

Abstract
Glaucoma causes loss of vision through degeneration of the retinal ganglion cell (RGC) projection to the brain. The disease is characterized by sensitivity to intraocular pressure (IOP) conveyed through the optic nerve head, through which RGC axons pass unmyelinated to form the optic nerve. From this point, a pathogenic triumvirate comprising inflammatory, oxidative, and metabolic stress influence both proximal structures in the retina and distal structures in the optic projection. This review focuses on metabolic stress and how the optic projection may compensate through novel adaptive mechanisms to protect excitatory signaling to the brain. In the retina and proximal nerve head, the unmyelinated RGC axon segment is energy-inefficient, which leads to increased demand for adenosine-5′-triphosphate (ATP) at the risk of vulnerability to Ca2+-related metabolic and oxidative pressure. This vulnerability may underlie the bidirectional nature of progression. However, recent evidence highlights that the optic projection in glaucoma is not passive but rather demonstrates adaptive processes that may push back against neurodegeneration. In the retina, even as synaptic and dendritic pruning ensues, early progression involves enhanced excitability of RGCs. Enhancement involves depolarization of the resting membrane potential and increased response to light, independent of RGC morphological type. This response is axogenic, arising from increased levels and translocation of voltage-gated sodium channels (NaV) in the unmyelinated segment. During this same early period, large-scale networks of gap-junction coupled astrocytes redistribute metabolic resources to the optic projection stressed by elevated IOP to slow loss of axon function. This redistribution may reflect more local remodeling, as astrocyte processes respond to focal metabolic duress by boosting glycogen turnover in response to axonal activity in an effort to promote survival of the healthiest axons. Both enhanced excitability and metabolic redistribution are transient, indicating that the same adaptive mechanisms that apparently serve to slow progression ultimately may be too expensive for the system to sustain over longer periods.
Keywords: glaucoma, neurodegeneration, axon degeneration, astrocytes, metabolic stress, oxidative stress, adaptive remodeling, gap junctions
1. Introduction
1.1. Glaucoma and IOP
Glaucoma is a group of optic neuropathies that together represent the world’s principal cause of irreversible blindness – that is, permanent blindness arising from degeneration of neural tissues in the optic projection from retina to brain. Incidence of glaucoma is increasing as the population ages; approximately 112 million will be afflicted worldwide by 2040 (Tham et al., 2014). Vision loss in glaucoma follows a characteristic pattern of field deficits that progress in wedges from one retinotopic sector to the next (Elze et al., 2015). These deficits are linked to sensitivity to intraocular pressure (IOP), which reflects the production and drainage of aqueous fluid in the anterior eye. This sensitivity is the hallmark feature of glaucoma, which we probe experimentally through induced elevations in IOP using animal models ranging from mice to non-human primates (Bouhenni et al, 2012). We elevate IOP in such models to identify mechanisms that are sensitive to IOP and determine whether modulating their activity or expression, either pharmacologically or genetically, slows progression in one or more models. In doing so we attempt to distinguish elements that are truly causal versus those that result secondarily from neurodegeneration. Importantly, models rarely capture variation in types of glaucoma and in afflicted patient populations, both of which lead to great diversity in IOP and in response to treatment (Susanna et al., 2015).
Glaucoma often (but not always) includes morphological changes in the appearance of the optic nerve head that may indicate nerve atrophy along the perimeter of the optic disc. These changes are associated with optic nerve degeneration but are not necessarily unique to glaucoma (Piette and Sergott, 2006). Rather, they likely reflect a collection of pathophysiological factors that biomechanically influence the structure and morphology of the posterior segment in glaucoma (Burgoyne, 2011). Besides age and IOP, other prominent risk factors are race, severe myopia, central corneal thickness, and familial history along with several gene variants linked to either congenital glaucoma or to greater susceptibility to the disease (Coleman and Miglior, 2008; Weinreb et al., 2014).
The diversity of risk factors for glaucoma speaks to the etiological complexity of the disease. Glaucoma comprises several types that each in some way involve IOP. The different forms of glaucoma tend to fall into two main categories depending on the geometry of the iridocorneal angle where the iris and cornea meet (King et al., 2013). In open-angle glaucoma, the angle’s width allows normal outflow of aqueous fluid from the anterior chamber to the drainage canals in the trabecular meshwork. Primary open-angle glaucoma (or POAG) is the most common type and is associated with increasing resistance within the aqueous outflow pathways. POAG encompasses a large IOP range and includes a large proportion of what is known as normal pressure (or normal tension) glaucoma, which occurs in the absence of elevated IOP. The caveat on this point is that some normal pressure glaucoma may present with an etiology separate from that of POAG, involving one or more autoimmune components (Rieck, 2013). In closed-angle glaucoma, narrowing or obstruction of the outflow path challenges drainage of aqueous fluid, which can lead to acute elevations in IOP or even severe ocular pain.
The relative incidence of glaucoma varies widely from population to population, reflecting variation in genetic and other factors that influence anterior segment morphology and susceptibility (Coleman and Miglior, 2008; Weinreb et al., 2014). For all types of glaucoma, clinical treatments attempt to lower IOP through a regimen of topical hypotensive drugs, surgery to facilitate aqueous fluid outflow, or both. Despite this investment in time and resources, many patients continue to lose vision, though there is great variability in the number of estimated non-responders across various studies (Susanna et al., 2015). For patients whose optic projection continues to degenerate despite efforts to lower IOP, we can do little. That we do not understand fully the mechanisms by which sensitivity to IOP exerts its destructive influence emphasizes the need for new therapies that target neurodegeneration directly, whether by protection, repair or outright regeneration (Calkins et al., 2017). Our hope is that such therapies will arise by pinpointing ever-earlier events and markers for progression within the optic projection that could underlie sensitivity to IOP and predispose the system to degeneration (Beykin et al., 2020). Importantly, glaucoma is not linear – the same level of IOP may not have the same influence on the optic projection at different points in progression, even within the same patient. In any case, the discussion of neurodegeneration that follows is agnostic in terms of anterior segment etiology – it focuses on what comes after the threshold for IOP-related pathogenesis is reached – with the following caveat. This review focuses on progressive neurodegeneration, which is typically associated with POAG, not on acute injury such as might arise from sharp IOP elevations or dramatic changes in systemic blood pressure or flow to the eye.
1.2. Neurodegeneration in Glaucoma is Bidirectional
The optic projection is formed by the 1.5 million or so axons of retinal ganglion cell (RGC) neurons. Due to the necessity of transparency for the path of light, RGC axons are unmyelinated in the retina and remain so in exiting the eye and entering the pre-laminar region of the optic nerve head (Figure 1). There, the axons form well-defined bundle-like fascicles that penetrate a dense plexus of capillaries, connective tissues, and ramifying processes of astrocyte glia. In forming the optic nerve, these fascicles pass through the lamina cribrosa, which is a porous, multi-layered collagenous structure that couples to the peripapillary sclera (Girard et al., 2011; Quigley, 2015). Because of their architectural orientation and support they provide through interactions with the scleral wall these collagen layers are often called “beams”. Astrocyte processes line the cribrosa pores and, with the cribrosa cells themselves, form a glial membrane that sheathes the passing axon fascicles. In leaving the lamina cribrosa RGC axons finally become myelinated in the retrolaminar region through interactions with oligodendrocytes, as the collection of fascicles form the optic nerve proper. The rodent optic nerve lacks a lamina cribrosa but instead demonstrates a specialized glial lamina in which astrocyte processes ramify transversely to form a honeycomb-like structure of tubes through which unmyelinated axon bundles pass (Sun et al., 2009; Wang et al., 2017). Like the retrolaminar region in humans and non-human primates, RGC axons become myelinated in a transition zone posterior to the glia lamina. Both the central retinal artery and central retinal vein penetrate the disc of the optic nerve head, serving the surrounding tissues through a network of small lateral capillaries.
Figure 1. Basic structure of the optic nerve head.
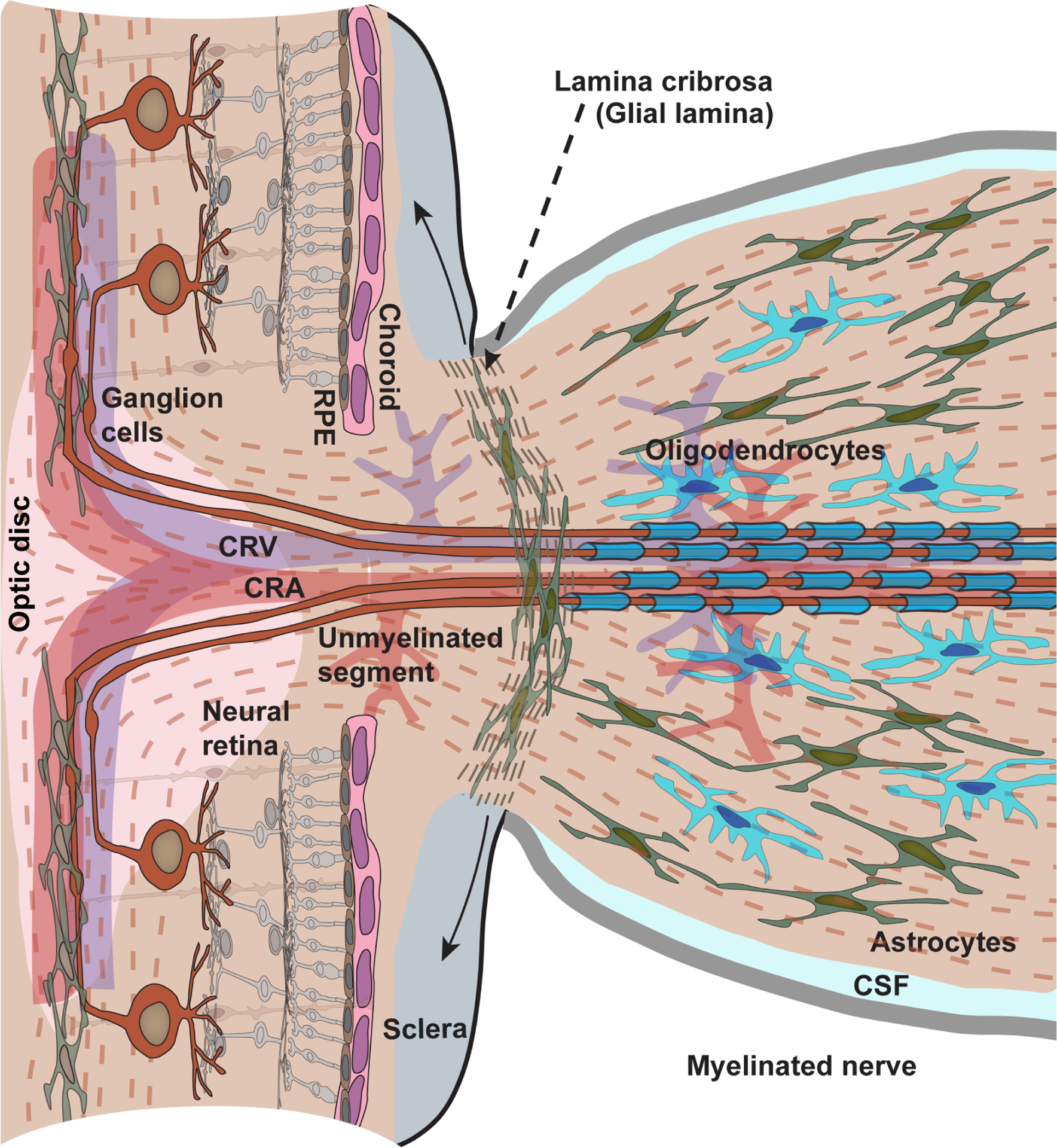
The vertebrate retina is inverted with respect to the path of light; the photoreceptor-renewing retinal pigment epithelium (RPE) and choroid are most proximal to the sclera. Retinal ganglion cell axons are unmyelinated in the retina and in entering the nerve head through to the prelaminar region. In passing through the lamina cribrosa, axons become myelinated by oligodendrocytes, first in the retrolaminar region and then throughout the optic nerve, which is bathed in cerebrospinal fluid (CSF). The cribrosa is a critical site of biomechanical coupling to ocular stress through attachments with the peripapillary region of the sclera (solid arrows); its normal backward bowing becomes dramatically more pronounced in glaucoma. In place of a well-defined cribrosa and retrolaminar region the rodent nerve head demonstrates a glial lamina of densely distributed astrocytes and a myelination transition zone, respectively. Astrocyte glia also interact with RGC axons in the retina and in the myelinated segment of the optic nerve. The central retina artery (CRA) and vein (CRV) enter the retina through the optic disc.
The optic nerve head is a critical site of pathogenesis in glaucoma, as studies using non-human primates and other mammals with a lamina cribrosa, human donor tissue, and non-invasive imaging of patients have demonstrated. There, multiple neuronal, vascular, glial and biomechanical components influence sensitivity of the optic projection to IOP and its susceptibility to degeneration for a given IOP (Sigal and Ethier, 2009; Burgoyne, 2011; Girard et al., 2011; Quigley, 2015; Wareham et al., 2021). This confluence means that the threshold for IOP-related damage will vary between patients and across different ages. Factors related to blood flow regulation and systemic inflammation in glaucoma influence the biophysical and physiological properties of the cells and tissues that comprise the nerve head. These properties and how they change with aging contribute to the accumulated effects of strain in connective tissues, diminished nutrient diffusion from capillaries to axon bundles, and variation in the capacity of the optic nerve to handle glaucomatous stress and therefore its sensitivity to IOP (Burgoyne, 2011; Downs, 2015).
From the nerve head, RGC degeneration in glaucoma progresses both in the anterograde direction towards central projection sites and in the retrograde direction back to the cell body and dendritic arbor in the inner retina (Figure 2). In the anterograde direction, the optic nerve projects to several subcortical structures, the most distal of which is the superior colliculus (SC). In rodents, nearly all RGC axons project to the colliculus with collaterals extending to the other more proximal nuclei. In primates and many other mammals, the lateral geniculate nucleus (LGN) is the primary projection; in all mammals, the LGN forms the direct projection into the primary visual cortex. An early marker of progression in rodents is degradation of anterograde axonal transport from the retina to central brain projection sites, followed by disassembly of the myelinated axon and degeneration of post-synaptic targets (Crish et al,, 2010;Calkins and Horner, 2012; Calkins, 2012; Crish and Calkins, 2015). In the retrograde direction, RGC dendritic arbors lose complexity as excitatory synapses are eliminated in a complement-dependent process (Agostinone and Di Polo, 2015; Berry et al., 2015; Williams et al., 2016). Pruning due to acute axonal injury in rodents involves loss of activity from mammalian target of rapamycin (or mTOR), which may have bearing in human glaucoma (Agostinone et al., 2018). Multiple types of RGC across vertebrate species reflect the diversity of functional and behavioral specialization. Susceptibility to dendritic pruning likely depends on RGC type, although variability between studies in mice is considerable (El-Danaf and Huberman, 2015; Ou et al., 2016; Risner et al., 2018).
Figure 2. Visual projection in glaucoma.
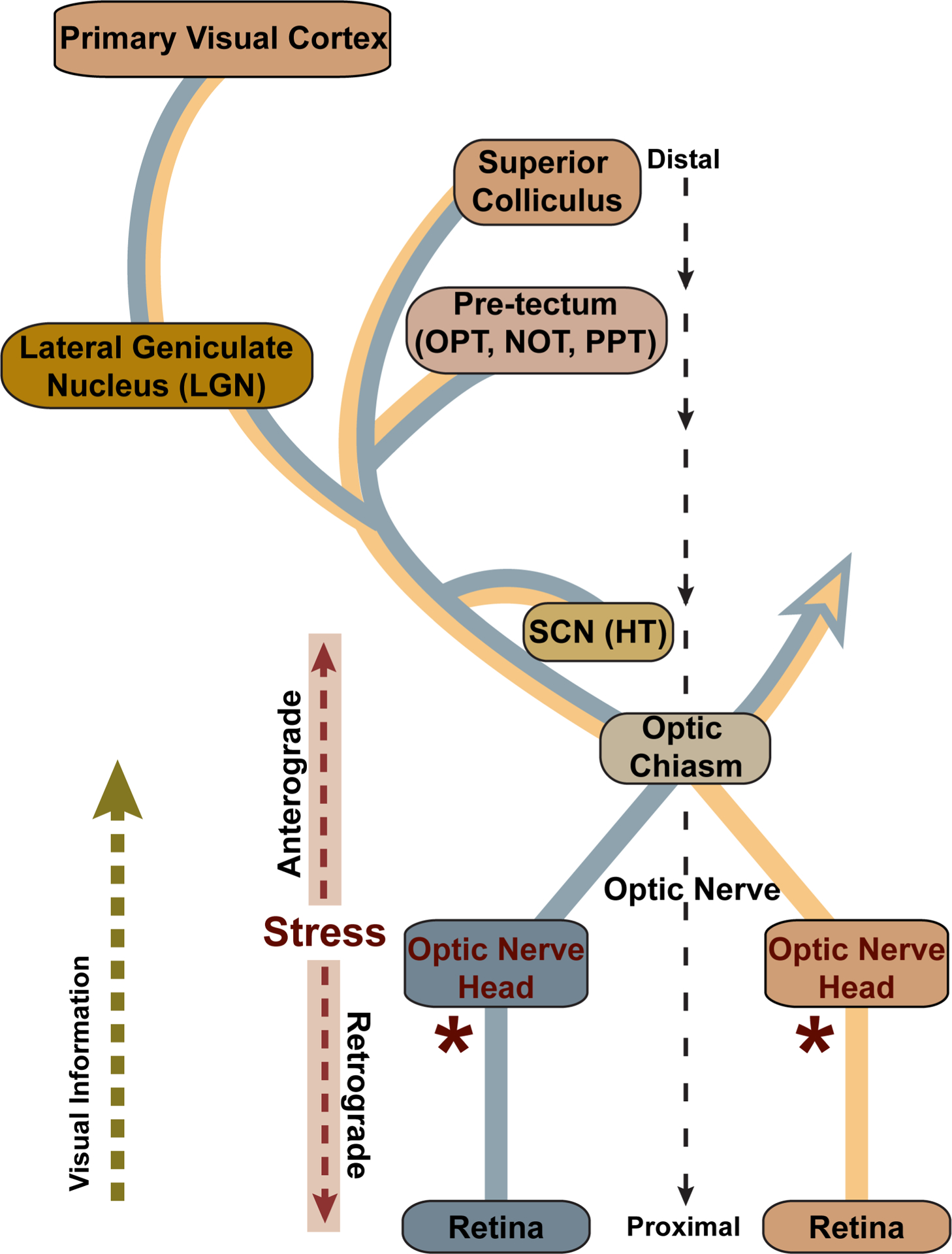
Retinal ganglion cell axons project through the optic nerve head and at the optic chiasm join either the ipsilateral or contralateral optic tract to central brain targets. The strength of each projection varies by species; the rodent retina projects primarily contralaterally, while the primate retina projects equally to both. Central targets include the suprachiasmatic nucleus (SCN) of the hypothalamus (HT) and the olivary pretectal nucleus (OPT), nucleus of the optic tract (NOT), and posterior pretectal (PPT) nucleus of the pretectum. The lateral geniculate nucleus (LGN) of the thalamus is the primary RGC recipient in primates, while in rodents all or nearly all ganglion cells project to the superior colliculus (SC), while extending axon collaterals to nuclei more proximal to the retina. In all mammals the SC is the most distal direct target, while the LGN provides all direct projection to the primary visual cortex. In the glaucoma, stress at the optic nerve head (*) is conveyed along the ganglion cell axon both towards the brain, in the anterograde direction, and towards the retina, in the retrograde direction. Modified from Crish and Calkins (2015).
The nerve head is an important nexus for other reasons. As distal (anterograde) and proximal (retrograde) degenerative programs unfold, the RGC body persists much further into progression, long after axon disassembly in the optic nerve and pruning of the dendritic arbor in the retina (reviewed in Calkins, 2012). Importantly, the unmyelinated axon segment also remains intact along with the RGC body, a fact appreciated decades ago from post-mortem histological examination of the human glaucomatous nerve head (Vrabec, 1976). More recently, specific transgenes in mouse models that ameliorate either axonal (Wlds) or somatic (Bax−/−) degeneration demonstrate the persistence of the unmyelinated segment and that distal and proximal degenerative programs progress to some extent independently (Schlamp, et al, 2006; Beirowski et al., 2008; Howell et al, 2007, 2013). Thus, the junction in the nerve head between the unmyelinated and myelinated axon segments represents a critical locus in progression. The following section examines physiological properties that may contribute to its importance in glaucoma.
2. Axonal Metabolism
2.1. Axons are Expensive; RGC Axons are Worse
The visual system is highly energetically expensive (Niven and Laughlin, 2008; Wong-Riley, 2010). Levels of adenosine-5′-triphosphate (ATP) are actually higher in retina than in other prominent brain regions (Zhu et al., 2020). Axons in particular are metabolically demanding. They require a large supply of mitochondrial-generated ATP along their length from cell body to terminals. This supports anterograde transport of mitochondria and synthesized proteins from the cell body to axon terminals, retrograde transport from the terminals back to the cell body, the generation and propagation of action potentials to signal post-synaptic neurons, and reestablishment of the resting membrane potential (Harris and Attwell, 2012; Misgeld and Schwarz, 2017). Excellent recent reviews highlight the metabolic challenges facing the RGC in health and disease (Yu et al., 2013; Osborne et al., 2016; Inman and Harun-Or-Rashid, 2017; Casson et al., 2020).
The capacity to generate action potentials requires not only ATP, but a high density of voltage-gated sodium channels, or NaV, whose distribution along the axon tracks well with that of the mitochondria that provide them energy (Barron et al., 2004). Axons normally transport mitochondria to regions of high metabolic demand. Where axons are myelinated, both mitochondria and NaV cluster at nodes of Ranvier, which support saltatory conductance of action potentials over great distances. While lack of myelination for RGC axons in the retina is beneficial from the standpoint of optics, transparency comes at a price. Unmyelinated axon segments are far less efficient at maintaining ionic gradients necessary for propagating action potentials, about 10-fold less so than the myelinated segment (Perge et al., 2009). Due to the absence of saltatory conduction, the unmyelinated segment requires far greater energy. Thus, the unmyelinated portion of the RGC axon contains a greater abundance of both mitochondria and NaV subunits, as well as the mitochondrial enzymes COX (cytochrome c oxidase) and succinate dehydrogenase, necessary for ATP production (Andrews et al., 1999; Balaratnasingam et al., 2009). As they course through the nerve fiber layer of the retina on their way to the optic nerve head, RGC axons contain intermittent varicosities that are packed with mitochondria and mark points of inter-axonal and axonal-astrocyte contact (Wang et al., 2003). These also contain NaV subunits (Figure 3). In the nerve fiber layer and prelaminar nerve head, the RGC and its axon contain most of the mitochondria, unlike the optic nerve where astrocytes appear to be most rich (Perge et al., 2009).
Figure 3. RGC axonal ultrastructure.
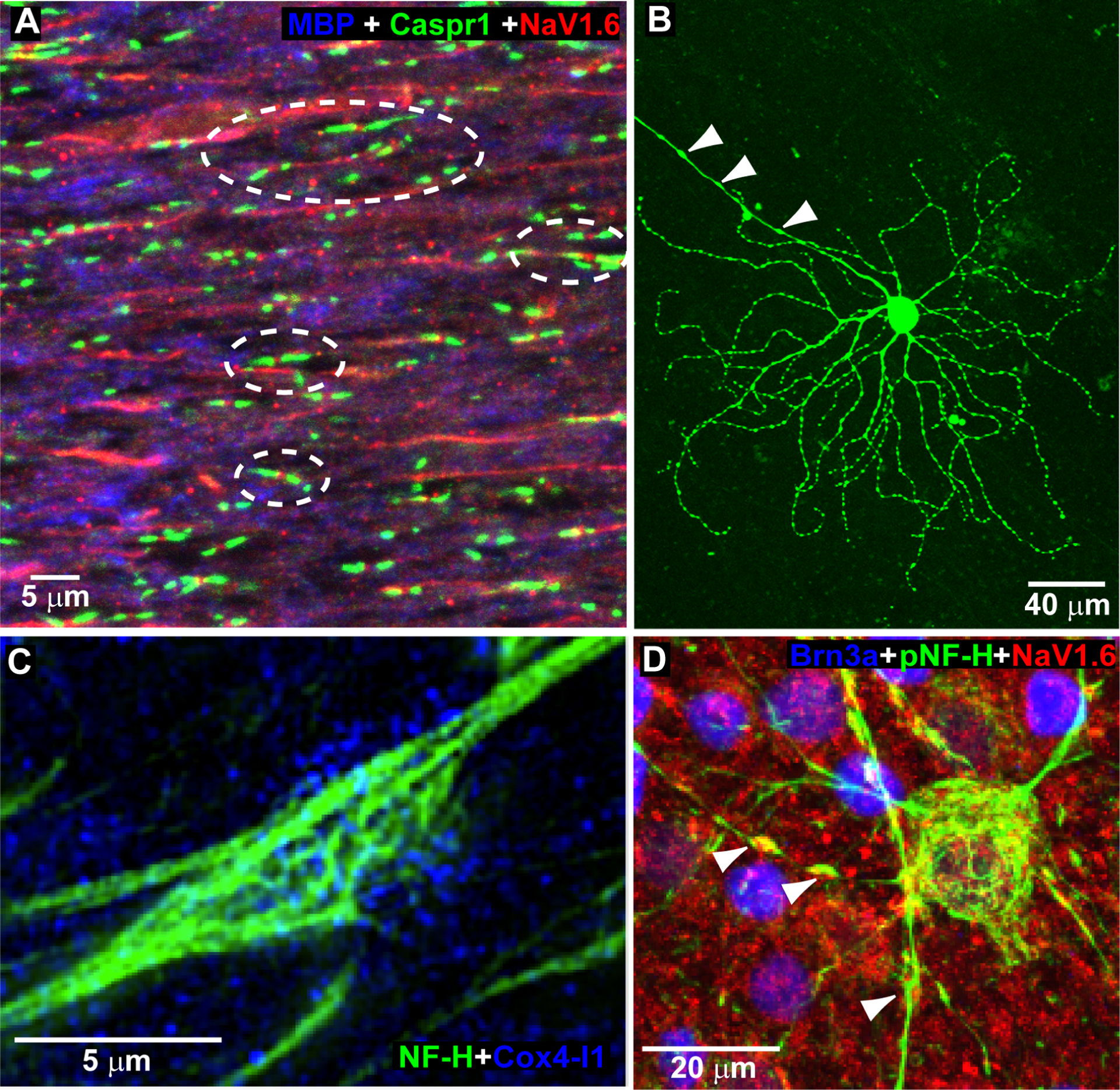
A. Mouse optic nerve labeled with antibodies against myelin basic protein (MBP) showing paranodal contactin-associated protein 1 (Caspr1) flanking nodes of Ranvier containing the voltage-gated sodium channel NaV1.6 subunit. B. Mouse ganglion cell in wholemount preparation following intracellular dye filling to illuminate dendritic arbor and unmyelinated axon segment rich in varicosities (arrowheads). C. High magnification micrograph of RGC axonal varicosity labeled for neurofilament heavy chains (NF-H) contains mitochondria rich in the enzyme cytochrome c oxidase subunit 4 isoform 1 (Cox4-I1). D. Phosphorylated neurofilament heavy chains (pNF-H) in Brn3a-labeled ganglion cell highlights varicosities in the unmyelinated axon segment containing NaV1.6 (arrowheads).
The caliber of the RGC axon does not increase at the transition between unmyelinated to myelinated segment, as it does for other axons (Perge et al., 2012). Rather, the ganglion cell axon is uniformly thin. To illustrate this point, though the unmyelinated segment is about 50-fold longer than the cone photoreceptor axon its mean diameter is only half (Perge et al., 2012). Thus, the difference of mitochondrial and NaV density between the unmyelinated and myelinated segment does not reflect a bottleneck at the nerve head, such as would occur at the transition from a narrow to wide opening, but rather constitutive differences in metabolic demand and therefore distribution (Bristow et al., 2002; Yu-Wai-Man et al., 2011). Indeed, like other small axons (Bechtold et al., 2005), thinness places the RGC axon at a natural disadvantage in conditions of metabolic stress due to low capacity to buffer of intraaxonal Ca2+ (Stys, 2004; Reeves et al., 2012), even more so for the unmyelinated segment due to higher need for mitochondrial ATP and Ca2+ buffering (Franke et al., 2006; Lee et al., 2011).
2.2. RGC Axonal Stress Challenges the Limited Supply of ATP
Levels of ATP in the optic nerve directly reflect its capacity to transmit action potentials (Trevisiol et al., 2017). This activity promotes signaling to post-synaptic neurons, which is necessary for focal secretion and retrograde transport of brain-derived neurotrophic factor (BDNF; Lu, 2003; Lou et al., 2005). Axons challenged by stress or injury require increased levels of ATP to support transport of proteins necessary for remodeling and maintenance of cytoskeletal architecture (Her and Goldstein, 2008; Bradke et al., 2012; Zhu et al., 2020). Where myelin is damaged or lost, local stationary mitochondria increase to compensate for diminished pools of ATP (Kiryu-Seo et al., 2010). The rapidity of this response is illustrated aptly for RGCs maintained in culture, in which acute (induced) axonal stress diminishes mitochondrial transport, with recovery of transport linked to greater cell survival (Yokota et al., 2015).
Even with mitochondrial compensation, chronic neuronal stress leads to reduced ATP and increased mitochondrial volume, both of which reduce motility in both anterograde and retrograde directions along microtubule and actin molecular tracks (Chang and Reynolds 2006). Diminished ATP in axon projections greatly reduce active transport and the capacity to maintain microtubule structures within the axon skeleton (De Vos et al., 2007, 2008; Prokop, 2013). The mitochondrial capacity of axons in the central nervous system reduces significantly during aging (Boumezbeur et al., 2010), which may contribute to the age-related loss of axons in the optic nerve that occurs for all mammalian species (Calkins, 2013).
Nerve aging and its associated bioenergetic ramification also influences glaucoma. A useful progressive animal model of glaucoma that has proven convenient for studying RGC axon transport is the DBA/2J mouse (Crish et al., 2010). This inbred strain demonstrates age-related elevations in IOP that correlate with RGC axon degeneration in the optic nerve (Inman et al., 2006; Cooper et al., 2016). In this model age reduces optic nerve mitochondrial COX (cytochrome c oxidase), necessary for ATP production, ATP itself, axon transport, and signaling capacity as measured by the compound action potential; elevated IOP exacerbates these reductions (Baltan, Inman et al. 2010). Similarly aging diminishes mitochondrial transport in non-glaucomatous mouse optic nerve and lengthens axonal segments depleted of mitochondria; induced elevations in IOP also accelerate these effects (Takihara et al., 2015).
Reduced mitochondrial motility and transport to distal neuronal processes contributes to pathogenesis in Alzheimer’s and other neurodegenerative diseases (Ebneth, Godemann et al. 1998, Chang and Reynolds 2006, Rintoul and Reynolds 2010). In rodent models of glaucoma, degradation of RGC axonal anterograde transport to central brain targets is an early harbinger of axon dysfunction, which precedes axon disassembly (Calkins, 2012; Cooper et al., 2020). As with depletion of ATP, age is the predominant factor influencing transport loss with IOP serving as an additional stressor (Crish et al, 2010; Fiedorowicz et al., 2018). Figure 4 summarizes data from the DBA/2J mouse model to demonstrate this point. Transport fails first within the distal-most termination in the SC, with degradation progressing distal-to-proximal from that point in the projection (Crish et al., 2010; 2013; Crish and Calkins, 2015; Ward et al., 2014). This pattern in which distal sites are affected first seems to be a general property of axon injury (Wang et al., 2012). As axons struggle, cytoskeletal transport of neurofilaments needed for repair becomes increasingly important. Transport is fastest in the unmyelinated axon segment and slows with distance from the retina (Li, Jung et al. 2012). Reductions in ATP likely lead to observed accumulations of phosphorylated neurofilaments at the myelination transition zone (Howell et al., 2007; Soto et al., 2008).
Figure 4. Age is predominating determinate of anterograde axonal transport.
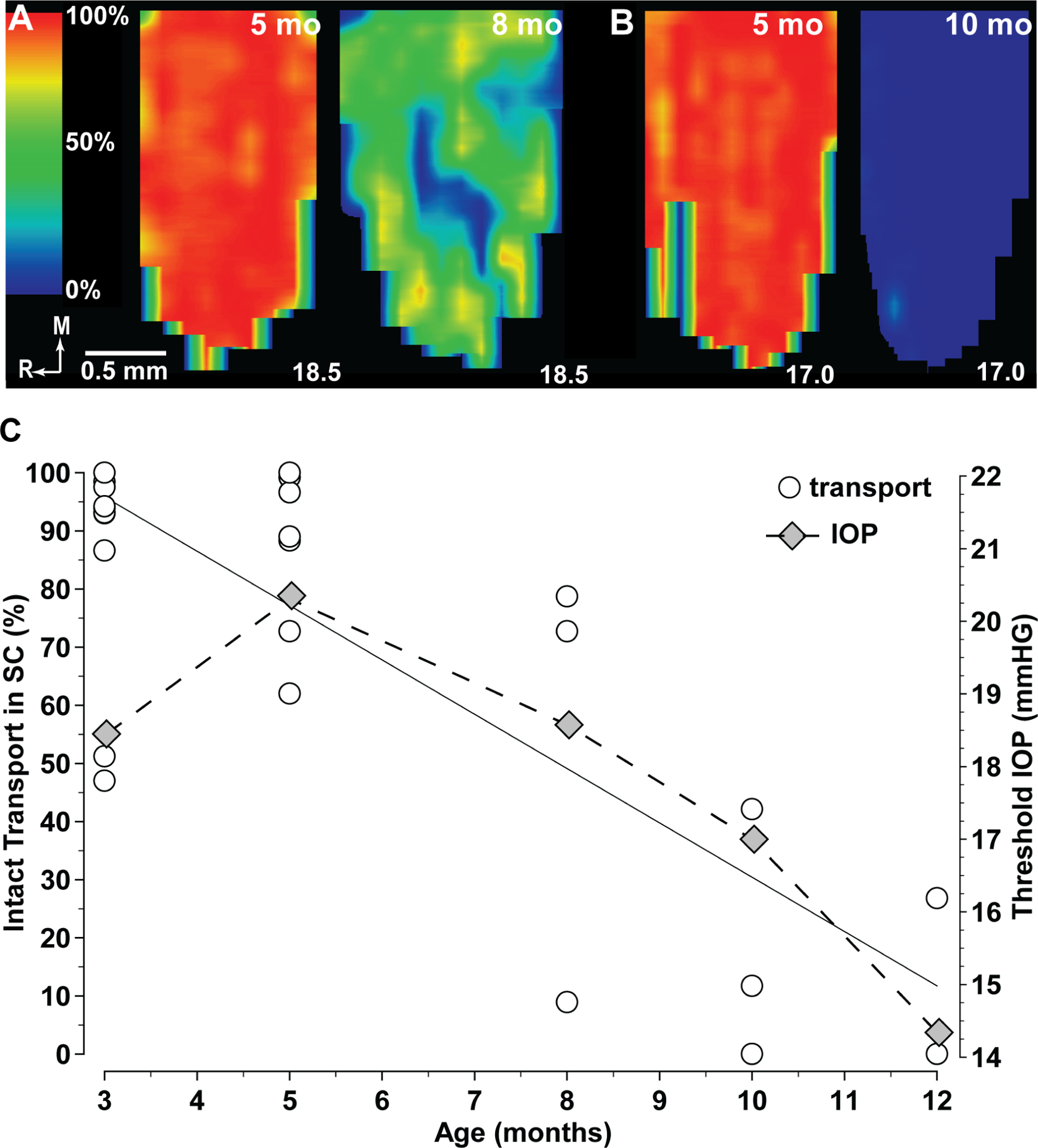
A. Retinotopic maps of superior colliculus (SC) from five-month (left) and eight-month (right) DBA/2J mice, each receiving retinal projections from eyes with mean lifetime IOP of 18.5 mm Hg. Maps show levels of cholera toxin B transported from the retina varying from 100% (red) to 50% (green) to 0% (blue). B. Maps of five-month (left) and 10-month (right) DBA/2J SC, with corresponding mean IOP of 17.0 mm Hg. C. Intact anterograde transport of cholera toxin b to the DBA/2J SC expressed as the fraction of the retinotopic map with ≥ 70% signal density vs. age in months (top). All SC received retinal projection from eyes with mean lifetime IOP < 20 mm Hg. Best-fitting regression shows a significant correlation (r2=0.64; p<0.001). Threshold mean lifetime IOP to induce a 50% loss of intact anterograde transport to the DBA/2J SC diminishes with age (bottom).
Age-related loss of anterograde transport in the DBA/2J is concomitant with malformed and reduced mitochondrial cristae and oxidative capacity and increased mitophagy (Ju et al., 2008; Coughlin et al., 2015; Kleesattel et al., 2015); enhancing mitochondrial function systemically reduces axon degeneration and prevents transport loss (Ju et al., 2010; Harun-Or-Rashid et al., 2018). The pattern of progressive failure in anterograde transport reflects prior depletion of metabolic resources available to the RGC axon, the necessity of intact mitochondrial motility, and the higher metabolic expense of anterograde vs. retrograde transport, which persists as long as the RGC axon itself (Calkins, 2012; Kleesattel et al., 2015; Inman and Harun-Or-Rashid, 2017).
2.3. Metabolic and Oxidative Stress: Two Sides of the Same Coin
Depletion of metabolic resources and the RGC intrinsic adaptation to counter this depletion is linked to two inter-related pathogenic arms. The first involves dysregulation of Ca2+ homeostasis (Crish and Calkins, 2011; Wang et al., 2012; Sappington et al., 2015), while the second relates to mitochondrial oxidative stress (reviewed in Tezel., 2006; Wareham and Calkins, 2020; Wareham et al., 2021). Diminished ATP and concurrent loss of signaling capacity in the glaucomatous optic nerve includes failure of Na+/K+ ion pumps (Ames, 2000). As the Na+/K+ ion exchange fails, Ca2+ accumulates within the axon as increased axoplasmic Na+ promotes reverse operation (Stirling and Stys, 2010). This imbalance leads to increased Ca+2-dependent protease activity, which degrades the axonal cytoskeleton (Mattson, 2007; Knöferle et al., 2010; Crish and Calkins, 2011). These effects are compounded by diminished anterograde transport of cytoskeletal components to distal axon segments (Her and Goldstein, 2008; Bradke et al., 2012; Zhu et al., 2020). Dysregulation of axonal Ca2+ is a major contributor to axonal disassembly and reducing excessive Ca2+ is efficacious in slowing degeneration in a variety of conditions (Wang et al., 2012; Vargas et al., 2015). This includes experimental glaucoma and other optic neuropathies (Govindarajan et al., 2008; Huang et al., 2008; Das et al., 2013). Excessive Ca2+ is also linked to and exacerbated by diminished mitochondrial motility (Rintoul and Reynolds, 2010; Liao et al., 2017). Interestingly, patients with ocular hypertension (elevated IOP) who do not develop optic neuropathy demonstrate a mitochondrial phenotype characterized by higher Ca2+ buffering capabilities compared with mitochondria from control patients (Lascaratos et al., 2015).
The optic nerve’s intrinsic attempts to restore functional levels of ATP are not without cost. During acute injury to the optic nerve, compensatory oxidative phosphorylation to boost mitochondrial ATP output leads to accelerated thinning of the RGC population (Zhu et al., 2020). This reduction is likely due to oxidative stress, which is broadly defined as imbalance between the production of reactive oxygen species (ROS) and the availability of antioxidants or radical scavengers to neutralize them. Reduced mitochondrial motility or increased pockets of stationary mitochondria increase local concentrations of Ca2+, free radicals and ROS to potentially toxic level (Wallace, 1999; Lee et al., 2010; Rintoul and Reynolds, 2010; Liao et al., 2017). In turn, chronic elevations in ROS in glaucoma cause not only oxidative damage, but also impede ATP generation (Chrysostomou et al, 2013). Progression of visual field deficits in glaucoma patients is associated with reduced blood serum levels of the antioxidant enzyme superoxide dismutase (SOD), a major cellular defense against ROS accumulation (Li et al., 2020). Inflammation in glaucoma is likely to exacerbate oxidative stress in the optic nerve head and retina by inhibiting mitochondrial motility via nitric oxide (NO)-dependent signalling pathways (Trivli et al., 2019).
Evidence from the DBA/2J mouse directly links oxidative stress to progressive degeneration of the RGC projection to the brain. Age and IOP both increase retinal levels of markers for lipid peroxidation and other genes and proteins related to oxidative stress; dietary treatment with the anti-oxdidant α-lipoic acid ameliorates these effects and improves RGC survival (Inman et al., 2013). The coenzyme Q10 (CoQ10) helps maintain mitochondrial membrane potential, ATP synthesis and intracellular Ca2+ homeostasis (Zhong et al., 2017); by inhibiting ROS formation, it also reduces oxidative stress in neurodegenerative conditions (Zhang et al., 2017). DBA/2J mice fed a dietary supplement of CoQ10 demonstrate increased retinal mitochondrial transcription factor A, which supports oxidative phosphorylation and ATP production, and reduced oxidative stress with improved axonal integrity and RGC survival (Lee et al., 2014). Interestingly, while α-lipoic acid increased astrocyte remodeling in the retina (Inman et al., 2013), CoQ10 reduced it in optic nerve head (Lee et al., 2014). It is worth noting that neither treatment completely abates progression in rodents. Additional evidence supporting a pathogenic role for oxidative stress in RGC degeneration in glaucoma is reviewed elsewhere (Almasieh et al., 2012).
Since dysregulation of Ca2+ homeostasis and mitochondrial stress are intimately linked, it is curious that the unmyelinated RGC axon segment – which is more susceptible to both – remains as long as it does in glaucoma, outlasting both the myelinated segment and dendritic arbor (Calkins, 2012). The next section examines a phenomenon associated with early progression in an animal model that could shed light on this persistence.
3. Adaptive Responses to Stress
3.1. The Unmyelinated Axon Segment and Enhanced Excitability
Persistence of the unmyelinated axon segment in glaucoma is not simply happenstance but may reflect instead an important functional role in promoting an adaptive RGC response in glaucoma. As with other locations of high mitochondrial density in the axon (Barron et al., 2004), varicosities in the unmyelinated RGC axon segment also contain pockets of localized NaV subunits (Risner et al., 2018; 2020; Figure 3), which comprise multiple types. The initiation and propagation of action potentials requires activation of the NaV1.6 voltage-gated sodium channel while the NaV1.2 subunit facilitates backpropagation of depolarization to the cell body and dendritic arbor (Hu et al., 2009; Spratt et al., 2019). In the retina, both subunits localize predominantly to RGC bodies and axons, consistent with their physiological role in neuronal excitation (Caldwell et al., 2000; Boico et al., 2003; Van Wart and Matthews, 2006; Van Hook et al. 2019).
Studies of progression in mice using microbead-induced elevations in IOP suggest that NaV subunits expressed in the RGC reorganize as part of an early adaptive role in glaucoma. We simplified and standardized microbead occlusion from earlier paradigms to provide an easy method to induce quick, sustainable, and moderate elevations in IOP in rodents (Sappington et al.,, 2010, Chen et al., 2011; Dapper et al., 2013; Calkins et al., 2018) and more recently in non-human primates (Lambert et al., 2019; 2020). In this model a small-volume injection of polystyrene microbeads into the anterior chamber impedes aqueous fluid outflow, causing an increase in IOP of about 35% that persists over several weeks. Mice demonstrate IOP-related deficits in spatial acuity concomitant with degradation of anterograde transport to the SC early, between one and two weeks (Risner et al., 2018; Cooper et al., 2020); axon degeneration in optic nerve follows deficits in axon transport (Ward et al., 2014; Weitlauf et al., 2014).
While distal axon function degrades early in microbead-induced elevations in IOP, at the proximal end of the axon other changes are taking place. Following two weeks of elevated IOP in mice, mitochondria-rich varicosities in the RGC unmyelinated axon segment enlarge and increase in density closer to the cell body (Risner et al., 2018). As they do, localization of NaV1.6 within the enlarged axonal varicosities increases, concomitant with enhanced excitability of multiple RGC types with increased spontaneous activity (Weitlauf et al., 2014; Risner et al., 2018; Tao et al., 2020). Excitability in this context includes increased depolarization of the resting membrane potential, which reduces the threshold for excitation. As a result, enhancement entails increased mean and integrated responses to light. Importantly, this increase occurs even for those cells demonstrating dendritic pruning (Figure 5). Enhancement is axogenic, dependent on NaV1.6 activation. Since axonal varicosities in the unmyelinated segment are rich in mitochondria (Wang et al., 2003), increased size likely accommodates more mitochondria to support extra NaV1.6 activity. Even so, both changes are transient, since by four weeks of elevation, levels of NaV1.6 diminish below control levels as does RGC excitability (Risner et al., 2018).
Figure 5. Enhanced excitability is independent of dendritic pruning.
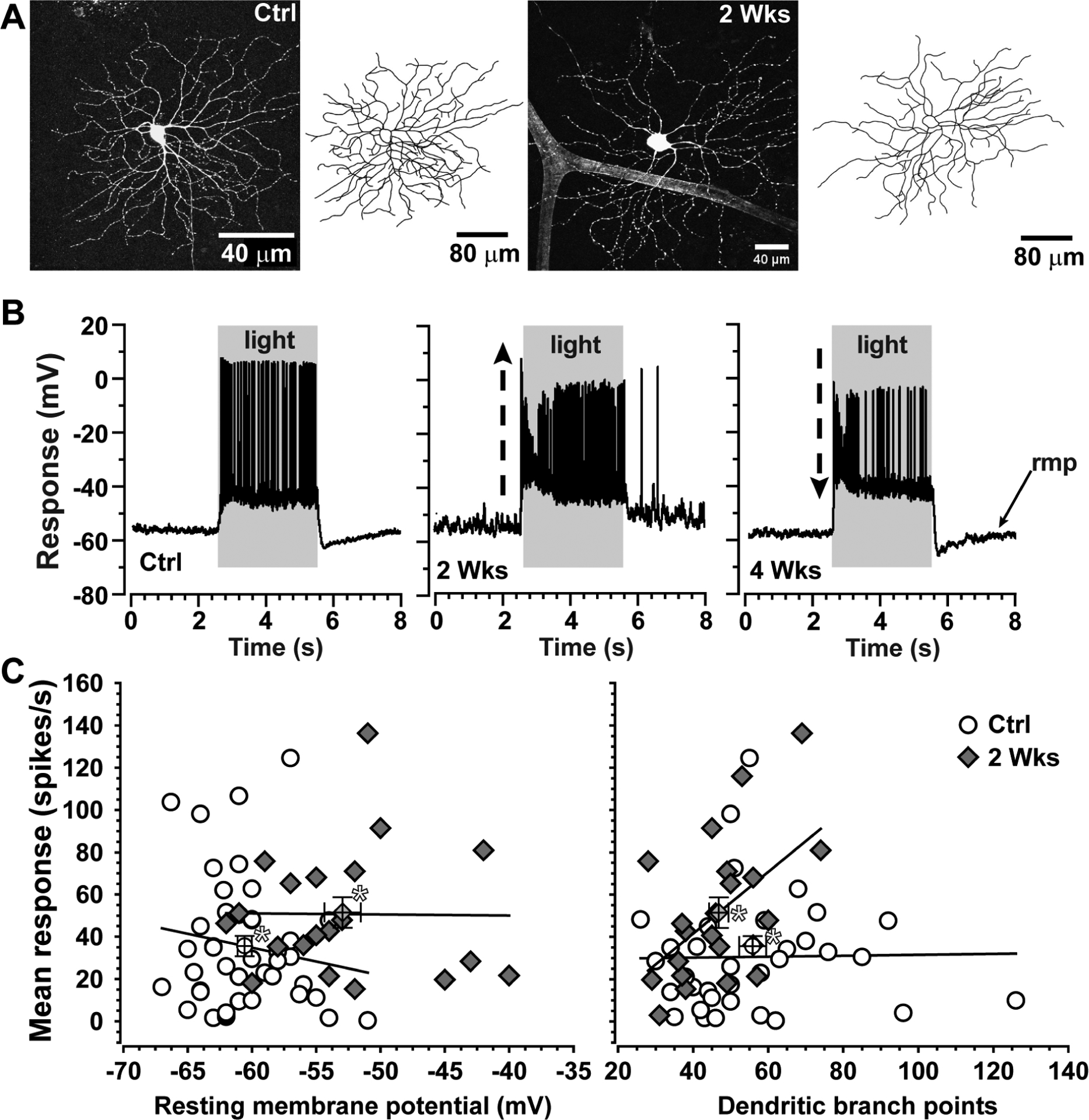
A. Dendritic arbor of an ON-Sustained RGC from control (left) mouse retina following intracellular filling during physiological recording. Same cell type following two weeks of microbead-induced IOP elevation (+35%) shows loss of dendritic branch points (right). B. Current-clamp recordings from single ON-Sustained RGCs from control mouse retina (left) and from retinas following two (middle) and four (right) weeks of elevated IOP. At two weeks, the RGC demonstrates enhanced response to light and a more depolarized resting membrane potential (rmp); by four weeks, response diminishes compared to control. C. Mean response to light after two weeks of elevated IOP increases as resting membrane potential becomes more depolarized (left) and dendritic branch points diminish (right) compared to control (* marks averages ± SEM). Despite having fewer branch points overall, the mean response of ON-Sustained RGCs at two weeks increases with dendritic complexity (p=0.01). Experiments described in Risner et al. (2018).
What could be the purpose of axogenic enhancement of RGC excitability? The answer may be linked to the physiological properties of the NaV1.6 subunit, which produces a persistent excitatory current that sustains higher rates of action potential generation compared to the NaV1.2 subunit (Rush et al., 2005). Even during brief periods of induced IOP elevation, certainly within the experimental window of enhanced excitability, some RGC dendritic arbors exhibit pruning with loss of conventional, excitatory glutamatergic synapses from bipolar cell axon terminals (Della Santina et al., 2013; El-Danaf et al., 2015; Ou et al., 2016; Risner et al., 2018). Increased NaV1.6 in the unmyelinated axon segment likely boosts the light response by facilitating excitation arising from the remaining intact dendritic arbor as a means to maintain signaling along the optic nerve to the brain. Two additional observations support this idea. First, enhanced excitability also involves a depolarizing shift in the RGC resting membrane potential (Figure 5), which facilities NaV1.6’s transition to an activated state (Hu et al., 2009), thereby necessitating less conventional depolarization from synapses in the dendritic arbor. Second, we also found that the compound action potential for mouse optic nerve during the period of enhanced excitability does not diminish (Cooper et al., 2020; McGrady et al., 2020). This means that whatever loss of excitation is occurring in the dendritic arbor during early progression does not translate to lost signaling in the nerve itself.
That NaV1.2 does not increase along with NaV1.6 likely protects already vulnerable RGC dendrites from excessive depolarization from backpropagation of the boosted axonal signal (Risner et al., 2020). In fact, during the period of enhancement, pruning appears to slow – many RGCs retain a full arbor and others demonstrate extensive dendritic remodeling (El-Danaf et al., 2015; Ou et al., 2016; Risner et al., 2018).The axogenic nature of enhanced excitability also reduces the chances of glutamate overload in the dendritic arbor, which could occur with an increase in conventional synaptic signaling. Excessive glutamate is associated with the transformation of mitochondria from elongated to round (Rintoul et al., 2003), which would exacerbate the challenge to mitochondrial transport within the unmyelinated axon segment (Rintoul and Reynolds, 2010).
3.2. Adaptation as a Catalyst for Degeneration
Enhanced excitability appears to be a general property that counters early neurodegenerative progression in glaucoma models. This phenomenon appears in both ganglion cells that respond to light onset or increments (ON cells) and those that respond to light offset or decrements (OFF cells), independent of the degree of dendritic pruning (Risner et al., 2018; Tao et al., 2020). This generality may help inform its purpose. From the standpoint of a single RGC, maintaining the capacity to generate action potentials and signal the brain in concert with neighbors must be a prerogative of survival in adulthood as it is in development. An increased rate of action potential firing improves correlated signaling between neurons (Barreiro and Ly, 2017); this may enable a struggling RGC to better encode information with retinotopic neighbors. Also, excitatory activity promotes Akt-dependent signaling and is necessary for maintaining synaptic strength, survival of post-synaptic neurons in central projection sites, and intact trophic signaling (Lu, 2003; Chiu et al., 2008; You et al., 2012; Rangaraju et al., 2014). In fact, Akt is one of a handful of protein kinases that can promote the neuroprotective influence of BDNF (Chen et al., 2013; Dong et al., 2018). In the SC of glaucomatous mice, both DBA/2J and microbead-induced, retinotopic regions of degraded RGC anterograde transport demonstrate focal upregulation of astrocyte-derived BDNF (Crish et al., 2013; Crish and Calkins, 2015). This injury-specific increase may explain the observed persistence of key synaptic structures in the SC and optic nerve even after anterograde transport is completely depleted and optic nerve degeneration has progressed considerably (Crish et al., 2010; Smith et al., 2016).
Apparently, whatever the adaptive benefit enhanced excitability has on maintaining RGC signaling, it is ultimately unsustainable, since the phenomenon is transient (Risner et al., 2018). After the period of enhancement, degradation of anterograde transport, visual acuity, and axonal integrity in the optic nerve resumes and accelerates (Risner et al., 2018). Similarly, while mitochondrial oxidative phosphorylation increases retinal ATP immediately following optic nerve crush, the excess eventually promotes RGC degeneration (Zhu et al., 2020). Even though enhanced activity could help the individual RGC survive, from the standpoint of the population functioning in a bioenergetic system, dwindling metabolic resources combined with oxidative stress in the unmyelinated axon segment could simply tip the balance from pro-survival to pro-degenerative. Increased neuronal activity drives ATP synthesis, which depletes local glucose reserves (Brown et al., 2003; Rangaraju et al., 2014). Thus, those RGCs tapping a limited supply of ATP to support both NaV in the unmyelinated segment for enhanced excitability and active transport to distal axon segments are simply fighting a losing battle. Finally, while excitation could help an individual RGC maintain concerted signaling with the population, ultimately too much of a good thing is, well, not so good. As activity and the demand for ATP pushes the limit of mitochondrial capacity to buffer Ca2+, the potential for excitotoxicity and oxidative damage increases (Arundine andTymianski, 2003). This could explain the beneficial effects of inhibition of Ca2+ channels following acute optic nerve injury (Knöferle et al., 2010).
3.3. Astrocyte Remodeling Redistributes Resources
Neurons, including the RGC, do not normally store substantial levels of glucose-derived glycogen for use during periods of high metabolic demands, since too much glycogen can be neurotoxic (Bélanger et al., 2011; Rai et al., 2018). Rather, like astrocytes elsewhere in the brain (Gjedde et al., 2002), those that blanket the nerve fiber layer and distribute densely throughout the optic projection store glycogen that is converted to bioenergetic substrates (particularly lactate) that can be transferred to the RGC and its axon in conditions of increased activity, stress or injury (Brown et al., 2003; Brown and Ransom, 2015; Brown et al., 2019; Mori et al., 2020). While neurons have an intrinsic capacity to generate energy from glucose directly through glycolysis (Díaz-García et al., 2017), indirect transfer via astrocytes may be a more expedient route to supply focal needs for ATP along lengthy axon tracts. Next, we consider whether astrocytes too, like RGCs, respond dynamically to early progression.
Enhanced RGC excitability entails increased initiation and propagation of action potentials along the optic nerve for a given level of light stimulation, which places additional metabolic burden on the myelinated segment (Trevisiol et al., 2017). Accordingly, just days following microbead-induced elevations in IOP, glycogen levels in the hypertensive nerve drop below levels in the control nerve (Cooper et al., 2020; Figure 6A–B). This is consistent with the influence of elevated IOP on ATP content in the DBA/2J mouse optic nerve (Baltan et al., 2010). However, quite unexpectedly, in the brief period of RGC enhanced excitability that follows, glycogen levels in the stressed optic nerve increase above those of the control nerve. In turn, the control optic nerve demonstrates diminished glycogen below levels in naïve optic nerve and a corresponding decrease in the control compound action potential. Interestingly, by four weeks of elevation – at which point enhanced excitability has ceased (Risner et al., 2018) – glycogen content in control and hypertensive nerves equilibrated and dropped below naïve levels (Cooper et al., 2020).
Figure 6. Astrocyte metabolism and remodeling in the optic nerve.
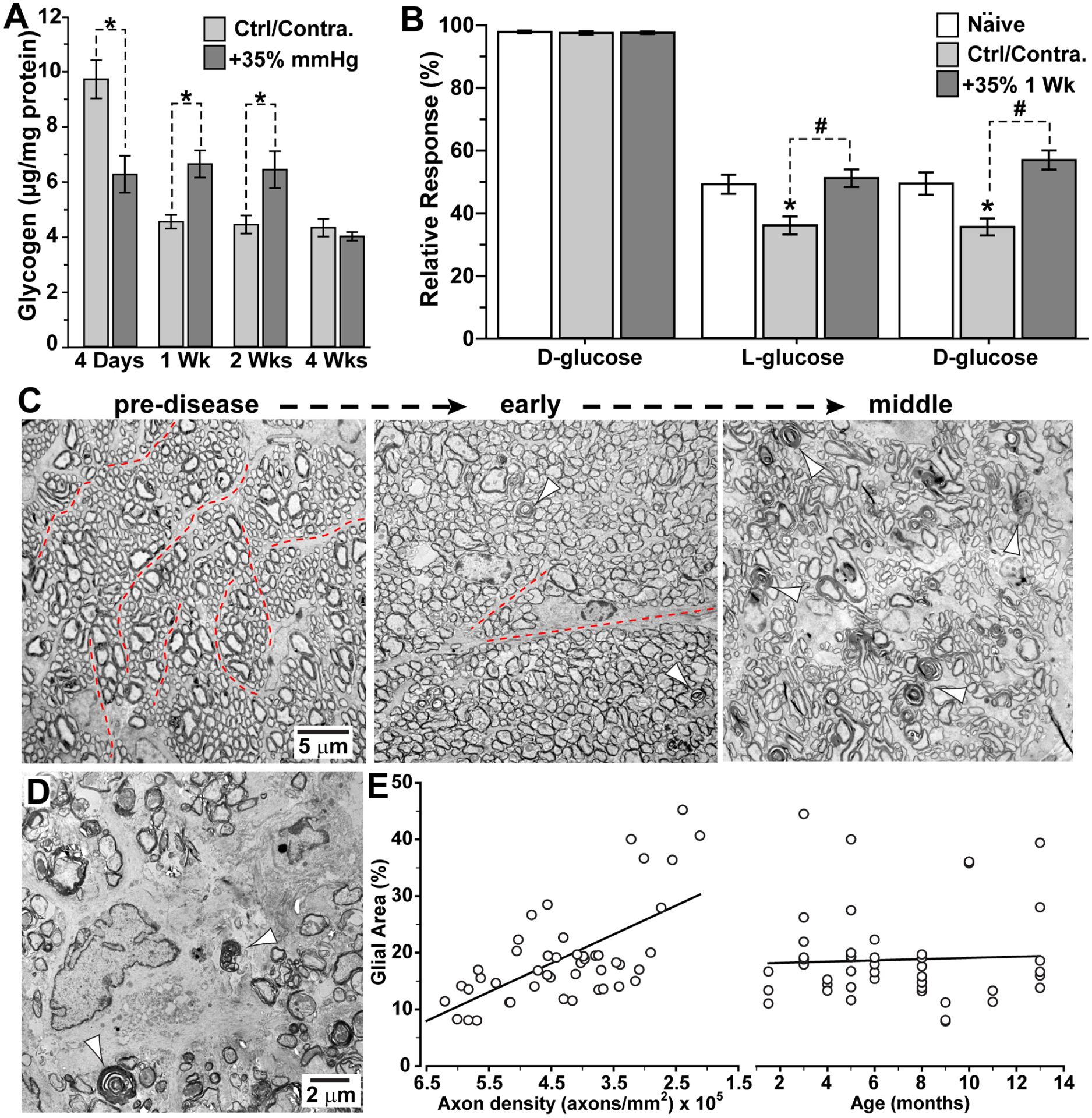
A. In C57 mice, microbead-induced elevations in IOP (+35%) initially diminish astrocyte-stored glycogen in the optic nerve compared to the control/contralateral control nerve; after one and two weeks, glycogen in the stressed nerve increases at the expense of the control nerve (*, p ≤ 0.05). By four weeks, glycogen equilibrates between the nerves. B. As a result of glycogen imbalance, compound action potential of C57 optic nerve following one week of elevated IOP responds stronger during and recovers faster from D-glucose deprivation than control/contralateral control nerve (#, p<0.001); control nerve also response more weakly than naïve nerve (*, p ≤ 0.002). C. Electron micrographs of cross-sections through DBA/2J optic nerve demonstrate intact axon fascicles bound by thin astrocyte processes (dotted lines) prior to progression (left). In the early stages, axons enlarge, and astrocyte processes withdraw with sparse examples of degenerating profiles (arrowheads). As progression continues (right), fascicles become disorganized as degeneration accelerates. D. In final stages, DBA/2J nerve demonstrates abundant degenerating profiles (arrowheads) as astrocyte hypertrophy predominates over intact axons. E. The density of intact RGC axons determines the extent of astrocyte hypertrophy in the DBA/2J more so than age (r2=0.42; p<0.001). A, B: modified from Cooper et al. (2020). C,D: see also Cooper et al., 2016; 2018.
The results from Cooper et al. (2020) indicate that the demand for energy exacted by enhanced excitability, along with myriad disease-relevant stressors, is met by an attempt by the optic projection to quickly distribute resources where needed, namely, to the hypertensive nerve. Importantly, the apparent transfer of metabolites occurs via an extensive, bilateral network of astrocytes formed by connexin 43-enriched gap junctions. When this protein is conditionally eliminated from astrocytes, the transfer of metabolites ceases, as does its compensatory effects to slow progression in the hypertensive optic projection. Finally, without intact gap junctions between astrocytes, anterograde transport to the SC and visual acuity degrade more rapidly with elevated IOP (Cooper et al., 2020). Similarly, in the DBA/2J optic nerve, diminished levels of connexin 43 is associated with more dramatic reductions in anterograde axon transport to the SC (Cooper et al., 2018).
Clearly, the idea of an astrocyte network in stasis unless oscillating between two extreme modes – quiescent or inflammatory – is far too simple. Other aspects of astrocyte organization in the optic nerve indicate that dynamic remodeling is an important aspect of progression. Injury or long-term exposure to disease-relevant stressors in the central nervous system results in replacement of neural tissue with an astrocytic scar, which impedes the possibility of systemic repair. Formation is preceded by a period of astrocyte reactivity, characterized by increased expression of intermediate filament proteins to support hypertrophy, inflammatory signaling, and proliferation or gliosis (Wang et al., 2018; reviewed in Lee et al., 2020). However, early progression in glaucoma involves a very different type of astrocyte remodeling (Figure 6C–E). In the optic nerve, prior to demonstrable degeneration of RGC axons, individual astrocyte processes undergo extensive motility within axon bundles that involves Ca2+-dependent signaling (Lye-Barthel et al., 2013; Ho et al., 2014; Wang et al., 2017).
This motility appears to be linked to metabolic redistribution. In the DBA/2J mouse, prior to outright axon degeneration, several important morphological changes take place; these are described in Cooper et al. (2016; 2018). First, RGC axons in the myelinated optic nerve enlarge, demonstrating disordered axoplasm that corresponds to increased phosphorylation of neurofilaments. As axons expand, astrocyte processes withdraw from the extra-axonal milieu, retreating towards the perimeter of the nerve, causing the local density of mitochondria for a given cross-sectional area of optic nerve to decrease. This withdrawal likely corresponds to or even causes elongation of astrocyte processes in the longitudinal plane of the optic nerve in early progression (Lye-Barthel et al., 2013; Wang et al., 2017). In later stages as RGC axons degenerate, glial coverage of the DBA/2J optic nerve – primarily from hypertrophic astrocyte processes – increases proportionally (Bosco et al., 2016). Even as IOP increases with age in this model, the key determinant of astrocyte hypertrophy is axon survival (Figure 6E). Finally, remodeling of the astrocyte network is important for promoting RGC survival. Attenuating reorganization by conditional knockout of the signal transducer and activator of transcription 3 (STAT3) from astrocytes accelerates degeneration following optic nerve injury (Sun et al., 2017).
4. Discussion
4.1. Adaptive Responses for the RGC and Astrocytes
That the RGC is simply a bystander in glaucoma, passively withstanding the buffets of degenerative pressures, is simply not a tenable concept, given the physiological evidence that tells a far different story. For example, in the retina, even as dendritic pruning progresses for multiple RGC types exposed to elevated IOP, remodeling of thin distal processes actually can expand the arbor (El-Danaf and Huberman, 2015). Interestingly, following acute axon injury in the optic nerve, mTOR complex 2 signaling when activated by exogenous insulin can extend RGC dendrites (Agostinone et al., 2018). Dendrite extension with elevated IOP appears to coincide with the period of enhanced excitability, which amplifies the RGC response to light and pauses dendritic pruning with some evidence of field enlargement (Risner et al., 2018). Early adaptation also involves increased spontaneous RGC activity, which makes sense given that the resting membrane potential becomes more depolarized during this time (Weitlauf et al., 2014; Risner et al., 2018; Tao et al., 2020). These physiological changes are accompanied by increased expression of functionally specialized excitatory channels, both voltage-gated (Risner et al., 2018) and non-voltage gated (Weitlauf et al., 2014).
We do not understand what initiates enhanced RGC excitability, though mechanistically we know the phenomenon itself is axogenic, involving voltage-gated sodium channels (Risner et al., 2018). The changes summarized here (section 3.1) loosely fall under the rubric of “intrinsic” to the RGC itself – properties we can assess by probing the RGC directly. This intrinsic response is only part of the story of adaptation, the other arising from elements extrinsic to the RGC. As we have seen (section 3.3), astrocytes undergo significant remodeling, well before the hypertrophic reactivity that we associate with significant axon loss in the optic projection (Cooper et al., 2016; 2018). The withdrawal of astrocyte processes prior to degeneration in the DBA/2J nerve is accompanied by diminished mitochondrial density, which likely contributes to reduced ATP and compound action potential (Cooper et al., 2016; Baltan et al., 2010). A similar process may be at play for laterally oriented astrocytes in the glial lamina, which also withdraw from axon bundles (Li et al., 2015). In any case, the idea that astrocytes in glaucoma are limited in their response to transitioning from a quiescent state, generally supportive of RGC function, to a reactive state, both inflammatory and prodegenerative, is an oversimplification. As we will see below, throughout progression astrocytes respond to local cues in ways that have broader implications for the optic projection as a system.
4.2. Adaptation in Progression: Survival of the Fittest
The transfer of astrocytic metabolites from unstressed to stressed optic nerve in experimental glaucoma induces a commensurate decrease in the unstressed nerve’s capacity to adapt to glucose deprivation (Cooper et al., 2020; Figure 6A,B). In neural tissue, focal increases in activity induce a transition from oxidative metabolism to increased reliance on astrocyte-supplied lactate (Kasischke et al., 2004). Indeed boosting RGC expression of the monocarboxylate transporter 2, which shuttles lactate and other metabolites in and out of cells, reduces degeneration in both DBA2J and microbead glaucoma (Harun-Or-Rashid et al., 2020). However, several critical functions of astrocytes are more energy-demanding than neurons; remodeling and reactivity ultimately may divert resources otherwise needed for survival of the optic projection (reviewed in Weber and Barros, 2015). As the transfer of metabolites from the unstressed to stressed retinal projection occurs during the period of enhanced excitability, the additional burden on astrocytes already taxed by remodeling and reactivity could ultimately cause widespread failure of the bioenergetic network, thereby increased metabolic and oxidative stress in the system. A similar cascade may contribute to pathogenesis in Alzheimer’s disease, in which astrocyte reactivity essentially syphons limited metabolic resources away from neurons (Steele and Robinson, 2012). The same reservoir of mitochondria that supports the metabolic needs of astrocytes also renders them susceptible to oxidative stress (Noh et al., 2013).
Astrocyte glia in the optic nerve respond to glutamate and potassium (K+) extruded by axons at nodes of Ranvier with Ca2+-dependent ATP release that stimulates glycogen turnover and inter-astrocyte communication within the gap-junction coupled network (reviewed in Butt et al., 2004; Kasischke et al., 2004). In effect, astrocytes receive a copy of the RGC signal as an index of activity that in turn can modulate availability of metabolites useful for the axon (Ransom and Orkand, 1996; Magistretti et al., 1999). As well, RGC axons also form intermittent connections to neighbors via gap junctions (Smedowski et al., 2020). These could serve to smooth focal activity, thereby reducing noise in the signal copied to astrocytes within a local network. It seems reasonable then that RGC enhanced excitability early in progression could serve as a catalyst to redirect resources through the astrocyte network to impede progression (Figure 7). This apparent redirection is extensive, from one optic projection to the other (Cooper et al., 2020; section 3.3). There is precedence for large-scale signaling. In optic nerve trauma, activation of the ATP-gated purinergic P2X7 receptor influences inflammation in the contralateral projection (Nadal-Nicolás et al., 2016).
Figure 7. Progression involves adaptive responses slow progression.
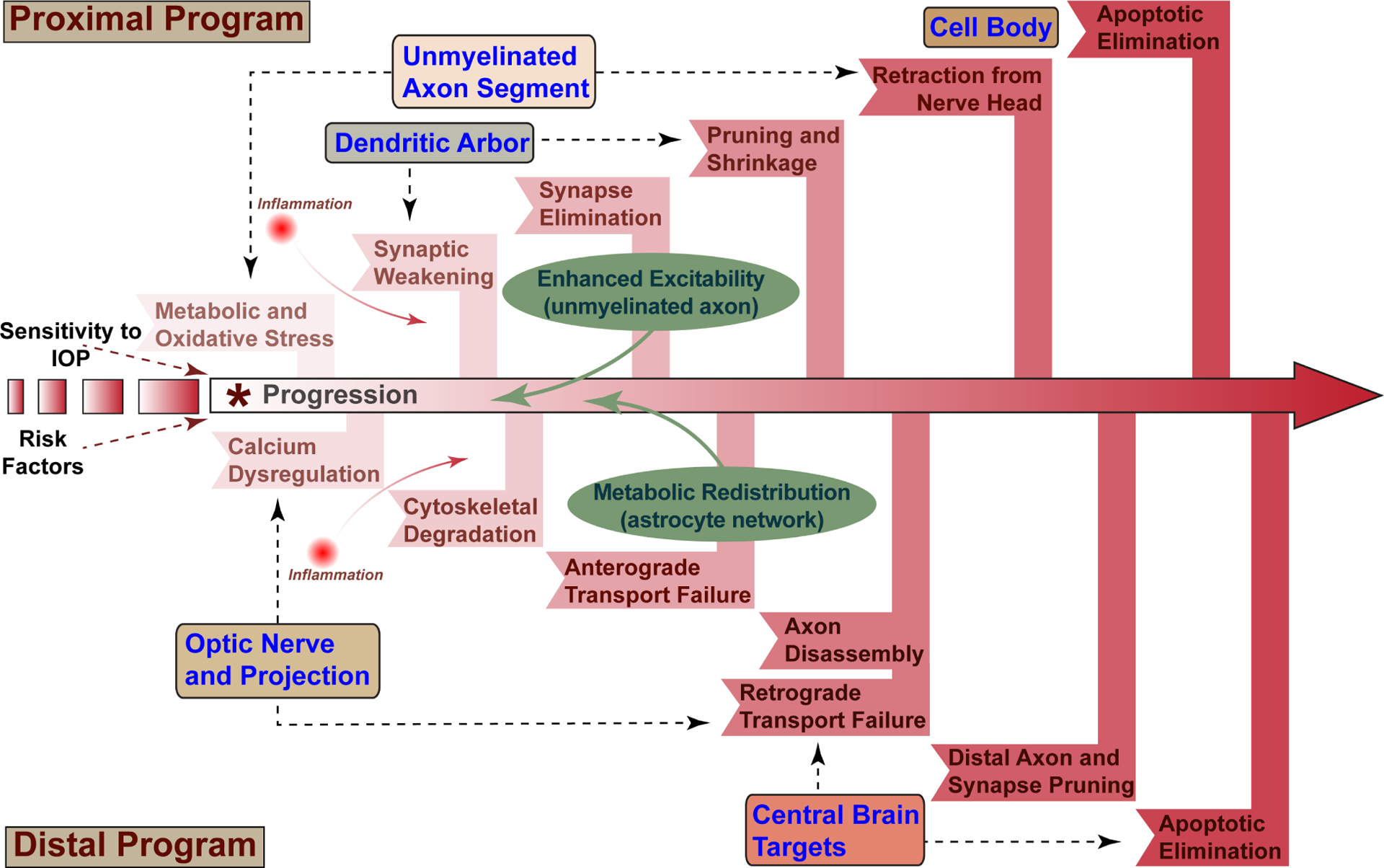
Sensitivity to IOP and other risk factors combine to increase susceptibility to glaucomatous neurodegeneration through stress conveyed to the RGC axon within the optic nerve head (*). Progression comprises a proximal program influencing the unmyelinated axon segment, RGC dendritic arbor, and cell body and a distal program affecting the myelinated axon and its projection into central brain targets. Each program includes inflammatory signaling, which contributes in multiple ways to pathogenesis. In this model, early metabolic and oxidative stress originating in the unmyelinated axon segment leads to both calcium dysregulation and axonopathy in the optic nerve and synaptic and dendritic pruning in the retina. Enhanced excitability originating in the unmyelinated segment and astrocyte-mediated metabolic redistribution represent adaptive responses that impede proximal and distal programs, respectively.
Conversely, bundles of axons struggling to maintain concerted firing early in progression could evoke the opposite response, eliciting the redirection of resources that causes rather than reflects enhanced excitability. A corollary of this hypothesis is that as astrocytes monitor axonal depolarization within an individual optic nerve, focal redirection could deprive specific bundles of axons necessary metabolites, should their signaling become erratic. Remodeling of the gap-junction coupled network could serve to define units of functional vs. dysfunctional axons (Weber and Barros, 2015). In the DBA/2J optic nerve, the high degree of pre-degenerative astrocyte remodeling we observed includes withdrawal from bundles of axons that have accumulated phosphorylated neurofilaments (Cooper et al., 2016; 2018; see also Li et al., 2015). Such a response could indicate a thresholding trigger, in which astrocytes monitoring axon health redirect limited supplies of metabolites to better functioning axon modules to maximize survival of the fittest throughout the optic projection.
4.3. Revisiting the Nerve Head in Progression
For the adaptive responses described (Figure 7), the unmyelinated segment is critical, as the likely locus of enhanced excitability (section 3.1). At the optic nerve head, near the junction between the unmyelinated and myelinated segments, failed transport within RGC axons subjected to glaucomatous stressors appears to cause accumulation of important intraaxonal cargo, such as amyloid precursor protein (Chidlow et al., 2011; Korneva et al., 2020; Maddineni et al., 2020), and reflects important cytoskeletal changes in axons that may influence functional transport (Balaratnasingam et al., 2007). Interestingly, a change in the gradient between IOP and cerebral spinal fluid pressure rather than absolute increases in pressure increases mitochondrial COX in the laminar region of the nerve head in pigs (Balaratnasingam et al., 2009). Thus, there is no doubt that this transition site demarcates physiological differences in how transport proceeds between the two segments. In this zone as well, astrocytes play a critical phagocytic role in clearing axonal debris, possibly from slowed transport of organelles (Nguyen et al., 2011). One idea is that RGC axons die back from or undergo axotomy at this juncture due to IOP-related insult (Soto et al., 2008; Li et al., 2015). Higher elevations in IOP that most certainly involve ischemic insult could lead to just such a scenario, along with rapid dendritic pruning in the retina and secondary loss of other retinal neurons through retrograde degeneration, akin to nerve crush or transection. However, progressive glaucoma typically does not involve such extreme IOP elevation (AGIS Investigators, 2000). Since some functional recovery is possible in patients undergoing treatment to lower IOP (Venture and Porciatti, 2005; Musch et al., 2014), dramatic mechanical separation of axonal segments is likely a special property of acute experimental models.
The nerve head is a hotbed for disease-relevant stressors that influence axon function and structure (section 1.2); progression from this point travels in both the anterograde and retrograde directions (Figure 2). Given the biomechanical strain associated with optic nerve head remodeling (Sigal and Ethier, 2009; Burgoyne et al., 2011; Girard et al., 2011; Quigley, 2015), one must consider whether spatially expansive signaling could arise from such strain and influence progression. An obvious candidate is ATP, working through purinergic receptors localized to RGC axons. Mechanical stimulation of many tissues elicits robust release of ATP, including conditions that incite inflammation (Mikolajewicz et al., 2018; 2019). Human optic nerve astrocytes exposed to mechanical stress similarly release ATP through pannexin channels (Beckel et al., 2014). In the retina, the RGC expresses the ATP-gated purinergic P2X7 receptor, the deletion of which delays degeneration following axotomy (Mitchell et al., 2008; Nadal-Nicolás et al., 2016). It seems reasonable that astrocyte remodeling due to glaucomatous strain, coupled with increased axonal demand for energy, could elicit changes in local ATP concentration. Since P2X7 receptors activate and gate influx of cations like Na+ and Ca2+ with very high concentrations of ATP (millimolar levels; Illes, 2020), astrocyte ATP could serve two purposes. Low concentrations of ATP extruded by astrocytes could increase glycogen turnover and inter-astrocyte communication (Butt et al., 2004; Kasischke et al., 2004), as physiological need demands, while higher concentrations would elicit cation dysregulation and Ca2+-dependent cytoskeletal degradation. In this way, increased bioenergic stress within the system transforms the protective nature of metabolic redistribution into one that ultimately accelerates system failure and wide-spread degeneration. The key to new protective therapies then could lie within modulating the delicate balance of astrocyte-axon interactions.
4.4. Inflammation – not a Third Wheel
Pathogenesis in glaucoma, which is taken here to indicate the neurodegenerative processes that lead to vision loss, is not just about metabolic and oxidative stress, which are closely related. Glaucoma also involves diverse inflammatory components spanning anterior segment to central optic projection sites in the brain; these are reviewed amply elsewhere and so are not included here (Baudouin et al., 2020; Wareham et al., 2020). Even so, it is remiss to believe that oxidative and metabolic stress are easily dissociated from inflammatory contributors to progression. They are not. Just as metabolic and oxidative stress are linked, inflammation and oxidative stress go hand in hand, as many critical intracellular and extracellular signaling components create a substrate for crosstalk between them. For example, just as ROS in neurons can activate pro-inflammatory transcription factors that induce local immune cells to secrete signals that amplify the immune response, immune cells can enhance ROS production that leads to oxidative stress (Herman et al., 2018). As we have seen, dysregulation of axonal Ca2+ contributes to oxidative stress through diminished mitochondrial motility (Rintoul and Reynolds, 2010; Liao et al., 2017). In a rat model of acute optic neuritis, an inhibitor of the Ca2+-activated protease calpain decreased inflammatory mediators, including cytokines, along with microglial and astrocyte reactivity (Das et al., 2013).
In the context of neurodegeneration, the term “inflammation” itself has evolved greatly and now is invoked – rightly or wrongly – in relation to a multitude of processes involving glial activity. These range from how we traditionally think of inflammation in terms of activation and recruitment of immune cells to remodeling of glial cytoskeletal components to neurovascular coupling and vascular permeability to dendritic pruning and synapse elimination. Indeed, the early remodeling of astrocyte process in the optic nerve we describe for DBA/2J mice could be considered a new facet of glial reactivity and therefore an aspect of inflammation (Figure 6; Cooper et al., 2016; 2018). As noted above (section 4.3), astrocyte remodeling likely modulates local concentrations of ATP, activating Ca2+-dependent cascades via P2X7 receptors localized to RGCs or their axons. However, microglia also express P2X7 receptors, activation of which triggers the release of inflammatory cytokines and ROS (Illes, 2020). In this case, we see that metabolic signaling and inflammation are linked.
Primary inflammatory diseases of the central nervous system such as multiple sclerosis are characterized by strong immune-mediated responses that elicit neurodegeneration (Degan et al., 2018). The neurodegenerative events described here and the adaptive responses that appear to counter them (Figure 7) involve combinations of glial reactivity, activation of complement-mediated pathways (e.g., synapse elimination), immune cell recruitment, and the production and secretion of inflammation mediators including cytokines and chemokines (Baudouin et al., 2020). In most types of glaucoma, but not all (Rieck, 2013), these responses are secondary to the primary injury to RGCs elicited by sensitivity to IOP. Astrocytes as well as microglia are key regulators of innate and adaptive immune responses (Colombo and Farina, 2016). Since metabolic redistribution and very likely enhanced excitability so intimately involve astrocyte remodeling, it is possible that they too elicit other components of neuroinflammation, including both pro-degenerative and reparative elements.
4.5. Relevance to Glaucoma Patients
Metabolic redistribution and enhanced excitability each represent a form of adaptative remodeling. Enhanced excitability involves reorganization of excitatory signaling substrates at the level of individual axons, while metabolic redistribution involves the movement of bioenergetic resources through astrocyte networks at the level of the optic projection. Each in different ways appears to counter progression, however transiently, by supporting RGC signaling along the optic nerve (Risner et al., 2018; Cooper et al., 2020). Given this protective element, it is reasonable to ask whether adaptive remodeling could be harnessed therapeutically for human patients.
As we have seen (section 4.2), the transient nature of adaptive remodeling may reflect limited bioenergetic resources for a system already taxed by primary pathogenic features of progression. There is evidence from animal studies that boosting such resources slows progression. In the DBA/2J mouse, levels of nicotinamide adenine dinucleotide (NAD+), a cofactor central to all cellular metabolism, decreases with age (Williams et al., 2017). Both oral administration of the NAD+ precursor nicotinamide (vitamin B3) and directed gene upregulation of Nmnat1, a key NAD+-producing enzyme, protects RGCs and slows progression was protective (Williams et al., 2017). Along these lines, oral supplementation with pyruvate, a product of glycolysis, is neuroprotective in both the DBA/2J mouse and in an inducible rat model of glaucoma, reducing both anterograde transport deficits and axon degeneration in the optic nerve (Harder et al., 2020).
These results may show a relatively simple path forward to slowing neurodegeneration in glaucoma patients, by providing additional metabolic resources to the optic projection. On the other hand, neuroprotective regimens that work in rodents do not necessarily translate to models involving larger mammals, including non-human primates, let alone to human patients. For example, inhibitors of p38 MAP kinases that attempt to reduce inflammatory signaling reduce degeneration in rats and mice but are far less effective in monkeys (Lambert et al., 2020). It is enticing to postulate that interventions involving bioenergetics are effective because they feed and extend adaptive remodeling. If so, a better way forward in glaucoma may identify targets within metabolic redistribution and enhanced excitability that enable their protective capacities while balancing oxidative byproducts that result from the use of additional fuel sources. The key lies in understanding collectively the adaptive pressures facing RGCs and astrocytes as local neuroexcitatory units that must respond to the overriding needs of the entire optic projection.
Acknowledgements.
The author would like to thank Research to Prevent Blindness, Inc., Glaucoma Research Foundation, and BrightFocus Foundation for their support over the years. Research described herein also supported by the Stanley Cohen Innovation Fund, and National Institutes for Health grants EY017427, EY024997, EY021453 and EY08126. The author would like to offer his sincere gratitude to colleagues both past and present for their immense contributions to the work described herein.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- AGIS Investigators. 2000. The Advanced Glaucoma Intervention study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration. Am J Ophthalmol, 130:429–440. [DOI] [PubMed] [Google Scholar]
- Agostinone J, Alarcon-Martinez L, Gamlin C, Yu WQ, Wong ROL, Di Polo A, 2018. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain 141, 1963–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostinone J, Di Polo A, 2015. Retinal ganglion cell dendrite pathology and synapse loss: Implications for glaucoma. Prog Brain Res 220, 199–216. [DOI] [PubMed] [Google Scholar]
- Almasieh M, Wilson AM, Morquette B, Cueva Vargas JL, Di Polo A, 2012. The molecular basis of retinal ganglion cell death in glaucoma. Prog Retin Eye Res 31, 152–181. [DOI] [PubMed] [Google Scholar]
- Ames A 3rd, 2000. CNS energy metabolism as related to function. Brain Res Brain Res Rev 34, 42–68. [DOI] [PubMed] [Google Scholar]
- Andrews RM, Griffiths PG, Johnson MA, Turnbull DM, 1999. Histochemical localisation of mitochondrial enzyme activity in human optic nerve and retina. Br J Ophthalmol 83, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arundine M, Tymianski M, 2003. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34, 325–337. [DOI] [PubMed] [Google Scholar]
- Balaratnasingam C, Morgan WH, Bass L, Matich G, Cringle SJ, & Yu DY 2007. Axonal transport and cytoskeletal changes in the laminar regions after elevated intraocular pressure. Invest Ophthalmol Vis Sci, 48(8), 3632–3644. [DOI] [PubMed] [Google Scholar]
- Balaratnasingam C, Pham D, Morgan WH, Bass L, Cringle SJ, & Yu DY 2009. Mitochondrial cytochrome c oxidase expression in the central nervous system is elevated at sites of pressure gradient elevation but not absolute pressure increase. J Neurosci Res, 87(13), 2973–2982. [DOI] [PubMed] [Google Scholar]
- Baltan S, Inman DM, Danilov CA, Morrison RS, Calkins DJ, Horner PJ, 2010. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J Neurosci 30, 5644–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro AK, Ly C, 2017. When do correlations increase with firing rates in recurrent networks? PLoS Comput Biol 13, e1005506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron MJ, Griffiths P, Turnbull DM, Bates D, Nichols P, 2004. The distributions of mitochondria and sodium channels reflect the specific energy requirements and conduction properties of the human optic nerve head. Br J Ophthalmol 88, 286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudouin C, Kolko M, Melik-Parsadaniantz S, Messmer EM 2020. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog Retin Eye Res. October17:100916. [DOI] [PubMed] [Google Scholar]
- Beckel JM, Argall AJ, Lim JC, Xia J, Lu W, Coffey EE, Macarak EJ, Shahidullah M, Delamere NA, Zode GS, Sheffield VC, Shestopalov VI, Laties AM, Mitchell CH. 2014. Mechanosensitive release of adenosine 5’-triphosphate through pannexin channels and mechanosensitive upregulation of pannexin channels in optic nerve head astrocytes: a mechanism for purinergic involvement in chronic strain. Glia. Sep;62(9):1486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtold DA, Yue X, Evans RM, Davies M, Gregson NA, Smith KJ, 2005. Axonal protection in experimental autoimmune neuritis by the sodium channel blocking agent flecainide. Brain 128, 18–28. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Coleman MP, Martin KR, 2008. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur J Neurosci 28, 1166–1179. [DOI] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ, 2011. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14, 724–738. [DOI] [PubMed] [Google Scholar]
- Berry RH, Qu J, John SW, Howell GR, Jakobs TC, 2015. Synapse Loss and Dendrite Remodeling in a Mouse Model of Glaucoma. PLoS One 10, e0144341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beykin G, Norcia AM, Srinivasan VJ, Dubra A, Goldberg JL, 2020. Discovery and clinical translation of novel glaucoma biomarkers. Prog Retin Eye Res, 100875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G, 2003. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23, 2306–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, Breen KT, Anderson SR, Steele MR, Calkins DJ, Vetter ML, 2016. Glial coverage in the optic nerve expands in proportion to optic axon loss in chronic mouse glaucoma. Exp Eye Res 150, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhenni RA, Dunmire J, Sewell A, Edward DP, 2012. Animal models of glaucoma. J Biomed Biotechnol 2012, 692609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumezbeur F, Mason GF, de Graaf RA, Behar KL, Cline GW, Shulman GI, Rothman DL, Petersen KF, 2010. Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J Cereb Blood Flow Metab 30, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradke F, Fawcett JW, Spira ME, 2012. Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci 13, 183–193. [DOI] [PubMed] [Google Scholar]
- Bristow EA, Griffiths PG, Andrews RM, Johnson MA, Turnbull DM, 2002. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol 120, 791–796. [DOI] [PubMed] [Google Scholar]
- Brown AM, Ransom BR, 2015. Astrocyte glycogen as an emergency fuel under conditions of glucose deprivation or intense neural activity. Metab Brain Dis 30, 233–239. [DOI] [PubMed] [Google Scholar]
- Brown AM, Rich LR, Ransom BR, 2019. Metabolism of Glycogen in Brain White Matter. Adv Neurobiol 23, 187–207. [DOI] [PubMed] [Google Scholar]
- Brown AM, Tekkök SB, Ransom BR, 2003. Glycogen regulation and functional role in mouse white matter. J Physiol 549, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne CF, 2011. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp Eye Res 93, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM, Pugh M, Hubbard P, James G, 2004. Functions of optic nerve glia: axoglial signalling in physiology and pathology. Eye (Lond) 18, 1110–1121. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR, 2000. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A 97, 5616–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ, 2012. Critical pathogenic events underlying progression of neurodegeneration in glaucoma. Prog Retin Eye Res 31, 702–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ, 2013. Age-related changes in the visual pathways: blame it on the axon. Invest Ophthalmol Vis Sci 54, Orsf37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ, Horner PJ, 2012. The cell and molecular biology of glaucoma: axonopathy and the brain. Invest Ophthalmol Vis Sci 53, 2482–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ, Lambert WS, Formichella CR, McLaughlin WM, Sappington RM, 2018. The Microbead Occlusion Model of Ocular Hypertension in Mice. Methods Mol Biol 1695, 23–39. [DOI] [PubMed] [Google Scholar]
- Calkins DJ, Pekny M, Cooper ML, Benowitz L, 2017. The challenge of regenerative therapies for the optic nerve in glaucoma. Exp Eye Res 157, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson RJ, Chidlow G, Crowston JG, Williams PA, Wood JPM, 2020. Retinal energy metabolism in health and glaucoma. Prog Retin Eye Res, 100881. [DOI] [PubMed] [Google Scholar]
- Chen A, Xiong LJ, Tong Y, Mao M, 2013. Neuroprotective effect of brain-derived neurotrophic factor mediated by autophagy through the PI3K/Akt/mTOR pathway. Mol Med Rep 8, 1011–1016. [DOI] [PubMed] [Google Scholar]
- Chen H, Wei X, Cho KS, Chen G, Sappington R, Calkins DJ, Chen DF, 2011. Optic neuropathy due to microbead-induced elevated intraocular pressure in the mouse. Invest Ophthalmol Vis Sci 52, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidlow G, Ebneter A, Wood JP, Casson RJ, 2011. The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol 121, 737–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu SL, Chen CM, Cline HT, 2008. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 58, 708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrysostomou V, Rezania F, Trounce IA, Crowston JG, 2013. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol 13, 12–15. [DOI] [PubMed] [Google Scholar]
- Coleman AL, Miglior S, 2008. Risk factors for glaucoma onset and progression. Surv Ophthalmol 53Suppl1, S3–10. [DOI] [PubMed] [Google Scholar]
- Colombo E, Farina C 2016. Astrocytes: key regulators of neuroinflammation. Trends Immunol. Sep;37(9):608–620. [DOI] [PubMed] [Google Scholar]
- Cooper ML, Crish SD, Inman DM, Horner PJ, Calkins DJ, 2016. Early astrocyte redistribution in the optic nerve precedes axonopathy in the DBA/2J mouse model of glaucoma. Exp Eye Res 150, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ML, Collyer JW, Calkins DJ, 2018. Astrocyte remodeling without gliosis precedes optic nerve Axonopathy. Acta Neuropathol Commun 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ML, Pasini S, Lambert WS, D’Alessandro KB, Yao V, Risner ML, Calkins DJ, 2020. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc Natl Acad Sci U S A 117, 18810–18821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin L, Morrison RS, Horner PJ, Inman DM, 2015. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Invest Ophthalmol Vis Sci 56, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crish SD, Calkins DJ, 2011. Neurodegeneration in glaucoma: progression and calcium-dependent intracellular mechanisms. Neuroscience 176, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crish SD, Calkins DJ, 2015. Central visual pathways in glaucoma: evidence for distal mechanisms of neuronal self-repair. J Neuroophthalmol 35Suppl 1, S29–37. [DOI] [PubMed] [Google Scholar]
- Crish SD, Dapper JD, MacNamee SE, Balaram P, Sidorova TN, Lambert WS, Calkins DJ, 2013. Failure of axonal transport induces a spatially coincident increase in astrocyte BDNF prior to synapse loss in a central target. Neuroscience 229, 55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ, 2010. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A 107, 5196–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapper JD, Crish SD, Pang IH, Calkins DJ, 2013. Proximal inhibition of p38 MAPK stress signaling prevents distal axonopathy. Neurobiol Dis 59, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Guyton MK, Smith A, Wallace G.t., McDowell ML, Matzelle DD, Ray SK, Banik NL, 2013. Calpain inhibitor attenuated optic nerve damage in acute optic neuritis in rats. J Neurochem 124, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CCJ, Grierson AJ, 2007. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet 16, 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC, 2008. Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 31, 151–173. [DOI] [PubMed] [Google Scholar]
- Degan D, Ornello R, Tiseo C, Carolei A, Sacco S, & Pistoia F (2018). The Role of Inflammation in Neurological Disorders. Current pharmaceutical design, 24(14), 1485–1501. [DOI] [PubMed] [Google Scholar]
- Della Santina L, Inman DM, Lupien CB, Horner PJ, Wong RO, 2013. Differential progression of structural and functional alterations in distinct retinal ganglion cell types in a mouse model of glaucoma. J Neurosci 33, 17444–17457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G, 2017. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26, 361–374.e364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Pu K, Duan W, Chen H, Chen L, Wang Y, 2018. Involvement of Akt/CREB signaling pathways in the protective effect of EPA against interleukin-1β-induced cytotoxicity and BDNF down-regulation in cultured rat hippocampal neurons. BMC Neurosci 19, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JC, 2015. Optic nerve head biomechanics in aging and disease. Exp Eye Res 133, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Danaf RN, Huberman AD, 2015. Characteristic patterns of dendritic remodeling in early-stage glaucoma: evidence from genetically identified retinal ganglion cell types. J Neurosci 35, 2329–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elze T, Pasquale LR, Shen LQ, Chen TC, Wiggs JL, Bex PJ, 2015. Patterns of functional vision loss in glaucoma determined with archetypal analysis. J R Soc Interface 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorowicz M, Orzel J, Kossowski B, Welniak-Kaminska M, Choragiewicz T, Swiatkiewicz M, Rejdak R, Bogorodzki P, Grieb P, 2018. Anterograde Transport in Axons of the Retinal Ganglion Cells and its Relationship to the Intraocular Pressure during Aging in Mice with Hereditary Pigmentary Glaucoma. Curr Eye Res 43, 539–546. [DOI] [PubMed] [Google Scholar]
- Girard MJ, Suh JK, Bottlang M, Burgoyne CF, Downs JC, 2011. Biomechanical changes in the sclera of monkey eyes exposed to chronic IOP elevations. Invest Ophthalmol Vis Sci 52, 5656–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan B, Laird J, Sherman R, Salomon RG, Bhattacharya SK, 2008. Neuroprotection in glaucoma using calpain-1 inhibitors: regional differences in calpain-1 activity in the trabecular meshwork, optic nerve and implications for therapeutics. CNS Neurol Disord Drug Targets 7, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder JM, Guymer C, Wood JPM, Daskalaki E, Chidlow G, Zhang C, Balasubramanian R, Cardozo BH, Foxworth NE, Deering KE, Ouellette TB, Montgomery C, Wheelock CE, Casson RJ, Williams PA, John SWM 2020. Disturbed glucose and pyruvate metabolism in glaucoma with neuroprotection by pyruvate or rapamycin. Proc Natl Acad Sci U S A. December29;117(52):33619–33627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Attwell D, 2012. The energetics of CNS white matter. J Neurosci 32, 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harun-Or-Rashid M, Pappenhagen N, Palmer PG, Smith MA, Gevorgyan V, Wilson GN, Crish SD, Inman DM, 2018. Structural and Functional Rescue of Chronic Metabolically Stressed Optic Nerves through Respiration. J Neurosci 38, 5122–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harun-Or-Rashid M, Pappenhagen N, Zubricky R, Coughlin L, Jassim AH, Inman DM, 2020. MCT2 overexpression rescues metabolic vulnerability and protects retinal ganglion cells in two models of glaucoma. Neurobiol Dis 141, 104944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Her LS, Goldstein LS, 2008. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci 28, 13662–13672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman F, Westfall S, Brathwaite J, Pasinetti GM 2018. Suppression of presymptomatic oxidative stress and inflammation in neurodegeneration by grape-derived polyphenols. Front Pharmacol. August28;9:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho KW, Lambert WS, Calkins DJ, 2014. Activation of the TRPV1 cation channel contributes to stress-induced astrocyte migration. Glia 62, 1435–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, Barbay JM, Marchant JK, Mahesh N, Porciatti V, Whitmore AV, Masland RH, John SW, 2007. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol 179, 1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Soto I, Libby RT, John SW, 2013. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp Neurol 246, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, Shu Y, 2009. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci 12, 996–1002. [DOI] [PubMed] [Google Scholar]
- Huang W, Fileta J, Rawe I, Qu J, Grosskreutz CL, 2010. Calpain activation in experimental glaucoma. Invest Ophthalmol Vis Sci 51, 3049–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illes P 2020. P2X7 receptors amplify CNS damage in neurodegenerative diseases Int. J. Mol. Sci 21(17), 5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Harun-Or-Rashid M, 2017. Metabolic Vulnerability in the Neurodegenerative Disease Glaucoma. Front Neurosci 11, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Lambert WS, Calkins DJ, Horner PJ, 2013. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS One 8, e65389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Sappington RM, Horner PJ, Calkins DJ, 2006. Quantitative correlation of optic nerve pathology with ocular pressure and corneal thickness in the DBA/2 mouse model of glaucoma. Invest Ophthalmol Vis Sci 47, 986–996. [DOI] [PubMed] [Google Scholar]
- Ju WK, Kim KY, Duong-Polk KX, Lindsey JD, Ellisman MH, Weinreb RN, 2010. Increased optic atrophy type 1 expression protects retinal ganglion cells in a mouse model of glaucoma. Mol Vis 16, 1331–1342. [PMC free article] [PubMed] [Google Scholar]
- Ju WK, Kim KY, Lindsey JD, Angert M, Duong-Polk KX, Scott RT, Kim JJ, Kukhmazov I, Ellisman MH, Perkins GA, Weinreb RN, 2008. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci 49, 4903–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW, 2004. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305, 99–103. [DOI] [PubMed] [Google Scholar]
- King A, Azuara-Blanco A, Tuulonen A, 2013. Glaucoma. Bmj 346, f3518. [DOI] [PubMed] [Google Scholar]
- Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD, 2010. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci 30, 6658–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleesattel D, Crish SD, Inman DM, 2015. Decreased Energy Capacity and Increased Autophagic Activity in Optic Nerve Axons With Defective Anterograde Transport. Invest Ophthalmol Vis Sci 56, 8215–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöferle J, Koch JC, Ostendorf T, Michel U, Planchamp V, Vutova P, Tönges L, Stadelmann C, Brück W, Bähr M, Lingor P, 2010. Mechanisms of acute axonal degeneration in the optic nerve in vivo. Proc Natl Acad Sci U S A 107, 6064–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneva A, Schaub J, Jefferys J, Kimball E, Pease ME, Nawathe M, Johnson TV, Pitha I, Quigley H, 2020. A method to quantify regional axonal transport blockade at the optic nerve head after short term intraocular pressure elevation in mice. Exp Eye Res 196, 108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert WS, Carlson BJ, Formichella CR, Sappington RM, Ahlem C, Calkins DJ, 2017. Oral Delivery of a Synthetic Sterol Reduces Axonopathy and Inflammation in a Rodent Model of Glaucoma. Front Neurosci 11, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert WS, Carlson BJ, Ghose P, Vest VD, Yao V, Calkins DJ, 2019. Towards A Microbead Occlusion Model of Glaucoma for a Non-Human Primate. Sci Rep 9, 11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert WS, Pasini S, Collyer JW, Formichella CR, Ghose P, Carlson BJ, Calkins DJ, 2020. Of Mice and Monkeys: Neuroprotective Efficacy of the p38 Inhibitor BIRB 796 Depends on Model Duration in Experimental Glaucoma. Sci Rep 10, 8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lascaratos G, Chau KY, Zhu H, Gkotsi D, King R, Gout I, Kamal D, Luthert PJ, Schapira AHV, Garway-Heath DF, 2015. Resistance to the most common optic neuropathy is associated with systemic mitochondrial efficiency. Neurobiol Dis 82, 78–85. [DOI] [PubMed] [Google Scholar]
- Lee D, Kim KY, Shim MS, Kim SY, Ellisman MH, Weinreb RN, Ju WK, 2014a. Coenzyme Q10 ameliorates oxidative stress and prevents mitochondrial alteration in ischemic retinal injury. Apoptosis 19, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Shim MS, Kim KY, Noh YH, Kim H, Kim SY, Weinreb RN, Ju WK, 2014b. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Invest Ophthalmol Vis Sci 55, 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Han JC, Park DY, Kee C, 2020. A neuroglia-based interpretation of glaucomatous neuroretinal rim thinning in the optic nerve head. Prog Retin Eye Res 77, 100840. [DOI] [PubMed] [Google Scholar]
- Lee S, Van Bergen NJ, Kong GY, Chrysostomou V, Waugh HS, O’Neill EC, Crowston JG, Trounce IA, 2011. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp Eye Res 93, 204–212. [DOI] [PubMed] [Google Scholar]
- Li S, Shao M, Li Y, Li X, Wan Y, Sun X, Cao W, 2020. Relationship between Oxidative Stress Biomarkers and Visual Field Progression in Patients with Primary Angle Closure Glaucoma. Oxid Med Cell Longev 2020, 2701539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Li D, Ying X, Khaw PT, Raisman G, 2015. An energy theory of glaucoma. Glia 63, 1537–1552. [DOI] [PubMed] [Google Scholar]
- Liao PC, Tandarich LC, Hollenbeck PJ, 2017. ROS regulation of axonal mitochondrial transport is mediated by Ca2+ and JNK in Drosophila. PLoS One 12, e0178105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Kim SK, Zaitsev E, Snell CR, Lu B, Loh YP, 2005. Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase e. Neuron 45, 245–255. [DOI] [PubMed] [Google Scholar]
- Lu B, 2003. BDNF and activity-dependent synaptic modulation. Learn Mem 10, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye-Barthel M, Sun D, Jakobs TC, 2013. Morphology of astrocytes in a glaucomatous optic nerve. Invest Ophthalmol Vis Sci 54, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddineni P, Kasetti RB, Patel PD, Millar JC, Kiehlbauch C, Clark AF, & Zode GS 2020. CNS axonal degeneration and transport deficits at the optic nerve head precede structural and functional loss of retinal ganglion cells in a mouse model of glaucoma. Mol Neurodegen, 15(1), 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, 2007. Calcium and neurodegeneration. Aging Cell 6, 337–350. [DOI] [PubMed] [Google Scholar]
- McGrady NR, Risner ML, Vest V, Calkins DJ, 2020. TRPV1 Tunes Optic Nerve Axon Excitability in Glaucoma. Front Physiol 11, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolajewicz N, Mohammed A, Morris M, Komarova SV, 2018. Mechanically stimulated ATP release from mammalian cells: systematic review and meta-analysis. J Cell Sci 131. [DOI] [PubMed] [Google Scholar]
- Mikolajewicz N, Sehayek S, Wiseman PW, Komarova SV, 2019. Transmission of mechanical information by purinergic signaling. Biophys J 116, 2009–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Schwarz TL, 2017. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 96, 651–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S, Kurimoto T, Miki A, Maeda H, Kusuhara S, Nakamura M, 2020. Aqp9 Gene Deletion Enhances Retinal Ganglion Cell (RGC) Death and Dysfunction Induced by Optic Nerve Crush: Evidence that Aquaporin 9 Acts as an Astrocyte-to-Neuron Lactate Shuttle in Concert with Monocarboxylate Transporters To Support RGC Function and Survival. Mol Neurobiol 57, 4530–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musch DC, Gillespie BW, Palmberg PF, Spaeth G, Niziol LM, Lichter PR. 2014. Visual field improvement in the collaborative initial glaucoma treatment study. Am J Ophthalmol. 158(1):96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal-Nicolás FM, Galindo-Romero C, Valiente-Soriano FJ, Barberà-Cremades M, deTorre-Minguela C, Salinas-Navarro M, Pelegrín P, & Agudo-Barriuso M 2016. Involvement of P2X7 receptor in neuronal degeneration triggered by traumatic injury. Sci Rep 6, 38499, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JV, Soto I, Kim KY, Bushong EA, Oglesby E, Valiente-Soriano FJ, Yang Z, Davis CH, Bedont JL, Son JL, Wei JO, Buchman VL, Zack DJ, Vidal-Sanz M, Ellisman MH, Marsh-Armstrong N, 2011. Myelination transition zone astrocytes are constitutively phagocytic and have synuclein dependent reactivity in glaucoma. Proc Natl Acad Sci U S A 108, 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niven JE, Laughlin SB, 2008. Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol 211, 1792–1804. [DOI] [PubMed] [Google Scholar]
- Noh YH, Kim KY, Shim MS, Choi SH, Choi S, Ellisman MH, Weinreb RN, Perkins GA, Ju WK, 2013. Inhibition of oxidative stress by coenzyme Q10 increases mitochondrial mass and improves bioenergetic function in optic nerve head astrocytes. Cell Death Dis 4, e820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Núñez-Álvarez C, Joglar B, Del Olmo-Aguado S, 2016. Glaucoma: Focus on mitochondria in relation to pathogenesis and neuroprotection. Eur J Pharmacol 787, 127–133. [DOI] [PubMed] [Google Scholar]
- Ou Y, Jo RE, Ullian EM, Wong RO, Della Santina L, 2016. Selective Vulnerability of Specific Retinal Ganglion Cell Types and Synapses after Transient Ocular Hypertension. J Neurosci 36, 9240–9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perge JA, Koch K, Miller R, Sterling P, Balasubramanian V, 2009. How the optic nerve allocates space, energy capacity, and information. J Neurosci 29, 7917–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perge JA, Niven JE, Mugnaini E, Balasubramanian V, Sterling P, 2012. Why do axons differ in caliber? J Neurosci 32, 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piette SD, Sergott RC, 2006. Pathological optic-disc cupping. Curr Opin Ophthalmol 17, 1–6. [DOI] [PubMed] [Google Scholar]
- Prokop A, 2013. The intricate relationship between microtubules and their associated motor proteins during axon growth and maintenance. Neural Dev 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, 2015. The contribution of the sclera and lamina cribrosa to the pathogenesis of glaucoma: Diagnostic and treatment implications. Prog Brain Res 220, 59–86. [DOI] [PubMed] [Google Scholar]
- Rai A, Singh PK, Singh V, Kumar V, Mishra R, Thakur AK, Mahadevan A, Shankar SK, Jana NR, Ganesh S, 2018. Glycogen synthase protects neurons from cytotoxicity of mutant huntingtin by enhancing the autophagy flux. Cell Death Dis 9, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Calloway N, Ryan TA, 2014. Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom BR, Orkand RK, 1996. Glial-neuronal interactions in non-synaptic areas of the brain: studies in the optic nerve. Trends Neurosci 19, 352–358. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Smith TL, Williamson JC, Phillips LL, 2012. Unmyelinated axons show selective rostrocaudal pathology in the corpus callosum after traumatic brain injury. J Neuropathol Exp Neurol 71, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck J 2013. The pathogenesis of glaucoma in the interplay with the immune system. Invest Ophthalmol Vis Sci March, Vol. 54, 2393–2409. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ, 2003. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23, 7881–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintoul GL, Reynolds IJ, 2010. Mitochondrial trafficking and morphology in neuronal injury. Biochim Biophys Acta 1802, 143–150. [DOI] [PubMed] [Google Scholar]
- Risner ML, McGrady NR, Pasini S, Lambert WS, Calkins DJ, 2020. Elevated ocular pressure reduces voltage-gated sodium channel NaV1.2 protein expression in retinal ganglion cell axons. Exp Eye Res 190, 107873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risner ML, Pasini S, Cooper ML, Lambert WS, Calkins DJ, 2018. Axogenic mechanism enhances retinal ganglion cell excitability during early progression in glaucoma. Proc Natl Acad Sci U S A 115, E2393–e2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Dib-Hajj SD, Waxman SG, 2005. Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol 564, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington RM, Carlson BJ, Crish SD, Calkins DJ, 2010. The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci 51, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington RM, Sidorova T, Ward NJ, Chakravarthy R, Ho KW, Calkins DJ, 2015. Activation of transient receptor potential vanilloid-1 (TRPV1) influences how retinal ganglion cell neurons respond to pressure-related stress. Channels (Austin) 9, 102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp CL, Li Y, Dietz JA, Janssen KT, Nickells RW, 2006. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci 7, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal IA, Ethier CR. 2009. Biomechanics of the optic nerve head. Exp Eye Res. 88:799–807. [DOI] [PubMed] [Google Scholar]
- Smedowski A, Akhtar S, Liu X, Pietrucha-Dutczak M, Podracka L, Toropainen E, Alkanaan A, Ruponen M, Urtti A, Varjosalo M, Kaarniranta K, Lewin-Kowalik J. Electrical synapses interconnecting axons revealed in the optic nerve head - a novel model of gap junctions’ involvement in optic nerve function. Acta Ophthalmol. 2020June;98(4):408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Xia CZ, Dengler-Crish CM, Fening KM, Inman DM, Schofield BR, Crish SD, 2016. Persistence of intact retinal ganglion cell terminals after axonal transport loss in the DBA/2J mouse model of glaucoma. J Comp Neurol 524, 3503–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto I, Oglesby E, Buckingham BP, Son JL, Roberson ED, Steele MR, Inman DM, Vetter ML, Horner PJ, Marsh-Armstrong N, 2008. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J Neurosci 28, 548–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ Jr., Clarkson RL, Sanders SJ, Bender KJ, 2019. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 103, 673–685.e675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele ML, Robinson SR, 2012. Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol Aging 33, 423.e421–413. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Stys PK, 2010. Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation. Trends Mol Med 16, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Lye-Barthel M, Masland RH, Jakobs TC, 2009. The morphology and spatial arrangement of astrocytes in the optic nerve head of the mouse. J Comp Neurol 516, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Moore S, Jakobs TC, 2017. Optic nerve astrocyte reactivity protects function in experimental glaucoma and other nerve injuries. J Exp Med 214, 1411–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susanna R Jr., De Moraes CG, Cioffi GA, Ritch R, 2015. Why Do People (Still) Go Blind from Glaucoma? Transl Vis Sci Technol 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, Takamura Y, Sakamoto H, Satoh K, Kondo M, Sakamoto T, Goldberg JL, Nabekura J, Tanihara H, 2015. In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proc Natl Acad Sci U S A 112, 10515–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Sabharwal J, Wu SM, Frankfort BJ, 2020. Intraocular Pressure Elevation Compromises Retinal Ganglion Cell Light Adaptation. Invest Ophthalmol Vis Sci 61, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, 2006. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res 25, 490–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY, 2014. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 121, 2081–2090. [DOI] [PubMed] [Google Scholar]
- Trevisiol A, Saab AS, Winkler U, Marx G, Imamura H, Möbius W, Kusch K, Nave KA, Hirrlinger J, 2017. Monitoring ATP dynamics in electrically active white matter tracts. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivli A, Koliarakis I, Terzidou C, Goulielmos GN, Siganos CS, Spandidos DA, Dalianis G, Detorakis ET, 2019. Normal-tension glaucoma: Pathogenesis and genetics. Exp Ther Med 17, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hook MJ, Nawy S, Thoreson WB, 2019. Voltage- and calcium-gated ion channels of neurons in the vertebrate retina. Prog Retin Eye Res 72, 100760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Matthews G, 2006. Expression of sodium channels Nav1.2 and Nav1.6 during postnatal development of the retina. Neurosci Lett 403, 315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas ME, Yamagishi Y, Tessier-Lavigne M, Sagasti A, 2015. Live Imaging of Calcium Dynamics during Axon Degeneration Reveals Two Functionally Distinct Phases of Calcium Influx. J Neurosci 35, 15026–15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura LM, Porciatti V 2005. Restoration of retinal ganglion cell function in early glaucoma after intraocular pressure reduction: a pilot study. Ophthalmology. 2005;112(1):20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrabec F Glaucomatous cupping of the human optic disk: a neuro-histologic study. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1976March3;198(3):223–34. [DOI] [PubMed] [Google Scholar]
- Wallace DC, 1999. Mitochondrial diseases in man and mouse. Science 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- Wang H, Song G, Chuang H, Chiu C, Abdelmaksoud A, Ye Y, Zhao L, 2018. Portrait of glial scar in neurological diseases. Int J Immunopathol Pharmacol 31, 2058738418801406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA, 2012. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 196, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Dong J, Cull G, Fortune B, Cioffi GA, 2003. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Invest Ophthalmol Vis Sci 44, 2–9. [DOI] [PubMed] [Google Scholar]
- Wang R, Seifert P, Jakobs TC, 2017. Astrocytes in the Optic Nerve Head of Glaucomatous Mice Display a Characteristic Reactive Phenotype. Invest Ophthalmol Vis Sci 58, 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NJ, Ho KW, Lambert WS, Weitlauf C, Calkins DJ, 2014. Absence of transient receptor potential vanilloid-1 accelerates stress-induced axonopathy in the optic projection. J Neurosci 34, 3161–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareham LK, Calkins DJ, 2020. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front Cell Dev Biol 8, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareham LK, Cooper ML, Calkins DJ, 2021. Glaucoma: mechanisms of neurodegeneration, in: Fritzsch B (Ed.), The Senses: A Comprehensive Reference 2nd ed, pp. 567–589. [Google Scholar]
- Weber B, Barros LF, 2015. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb Perspect Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN, Aung T, Medeiros FA, 2014. The pathophysiology and treatment of glaucoma: a review. Jama 311, 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitlauf C, Ward NJ, Lambert WS, Sidorova TN, Ho KW, Sappington RM, Calkins DJ, 2014. Short-term increases in transient receptor potential vanilloid-1 mediate stress-induced enhancement of neuronal excitation. J Neurosci 34, 15369–15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, Tribble JR, Pepper KW, Cross SD, Morgan BP, Morgan JE, John SW, Howell GR, 2016. Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol Neurodegener 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies O, & John SW (2017). Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science (New York, N.Y.), 355(6326), 756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley MT, 2010. Energy metabolism of the visual system. Eye Brain 2, 99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota S, Takihara Y, Arimura S, Miyake S, Takamura Y, Yoshimura N, Inatani M, 2015. Altered Transport Velocity of Axonal Mitochondria in Retinal Ganglion Cells After Laser-Induced Axonal Injury In Vitro. Invest Ophthalmol Vis Sci 56, 8019–8025. [DOI] [PubMed] [Google Scholar]
- You Y, Gupta VK, Graham SL, Klistorner A, 2012. Anterograde degeneration along the visual pathway after optic nerve injury. PLoS One 7, e52061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu DY, Cringle SJ, Balaratnasingam C, Morgan WH, Yu PK, Su EN Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog Retin Eye Res. 2013September;36:217–46. [DOI] [PubMed] [Google Scholar]
- Yu-Wai-Man P, Griffiths PG, Chinnery PF, 2011. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res 30, 81–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tohari AM, Marcheggiani F, Zhou X, Reilly J, Tiano L, Shu X, 2017. Therapeutic Potential of Co-enzyme Q10 in Retinal Diseases. Curr Med Chem 24, 4329–4339. [DOI] [PubMed] [Google Scholar]
- Zhong X, Yi X, da Silveira ESRC, Zhang Y, Liu K, Xiao F, Zhong C, 2017. CoQ10 Deficiency May Indicate Mitochondrial Dysfunction in Cr(VI) Toxicity. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Li P, Zhou YG, Ye J, 2020. Altered Energy Metabolism During Early Optic Nerve Crush Injury: Implications of Warburg-Like Aerobic Glycolysis in Facilitating Retinal Ganglion Cell Survival. Neurosci Bull 36, 761–777. [DOI] [PMC free article] [PubMed] [Google Scholar]