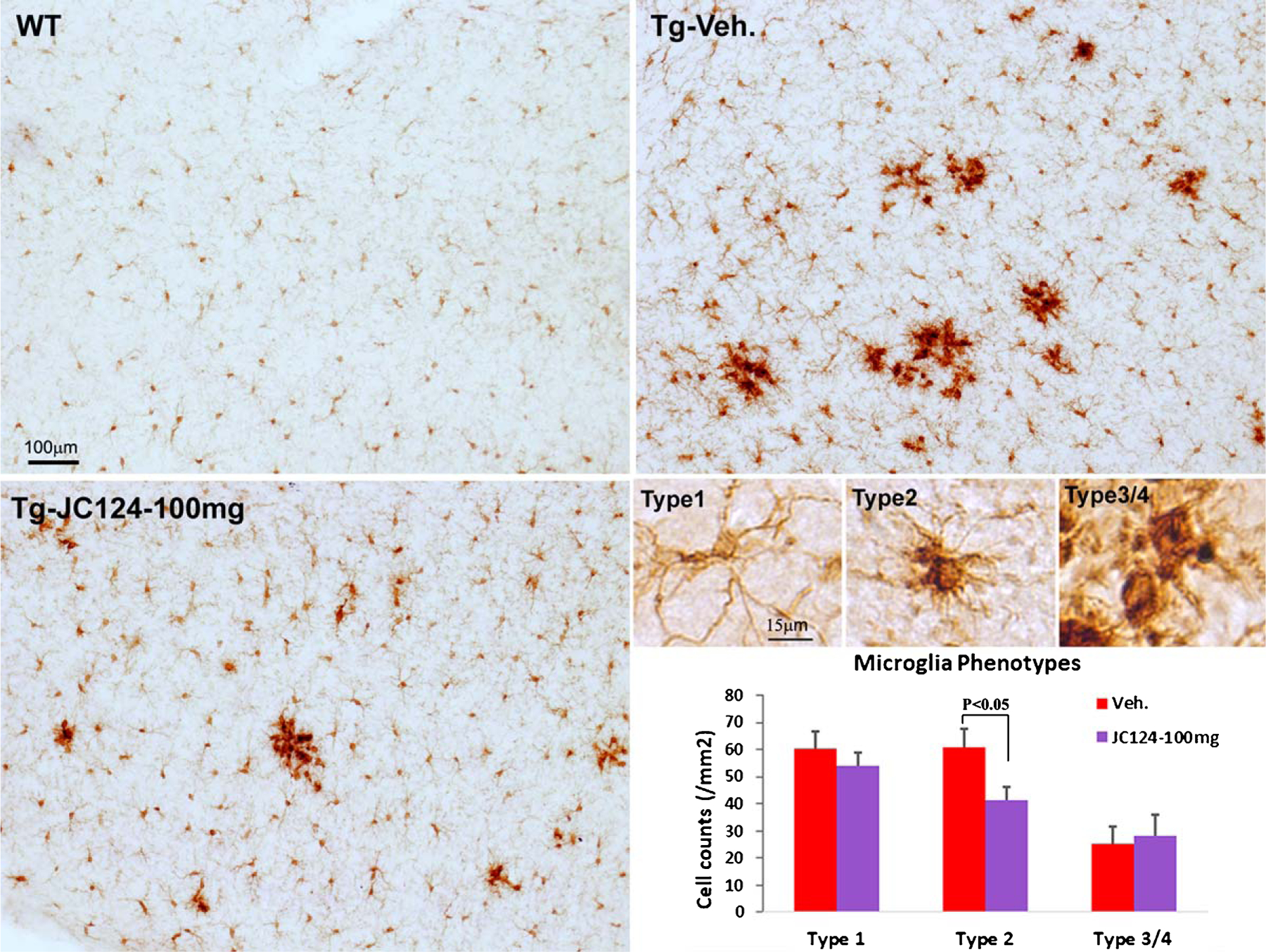
NLRP3 inflammasome inhibition by JC 124 reduces the number of activated microglia. Immunostaining for microglia with anti-Iba1 showed that in the wild type mice brain, Iba1 + microglia cells were at resting state with small cell bodies and slender ramified morphology (type 1 cell). In Tg brains, more microglia cells transformed into the activated state with increased cell body size, and bushy and stubby processes (type 2), and some cells became amoeboid with large irregular cell bodies (type 3/4 type) located close to plaques. Tg mice with vehicle treatment showed more dense Ib1 staining than Tg mice treated with JC124. When quantifying the number of Iba1 + cells with three distinct morphologies in the cortices of Tg mice, the number of type 2 microglia was significantly lower in Tg mice following JC124 treatment. No difference was found in the number of type 1 (ramified) and type 3/4 (amoeboid) microglia.
Western blotting analysis showed that the expression of Iba1 was significantly higher in Tg mice treated with vehicle compared to the wild type mice treated with vehicle. Treatment of Tg mice with JC124 at both 50 and 100 mg/kg resulted in significant reduction of Iba1 (Fig. 4A, B). A similar pattern for HMGB1 expression was observed after normalization with total protein load, with a higher expression in the Tg mice with vehicle treatment compared to wild type control, and reduced expression following JC124 treatment, especially at 100 mg/kg (Fig. 4C, D).
Fig. 4.

NLRP3 inflammasome inhibition by JC 124 reduces inflammatory response. Using western blotting, the protein expression level of inflammatory markers (Iba1 and HMGB1) was measured to assess inflammatory cell responses. A, C). Representative western blotting images showing the 20 KD band of Iba1 (A) and band of 29 KD HMGB1 (C) detected in brain lysates from WT, Tg-vehicle, Tg-JC124 50 mg/kg and 100 mg/kg groups. B, D). The densitometry values in the bar graphs showed that Tg-vehicle animals had significantly higher expression of Iba1 and HMGB1 compared to WT and Tg-JC124-treated groups. No difference was found between WT and Tg-JC124-treated groups in Iba1 expression, whereas the HMGB1 level was lower in Tg mice treated with 100 mg/kg versus 50 mg/kg JC124.
Using immunostaining and western blotting, we also assessed astrogliosis by GFAP expression. As revealed by GFAP staining, in vehicle treated Tg animals, patches of astrocytes with strong GFAP expression in hypertrophic morphology were present throughout the cortex and hippocampus, and this staining pattern was largely diminished in Tg JC124 treated brains (Fig. 5). Western blotting analysis showed that a much higher level of GFAP in Tg vehicle-treated mice was found as compared to the wild type and JC124 treated Tg groups, and JC124 treatment at both doses was equally effective in reducing astrogliosis (Fig. 5).
Fig. 5.
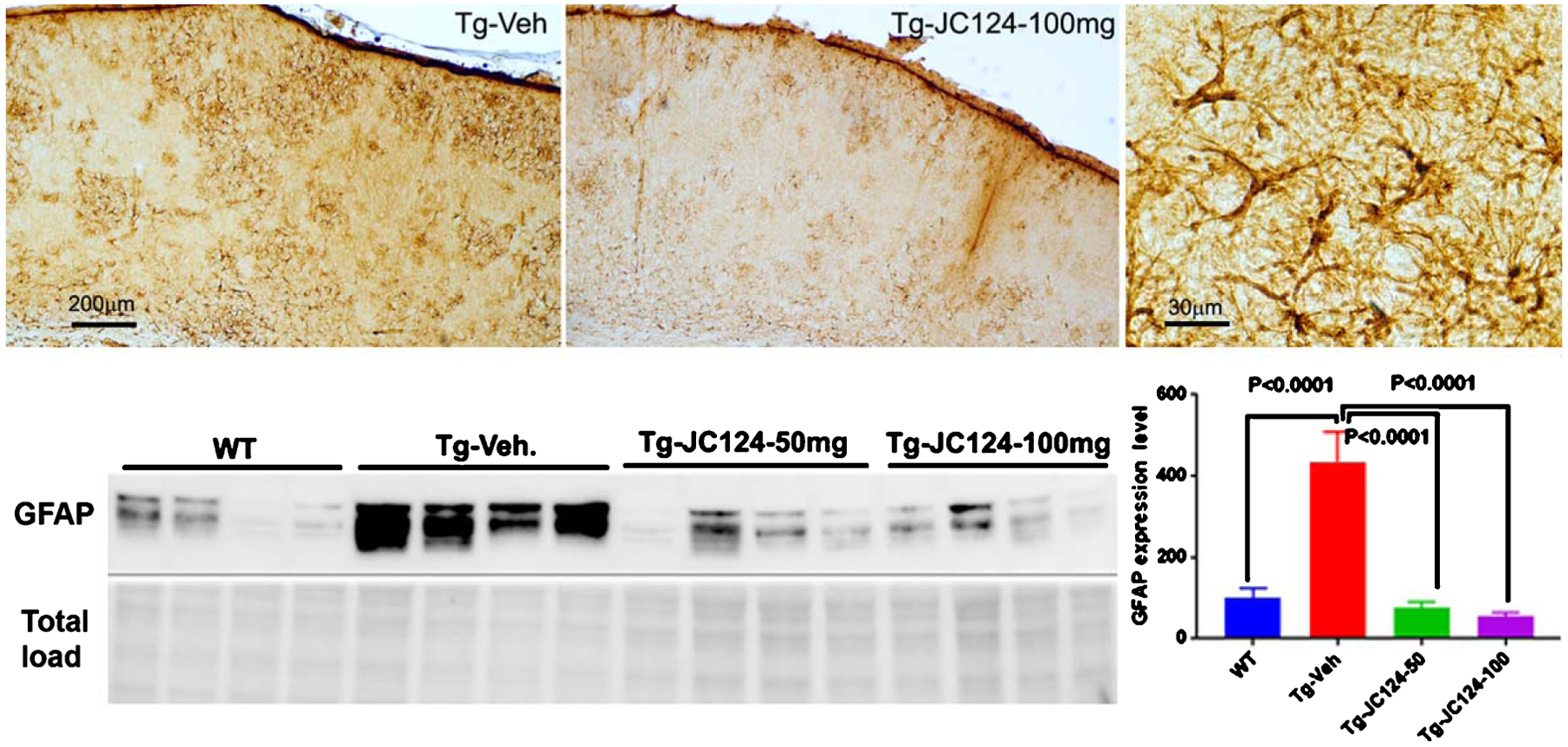
NLRP3 inflammasome inhibition by JC 124 reduces astrogliosis. Representative GFAP staining to show astrocytes response in the Tg brains. Strong GFAP expression and astrocytes hypertrophy were observed in regions associated with plaques in the Tg vehicle treated animal in comparison to the Tg JC124 treated animal, typical astrocytes hypertrophy was shown in the higher magnification micrograph. Representative western blotting images showing the 55 KD band of GFAP were detected in brain lysates from WT, Tg-vehicle, Tg-JC124 50 mg/kg and 100 mg/kg groups. The densitometry values in the bar graphs showed that Tg-vehicle animals had significantly higher expression level of GFAP compared to WT and Tg-JC124 treated groups. No difference was found between WT and Tg-JC124 treated groups.
JC124 treatment decreases cell cycle re-entry and increases hippocampal neurogenesis
Studies have shown that cortical neurons in AD brain exhibit ectopic cell cycle re-entry that ultimately leads to cell death [50–54]. To assess if inhibition of the NLRP3 inflammasome with JC124 affects cell cycle re-entry, we examined the expression of cyclin D1, a marker of G1 of the cell cycle. Western blotting of brain lysates showed that in the vehicle-treated Tg mice, the level of cyclin D1 was much higher when compared to the wild type mice, and JC124 treatment reduced the expression of cyclin D1 in the Tg mice to a level similar to that found in wild type mice (Fig. 6A, B). Because the western blotting results reported total Cyclin D1 levels in neurons and glia combined, we used immunofluorescence to highlight expression in neurons alone by double labeling with antibodies to Cyclin D1 and the mature neuron-specific marker, NeuN. This approach revealed that in the cerebral cortex of the Tg mice, a high proportion of neurons in vehicle treated Tg mice were Cyclin D1-positive, and that JC124 treatment in both doses substantially reduced the numbers (Fig. 6C, D), indicating that JC124 suppresses neuronal cell cycle re-entry.
Fig. 6.
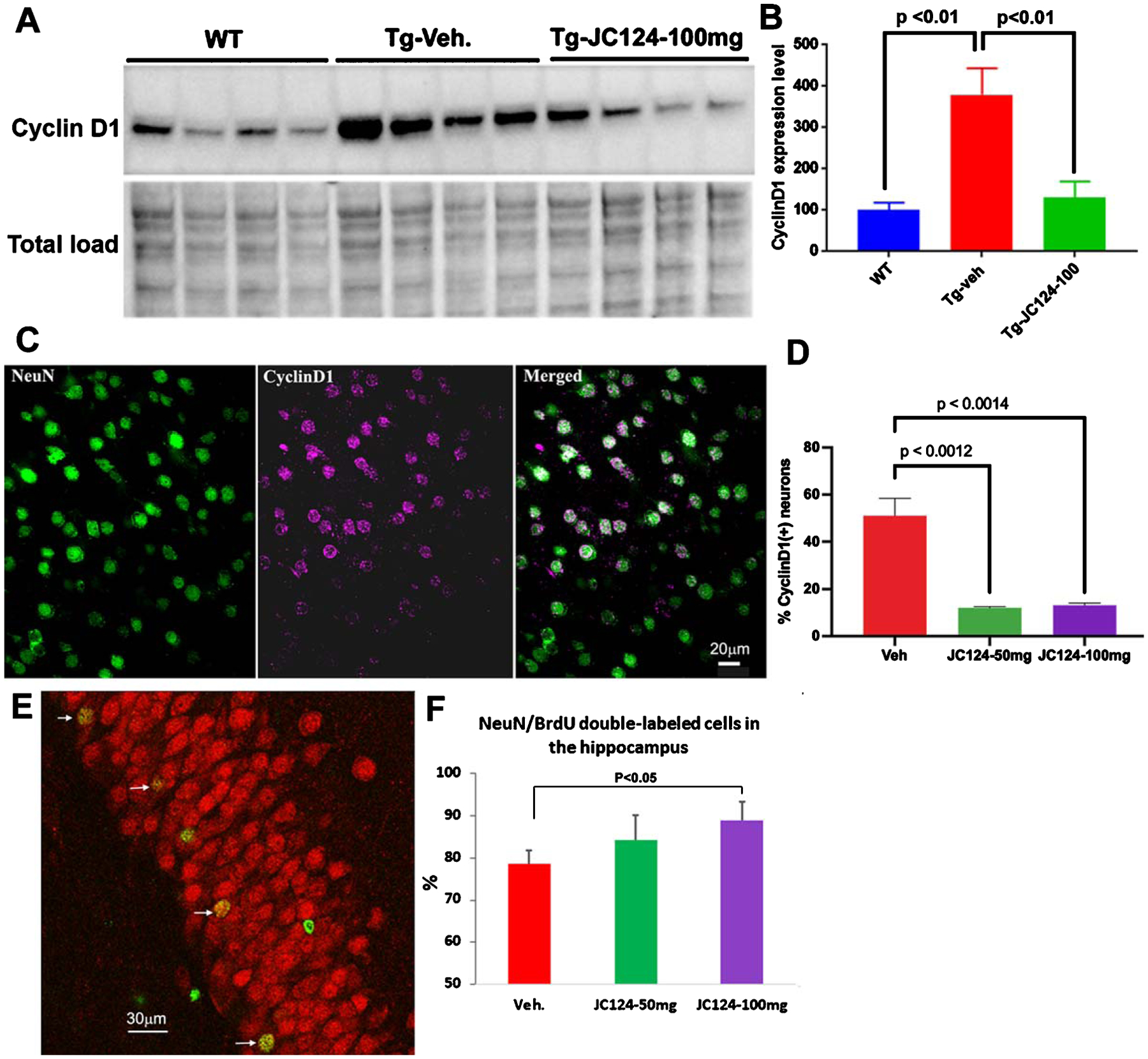
NLRP3 inflammasome inhibition by JC 124 decreases cell cycle re-entry and increases hippocampal neurogenesis. A) Representative western blotting image showing the 34 KD band of cyclin D1 detected in brain lysates from WT, Tg-vehicle, and Tg-JC124 100 mg/kg treated animals. B) The densitometry values in the bar graph showed that Tg-vehicle vehicle group had significantly higher expression of cyclin D1 as compared to WT and Tg-JC124 treated groups. C) Confocal images taken from cerebral cortex of Tg mice showing co-localization of cyclin D1 with NeuN. D) Quantification showed the percentage of NeuN/CyclinD1 double-labeled cells was significantly lower in the Tg-JC124 treated mice at both doses compared to Tg-vehicle treated mice. E) Representative confocal images of part of the granular cell layer of the DG double labeled for NeuN and BrdU, with arrows indicating NeuN-labeled neurons co-labeled with BrdU. F) Quantification showed that the percentage of BrdU/NeuN double-labeled cells was significantly higher in Tg-JC124 100 mg/kg treated group but not in the JC124 50 mg/kg group, as compared to Tg-vehicle treated group.
Endogenous neurogenesis persists throughout life in mammalian brain in primary neurogenic regions, including the subventricular zone and the hippocampal dentate gyrus (DG), and adult neurogenesis in the hippocampus is associated to cognitive function [36]. Decreased hippocampal neurogenesis has been observed in AD patients [37, 38]. Therefore, we also assessed the effects of JC124 treatment on neurogenesis in APP/PS1 mice. Using BrdU to label dividing cells, we assessed the neurogenesis during a 5-day window while under JC124 or vehicle treatment in Tg animals by quantifying the percentage of BrdU/NeuN double-labeled cells in the DG using confocal microscopy. In the DG of the Tg mice, BrdU + cells were scattered in the granular cell layer, and the average number of BrdU + cells per section was about 25 ± 5. No difference was found between JC124 or vehicle treated groups. When BrdU and NeuN double fluorescent staining was analyzed, 78 ± 3% of BrdU + cells were NeuN + in Tg mice treated with vehicle. The percentage of BrdU/NeuN double-labeled cells in the DG of Tg mice treated 5% and with JC124 at 50 or 100 mg/kg was 84 ± 5% and 89 ± 4%, respectively (Fig. 6E, F). The results suggest that inhibition of the NLRP3 inflammasome by JC124 promotes generation and survival of new neurons in the hippocampus. As we did not do BrdU pulse labeling, we cannot tell if cell proliferation was affected by JC124.
JC124 treatment increases synaptic plasticity
To examine whether treatment with JC124 has any effects in synaptic plasticity, using western blotting, we assessed the expression of pre- and post-synaptic proteins in the cerebral cortex. Compared to the wild type control, vehicle treated Tg mice showed lower expression of the pre-synaptic proteins, synapsin-1 and synaptophysin, as well as of the post-synaptic protein, PSD-95. In contrast, JC124-treated Tg mice at both doses had significantly higher expression of all three synaptic proteins, to levels comparable to or higher than levels found in the wild type control group (Fig. 7).
Fig. 7.
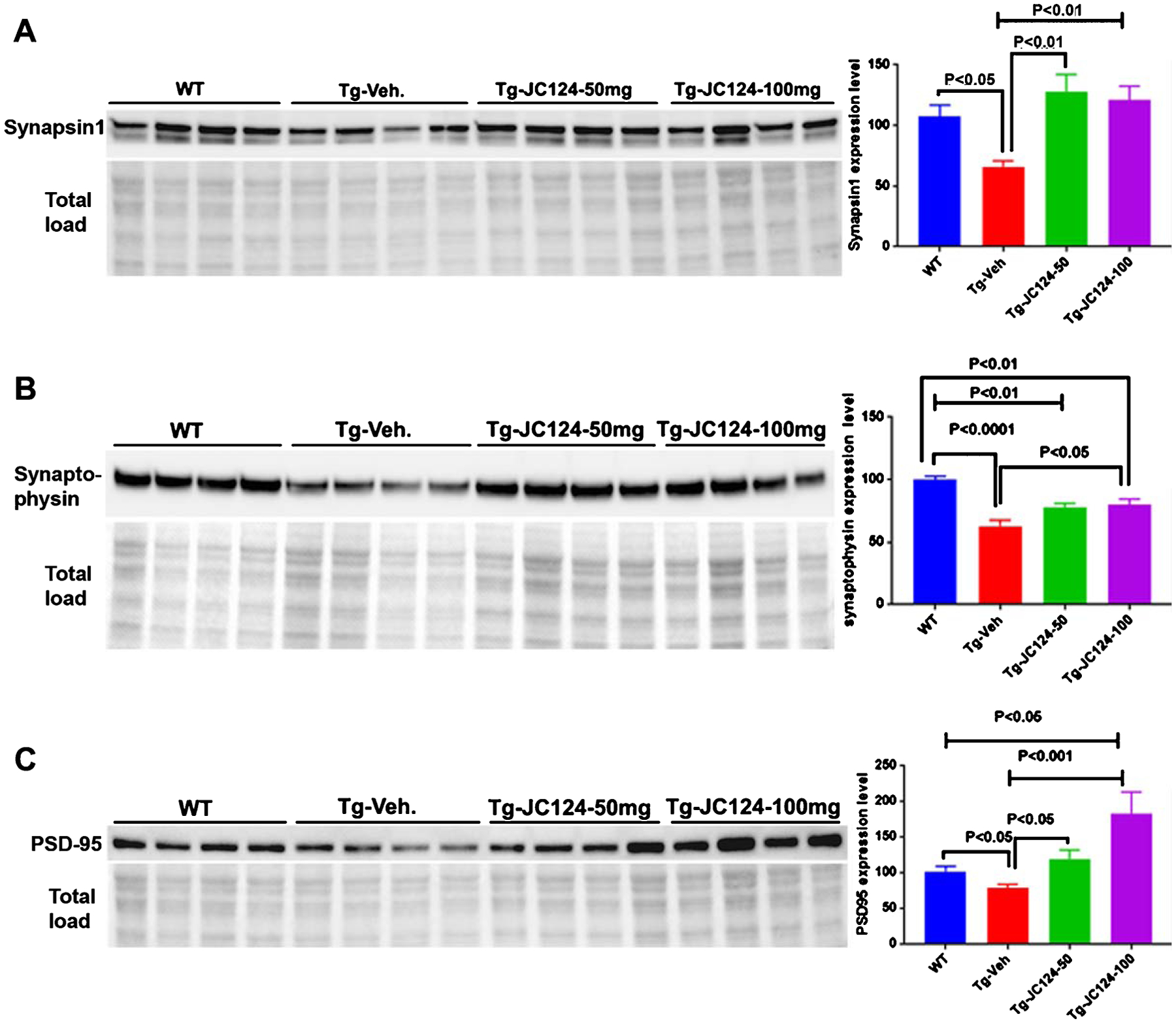
NLRP3 inflammasome inhibition by JC 124 increases synaptic plasticity. A-C). Representative western blotting image showing the 80–85 KD band of synapsin 1, 42 KD band of synaptophysin, and 95 KD band of PSD95 detected in the brain lysates from WT, Tg-vehicle, and Tg-JC124 animals. The densitometry values in the bar graph showed that Tg-vehicle groups had significantly reduced expression of all three synaptic proteins compared to WT, whereas higher expression levels of these proteins were found in Tg-JC124 treated animals.
DISCUSSION
AD is an increasing worldwide health issue. Recent evidences suggest that inflammasomes are involved in the pathologenesis of AD [20, 21]. The current study has shown that targeting NLRP3 inflammasome with our small molecule inhibitor, JC124, is beneficial in the AD transgenic animal model of APP/PS1 mice. Specifically, treatment with JC124 significantly decreased Aβ accumulation in the brain and improved cognitive function of the transgenic mice. The ameliorated AD pathology and behavior were accompanied by reductions in neuroinflammation, astrogliosis and neuronal cell cycle re-entry, as well as enhanced synaptic plasticity and neurogenesis in the hippocampus. Our results emphasize the importance of NLRP3 inflammasome in pathological development of AD, as inhibition of NLRP3 inflammasome can significantly alleviate AD progression in the AD transgenic mice model.
Neuroinflammation is an essential player driving disease development in many neurological diseases including AD, a notion supported by microglial activation, reactive astrocytes and elevated cytokine expression in AD patients. Examination of postmortem AD patient brain samples has revealed upregulation of inflammatory molecules and activated glial cells surrounding senile plaques [39–41], along with downregulation of anti-inflammatory molecules [42]. Furthermore, heightened expression of multiple inflammatory cytokines was found even during the early stages of AD [43]. These observations clearly suggest the involvement of inflammatory responses in the pathogenesis of AD. In the CNS, activated microglia and reactive astrocytes are the major cells responsible for production of inflammatory molecules including cytokines, chemokines, reactive oxygen species, etc. [44]. Interleukins, particularly IL-1β and IL-18, mediate expression of an array of inflammatory genes leading to a cascade of inflammatory process [45]. Furthermore, both IL-1β and IL-18 play essential roles in AD, such as synaptic plasticity, amyloidogenesis, and tauopathy [10, 46, 47]. Inflammasomes, including the NLRP3 inflammasome, play essential roles in regulating the maturation and production of IL-1β and IL-18, both of which are upregulated in AD brains [48]. Studies analyzing peripheral immune cells of AD patients have found that NLRP3 and NLRP1 inflammasomes are activated with increased production of IL-1β and IL-18 in both mild and severe AD [20]. Studies in vitro and in vivo have demonstrated that the NLRP3 inflammasome can sense Aβ aggregates and mediate the recruitment of microglia to Aβ [19, 21]. Notably, NLRP3−/− and Casp−/− mice carrying mutations associated with familial AD exhibited reduced Aβ burden, decreased synaptic plasticity loss, and improved spatial memory functions [21]. A similar genetic study employing 5xFAD mice carrying the ASC+/− genotype also rescued spatial reference memory deficits and Aβ plaque in the AD transgenic mice [22]. A recent study also found that NLRP3 activation induces tau hyperphosphorylation and aggregation [49]. These studies strongly imply critical roles for the NLRP3 inflammasome in AD development and its potential as a therapeutic target for AD.
Our laboratories have recently designed and developed a sulfonamide analogue, JC124, based on the structure of glyburide. We established that JC124 is an active and selective NLRP3 inhibitor by blocking ASC aggregation, activation of caspase-1 and release of IL-1β in macrophages that constitutively express active NLRP3 [24]. Our studies have also demonstrated the protective effect of this compound in a mouse acute myocardial infarction model [25], and rat traumatic brain injury model [27]. In AD animal models, we previously found that treatment of CRND8 APP transgenic mice (TgCRND8) with JC124 at the age when AD pathology was already present resulted in the inhibition of NLRP3 inflammasome signaling, decreased levels of plaque-associated soluble and insoluble forms of Aβ, decreased oxidative stress, microglia activation, and enhanced expression of GFAP and synaptophysin [28]. In the current study, we further demonstrate the beneficial effect of inhibition of NLRP3 inflammasome with JC124 in AD when starting the treatment before the onset of AD pathology in APP/PS1 mice, another widely used AD transgenic mouse model. The results of our current study have demonstrated that following JC124 treatment, APP/PS1 mice had dose-dependent lower level of Aβ plaques deposits and microglia activation, similar to what have observed in our previous studies in TgCRND8 mice. Interestingly, JC124 treatment significantly suppressed astrogliosis in APP/PS1 mice under the current experimental settings, while our studies in TgCRND8 mice showed increased astrogliosis upon JC124 treatment. This could be due to the different strains of transgenic mice used in these studies. This might also be due to the difference in the treatment regimen, as we employed a preventive treatment strategy with longer treatment time in the current study, while a therapeutic strategy with shorter treatment time was used in the TgCRND8 mice studies. Further studies are warranted to confirm the effects of JC124 or its analogs on astrogliosis. JC124 treatment also led to a dose-dependent higher expression level of both pre- and post-synaptic proteins, to the normal levels found in wild type mice. Although these results of the current study are inconsistent with our previous studies in TgCRND8 mice, the targeting aspect may be different. The animals in the current study started to receive drug treatment before the onset of AD pathology, and the observed lower levels of Aβ plaques and associated microglia activation and astrogliosis, as well as higher level of synaptic plasticity proteins may due to the delayed onset or reduced severity of AD pathology with this preventative regime. In our previous TgCRND8 mice study, the drug was given to older mice when AD pathology was already present, and the reduced Aβ level in that case may be due to the clearance rather production. Nevertheless, our studies have demonstrated a clear beneficial effect of JC124 in two AD models with two treatment regimes.
In adult brain, nearly all neurons are terminally differentiated and have exited the cell cycle into G0. However, studies have shown that cortical neurons in AD brain exhibit cell cycle re-entry that ultimately leads to cell death [50–54]. An exception is the primary neurogenic regions including the subventricular zone and the DG region of the hippocampus, where a very low level of neurogenesis persists throughout life and contributes to cognitive function [55]. However, this neurogenic response is decreased in AD [37]. Therefore, we further evaluated the effects of JC124 treatment on cell cycle re-entry in the cortex and neurogenesis in the DG. We found increased expression of the G1 marker, cyclin D1, in cortical neurons in APP/PS1 mice and this was suppressed by JC124 treatment. Our data therefore indicate that inhibition of NLRP3 activation blocks neuronal cell cycle re-entry. For hippocampal neurogenesis, the results revealed a higher percentage of the BrdU+/NeuN + neurons in a dose-dependent manner upon JC124 treatment in APP/PS1 mice, demonstrating the ability of JC124 to promote neurogenesis in the hippocampus region. Collectively we have demonstrated the efficacy of JC124 on Aβ plaques, glial cell response, cell cycle re-entry, hippocampal neurogenesis, and synaptic plasticity, which collectively contribute to the improved cognitive function in APP/PS1 mice. In the current study, we explored two doses of JC124, the higher dose had more significant effect in Aβ plaques and neurogenesis, but both doses were equally beneficial for memory function, glial cell response and synaptic plasticity. In cognitive functional assessment, deficits were only observed in fear memory, not in recognition memory in the Tg mice, and JC124 improved fear memory. As deficits in recognition memory in APP/PS1 mice were reported by others, the failure in the current study may be due to errors in selection of testing objects for NOR test. Further studies are warranted.
The effect of our NLRP3 inhibitor in reducing neuroinflammation and AD pathology and improving cognitive function is in agreement with other published studies. Studies have found that several clinically approved non-steroidal anti-inflammatory drug of the fenamate class can selectively inhibit NLRP3 activation via inhibition of the volume-regulated anion channel in macrophages, and show efficacy in reducing microglia activation and IL-1β expression, as well as improving memory function in 3xTg AD mice [56]. Treatment with dihydromyricetin, a plant flavonoid compound, was reported to ameliorate microglial activation and memory deficits in APP/PS1 mice via its function in reducing NLRP3 activation [57]. MCC950, a recently developed NILPR inhibitor blocking canonical and noncanonical NLRP3 activation [58, 59], reduced microglia activation and Aβ pathology, and improved cognitive function in NOR tests when administrated to APP/PS1 mice [60]. Inhibition of NLRP3 with MCC950 also showed abrogated synaptic plasticity disruption in transgenic rats overexpressing AD-associated APP at the pre-plaque stage of amyloidosis [61]. This is in line with our results of enhanced synaptic protein expression in the APP/PS1 mice following JC124 treatment. Overall, these published studies, together with our current study, strongly suggest the therapeutic benefit of targeting NLRP3 for AD.
CONCLUSIONS
Recent studies have suggested an important role of the NLRP3 inflammasome in AD pathological development. The current study demonstrated that targeting NLRP3 inflammasome with a small molecule inhibitor, JC124, exhibited beneficial effects in APP/PS1 mice. Specifically, treatment with JC124 significantly decreased Aβ accumulation and improved cognitive function. This improved AD pathology and behavior performance were accompanied by reduction of neuroinflammatory responses, astrogliosis, and neuronal cell cycle re-entry. Upon JC124 treatment, improved synaptic plasticity and endogenous neurogenesis in the hippocampus were also observed. Our results suggest that targeting NLRP3 inflammasome represents a viable strategy to develop potential effective treatments for AD.
ACKNOWLEDGMENTS
This work was supported by NIA/NIH R01 AG 058673 (SZ, DS), RF1 AG051085 (GSB); Alzheimer’s Drug Discovery Foundation (20150601, SZ), Commonwealth of Virginia Center on Aging - Alzheimer’s and Related Disease Research Award Fund (18-2, SZ; 21-1, DS); The Owens Family Foundation (GSB); and the Cure Alzheimer’s Fund (GSB).
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0400r2).
REFERENCES
- [1].Krstic D, Knuesel I (2013) Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol 9, 25–34. [DOI] [PubMed] [Google Scholar]
- [2].Pimplikar SW (2014) Neuroinflammation in Alzheimer’s disease: From pathogenesis to a therapeutic target. J Clin Immunol 34, 64–69. [DOI] [PubMed] [Google Scholar]
- [3].Takeda S, Sato N, Morishita R (2014) Systemic inflammation, blood-brain barrier vulnerability and cognitive / non-cognitive symptoms in Alzheimer disease: Relevance to pathogenesis and therapy. Front Aging Neurosci 6, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bibi F, Yasir M, Sohrab S, Azhar E, Al-Qahtani M, Abuzenadah A, Kamal M, Naseer M (2014) Link between chronic bacterial inflammation and Alzheimer disease. CNS Neurol Disorder 13, 1140–1147. [DOI] [PubMed] [Google Scholar]
- [5].Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JCS, Goodman SN (2004) Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: A systematic review. Neuroepidemiology 23, 159–169. [DOI] [PubMed] [Google Scholar]
- [6].Vlad SC, Miller DR, Kowall NW, Felson DT (2008) Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70, 1672–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Leoutsakos JMS, Muthen BO, Breitner JCS, Lyketsos CG (2012) Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: Findings from the randomized controlled Alzheimer’s Disease Anti-inflammatory Prevention Trial. Int J Geriatr Psychiatry 27, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Heneka MT, Carson MJ, Khoury J, El Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rosenthal SL, Kamboh MI (2014) Late-onset Alzheimer’s disease genes and the potentially implicated pathways. Curr Genet Med Rep 2, 85–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25, 181–213. [DOI] [PubMed] [Google Scholar]
- [11].Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB (2008) Neuroinflammation: Implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res 39, 1–16. [DOI] [PubMed] [Google Scholar]
- [12].Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TCC, Maeda J, Suhara T, Trojanowski JQ, Lee VMY (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
- [13].Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10, 210–215. [DOI] [PubMed] [Google Scholar]
- [15].Song L, Pei L, Yao S, Wu Y, Shang Y (2017) NLRP3 inflammasome in neurological diseases, from functions to therapies. Front Cell Neurosci 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M (2015) NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol 4, 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci 20, 3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Elliott EI, Sutterwala FS (2015) Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 265, 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol 9, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R, Mancuso R, Clerici M (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Heneka MT, Kummer MP, Stutz A, Delekate A, Saecker A, Griep A, Axt D, Remus A, Tzeng T, Gelpi E, Halle A, Korte M, Latz E, Golenbock D (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Couturier J, Stancu IC, Schakman O, Pierrot N, Huaux F, Kienlen-Campard P, Dewachter I, Octave JN (2016) Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J Neuroinflammation 13, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JPY (2010) NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 185, 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Marchetti C, Toldo S, Chojnacki J, Mezzaroma E, Liu K, Salloum FN, Nordio A, Carbone S, Mauro AG, Das A, Zalavadia AA, Halquist MS, Federici M, Van Tassell BW, Zhang S, Abbate A (2015) Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J Cardiovasc Pharmacol 66, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fulp J, He L, Toldo S, Jiang Y, Boice A, Guo C, Li X, Rolfe A, Sun D, Abbate A, Wang XY, Zhang S (2018) Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: Design, synthesis, and biological characterization. J Med Chem 61, 5412–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gaspar JA, Doss MX, Hengstler JG, Cadenas C, Hescheler J, Sachinidis A (2014) Unique metabolic features of stem cells, cardiomyocytes, and their progenitors. Circ Res 114, 1346–1360. [DOI] [PubMed] [Google Scholar]
- [27].Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, Zhang S, Sun D (2019) A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflammation 16, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, Zhu X (2018) NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Mol Neurobiol 55, 1977–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tramutola A, Pupo G, Di Domenico F, Barone E, Arena A, Lanzillotta C, Broekaart D, Blarzino C, Head E, Butter-field DA, Perluigi M (2016) Activation of p53 in Down syndrome and in the Ts65Dn mouse brain is associated with a pro-apoptotic phenotype. J Alzheimers Dis 52, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sun D, Bullock MR, Altememi N, Zhou Z, Hagood S, Rolfe A, McGinn MJ, Hamm R, Colello RJ (2010) The effect of epidermal growth factor in the injured brain after trauma in rats. J Neurotrauma 27, 923–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gerenu G, Liu K, Chojnacki JE, Saathoff JM, Martínez-Martín P, Perry G, Zhu X, Lee HG, Zhang S (2015) Curcumin/melatonin hybrid 5-(4-Hydroxy-phenyl)-3-oxo-pentanoic acid [2-(5-Methoxy-1H-indol-3-yl)-ethyl]-amide ameliorates AD-like pathology in the APP/PS1 mouse model. ACS Chem Neurosci 6, 1393–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. [DOI] [PubMed] [Google Scholar]
- [33].Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT (2010) HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim JB, Joon SC, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK (2006) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci 26, 6413–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee S, Nam Y, Koo JY, Lim D, Park J, Ock J, Kim J, Suk K, Park SB (2014) A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat Chem Biol 10, 1055–1060. [DOI] [PubMed] [Google Scholar]
- [36].Sun D (2014) The potential of endogenous neurogenesis for brain repair and regeneration following traumatic brain injury. Neural Regen Res 9, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, Ávila J, Llorens-Martín M (2019) Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25, 554–560. [DOI] [PubMed] [Google Scholar]
- [38].Moon M, Cha MY, Mook-Jung I (2014) Impaired hippocampal neurogenesis and its enhancement with ghrelin in 5XFADl. J Alzheimers Dis 41, 233–241. [DOI] [PubMed] [Google Scholar]
- [39].Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, Yolk B, Berger M (1991) Interleukin-6 and α-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett 285, 111–114. [DOI] [PubMed] [Google Scholar]
- [40].Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB (2001) In-vivo measurement of activated microglia in dementia. Lancet 92, 319–322. [DOI] [PubMed] [Google Scholar]
- [41].Fillit H, Ding W, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G (1991) Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett 129, 318–320. [DOI] [PubMed] [Google Scholar]
- [42].Walker DG, Tang TM, Lue LF (2017) Studies on colony stimulating factor receptor-1 and ligands colony stimulating factor-1 and interleukin-34 in Alzheimer’s disease brains and human microglia. Front Aging Neurosci 9, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM (2013) Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging 34, 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Johnstone M, Gearing AJH, Miller KM (1999) A central role for astrocytes in the inflammatory response to β- amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol 93, 182–193. [DOI] [PubMed] [Google Scholar]
- [45].Weber A, Wasiliew P, Kracht M (2010) Interleukin-1 (IL-1) pathway. Sci Signal 3, 1. [DOI] [PubMed] [Google Scholar]
- [46].Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, O’Banion KM (2013) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an alzheimer’s mouse model. J Neurosci 33, 5053–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cumiskey D, Curran BP, Herron CE, O’Connor JJ (2007) A role for inflammatory mediators in the IL-18 mediated attenuation of LTP in the rat dentate gyrus. Neuropharmacology 52, 1616–1623. [DOI] [PubMed] [Google Scholar]
- [48].Rubio-Perez JM, Morillas-Ruiz JM (2012) A review: Inflammatory process in Alzheimer’s disease, role of cytokines. ScientificWorldJournal 2012, 756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stun-den J, Merten M, Kayed R, Golenbock DT, Blum D, Heneka MT (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yang Y, Mufson EJ, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci 23, 2557–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nagy Z, Esiri MM, Cato AM, Smith AD (1997) Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol 94, 6–15. [DOI] [PubMed] [Google Scholar]
- [52].Arendt T, Brückner MK, Mösch B, Losche A (2010) Selective cell death of hyperploid neurons in Alzheimer’s disease. Am J Pathol 177, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Seward ME, Swanson E, Norambuena A, Reimann A, Nicholas Cochran J, Li R, Roberson ED, Bloom GS (2013) Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in alzheimer’s disease. J Cell Sci 126, 1278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Norambuena A, Wallrabe H, McMahon L, Silva A, Swanson E, Khan SS, Baerthlein D, Kodis E, Oddo S, Mandell JW, Bloom GS (2017) mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease. Alzheimers Dement 13, 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Patel K, Sun D (2016) Strategies targeting endogenous neurogenic cell response to improve recovery following traumatic brain injury. Brain Res 1640, 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Daniels MJD, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, Booth SJ, White CS, Baldwin AG, Freeman S, Wong R, Latta C, Yu S, Jackson J, Fischer N, Koziel V, Pillot T, Bagnall J, Allan SM, Brough D (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun 11, 12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Feng J, Wang JX, Du YH, Liu Y, Zhang W, Chen JF, Liu YJ, Zheng M, Wang KJ, He GQ (2018) Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci Ther 24, 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA, Jennings MP, Robertson AAB, Schroder K (2019) MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15, 556–559. [DOI] [PubMed] [Google Scholar]
- [59].Jiao J, Zhao G, Wang Y, Ren P, Wu M (2020) MCC950, a selective inhibitor of NLRP3 inflammasome, reduces the inflammatory response and improves neurological outcomes in mice model of spinal cord injury. Front Mol Biosci 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AAB, Cooper MA, O’Neill LAJ, Lynch MA (2017) Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun 61, 306–316. [DOI] [PubMed] [Google Scholar]
- [61].Qi Y, Klyubin I, Cuello CA, Rowan MJ (2018) NLRP3-dependent synaptic plasticity deficit in an Alzheimer’s disease amyloidosis model in vivo. Neurobiol Dis 114, 24–30. [DOI] [PubMed] [Google Scholar]