

Graphical abstract
Keywords: Lactylation, Post-translational modification, Few-shot learning, Deep neural network, Multi-feature hybrid system, Ensemble learning
Abstract
As a novel lactate-derived post-translational modification (PTM), lysine lactylation (Kla) is involved in diverse biological processes, and participates in human tumorigenesis. Identification of Kla substrates with their exact sites is crucial for revealing the molecular mechanisms of lactylation. In contrast with labor-intensive and time-consuming experimental approaches, computational prediction of Kla could provide convenience and increased speed, but is still lacking. In this work, although current identified Kla sites are limited, we constructed the first Kla benchmark dataset and developed a few-shot learning-based architecture approach to leverage the power of small datasets and reduce the impact of imbalance and overfitting. A maximum 11.7% (0.745 versus 0.667) increase of area under the curve (AUC) value was achieved in contrast to conventional machine learning methods. We conducted a comprehensive survey of the performance by combining 8 sequence-based features and 3 structure-based features and tailored a multi-feature hybrid system for synergistic combination. This system achieved >16.2% improvement of the AUC value (0.889 versus 0.765) compared with single feature-based models for the prediction of Kla sites in silico. Taken few-shot learning and hybrid system together, we present our newly designed predictor named FSL-Kla, which is not only a cutting-edge tool for Kla site profile but also could generate candidates for further experimental approaches. The webserver of FSL-Kla is freely accessible for academic research at http://kla.zbiolab.cn/.
1. Introduction
The Warburg effect, originally known for the ravenous consumption of glucose leading to lactate production, even in the presence of oxygen, is involved in diverse cellular processes such as cell signaling, macrophage polarization, immunological response and participates in human tumorigenesis [1], [2], [3]. Lactate, the end metabolite produced during fermentative glycolysis which is a phenotype described as part of the Warburg effect, not only serves metabolic functions but also acts as non-metabolic roles [4], [5]. Although the former has been extensively studied, its non-metabolic functions in physiology and disease remain largely unknown. Recently, a novel lactate-derived post-translational modification (PTM), lysine lactylation (Kla) was discovered, which belongs to metabolite-derived PTMs similar to lysine acetylation (Kac) [6]. Biochemically, Kac introduces a small acetyl group on the ε amine group of the lysine residue, with a mass of 42.0106 Daltons (Da) [7]. Kla attaches a lactyl group to the ε amino group of a lysine residue, with a much larger mass of 72.021 Da [6]. Similar to Kac, Kla occurs in both histone and non-histone proteins, and faithfully orchestrates numerous biological processes, such as signal transduction, metabolism and inflammatory responses [6], [8]. In addition, dysregulation of lactylation contributes to tumorigenesis [9]. Kla represents a typical non-metabolic role of lactate and illuminates a new avenue to study the diverse physiological functions of lactate. Although the biological importance of protein lactylation has been rapidly recognized in recent years, its underlying mechanisms are still largely unclear.
Identification of Kla targets and their precise sites is fundamental for understanding the molecular mechanisms and regulatory roles of Kla. Conventionally, Kla was initially detected by mass spectrometry-based approaches, with a mass shift of 72 Da and further confirmed by chemical and biochemical methods, such as peptide synthesis and isotopic labeling [10]. In 2019, Zhang et al. first identified 26 and 16 histone Kla sites from human HeLa cells and mouse bone marrow-derived macrophages, respectively. Subsequent study by Gao identified 273 Kla sites in 166 proteins using LC-MS/MS [10]. In contrast to labor-intensive and time-consuming experiments, computational prediction of Kla sites from protein sequences is an alternative approach to efficiently prioritize highly potential candidates for further experimental consideration. Although lactylation is comparable with other major PTMs for which a variety of computational approaches were explored, the dedicated computational resource for Kla remains to be developed.
However, for computational prediction of Kla, three important challenges remain, including few-shot samples, extreme imbalance of benchmark dataset and overfitting in deep learning models. Previously, we had integrated seven types of sequence features and designed the hybrid-learning framework HybridSucc for lysine succinylation sites prediction with an accuracy of 17.8%-50.6% higher than the existing method [11]. Moreover, we developed hybrid learning-based graphic presentation system GPS-Palm to accurately predict S-palmitoylation sites and achieved a much higher accuracy of 31.3% than the second-best tool [12]. In addition, the hybrid-learning architecture performed outstanding in quite different types of datasets. We had designed HUST-19, an artificial intelligence diagnostic software to automatically achieve the diagnosis of CT imaging and clinical features, which had significantly improved the accuracy for predicting the prognosis of patients with COVID-19 [13]. Although hybrid-learning architectures have achieved fairly promising performance, especially on large datasets, it easily tends to overfit because of a huge number of model parameters but few samples of Kla sites. This phenomenon is common for many deep learning-based architectures in such a scenario. Notably, the few-shot learning, which was proposed to predict unseen classes with a few training examples, has attracted a lot of attention and was successfully implemented in biological context such as drug response in recent years [14]. In this regard, an interesting question has emerged: can we introduce the intriguing possibility of the few-shot learning-based multi-feature hybrid architecture for Kla sites prediction?
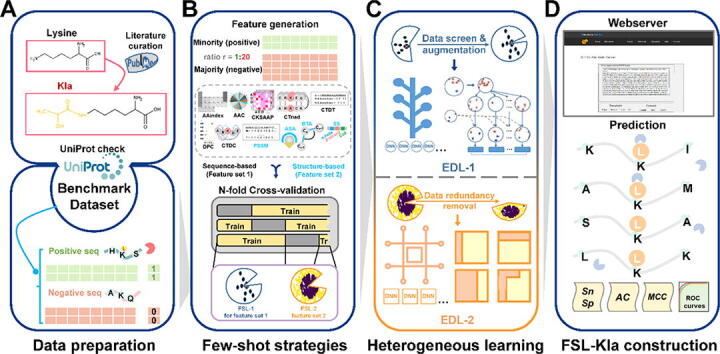
In this work, we manually collected 343 unique lactylation sites across 191 unique proteins from 3 species to construct the first Kla benchmark dataset (Fig. 1A, Table S1). We comprehensively analyzed and assessed 11 types of feature encoding schemes, consisting of 8 types of sequence-derived features including amino acid composition (AAC), amino acid index (AAindex), composition of k-spaced amino acid pairs (CKSAAP), composition/transition (CTDC, CTDT), conjoint triad (CTriad), di-peptide composition (DPC) as well as position specific scoring matrix (PSSM), and the three additional types of structural features including accessible solvent accessibility (ASA), backbone torsion angles (BTA) and secondary structure (SS) (Fig. 1B) [15], [16], [17], [18], [19], [20]. In this study, heterogeneous few-shot strategies were developed for balancing dataset and reducing overfitting (Fig. 1C). A multi-feature hybrid system was designed for integrating and combining up to 11 individual features synergistically. Taken together, we developed a novel tool named FSL-Kla for computational prediction of Kla sites (Fig. 1D). We hope that FSL-Kla might be a helpful tool to analyze Kla systematically and could inspire approaches for predicting many types of PTM sites. A webserver for FSL-Kla was implemented for free access at http://kla.zbiolab.cn/ to facilitate academic research.
Fig. 1.

The protocol of this study. A. From the latest literature, we obtained and collected 343 unique Kla sites as the dataset. All the entries must have a PMID and specify a clear position for Kla. Then we corrected the above dataset by UniProt and formed the benchmark dataset. All the entries are labeled and the non-Kla sites in the same protein or peptide are regarded as negative samples. B. The feature encoding schemes for both feature set 1 and set 2 with their corresponding imbalance strategy. The generated feature encodings including AAC, CTriad, AAindex, DPC, CTDT, CTDC, CKSAPP are grouped in feature set 1 while other four encodings ASA, BTA, PSSM and SS are grouped in feature set 2. Stratified cross-validations with few-shot strategies were conducted. FSL-1 was applied in feature set 1 while only FSL-2 was applied in feature set 2 because of some principles of few-shot strategies. C. The diagram of EDL-1 for feature set 1 and EDL-2 for feature set 2. The upper cell shows the strategic combination for both major and minor class and then a vote determines the combinatory results. The lower cell shows the process of ensemble. Samples that are wrongly classified will attain higher weight values in the next iteration. The final box is an optimized box based on many base classifiers. An adaptive decision boundary was also shown in the final box. D. The construction of FSL-Kla webserver and some evaluation metrics of Kla sites prediction.
2. Methods
2.1. Data collection
In FSL-Kla, all references were obtained from PubMed and were searched according to the following keywords: “lactylation” or “post-translational modifications and lactate” or “PTMs and lactate”. Since 2013, the researches on lactylation have been accelerated. We searched experimentally identified Kla sites by carefully checking abstracts or full texts of the scientific literature published. Currently, our benchmark dataset covers 343 unique lactylation sites across 191 unique proteins from 3 species including Homo sapiens, Mus musculus and Botryotinia fuckeliana (Table S1). Here, we defined a Kla site peptide KSP(m, n) as a lysine residue flanked by m residues upstream and n residues downstream. As previously described [17], we adopted KSP(10, 10) for model training and parameter optimization in a rapid manner. For KSPs located at N- or C-terminals, we added one or multiple special characters “*” to complement the full KSP(10, 10) entries. To obtain the benchmark data set for the initial model training, KSP(10, 10) peptides around known Kla sites were regarded as positive data, whereas KSP(10, 10) items derived from the remaining non-lactylated lysine residues in the same proteins were taken as negative data. The redundancy at the peptide level was cleared for positive and negative data, respectively, and only one KSP(10, 10) was reserved if multiple identical peptides were detected.
2.2. Feature encoding schemes
Sequence representation has been widely adopted in various computational methods for proteins. It has been proved that comprehensive and effective feature encodings are of great importance in producing a high-performance predictor [15]. A structural or physiochemical property could be extracted from protein or peptide sequences by a descriptor [12], [21], [22]. To make robust and accurate prediction, we adopted two sets of features including 7 amino acid composition-based features (feature set 1) as well as position specific scoring matrix (PSSM) profile [16] and 3 structure-based features (feature set 2) [23]. In the past study, these two groups of features were regarded as independent but highly complementary features. Advances of multiple feature encodings are obvious and facilitate the in silico PTM site prediction to a large extent [12], [17]. Reproduceable and stable extraction is important for feature encodings [12]. Automatic feature encoding is considered in this work. Hereby, we used iFeature, a state-of-the-art toolkit for protein and peptide sequence encoding [15] to generate features for feature set 1 (Fig. 2A). As for the feature set 2, we generated a sequence-based feature, including PSSM by PSI-BLAST [18] as well as three structure-based features such as ASA, BTA and SS by SPIDER2 [19] (Fig. 2A). Finally, a peptide-to-vector transformation (PVT) approach was adopted to generate the input matrix of the two sets of features (Fig. 2B).
Fig. 2.

Architecture and implemental steps of the ensemble method with imbalance strategy. A. Data collection and feature encoding schemes for both amino acid composition-based features as well as PSSM profile and structure-based features. B. Generating encodings for labeled proteins and peptides: A procedure for peptides to vectors transformation (PVT). C. Implementation of FSL-1 with EDL-1 for feature set 1 while FSL-2 with EDL-2 for feature set 2. Probability calibration performs an important role in correcting prediction after primary ensemble. Base learners’ outputs after probability calibration are stacked as information for the input of mFHS. The 4, 6, 8 and 10-fold stratified cross-validations were executed to evaluate the performance. Conducting few-shot strategies in training data and keeping testing data untouched provides a reasonable evaluation approach from the data aspect. ROC curves with some evaluation metrics including Sn, Sp, Ac, PPV, NPV and MCC are utilized.
Feature set 1: Amino acid composition-based features
2.2.1. AAC
The feature AAC reflects the amino acid frequencies of the sequence fragments surrounding the PTM sites [20]. As one of the basic methods to analyze the sequence, there are also some places to be modified. Because a number of Kla sites are located at the N- or C-terminus of proteins, special characters such as “*” should be added to complement the full peptides [24]. In addition, there might be some atypical amino acids in protein or peptide sequences. Thus, the additional pseudo amino acid “*” was also considered to encode such unusual residues. In this work, we calculated the frequencies of 21 types of amino acids in alphabetic order (A, C, D, …, Y, *) from Kla peptides, and each peptide i was encoded into a 21-dimensional vector as follows:
2.2.2. AAindex
AAindex is a database of amino acid physiochemical properties containing up to 566 amino acid indices [25], [26], [27]. This database is widely used to generate amino acid index-based physicochemical properties for a protein or peptide. In this study, each position of residue in Kla peptide (with a fixed length of 21 residues) was substituted by corresponding indices according to adopted physiochemical properties. To consider all the amino acid indices comprehensively, we concatenated all the numerically applicable indices together for next feature processing. Then, the dimension of generated feature vector of AAindex is 21 × 531 = 11,151.
2.2.3. CKSAAP
CKSAAP is short for Composition of k-spaced Amino Acid Pairs. As the supplement of AAC, this feature encoding contains the frequency of amino acids pairs with k spaces separation [28]. CKSAAP provides local context information of Kla sites at the scale of given distance k which is quite flexible. For instance, if the value of k is 0, there are 20 × 20 = 400 possible residue pairs (from AA, AC, AD, … to YY). Then again, we must consider rare amino acids or “place filling” virtual amino acid “*” [24], so the ultimate dimension of CKSAAP feature vector of Kla peptides is 21 × 21 = 441. We could also count the values of arbitrary k-space amino acid pairs and define the feature vector as follows:
The values of numerators denote how many times a corresponding residue pair appears in a protein or peptide for the given space k. And the values of denominators are the total number of k-space residue pairs in the protein or peptide. It was noteworthy that the values of Ntotal are not the same. For instance, the values of Ntotal are l-1, l-2, l-3, l-4 for a protein of sequence length l for k = 0, 1, 2, 3 respectively.
2.2.4. CTDC
CTDC is one sub conception of the method of composition, transition, and distribution (CTD). Three descriptors are composition (C) descriptor, transition (T) descriptor, and distribution (D) descriptor, which help to define the status and its change of different amino acid groups [29]. CTD is a well-known and classic sequence feature generation method proposed by Dubchak et al. [30].
As the first part of CTD method, CTDC was used to generate 39 features from each protein or peptide referring to the ratio of the number of single amino acid with specific properties [31], [32]. The formulation is expressed as follows:
where N(r) is the number of amino acids of type r in the encoded sequence and N is the length of the sequence. Hydrophobicity is the studied property r and amino acids are categorized into three different groups including polar, neutral or hydrophobic regarding.
2.2.5. CTDT
CTDT is the second part of the CTD method. This part describes the transition from one subgroup to another subgroup in the studied property and summaries the percentage frequency of transition [30]. Still taking hydrophobicity as an example, transitions between the hydrophobic group and the neutral group and those between the hydrophobic group and the polar group are counted, organized and formulated as below:
where N(r,s) and N(s,r) are the respective dipeptides numbers encoded as “rs” and “sr” in the sequence, while N is the full length of the sequence.
2.2.6. CTriad
The Conjoint Triad descriptor (CTriad) is the property of one residue and its vicinal amino acids by regarding three adjacent amino acids as a single unit [33]. Compared with AAC and CKSAAP, CTraid provides additional information of amino acid composition not by offering Tripeptide composition (TPC) but follows the below manners. First, the protein sequence is represented in a binary space (V, F), where V denotes the sequence features’ vector space, and each feature (Vi) represents a kind of triad type; F is the corresponding number vector for V, where fi, the value of the i-th dimension of F, is the number of types Vi appearing in the protein sequence. Here, all amino acids have been catalogued into eight classes, the size of V should be equal to 8 × 8 × 8 = 512. Accordingly, i = 1, 2, 3, …, 512. It is worth noting that protein sequences with longer full length tend to have larger f values which confounds the comparison of proteins with different lengths. So, we need to normalize fi by a following standardization.
The di, a newly standardized parameter, is calculated by the equation above.
2.2.7. DPC
DPC is short for the dipeptide composition [34], which gives a 20 × 20 = 400 dimension vector. Although DPC is a special case of CKSAAP where the value of k is 0, it is still an indispensable sequence statistic counting the dipeptide composition of a protein or peptide. We use the DPC as an independent feature which’s formula is defined as follows:
where Nrs is the number of dipeptides represented by two amino acids r and s.
Feature set 2: PSSM profile and structure-based features
2.2.8. PSSM
PSSM (position-specific scoring matrix) measures the evolutionary information of Kla sites, by calculating the probability that an amino acid will appear at a specific position. As previously described, the position-specific iterative BLAST (PSI-BLAST) [18] program in the BLAST package, was adopted to align all Kla peptides of each data set to Swiss-Prot protein sequences downloaded from the UniProt database [35]. Evolutionary information for each amino acid was encapsulated in a row vector of 20 dimensions and the size of PSSM for a peptide with n residues is 20 × n. A unique PSSM was returned for each Kla peptide, and the probability values of the 20 types of typical amino acids at 21 positions (Pi, i = 1, 2, 3, …, 21) were obtained. Then we encoded each Kla peptide i into a 420-dimensional number vector as:
For instance, was the PSSM value for amino acid A at position 1 for the given peptide.
2.2.9. ASA
ASA (accessible surface area) indicates the approximately exposed area of an amino acid residue to solvent [36], [37]. ASA also takes the residue’s position in the 3D configuration of a protein into account [38]. By representing different surface areas among 21 types of amino acids, ASA was also encapsulated in the form of vector. The SPIDER2 tool [19] was adopted to compute potential ASA values Ai (i = 1, 2, 3, …, 21) for each amino acid in the peptides as we mentioned previously. Then, Kla peptides were characterized by 21-dimensional digital vectors regrading feature ASA as:
2.2.10. SS
SS is short for Secondary Structure. If the secondary structural information of each amino acid can be provided in an explicit and numerical form, we could make the prediction more accurate by introducing the feature SS. As previously described [36], [37], [39], the probability score Si (i = 1, 2, 3, …, 21) of α-helix, β-strand or coil was computed by SPIDER2 [19] for each amino acid in the sequences because the protein structure may also play an indispensable role in the prediction of Kla sites as we hypothesized in most PTMs [12], [39]. Then each Kla peptide was transformed into a 3 × 21 = 63-dimension vector as:
where the probability score Si (i = 1, 2, 3, …, 21) of α-helix, β-strand or irregular coiled coil was calculated by SPIDER2.
2.2.11. BTA
BTA is the abbreviation of the backbone torsion angles. Including the backbone torsion angles φ and Ψ, the angle between Cαi-1-Cαi-Cαi+1 (θ) as well as the dihedral angle rotated around the Cαi-Cαi+1 bond (τ), four parameters were computed by SPIDER2 [19]. The BTA reflected more detailed geometric information at specific positions. Four classes of angles provided exact space extension of peptides to facilitate the Kla site prediction. Then, each Kla peptide was transformed into a 4 × 21 = 84-dimension vector as:
where Li is the BTA angle value for the i-th residue (i = 1, 2, …, 21). The 84-dimensional vector was constructed by these values.
2.3. Heterogeneous few-shot strategies in FSL-Kla
Many types of PTMs have rich datasets which lead to successful prediction with an ensemble learning approach [12], [22], [40], [17], [17]. However, as a sort of PTMs without abundant benchmark datasets (because of the limited number of identified Kla sites/sequences), a natural approach is to use few-shot learning along with ensemble learning. Meanwhile, a reasonable and readily applicable way is to conduct data augmentation. However, for biomedical data, pipelines should be carefully designed since some augmentation methods might lead to ridiculous samples [41]. Accordingly, in FSL-Kla, we adopted a set of coherent few-shot learning approaches to enrich our training resources as well as reduce the dataset imbalance. We manually designed a two-way data screen and augmentation method based on the heterogenous feature encoding schemes’ intrinsic properties and then corresponding algorithms were designed as well. A brief work flow of FSL-Kla was as mentioned below. For feature encodings in feature set 1, we designed FSL-1 for data augmentation and screening, a cascade ensemble deep learning architecture EDL-1 was then incorporated for processing FSL-1’s outcomes (Fig. 2). Similarly, a parallel ensemble deep learning architecture EDL-2 for feature set 2 was also adopted along with the few-shot strategy FSL-2 for feature set 2 (Fig. 2C). Since upstream EDL-1 and EDL-2 were efficient and complementary to each other, we further speculated that the ensemble of EDL-1 and EDL-2 could improve the accuracy. We used PLR to play the role of stacking [42] in the hybrid model mFHS of EDL-1 and EDL-2. The detailed pipeline for FSL-1, FSL-2, EDL-1, EDL-2 and mFHS was shown in Fig. 3.
Fig. 3.

The detailed pipline of FSL-1, FSL-2, EDL-1, EDL-2 and mFHS. The pipeline treats feature encodings from feature set 1 and feature set 2 differently. FSL-Kla introduces new samples and screening samples out for feature set 1. For feature set 2, the pipeline reduces negative samples’ redundancy to make the data set more balance. The mFHS incorporates intermediate results from EDL-1 and EDL-2 which were calibrated by the mentioned method for probability calibration. The binary cross-entropy loss was measured. Back propagation of loss was utilized to train base learners following an end-to-end way.
Because of the deficiency and imbalance of benchmark dataset, we carried out two-way FSL methods for both feature set 1 and feature set 2. For feature set 1, we refined FSL-1 for the augmentation of original data to enrich data and reduce imbalance. Synthetic minority over-sampling technique (SMOTE) [43] is a data augmentation approach in which the minority class is over-sampled by creating “synthetic” examples instead of over-sampling with replacement. Briefly, the idea is interpolation, which generates additional samples in the minority class. Specifically, for a minority class p sample xi, SMOTE uses its k neighbors (specifying the value if k in advance) and calculates the k minority class samples with the nearest distance to xi (the distance is usually defined as the n-dimensional feature space of the Euclidean distance between samples). Then we randomly selected one sample from k neighbors and then generated a new sample by the following formula:
where xi is the neighbor selected, and δ is a random value between 0 and 1.
Although this augmentation step in FSL-1 will randomly select a minority of samples to synthesize the new samples, it is likely to generate samples which provide redundant information or less information. Another trick here is that a minority of samples may be noisy if the selected minority samples are surrounded by majority samples. The newly synthesized samples may overlap the surrounding majority samples to a large extent, which will make the classification difficult [44]. The first step in FSL-1 was to make the extremely imbalanced feature encodings in feature set 1 more balanced as it was shown in Fig. 2C. In order to help distinguish the Kla sites and non-Kla sites in the feature space, after the first step, we introduced a heuristic step in FSL-1 to remove Tomek links [45], [46]. A Tomek link is defined as such a sample pair that is from two different categories but sharing the nearest distance in feature space. For instance, sample p's (from category P) nearest neighbor is q (from category Q), and q's nearest neighbor is p as well. Then p, q is defined as a Tomek link. The key idea for data screening and cleaning was to remove redundant or overlapped data by specific rules, which also achieved the goal of down sampling (Fig. 2C). In this step, FSL-1 distinguished all Tomek links and deleted them by removing the majority samples in a Tomek link so that the Tomek links didn’t exist after deletion. As we mentioned above, it was hard to discriminate against newly generated minority samples with their majority neighbors while a data cleansing method can handle it by removing most of the redundant samples. The combination of the two aforementioned steps in FSL-1 achieved the goal of data enrichment and reduction of imbalance.
As for feature encodings in feature set 2, synthesis methods were not applicable, which means the data augmentation was hard to carry out. Thus, different from feature set 1, we didn’t perform FSL-1 in feature set 2 because these four features are not continuous. In other words, for example, if we found a high-level accessible surface area amino acid residue and another rather lower accessible surface area residue, we could not generate a “novel” peptide based on the two samples although one of these ideas of FSL-1 was synthesis. Besides, feature encodings in features set 1 were comprehensive statistics features with aligned feature vector length naturally. In addition, because the design space of protein sequence was also exponential growth based on the sequence length, it was reasonable to generate “novel” peptides in FSL-1. Almost continuous changes in sequence made the interpolation possible [47]. On the contrary, feature set 2’s encodings were tightly related to proteins or peptides’ full length. Here, we tailored another few-shot strategy FSL-2 to achieve the same two major goals mentioned in FSL-1 as well, data augmentation and imbalance reduction. Since it was not proper to generate “novel” peptides as we did in FSL-1, we introduced basic random under sampling (RUS) to reduce imbalance while maintaining the key structural components or contexts in proteins or peptides with Kla sites, which reduced the number of examples in the majority class (non-Kla proteins or peptides) in the transformed data [48]. FSL-2 was effective in situations where the minority class had a limited but relatively sufficient number of samples despite the severe imbalance. FSL-2 mainly performed a role in reducing the imbalance of feature encodings in feature set 2 (Fig. 2).
2.4. Ensemble deep learning models as components of hybrid system
As a cutting-edge branch in machine learning, ensemble learning has been utilized the bioinformatics prediction of PTMs successfully [22]. Based on the previous attempts [11], [12], we combined multiple deep learning models by ensemble method. The screened data with less imbalance after FSL-1 and FSL-2 made it possible to avoid overfitting in deep neural network (DNN)-based base learners. Then, the ensemble methods would achieve synergistic performance among multiple base models, which facilitated the prediction power of FSL-Kla via EDL-1 and EDL-2. Based on the distribution, pattern and size of refined data from FSL-1 and FSL-2, we manually tested and adopted heterogeneous approaches in EDL-1 and EDL-2 respectively. Both the absolute number and features dimensions were higher after FSL-1 compared to dataflow after FSL-2. We designed EDL-2 as the downstream ensemble deep learning method with serial ensemble ideas regarding the data pattern and complexity after FSL-2, while a more adaptive model EasyEnsemble with more sophisticated ensemble idea (both serial and parallel) was suitable for FSL-1's downstream method, EDL-1. We incorporated both AdaBoost for EDL-2 and its unbalanced application variant, EasyEnsemble for EDL-1 to leverage the whole samples after FSL-1 and FSL-2 by reusing them circularly. We also organized all intermediate results by specific ensemble rules to improve the performance as Dvornik et al. did [49].
The base learners in this study for EDL-1 and EDL-2 were DNNs. The architecture of the designed DNN included the input layer with equal nodes to feature dimension, three hidden layers with proper layer width (128, 64, 48) and one output layer. One of the key components of the few-shot learning was avoiding overfitting, so for feature encodings with dimension larger than 1000, we utilized the principal component analysis (PCA) to reduce the complexity and examine the first 200 principal components (PCs). Hence, the transformed input fitted the proper layer sizes, which didn’t have extremely large parameters that might lead to overfit. In addition, when a rather complex feedforward neural network was trained on small data sets, it was easy to cause overfitting. In order to prevent overfitting, the performance of the neural network could be improved by preventing the co-adaptation of the feature detectors [50]. Then dropout was proposed to avoid overfitting and reduce some computational load. In EDL-1 and EDL-2, dropout layers have dropout rate of 0.5 to avoid overfitting in base DNNs. As for the activation, we used the non-learner function rectified linear unit (ReLu) followed linear layers. The formula was defined as below:
where x was the weighted sum of a neuron. In the first layer, the input layer received data matrices after FSL-1 or FSL-2. The next three hidden layers played roles for feature extraction and representation, in which each neuron contained the unique feature patterns as feature reservoirs. The last layer performed the role of generating predictions. And the sigmoid activation function was adopted to predict Kla sites with probabilities [51].
A multi-feature hybrid system for synergistic prediction was designed for integrating and combining up to 11 individual features synergistically. This second ensemble of EDL-1 and EDL-2 was conducted to form the multi-feature hybrid architecture mFHS which was proved to enhance accuracy of Kla sites prediction by incorporating complementary information from EDL-1 and EDL-2 to facilitate synergistic prediction. Apart from the ensemble step previously, this was a second ensemble step as well as a multi-feature incorporation and combined following the stacking idea [12], [42]. A refined PLR method algorithm was adopted as a stacking method for hybrid multiple features. There were two steps, including random mutation and random zeroing [52]. The weight values of different intermediate outcomes (ensemble outcomes) of EDL-1 and EDL-2 after probability calibration were calculated by PLR with l2 penalty (Fig. 3).
2.5. Probability calibration
Due to the ensemble approaches for Kla sites prediction, there were some transformations of the original outputs of base DNNs in EDL-1 and EDL-2. However, we often wanted to predict not only the final label (Yes/No), but also the associated probability. This probability gave some kind of confidence in the prediction. Since in FSL-Kla, our base models were multiples neural networks but outer ensemble ideas were boosting, it was necessary to use probability calibration to obtain reasonable labels with quantitative consistent confidence [53]. Then, quantitative confidence could be used as the input for train ensemble models. In this study, Platt’s logistic model [54] was used to calibrate outcomes of EDL-1 and EDL-2.
Fitting a calibrator that mapped the output of the classifier was to a calibrated probability in [0, 1]. Denoting the output of the classifier for a given sample by fi, the calibrator tried to predict . Here we adopted the sigmoid regressor based on Platt’s logistic model [54].
where yi is the true label of sample i and fi is the output of the un-calibrated classifier for sample. A and B are real numbers to be determined when fitting the regressor via maximum likelihood. In general, the sigmoid method was effective when the un-calibrated model was under-confident and had similar calibration errors for both high and low outputs [55].
Brier score is a measure of probability calibration in simple terms. Brier score is a measure of the calibration of probability prediction, or the cost function. This set of probability must have the sum of probabilities to be 1 and have mutual exclusivity as well. The lower Brier score for a set of predicted values is, the better probability calibration will be. The definition for binary classification is originally formulated as follows:
where ft is the predicted probability, ot is the actual probability of event t (0 if it does not occur), and N is the number of predicted events.
We evaluated our probability calibration by recalculating the classification prediction probability from original classific function and then calculated the Brier score. We further judged whether to support or oppose the initial prediction result according to the Brier score. It was worth noting that we didn’t have to apply the probability calibration for the final ensemble model’s output since the PLR itself in mFHS had the ability to return well calibrated prediction as it directly optimized log-loss.
2.6. Performance evaluation
Here, including sensitivity (Sn), specificity (Sp), accuracy (Ac), positive predictive value (PPV), negative predictive value (NPV), and Mathew Correlation Coefficient (MCC), some widely used evaluation metrics were used for the prediction assessment. The definitions of these six metrices are as below.
where TP is the number of positive samples with a true predicted label. TN is the number of negative samples with correct classification. FP and FN indicate the numbers of positive and negative samples that are predicted incorrectly.
Evaluation of the prediction performance was conducted by 4, 6 8, 10-fold cross-validations [51], [56], [57], [58], [59]. The receiver operating characteristic curve (ROC) was obtained and plotted at different thresholds of sensitivities. It was worth noting that ROC was used to evaluate the performance at multiple sensitivities and could represent the performance for imbalanced dataset. In this study, aforementioned imbalance strategies were incorporated to reduce the imbalance with stratified cross-validations. So, we conducted resampling in training sets and keep testing sets untouched. In other words, taking 10-fold stratified cross-validation as an instance, we had resampled the 9-fold training dataset and made it less imbalanced. And then, we evaluated on the testing dataset that was still as imbalanced as the original benchmark dataset which was exactly what ROC and AUC work for. We finally concatenated every single 1-fold testing result and formulated the final ROCs. The Platt’s logistic model for probability calibration was adopted in such a manner that after training models with corresponding training folds in 4-, 6-, 8-, 10-fold stratified cross-validations and the Brier scores on the left testing fold were obtained. By this way, we maintained the power of imbalance algorithm but also evaluated performance without inducing more “new” data in the testing dataset. This inducing process would significantly improve performance but was not correct because the testing data was going to be more balanced artificially and changed the original distributions.
2.7. Implementation details
For model training, we used a lab computer with an Intel(R) Core™ i7-6700 K@ 4.00 GHz central processing unit (CPU), 32 GB of RAM and a NVIDIA GeForce GTX 1070 core. The Pytorch version 1.7.1 (http://pytorch.org), a highly useful deep learning API that was written with Python and developed for auto gradient computing and rapid parallel computing was adopted. The imbalanced-learn which offered a number of re-sampling techniques commonly used in datasets showing strong between-class imbalance was adopted for the application of imbalance learning. The Adam optimizer in Pytorch was adopted, by using parameters of 0.0005 for learning rate, 0.98 for the first exponential decay rate, 0.998 for the second exponential decay rate and 512 for minibatch size. Other hyperparameters such as number of base estimators and number of iterations for AdaBoost and EasyEnsemble and boosting algorithm were set as default values. The high, medium and low thresholds were adopted with false positive rate (FPR) of 5, 10 and 15%. We also implemented an “All” option to output all predictions. FSL-Kla was extensively tested on various web browsers including Internet Explorer, Mozilla Firefox, and Google Chrome to ensure its usability.
3. Results
3.1. Construction of the benchmark dataset and analysis of Kla context
Through the literature biocuration, we obtained 343 non-redundant Kla sites in 191 known substrates. The distribution of proteins with different Kla sites was summarized in Fig. 4A. In addition, we also described the proportion of sites in proteins with different numbers of Kla sites. We found that up to 107 proteins had 1 Kla site, whereas only 3 proteins had more than 9 Kla sites. The number of proteins with 2, 3, 4, 5, 6, 7 Kla sites were 44, 18, 10, 3, 2 and 1, respectively. It was worth noting that most proteins tended to have a few Kla sites. In addition, we found that about half (56.02%) of Kla sites existed in proteins with only one Kla site, suggesting that the more Kla sites are there in a single protein, the rarer these kinds of proteins are. Furthermore, we used pLogo (http://plogo.uconn.edu/) [60], a sequence logo generator to analyze amino acid preferences around the Kla sites and non-Kla sites (Fig. 4B). For the Kla sites, lysine residues were also enriched at positions −4 and +4, while for the non-Kla sites, lysine resides preferred to occur at position −1. Other amino acids residues such as A, G, R, Q, S, F tended to enrich within the position −4 and +4. On the contrary, amino acids P, L, T, I, D, E were more abundant in the same positions in the non-Kla sites’ ±4 flanks. All these enrichments didn’t lead to a defined motif or pattern but showed much difference context between Kla sites and non-Kla sites. Taken together, our results demonstrated the latest profile of currently collected Kla sites’ context and provided the support to perform pattern recognition by few-shot learning and ensemble learning.
Fig. 4.

Data pattern and distribution analysis. A. A pie plot combo of the number of Kla sites on logarithm scale and the proportion of sites. B. The pLogo graph indicates the potential pattern difference in positive and negative data. C. A bar chart of the predicted secondary structure probabilities in Kla lysine and Non-Kla lysine. The symbol “**” means statistically significant difference at level p-value = 0.05 and “***” means the same but at level of p-value = 0.01. D. A combo of violin and heatmap plot with position-specific BTA distribution and the aligned heat map for amino acid distribution in Kla sites’ flanks. A reference bar is located in the right region for quantitative measurement.
To clarify whether there is a structure preference for Kla sites, the SPIDER2 [19] was also implemented for structure analysis among the positive and negative datasets. The result showed that approximately 38.45% of Kla sites were found in α helices, 10.35% were located in β strands and the remaining 51.20% were seen in disordered coils (Fig. 4C), while the non-Kla sites were found more in β strands and α helices but less in disordered coils. As for the statistical comparison of secondary structure pattern, β strands and α helices showed a significant difference in Kla sites’ flanks and non-Kla sites’s flanks at test level p-value = 0.05. There was a significant difference in coils at test level p-value = 0.01. These mentioned results suggested that the structural information such as SS and BTA of the Kla proteins should not be treated as same as feature encodings in feature set 1. Thus, we developed FSL-2 and EDL-2 for further training and evaluation. We also presented our above analysis in a violin plot (Fig. 4D) for BTA distributions with the aligned heat map for amino acids distribution in Kla sites’ flanks. The most BTA for Kla sites and non-Kla sites ranged from 0° to 200°, while distribution of Kla sites and non-Kla sites themselves were contracted compared to flank sites. This might be caused by the space configuration of lysine residues in proteins. From the BTA distribution, we could also find the range of Kla sites’ flank and non-Kla sites’ flank is almost the same but some hidden patterns are concealed. The corresponding heat map showed the amino acid composition in Kla peptides (Fig. 4D). The heat map could be regarded as a graphic feature representation of AAC. And the pattern in heat map was also coincident with the pLogo pattern, which supported our analysis for heterogenous information density in feature set 1 and feature set 2.
3.2. Performance comparison shows the superiority of few-shot learning strategies
We first evaluated our heterogenous few-shot learning approaches FSL-1 and FSL-2. As shown in Fig. 5A and B, some representative conventional machine learning methods as well as deep learning methods without few-shot strategy were performed to compare with FSL-1 and FSL-2. In other words, we first controlled the existence of imbalance strategy (or None) and ensemble deep learning (or representative conventional machine learning methods such as PLR and RF). Single DNN without any strategy was also performed as a comparison. Considering single set of feature encodings, DNN, RF and PLR achieved a median AUC of 0.702, 0.685 and 0.667 in feature set 1 while 0.658, 0.662, 0.652 in feature set 2, respectively. EDL-1 and EDL-2 achieved significant higher performance compared to DNN, RF and PLR. Along with few-shot methods FSL-1 and FSL-2, we obtained EDL-1 and EDL-2 with upgrading values of AUC. We conducted a rank sum test and found statistical difference at level p-value = 0.05 for both FSL-1 + EDL-1 vs EDL-1 and FSL-2 + EDL-2 vs EDL-2. Then, median AUC values for FSL-1 + EDL-1, EDL-1, FSL-2 + EDL-2, EDL-2 are 0.745, 0.705, 0.696 and 0.674 in two feature sets respectively. With few-shot learning strategies, a maximum 11.7% (0.745 versus 0.667) increase of AUC value was achieved compared with conventional machine learning methods and a maximum 5.7% (0.745 versus 0.705) increase compared with the same EDL-1 algorithm but without few-shot approaches. It was also worth noting that ensemble approaches with few-shot strategy had shorter whiskers indicating few-short learning with ensemble ideas not only upgraded accuracy and robustness. The detailed values of performance were summarized in Table S2. We also merged feature encodings in both feature set 1 and feature set 2 together for profiling the general performance (Fig. 5C). These violins included all feature encodings with corresponding methods. Some double belt peaks in some violins indicated that the performance of feature set 1 and feature set 2 centered at different AUC values because of the different accuracies.
Fig. 5.

Evaluation of performance for heterogeneous few-shot learning approaches with conventional machine learning methods. A. The performance for multiple feature encodings of individual models in feature set 1 was summarized into a single box in the box plot to evaluate multiple methods’ performance. B. The same comparison in box diagram A was also conducted for feature set 2. C. To profile the performance of tailored algorithm pipelines for feature set 1 and feature set 2 with conventional machine learning method, a violin plot was drawn.
3.3. Eleven types of sequence and structural features are efficient and informative
We then evaluated the performance of 11 aforementioned feature encodings (including feature set 1 and feature set 2). For feature set 1, the 4-, 6-, 8-, 10-fold stratified cross validation tests (Fig. 6) were conducted by FSL-1 and EDL-1. As for the feature set 2, we still performed the 4-, 6-, 8-, 10-fold stratified cross validation tests but applied the FSL-2 and EDL-2. Final performance was obtained by mFHS, combining the information from EDL-1 and EDL-2.
Fig. 6.

Evaluation of the performance for multiple feature encodings. A. Multiple box plot for the ROC curves and corresponding mean AUC values with standard deviations for different feature encodings’ base models. Amino acid composition-based models are colored in light blue in curves. PSSM profile and structure-based models are colored in orange in curves. The final model’s curve is colored in pink. The lightly colored regions up and down solid lines are calculated regions with standard deviations for Sp and Sn from 4, 6, 8 and 10-fold stratified cross-validations at different thresholds. B. FSL-Kla integrated all these feature encodings and achieved superior and synergistic accuracy in Kla site prediction. C. A line chart of performance for FSL-Kla from 4-, 6-, 8- and 10-fold stratified cross-validations. The error bars for different folds were too minor to observe in plot. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The performance for seven feature encoding schemes in feature set 1 was shown in Fig. 6A with light-blue curve. The mean AUCs and their standard deviations (4-, 6-, 8-, 10-fold stratified cross validations) from the highest to the lowest for the seven features were 0.765 ± 0.013, 0.749 ± 0.014, 0.747 ± 0.007, 0.745 ± 0.025, 0.711 ± 0.008, 0.705 ± 0.019 and 0.692 ± 0.018 for AAC, CTriad, AAindex, DPC, CTDT, CTDC and CKSAPP, respectively (Table S2). The amino acid composition-based features AAC, DPC and CTriad along with physicochemical property-based features and AAindex contributed most and produced rather satisfactory performance. This result supported that amino acid composition was still one group of simple but irreplaceable features schemes [17], [21]. Two CTD-based features performed slightly poorer possibly because we haven’t found much informative global composition, transition and distribution pattern of Kla proteins or peptides. CKSAAP is an important feature scheme when considering the amino acid pairs with separated positions and has proved to be very useful in many PTMs prediction. However, in this study, CKSAAP didn’t contribute as much as it in other PTM prediction studies [23].
In feature set 2, we obtained the performance of AUC with the secondary structure (0.710) while the worst performance of AUC with PSSM (0.653) (Table S2). This indicated the necessity and importance of exploiting the structure-based features. Although PSSM was not a structural but sequence-based feature, we took PSSM into account in feature set 2 because PSSM shared some common peculiarity with SS, BTA and ASA, not like in Zhang et al.’s study [22]. It was also noteworthy that the cascade utilization of FSL-2 and EDL-2 maximized the prediction power of feature sets 2, although, in general, there was no single feature encoding to outperform others significantly. However, three structural features still contributed more comparing with PSSM with constant and lower standard deviation in intra feature set 2 or even inter feature sets (0.007 for SS, BTA and 0.006 for BTA vs. 0.021 for PSSM), which indicated that structure-based features were more robust (Fig. 6A).
3.4. Multi-feature hybrid-system achieved superior and synergistic accuracy
Above performance of 11 different features both in feature set 1 and feature set 2 implied the necessity to combine these effective features comprehensively from various aspects instead of using each of them individually. Thus, the application of ensemble methods broke the limitation of single prediction model (base model) and leveraged the hybrid predictive power by ensemble intelligence [61]. This was mainly reflected in two aspects as below. Firstly, the EDL-1 and EDL-2 leveraged the predictive power of single feature scheme in corresponding feature sets which achieved rather plausible performance by training the base DNN learners. Secondly, the stacking-based multi-feature hybrid idea in the final mFHS method [11] was adopted to upgrade the base learner performance to form a strong learner by training a downstream PLR afterwards as the final model. The experiment results in Fig. 5A, B showed the performance increase of the first aspect (EDL-1 with DNN as base learner vs. single DNN, EDL-2 with DNN as base learner vs. DNN) and the ROC curve with pink solid curve in Fig. 6A, the boxes in Fig. 6B and line chart in Fig. 6C showed the synergistic promotion of the second aspect. The final strong model FSL-Kla achieved a mean AUC of 0.889 with the corresponding standard deviation of 0.023 (Fig. 6B). Compared to the best individual model (AAC, 0.765) and the worst individual model (PSSM, 0.653), the final model achieved a 16.2% and a 36.1% increase respectively, indicating the hybrid algorithm mFHS successfully combined the supplementary support of single outputs from EDL-1 and EDL-2 and produced significant synergistic promotion of performance. Lastly, we carried out a profile for the performance of FSL-Kla at 4-, 6-, 8-, 10-fold stratified cross-validations (Fig. 6C). The mean values of AUC and standard deviations were 0.882 ± 0.005, 0.880 ± 0.006, 0.909 ± 0.003, 0.891 ± 0.002, respectively. Based on the above analysis, we believed that the FSL-Kla effectively implemented the mFHS to achieve the performance superiorly and synergistically.
3.5. Performance comparison after probability calibration
The results after probability calibration showed precise prediction with aligned confidence because it directly optimized the log-loss instead of returning biased probabilities which were widely observed in many common models. For instance, a well-calibrated (binary) probabilistic classifier should classify samples and give a sample with precise predicted probability value. A sample set with a confidence level close to 0.8, should indicate about 80% actually belong to the positive class. Brier scores before and after probability calibration in this study were obtained and shown in Table S3.
There was a significant decrease in the Brier score after probability calibration. For feature set 1, the average Brier score decreased from 0.2376 to 0.0496, while the mean Brier score dropped from 0.2459 to 0.0502 for feature set 2. Although the average Brier score showed that feature set 2 seemed to have more biased probabilities than feature set 1, this result was not supported by statistics since there was no significant difference based on unpaired t test. The sharp drop before and after probability calibration was possible related to the extreme imbalance of benchmark dataset. Probability calibration might correct biased probabilities in such circumstances to a large extent. As we mentioned in the method section, we didn’t have to conduct the probability calibration for the final hybrid model’s output, since the method (PLR) itself had the unbiased predicted probabilities. The final model’s Brier score for concatenated 10-fold cross-validation was 0.0181, which was much lower than any Brier score for the base learners’ results. In other words, the ultimate results could be regarded as a series of automatic well-calibrated probabilities.
3.6. Development of online service for predicting Kla sites
To facilitate the community-wide prediction of lactylation sites, we implemented an online web server. FSL-Kla is a user-friendly application available at and hosted by extensible Alicloud computing facility with 8-core processors, 32 GB memory and 2 TB disk. It is publicly accessible at http://kla.zbiolab.cn/. The user submission interface (Fig. 1D) allows users to directly input the query protein or peptide sequences or upload the data set by clicking the Browse button (both in the FASTA format). The high, medium and low thresholds were adopted with FPR of 5, 10 and 15%. We also implemented an “All” option to output all predictions (Table S4). After specifying the prediction cutoff value, users can click the Submit button to initiate processing of their tasks. Users can then check the processing status of the submitted jobs using a unique URL link. From the result web page, users can download the prediction result in multiple formats, allowing subsequent in-depth analysis on their local computers or incorporate this into their customized pipelines. Using FSL-Kla, we conducted a large-scale prediction to computationally annotate potential Kla sites in human proteomic data set, and observed that there were 13,988 potential Kla sites of 7361 proteins under the high threshold. Furthermore, we performed an enrichment analysis based on gene ontology (GO) annotations with the hypergeometric test [62] (Table S5, p-value <0.05). Top 20 mostly enriched GO terms were selected and revealed that lactylated proteins are mainly involved in microtubule-based movement (GO:0007018), RRNA processing (GO:0006364), cell division (GO:0051301) and nucleosome assembly (GO:0006334). In addition, a number of enriched GO molecular functions and cellular components, such as ATP binding (GO:0005524), RNA binding (GO:0003723), DNA binding (GO:0003677), nucleus (GO:0005634) and nucleoplasm (GO:0005654), supported an impact of Kla in metabolic functions.
4. Discussion
PTMs enrich the functional diversity of most eukaryote proteins and play essential roles in almost all biological processes. Dysregulation of protein PTMs is associated with numerous human diseases such as cancer of which Warburg effect is one of the hallmarks [63], [64]. Lactate is the major metabolite generated during the Warburg effect in cancer cells, which can not only serve as an energy source, but also exert a variety of surprising and important physiological functions, such as a signaling function [4], [5]. However, the physiological mechanisms by which lactate exerts its diverse effects remain to be explored. Recently, a novel lactate-derived PTM, lysine Kla was discovered, which is a new type of histone mark and couples metabolism to gene expression, representing a novel adapted mechanism for cellular signaling. Notably, emerging evidence suggested elevated lactylation level in tumor tissues can lead to poor prognosis of ocular melanoma [9]. In this regard, the identification of new lactylated substrates with exact sites is the foundation of understanding the molecular mechanisms and regulatory roles of lactylation.
In contrast with time-consuming experimental assays, computational prediction of lactylation sites in proteins can greatly narrow down potential candidates for further experimental consideration. Recently, there were some researchers trying to combine the advantages of both few-shot learning and deep learning in biological context such as drug response [13]. The few-shot deep learning models can not only bridge the application of deep neural networks from large samples to small samples, but also successfully avoid the overfitting of deep learning models in small samples.
In this work, we conducted a comprehensive survey of the performance by combining sequence-based features, physicochemical properties and structure-based features, in which deep neural network was adopted as component learners, FSL-1 and FSL-2 as the few-shot strategies, EDL-1 or EDL-2 as the ensemble method for based learners and mFHS as the multi-feature hybrid approach for synergistic prediction. Results showed that our newly designed predictor, FSL-Kla, achieved at least 16.2% improvement of the AUC value (0.889 versus 0.765) for the ensemble prediction of Kla sites.
Usually, the success of DNNs relies on a great number of samples, which is expensive to collect or the collection is still in process. To tackle this issue, much effort has been taken by training the sophisticated model. However, this approach might lead to overfit because of the extremely limited data but high dimensional parameter space. The ideal way required an adequate number of novel examples which can be sampled and evaluated based on real samples. Establishing a comprehensive and well-annotated Kla database required much continuous effort with high-throughput approaches and careful reviews. Currently, we believed that few-shot learning could best leverage the tiny but useful dataset of Kla sites although some issues remain to be addressed. For example, it was always important to consider the prospect of valuable information being deleted when we randomly removed them from our data set in FSL-2, but we had no way to detect or preserve information-rich examples in the non-Kla samples [43], [44], [46].
In the future, we will continuously maintain FSL-Kla by curating more experimentally identified Kla sites if new datasets are available. The computational models in FSL-Kla will be updated by integrating other sequence-based features. Besides DNN, other algorithms for base learners will be tested and integrated into FSL-Kla if the accuracy can be improved. We anticipate that FSL-Kla can be a useful tool for further exploration of Kla sites.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81872335), China Postdoctoral Science Foundation (2021M692936), National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (2018ZX09711002).
CRediT authorship contribution statement
Peiran Jiang: Conceptualization, Writing - original draft, Writing - review & editing, Formal analysis, Data curation, Investigation, Visualization, Methodology. Wanshan Ning: Software, Writing - original draft, Writing - review & editing, Conceptualization, Formal analysis, Data curation, Investigation, Visualization, Methodology. Yunshu Shi: Data curation, Investigation. Chuan Liu: Resources. Saijun Mo: Data curation. Haoran Zhou: Data curation. Kangdong Liu: Writing - original draft, Supervision, Data curation, Formal analysis, Resources, Funding acquisition. Yaping Guo: Writing - review & editing, Supervision, Data curation, Formal analysis, Conceptualization, Resources, Visualization, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.08.013.
Contributor Information
Kangdong Liu, Email: kdliu@zzu.edu.cn.
Yaping Guo, Email: guoyaping@zzu.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science:123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science:1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D., Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Brooks G.A. Lactate as a fulcrum of metabolism. Redox Biol. 2020;35:101454. doi: 10.1016/j.redox.2020.101454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palsson-McDermott E., Curtis A., Goel G., Lauterbach M.R., Sheedy F., Gleeson L. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65–80. doi: 10.1016/j.cmet.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575–580. doi: 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabari B.R., Zhang D.i., Allis C.D., Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 2017;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irizarry-Caro R.A., McDaniel M.M., Overcast G.R., Jain V.G., Troutman T.D., Pasare C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc Natl Acad Sci U S A. 2020;117:30628–30638. doi: 10.1073/pnas.2009778117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J., Chai P., Xie M., Ge S., Ruan J., Fan X. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22 doi: 10.1186/s13059-021-02308-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Systematic analysis of lysine lactylation in the plant fungal pathogen Botrytis cinerea - PubMed n.d. https://pubmed.ncbi.nlm.nih.gov/33193272/ (accessed April 7, 2021). [DOI] [PMC free article] [PubMed]
- 11.Ning W., Xu H., Jiang P., Cheng H., Deng W., Guo Y. HybridSucc: a hybrid-learning architecture for general and species-specific succinylation site prediction. Genomics Proteomics Bioinformatics. 2020;18:194–207. doi: 10.1016/j.gpb.2019.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ning W, Jiang P, Guo Y, Wang C, Tan X, Zhang W, et al. GPS-Palm: a deep learning-based graphic presentation system for the prediction of S-palmitoylation sites in proteins. Brief Bioinform 2021;22:1836–47. 10.1093/bib/bbaa038. [DOI] [PubMed]
- 13.Ning W., Lei S., Yang J., Cao Y., Jiang P., Yang Q. Open resource of clinical data from patients with pneumonia for the prediction of COVID-19 outcomes via deep learning. Nat Biomed Eng. 2020;4:1197–1207. doi: 10.1038/s41551-020-00633-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma J., Fong S.H., Luo Y., Bakkenist C.J., Shen J.P., Mourragui S. Few-shot learning creates predictive models of drug response that translate from high-throughput screens to individual patients. Nat Cancer. 2021;2:233–244. doi: 10.1038/s43018-020-00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z, Zhao P, Li F, Leier A, Marquez-Lago TT, Wang Y, et al. iFeature: a Python package and web server for features extraction and selection from protein and peptide sequences. Bioinforma Oxf Engl 2018;34:2499–502. 10.1093/bioinformatics/bty140. [DOI] [PMC free article] [PubMed]
- 16.Zou L., Nan C., Hu F. Accurate prediction of bacterial type IV secreted effectors using amino acid composition and PSSM profiles. Bioinformatics. 2013;29:3135–3142. doi: 10.1093/bioinformatics/btt554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Y., Ning W., Jiang P., Lin S., Wang C., Tan X. GPS-PBS: a deep learning framework to predict phosphorylation sites that specifically interact with phosphoprotein-binding domains. Cells. 2020;9:1266. doi: 10.3390/cells9051266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Heffernan R, Paliwal K, Lyons J, Dehzangi A, Sharma A, et al. SPIDER2: a package to predict secondary structure, accessible surface area, and main-chain torsional angles by deep neural networks. In: Zhou Y, Kloczkowski A, Faraggi E, Yang Y, editors. Predict. Protein Second. Struct., vol. 1484, New York, NY: Springer New York; 2017, p. 55–63. 10.1007/978-1-4939-6406-2_6. [DOI] [PubMed]
- 20.Chou K.-C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinforma Oxf Engl. 2005;21:10–19. doi: 10.1093/bioinformatics/bth466. [DOI] [PubMed] [Google Scholar]
- 21.Lv H, Dao F-Y, Guan Z-X, Yang H, Li Y-W, Lin H. Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method. Brief Bioinform 2020:bbaa255. 10.1093/bib/bbaa255. [DOI] [PubMed]
- 22.Zhang Y, Xie R, Wang J, Leier A, Marquez-Lago TT, Akutsu T, et al. Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework. Brief Bioinform 2019;20:2185–99. 10.1093/bib/bby079. [DOI] [PMC free article] [PubMed]
- 23.Wang J, Yang B, An Y, Marquez-Lago T, Leier A, Wilksch J, et al. Systematic analysis and prediction of type IV secreted effector proteins by machine learning approaches. Brief Bioinform 2019;20:931–51. 10.1093/bib/bbx164. [DOI] [PMC free article] [PubMed]
- 24.Xu Y., Ding Y.-X., Ding J., Lei Y.-H., Wu L.-Y., Deng N.-Y. iSuc-PseAAC: predicting lysine succinylation in proteins by incorporating peptide position-specific propensity. Sci Rep. 2015;5:10184. doi: 10.1038/srep10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong W., Zhou D., Ren Y., Wang Y., Zuo Z., Shen Y. PepCyber:P~PEP: a database of human protein protein interactions mediated by phosphoprotein-binding domains. Nucleic Acids Res. 2008;36:D679–D683. doi: 10.1093/nar/gkm854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinkel H., Chica C., Via A., Gould C.M., Jensen L.J., Gibson T.J. Phospho.ELM: a database of phosphorylation sites–update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obenauer J.C., Cantley L.C., Yaffe M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z., Chen Y.-Z., Wang X.-F., Wang C., Yan R.-X., Zhang Z. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLoS ONE. 2011;6:e22930. doi: 10.1371/journal.pone.0022930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomii K., Kanehisa M. Analysis of amino acid indices and mutation matrices for sequence comparison and structure prediction of proteins. Protein Eng. 1996;9:27–36. doi: 10.1093/protein/9.1.27. [DOI] [PubMed] [Google Scholar]
- 30.Dubchak I., Muchnik I., Holbrook S.R., Kim S.H. Prediction of protein folding class using global description of amino acid sequence. Proc Natl Acad Sci U S A. 1995;92:8700–8704. doi: 10.1073/pnas.92.19.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao Z., Li Y., Teng Z., Zhao Y., Ding H. A Method for identifying vesicle transport proteins based on LibSVM and MRMD. Comput Math Methods Med. 2020;2020:1–9. doi: 10.1155/2020/8926750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan J.-X., Li S.-H., Zhang Z.-M., Chen C.-X., Chen W., Tang H. Identification of hormone binding proteins based on machine learning methods. Math Biosci Eng. 2019;16:2466–2480. doi: 10.3934/mbe.2019123. [DOI] [PubMed] [Google Scholar]
- 33.Shen J., Zhang J., Luo X., Zhu W., Yu K., Chen K. Predicting protein–protein interactions based only on sequences information. Proc Natl Acad Sci U S A. 2007;104:4337–4341. doi: 10.1073/pnas.0607879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saravanan V., Gautham N. harnessing computational biology for exact linear B-cell epitope prediction: a novel amino acid composition-based feature descriptor. Omics J Integr Biol. 2015;19:648–658. doi: 10.1089/omi.2015.0095. [DOI] [PubMed] [Google Scholar]
- 35.UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.López Y., Dehzangi A., Lal S.P., Taherzadeh G., Michaelson J., Sattar A. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Anal Biochem. 2017;527:24–32. doi: 10.1016/j.ab.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 37.López Y., Sharma A., Dehzangi A., Lal S.P., Taherzadeh G., Sattar A. Success: evolutionary and structural properties of amino acids prove effective for succinylation site prediction. BMC Genomics. 2018;19 doi: 10.1186/s12864-017-4336-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lins L., Thomas A., Brasseur R. Analysis of accessible surface of residues in proteins. Protein Sci Publ Protein Soc. 2003;12:1406–1417. doi: 10.1110/ps.0304803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dehzangi A., López Y., Lal S.P., Taherzadeh G., Sattar A., Tsunoda T. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PLoS ONE. 2018;13:e0191900. doi: 10.1371/journal.pone.0191900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y.-C., Peterson S.E., Loring J.F. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24:143–160. doi: 10.1038/cr.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z., Pan Z., Ying Y.i., Xie Z., Adhikari S., Phillips J. Deep-learning augmented RNA-seq analysis of transcript splicing. Nat Methods. 2019;16:307–310. doi: 10.1038/s41592-019-0351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim C, You SC, Reps JM, Cheong JY, Park RW. Machine-learning model to predict the cause of death using a stacking ensemble method for observational data. J Am Med Inform Assoc JAMIA 2020. 10.1093/jamia/ocaa277. [DOI] [PMC free article] [PubMed]
- 43.Chawla N.V., Bowyer K.W., Hall L.O., Kegelmeyer W.P. SMOTE: synthetic minority over-sampling technique. J Artif Intell Res. 2002;16:321–357. doi: 10.1613/jair.953. [DOI] [Google Scholar]
- 44.Han H, Wang W-Y, Mao B-H. Borderline-SMOTE: a new over-sampling method in imbalanced data sets learning. In: Huang D-S, Zhang X-P, Huang G-B, editors. Adv. Intell. Comput., Berlin, Heidelberg: Springer; 2005, p. 878–87. 10.1007/11538059_91.
- 45.Elhassan A, Al-Mohanna. Classification of imbalance data using Tomek Link (T-Link) Combined with random under-sampling (RUS) as a data reduction method, 2017. 10.21767/2472-1956.100011.
- 46.Boardman J, Biron K. Mitigating the effects of class imbalance using smote and Tomek link undersampling in SAS, 2018.
- 47.Dhall A., Patiyal S., Sharma N., Usmani S.S., Raghava G.P.S. Computer-aided prediction and design of IL-6 inducing peptides: IL-6 plays a crucial role in COVID-19. Brief Bioinform. 2021;22:936–945. doi: 10.1093/bib/bbaa259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He H, Ma Y, editors. Imbalanced learning: foundations, algorithms, and applications. Hoboken, New Jersey: John Wiley & Sons, Inc; 2013.
- 49.Dvornik N., Mairal J., Schmid C. Diversity with cooperation: ensemble methods for few-shot classification. IEEE Comput Soc. 2019:3722–3730. doi: 10.1109/ICCV.2019.00382. [DOI] [Google Scholar]
- 50.Hinton GE. Improving neural networks by preventing co-adaptation of feature detectors. CoRR 2012;abs/1207.0580.
- 51.Manavalan B, Basith S, Shin TH, Wei L, Lee G. mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinforma Oxf Engl 2019;35:2757–65. 10.1093/bioinformatics/bty1047. [DOI] [PubMed]
- 52.Wang C., Xu H., Lin S., Deng W., Zhou J., Zhang Y. GPS 5.0: an update on the prediction of kinase-specific phosphorylation sites in proteins. Genomics Proteomics Bioinformatics. 2020;18:72–80. doi: 10.1016/j.gpb.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leathart T, Frank E, Holmes G, Pfahringer B. Probability Calibration Trees n.d.:16.
- 54.Platt J. Probabilistic outputs for support vector machines and comparisons to regularized likelihood methods. Adv Large Margin Classif. 2000;10 [Google Scholar]
- 55.Kull M., Silva Filho T., Flach P. Beyond sigmoids: How to obtain well-calibrated probabilities from binary classifiers with beta calibration. Electron J Stat. 2017;11:5052–5080. doi: 10.1214/17-EJS1338SI. [DOI] [Google Scholar]
- 56.Hasan Md., Ben Islam Md., Rahman J., Ahmad S. Citrullination site prediction by incorporating sequence coupled effects into PseAAC and resolving data imbalance issue. Curr Bioinforma. 2020;15:235–245. [Google Scholar]
- 57.Basith S., Manavalan B., Shin T.H., Lee G. SDM6A: a web-based integrative machine-learning framework for predicting 6mA sites in the rice genome. Mol Ther Nucleic Acids. 2019;18:131–141. doi: 10.1016/j.omtn.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. Meta-4mCpred: a sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Mol Ther Nucleic Acids. 2019;16:733–744. doi: 10.1016/j.omtn.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu K, Chen W. iMRM: a platform for simultaneously identifying multiple kinds of RNA modifications. Bioinforma Oxf Engl 2020;36:3336–42. 10.1093/bioinformatics/btaa155. [DOI] [PubMed]
- 60.O'Shea J.P., Chou M.F., Quader S.A., Ryan J.K., Church G.M., Schwartz D. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods. 2013;10:1211–1212. doi: 10.1038/nmeth.2646. [DOI] [PubMed] [Google Scholar]
- 61.Li F, Chen J, Ge Z, Wen Y, Yue Y, Hayashida M, et al. Computational prediction and interpretation of both general and specific types of promoters in Escherichia coli by exploiting a stacked ensemble-learning framework. Brief Bioinform 2021;22:2126–40. 10.1093/bib/bbaa049. [DOI] [PMC free article] [PubMed]
- 62.The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–D338. doi: 10.1093/nar/gkw1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ordway B., Swietach P., Gillies R.J., Damaghi M. Causes and consequences of variable tumor cell metabolism on heritable modifications and tumor evolution. Front Oncol. 2020;10:373. doi: 10.3389/fonc.2020.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hitosugi T, Chen J. Post-translational modifications and the Warburg effect. Oncogene. 2014;33:4279–4285. doi: 10.1038/onc.2013.406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.