

Abstract
The only current curative treatment for chronic lymphocytic leukemia (CLL) is allogenic hematopoietic stem cell transplantation. Chimeric antigen receptor treatment targeting CD19 for CLL achieved some complete responses, suggesting the need for alternative or combinational therapies to achieve a more robust response. In this work, we evaluated CAR-T cells specific for Siglec-6, an antigen expressed in CLL, as a novel CAR-T cell treatment for CLL. We found that detection of SIGLEC6 mRNA and Siglec-6 protein is highly restricted to placenta and immune cells in other tissues and it is not expressed in hematopoietic stem cells. We generated CAR-T cells specific for Siglec-6 based on the sequence of the fully human anti-Siglec-6 antibody (JML1), which was identified in a CLL patient that was cured after allo-hematopoietic stem cell transplantation (alloHSCT), and observed that it specifically targeted CLL cells in vitro and in a xenograft mouse model. Interestingly, a short hinge region increased the activity of CAR-T cells to target cells expressing higher Siglec-6 levels but similarly targeted CLL cells expressing lower Siglec-6 levels in vitro and in vivo. Our results identify a novel CAR-T cell therapy for CLL and establish Siglec-6 as a possible target for immunotherapy.
Introduction
CLL is a clinically heterogeneous B-cell malignancy that typically has a CD5+/CD19+/CD20+/CD23+/IgD+/IgM+ immunophenotype yet lacks common genetic abnormalities (1, 2). The leukemia is typified by the gradual accumulation of monoclonal B cells, which can rise up to 500,000 cells/μL. CLL cells infiltrate and proliferate in bone marrow, spleen, lymph nodes, and other tissues.
Three anti-CD20 mAbs in combination with chemotherapy; and two small molecule kinase inhibitors (SMKIs) that target BTK and PIK3CD in the B-cell receptor (BCR) signaling pathway, are approved treatments for CLL (3). However, the disease is not cured, and interruption of treatment leads to re-emergence of the driver B-cell clone. In addition, mutations of SMKI targets and activating downstream mutations may lead to resistance to the treatment (4, 5). Allogeneic hematopoietic stem cell transplantation (alloHSCT) is the only treatment able to achieve a cure, but associated morbidity and mortality limit its application (1, 6). CD19-specific recombinant chimeric antigen receptors (CARs) have shown overall positive and complete responses in CLL, underscoring the potency of CAR-T cell therapy in eradicating CLL (7). However, treatment of CLL with CD19-CAR T-cells has achieved only 15-30% complete responses (8). Antigen escape by loss of the CD19 antigen in tumor cells was shown in 18 to 24% of relapses from complete responders (9–11). Therefore, the identification of additional CAR T-cell therapies targeting novel molecules in CLL may complement CD19-CAR immunotherapy.
Some sialic acid-binding immunoglobulin-like lectins (Siglecs) are present in only a few immune cell types making them potential immunotherapeutic targets (12). CAR-T cells directed against Siglec-2/CD22 for the treatment of pediatric and adult ALL as well as diffuse large B-cell lymphoma (DLBCL) have shown a morphologic complete response and remission in 57% and 43% of patients, respectively (13), representing an alternative to CD19-CAR T-cells for acute B-cell malignancies.
Siglec-6 is an immune inhibitory protein that shows restricted expression in the placenta (14) and mast cells (15), in tissue-like memory B-cells and exhausted B-cells but not in naïve B-cells (16). Primary CLL cells express Siglec-6, suggesting that Siglec-6 may represents a novel target for immunotherapy to CLL without affecting naïve B-cells (17).
In this work, we have developed a novel fully human anti-Siglec-6-CAR-T for CLL based on the sequence of a fully human monoclonal antibody against Siglec-6 isolated from a CLL patient, who was cured by alloHSCT (17, 18). We show that anti-Siglec-6-CAR-T cells effectively target CLL in vitro and in a xenograft mouse model.
Methods
Human blood samples and cells
Healthy blood from anonymous donors were obtained with written consent from the Department of Transfusion Medicine, NIH Clinical Center, and analyzed with the approval of the ethical review committee of the NIH. CLL patient samples were obtained after written informed consent in accordance with the Declaration of Helsinki, applicable federal regulations, and requirements from the National Heart, Lung, and Blood Institute Institutional Review Board. The clinical study is registered at clinicaltrials.gov as NCT00923507 and NCT00071045. Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats using Ficoll-Hypaque (GE Healthcare Biosciences, Pittsburgh, PA) by gradient centrifugation. The following cell lines were cultured in complete media and used as a target in assays: CCRF-CEM, MEC1, Nalm6 and U937. MEC1-001 and MEC1-002 were descripted previously (17), and were tested negative for mycoplasma contamination. Both B- and T- cells were isolated from buffy coat using human CD19 microbeads and pan T cell isolation kit (Miltenyi Biotech, Auburn, CA). Human CD34+ hematopoietic stem cells (HSCs) were purchased from StemCell Technologies (Vancouver, BC, Canada). MEC1-001-Siglec-6 Transgenic (TG) cells we generated by retroviral transduction with pEV-Thy1.1-hSiglec-6. The CD19-CAR construct corresponds to the previously described MSGV-FMC63-28Z (19).
RNA preparation and quantitative real-time PCR
Total RNA was extracted from enriched B-, T- cells and immortalized cell lines using RNeasy Miniprep kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized with High-Capacity RNA-to-cDNA kit (Applied Biosystems, Carlsbad, CA) using 1 μg of total RNA in a 20-μl reaction volume, one-hundredth of which was used as a template for real-time PCR reaction in a QuantStudio 3 Real-Time PCR system (Applied Biosystems) using the Taqman probe (Applied Biosystems) according to the manufacturer’s instructions. Human normal tissue cDNA array was purchased from OriGene Technologies (Rockville, MD). Each PCR procedure included a non-template negative control reaction. The level of 18s rRNA expression was used as the internal control, and ΔC(t) values were calculated according to the 2−ΔΔC(t) method. Each sample subjected to quantitative real-time PCR was analyzed in triplicate and repeated 3 times.
Immunohistochemistry and tissue arrays
Immunohistochemistry (IHC) analysis was performed using IHC protocols on commercial bone marrow, lymphoma, normal lymph node and normal tissue arrays BM241, LM721 and BCN921 US Biomax, Rockville, MD) using rabbit anti-human Siglec-6 polyclonal antibodies (Ab38581, Abcam, Cambridge, MA).
Generation of the Siglec 6-specific JML1 CAR
Fragments corresponding to JML1-CAR scFv constructs were generated as JML1VL-linker-JML1VH (linker amino acid sequence= GSTSGSGKPGSGEGSTKG) and cloned onto the CD19-CAR MSGV FMC63-28Z retroviral vector by replacing the fragments including CD19 scFv via XhoI and BmgBI restriction enzymes and T4 ligation. The resulting constructs contained a JML1 scFv plus a longer hinge region derived from CD28 (amino acid sequence= ALSNSIMYFSHFVPVFLPAKPTTTPAPRPPTPAPTIASQPLSLRPEASRPAAGGAVHTRGLDFACD) or a shorter hinge region derived from IgG4 (amino acid sequence= ESKYGPPCPPCP), followed by the CD28 transmembrane and co-stimulatory domains and by the CD3 zeta stimulatory cytoplastic domain.
Retrovirus was produced using a 293GP packaging cell line by transient co-transfection with retroviral vector plasmid (JML1 CAR and CD19 CAR construct) and a plasmid encoding the RD114 envelope protein. Culture supernatant containing retroviral particles was harvested after 48-72hs and stored at −80°C. To prepare CAR transduced T cells, cells were prestimulated with an anti-CD3ε monoclonal antibody (clone OKT-3, 50 ng/mL) for 48 hours and centrifuged at 32°C for 2 hours with viral particles onto retronectin (10 μg/mL, Takara Bio, Mountain View, CA)-coated multi-well plates. After transduction, cells were expanded for 3-4 weeks in AIM-V media containing human AB serum and IL-2 (300 IU/ml). Experiment were performed with transduced T cells, with least 50% efficiency, generated from a single donor, and repeated >3 times with PBMCs from different donors.
Flow cytometry analysis and antibodies
Single cell suspensions (PBS 2%FBS), were stained with fluorochrome-conjugated monoclonal antibodies against CD3 (OKT3 and SK7), CD4 (RPA-T4), CD8 (RPA-T8), CD19 (HIB19), CD5 (UCHT2), CD107a (H4A3), IFNγ (4S.B3), (BioLegend, San Diego, CA), CD327 (Siglec-6, clone REA852, Miltenyi Biotec, Bergisch Gladbach, Germany), biotinylated-human Siglec-6/Fc Chimera (R&D Systems, Minneapolis, MN) and bio-Protein L (Thermo Fisher Scientific, Waltham, MA) for 20 min at 4°C. For intracellular staining, cells were fixed and permeabilized with Foxp3 Fixation kit (eBioscience, San Diego, CA). Stained cells were acquired on an LSRII instrument (BD) or NovoCyte (ACEA Bioscience, San Diego, CA) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Cell proliferation and cytotoxicity assay
CAR-transduced T cells were labeled with 10 μM eFluor 450 cell proliferation dye (CPD, eBioscience) according to the manufacturer’s instructions. 5 × 105 CPD-labeled transduced CAR T cells were plated on 24 well/plates coated with recombinant human Siglec-6/Fc chimera protein (R&D System, Minneapolis, MN). Proliferation of T cells was analyzed by flow cytometry.
CAR T cell cytotoxicity assay was performed as described (20) with some modifications. Briefly, 2-week expanded, CAR T cells were co-cultured with target cells at the indicated ratios for 4-6 hours and analyzed by flow cytometry. %specific lysis=[(negative target-experimental target)/negative target x100]. The cytotoxic assay on primary CLL cells was performed by mixing a similar number of CAR-transduced T cells and primary CLL cells followed by co-culture for 5hs. Remaining CLL cells in culture were identified by flow cytometry as CD5+CD19+ cells.
Cytokine analyses
For in vitro cytokine secretion experiments, 5×104 CAR T cells and 5×104 target cells (MEC1-001, MEC1-002, U937) were cocultured for 16-24 hours. 200ul of supernatant was collected and sent to Eve Technologies (Calgary,AB, Canada).
In vivo tumor growth
All animal experiments were done after approval by the NCI ACUC to follow ethical guidelines. NSG mice were obtained from the NCI repository. We injected subdermally 200 μl per mouse of a 1:1 mixture of PBS:Matrigel (Corning, Corning NY) containing 2×107 MEC1-002 or MEC1-001-Siglec6TG cells stably expressing luciferase. Seven days later 5×106 effector T cells were injected i.v. in 200 μl of PBS. Luminescence was read 15 minutes after i.p. injection of 200 μl of luciferase (15mg/ml) in a Xenogen IVIS system (Perkin Elmer, Hopkinton, MA). Randomization and blinding was performed as follows: transduction of T-cells was performed by a technician and all in vivo procedures and analysis was performed by the investigator without knowledge of the specific group conditions.
Statistics
All statistical analyses were performed using Prism software (v8.0, GraphPad Software, La Jolla, CA). Two-tailed T-test were performed to compare between two samples, mean plus SD or individual data points are shown
Results
Siglec-6 expression is highly restricted in normal tissue
To establish the levels of expression of SIGLEC6 mRNA in healthy human tissue we performed qPCR for Siglec-6 in human tissue arrays representing 48 healthy organs and cells. In correlation with reported data, placenta was the tissue with the highest expression level, which was approximately 30 times higher than spleen and 50 times higher than the small intestine, the second and third highest expressing organs, respectively. Other tissues known to contain immune cells such as lung, liver, lymph nodes, colon and PBMCs showed low expression levels. Prostate, heart right ventricle, brain and bone marrow showed minimal SIGLEC6 mRNA levels (Figure 1A).
Figure 1. Siglec-6 expression in normal tissue and cells.
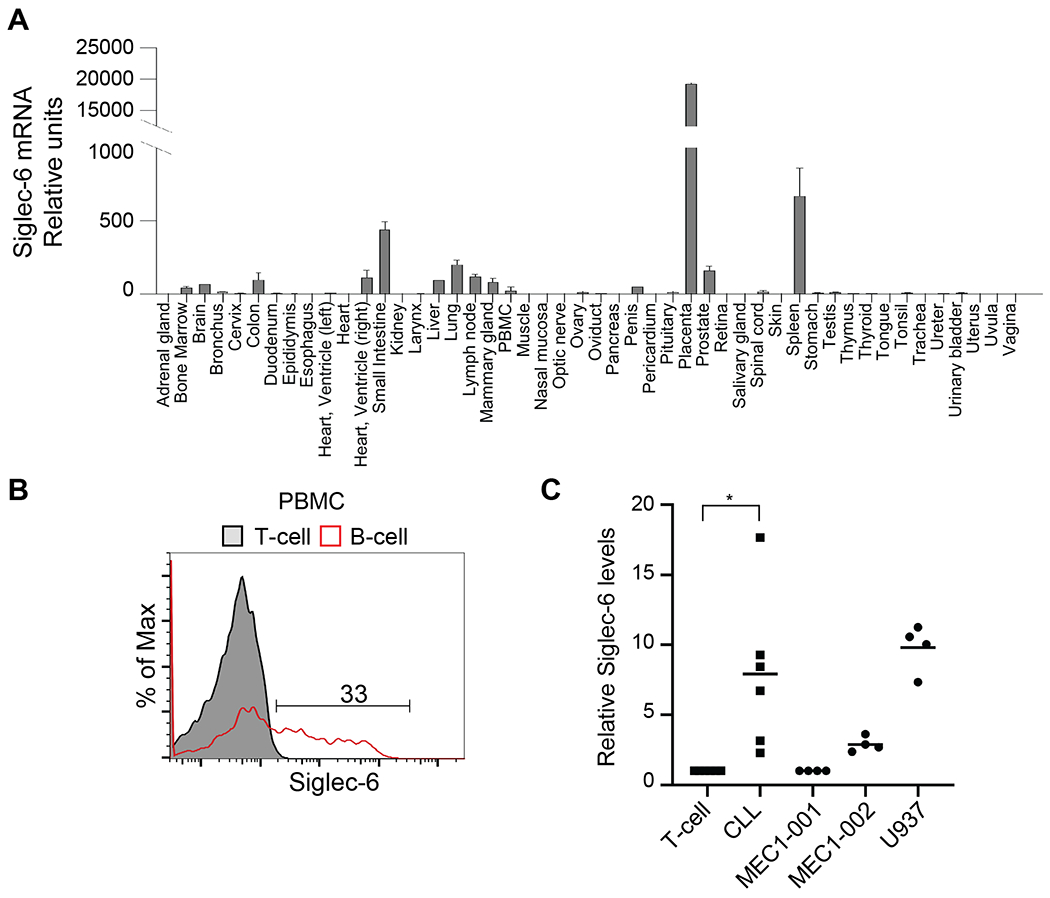
A)SIGLEC6 mRNA qPCR analysis in tissue arrays, data is derived from three independent experiments. B) Representative flow cytometry analysis of Siglec-6 levels in primary B-cells (CD19+) and T-cells (CD3+) from healthy PBMCs. Data is representative of eight healthy donor samples. C) Relative siglec-6 MFI levels in CLL (CD5+CD19+) relative to T-cells (CD3+) from the same patient and Siglec-6 MFI levels in the indicated cell lines relative to MEC1-001 cells. T-test, n=6 p<0.05.
Siglec-6 is not expressed in CD34+ hematopoietic stem cells
As a condition of immunotherapy is that the target antigen is not expressed in HSCs, we performed qPCR to detect SIGLEC6 mRNA in primary CD34+ HSCs in relation to primary T-cells and B-cells, the acute lymphoblastic leukemia (ALL) cell line Nalm6 and the myeloid cell line U937. SIGLEC6 mRNA expression was observed in primary B-cells, at approximately 5 times lower levels than in U937 cells, which are known to express cell surface Siglec-6 (21). Primary T-cells, Nalm-6 and primary CD34+ HSCs did not present SIGLEC6 mRNA (Supplemental Figure 1A).
The CLL cell line MEC1, is heterogeneous and approximately 10% of the cells are positive for cell surface Siglec-6 staining (17). A subset of this cell line that is positive for Siglec-6 was sorted by FACS and called MEC1-002. Sorted MEC1 cells that are negative for Siglec-6 were called MEC1-001 (17). We corroborated that MEC1-002 expresses Siglec-6 by qPCR and the levels were three times lower than those from U937 cells. Both B-ALL (Nalm6) and T-ALL (CCRF-CEM) cells did not present detectable SIGLEC6 mRNA (Supplemental Figure 1A).
SIGLEC6 mRNA levels correlated with Siglec-6 protein as analyzed by flow cytometry. MEC1-001 cells were negative for Siglec-6 and MEC1-002 presented lower amounts of Siglec-6 than U937 cells (Supplemental Figure 1B). The percentage of Siglec-6+ B-cells on PBMCs was highly varied among different samples and ranged from 13% to 57% with a mean value of 34.4% B-cells being Siglec6+ (Figure 1B and Supplemental Figure 1C). All other cells in PBMCs, including myeloid (CD11b+), granulocyte (CD66b+) and T-cells (CD3+) were Siglec-6 negative (data not shown). Primary and relapsed CLL were Siglec-6 positive as compared to T-cells, but Siglec-6 levels were highly varied among different individuals (Supplemental Figure 1D), in agreement with previous studies (17, 18). Despite the diverse Siglec-6 levels on different CLL samples, CLL cells were Siglec-6 positive and significantly different to Siglec-6 levels in the T-cells from the same patient (Figure 1C). Relative Siglec-6 levels in CLL cells were similar to those observed in MEC1-002 and U937 cell lines (Figure 1C)
Collectively, these data show that expression of SIGLEC6 mRNA is highly restricted to CLL cells and B-cells and not detected in HSCs.
Siglec-6 protein is detected within immune cells in healthy tissues.
Expression of Siglec-6 was previously shown in placenta, mast cells and activated B-cells (14–16), and the Human Protein Atlas identifies Siglec-6 protein levels only in placenta (www.proteinatlas.org/ENSG00000105492-SIGLEC6/tissue). To analyze Siglec-6 protein levels in healthy human tissues, we performed IHC on normal tissue arrays, including bone marrow, lymph nodes, stomach, kidney, heart, liver, spleen, lung, brain, colon, testis and ovary tissues. Our analysis showed that positive staining was observed in circulating mononuclear cells in some tissues and that no positive staining was observed in any other cell types. Positive staining of mononuclear cells was seen in spleen, lung and stomach; all other tissues analyzed showed no positive staining (Figure 2A and B). In the stomach, we observed some uptake of the Siglec-6 antibody, but it is granular and not specific as zymogen granules pick up stains in a non-specific pattern. In addition, few plasma cells were intensively positive, which we have seen with multiple different antibodies and also consider a non-specific pattern. These results indicate that detectable endogenous Siglec-6 protein is highly restricted to the immune compartment in healthy tissues.
Figure 2. Siglec-6 Immunohistochemistry in healthy human tissue.
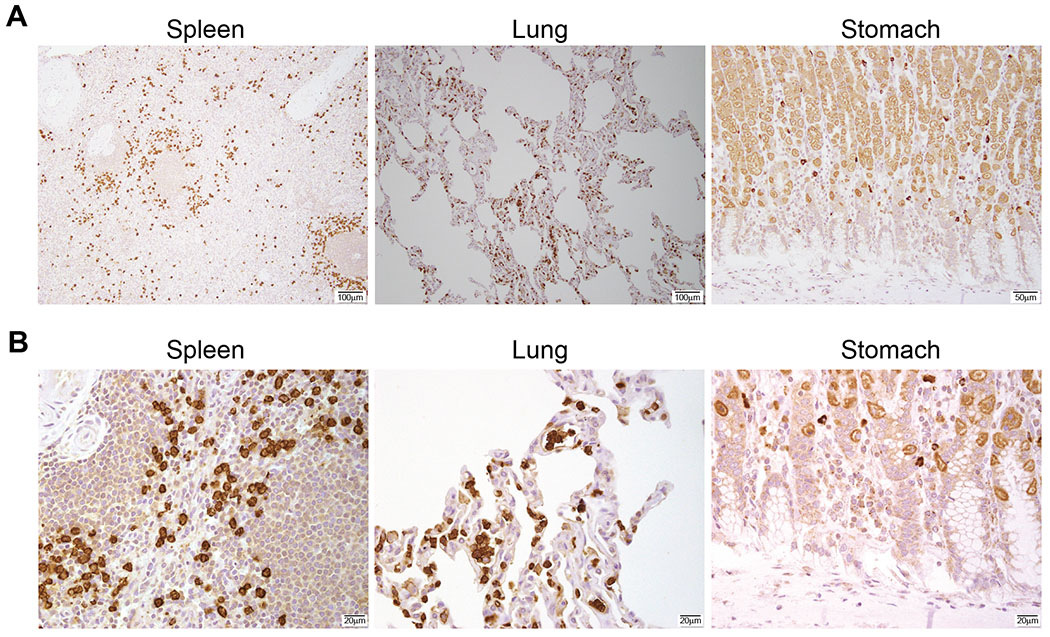
A) Representative tissues where Siglec-6 positive staining was detected in tissue arrays (BCN921 Biomax). B) Higher magnification of the tissues shown in (A). Data are representative of 10 independent tissues.
Anti-Siglec-6 JML1-CAR T-cells recognize purified Siglec-6-Fc protein
JML1 is a fully human, human-derived monoclonal antibody that recognizes Siglec-6 and was identified by phage display technology from post-alloHSCT PBMCs of a CLL patient (18). To specifically target CLL, we decided to develop a CAR based on the JML1 heavy and light chain variable domain sequences and cloned it into the MSGV retroviral vector. We generated two CAR constructs. JML1-CAR contained a hinge extracellular, transmembrane and co-stimulatory domain derived from CD28 and JML1-CAR-short contained a shorter hinge extracellular region derived from human IgG4 (Figure 3A).
Figure 3. JML1-CAR constructs recognize Siglec-6.
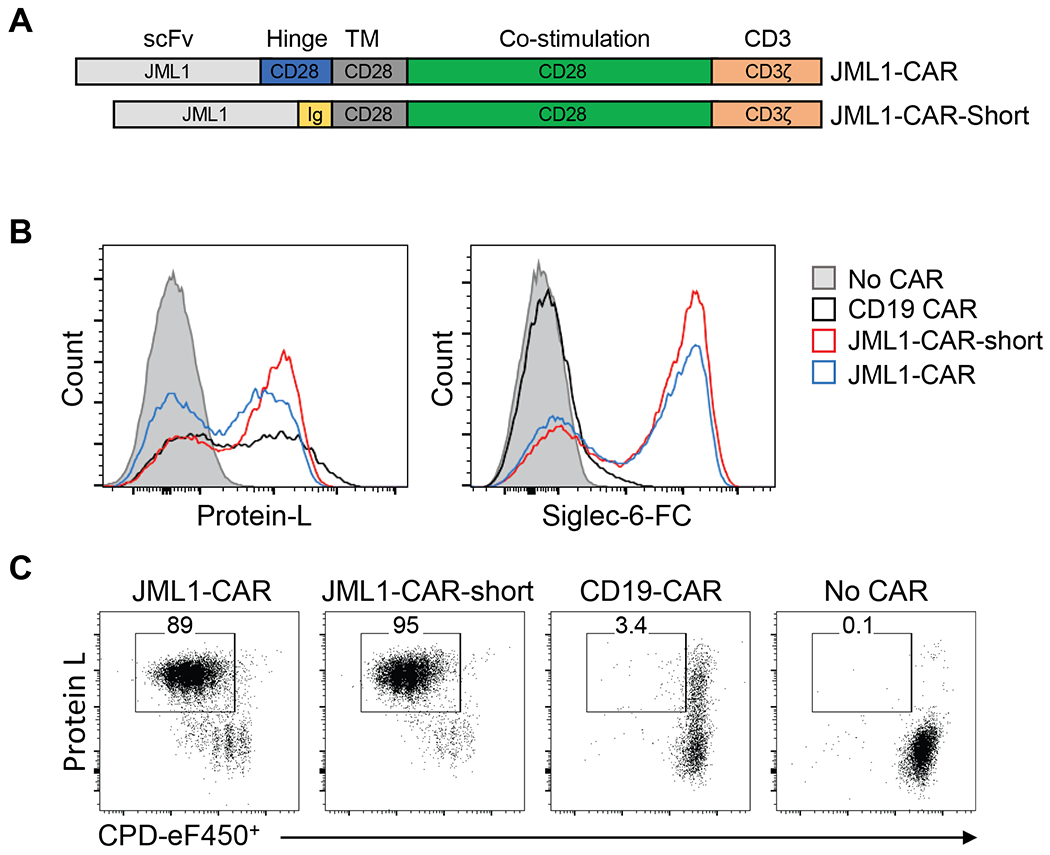
A) Diagram representing the domains of JML1-CAR and JML1-CAR-short used in this study. Ig corresponds to IgG4 hinge domain. B) Flow cytometry analysis of CAR-T cells transduced with different CAR constructs and stained with biotinylated Protein-L or a biotinylated recombinant Siglec-6-Fc chimera protein. C) Flow cytometry analysis of CAR-T cell proliferation by CPD dilution after three days culture in multiwell plates coated with recombinant Siglec-6-Fc. Data is representative of four independent experiments.
Analysis of Protein-L staining, which binds to the kappa light chain variable domain of the single-chain variable fragment (scFv) in CARs, showed that cell surface JML1-CARs levels were similar to the CD19-CARs. We then tested the ability of the JML1-CAR to bind biotinylated recombinant human Siglec-6-Fc by flow cytometry and observed that Siglec-6-Fc was able to bind JML1-CAR T-cells but not CD19-CAR T-cells, confirming the specificity of the JML1-CAR for Siglec-6 (Figure 3B).
To evaluate if Siglec-6 was able to trigger the proliferation of JML1-CAR T-cells in vitro we cultured them in plates coated with recombinant human Siglec-6-Fc and analyzed their proliferation by dilution of cell proliferation dye (CPD) after 3 days. We observed that both JML1-CAR and JML1-CAR-short T cells similarly proliferated in the presence of the antigen, while T-cells containing no CAR or a CD19-CAR did not proliferate (Figure 3C). The proliferation elicited by JML1-CAR and JML1-CAR-short did not show any significant difference as a similar proportion of cells were found in each cycle (Supplemental Figure 2).
These data show that the JML1-CAR is expressed on the cell surface of transduced T-cells, and that JML1-CAR-T cells recognize Siglec-6 and proliferate in response to the antigen.
JML1-CARs secrete diverse cytokines in response to Siglec-6+ target cells.
To test the ability of JML1-CAR-T-cells to secrete cytokines, we measured the intracellular proportion of IFNγ+ CD8 T-cells after co-culture with target cells. The frequency of IFNγ+ cells significantly increased in both JML1-CAR and JML1-CAR-short after co-culture with MEC1-002 cells. However, this proportion seemed to be higher in response to Siglec-6hi U937 cells than to Siglec-6lo MEC1-002 cells. Cytokine secretion by JML1-CARs was approximately 10 times lower than that of CD19-CARs in response to MEC1-002 (Figure 4A). JML1-CAR-short presented a significantly higher IFNγ secretion than JML1-CAR in response to Siglec-6hi U937 cells, but a similar activity in response to Siglec-6lo MEC1-002 cells (Figure 4B, 4D and 4E). A similar tendency was observed when analyzing cytokine secretion for different cytokines. While secretion of cytokines was similar between JML1-CAR-short and JML1-CAR (ratio approximately 1) when cells were co-cultured with Siglec-6lo MEC1-002 cells, co-culture with Siglec-6hi U937 cells led to a ratio of approximately 2-4 among these cytokines (Figure 4C). However significant differences between the two JML CARs were observed only on IFN-γ secretion.
Figure 4. JML1-CAR-short-T-cells elicit higher activation than JML1-CAR-T-cells.
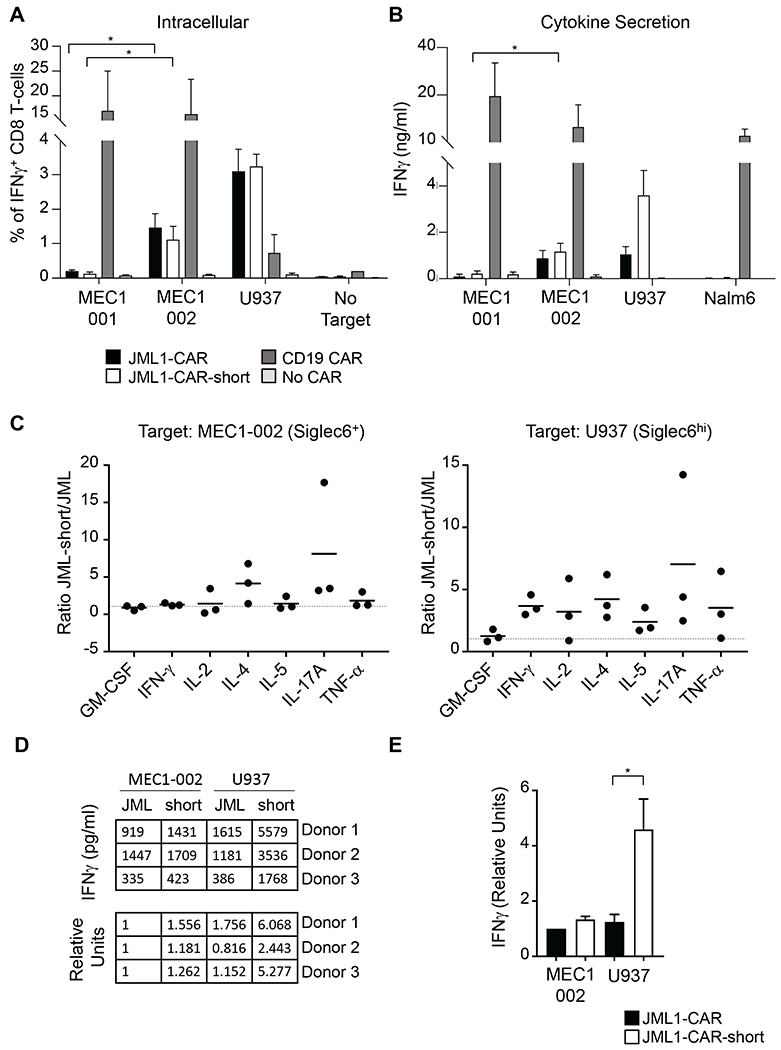
A) Frequency of IFNγ +CD8 CAR-T cells after co-culture with the indicated target cell lines. B) Analysis of IFNγ secretion to 200μl of the cell supernatant after co-culture of 5×104 CAR-T cells with target cells for 16-24 h. Data corresponds to 3 independent experiments. Significance denotes t-test n=3 p<0.05. C) Comparative analysis of the ratio of cytokine secretion between JML1-CAR-short and JML1-CAR after co-culture with the indicated target cells. Each dot indicates an independent sample measured in duplicate. D) Representative IFN-γ secretion of different experiments after co-culture with the indicated target cell lines. E) Quantitation of IFN-γ secretion, t-test, n=3 p<0.05.
JML1-CAR T-cells have cytotoxic activity against Siglec-6+ CLL cells in vitro.
We then tested the ability of CAR-T cells to upregulate CD107a, which is a marker of degranulation and cytotoxic activity, after co-culture with target cells. We observed that both JML1 and JML1-CAR-short constructs upregulated CD107a in response to Siglec-6+ target cells (MEC1-002 and U937) but did not respond when co-cultured with Siglec-6− cells (MEC1-001) (Figure 5A and B).
Figure 5. JML1-CAR-short and JML1-CAR T-cells have cytotoxic activity against CLL cells in vitro.
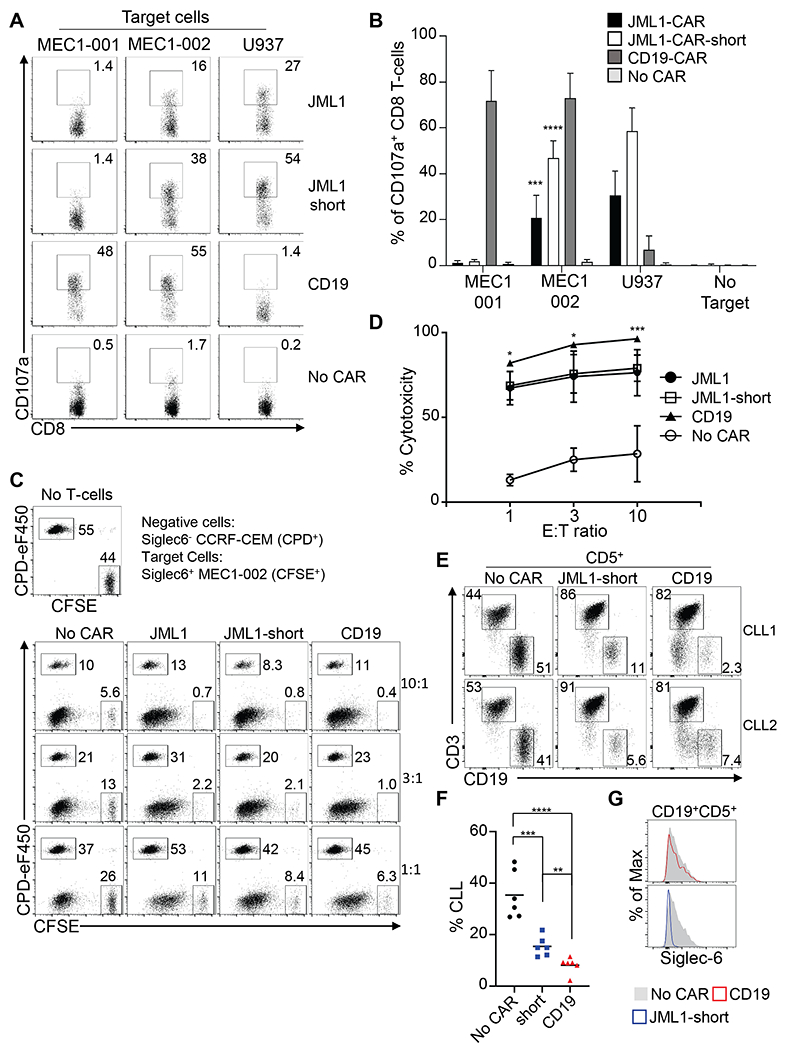
A) Flow cytometry analysis showing CD107a levels of CAR-T cells after co-culture with the indicated target cells at an effector:target ration of 1:1. B) Quantification of the percentage of CD107a+CD8 T-cells in each condition. Data correspond to three independent experiments performed in duplicates. T-test against MEC1-001, n=3. p<0.001 (***) or p<0.0001 (****). C) In vitro cytotoxic analysis after incubation of Siglec-6− internal negative cells (CPD+), Siglec-6+ MEC1-002 cells (CFSE+) with the indicated CAR-T cells for 4–6hs at the indicted effector:target ratios of 1:1, 3:1 and 10:1. D) Quantification of cytotoxicity at the indicated effector:target cell ratios. Cytotoxic activity is derived as described in the Methods section. T-test of JML1-CAR and JML1-short-CAR against No CAR at all effector:target ratios is p<0.0001, n=6 (not shown). T-test of JML1-short-CAR vs CD19-CAR is shown p<0.001 (***) or p<0.05 (*), n=6. E) In vitro cytotoxic assay after incubation of primary CLL samples with CAR-T cells for 4-6hs at a 1:1 effector:target ratio. F) Quantitation of percentage of remaining CLL cells after co-culture obtained in three independent experiments, each dot corresponds to a primary CLL sample. T-test, n=6 p<0.01 (**), p<0.001 (***) or p<0.0001 (****). G) Representative histogram showing Siglec-6 levels in CLL samples (CD19+CD5+) after co-culture with effector cells. Data is representative of three independent experiments and 6 CLL samples analyzed.
We then analyzed the ability of JML1-CAR, JML1-CAR-short and CD19-CAR T-cells to eliminate Siglec-6+ MEC1-002 cells in vitro. We used the T-cell line CCRF-CEM, as a non-specific target control and performed the co-culture at different effector to target cell ratios. We observed that both JML1-CAR and JML1-CAR-short T-cells were similarly able to eliminate MEC1-002 cells, with a significantly higher cytotoxic activity than effector cells without CARs (No CAR). CD19-CAR T-cells, however, had a significantly higher cytotoxic activity than JML1-CAR T-cells (Figure 5C and D). In correlation with a significantly higher IFN-γ secretion of JML1-CAR-short when co-cultured with Siglec-6hi U937 cells, we found that JML1-CAR-short T-cells had a significantly increased cytotoxic activity against U937 cells (Supplemental Figure 3). This increased activity of JML1-CAR-short correlates with JML1 recognizing a membrane-distal epitope of Siglec-6, which may favor CAR-T cells with a shorter hinge region for efficient synapse formation (Supplemental Figure 4).
Taken together, these data show that JML1-CAR T-cells effectively target and eliminate Siglec-6+ cells in vitro, and that JML1-CAR-short with a shorter hinge region is the most efficient construct for targeting cells expressing higher Siglec-6 levels.
JML1-CARs have cytotoxic activity against primary CLL cells in vitro
We next evaluated that ability of JML1-CAR-short and CD19-CAR to target primary CLL cells in vitro. For this purpose, we co-cultured primary CLL cells with effector cells derived from healthy PBMCs in a 1:1 effector to target cell ratio and analyzed the presence of CLL cells after the co-culture identified as CD5+CD19+ cells. We observed that both JML1-CAR-short and CD19-CAR T-cells were able to significantly eliminate primary CLL cells in vitro, although CD19-CAR T-cells revealed a significantly stronger cytotoxic activity (Figure 5E and F). Siglec-6 levels in CLL1 and CLL2 samples are shown in Supplemental Figure 5A. As expected, remaining CLL cells after co-culture with CD19-CAR T-cells were Siglec-6+ while remaining CLL cells after co-culture with JML1-CAR-short T-cells were negative (Figure 5G and Supplemental Figure 5B). This finding suggests that JML1-CAR-short T-cells may be able to eliminate surviving CLL cells after CD19-CAR T-cell treatment.
JML1-CARs T-cells eliminate CLL cells in vivo
In order to establish a suitable experimental model to test the activity of JML1-CARs in xenografts we generated MEC1-001 cells stably expressing Siglec-6 and luciferase, named MEC1-001-Siglec-6-TG. We corroborated the presence of transgenic Siglec-6 on the cell surface by flow cytometry (Supplemental Figure 6A). We next evaluated the ability of these cell lines to engraft NOD-severe combined immunodeficient γ-chain deficient (NSG) mice under the skin via subdermal injection and quantitated their tumor burden by luminescence and IVIS imaging. We observed that the rate of tumor growth was different among the different cell lines used. While MEC1-002 tumors, with endogenous Siglec-6, grew very slowly, both MEC1-001 and MEC1-001-Siglec-6-TG tumors grew faster (Supplemental Figure 6B and C). These data suggest that the lower growth of MEC1-002 cells is not due to Siglec-6 expression, and other genetic differences in MEC1-002, which were previously analyzed by differential RNA-seq (17), might be responsible for their slow growth.
We next evaluated if JML1-CAR T-cells could eliminate MEC1-002 cells in vivo which expresses Siglec-6 endogenously. Seven days after injection of cells to generate subdermal tumors, CAR-T or control untransduced (No CAR) T-cells were injected intravenously and luminescence was analyzed at different time intervals. We observed that at day 21 both JML1-CAR and JML1-CAR-short groups significantly reduced the tumor burden when compared to mice that either received untransduced or no T-cells (PBS) (Supplemental Figure 6D and E). However, untransduced T-cells had a cytotoxic effect that started to be observed later on day 28 as the tumor luminescence was reduced by T-cells having no CARs compared to PBS. Treatment of mice with an unrelated CAR (SP6) showed a similar response as the No CAR group (data not shown). These data indicate that JML1-CARs have a specific activity against Siglec-6+ MEC1-002 cells but that T-cells without CARs can also eliminate target cells at a later time point.
As the cytotoxic effect of T cells without CARs may mask the JML1-CAR T-cell specific reduction of tumor burden, and to more clearly evaluate the in vivo effect of JML1-CAR T-cells, we decided to use the faster growing MEC1-001-Siglec-6-TG cells as target.
We observed that MEC1-001-Siglec-6-TG tumors grew over time in the absence of T-cells. Both JML1-CAR and JML1-short-CAR-T cells eliminated the tumor, which was already undetectable at day 22 post-injection. T-cells not transduced with any CAR also reduced the tumor burden but at a later time point, which was observed at day 39 post-injection (Figure 6A and B).
Figure 6. JML1-CAR-short and JML1-CAR have antitumor activity in a xenograft mouse model of CLL.
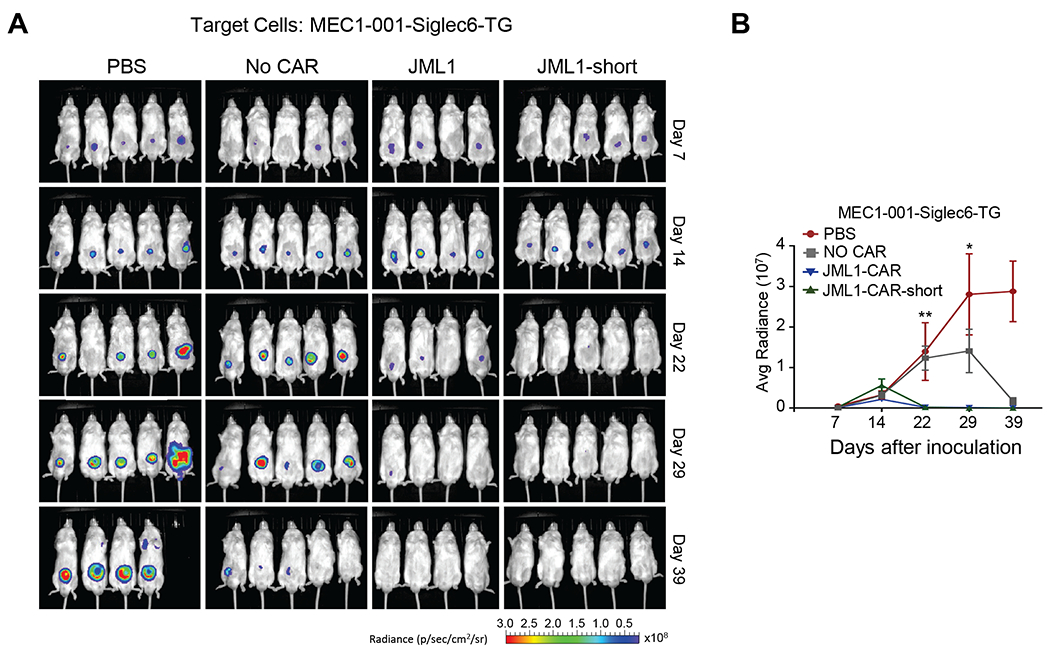
A) Image of in vivo luminescence at different times after injection of MEC1-001-Siglec-6-TG cells and the indicated CAR-T cells. B) Quantitative analysis of luminescence observed in (A) at the indicated times. Average radiance corresponds to [p/s/cm2/sr]. T-test of JML1-CAR against No CAR n=5. p<0.01 (**) or p<0.05 (*). Experiments of groups of 5 mice shown were repeated two times.
Altogether, these results show that both JML1-CAR and JML1-CAR-short are equally efficient with respect to eliminating CLL tumors expressing Siglec-6 in xenograft mouse models.
In summary, we have identified that Siglec-6, an antigen previously shown to be expressed in primary CLL cells, present restricted expression in cells of the immune system and that Siglec-6 protein is not present in healthy tissues or HSCs, making it a suitable target for immunotherapy. We further developed a fully human, human-derived CAR-T construct against Siglec-6 and characterized the ability of Siglec-6-specific CAR-T cells to eliminate Siglec-6 positive CLL cells in vitro and in vivo.
Discussion
We developed and evaluated CAR-T cells specific for Siglec-6, a lectin expressed by CLL cells (17), as a possible novel therapy for CLL. High level of Siglec-6 expression has been reported in placenta, activated B-cells and mast cells (12, 21). We observed that Siglec-6 was not expressed in HSCs or in normal healthy tissues except for placenta. Primary and relapsed CLL presented homogenous and highly varied Siglec-6 levels among different individuals.
The anti-Siglec-6-CAR design was based on the human monoclonal antibody JML1, which was identified in a patient that was cured after alloHSCT (17). We observed that a shorter hinge region in JML1-CAR led to higher activation of CAR T-cells and cytotoxic activity when co-cultured with target cells expressing high levels of Siglec-6. This differential activity correlated with JML1 recognition of a membrane-distal epitope in Siglec-6. A short hinge region has been associated with increased activity of CARs that target membrane-distal epitopes (22), and the length of the hinge region determining an optimal synapse for T-cell activation has been reported (23, 24). As a possible mechanism for these effects it was postulated that a too long inter-space would not exclude the phosphatase CD45 from the synapse leading to inefficient activation of T-cells (25).
JML1-CAR T-cells efficiently eliminated CLL cell lines in vitro and in vivo, and T-cells not containing the JML1-CARs were able to eliminate the tumor but with slower kinetics. This non-specific antitumor response is most likely due to the allogenic nature of cell lines and not specific to the MEC1 model, as we observed a similar effect on the cytotoxicity against U937 cells. Importantly, JML1-CARs were able to significantly reduce primary CLL cells in vitro, and the remaining CLL cells were Siglec-6 negative. As expected, CLL cells remaining after CD19-CAR treatment were Siglec-6 positive, suggesting that they could still be susceptible to JML1-CAR cytotoxicity.
Several points should be considered before translating our findings to the clinic. Although we have not detected target expression in HSCs, additional safety controls such as possible on-target/off-cancer cytotoxicity against immune progenitors should be evaluated. As JML1-CAR exerted a lower effector function than CD19-CAR, JML1-CARs should be first considered as complementary to CD19-CAR therapy. Finally, the high heterogeneity of Siglec-6 expression between different CLL samples, the higher activity of JML1-CAR against cells expressing high Siglec-6 levels, and a possible downregulation of Siglec-6 after CAR treatment may negatively affect efficacy and selection of high Siglec-6 expressing CLL may be required.
Altogether, we have identified Siglec-6 as a possible target to CLL and showed its restricted expression in healthy tissue. We further generated a fully human human-derived anti-Siglec-6 CAR and showed it effectively eliminated CLL cells in vitro and in xenograft models.
Supplementary Material
Acknowledgments
We thank Dr. James N. Kochenderfer for providing the CD19-CAR construct.
This work was supported by an NIH, Bench to Bedside and back Program (Grant ID: 600897) to DK, REG, SP and CR. CR also acknowledges funding by NIH grant R21 CA229961.
Footnotes
Conflict of Interest Disclosures: CR is an inventor of U.S. Patent 8,877,199 which claims monoclonal antibody JML1. All other authors declare no conflict of interests
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–15. [DOI] [PubMed] [Google Scholar]
- 2.Kipps TJ, Stevenson FK, Wu CJ, Croce CM, Packham G, Wierda WG, et al. Chronic lymphocytic leukaemia. Nat Rev Dis Primers. 2017;3:17008. [DOI] [PubMed] [Google Scholar]
- 3.Wiestner A The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica. 2015;100(12):1495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood. 2017;129(11):1469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370(24):2286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyiadzis M, Foon KA, Pavletic S. Hematopoietic stem cell transplantation for chronic lymphocytic leukemia: potential cure for an incurable disease. Expert Opin Biol Ther. 2007;7(12):1789–97. [DOI] [PubMed] [Google Scholar]
- 7.Mato A, Porter DL. A drive through cellular therapy for CLL in 2015: allogeneic cell transplantation and CARs. Blood. 2015;126(4):478–85. [DOI] [PubMed] [Google Scholar]
- 8.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019;25(9):1341–55. [DOI] [PubMed] [Google Scholar]
- 9.Ruella M, Maus MV. Catch me if you can: Leukemia Escape after CD19-Directed T Cell Immunotherapies. Comput Struct Biotechnol J. 2016;14:357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018;8(10):1219–26. [DOI] [PubMed] [Google Scholar]
- 12.O’Reilly MK, Paulson JC. Siglecs as targets for therapy in immune-cell-mediated disease. Trends Pharmacol Sci. 2009;30(5):240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pehlivan KC, Duncan BB, Lee DW. CAR-T Cell Therapy for Acute Lymphoblastic Leukemia: Transforming the Treatment of Relapsed and Refractory Disease. Curr Hematol Malig Rep. 2018;13(5):396–406. [DOI] [PubMed] [Google Scholar]
- 14.Rumer KK, Uyenishi J, Hoffman MC, Fisher BM, Winn VD. Siglec-6 expression is increased in placentas from pregnancies complicated by preterm preeclampsia. Reprod Sci. 2013;20(6):646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Y, Blokhuis BRJ, Diks MAP, Keshavarzian A, Garssen J, Redegeld FA. Functional Inhibitory Siglec-6 Is Upregulated in Human Colorectal Cancer-Associated Mast Cells. Front Immunol. 2018;9:2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kardava L, Moir S, Wang W, Ho J, Buckner CM, Posada JG, et al. Attenuation of HIV-associated human B cell exhaustion by siRNA downregulation of inhibitory receptors. J Clin Invest. 2011;121(7):2614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang J, Peng H, Shaffer BC, Baskar S, Wecken IC, Cyr MG, et al. Siglec-6 on Chronic Lymphocytic Leukemia Cells Is a Target for Post-Allogeneic Hematopoietic Stem Cell Transplantation Antibodies. Cancer Immunol Res. 2018;6(9):1008–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baskar S, Suschak JM, Samija I, Srinivasan R, Childs RW, Pavletic SZ, et al. A human monoclonal antibody drug and target discovery platform for B-cell chronic lymphocytic leukemia based on allogeneic hematopoietic stem cell transplantation and phage display. Blood. 2009;114(20):4494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32(7):689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hermans IF, Silk JD, Yang J, Palmowski MJ, Gileadi U, McCarthy C, et al. The VITAL assay: a versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J Immunol Methods. 2004;285(1):25–40. [DOI] [PubMed] [Google Scholar]
- 21.Patel N, Brinkman-Van der Linden EC, Altmann SW, Gish K, Balasubramanian S, Timans JC, et al. OB-BP1/Siglec-6. a leptin- and sialic acid-binding protein of the immunoglobulin superfamily. J Biol Chem. 1999;274(32):22729–38. [DOI] [PubMed] [Google Scholar]
- 22.Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–11. [DOI] [PubMed] [Google Scholar]
- 23.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng H, Nerreter T, Chang J, Qi J, Li X, Karunadharma P, et al. Mining Naive Rabbit Antibody Repertoires by Phage Display for Monoclonal Antibodies of Therapeutic Utility. J Mol Biol. 2017;429(19):2954–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015;36(8):494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.