

Abstract
In contrast to humans and dogs, the skin microbiota of wolves is yet to be described. Here, we investigated the skin microbiota of dogs and wolves kept in outdoor packs at the Wolf Science Center (WSC) via 16S rRNA gene amplicon sequencing. Skin swab samples were also collected from human care takers and their pet dogs. When comparing the three canine groups, representing different degrees of human contact to the care takers and each other, the pet dogs showed the highest level of diversity. Additionally, while human skin was dominated by a few abundant phylotypes, the skin microbiota of the care takers who had particularly close contact with the WSC animals was more similar to the microbiota of dogs and wolves compared to the humans who had less contact with these animals. Our results suggest that domestication may have an impact on the diversity of the skin microbiota, and that the canine skin microbiota can be shared with humans, depending on the level of interaction.
Subject terms: Ecology, Microbiology
Introduction
As domestic dogs (Canis familiaris) are among the most popular companion animals in Western societies, their skin microbiome, to which humans are exposed to, is of general interest1–3. Since domestication from wild grey wolves began more than 30,000 years ago4, domesticated dogs have undergone dramatic phenotypic and genotypic changes5–8 that are linked to having switched to living close to humans and feeding on human waste. Even though this new ecological niche of dogs has likely affected their microbiome as well, direct comparisons of the microbiota of domestic dogs and wolves (Canis lupus) remain at present scarce9,10. A few more independent investigations into taxonomic, metabolic and nutritional aspects of the canine gut microbiota and the composition of the wolf fecal microbiome have recently been published9–13, while that of the domesticated dogs has been studied for over a decade14–17 and remains of interest18,19. However, no information on the skin microbiota of wolves has been published and the skin microbiota of dogs has also been investigated only to a limited extent1,3,20–23. Importantly, the skin microbiota of dogs is of special interest, as, given that dogs and humans are frequently in physical contact and often share the same living environment, it may have a large impact on the microbiota of cohabiting humans2.
The skin is the interface between body and environment, is involved in regulating body temperature, and enables the sensation of touch and temperature. The skin microbiota is considered to play an important role in the prevention of disease via cross-talk with host cells, influencing cellular function and immunity24. It has been shown to differ greatly among body sites and individuals1–3,22. This has also been largely confirmed in human individuals25–27. Additionally, there are several additional factors known to affect the composition of the skin microbiota of humans, such as age28,29, birth delivery mode30, sex31,32, hygiene, geography33–35 and urbanization32,36. This is also true for the canine skin microbiota composition20,22. Moreover, cohabitation2,38 seems to be among the most important factors shaping the skin microbiota of both humans39 and dogs21,22,35, even if these changes are mostly driven by low abundant phylotypes37. Several studies have demonstrated that cohabitation with dogs leads to an exchange of gut as well as skin microbes between dogs and humans2,38,40. Sharing skin microbiota between dogs and humans has also been reported at the level of individual correlations specific to dyads living in the same household, and this process even affects the microbial exchange between cohabiting humans2,38. Interestingly, one study that included not only dogs but also cats as pets found weaker effects in this regard41, raising the possibility that dogs may have an especially strong impact on the microbiota of cohabiting humans. Given that pet owners tend to establish especially close relationships and engage in the most diverse activities with their dogs, this closeness as well as the long evolutionary history of dog–human cohabitation may contribute to the successful establishment of exchanged microbes between dogs and humans.
In the current study, we aimed to investigate the skin microbiota of wolves, dogs and humans that all had a varying amount of contact with each other and partly inhabited the same environment. To do so, we sampled dogs and wolves that had been raised by human care takers and were kept in a game park setting at the Wolf Science Center (WSC) for the purpose of behavioural and cognitive research. As such, both groups of animals had a similar but limited amount of contact to humans as compared to pets. Furthermore, the care takers of these animals and the pet dogs belonging to them were also sampled, in order to investigate whether various levels of contact with humans corresponds to different microbial compositions inhabiting the skin of composition in dogs. Indeed, the human participants and the pet dogs had varying amounts of contact to the wolves and dogs kept at the game park (WSC dogs and wolves), ranging from frequent direct contact (animal trainers) to medium or low contact (researchers studying animal behaviour). By comparing these groups of humans and canines we were able to show that the pet dogs had the highest level of diversity in their skin microbiomes. Additionally, the trainers that had particularly close contact with the animals had a more similar skin microbiome to the wolves and dogs, which was observed when comparing the distribution of various taxonomic levels. Lastly, the human skin was dominated by a few highly abundant phylotypes. Overall, our results suggest that exposure to various environments can have a large impact on the diversity of the skin microbiome and that the canine skin microbiome can be shared with humans to a degree that depends on the level of interaction.
Results and discussion
Humans have the least and pet dogs have the most diverse skin microbiota
Out of all four groups, species richness and diversity were lowest in human skin microbiota, whereas the pet dog group had the highest species richness and diversity. All four groups differed significantly to each other (Kruskal–Wallis; Chao1, chi-squared = 20.828, df = 3, p < 0.001; Shannon, chi-squared = 23.332, df = 3, p < 0.001). In the following pairwise comparisons all groups differed significantly from each other (p < 0.005), except for the WSC dog group vs. the WSC wolf group, that showed a very similar species richness and diversity (Dunn's-test for multiple comparisons; Chao1, p = 0.574; Shannon, p = 0.579; Fig. 1.). The low species richness and diversity in the human skin microbiota has also been shown in previous studies42,43 and might be driven by physiological differences of the skin between humans and canines, such as pH and hair covering, as well as by skin hygiene practices and differences in regular environmental contact44,45. For humans, protection from invasion by microorganisms is controlled by microbial desiccation, competition with resident microbiota, and an acidic pH. The average reported cutaneous pH of humans is 4.8, while the average cutaneous pH of dogs is 7.4, suggesting that the higher pH might support a more complex skin microbiota composition, as compared to humans45. Moreover, Clemente et al.33 showed that the skin microbiome of uncontacted humans living outdoors is significantly more diverse than that of westernized people, supporting the assumption that modern habits, including personal and environmental hygiene, lead to a decrease in skin microbiota species richness and diversity33,46. With respect to the high diversity of the pet dogs’ microbiota, we suggest that the pet dog skin was regularly exposed to a diverse set of environmental microbiomes, both indoors and outdoors. The WSC animals (wolves and dogs), in contrast, are restricted to the game park environment.
Figure 1.
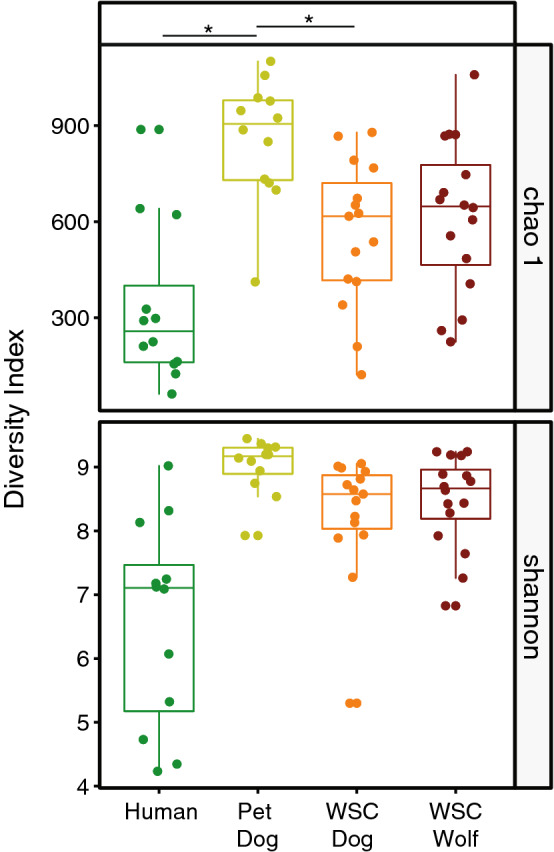
Species richness of microbiota of humans, pet dogs, WSC dogs, and WSC wolves are displayed as Chao1 index and alpha diversity as Shannon index. An asterisk indicates a p value below 0.05 when conducted pairwise Kruskal–Wallis test for both indices. All four groups differed significantly among each other in the pairwise comparison for the Chao1 and Shannon indices (all p values ≤ 0.05), except for the WSC dog group versus the WSC wolf group (both p values ≥ 0.05).
Humans and pet dogs are less similar compared to WSC dogs and wolves, which are more similar to each other in their skin microbiota composition
The analysis of independent variable influence, i.e. sex (male vs. female), age class (sub adult vs. adult), last antibiotic treatment (early vs. late) and human contact did not reveal significant effects on the canine skin microbiota composition (multivariate PERMANOVA in Adonis; diet, p = 0. 387; age, p = 0.136; sex, p = 0.114; last antibiotic treatment, p = 0.728, human contact, p = 0.539), but the groups differed significantly (multivariate PERMANOVA in Adonis; group, R2 = 0.074, F model = 1.622, p = 0.0228). Also in beta diversity significant differences were detected between groups (PERMANOVA; Bray–Curtis dissimilarity; pseudo R2 = 0.180; F model = 3.741; p = 0.001). In the pairwise comparison, all groups differed significantly from each other (R2 = 0.076–0.209, F model = 1.468–6.598, p = 0.006). In the visual inspection of tSNE plots which were based on Jensen-Shannon divergence and Bray–Curtis dissimilarity (Fig. 2.), humans and pet dogs appear less similar to each other and the other groups, while WSC dogs and wolves appear more similar to each other in their skin microbiota composition. Not surprisingly, among the four groups humans appeared most distinct to all canines that grouped more closely together.
Figure 2.
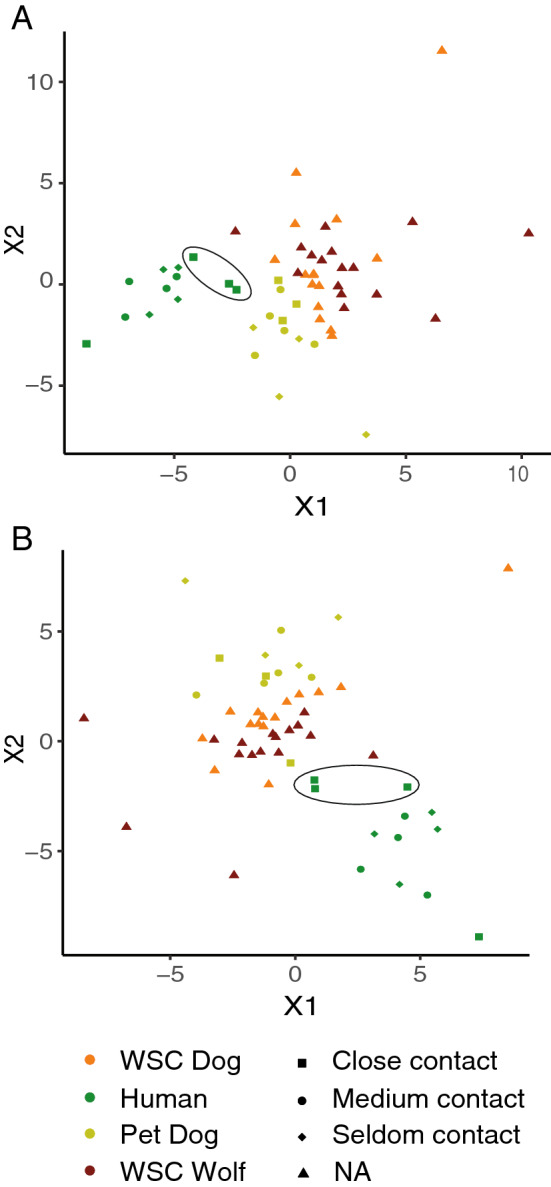
t-distributed stochastic neighbor embedding (tSNE) plots. Humans that have very close and frequent physical interactions with the WSC animals (dog and wolf group) are circled and more similar to the WSC animals. (A) Jensen–Shannon divergence. (B) Bray–Curtis dissimilarity.
Physiological differences between canine species affecting their microbiota may be relatively small. DeCandia et al. (2019)47 found out that coyotes, red and grey foxes living in the wild in North-America responded with a similar microbial community shift to a Sarcoptes scabiei mite infection. However, the environment and living conditions of individuals do lead to a difference in microbial composition within species48 suggesting that environmental effects on microbiota are significant. These observations are also consistent with the pairwise alpha diversity comparisons above, where the only non-significant difference between groups was the comparison of WSC dogs to wolves.
While physiological differences of the skin are likely to contribute to the differences observed between humans and canines, in this study mutual contact and living in the same environment have likely reduced difference between the canine groups44,45. This is probably an important factor that has made the WSC dog and wolf samples most similar to each other (Fig. 2.). This is consistent with the species richness and alpha diversity, and suggests that environmental exposure has a similar or stronger impact on shaping the skin microbiota than the evolutionary segregation of wolves and dogs due to domestication (see also21,22,35).
It has further been suggested that diet shapes the skin microbiome49. Here, diet does not explain the difference between the pet dog group and the WSC animals (PERMANOVA; diet, R2 = 0.046, F model = 1.007, p = 0.386). Both dog groups, WSC dogs and pet dogs were fed a similar diet. However, the WSC dog and wolf groups microbial community structure was similar to each other despite being fed different diets (Supplementary Dataset 1).
The hologenome theory of evolution supports the idea that specific groups or species evolved together with their microbiomes, and that this symbiosis greatly affects their health status50,51. A loss of microbiome diversity can be caused by several factors and may impact health. For example, a decrease in species richness and diversity can be caused by specific living conditions over several generations, as seen in both humans and canines living in or close to urban environments, as compared to populations living under natural conditions33. Outside of these long-term shifts in the skin microbiomes of certain groups, similar changes in diversity that do not necessarily impact health, can also be caused within shorter time spans, as represented in our canine groups. The pet dogs within the current study were similar to each other in terms of beta diversity, but distinct from the WSC animals (Fig. 2). Of the pet dogs, four out of twelve were originally born at the WSC and later on adopted by the human caretakers, while remaining on the same diet; their origin however apparently did not leave remaining track in their microbiome as they were not more similar to the WSC dogs than the other pet dogs. Lastly, three of the four humans, that were a priory classified as having close physical interactions with the WSC dogs and wolves were more similar to the WSC animals than the other human participants with less animal contact (PERMANOVA; R2 = 0.167, F = 1.967, p = 0.077; Fig. 2). This was also found when conducting a PCoA (Supplementary Fig. 1). Based on this finding, a new group named “human close” was used to label downstream analyses and included the three human samples that clustered closer to the animals.
The skin microbiota of humans with close contact to WSC animals is more similar to the microbiota of the WSC animals
LEfSe analysis revealed an enrichment of Acidobacteria, Verrucomicrobia and Proteobacteria (in particular Alphaproteobacteria and Gammaproteobacteria) in wolves, and Bacteroidetes and Proteobacteria (in particular Betaproteobacteria) in WSC dogs, whereas Firmicutes were enriched in human samples (Fig. 3A). Interestingly, not only the three canine groups, but also the humans with close contact to the WSC animals, had a microbiome that showed higher proportions of Proteobacteria, Acidobacteria, Verrucomicrobia, and Bacteroidetes (Fig. 4). The proportions of Firmicutes and Actinobacteria were lower in these groups compared to the humans without close contact to the WSC animals. This suggests that contact with the animals increased the ratio of gram negative to gram positive microorganisms on the skin, and the phylum level diversity. The Proteobacteria appeared to be particularly shared between humans with close animal contact and pet dogs (Fig. 4). The patterns observed at phylum level could also be seen on lower taxonomic levels in the LEfSe analysis, with a significant enrichment of f.e. Sphingomonadaceae in the wolf group (Fig. 3B), and f.e. Pseudomonadaceae in the WSC dog group (Fig. 3C). Consistent with this observation, humans with close contact to the WSC animals had microbial community shifts mainly caused by an increase in Pseudomonadaceae, Sphingomonadaceae and Flavobacteriaceae abundance and a decrease of Staphylococcacceae and Corynebacteriaceae abundance (Supplementary Fig. 2).
Figure 3.

(A) LEfSe (Linear discriminant analysis Effect Size) calculation which determines features most likely to explain differences between groups, (B–D) LEfSe plot on differential features. H = human, PD = pet dog, D = WSC dog, W = WSC wolf.
Figure 4.

Phylum level taxonomic distributions. (A) Each individual sample comparison ordered by group. Humans are additionally ordered by level of animal contact, as classified based on the tSNE plots (Fig. 2). (B) Average distribution across groups, including humans with close contact. (C) Proportion of ASVs that are exclusively observed more than 200 times in only non-close contact Humans (not shared) or in both close contact Humans and Pet Dogs (shared).
Lehtimaki et al.39 found that intensive contact to forest and arable land (which was the case for our canine groups compared to the human individuals) correlated with a higher diversity of soil-based Proteobacteria and other soil-related taxa on skin, while in urban areas, skin-based Actinobacteria were more abundant. This was also the case in our human samples, with the exception of those from humans with very close contact to WSC animals. This supports the importance of human-animal interactions in providing exposure to environmental microbes and affecting the skin microbiome composition2,23. These findings also indicate that not only the pet dog skin microbiota seems to be greatly affected by their surroundings, but that the human skin microbiota can also show strong shifts when being exposed to canine animals and their environment. Coelho et al.53 recently indicated that the similarities in the gut microbiome of dogs and humans might not be solely explained by direct transmission, but rather a function of similar physiology and lifestyle53. Lehtimaki et al.54 also examined that a shared living environment as well as lifestyle patterns correlate with microbial similarities in dog-owner pairs and that they influence the structure of skin microbiota in both species. In our study, direct transmission might have also contributed to the similarities observed, as the skin was not cleaned before sampling thereby washing off allochthonous microorganisms, as applied in skin microbiota sampling of amphibians43,55. However, different to former studies on pets2,38, we found no evidence that owners and pet dogs living in the same household would have more similar skin microbiota to each other than to other pet dogs or other owners, respectively.
A rural environment or countryside lifestyle are known to boost protection against allergic diseases39. This protection has suggested to be mediated by acquiring a more diverse microbiome or by exposure to environmental microbes that support immune tolerance39. In this study, the three humans with close contact to the WSC animals had a microbiota structure with altered composition (PERMANOVA; R2 = 0.167, F = 1.967, p = 0.077) but without higher species richness and/or diversity. Whether these changes might benefit their hosts in terms of immune responses or allergies, as suggested previously39,46,54, needs further investigation.
The human skin microbiota
A total of 15,181 ASVs were detected in all skin samples from the four groups. In the human group, 4,676 ASVs were found, of which 581 (12%) were shared with the pet dogs. Firmicutes, in particular Staphylococcaceae were identified as significantly enriched in humans in the LefSe analysis (Fig. 3A,D). The six most abundant ASVs in the human group were classified as Staphylococcus with relative abundances from 7.22 to 2.39%. The dominance of Staphylococcaceae is in line with previous literature56. Staphylococcus species are known to especially inhabit the human skin as commensals and opportunistic pathogens57. Other highly abundant phylotypes detected in the current dataset have also been described as normal inhabitants of the human skin, such as Corynebacteria, that show a higher sensitivity to environmental factors compared to staphylococci58. Cutibacterium has also been found on human skin samples. Cutibacterium acne is seen as an important species within this genus, which is linked to acne and balanced by specific Staphylococcus strains59. Several ASVs among the 50 most abundant, which were classified as Staphylococcus, Corynebacterium and Cutibacterium were significantly higher abundant in human samples in the pairwise comparison with canine skin samples (Table 1).
Table 1.
The statistically significant differences in relative abundance among the 50 most abundant ASVs are shown for the human, pet dog, WSC dog und wolf group.
| Taxonomy | ASV number | Relative abundance ± standard deviation [%] | |||
|---|---|---|---|---|---|
| Human | Pet Dog | WSC Dog | WSC Wolf | ||
| Staphylococcus | 1 | 7,22 ± 6,700a | 0,27 ± 0,173b | 0,09 ± 0,345c | 0,00 ± 0,000c |
| Staphylococcus | 2 | 5,19 ± 4,930a | 0,18 ± 0,130b | 0,01 ± 0,055c | 0,03 ± 0,132c |
| Staphylococcus | 3 | 4,58 ± 4,227a | 0,12 ± 0,154b | 0,00 ± 0,000b | 0,00 ± 0,000b |
| Staphylococcus | 4 | 4,12 ± 3,960a | 0,08 ± 0,090b | 0,00 ± 0,000c | 0,00 ± 0,000c |
| Rhodococcus | 5 | 0,50 ± 0,759b | 0,48 ± 0,522ab | 1,26 ± 0,892a | 0,83 ± 0,849ab |
| Pseudomonas | 6 | 0,75 ± 1,455 | 0,52 ± 1,225 | 0,31 ± 0,469 | 1,10 ± 2,541 |
| Staphylococcus | 7 | 2,46 ± 2,434a | 0,03 ± 0,051b | 0,00 ± 0,000b | 0,00 ± 0,000b |
| Staphylococcus | 8 | 2,39 ± 2,409a | 0,01 ± 0,034b | 0,00 ± 0,000b | 0,00 ± 0,000b |
| Cutibacterium | 9 | 2,21 ± 2,576a | 0,00 ± 0,000b | 0,02 ± 0,067b | 0,00 ± 0,000b |
| Sphingomonas | 10 | 0,14 ± 0,208b | 0,46 ± 0,401ab | 0,63 ± 0,370a | 0,60 ± 0,530a |
| Streptococcus | 11 | 1,94 ± 3,045a | 0,14 ± 0,125b | 0,01 ± 0,036c | 0,01 ± 0,024c |
| Staphylococcus | 12 | 1,78 ± 1,848a | 0,01 ± 0,018b | 0,00 ± 0,000b | 0,00 ± 0,000b |
| Unclassified Micrococcaceae | 13 | 0,06 ± 0,119b | 0,35 ± 0,213a | 0,60 ± 0,386a | 0,47 ± 0,359a |
| Unclassified Xanthomonadaceae | 14 | 0,11 ± 0,230b | 0,35 ± 0,342ab | 0,39 ± 0,433ab | 0,59 ± 0,498a |
| Rhodococcus | 15 | 0,12 ± 0,274b | 0,21 ± 0,335ab | 0,64 ± 0,581a | 0,43 ± 0,603ab |
| Rhodococcus | 16 | 0,14 ± 0,255 | 0,21 ± 0,270 | 0,59 ± 0,500 | 0,42 ± 0,549 |
| Micrococcus | 17 | 1,09 ± 1,244a | 0,49 ± 0,492a | 0,03 ± 0,109b | 0,02 ± 0,064b |
| Pseudomonas | 18 | 0,33 ± 0,821 | 0,22 ± 0,510 | 0,14 ± 0,263 | 0,67 ± 1,451 |
| Cutibacterium | 19 | 1,54 ± 1,745a | 0,01 ± 0,013b | 0,00 ± 0,000b | 0,00 ± 0,000b |
| Pseudomonas | 20 | 0,33 ± 0,734 | 0,20 ± 0,506 | 0,10 ± 0,193 | 0,63 ± 1,433 |
| Staphylococcus | 21 | 0,00 ± 0,000 | 0,07 ± 0,187 | 1,11 ± 4,252 | 0,01 ± 0,053 |
| Gp4 | 22 | 0,02 ± 0,086c | 0,06 ± 0,131bc | 0,74 ± 0,549a | 0,31 ± 0,318ab |
| Janibacter | 23 | 0,11 ± 0,282b | 0,06 ± 0,152b | 0,54 ± 0,591a | 0,40 ± 0,703ab |
| Rhodococcus | 24 | 0,15 ± 0,289 | 0,20 ± 0,273 | 0,50 ± 0,439 | 0,31 ± 0,451 |
| Streptococcus | 25 | 1,31 ± 1,875a | 0,03 ± 0,079b | 0,02 ± 0,077b | 0,00 ± 0,000b |
| Cutibacterium | 26 | 1,36 ± 1,472a | 0,00 ± 0,000b | 0,00 ± 0,000b | 0,00 ± 0,000b |
| Unclassified Microbacteriaceae | 27 | 0,08 ± 0,209b | 0,41 ± 0,242a | 0,43 ± 0,313a | 0,24 ± 0,294ab |
| Massilia | 28 | 0,13 ± 0,218a | 0,19 ± 0,223ab | 0,54 ± 0,489a | 0,25 ± 0,227ab |
| Pseudomonas | 29 | 0,28 ± 0,690 | 0,15 ± 0,417 | 0,15 ± 0,233 | 0,51 ± 1,230 |
| Corynebacterium | 30 | 1,19 ± 2,431a | 0,09 ± 0,143b | 0,01 ± 0,020b | 0,00 ± 0,000b |
| Nocardioides | 31 | 0,07 ± 0,226 | 0,36 ± 0,421 | 0,45 ± 0,447 | 0,23 ± 0,290 |
| Unclassified Intrasporangiaceae | 32 | 0,19 ± 0,299 | 0,48 ± 0,501 | 0,27 ± 0,537 | 0,18 ± 0,221 |
| Cutibacterium | 33 | 1,22 ± 1,338a | 0,01 ± 0,017b | 0,00 ± 0,000b | 0,00 ± 0,000b |
| Spartobacteria_genera_incertae_sedis | 34 | 0,08 ± 0,195 | 0,03 ± 0,079 | 0,47 ± 0,587 | 0,34 ± 0,666 |
| Sphingomonas | 35 | 0,05 ± 0,190b | 0,35 ± 0,555ab | 0,42 ± 0,434a | 0,14 ± 0,263ab |
| Pedobacter | 36 | 0,00 ± 0,000c | 0,45 ± 0,379a | 0,43 ± 0,479ab | 0,08 ± 0,138bc |
| Unclassified Actinomycetales | 37 | 0,96 ± 2,368a | 0,11 ± 0,124a | 0,01 ± 0,032b | 0,00 ± 0,000b |
| Massilia | 38 | 0,13 ± 0,192 | 0,20 ± 0,219 | 0,29 ± 0,301 | 0,28 ± 0,360 |
| Gp16 | 39 | 0,06 ± 0,206 | 0,28 ± 0,367 | 0,32 ± 0,408 | 0,22 ± 0,330 |
| Unclassified Pasteurellaceae | 40 | 0,12 ± 0,338b | 0,34 ± 0,237a | 0,24 ± 0,274ab | 0,19 ± 0,313ab |
| Acinetobacter | 41 | 0,30 ± 0,711 | 0,15 ± 0,263 | 0,16 ± 0,245 | 0,27 ± 0,258 |
| Polaromonas | 42 | 0,00 ± 0,000 | 0,32 ± 0,440 | 0,21 ± 0,400 | 0,30 ± 0,374 |
| Sphingosinicella | 43 | 0,02 ± 0,082c | 0,09 ± 0,208bc | 0,30 ± 0,358ab | 0,37 ± 0,377a |
| Sphingomonas | 44 | 0,04 ± 0,087b | 0,24 ± 0,146a | 0,36 ± 0,317a | 0,17 ± 0,230ab |
| Unclassified micrococcaceae | 45 | 0,16 ± 0,255 | 0,22 ± 0,198 | 0,12 ± 0,157 | 0,31 ± 0,210 |
| Unclassified intrasporangiaceae | 46 | 0,10 ± 0,332 | 0,25 ± 0,336 | 0,23 ± 0,363 | 0,21 ± 0,422 |
| Cryobacterium | 47 | 0,15 ± 0,284 | 0,29 ± 0,328 | 0,09 ± 0,180 | 0,27 ± 0,296 |
| Sphingomonas | 48 | 0,00 ± 0,000c | 0,27 ± 0,361ab | 0,08 ± 0,195bc | 0,38 ± 0,404a |
| Acinetobacter | 49 | 0,06 ± 0,146 | 0,10 ± 0,174 | 0,25 ± 0,519 | 0,29 ± 0,570 |
| Janibacter | 50 | 0,10 ± 0,210 | 0,06 ± 0,141 | 0,30 ± 0,504 | 0,25 ± 0,347 |
Different superscripts in same row indicate statistical significance (p value ≤ 0.05).
The canine skin microbiota
This is the first study to describe the bacterial composition on the skin of wolves. Overall, the wolf skin was inhabited by similar abundant phylotypes compared to the dog skin. The most abundant ASVs in the three canine groups were classified as Pseudomonas, Rhodococcus, Staphylococcus, Micrococcus and Sphingomonas with relative abundances between 1.26 to 0.60% (Table 1, Supplementary Dataset 2). Several phylotypes were enriched in the wolf group in the LEfse analysis, among them Alpha- and Gammaproteobacteria, Sphingomonadaceae and Pseudomonadales. In the WSC dog group, Bacteroidetes, Betaproteobacteria and Flavobacteriaceae were enriched, while in the pet dog group Actinobacteria, Pseudomonadaceae and Sphingobacteria showed significant enrichment (Fig. 3). Only one of the 50 most abundant ASVs that differed significantly between the WSC wolf and WSC dog group (Sphingomonas ASV 48, enriched in WSC wolf skin samples compared to WSC dog skin samples), while seven ASVs differed significantly between the WSC wolf and pet dog groups (three Staphylococcus-ASVs, Streptococcus, Pedobacter, unclassified Actinomycetales and Sphingosinicella, Table 1). Except of Sphingosinicella all of these ASVs were enriched in the pet dog skin samples, compared to the WSC wolf skin samples. All highly abundant phylotypes have been found on the skin of healthy dogs before2. Overall, these findings indicate that the wolf skin microbiota is similar to the dog skin microbiota, but several phylotypes are more dominant in the WSC animals. This might be largely affected by living environment21, but whether and to what extent the similar skin microbiota is due to the wolf and dog subjects of the current study sharing the same environment is difficult to tell. A former study on red wolves has shown that living in a captive environment significantly affects the gut microbiota of the animals, and this effect is apparent even if the animals are fed with a diet reflective of their natural environment60. Given that our wolves also live in captivity and do share some of the facilities with the dogs, it is not surprising that the highly abundant ASVs on the WSC wolf skin were also present on the WSC dog skin. However, the fact that they were detected also on the skin of the pet dogs in this study suggests fundamental similarities in the skin microbiota of wolves and dogs, although the abundances of these phylotypes differed among groups. In this study, the shared environment of the wolves and WSC dogs seem to have impacted at least the profile of highly abundant phylotypes of the animals’ skin microbiota (Table 1, just 1 ASV was significantly different between WSC dogs and wolves) and the WSC groups had a similar alpha diversity (WSC dog group vs. the wolf group; Dunn's-test for multiple comparisons; Chao1, p = 0.574; Shannon, p = 0.579). The shared environment might have had more impact on the alpha diversity than their differential diet and their divergent evolution, as also indicated by the lower similarity between the wolf and pet dog group (Dunn’s test; Chao1, p = 0.025; Shannon, p = 0.041) compared to the wolf and WSC dog group (Dunn’s test; Chao1, p = 0.574; Shannon, p = 0.579). The number of shared ASVs within the groups is shown in Supplementary Fig. 3. A huge amount of low abundant organisms was uniquely found in each canine group. The number of unique ASV was more than double in canine groups compared to the humans. The importance of a high skin microbiota diversity in canines, the function of unique low abundant phylotypes, as well as the importance of specific microbial enrichments in groups has not been investigated until now and has to be examined in future research.
Conclusion
Overall, the current results suggest that exposure to various environments and cohabitation affect the skin microbiota more than ecological and physiological changes that took place during the course of dog domestication and can lead to a significant shift of the skin microbiome both in dogs and in humans. Close contact with dogs and wolves shape the human skin microbiome by causing large shifts in the microbiota composition at the phylum and family level, leading to an increase in the ratio of gram negative to positive bacteria.
Material and methods
Ethics statement
The study was discussed and approved by the institutional ethics and animal welfare committee of the University of Veterinary Medicine, Vienna in accordance with good scientific practice guidelines and Austrian national legislation TVG 2012 (ETK-32/02/2019). The study was discussed and approved by the ethics committee of the Medical University, Vienna in accordance with good scientific practice guidelines (EK Nr.: 1058/2020). Informed consent was obtained from all human participants.
Sample collection
Skin swab samples were collected from 16 wolves and 15 dogs kept in large enclosures at the Wolf Science Center (WSC) in Ernstbrunn, Austria. In addition, skin swabs were also taken from 12 humans working at the WSC and 12 pet dogs living in the households of ten of these 12 human individuals. The WSC animals (wolf and dog groups) were hand-raised from their age of 1 week to 5 months of age when they were introduced in packs of adult animals. This period included 20–24 h of close contact to humans daily, including bottle- and hand-feeding, and sleeping together (for more see Virányi and Range (2011)64). Later on, the WSC animals participated in daily sessions of training, along with cognitive and behavioral testing, that included direct physical contact with their trainers. The pet dogs lived in the households of ten of the human participants of the study, had regular access to the WSC environment and were, to varying degrees, in direct contact with the WSC dogs and wolves. A complete and detailed list of all individuals including information about diet, age, sex, origin, health status of the sampled individuals, dog-owner relationship and degree of contact with WSC animals is described in Supplementary Dataset 1, where sample IDs were assigned randomly. The skin swab samples from all individuals (humans, pet dogs, WSC dogs and WSC wolves) were taken from the chest region by rotating the swab (Raucotupf Cotton tipped applicators “S”, Lohmann & Rauscher GmbH, Austria, Vienna) ten seconds on the skin. Blinding during sample collection was not possible. Sampling of all groups happened within the same 3 month period in autumn. Samples were frozen at − 20 °C within 15 min after sampling and stored at − 20 °C until further processing.
DNA extraction and illumina 16S rRNA gene amplicon sequencing
The samples were thawed at room temperature immediately before DNA extraction. For DNA isolation the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) was used according to the manufacturers ‘ protocol, with the exception of mechanical lysis, which was conducted for 20 min instead of 10 min. DNA was eluted in 50 µl DEPC-treated water preheated to 70 °C. DNA concentration was determined using the Qubit 2.0 Fluorimeter (Qubit dsDNA BR Assay Kit, Thermo Fisher Scientific, Vienna, Austria). The samples were all processed at the same time in two batches and a negative extraction control was isolated and processed together with each batch of skin swab samples. For sequencing of 56 samples (skin swab samples plus one negative extraction control), the hypervariable region V4 of bacterial and archaeal 16S rRNA genes was targeted using the primer set 515F-GTGYCAGCMGCCGCGGTAA-modified65 and 806R-GGACTACNVGGGTWTCTAAT-modified66 to generate an approximate amplicon size of 291 bp. Libraries were constructed by ligating sequencing adapters and indices onto purified PCR products using the Nextera XT Sample Preparation Kit (Illumina) according to the recommendations of the manufacturer (including negative controls for the PCR). Equimolar amounts of each of the libraries were pooled and submitted for sequencing on an Illumina MiSeq Personal Sequencer using a 300 bp read length paired-end protocol (MiSeq Reagent Kit v3). Sequencing was performed at the Core Facility Molecular Biology of the Medical University Graz, Austria and sequencing data was provided as demultiplexed forward and reverse fastq files.
Read processing
For all analyses, the forward reads from the dataset were processed into amplicon sequence variants (ASV) using DADA267 (version 1.9.1) with a forward cutoff of 30 and a length cutoff of 290 within the QIIME268 (version 2019.1) environment. All ASVs present in the negative control above 3% relative abundance were removed, leading to the removal of 10 ASVs. Before all beta-diversity and taxonomic comparisons were made, sequences that were classified at the class level as ‘Chloroplast’ were removed. A full overview on the absolute abundances per sample is shown in Supplementary Dataset 3. This data was used for all alpha and beta- diversity metrics, in addition to phylum level comparisons. Phylum level taxonomic assignment was conducted using the RDP package within R (version 1.20.0).
The python and R code used in the analysis is available at [https://github.com/cameronstrachan/WolfSkinCommunity2020].
Statistical analysis
Only samples with a total of more than 10,000 reads after quality control were kept. Relative abundances were then calculated and used in downstream statistical analysis. Permutational multivariate analysis of variance (PERMANOVA) was used to examine the association between the microbial communities and independent variables including sex, diet, age, last antibiotic treatment and human contact. Species richness were calculated using the Chao1 index and Shannon index. The normality distribution of alpha diversity and ASVs was checked in semi-parametric factorial designs with corresponding histograms, Q–Q plots, residual versus fitted plots, the Shapiro–Wilk Test and skewness and kurtosis test for normality as well as for different transformations techniques such as log transformation. Additionally, the homogeneity of variances across samples was tested with the Levense’s test. Due to the non-normal distribution of diversity data and ASV abundances, the Kruskal–Wallis test was applied followed by a Dunn’s test for multiple comparisons. Further, means, standard deviations, and standard errors were calculated. The results were considered statistically significant at p value < 0.05 and the p value was adjusted for multiple comparison with the p-adjusted method Benjamini & Hochberg. The statistical analysis was done using the R statistical computing environment (Version 4.0.5 R Foundation for Statistical Computing, Vienna, Austria) using the package normtest, and stats and vegan. For beta diversity analysis, T-distributed stochastic neighbor embedding (tSNE) plots, based on Jensen-Shannon divergence and Bray–Curtis dissimilarity, were calculated with the package tsne. To find biomarkers that explain the differences between the groups, LEfSe69 was applied by using default values, except of the threshold on the logarithmic LDA score for discriminative features, which was set to 4.
Accession number
Illumina MiSeq sequencing data are available in BioProject SRA database under the accession number PRJEB36827.
ARRIVE guidelines
The study was carried out in compliance with the ARRIVE guidelines (http://www.nc3rs.org.uk/page.asp?id=1357).
Supplementary Information
{kind=link}
Acknowledgements
The Wolf Science Centre was established by Friederike Range, Kurt Kotrschal and Zsófia Virányi, and we thank all the helpers who made this possible hence indirectly supporting this research. We thank all animal trainers at the WSC for raising and caring for the animals, and all human participants of this study for providing samples of their own and their pet dogs. We further thank Kurt Kotrschal and Friederike Range for comments on a previous version of the manuscript. Finally, we greatly thank Monika Dzieciol, Sarah Thalguter and Sara Ricci for their support during sample processing.
Author contributions
E.S., Z.V., I.A.B., M.W. and S.U.W. designed the study. S.U.W. and Z.V. collected and processed the samples. S.U.W., C.S., B.C. and E.S. analyzed and interpreted the data. S.U.W., Z.V., C.S. and E.S. wrote the draft manuscript, which was approved by all authors.
Funding
The competence centre FFoQSI was funded by the Austrian ministries BMVIT, BMDW, and the Austrian provinces Niederoesterreich, Upper Austria, and Vienna within the scope of COMET—Competence Centers for Excellent Technologies. The program COMET is handled by the Austrian Research Promotion Agency FFG. The research work of the authors was also financially supported by the JPI HDHL KP (diet, microbiome, health) co-financed by the Austrian Research Promotion Agency FFG (to ES). The staff of the Wolf Science Center collecting the samples was funded by the Austrian Science Fund (FWF project 30704-B29 to Friederike Range) and by the Vienna Science and Technology Fund (WWTF project CS15-018 to Sarah Marshall-Pescini). The authors further thank many private sponsors including Royal Canin for financial support and the Game Park Ernstbrunn for hosting the Wolf Science Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors contributed equally: Zsófia Virányi and Evelyne Selberherr
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-96160-7.
References
- 1.Rodrigues Hoffmann A, et al. The skin microbiome in healthy and allergic dogs. PLoS ONE. 2014;9:e83197. doi: 10.1371/journal.pone.0083197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Song SJ, et al. Cohabiting family members share microbiota with one another and with their dogs. Elife. 2013;2:1458. doi: 10.7554/eLife.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weese JS. The canine and feline skin microbiome in health and disease. Vet. Dermatol. 2013;24:137–145.e131. doi: 10.1111/j.1365-3164.2012.01076.x. [DOI] [PubMed] [Google Scholar]
- 4.Frantz LA, et al. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science (New York, N.Y.) 2016;352:1228–1231. doi: 10.1126/science.aaf3161. [DOI] [PubMed] [Google Scholar]
- 5.Axelsson E, et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 6.Jardim-Messeder D, et al. Dogs have the most neurons, though not the largest brain: trade-off between body mass and number of neurons in the cerebral cortex of large carnivoran species. Front. Neuroanat. 2017;11:118. doi: 10.3389/fnana.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostrander EA, et al. Dog10K: an international sequencing effort to advance studies of canine domestication, phenotypes and health. Natl. Sci. Rev. 2019;6:810–824. doi: 10.1093/nsr/nwz049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Savolainen P, Zhang YP, Luo J, Lundeberg J, Leitner T. Genetic evidence for an East Asian origin of domestic dogs. Science (New York, NY) 2002;298:1610–1613. doi: 10.1126/science.1073906. [DOI] [PubMed] [Google Scholar]
- 9.Alessandri G, et al. Metagenomic dissection of the canine gut microbiota: insights into taxonomic, metabolic and nutritional features. Environ. Microbiol. 2019 doi: 10.1111/1462-2920.14540. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, et al. Analysis and comparison of the wolf microbiome under different environmental factors using three different data of next generation sequencing. Sci. Rep. 2017;7:11332. doi: 10.1038/s41598-017-11770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goncalves A, et al. Iberian wolf as a reservoir of extended-spectrum beta-lactamase-producing Escherichia coli of the TEM, SHV, and CTX-M groups. Microb. Drug Resist. (Larchmont, NY) 2012;18:215–219. doi: 10.1089/mdr.2011.0145. [DOI] [PubMed] [Google Scholar]
- 12.Lyu T, et al. Changes in feeding habits promoted the differentiation of the composition and function of gut microbiotas between domestic dogs (Canis lupus familiaris) and gray wolves (Canis lupus) AMB Express. 2018;8:123. doi: 10.1186/s13568-018-0652-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, Chen L. Phylogenetic analysis of 16S rRNA gene sequences reveals distal gut bacterial diversity in wild wolves (Canis lupus) Mol. Biol. Rep. 2010;37:4013–4022. doi: 10.1007/s11033-010-0060-z. [DOI] [PubMed] [Google Scholar]
- 14.Greetham HL, Giffard C, Hutson RA, Collins MD, Gibson GR. Bacteriology of the Labrador dog gut: a cultural and genotypic approach. J. Appl. Microbiol. 2002;93:640–646. doi: 10.1046/j.1365-2672.2002.01724.x. [DOI] [PubMed] [Google Scholar]
- 15.Hooda S, Minamoto Y, Suchodolski JS, Swanson KS. Current state of knowledge: the canine gastrointestinal microbiome. Anim. Health Res. Rev. 2012;13:78–88. doi: 10.1017/s1466252312000059. [DOI] [PubMed] [Google Scholar]
- 16.Suchodolski JS. Intestinal microbiota of dogs and cats: a bigger world than we thought. Vet. Clin. Am-Small. 2011;41:261–272. doi: 10.1016/j.cvsm.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suchodolski JS, Camacho J, Steiner JM. Analysis of bacterial diversity in the canine duodenum, jejunum, ileum, and colon by comparative 16S rRNA gene analysis. FEMS Microb. Ecol. 2008;66:567–578. doi: 10.1111/j.1574-6941.2008.00521.x. [DOI] [PubMed] [Google Scholar]
- 18.Alessandri G, et al. Deciphering the bifidobacterial populations within the canine and feline gut microbiota. Appl. Environ. Microb. 2020 doi: 10.1128/AEM.02875-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jha AR, et al. Characterization of gut microbiomes of household pets in the United States using a direct-to-consumer approach. PLoS ONE. 2020 doi: 10.1371/journal.pone.0227289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cusco A, et al. Individual signatures and environmental factors shape skin microbiota in healthy dogs. Microbiome. 2017;5:139. doi: 10.1186/s40168-017-0355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meason-Smith C, et al. What is living on your dog's skin? Characterization of the canine cutaneous mycobiota and fungal dysbiosis in canine allergic dermatitis. FEMS Microbiol. Ecol. 2015 doi: 10.1093/femsec/fiv139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-Campos S, et al. Impact of the early-life skin microbiota on the development of canine atopic dermatitis in a high-risk breed birth cohort. Sci. Rep. 2020;10:1044. doi: 10.1038/s41598-020-57798-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Torres S, et al. Diverse bacterial communities exist on canine skin and are impacted by cohabitation and time. PeerJ. 2017;5:e3075. doi: 10.7717/peerj.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanford JA, Gallo RL. Functions of the skin microbiota in health and disease. Semin. Immunol. 2013;25:370–377. doi: 10.1016/j.smim.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat. Immunol. 2013;14:646–653. doi: 10.1038/ni.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science (New York, NY) 2014;346:954–959. doi: 10.1126/science.1260144. [DOI] [PubMed] [Google Scholar]
- 27.Grice EA, et al. A diversity profile of the human skin microbiota. Genome Res. 2008;18:1043–1050. doi: 10.1101/gr.075549.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capone KA, Dowd SE, Stamatas GN, Nikolovski J. Diversity of the human skin microbiome early in life. J. Invest. Dermatol. 2011;131:2026–2032. doi: 10.1038/jid.2011.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh J, Conlan S, Polley EC, Segre JA, Kong HH. Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 2012;4:77. doi: 10.1186/gm378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dominguez-Bello MG, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc. Natl. Acad. Sci. USA. 2008;105:17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ying S, et al. The influence of age and gender on skin-associated microbial communities in urban and rural human populations. PLoS ONE. 2015;10:e0141842. doi: 10.1371/journal.pone.0141842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clemente JC, et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015 doi: 10.1126/sciadv.1500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hospodsky D, et al. Hand bacterial communities vary across two different human populations. Microbiology. 2014;160:1144–1152. doi: 10.1099/mic.0.075390-0. [DOI] [PubMed] [Google Scholar]
- 35.Leung MHY, Wilkins D, Lee PKH. Insights into the pan-microbiome: skin microbial communities of Chinese individuals differ from other racial groups (vol 5, 11845, 2015) Sci. Rep. Uk. 2016 doi: 10.1038/srep21355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehtimaki J, et al. Patterns in the skin microbiota differ in children and teenagers between rural and urban environments. Sci. Rep. Uk. 2017 doi: 10.1038/srep45651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fierer N, et al. Forensic identification using skin bacterial communities. Proc. Natl. Acad. Sci. USA. 2010;107:6477–6481. doi: 10.1073/pnas.1000162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Misic AM, et al. The shared microbiota of humans and companion animals as evaluated from Staphylococcus carriage sites. Microbiome. 2015 doi: 10.1186/s40168-014-0052-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehtimaki J, et al. Skin microbiota and allergic symptoms associate with exposure to environmental microbes. Proc. Natl. Acad. Sci. USA. 2018;115:4897–4902. doi: 10.1073/pnas.1719785115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Azad MB, et al. Infant gut microbiota and the hygiene hypothesis of allergic disease: impact of household pets and siblings on microbiota composition and diversity. Allergy Asthma Clin. Immunol. 2013;9:15. doi: 10.1186/1710-1492-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kates AE, et al. Household pet ownership and the microbial diversity of the human gut microbiota. Front. Cell Infect. Microbiol. 2020;10:73. doi: 10.3389/fcimb.2020.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Council SE, et al. Diversity and evolution of the primate skin microbiome. Proc. Biol. Sci. 2016 doi: 10.1098/rspb.2015.2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ross AA, Hoffmann AR, Neufeld JD. The skin microbiome of vertebrates. Microbiome. 2019 doi: 10.1186/s40168-019-0694-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fyhrquist N. The human microbiota and its relationship with allergies. Gastroenterol. Clin. N. 2019;48:377. doi: 10.1016/j.gtc.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 45.Matousek JL, Campbell KL. A comparative review of cutaneous pH. Vet. Dermatol. 2002;13:293–300. doi: 10.1046/j.1365-3164.2002.00312.x. [DOI] [PubMed] [Google Scholar]
- 46.Mills JG, et al. Relating urban biodiversity to human health with the 'holobiont' concept. Front. Microbiol. 2019 doi: 10.3389/fmicb.2019.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeCandia AL, Leverett KN, vonHoldt BM. Of microbes and mange: consistent changes in the skin microbiome of three canid species infected with Sarcoptes scabiei mites. Parasit. Vectors. 2019;12:488. doi: 10.1186/s13071-019-3724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sugden S, St Clair CC, Stein LY. Individual and site-specific variation in a biogeographical profile of the coyote gastrointestinal microbiota. Microb. Ecol. 2021;81:240–252. doi: 10.1007/s00248-020-01547-0. [DOI] [PubMed] [Google Scholar]
- 49.Divya S, Sriharsha M, Narotham RK, Krupa SN, Siva TRK. Role of diet indermatological conditions. Nutr. Food Sci. (2015).
- 50.Strachan DP. Hay-fever, hygiene, and household size. Br. Med. J. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wold AE. The hygiene hypothesis revised: Is the rising frequency of allergy due to changes in the intestinal flora? Allergy. 1998;53:20–25. doi: 10.1111/j.1398-9995.1998.tb04953.x. [DOI] [PubMed] [Google Scholar]
- 52.Rossi CC, Andrade-Oliveira AL, Giambiagi-deMarval M. CRISPR tracking reveals global spreading of antimicrobial resistance genes by Staphylococcus of canine origin. Vet. Microbiol. 2019;232:65–69. doi: 10.1016/j.vetmic.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 53.Coelho LP, et al. Similarity of the dog and human gut microbiomes in gene content and response to diet. Microbiome. 2018;6:72. doi: 10.1186/s40168-018-0450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lehtimaki J, et al. Simultaneous allergic traits in dogs and their owners are associated with living environment, lifestyle and microbial exposures. Sci. Rep. Uk. 2020 doi: 10.1038/S41598-020-79055-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Culp CE, Falkinham JO, Belden LK. Identification of the natural bacterial microflora on the skin of eastern newts, bullfrog tadpoles and redback salamanders. Herpetologica. 2007;63:66–71. doi: 10.1655/0018-0831(2007)63[66:Iotnbm]2.0.Co;2. [DOI] [Google Scholar]
- 56.Oh J, et al. Temporal stability of the human skin microbiome. Cell. 2016;165:854–866. doi: 10.1016/j.cell.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coates R, Moran J, Horsburgh MJ. Staphylococci: colonizers and pathogens of human skin. Future Microbiol. 2014;9:75–91. doi: 10.2217/fmb.13.145. [DOI] [PubMed] [Google Scholar]
- 58.Kwaszewska A, Sobis-Glinkowska M, Szewczyk EM. Cohabitation-relationships of corynebacteria and staphylococci on human skin. Folia Microbiol. 2014;59:495–502. doi: 10.1007/s12223-014-0326-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Sullivan JN, Rea MC, O'Connor PM, Hill C, Ross RP. Human skin microbiota is a rich source of bacteriocin-producing staphylococci that kill human pathogens. FEMS Microb. Ecol. 2019 doi: 10.1093/femsec/fiy241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bragg M, Freeman EW, Lim HC, Songsasen N, Muletz-Wolz CR. Gut microbiomes differ among dietary types and stool consistency in the captive red wolf (Canis rufus) Front. Microbiol. 2020 doi: 10.3389/Fmicb.2020.590212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bradley CW, et al. Longitudinal evaluation of the skin microbiome and association with microenvironment and treatment in canine atopic dermatitis. J. Invest. Dermatol. 2016;136:1182–1190. doi: 10.1016/j.jid.2016.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dimitriu PA, et al. New insights into the intrinsic and extrinsic factors that shape the human skin microbiome. MBio. 2019 doi: 10.1128/mBio.00839-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hui N, et al. Diverse environmental microbiota as a tool to augment biodiversity in urban landscaping materials. Front. Microbiol. 2019 doi: 10.3389/fmicb.2019.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Viranyi Z, Range F. Evaluating the logic of perspective-taking experiments. Learn. Behav. 2011;39:306–309. doi: 10.3758/s13420-011-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016;18:1403–1414. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 66.Apprill A, McNally S, Parsons R, Weber L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015;75:129–137. doi: 10.3354/ame01753. [DOI] [Google Scholar]
- 67.Callahan BJ, et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bolyen E, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2 (vol 37, pg 852, 2019) Nat. Biotechnol. 2019;37:1091–1091. doi: 10.1038/s41587-019-0252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segata N, et al. Metagenomic biomarker discovery and explanation. Gen. Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.