

Summary
Angelman syndrome (AS) is a neurodevelopmental disorder caused by the loss of maternal UBE3A, a ubiquitin protein ligase E3A. Here, we study neurons derived from patients with AS and neurotypical individuals, and reciprocally modulate UBE3A using antisense oligonucleotides. Unbiased proteomics reveal proteins that are regulated by UBE3A in a disease-specific manner, including PEG10, a retrotransposon-derived GAG protein. PEG10 protein increase, but not RNA, is dependent on UBE3A and proteasome function. PEG10 binds to both RNA and ataxia-associated proteins (ATXN2 and ATXN10), localizes to stress granules, and is secreted in extracellular vesicles, modulating vesicle content. Rescue of AS patient-derived neurons by UBE3A reinstatement or PEG10 reduction reveals similarity in transcriptome changes. Overexpression of PEG10 during mouse brain development alters neuronal migration, suggesting that it can affect brain development. These findings imply that PEG10 is a secreted human UBE3A target involved in AS pathophysiology.
Keywords: UBE3A, Angelman syndrome, hiPSC neurons, PEG10, retroviral GAG, RNA-binding protein, stress granules, extracellular vesicles
Graphical abstract
Highlights
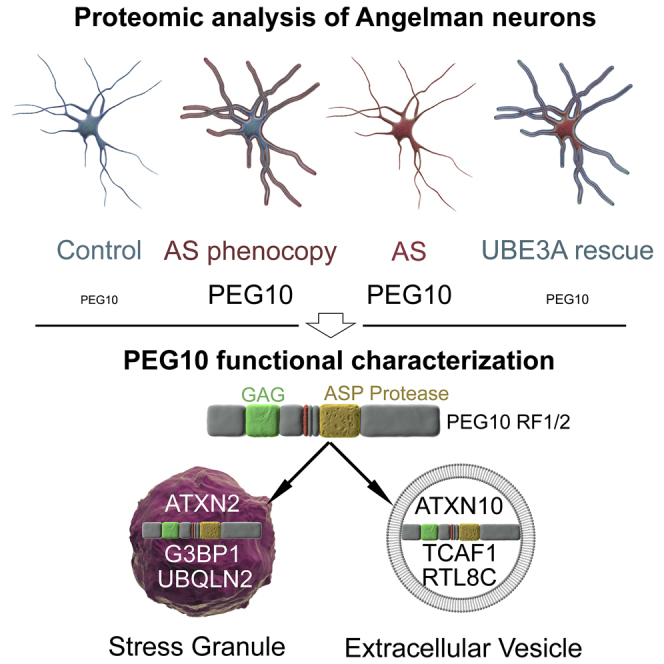
Proteomic analysis of Angelman hIPSC-derived neurons reveals UBE3A targets
PEG10 protein is reciprocally modulated by UBE3A using antisense oligonucleotides
PEG10 regulates extracellular vesicle proteome and neuronal transcriptome
Pandya et al. use proteomic analysis on antisense oligonucleotides-treated control and Angelman syndrome pluripotent stem-cell-derived neurons to reciprocally modulate UBE3A levels and identify PEG10. PEG10 is a GAG domain-containing extracellular vesicle protein involved in modulating EV proteome and neuronal transcriptome downstream of UBE3A.
Introduction
Dysfunction of the ubiquitin ligase gene UBE3A, which is paternally silenced in most neurons, is linked to two severe human neurodevelopmental disorders (NDDs): Angelman syndrome (AS), which are caused by the loss of neuronal UBE3A expression, and the 15q11.2-q13.3 duplication (Dup15q) syndrome, resulting from UBE3A overexpression.1, 2, 3 AS is characterized by severe developmental delay, lack of speech, ataxia, microcephaly, epilepsy, sleep disturbances, and a “happy” demeanor.4,5 It remains largely unclear how UBE3A contributes to the pathophysiology of these two different NDDs. There are no treatments for either AS or Dup15q syndrome.
UBE3A expression in neurons is uniquely regulated by the paternally cis-acting long non-coding antisense transcript (UBE3A-ATS), which represses the paternal copy of UBE3A.6 Recent work has demonstrated that blocking the expression of UBE3A-ATS, either with small-molecule topoisomerase inhibitors or with antisense oligonucleotides (ASOs), can reactivate the paternal UBE3A allele.7,8 These approaches have the potential to be disease-modifying therapies. However, there is still a paucity of knowledge of downstream molecular and cellular dysfunction caused by neuronal UBE3A loss. Notably, AS mouse models only recapitulate part of the clinical manifestations; for example, the cognitive deficits are not captured.9,10 These studies highlight the need to determine human-specific downstream targets of UBE3A.
As UBE3A is a HECT domain ubiquitin E3 ligase, which regulates the proteasomal degradation of its substrates, we aimed to define the UBE3A-driven alterations in the proteome of AS patient-derived neurons. Patient-specific human-induced pluripotent stem cells (hiPSC)-derived neurons provide a unique opportunity to study the UBE3A pathophysiology during the early stages of human neuronal development against the backdrop of the complexity of human genetics.11,12 For example, AS individuals with a maternal deletion of the 15q11-q13 locus lack multiple genes that can influence cellular phenotypes as well as disease trajectories.13 AS mice also exhibit different penetrance of phenotypes based on the genetic background.9,14 To circumvent this, we devised an approach to identify UBE3A-driven molecular alterations.
To identify neuronal proteins whose abundance depends on UBE3A expression, we generated ASOs that knocked down (KD) UBE3A levels in hiPSC-derived neurons from neurotypical individuals (control neurons) to phenocopy AS. Conversely, we restored UBE3A levels in hiPSC-derived AS neurons by knocking down the UBE3A-ATS transcript, thus reinstating UBE3A expression by activating the paternal allele. This potential disease-modifying therapy is under development for AS (ClinicalTrials.gov: NCT04259281). We hypothesized that neuronal protein substrates of UBE3A should be elevated both in AS neurons and, upon UBE3A KD, in control neurons, whereas they should be normalized upon UBE3A reinstatement in AS neurons. We used tandem mass tag (TMT)-MS3-based15 deep proteomic profiling to identify downstream UBE3A targets. Among the most significant and consistently regulated proteins were paternally expressed gene 10 (PEG10) and its reported binding partners TCAF1 and RTL8A, RTL8B, and RTL8C.16 The long isoform of PEG10 (reading frame 1/2 [RF1/2]) was specifically upregulated in AS neurons. Using a combination of imaging, immunoprecipitation (IP) for PEG10, liquid chromatography-mass spectrometry (LC-MS), and RNA sequencing (RNA-seq) approaches, we characterize PEG10 as a component of stress granules (SGs) and extracellular vesicles (EVs), identify its binding partners, and reveal a role as an endogenous retrovirus-like GAG domain-containing protein in modulating AS neuronal molecular pathophysiology. Finally, we demonstrate that the in vivo expression of PEG10 alters neuronal migration during mouse brain development.
Results
AS (del. and pt. mut.) patient-derived hiPSC neurons recapitulate UBE3A loss and have an altered molecular profile
AS patients with deletions in the 15q11 locus show more severe phenotypes compared with patients with UBE3A mutations, possibly due to hemizygosity in several GABA receptor subunits, which could contribute to AS pathophysiology.13 To capture this genetic heterogeneity, we derived hiPSC lines from two AS individuals harboring deletions of the 15q11-13 locus (AS del. 1, AS del. 2) and one from an individual harboring a point mutation in UBE3A (AS pt. mut.) (Table S1). Two lines were generated from neurotypical controls (control 1, 2), one of which (control 2) was the mother of AS del. 2. Control and AS hiPSC-derived neural progenitor cells (NPCs) were obtained using a slightly modified dual SMAD inhibition protocol as previously described.17,18 To model a physiologically relevant neuronal network with excitatory and inhibitory neuronal connectivity, we used a protocol that differentiates NPCs into a mixture of both glutamatergic and GABAergic neurons, which developed significant synaptic maturation as evidenced by synchronous network activity by day 42 of neuronal differentiation.17,18
Loss of UBE3A expression and expression of the UBE3A-ATS transcript in mature neurons by week 6 (day 42) in all AS lines was confirmed by qPCRs (Figures S1A and S1B) and at the protein level by immunoblotting analysis (Figure 1A). The selective reaction monitoring (SRM) assay for UBE3A protein over the course of neuronal maturation showed that AS NPCs exhibit significantly reduced levels of UBE3A protein (AS versus control, log2 fold change [FC] = −0.8735, adjusted p < 0.001) (Figure 1B), with mature neurons at day 42 exhibiting the largest differences (AS versus WT, log2 FC = −2.1702, adjusted p < 0.0001). This decrease is accompanied by the upregulation of the paternally expressed cis-acting long non-coding transcript (UBE3A-ATS) (Figures S1C and S1D), resulting in a near-complete loss of UBE3A expression in AS neurons by day 42 in culture. We concluded that our AS hiPSC-derived neuronal model recapitulates the loss of UBE3A during neuronal development. We also validated the hiPSC-derived neurons from AS del. 1 and del. 2 (AS neurons) by RNA-seq analysis and confirmed the loss of UBE3A mRNA and a robust downregulation of HERC2, GABA receptor subunits GABRB3, GABRG3, GABRA5, OCA2, and ATP10A transcripts in the 15q11-13 locus, compared with controls (Figure S1F).
Figure 1.

Angelman syndrome (del. and pt. mut.) patient-derived iPSC neurons recapitulate UBE3A loss and have an altered molecular profile
(A) Left: immunoblot analysis of UBE3A in hiPSC-derived neurons from controls and patients with AS (deletions: AS del. 1 and AS del. 2; point mutation: AS pt. mut.). Right: quantification of UBE3A protein expression using immunoblotting. Values represent UBE3A signal intensities normalized to total protein using stain-free gel (n = 4 independent neuronal differentiations, p values: Holm-Sidak’s multiple comparison test).
(B) Top: scheme of neuronal differentiation from hiPSC-derived NPCs to mature neurons. Bottom: SRM for UBE3A peptide peak areas in control and AS lines normalized to a peptide for β-actin in NPCs. Results for neurons at day 0, day 19, and mature neurons at day 42 (n = 3 independent differentiations with 2 control and 3 AS cell lines; p values: Holm-Sidak’s multiple comparison test).
(C) Volcano plot showing differential protein expression as adjusted p value versus log2-fold change (FC) of AS del. versus control lines (n = 4 independent differentiations). All proteins of the mitochondrial complex 1 are highlighted, with NDUFB7 and NDUFA9 marked with largest effect sizes by p value. Dotted lines indicate adjusted p value cutoff at 0.05.
(D) Left: heatmap of top 10 upregulated and downregulated proteins in AS del. versus control neurons. Right: SRM validation of subset of top 10 dysregulated proteins in AS del. and pt. mut. (n = 3 independent differentiations. Colors: row sum normalized intensities per protein).
We next set out to identify protein level dysregulation in AS. We performed protein expression profiling using TMT-MS315 (Figure S1E) on the two AS del. lines and the two control neuronal lines and quantified >7,000 proteins (Table S2). UBE3A protein was significantly downregulated in AS del. (Figure 1C, log2 FC = −1.7575, adjusted p = 5.50E−9) compared with controls. No significant alterations were observed in neuronal precursor markers, pan-neuronal, synaptic, and astrocytic markers. The protein expression levels of GABAergic neuronal markers GAD65, GAD67, and VGAT (GAD1, GAD2, and SLC32A1) were significantly reduced in AS del. compared with controls (Figures S1G and S1H), both at the protein and transcript levels. Analysis of the top 20 modulated proteins ranked by adjusted p value revealed that several subunits of the mitochondrial electron transport chain (NDUFB7, NDUFA10, NDUFA9) and the microtubule binding protein SOGA19 were consistently upregulated in both AS lines. Metabolism-related proteins (COMT, TXNRD2, and CRYZ) were consistently downregulated in AS del. lines. RNA-seq analysis showed that UBE3A, CRYZ, TXNRD2, and COMT transcripts were selectively downregulated in AS del. (Figures S1I and S1J). Pathway analysis (Tables S2 and S3) of the proteomic data revealed a significant upregulation of several mitochondrial complex 1 proteins, whose mRNA transcripts remained largely unaltered (Figures 1C, S1I, and S1K), a finding possibly in line with the mitochondrial association of UBE3A20 and the mitochondrial defects observed in AS mice.21, 22, 23 To investigate whether the observed changes could be specifically attributed to the loss of UBE3A, SRM was performed with UBE3A pt. mut. neurons, in addition to the AS del. lines, on a subset of altered proteins identified in the TMT-MS3 analysis. This independent experiment confirmed the altered expression of CRYZ, TCAF1, PEG10, and TXNRD2, while other mitochondrial protein changes exhibited line-to-line variability, indicating that genetic background could influence the dysregulation of mitochondrial complex 1 proteins (Figure 1D, right panel).
UBE3A modulation reveals potential human UBE3A targets in AS neurons
To further delineate UBE3A-specific effects in hiPSC-derived neurons during neuronal maturation, we used ASOs that target the UBE3A-ATS transcript (“UBE3A ATS KD ASO”) to reinstate the paternal allele of UBE3A and rescue its expression back to control levels (Figure S1E). RNA-seq analysis revealed that reads mapping to UBE3A exons were absent in AS patient neurons (Figure S2A), and that these reads were virtually indistinguishable between control and AS patient neurons treated with UBE3A ATS KD ASO (Figure S2A). This was accompanied by an absence of reads mapping to the cis-acting ATS transcript on the UBE3A gene body (Figure S2B). Importantly, we observed reads mapping to exons of all three human UBE3A isoforms, ruling out any isoform specificity in UBE3A reinstatement (Figure S2C).24,25 To induce the loss of UBE3A in control lines, we generated ASOs to knock down the UBE3A transcript (“UBE3A KD”), thereby mimicking UBE3A loss in AS. qPCR analysis revealed robust KD of UBE3A transcript in control neurons using a UBE3A-targeted ASO (UBE3A KD), and UBE3A reinstatement using UBE3A-ATS ASO coincided with a reduction in the UBE3A-ATS transcript levels (Figure S2D). Immunocytochemical and immunoblotting analyses demonstrated that the UBE3A KD and the UBE3A ATS KD ASO dramatically reduced or restored UBE3A protein levels, respectively (Figures 2A and 2B, quantified at right).
Figure 2.

UBE3A modulation reveals human UBE3A targets in AS neurons
(A) Left: representative immunostaining UBE3A (green) and MAP2 (magenta) in control and AS neurons, in control cells with UBE3A knockdown (control + UBE3A KD), and UBE3A reinstatement in AS cells at day 42 with 1 μM antisense oligonucleotide (ASO) treatment (AS + UBE3A ATS). Scale bar: 15 μm. Right: quantification of the UBE3A signal in neuronal cell bodies of control and AS neuronal lines (AS del. and AS pt. mut.) treated with either non-targeting (NT) ASO or UBE3A KD ASO (for controls) or UBE3A-ATS KD ASO (for AS) (pooled results for 2 independent differentiations; each data point is represented by 1 neuron; p values adjusted for multiple comparisons by Kruskal-Wallis test; error bars are means ± SEMs).
(B) Immunoblotting analysis of UBE3A expression in control and AS neurons at day 42 of differentiation upon chronic ASO treatment (42-day treatment, 1 and 5 μM ASO treatment).
(C) Experimental design for proteomics analysis using TMT-MS3 for the identification of UBE3A targets in control and AS neurons. Neuronal lines: 1 control, 2 AS (deletion: del. 2; point mutation: pt. mut.). Neural progenitor cells (NPCs) were differentiated in 2 independent biologic replicates and treated with either NT ASO or UBE3A KD ASO for controls and NT ASO or UBE3A-ATS KD ASO for AS lines. ASO treatment was either for 2 weeks starting day 28 of neuronal differentiation or for 42 days starting from day 0 of neuronal differentiation. ASO were used at the 1 or 5 μM, resulting in a total of 48 samples (n = 2 independent differentiations per condition). Samples were randomly assigned to 6× TMT10s, with each TMT10 plex experiment containing 2 pooled samples, which were used for normalization across the different runs.
(D) Heatmap of proteins significantly and inversely altered in a UBE3A and AS manner (adjusted p < 0.05 for control versus UBE3A KD and AS versus AS UBE3A-ATS KD; FC > 0). Data represent mean row sum normalized intensities per protein, n = 2). NT, NT ASO; KD, UBE3A KD ASO; ATS, UBE3A-ATS ASO.
(E) Validation of UBE3A targets identified in (D) with SRM in control and AS lines with chronic treatment (starting at day 0) (n = 3, 3 independent differentiations per line; data represent mean row sum normalized intensities per protein).
Next, we aimed to identify proteins whose abundance was affected by changing UBE3A expression levels. Using protein expression profiling by TMT-MS3 on one control and two AS lines (AS del. 2 and AS pt. mut.; Figure 2C), >7,000 proteins were quantified (Table S4). We focused on proteins that were upregulated in the AS neuronal and control line upon UBE3A KD (adjusted p < 0.05), and which were inversely modulated upon UBE3A reinstatement in AS neurons (Figure 2D). Among these proteins, we identified UCHL5, a deubiquitinase associated with the 19S proteasome regulatory subunit26 and the giant E3 ligases HERC2, which is part of the 15q11 locus, and HERC1, both of which are associated with intellectual disability.27, 28, 29 HERC2 has been reported previously to interact with and modulate UBE3A.30,31 Interestingly, we identified several UBE3A-regulated targets, including the cytoskeletal proteins Girdin (encoded by CCDC88A) and Dystonin, which are associated with dystonia and have reported roles in neuronal migration,32,33 peptidylprolyl isomerase D (PPID; cyclophilin 40), which has been reported to clear tau aggregates by interacting with polyproline residues on tau,34 and a less-studied guanine exchange factor, MCF2l, which is expressed selectively in brain tissue.35 Notably, PEG10 and its reported binding partners TCAF1 and retrotransposon-like 8A, 8B, and 8C (RTL8A, RTL8B, and RTL8C)16 were the most robust and significantly changed proteins upon modulating UBE3A levels (adjusted p < 0.01).
In an independent experiment involving three AS lines (AS del. 1, AS del. 2, AS pt. mut.) and two neurotypical lines, SRM confirmed that these targets are modulated by UBE3A levels (Figure 2E, Table S5). Immunoblotting analysis for UCHL5 in AS pt. mut. and control neurons treated with both sense oligonucleotides and ASOs further confirmed that its regulation was UBE3A dependent (Figure S3A, top). All of the additional proteins that were modulated in the UBE3A KD lines or only rescued upon UBE3A reinstatement in AS neurons are listed in Table S4. Overall, these data represent a comprehensive proteomic set for mining UBE3A-regulated proteins in human neuronal cultures.
To determine whether UBE3A reinstatement is also able to modulate PEG10 levels in mature neuronal networks, and to validate our findings using a different UBE3A ATS-targeting ASO, we treated AS neurons acutely with UBE3A ATS KD 2 (Figure S1E). Treatment for 1 week was sufficient to reinstate UBE3A to control levels, which again resulted in reduced PEG10 levels. PEG10 appeared to be upregulated at the post-translational level, as analysis of the RNA-seq data revealed that PEG10 mRNA was unaltered in AS and not affected by UBE3A reinstatement with ASOs (Figure S2E).
UBE3A regulates the expression of retrotransposon-derived GAG protein PEG10 via the proteasome
We next set out to characterize how UBE3A modulates PEG10 levels. PEG10 is an imprinted gene belonging to the Ty3-gypsy retrotransposon family; it retains the retroviral GAG and GAG-Pol-like protein architecture36,37 and harbors a CCHC-zinc finger RNA-binding motif (Figure 3A). Furthermore, PEG10 mRNA gives rise to two protein isoforms via a −1 ribosomal frameshifting mechanism leading to the generation of a longer PEG10-RF1/2 protein and a shorter PEG10-RF1 protein.38 While both isoforms harbor the GAG and zinc finger motif, only the longer RF1/2 isoform retains an aspartyl protease motif (Figure 3A). Immunoblotting for both PEG10 isoforms revealed a high-molecular-weight band (∼100 kDa) corresponding to PEG10-RF1/2 and a lower molecular weight band (∼50 kDa) corresponding to RF1. PEG10 targeting ASOs (PEG10 KD) reduced both isoforms (Figure S3A, bottom). Immunoblotting data revealed that PEG10-RF1/2 is primarily modulated in a UBE3A- and AS-specific manner, while the shorter isoform remains largely unaffected (Figure 3B). Immunocytochemical analysis using antibody specific for PEG10-RF1/2 (Figure S3B) showed that PEG10-RF1/2 localization was largely diffused in neuronal cytosol and proximal dendrites (Figure 3C, quantification on right). We confirmed the elevated expression of PEG10-RF1/2 in UBE3A KD and AS (HuCD+) neurons, which could be rescued by UBE3A reinstatement in AS neurons (Figure 3C, right; control versus sense: 614.2 ± 82.02 versus 2,461 ± 229.4, 75.05% increase, p < 0.0001; AS versus ATS: 4,464 ± 667.9 versus 2,021 ± 192.7, 54.72% decrease, p < 0.0001). To further confirm whether the UBE3A-dependent regulation of PEG10 occurs in neurons, we treated iCell-glutamatergic post-mitotically differentiating neurons with UBE3A sense ASO and observed a similar robust upregulation of PEG10-RF1/2 (Figure S3C).
Figure 3.

UBE3A regulates the expression of retrotransposon-derived GAG protein PEG10 via the proteasome
(A) PEG10 domain architecture: 2 isoforms, RF1/2 and RF1, are generated from the PEG10 transcript by a −1 ribosomal frameshift. Labels: GAG-like domain (blue), CCHC-zinc finger motif (red), and the aspartyl protease motif (ASP; yellow). Numbers indicate amino acid positions.
(B) Immunoblotting analysis of UBE3A and PEG10 expression in control and AS deletion neurons. Saline: PBS treatment; NT, non-targeting ASO treatment; UBE3A KD, UBE3A KD ASO treatment; UBE3A-ATS KD, UBE3A reinstatement.
(C) Left: representative immunostaining for PEG10-RF1/2 in control neurons without (+NT) and with UBE3A KD (+UBE3A KD) and in AS neurons without (+NT) and with UBE3A reinstatement (UBE3A ATS). Scale bar: 30 μm.
Right: quantification of PEG10 fluorescence intensity in control and AS HuC/D+ neurons (data points are individual neurons from 2 independent neuronal differentiations; p values are adjusted for multiple comparisons based on Dunn’s multiple comparison test).
(D) Left: immunoblotting analysis of UBE3A and PEG10 expression in control neurons at 0, 4, and 8 h after addition of proteasome inhibitor (MG132, 10 mM) with (UBE3A KD) and without (NT) UBE3A KD.
Right: quantification of UBE3A and PEG10-RF1/2 expression after proteasome inhibition (n = 3 independent experiments, p values: Dunn’s multiple comparison test).
(E) Immunoblotting analysis of PEG10 ubiquitination with PEG10 IP in control, AS, and AS+ATS treatment with proteasome inhibition (MG132, 10 mM, 6 h).
As RNA-seq analysis revealed that UBE3A levels do not affect PEG10 mRNA levels, we sought to examine other possible routes of PEG10 protein regulation by UBE3A. To this end, we assessed whether PEG10 was regulated by UBE3A via proteasome-dependent degradation. PEG10 protein levels were measured over a time course of proteasome inhibition (MG132) in control neurons and upon UBE3A KD (UBE3A KD; Figure 3D). Immunoblotting with anti-K48 UB revealed a robust increase in polyubiquitinated proteins in a time-dependent manner upon proteasome inhibition (MG132, 0–8 h; Figure 3D, left). UBE3A protein remained unaltered during the 8-h exposure to MG132 treatment, which is consistent with its long half-life.39 In contrast, a robust increase in both PEG10 isoforms was observed. No significant increase in PEG10-RF1/2 was detected when MG132 treatment was combined with UBE3A KD, suggesting that PEG10-RF1/2 proteasomal degradation in hiPSC neurons requires UBE3A (Figure 3D, right quantified). We next explored whether UBE3A and PEG10 can interact with each other and whether UBE3A can directly ubiquitinate PEG10. A yeast two-hybrid (Y2H) interaction analysis of UBE3A and PEG10-RF1/2 confirmed a weak interaction (Figure S3D). In addition, Y2H analysis revealed a strong PEG10 interaction with previously established interacting proteins RTL8C and TCAF1 (Figure S3E).40 UBE3A IP followed by immunoblotting (IP-WB) analysis under native conditions, confirmed the interaction between PEG10 and UBE3A, which could be stabilized by proteasomal inhibition (MG132) (Figure S3F, red stars).
We next tested whether UBE3A can directly ubiquitinate PEG10 in a bacterial ubiquitination assay system (Figure S3G).41 Neither PEG10-RF1 nor PEG10-RF1/2 were directly ubiquitinated by UBE3A, contrary to the validated positive control RING1B (Figure S3G).42 To investigate this, we tested whether PEG10 ubiquitination in human neurons requires UBE3A. IP-WB revealed an increase in the PEG10-RF1/2 polyubiquitination in control neurons upon MG132 treatment, which was dramatically diminished in AS neurons, despite high PEG10 levels, and could be rescued by UBE3A reinstatement with UBE3A-ATS-targeting ASOs (Figure 3E). Although we were unable to determine whether UBE3A directly ubiquitinates PEG10 in our bacterial ubiquitination assay, its ubiquitination and subsequent degradation in neurons appears to be UBE3A dependent.
To determine whether PEG10 levels are expressed in postmortem AS patient brains, we performed an automated immunohistochemical (IHC) analysis on one high-quality AS and control postmortem formalin-fixed paraffin-embedded (FFPE) cortical tissue (Figure S4A, STAR Methods: Biological samples). IHC confirmed the absence of UBE3A within neuronal nuclei in AS when compared with two controls (one age-matched [40 years] and one older [80 years]; Figure S4B). IHC of both PEG10 isoforms confirmed high specific expression of PEG10 in placental trophoblast cells (Figure S4C)36 and revealed no major differences between controls and AS, with PEG10+ cells found in all of the samples (Figure S4D). Moreover, fluorescence confocal microscopy showed cytosolic distribution of PEG10 in the neuronal cell bodies of both control and AS patient brains (Figure S4E). IHC analysis of PEG10-RF1/2 showed strong immunoreactivity in neurons from the AS patient (Figures S4G and S4H). Specifically, PEG10-RF1/2 was found to be enriched in neuronal cell bodies in layers II, III, and V/VI. We further obtained fresh-frozen tissue from the temporal cortex of two AS individuals and an age-matched control sample from the NIH NeuroBioBank (formerly NICHD BTB). Immunoblotting analysis confirmed the loss of UBE3A expression in AS tissue and expression of PEG10-RF1/2 (Figure S4F). Future work will be required to determine whether it is persistently upregulated in the AS brain.
In summary, these data indicate that UBE3A regulates PEG10-RF1/2 in a proteasome-dependent manner in AS hiPSC-derived neurons and suggests that PEG10-RF1/2 expression is also present in AS patient brains.
PEG10 binds to RNAs and RNA-binding proteins and localizes to SGs and EVs in AS
To better understand the consequences of increased PEG10 levels in AS neurons, we set out to identify its binding partners. We performed a PEG10 IP combined with LC-MS (IP-MS) in AS hiPSC-derived neurons, by using two different antibodies against PEG10 (Ab 1: recognizing RF1/2 and Ab 2: recognizing RF1/2 and RF1). AS neurons in which PEG10 was knocked down served as negative control. A total of 35 proteins were significantly enriched in PEG10 IPs (adjusted p < 0.001) with both Ab 1 and Ab 2, 13 of which were tightly co-regulated with PEG10 (Figure 4A; Table S6). We next tested whether we could identify the same binding partners in neurotypical control neurons. A subset of proteins, including TCAF1, RBM14, NONO, MAP6, ATXN2, and ATNX10, were enriched along with PEG10 upon UBE3A KD (Figure S5A). Reverse IP-WB for ATXN2 and ATNX10 confirmed the interaction of PEG10 with both proteins in AS neurons (Figure 4B). ATXN2 is a known component of SGs, while ATXN10 has been reported as a resident EV protein in several cell-type EV proteomic analyses.43,44 Therefore, we sought to determine PEG10 localization to SGs and EVs and to identify the role of PEG10 in each of these processes.
Figure 4.

PEG10 binds to RNAs and RNA-binding proteins, localizes to stress granules and extracellular vesicles (EVs) in AS
(A) IP-MS analysis of PEG10 binding partners in AS versus AS + PEG10 KD using 2 PEG10-specific antibodies (Ab 1, Ab 2). n = 4 independent IPs, values in heatmap represent average log10 label-free quantification (LFQ) intensities from MaxQuant; hits selected are enriched in PEG10 IPs with an adjusted p value cutoff of 0.001. NA, saline treatment; P10, PEG10 KD.
(B) Validation of ATXN2 and ATXN10 binding to PEG10 in AS by IP-immunoblotting.
(C) Representative immunostainings for PEG10-RF1-GFP and RF1/2-GFP expressed in H4 cells co-stained with G3BP1 after PBS treatment or treatment with 0.5 mM sodium arsenite (45 min; scale bar: 20 μm).
(D) Representative immunostainings for PEG10-RF1/2 in AS neurons along with ATXN2 and MAP2 upon PBS or 0.5 mM sodium arsenate treatment (45 min; scale bar: 20 μm).
(E) Left: volcano plot (adjusted p value versus log2 FC AS versus control) of PEG10 RNA immunoprecipitation sequencing (RIP-seq) in control and AS neurons representing the 1,000 most abundant transcripts in PEG10 IPs. Horizontal line represents adjusted p value cutoff of 0.05. Vertical line represents FC > 0. Right: the 20 most abundant transcripts in PEG10 IPs. Data are expressed as raw counts per million and are represented in descending order. Stars represent statistically significant enrichment in AS neurons versus control, adj. p < 0.05.
(F) Schematic of EV isolation from human iPS-derived neurons.
(G) Representative transmission electron microscope (TEM) images after uranyl acetate negative staining (magnification: 15,000×; scale bar: 200 nm).
(H) Hydrodynamic diameter (Dh) of vesicles isolated from control and AS neurons analyzed by dynamic light scattering. Results are shown as means ± SEMs, n = 3 independent replicates for each line.
(I) EV particle concentration (per mg EV protein) measurements from control and AS EVs analyzed by multi-angle dynamic light scattering (MALDS). Results are shown as means ± SEMs, n = 3 independent replicates from each line.
(J) Representative immuno-EM measurements for PEG10-RF1/2 and TSG101 in EVs from AS cells (magnification: 15,000×; insert 4× magnification; scale bar: 200 nm).
(K) Quantification of PEG10-RF1/2-positive EVs from control and AS cells (n = 3 independent EV preparations, p value: Mann-Whitney test).
(L) LC-MS heatmap for PEG10 and its binding proteins and selected EV markers in control and AS lysates (values are protein-level intensities obtained from Spectronaut and are averages of 3 independent lysate and EV preparations).
(M) Immunoblotting analysis for PEG10-RF1/2 and ATXN10 along with EV markers with equal total protein loaded for lysates and EVs (for quantification, see Figure S5D).
One of the neuronal interactors of PEG10 is ATXN2, which is a constituent of SGs and interacts with other RNA-binding proteins, such as A2BP1, TDP-43, and PABP1.45, 46, 47 PEG10 contains a GAG-like domain, which has been shown to subvert SG formation in retroviruses to enable the translation of viral mRNAs.48,49 We hypothesized that PEG10 could either be recruited to SGs or modulate their formation. To explore this, we expressed different PEG10 isoforms, PEG10-RF1-GFP or PEG10-RF1/2-GFP, initially in the glioblastoma H4 cells because of their large cytoplasm and their neuronal lineage, and co-stained with G3BP1, a bona fide marker for SGs, which were induced using sodium arsenite (Figure 4C).50 In H4 cells, G3BP1 showed diffused cytosolic staining. Sodium arsenite treatment robustly induced formation of G3BP1 foci (SGs) in the cytosol (Figure 4C). PEG10-RF1 showed diffuse cytosolic as well as nuclear distribution under non-stress conditions, which was not influenced by treatment with sodium arsenite. PEG10-RF1/2 showed exclusive cytosolic localization and was robustly recruited to G3BP1 foci upon treatment with sodium arsenite (Figure 4C). The presence of the green fluorescent protein (GFP) tag on PEG10-RF1/2 had no influence on SG localization (Figure S5B). Non-transfected cells also formed G3BP1 SGs foci to a similar degree, suggesting that PEG10 does not influence SG assembly. We tested whether PEG10-RF1/2 also localized to SGs in AS neurons. PEG10-RF1/2 and ATXN2 showed diffuse cytosolic staining in AS neurons under basal conditions. Upon SG induction with sodium arsenite, PEG10-RF1/2, ATXN2, and UBQLN2 (another canonical marker of SGs) localized in SG foci in AS neurons (Figures 4D and S5C).
As PEG10 harbors a CCHC-type zinc finger RNA-binding domain (Figure 3A) and localizes to SGs in AS neurons, we aimed to identify the transcripts bound to PEG10, using PEG10 IP followed by RNA-seq (IP-RNA-seq) on control and AS neurons. We identified 3,776 that were enriched in PEG10 IPs in AS neurons (Figure 4E; Table S7). The most abundant AS-specific transcripts included PEG10 mRNA (cutoff by log2 CPM (counts per million), top 1,000) (Figure 4E, right), confirming that PEG10 retains its ability to bind to its own mRNA.36 PEG10 complexes, as opposed to retroviral GAG proteins, seem to contain several thousand transcripts, suggesting that either PEG10 binding specificity has gained promiscuity in evolution after inserting into the host genome or its interactions with other RNA-binding proteins such as ATXN2 facilitate indirect binding to neuronal transcripts in AS neurons. Thus, PEG10 could play a broad role in binding many neuronal transcripts.
Given that PEG10 contains a GAG-like domain and PEG10 GAG is sufficient to make virus-like capsids and interacts with the known EV protein ATXN10 (Figure 4A),36 we sought to determine whether PEG10 is also secreted in EVs isolated from hiPSC-derived control and AS neurons. Using sequential ultracentrifugation (Figures 4F and 4G), we observed a significant enrichment of EV marker proteins such as TSG101 and PDCD6IP (Alix) in the purified EV fraction, but not of other proteins such as β-actin, compared with neuronal lysates (Figure 4M). EV hydrodynamic radius measurements confirmed the presence of 160-nm particles and showed no difference between control and AS (Figure 4H). EV particle concentration normalized to protein levels remained unaltered between control and AS (Figure 4I). Immunoelectron microscopy (immuno-EM) analysis of secreted vesicles confirmed the presence of the canonical EV marker TSG101 in EVs from AS neurons. Importantly, the same analysis revealed the presence of PEG10-RF1/2. Quantification of EVs from control and AS neurons using immuno-EM revealed that 20.73% (±1.27 SEM) of AS EVs were positive for PEG10, as opposed to 6.26% (±1.68 SEM) in controls (Figures 4J and 4K).
EV and neuron proteome were quantified using data-independent acquisition (DIA) MS on control and AS neuronal lysates and the corresponding EV fractions (Table S8). DIA analysis confirmed significant upregulation (adjusted p < 0.05) of core EV markers, including TSG101, PDCD6IP (Alix), CD81, and CD63 in the EV fraction compared with total protein lysates, further confirming the quality of the EV isolation (Figure 4L). Consistent with the immuno-EM, PEG10 was upregulated in AS cell lysates and EVs (log2 FC = 0.99 and 0.80; adjusted p = 0). In addition, the EV proteome revealed a significant elevation of PEG10 binding partners TCAF1 and ATXN10 in AS EVs. Immunoblotting analysis in an independent sample set confirmed the DIA results (Figure 4M, quantified in Figure S5D). PEG10-RF1/2 was also detected in AS EVs using immunoblotting, and fragmentation of PEG10 was observed in EVs, a property that is reminiscent of fragmented GAG domains in mature viral particles, driven by cleavage via the aspartyl protease motif (Figure 4M, PEG10 fragments at 80–90 kDa). Immunoblotting showed that ATXN10 was selectively increased in AS neuronal EVs (Figure 4M, quantified in Figure S5D).
PEG10 regulates the neuronal transcriptome in a UBE3A-dependent manner and alters neuronal migration in vivo
To gain more insight into the importance of the PEG10-UBE3A relationship in AS pathophysiology, we performed epistasis experiments to compare the effects of independently manipulating UBE3A levels and PEG10 levels. Specifically, we compared UBE3A ATS KD (to reinstate UBE3A expression) and PEG10 KD in AS neurons, and UBE3A KD in control neurons. These cells were then harvested, followed by DIA proteomics on neurons and EVs and RNA-seq analysis on neurons (Figures 5A and S6A). Notably, proteomic analysis of AS neurons revealed a modest correlation between reducing PEG10 levels (PEG10 KD) and increasing UBE3A levels (UBE3A ATS KD) (R = 0.54; Figure 5B). PEG10 expression alterations in control and AS neurons led to concomitant changes in neurons and neuronal EVs for ATXN10, TCAF1, and RTL8C, indicating that PEG10-containing complexes from neurons are secreted in EVs (Figure S6B, Table S9). We confirmed PEG10-dependent alterations in ATXN10 in cell lysates and EVs with immunoblot in an independent set of samples (Figure S6C). These observations suggest that PEG10 recruits a subset of cellular proteins (ATXN10, TCAF1, and RTL8C) to EVs, which alters the neuronal and EV proteome in AS.
Figure 5.

PEG10 regulates the neuronal transcriptome in a UBE3A-dependent manner and alters neuronal migration in vivo
(A) Immunoblotting analysis for PEG10 and UBE3A in control neurons without (+NT) and with UBE3A KD (+UBE3A KD), and in AS neurons without (+NT) and with UBE3A reinstatement (UBE3A ATS KD), and with PEG10 KD. PEG10-RF1/2: 100 kDa; PEG10-RF1: 50 kDa.
(B) Correlation analysis of FCs of proteins quantified using data-independent acquisition (DIA) upon PEG 10 KD versus UBE3A reinstatement (UBE3A-ATS KD) in AS neurons. R = 0.54, linear regression.
(C) Correlation analysis of FCs of transcripts quantified using RNA-seq upon PEG 10 KD versus UBE3A reinstatement (UBE3A-ATS KD) in AS neurons. R = 0.8, linear regression.
(D) Venn diagram depicting overlap of significantly (adjusted p < 0.01) altered transcripts upon PEG10 KD and UBE3A reinstatement (UBE3A ATS KD) in AS neurons using RNA-seq.
(E) Pathways significantly increased (adjusted p < 0.05) upon PEG10 KD (PEG10 KD versus NT) and UBE3A reinstatement (UBE3A ATS KD versus NT) in AS neurons and downregulated upon UBE3A KD in control neurons (UBE3A KD versus NT).
(F) Heatmap of T statistic of transcripts of the cell adhesion-cell matrix interaction network significantly downregulated upon UBE3A KD in control neurons (UBE3A KD versus NT) and increased upon PEG10 KD (PEG10 KD versus NT) and UBE3A reinstatement (UBE3A ATS KD versus NT) in AS neurons.
(G) Single-cell RNA-seq data for PEG10 in human and mouse brains in clusters of excitatory neurons, inhibitory neurons, and astrocytes. Colors represent log2 FPKM (fragments per kilobase of transcript per million mapped reads) values.
(H) Representative images of P1 WT and AS pups in utero electroporated at E14.5 with empty control vector or PEG10-RF1/2. tdTOMATO+ cells indicate the successfully targeted neurons. Arrowheads indicate the cortical plate (CP) of the somatosensory cortex, while the arrow indicates the subventricular zone (SVZ). Scale bar: 1,000 μm.
(I) Quantification of percentage of electroporated cells (tdTomato expressing) in bins from SVZ to the CP for WT (black) and AS (red) pups with tdTOMATO empty vector and WT pups with PEG10-RF1/2_tdTOMATO construct (blue).
(J) Quantitation of sum of percentage of cells in bins 1–4 (from the CP) (p values: Holm-Sidak multiple test).
Since we found that PEG10 binds to several neuronal transcripts and is localized to SGs in AS neurons, we asked whether the AS transcriptome changed in a similar fashion by manipulating either PEG10 or UBE3A levels. RNA-seq analysis (Table S10) showed that UBE3A reinstatement (UBE3A ATS KD) or PEG10 KD dramatically and similarly altered the AS transcriptome (R = 0.8; Figure 5C). There was a significant overlap of transcripts in AS neurons modulated by UBE3A reinstatement and PEG10 KD (hypergeometric test, p = 0; Figure 5D). We focused on pathways that were PEG10 (modulated with PEG10 KD) and UBE3A dependent (modulated with UBE3A ATS KD and UBE3A KD). We observed alterations in cell-adhesion and cell-matrix interactions consistently in control as well as in AS neurons (Figure 5E). Analysis of the leading-edge transcripts of these pathways confirmed these alterations in key genes involved in neuronal migration and development such as laminin subunits (LAMB1, LAMA1, LAMA2, and LAMA4).51, 52, 53 Extracellular matrix components (ADAM12, ITGB5), also belonging to cell adhesion and migration pathways, were altered in a UBE3A- and PEG10-dependent manner (Figure 5F).
We next assessed the functional effects of the aberrant PEG10 expression in vivo in the context of neuronal adhesion and migration. Analysis of single-cell RNA-seq data from mouse54 and human brains55 revealed that PEG10 is highly expressed in humans, but almost undetectable in mice (Figures 5G and S7A). In addition, bulk RNA-seq analysis of human versus mouse brains revealed a similar pattern, with high PEG10 expression in human neurons, but close to the limit of detection in mouse brains (Figure S7B).56,57 In the human brain, PEG10 expression peaks during fetal brain development across all brain regions and remains constant after birth58 (Figure S7C). We hypothesized that increased PEG10 expression during brain development could affect neuronal function at this critical time of brain development. Since mouse cortical neurons express low levels of PEG10, we took advantage of the in utero electroporation technique to establish the effect of PEG10 overexpression.59 This assay makes use of the ability of neurons to migrate from the subventricular zone (SVZ) to layer II/III of the somatosensory cortex. This intricate process is highly sensitive to changes that perturb normal neuronal function, resulting in migration delay. A vector that drives the overexpression of PEG10-RF1/2 was delivered via in utero electroporation to wild-type (WT) and AS mice at embryonic day 14.5 (E145.5), along with a control vector expressing only the tdTomato reporter gene. Whereas the control vector showed no changes in the migration of AS neurons versus WT neurons, the overexpression of PEG10-RF1/2 resulted in a severe deficit in the ability of targeted precursor neurons to migrate to the cortical plate (CP), with the majority of precursor cells remaining close to the SVZ (Figure 5H, quantified in Figures 5I and 5J). These results indicate that the overexpression of PEG10 interferes with normal neuronal function and could affect overall brain development.
Discussion
Using proteomic analysis combined with the specific modulation of UBE3A expression in control and AS patient-derived neurons, we identified retrotransposon-derived GAG domain-containing PEG10 protein as a target that is regulated by UBE3A. We confirmed that PEG10-RF1/2 protein is expressed in postmortem AS patient brain tissue. We characterized PEG10 as a multifunctional RNA-binding protein localizing to SGs in complex with other RNA-binding proteins, and secreted in neuronal EVs. We discovered that the increased expression of PEG10 in AS leads to the altered expression of pathways associated with cell migration and adhesion. Although mice have almost undetectable levels of PEG10, its forced overexpression in vivo disrupts neuronal migration during cortical development.
AS mouse models with deletion of the maternal Ube3a allele have been valuable to understand UBE3A-driven disease mechanisms. However, these mice do not recapitulate all of the phenotypes observed in patients; there is a notable lack of a clear cognitive phenotype.10 These observations led us to the hypothesis that there could be human-specific UBE3A pathways in neuronal development and physiology. Transcriptomic and proteomic experiments in control and AS patient iPSC-derived neurons identified alterations in AS-related molecular pathways. This analysis revealed both potential transcriptional and post-transcriptional alterations. For instance, transcript and protein levels of GAD2, an enzyme involved in GABA synthesis, were reduced in both AS deletion and point mutation lines, but not phenocopied or rescued with UBE3A modulation using ASOs. It is conceivable that these deficits could reflect a delayed GABAergic-specific neuronal maturation. Similarly, mitochondrial complex 1 proteins were modulated only at the protein level and showed no phenocopy or rescue with UBE3A expression modulation. Our dataset provides a rich resource for interrogating human disease-associated changes. However, as we only had a single point mutation patient in the present study, future studies will be required to determine whether there are altered molecular changes for UBE3A point mutation versus deletion, a question with relevant therapeutic implications.
To dissect the molecular pathways downstream of UBE3A in these AS hiPSC-derived neuronal models, we investigated the molecular changes upon UBE3A reinstatement and, conversely, we recapitulated the disease model in control neurons by reducing UBE3A levels using ASOs. This experimental system enabled a proteomic screen to identify human UBE3A targets elevated in AS and inversely modulated with respect to UBE3A expression upon ASO treatment. This screening identified UCHL5 (a proteasome-associated deubiquitinase [DUB]) and HERC2 (an E3 ligase), both of which could lead to downstream effects on the ubiquitin proteasome system, as previously reported.60 Future research to investigate the functional consequences of UCHL5 and HERC2 alterations will shed more light on UBE3A regulation of the ubiquitin proteasome system. Other targets discovered downstream of UBE3A included the cytoskeletal components Girdin and Dystonin; both have roles in neuronal development and therefore could be involved in AS pathophysiology.
The clusters of proteins most strongly regulated in a UBE3A-dependent manner were PEG10, TCAF1, and RTL8A, RTL8B, and RTL8C. PEG10 is a GAG domain-containing retrotransposon-derived gene, originating from the Ty3-gypsy family of retro-elements.61 In mammals, PEG10 has lost the elements necessary for transposition in the genome, although it has become indispensable for placental development.62 PEG10 gives rise to two protein isoforms via ribosomal-1 frame shifting, a mechanism that is used by retroviruses to generate GAG (from RF1) and GAG-Pol proteins (from RF1/2).38 During evolution, PEG10-RF1/2 lost the polymerase domain, but retained the CCHC-type RNA-binding motif and a potentially functional aspartyl protease motif.63 We hypothesized that the loss of UBE3A in AS neurons may dysregulate important mechanisms during nervous system development through increased PEG10 levels.
UBE3A reinstatement could normalize PEG10 levels both chronically and acutely in neuronal cultures. We observed UBE3A-dependent alterations in PEG10-RF1/2 protein levels, but not in the shorter PEG10-RF1 isoform. As PEG10-RF1/2 regulation by UBE3A is proteasome dependent, we asked whether UBE3A directly targets PEG10 for degradation. Using Y2H and IP-WB, we demonstrated that PEG10-RF1/2 and UBE3A interact and observed polyubiquitination of PEG10 using IP-WB analysis. However, in bacterial ubiquitination experiments, UBE3A did not directly ubiquitinate PEG10-RF1/2. Thus, the UBE3A-dependent regulation of PEG10-RF1/2 levels could be indirect via regulation of PEG10-binding partners such as TCAF1 and RTL8C, also identified downstream of UBE3A (Figure 2), or they would require additional UBE3A adaptor/regulatory proteins (HERC2 and NEURL4) in a cellular context. Alternatively, as proteins associated with the proteasome were regulated by UBE3A, including the DUB UCHL5, it is conceivable that the regulation of PEG10 by UBE3A occurs via alterations in the proteasome. Further experiments to study UBE3A-regulated PEG10 mechanisms, including the role of UBQLN264 in this pathway, may shed light on the complex biology of the direct and indirect targets of UBE3A.
Immunoblot analysis revealed the expression of PEG10-RF1/2 in postmortem brain tissue from three AS patients. IHC analysis of an independent sample showed a PEG10-RF1/2 signal in AS neurons, while the protein was not detectable by IHC in control tissue. However, given the small sample size and known variability, this observation will require independent validation with additional patient samples. Since we demonstrated that PEG10 is secreted in neuronal exosomes, analysis of PEG10 in cerebrospinal fluid (CSF) samples could provide a pharmacodynamic marker for UBE3A-targeting therapies in AS.
We found that PEG10 interacts with ataxia-associated proteins ATXN2 and ATXN10. PEG10-RF1/2 specifically localized to ATXN2+ and UBQLN2+ RNA granules or SGs upon sodium arsenite stress. Characterization of PEG10-bound transcripts revealed an enrichment of several thousand mRNAs, including its own, in AS neurons compared with control neurons. These interactions could be direct, as cross-linking IP (CLIP) experiments revealed that PEG10 protein directly binds to its own mRNA36 or indirectly, for example, via PEG10-interacting proteins including ATXN2 and UBQLN2.65 PEG10 reduction affected the levels of PEG10-interacting proteins, including ATXN10, TCAF1, and RTL8C in AS neurons, and slightly affected the cellular proteome. Reducing PEG10 expression in AS neurons altered the amount of a known EV-associated protein ATXN1043,44 in a UBE3A-dependent manner. This finding suggests that PEG10 influences the specific proteins and RNAs that are localized to EVs.
Under basal conditions, PEG10 was not preferentially secreted in EVs compared with canonical EV proteins such as TSG101 or CD63, nor did we detect any alterations in EV numbers from control and AS neurons. Future work should address whether PEG10 secretion and expression in EVs is activity dependent and whether it spreads trans-synaptically, as recently described for Arc, another RNA-binding GAG domain-containing protein66,67 that is sensitive to UBE3A levels.68 The secretion of PEG10 in EVs may have implications in AS pathophysiology; it will be interesting to determine whether EVs isolated from the CSF of AS patients are altered.
Transcriptome analysis showed that PEG10 downregulation to a large extent phenocopied UBE3A reinstatement, suggesting that PEG10 could be a significant driver of the molecular pathophysiology caused by the loss of UBE3A. We focused on the subset of transcripts that were reciprocally modulated in control versus AS neurons. These transcripts point toward a role of PEG10 in cell adhesion and migration by regulating the expression of key transcripts of the extracellular matrix, such as several laminin subunits.51, 52, 53 We hypothesized that alterations in these pathways via PEG10 could affect neuronal function. Therefore, by using in utero electroporation, we expressed human PEG10-RF1/2 during early cortical development in mice. Overexpression of PEG10-RF1/2 in neuronal precursors resulted in severely impaired migration, indicating a pronounced effect of increased PEG10 expression on neuronal development. It is interesting to note that even after extensive proteomic experiments of both AS mouse and rat models, we were unable to detect PEG10 protein in mouse brains (data not shown), and RNA expression analysis in mice from publicly available datasets indicated that in adult mouse neurons, Peg10 expression is nearly 5 times lower than that observed in human neurons. This probably indicates that PEG10 does not play a role in mouse brain development, and does not explain the AS mouse phenotypes. We cannot rule out that PEG10 does not have any biologic function in neuronal tissue, and that UBE3A fulfills a safeguarding mechanism to reduce its levels in humans. Alternatively, PEG10 could have acquired an additional function besides its role in placental development, which is specific for human brain function or development 69.
In summary, this study highlights the importance of using hiPSC-derived neuron models in studying neurodevelopmental disorders. We demonstrated that the levels of PEG10 protein are sensitive to rising UBE3A expression levels, and that overexpression of PEG10 remarkably phenocopies the transcriptional changes seen with UBE3A loss in AS, implying that PEG10 could be critically involved in AS pathophysiology. Given that PEG10 is a secreted protein, it could serve as a biomarker for monitoring UBE3A levels in ASO or gene therapy trials for Angelman syndrome.
Limitations of the study
A major focus of the work presented leverages hiPSC AS patient-derived neurons. Taking into account that not all steps of brain development and maturity are recapitulated, it is possible that UBE3A targets beyond those reported here may be altered in development and disease. Furthermore, the very limited AS brain samples presented here suggest that PEG10 is expressed in disease. Future efforts will be required to determine the altered human proteome from AS brain postmortem samples and to confirm that PEG10 is persistently upregulated in development and disease. In addition, analysis of patient CSF samples is required to determine whether PEG10 is secreted from neurons in disease and to validate it as a downstream UBE3A biomarker. Although PEG10 expression affects disease transcriptome, the precise role of PEG10 in human neurons remains unknown. Further characterization using independent cellular models and detailed developmental and physiological characterization in vitro and in vivo is important to fully define the function of PEG10 in AS.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-UBE3A (rb) | Sigma-Aldrich | Cat# SAB2102627; RRID:AB_10600687 |
| Anti-UBE3A (ms) | Sigma-Aldrich | Cat# SAB1404508; RRID:AB_10740376 |
| Anti-UBE3A (ms) | Sigma-Aldrich | Cat# E8655; RRID:AB_261956 |
| Anti-HuCD (rb) | Thermo Fisher Scientific | Cat# A21271; RRID:AB_221448 |
| Anti-NeuN (rb) | Merck | Cat# MAB377; RRID:AB_2298772 |
| Anti-PEG10 RF1/2 (rb) | Abcam | Cat# ab240392; RRID:AB_2889941 |
| Anti-PEG10 RF1 and RF1/2 (ms) | Abcam | Cat# ab250462 |
| Anti-PEG10 (ms) | Abcam | Cat# ab215035; RRID:AB_2891312 |
| Anti-MAP2 (ch) | Neuromics | Cat# CH22103; RRID:AB_2314763 |
| Anti-UBQLN2 (ms) | Abcam | Cat# ab190283; RRID:AB_2747782 |
| Anti-G3BP1 (ms) | Abcam | Cat# ab56574; RRID:AB_941699 |
| Anti-GFP (ms) | Abcam | Cat# ab6556; RRID:AB_305564 |
| Anti-ATXN2 (ms) | BD Biosciences | Cat# 611378; RRID:AB_398900 |
| Anti-ATXN10 (ms) | Abcam | Cat# ab153875; RRID:AB_2889942 |
| Anti-TSG101 (ms) | Abcam | Cat# ab83; RRID:AB_306450 |
| Anti-PDCD6IP (ms) | Abcam | Cat# ab117600; RRID:AB_10899268 |
| V5 Tag Monoclonal Antibody, HRP | Thermo Fisher Scientific | Cat# R961-25 |
| Anti-HA-Peroxidase, High Affinity from rat IgG1 | Roche | Cat# 12013819001 |
| Peroxidase-AffiniPure Goat Anti-Mouse IgG (H + L) antibody | Jackson ImmunoResearch Labs | Cat# 115-035-003; RRID:AB_10015289 |
| Biotin-SP (long spacer) AffiniPure Goat Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 115-065-166 RRID:AB_2338569 |
| Alexa Fluor® 488 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 715-545-151; RRID:AB_2341099 |
| Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (min X Hu,Ms,Rat Sr Prot) antibody | Jackson ImmunoResearch Labs | Cat# 111-035-144; RRID:AB_2307391 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat# A32723; RRID:AB_2633275 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat# A32731; RRID:AB_2633280 |
| Goat Anti-Mouse IgG (H+L) Highly Cross-adsorbed Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11031; RRID:AB_144696 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11011; RRID:AB_143157 |
| Goat anti-Chicken IgY (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 647 | Thermo Fisher Scientific | Cat# A32933; RRID:AB_2762845 |
| OmniMap anti-Rb HRP | Ventana Medical Systems | Cat# 760-4311 |
| Goat F(ab’)2 Anti-Rabbit IgG H&L (10nm Gold) | Abcam | Cat# ab39601; RRID: AB_954434 |
| Bacterial and virus strains | ||
| E. coli BL21 gold (DE3) | Agilent | Cat# #230132 |
| PEG10-RF1/2-GFP | Sirion (Custom) | N/A |
| PEG10-RF1-GFP | Sirion (Custom) | N/A |
| PEG10-RF1/2-IRES-GFP | Sirion (Custom) | N/A |
| Biological samples | ||
| Human brain tissues | the Netherlands Brain Bank | https://www.brainbank.nl |
| Human brain tissues | University of Pennsylvania Cooperative Human Tissue Network | https://www.chtneast.org |
| Human brain tissues | Analytical Biological Services | https://www.absbio.com |
| Human brain tissues | NeuroBioBank | https://neurobiobank.nih.gov/specimens/ |
| Chemicals, peptides, and recombinant proteins | ||
| MG-132 | Sigma-Aldrich | Cat# M7449 |
| NP-40 | Thermo Fisher Scientific | Cat# 85124 |
| Laemmli sample buffer | Sigma-Aldrich | Cat# S3401 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23227 |
| iST sample preparation kit | PreOmics | Cat# P.O.00001 |
| Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat# 88802 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| TruSeq Stranded Total RNA Sample Prep kit with Ribo-Zero Gold | Illumina | Cat# RS-122-2301/02 |
| TaqMan® RNA-to-Ct 1-Step Kit | Thermo Fisher Scientific | Cat# 4392656 |
| DISCOVERY DAB Map Detection Kit | Ventana Medical Systems | Cat# 760-124 |
| DISCOVERY OmniMap anti-Ms HRP detection kit | Ventana Medical Systems | Cat# 760-4310 |
| Heavy labeled peptides for SRM | JPT Peptide Technologies | Table S5 |
| Deposited data | ||
| RNA-Seq data generated in this study | https://www.ncbi.nlm.nih.gov/geo | PRJNA712999 |
| Proteomics data generated in this study | https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp | MSV000087645 |
| Experimental models: cell lines | ||
| Human: CRA1302 | This study | Table S1 |
| Human: CRA 902 | This study | Table S1 |
| Human: CRA1301 | This study | Table S1 |
| Human: CRA1501 | This study | Table S1 |
| Human: CRA1101 | This study | Table S1 |
| Human: H4 | ATCC | HTB-148 |
| Human: iCell® GlutaNeurons | Fujifilm | Cat # R1034 |
| Experimental models: organisms/strains | ||
| Yeast strain: Y187 | Clontech | Cat# 630457 |
| Yeast strain: Y2H Gold | Clontech | Cat# 630498 |
| FVB/NHsd mice | Erasmus MC Rotterdam | N/A |
| C57Bl6/J mice | Erasmus MC Rotterdam | N/A |
| Oligonucleotides | ||
| ASO-UBE3A KD: TTTAcacctacttcttaaCA | This study | N/A |
| ASO-UBE3A ATS KD 1: CTttccatttatttccATTT | This study | N/A |
| ASO-UBE3A ATS KD 2: GATtaggcacattAAT | This study | N/A |
| ASO-NT: TTGaataagtggaTGT | This study | N/A |
| ASO-PEG10 KD: ATTtctattccacaaacaCA | This study | N/A |
| Recombinant DNA | ||
| TCAF1-V5 | Genscript (Custom) | N/A |
| RTL8C-V5 | Genscript (Custom) | N/A |
| UBE3A | ErasmusMC (Custom) | N/A |
| Software and algorithms | ||
| Proteome Discoverer Software | Thermo Fisher Scientific | Cat# OPTON-20141 |
| R | R Core Team | https://www.r-project.org/ |
| limma (R package) | R | https://bioconductor.org/packages/release/bioc/html/limma.html |
| multcomp (R package) | R | https://cran.r-project.org/web/packages/multcomp/index.html |
| Emmeans (R package) | R | https://cran.r-project.org/web/packages/emmeans/index.html |
| Skyline v 4.2 | MacCoss Lab Software | https://skyline.ms/project/home/software/Skyline/begin.view |
| Spotfire | Tibco | https://www.tibco.com/products/tibco-spotfire |
| FIJI | FIJI | https://fiji.sc/ |
| Spectronaut | Biognosys | https://biognosys.com/software/spectronaut/ |
| FastQC 0.11.5 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/ |
| STAR 2.5.2a | N/A | https://github.com/alexdobin/STAR |
| Other | ||
| Easy LC 1200 system | Thermo Fisher Scientific | Cat# LC140 |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | Thermo Fisher Scientific | Cat# IQLAAEGAAPFADBMBHQ |
| NextSeq 500 | Illumina | Cat# SY-415-1001 |
| Dulbecco’s Modified Eagle’s Medium (DMEM) | ATCC | Cat# 30-2002 |
| Zetasizer Ultra | Malvern Panalytical | N/A |
| BenchMark ULTRA system | Ventana Medical Systems/Roche Diagnostics | Cat# N750-BMKU-FS 05342716001 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ravi Jagasia (ravi.jagasia@roche.com)
Materials availability
All reagents generated in this study are available upon request from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Patient recruitment and reprogramming
The subjects enrolled in this study were recruited through Boston Children’s Hospital. Protocol was approved by Boston Children’s Hospital (Boston, USA) IRB (P00000219). Research performed on samples of human origin was conducted following informed consent, as approved by the institutional review board of Boston Children’s Hospital. Briefly, hiPSC lines were derived from 3 individuals with Angelman’s 2 deletion (CRA1301, CRA1501) and 1 point mutation (line CRA1101). hiPSC reprogramming was done from blood using Sendai virus at Harvard Stem Cell Institute. 80% confluent were transduced with each of the four viruses at a multiplicity of infection (MOI) of 3. Cells were fed every other day, and 50 k, 100 k and 200 k cells were replated on day 7 onto 0.1% gelatin-coated, 10-cm dishes containing. Medium was switched to hESC medium Colonies were picked by mechanical dissection, were transferred to fresh feeders, and were expanded using) clump passaging methods Control hiPSCs were derived either from the parents of the AS-patients and gender matched individuals without UBE3A deletion/mutations (line CRA1302 and line CRA902).
Cell culture and neuronal differentiation
Cell culture of NPCs and neurons was performed as described in Costa et al.18 NPCs between passage numbers 15–25 were used for the entire analysis. Neuronal cell culture media: N2B27 medium is a 1:1 mixture of DMEM/F12 (1:1) medium with GlutaMAX I and Neurobasal medium, containing 1 × N2 supplement, 1 × B27 supplement minus Vitamin A, and 50 μM βmercaptoethanol (all from LifeTechnologies). NEP medium is N2B27 medium containing 5 ng/ml FGF-2, 250 ng/ml noggin (R&D Systems or Peprotech), and 20 μM SB 431542 (Tocris). FEB medium is N2B27 medium containing 10 ng/ml FGF-2 (Peprotech), 10 ng/ml EGF (R&D Technologies), and 20 ng/ml BDNF (Peprotech). SFA medium is N2B27 medium containing 100 ng/ml FGF-8 (Peprotech), 200 ng/ml sonic hedgehog (Peprotech) and 100 μM ascorbic acid 2-phosphate (Sigma). BGAA medium is N2B27 medium containing 20 ng/ml BDNF, 10 ng/ml GDNF (Peprotech), 500 μM dibutyryl cyclic AMP (Sigma) and 100 μM ascorbic acid 2- phosphate. Generation of neural precursor cells (NPCs). NPCs were generated from hESCs using a modified dual SMAD inhibition protocol.70 hESCs were dissociated to single cells and plated into AggreWell800 plates (STEMCELL Technologies) at a density of 5,000 cells per microwell in NEP medium supplemented with 10 μM Y-27632. After five days, aggregates were recovered, plated on polyornithine-/laminin-coated (PL) dishes in NEP medium and cultured for an additional three days to form neural rosettes. Rosettes were isolated manually and replated on PL dishes in NEP medium. Upon reaching confluence, cells were dissociated with 0.05% trypsin/EDTA solution (LifeTechnologies) and plated at 100,000 cells/cm2 on PL dishes in FEB medium. Cells were cultured under these conditions with passaging every 2 – 3 days for ca. 15 population doublings (PDs), followed by a stepwise decrease in plating density to 25,000 cells/cm2 within ca. 10 PDs. Medium was replaced daily throughout the entire NPC derivation procedure. NPC lines were characterized by FACS and immunocytochemical analyses. Neuronal differentiation. NPCs were dissociated with trypsin/EDTA, plated on PL dishes at 10,000 – 15,000 cells/cm2 in SFA medium, and cultured for one week with medium replacement after four days. The resultant progenitors were dissociated with trypsin/EDTA, plated on PL dishes at 35,000 – 50,000 cells/cm2 in BGAA medium and differentiated for up to 10 weeks with biweekly medium replacement.
Human postmortem brain samples for western blot analysis
Frozen (–80°C) human postmortem samples from the NICHD Brain and Tissue Bank (http://medschool.umaryland.edu/BTBank/ (now part of the NeuroBioBank: https://neurobiobank.nih.gov/specimens/) used. The case number and patient control sample data are presented below. All samples were treated with informed consents permissions and ethical considerations of the University of Maryland Brain and Tissue Bank and the Netherlands Brain Bank.
| Sample ID | Diagnosis | Age | Gender | Brain region | Application |
|---|---|---|---|---|---|
| 1754 | AS | 4 years | Male | Temporal cortex | Western blotting |
| 1824 | AS | 4 years | Male | Temporal cortex | Western blotting |
| 1185 | Unaffected control | 4 years | Male | Temporal cortex | Western blotting |
Method details
Immunoblotting analysis
Cell pellets were reconstituted in RIPA buffer (Thermo) with protease and phosphatase inhibitor cocktail (Roche). Protein quantification was performed using BCA assay (Pierce BCA Protein Assay Kit) followed by reconstitution of samples in 4X SDS lamelli buffer (BioRad) and 10X sample reducing agent (NuPAGE Sample Reducing Agent (10X), boiled at 95°C. Post sample prep immunoblotting analysis was performed as per Pandya et al.71
TMT-MS3 Analysis
Samples came from four independent differentiations for Control versus AS deletion comparison and two independent biological replicates for Control versus AS with treatments. Sample preparation TMT labeling was randomized across each sample. In order to normalize across multiple TMT runs, two pooled samples were included in each TMT-MS3 run. Sample preparation, data acquisition and analysis was performed by TMT-MS3 on the Thermo Orbitrap Fusion performed as described in Pandya et al.15
TMT-MS3 data analysis and data normalization
Samples were analyzed in multiple TMT runs with pooled sample(s) in each plex. Data were annotated and normalized with Proteome Discoverer (Thermo Fisher Scientific). Normalization was done on the peptide level to the maximum of summed intensities for each channel. The common pooled samples were used to normalize across the TMT-plexes with the internal reference scaling (IRS) method: scaling factors were calculated for each protein to adjust their reference value to the geometric mean of the pooled samples, and these were then used to scale the abundances for each protein in the remaining samples in each TMT experiment.72 Differential abundances of proteins were calculated by fitting linear models for each protein with the limma R package73 and applying an Empirical Bayes method to moderate the variances.74 Different conditions were compared by calculating contrasts with multcomp75 and emmeans76 packages. The computed p values were adjusted for multiple testing by controlling the false discovery rate.77 All calculations were performed in R.78
Selective reaction monitoring (SRM) assays
A target list of 35 proteins (Table S2) was selected for selective reaction monitoring analysis as described in Dunkley et al.17 In brief, sample preparation for LC-MS analysis was performed using the iST kit (PreOmics) as per manufacturer’s instructions. Heavy labeled peptides were used for normalization as described in Dunkley et al.17 Skyline v 4.2 was used to generate a target peptide list based on shotgun MS analysis of human IPSC derived neurons.Isotope-labeled peptides (unpurified), containing either L-[U-13C, U-15N]R or L-[U-13C, U-15N]K, corresponding to the 56 unique target peptides were synthesized (JPT Peptide Technologies) and their sequences confirmed by LC-MS/MS.
Peptide samples from independent triplicates from neural precursor cells (NPCs) and neurons at Day 0, Day 19, Day 42 from Control and AS lines with either no treatment (NA), Non-targeting ASO (NT) and UBE3A KD ASO (on Control neurons) and UBE3A ATS ASO (on Angelman neurons) were prepared using the Preomics iST technology kit using manufacturor’s instructions. Sample preperation for 90 samples was completely randomized to avoid biases.
For MS analysis, digests were diluted to 200 ng/μL with 0.1% v/v formic acid, 2% v/v ACN containing the pooled isotope-labeled peptides at a final concentration of approximately 3 fmole/μL. SRM analyses were performed on an Ultimate RSLCnano LC coupled to a TSQ Quantiva triple quadrupole MS (Thermo Scientific). Samples (5 μl) were loaded at 3 μl/min for 6 min onto a 2 cm × 75 μm C18 trap column (Acclaim Pepmap 100, 3 μm, 300 Å, Thermo Scientific) in loading buffer (0.5% v/v formic acid, 2% v/v ACN). Peptides were then resolved on a 50 cm × 75 μm C18 analytical column with integrated electrospray emitter heated to 40°C (Easy-SPRAY, 2 μm, 100 Å, Thermo Scientific) using the following gradient at a flow rate of 250 nL/min: 6 min, 98% buffer A (2% ACN, 0.1% formic acid), 2% buffer B (ACN + 0.1% formic acid); 90 min, 30% buffer B; 96 min, 60% buffer B; 98 min, 80% buffer B; 114 min, 80% buffer B; 115 min, 2% buffer B; 138 min, 2% buffer B. The TSQ Vantage was operated in 2 min retention time windows; cycle time, 1.5 s; spray voltage, 2600 V; collision gas pressure, 1.5 mTorr; Q1 and Q3 resolution, 0.7 FWHM; capillary temperature 240°C.
Skyline version 4.2 was used for automated peak integration. In a small number of cases, the peak selection was manually corrected based on the isotope-labeled peptide elution time and transition ratio as well as the expected transition ratios based on the chromatogram library. A transition was removed from a peptide if obvious interference was identified (through comparison to the chromatogram library and the isotope-labeled peptides). Post data acquisition, total precursor peak areas were exported and further analysis was performed on Spotfire (Tibco). The endogenous peptide peak areas were corrected for using spiked-in heavy labeled peptides by applying a correction factor (Median heavy peptide intensity / heavy peptide area per sample). Further, the corrected light peptide areas were corrected for Actin peptide using an actin correction factor (Median Actin abundance / Actin abundance). The protein abundance was then calculated as a sum of Actin corrected peptide abundances for each protein. Corresponding raw data for each of the proteins is supplied in Table S2.
Immunoprecipitation-immunoblotting (IP-WB)
All steps were performed on ice or 4°C. IP analysis was performed as described in Pandya et al.71 Briefly, frozen neuronal cell pellets (∼0.5 mg) were suspended in 500 μL of IP extraction buffer. Frozen neuronal cell pellets were suspended in 500 μl of IP extraction buffer (1% n-Dodecyl-B-D-maltoside, 150 mM NaCl, 25 mM HEPES, pH 7.4, Roche protease inhibitor cocktail) and incubated 30 min on a rotor at 8 rpm. Next the lysate was clarified by centrifugation at 20,000 x g for 20 min. The supernatant was then incubated with 5 μg of antibody at 8 rpm for 2 hr. Next 50 μg of protein A/G magnetic beads (Thermo Scientific) were washed with lysis buffer 2X and the cell lysate with antibody was further incubated for 1 hr at 8 rpm. Post bead incubation, the beads were washed thoroughly with lysis buffer 4X, followed by elution of proteins for immunoblotting analysis using lamelli buffer with reducing agent. Immunoblotting analysis was performed as described previously.
Immunoprecipitation-mass spectrometry
Immunoprecipitation for PEG10 was performed using two antibodies against PEG10 (ab215035 and ab255695) as before. Post IP, samples were eluted by boiling at 95°C SDS-PAGE lamelli buffer with reducing agent. The samples were further loaded on precast gels (Biorad) and processed for in-gel digestion as described in Pandya et al.71
Immunoprecipitation-RIP-seq for PEG10
All steps were performed on ice or at 4°C. Immunoprecipitation for PEG10 was performed on four control and four AS neuron independent cell pellets from 10 cm dishes.
Cell pellets were lysed in 1 mL of IP lysis buffer (1% n-Dodecyl-B-D-maltoside, 150 mM NaCl, 25 mM HEPES, pH 7.4, Roche protease inhibitor cocktail, 100 U/mL Invitrogen RNaseOUT Recombinant Ribonuclease Inhibitor) and extracted for 1 hr on a rotor at 8 rpm. The lysates were then spun at 20,000 x g to get rid of insoluble cell debris and the supernatant collected. BCA assay was performed to determine protein concentration in the samples and equal amount of extracted total protein (1 mg) was incubated with 50 μg PEG10 antibody (ab255695) on rotor for 1 hr followed by incubation with pre-equilibrated 100 μl of Protein A/G beads for 1 hr. Post IP, the beads were thoroughly washed with lysis buffer (4X) and once with 50 mM Tris-HCl pH 7.5. Elution of RNA was performed by resuspending the beads in proteinase K buffer (50 mM Tris-HCl pH 7.5, 1 mM EDTA, 1% SDS) containing 0.5 mg/mL proteinase K (Roche) followed by incubating for 30 min at 55°C with shaking. The RNA purification was carried out using the QIAGEN RNeasy Kit according to the manufacturer’s protocol.
RIP-seq and total RNA-seq
Total RNA from cell pellets was extracted using RNeasy Mini Kit (QIAGEN) following the manufacturer’s instructions. RIP-Seq and Total RNA-Seq libraries were constructed and further sequenced at Fasteris (Switzerland). Sequencing libraries were generated using TruSeq stranded total RNA library preparation protocol with Ribozero Gold kits to remove rRNA (Illumina). Libraries were subjected to single-end sequencing on a NextSeq 500 sequencer (Illumina) with 50-bp read length.
qPCR analysis of UBE3A sense and ATS transcript
RNA for qPCR was extracted using the QIAGEN RNeasy Kit according to the manufacturer’s protocol. Expression of target genes was quantified using TaqMan RNA-to-CT 1-Step Kit and TaqMan Gene Expression Assays (Thermo Fisher Scientific). qRT-PCR was done on Applied Biosystem QuantStudio 12K Flex Real-Time PCR System. The following TaqMan gene expression assays were used: UBE3A (Hs00166580_m1), UBE3A-ATS (Hs01372957_m1), MAPT (Hs00902194_m1), and PPIA (4326316E). Relative expression levels of UBE3A and UBE3A-ATS were calculated using the delta-delta Ct method.
Proteasome inhibition assay
Control neurons were incubated with the 5 μM ASOs at of UBE3A sense ASO at Day 28 and ASO was maintained in the cell culture media. At Day 42, cells were either treated with DMSO or 10 mM MG-132 (M7449, Sigma) for 0, 4, and 8 hr. The cells were harvested and immediately frozen on dry ice by scraping in phosphate buffer saline without Mg2+ and Ca2+. The cell pellets were suspended in RIPA lysis buffer at 4°C (with Protease and phosphatase inhibitors; Roche), BCA performed and equal amounts of total protein was suspended in SDS-PAGE lamelli buffer with reducing agent for immunoblotting analysis with UBE3A, PEG10, and poly-ubiquitin.
Bacterial ubiquitination assay and immunoblotting
The bacterial ubiquitination assay was performed as described previously in Avagliano Trezza et al.41 Briefly, the assay consists of a polycistronic plasmid expressing rabbit E1, E2 (UbcH5c) and ubiquitin or a plasmid in which the ubiquitin gene was deleted, a second plasmid expressing HA-UBE3A (N-terminally tagged) and a third plasmid expressing either V5-PEG10 RF1 or V5-PEG10 RF1/2 (N-terminally tagged). A plasmid expressing the UBE3A target V5-RING1B (N-terminally tagged) served as a positive control. These constructs were co-transformed into E. coli strain BL21-GOLD (DE3) [B F– ompT hsdS(rb- mb-) dcm+ Tetr gal λ(DE3) endA Hte] and selected on LB agar (1% (w/v) Bacto tryptone, 0.5% (w/v) Bacto yeast extract, 1% (w/v) NaCl, 1.5% agar) containing half the concentration of antibiotics as needed (ampicillin, 25 μg/mL; kanamycin, 15 μg/mL; streptomycin/spectinomycin, 25 μg/mL). Single transformants were grown overnight at 37°C in LB medium supplemented with 2% glucose, 50 mM Tris-HCl [pH 8.0] and appropriate antibiotics. The next day the culture was diluted to an OD600 of 0.2 and grown at room temperature until it reached an OD600 of 0.7. Protein expression was induced by adding 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma-Aldrich I6758) and the culture was incubated overnight at 16°C while shaking. The following morning, 20 OD600 units per culture were collected, and lysed in lysis buffer (50 mM Na-Pi buffer (pH: 8.0), 300 mM NaCl, 5% glycerol, 5 mM 2-mercaptoethanol, 1mM PMSF, Protease Inhibitor Cocktail (Sigma-Aldrich P8340), DNase (0.01 mg/mL) and RNase (0.01 mg/mL). The lysate was sonicated for 3 rounds of 10 s at 10 mAmp. Protein amount corresponding to 0.3 OD600-units was used for Immuno blot analysis. Validation and further specifics of this assay are described in Avagliano Trezza et al.41
Immunoblot analysis of bacterial ubiquitination
Protein samples corresponding to approximately 0.3 OD600 units were separated on 4%–12% SDS-PAGE gel and transferred onto nitrocellulose membranes. These were subsequently blocked in blocking solution (10 mM Tris-HCl [pH 8.0], 150 mM NaCl + 5% (w/v) powdered milk) for 1 hour at room temperature. The blots were probed over-night (at 4°C) with primary antibodies dissolved in TBST (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% Tween-20, (Sigma P1379) while rotating end-over-end. The next day, the blots were washed three times for 10 minutes with TBST and three times for 10 minutes with TBS, where after they were analyzed by measuring enhanced chemiluminescence (ECL) using an AI600 Chemiluminescent Imager. Primary antibodies used for Immunoblotting were: mouse monoclonal anti-V5 horseradish peroxidase (HRP)-conjugated (Thermo Fisher Scientific R961-25), rat monoclonal anti-HA HRP conjugated (Roche 12013819001).
Yeast two-hybrid assay
For the yeast two hybrid experiments the following S. cerevisiae strains were used: Y187 (MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, met–, gal80Δ, URA3::GAL1UAS-GAL1TATA-lacZ; Clontech) and Y2H Gold (MATa, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, gal80Δ, met–, LYS2:: GAL1UAS-Gal1TATA-His3,GAL2UAS-Gal2TATA-Ade2, URA3:: MEL1UAS-Mel1TATA-AUR1-C MEL1; Clontech). Cells were grown at 28°C in rich medium or in minimal glucose medium, according to the protocol previously published.79 Yeast strains were transfected, mated and, subjected to the yeast two-hybrid protocol, as published previously.80
PEG10 ubiquitination assay in neurons
Control and AS neurons treated with UBE3A ATS KD ASOs were treated with either DMSO or 10 mM MG132 (M1449, Sigma) for 6 hr. Post-treatment, cell pellets were lysed in IP lysis buffer, as described in immunoblotting analysis section, and IP for PEG10 was performed using ab255695. Post IP, immunoblots were performed as described for PEG10 and polyubiquitin.
Stress granule imaging in H4 cells
H4 cells (H4 (ATCC® HTB-148) or neurons were cultured in Dulbecco’s Modified Eagle’s Medium, Catalog No. 30-2002 media supplemented with 10% FBS (One Shot Fetal Bovine Serum, GIBCO). Cells were seeded in a 96 well plate (Falcon® 96-well Black/Clear Flat Bottom) at 1500 cells per well. On the next day, they were transduced using PEG10- RF1/2-GFP or PEG10- RF1-GFP or just GFP construct at an MOI of 2 overnight and cell culture media was replaced the next day prior to treatments. At day 3, cells were either treated with 5 mM sodium arsenate (Sigma) or PBS for 1 hr. Post treatment, cells were immediately fixed in 4% PFA for 15 min and stained for PEG10 (using anti-GFP antibody), anti-G3BP1 antibody and DAPI to label the nuclei as described.
Stress granule imaging in human iPSC derived neurons
Control and AS deletion neurons were cultured in BGAA media. Media was changed one day before treatment. Neurons were either treated with PBS or 5 mM Sodium Arsenate for 1 hr. Post treatment, cells were immediately fixed and stained for PEG10 RF1/2, MAP2, ATXN2/UBQLN2 and DAPI as described.
Cells cultured in 96-well (BD Falcon) were fixed in 4% paraformaldehyde in PBS−/− for 15 min, washed with PBS−/− 3 times, permeabilized with 0.2% (v/v) Triton in PBS−/− for 10 min at RT, incubated for 20 min in blocking buffer (5% BSA w/v, 0.2% Triton v/v in PBS−/−,filtered through 0.2 μm membrane). Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. Cells were washed in PBS−/− thrice. Secondary antibodies were diluted in blocking buffer and incubated for 2 h at room temperature. Cells were incubated with DAPI (1 μg/mL) in PBS−/− for 3 min. at room temperature. Wells were washed with PBS−/− thrice and stored at 4°C. Antibody dilutions and blocking buffer were spun down at 15,000 x g for 15 min
Immunocytochemistry in human iCell GlutaNeurons
Human iCell® GlutaNeurons (FUJIFILM Cellular Dynamics) were seeded at a density of 6 × 104 cells per well in 96-well plates (BD Falcon) according to the manufacturer’s instructions. Cells were treated with ASO to KD UBE3A from Day 3 and immunocytochemistry was performed at Day 21 as described above.
Imaging and imaging data analysis
Confocal images were obtained on Leica SP5. Maximum intensity projections were made for each image on FIJI (ImageJ). Quantification of UBE3A and PEG10 intensities was performed blinded. MAP2 or HuC/D staining was used to generate regions of interest (ROIs) of single neuronal somas with the magic wand tool of FIJI. Median intensity of PEG10 and UBE3A channels were measured. Two or three biological replicates were combined for data analysis.
Extracellular vesicle isolation
Extracellular vesicles were isolated according to recommendations of the International society of extracellular vesicles81 and as in Pastuzyn et al.66 with minor modifications (Figure 4I). Control and AS patient derived neurons were cultured in T175 flasks for 6 weeks. 25 mL of conditioned media from two flasks were pooled to obtain one replicate of extracellular vesicles. The corresponding cell pellets were pooled and aliquoted for protein and mRNA analysis. Post media harvest, cell debris was removed by centrifuging at 200 x g for 10 min. Clarified media then centrifuged at 10,000 x g for 30 mins to remove microvesicles. The supernatant was carefully removed and ultracentrifuged at 100,000 x g for 2 hr (Hitachi CP 100NX). The supernatant was discarded and the pellet suspended thoroughly in 2 mL cold PBS followed by another round of ultracentrifugation at 100,000 x g for 1 hr. The EV pellets were then suspended in PBS and stored at −80°C for immunoblotting and mass spectrometry analysis.
Multi-angle dynamic light scattering (MADLS)
Mean hydrodynamic diameter (Dh) was determined by dynamic light scattering (DLS) using a Zetasizer Ultra (Malvern Panalytical, Malvern, UK). Exosomes in PBS were placed in a quartz cuvette (Hellma, Müllheim, Germany) and Dh was measured in back scatter at an angle of 173° and at 25°C. Particle concentration was measured using multi-angle DLS at 13°, 90°, and 173° measurement angles and at 25°C. All measurements were performed in three technical replicates.
Electron microscopy
Glow discharged formvar-coated copper grids (Electron Microscopy Sciences, Hatfield, PA, USA) were incubated with EV suspensions for 2 min, washed 3x with PBS and fixed with 2% paraformaldehyde for 10 min.. Grids were washed 3x with ultrapure water, 1x with 3% uranyl-acetate and were negatively stained with 3% uranyl-acetate for 30 s at room temperature. Grids were allowed to dry at room temperature. For immuno-TEM, samples were mounted on formvar-coated copper grids as described above and were fixed with 2% paraformaldehyde for 15 min. Grids were then washed 3x with PBS and were incubated with blocking buffer 1 (1% BSA, 2.5% NGS in PBS) for 30 min. Grids were then incubated with primary antibodies (diluted 1:100) or control antibody in antibody buffer (1% BSA, 1% NGS, 0.25% saponin in PBS) over night at 4°C. Grids were then washed 3x with PBS and 3x with blocking buffer. Subsequently, grids were incubated with 10 nm gold-labeled secondary antibodies (1:40) in antibody buffer for 60 min and washed 3x with PBS. Samples were incubated with 3% glutaraldehyde for 10 min, washed 3x with ultrapure water and negatively stained as described above. Grids were allowed to dry at room temperature. Samples were analyzed on a 120 kV JEM-1400 electron microscope (JOEL, Tokyo, Japan) and images were acquired using a sCMOS camera.
Data independent acquisition proteomics of cell lysates and extracellular vesicles
Samples and cell pellets and extracellular vesicle samples were prepared as described previously. Cell pellets and EVs were suspended in iST kit lysis buffer (Preomics) and lysed according to manufacturer’s instructions. Post-lysis, protein amounts were estimated using the BCA assay (Pierce). Equal amounts of protein (20 μg) from cell lysates and EVs were loaded on SDS-PAGE gel, fixed, and in-gel digestion was performed as in Pandya et al.71 Post-digestion, peptides corresponding to 1 μg protein was injected for LC-MS analysis using data independent acquisition as in Pandya et al.71 Post-digestion, peptides corresponding to 1 μg protein was injected for LC-MS analysis using data independent acquisition.
Peptides were analyzed by nano LC MS/MS using an Easy LC system (Thermo Scientific) coupled to the Orbitrap Fusion Lumos instrument (Thermo Scientific). Peptides were trapped on a 5 mm Pepmap 100 C18 column (300 μm i.d., 5 μm particle size, Dionex) and fractionated on a 500 mm Alltima C18 column (300 μm i.d., 3 μm particle size). The acetonitrile concentration in the mobile phase was increased from 5 to 18% in 88 min, to 25% at 98 min, 40% at 108 min and to 90% in 2 min, at a flow rate of 200 nL/min. The eluted peptides were electro-sprayed into the Orbitrap Fusion Lumos MS. The nano-spray needle voltage was set to 2100 V. For the generation of spectral library, the mass spectrometer was operated in a data-dependent mode with a single MS full scan (m/z 350-1250, 150 msec) followed by a top 32 MS/MS (m/z 200–1800, 150 msec) at high sensitivity mode in UNIT resolution, precursor ion > 150 counts/s, charge state from +2 to +5) with an exclusion time of 16 s once the peptide was fragmented. Ions were fragmented in the collision cell using rolling collision energy, and a spread energy of 5 eV.
Nano-LC and DIA mass spectrometry
The conditions used for LC in DIA MS-based experiments were the same as those of the DDA experiments. DIA experiments consisted of a parent ion scan of 150 msec followed by DIA windows according to the following table and stepped through the mass range between 400–1200 m/z. The total cycle time was about 4.5 s, which yielded in general 5-6 measurement points across a typical peptide with an elution time of 30 s. Raw data was processed on Spectronaut Pulsar using default parameters and peak areas for each peptide was rolled-up to the protein group and gene level by the sum of top N most intense peptides.
Immunohistochemical analysis of human brain for UBE3A and PEG10 using DAB
The AS brain tissue sample was obtained from the Netherlands Brain Bank, Netherlands Institute for Neuroscience, Amsterdam (open access: https://www.brainbank.nl). All material has been collected from donors for or from whom a written informed consent for a brain autopsy and the use of the material and clinical information for research purposes had been obtained by the NBB.
Control brain tissues were obtained from the University of Pennsylvania Cooperative Human Tissue Network (CHTN; https://www.chtneast.org) and from Analytical Biological Services (https://www.absbio.com).
| Sample ID | Diagnosis | Age | Gender | Brain region | Application |
|---|---|---|---|---|---|
| 2006-009 | AS | 43 years | Female | Frontal cortex | IHC |
| H0578 | Unaffected control | 40 years | Female | Frontal cortex | IHC |
| HB39 | Unaffected control | 80 years | Male | Frontal cortex | IHC |
Immunohistochemical staining of 5 μm FFPE sections were performed on a Ventana BenchMark ULTRA system. For deparaffinization, the tissue was baked at 60°C for 8 min before heating 3 times to 69°C for 8 min each cycle.
Primary antibodies (diluted in Antibody Diluent D4 [Ventana, 760-108]): mouse anti-NeuN (Merck, MAB377; 1/200), mouse anti-Ube3a (Merck, E8655; 1/50), rabbit anti-PEG10 RF1/2 (Abcam, ab240392; 1/50), rabbit anti-PEG10 RF1 (Abcam, ab250462; 1/200).
Primary antibody labeling: Tissue sections were incubated at 95°C in Cell Conditioning solution 1 (CC1; Ventana, Cat. No. 950-500) for 36 min (for NeuN, Ube3a, and PEG10 RF1/2) or 12 min (for PEG10 RF1) to retrieve antigens. Blocking was performed by applying serum free Protein Block (Dako, Cat. No. X0909) for 12 min at room temperature (for NeuN) or 20% goat serum (Dako, Cat. No. X0907) for 60 min (for Ube3a) or 20% goat serum for 60 min (for PEG10 RF1) at room temperature followed by Blocker A (Ventana, Cat. No. 253-2030) and Blocker B (Ventana, Cat. No. 253-2031) for 12 min each. No blocking step was included for PEG10 RF1/2 detection. Primary antibodies were incubated for 12 hours at room temperature.
Secondary antibody labeling and detection: For NeuN labeling, secondary antibody Biotin-SP goat anti-mouse IgG (Jackson ImmunoResearch, Cat. No. 115-065-166; 1/100 in D4) was applied for 24 min at room temperature and detection was performed using the DISCOVERY DAB Map Detection Kit (RUO; Ventana, Cat. No. 760-124). DISCOVERY OmniMap anti-Ms HRP detection kit (Ventana, Cat. No. 760-4310) for Ube3a, or anti-Rb HRP (Ventana, 760-4311) for PEG10, was applied for 24 min at room temperature and detection was performed using the DISCOVERY ChromoMap DAB Kit (RUO; Ventana, 760-159). Counterstain of cell nuclei was perfomed by applying Hematoxylin II (Ventana, Cat. No. 790-2208) for 8 min, followed by Bluing reagent (Ventana, Cat. No. 760-2037) for 4 min.
Immunohistochemical analysis of human brain for UBE3A and PEG10 using fluorescence staining
Tissue sections were incubated at 95°C in Cell Conditioning solution 1 (CC1; Ventana, 950-500) for 36 min to retrieve antigens. Blocking was performed by applying 20% goat serum (Dako, X0907) in background-reducing antibody diluent (Dako, S3022) for 20 min at room temperature followed by Blocker A (Ventana, 253-2030) and Blocker B (Ventana, 253-2031) for 12 min each. Primary antibodies were diluted in background-reducing antibody diluent and incubated sequentially for 12 hours at room temperature (for PEG10) and 1 hour at 37°C (for NeuN).
Secondary antibody labeling and detection: DISCOVERY OmniMap anti-Ms and anti-Rb HRP detection kits were used sequentially for PEG10/NeuN co-labeling. Secondary antibodies were applied for 24 min at room temperature and detection was performed using the DISCOVERY FITC Kit (RUO; Ventana, 760-232) for PEG10 and DISCOVERY Cy5 Kit (RUO; Ventana, 760-238) for NeuN. Counterstain of cell nuclei was performed by applying DAPI solution (Roche, 10236276001) for 12 min.
Immunoblotting analysis of human brain samples
Human brain tissue was lysed in modified RIPA buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1mM EDTA, 0.5% NP-40 with protease inhibitor cocktail. The lyates were cleared of any IgGs by pretreatment with protein A/G beads (overnight 4°C). The eluate was boiled in laemmli buffer and prepared for immunoblotting analysis for PEG10 RF1/2 and UBE3A as described previously.
In utero electroporation and immunohistochemistry
For the in utero electroporation experiments female FvB/NHsD were crossed with male C57Bl6/J or 129/Bl6/J in the case of Angelman mice. The procedure was performed in pregnant mice at Day 14.5 of gestation, in order to target mainly the progenitor cells giving rise to pyramidal cells of the layer 2/3 as per Saito82 and Taniguchi et al.83 The DNA constructs (1.5–3 ug/uL) were diluted in fast green (0.05%) and injected in the lateral ventricle of the embryos, while still in uterus, using a glass pipette controlled by a Picospritzer III device. To ensure the proper electroporation of the injected DNA constructs (1–2 uL) into the progenitor cells, five electrical square pulses of 45V with a duration of 50ms per pulse and 150 ms inter-pulse interval were delivered using tweezer-type electrodes connected to a pulse generator (ECM830, BTX Harvard Apparatus). The electrodes were placed in such a way that the positive pole was targeting the developing somatosensory cortex. The pups were sacrificed one day after birth (P1) for histochemical processing.
Immuno histo-chemistry in utero electroporation
Mice were deeply anesthetized with an overdose of Nembutal and transcardially perfused with 4% paraformaldehyde (PFA). Brains were extracted and post-fixed in 4% PFA. Subsequently, the brains were embedded in gelatin and sectioned using a freezing microtome (50μm thick). Free-floating coronal sections were washed in 0.1 M phosphate buffer and selected sections were counterstained with 4′,6-diamidino-2-phenylindole solution (DAPI, 1:10,000, Thermo Fisher Scientific, D3571) before being mounted with Mowiol on glass. In the case of the AS mice and their WT littermates, brain slices first underwent blocking with 10% Normal Horse Serum (NHS) and 0,4% Triton-X in PBS, subsequently washed 3x with PBS and then incubated with primary antibody (mouse anti-UBE3A, Sigma Aldrich, E8655) over night at room temperature while shaking. Following day, slices were rinsed 3x with PBS and then incubated with secondary antibody (Alexa Fluor® 488 AffiniPure Donkey Anti-Mouse IgG (H+L), Jackson ImmunoResearch) for 3h, while shaking at RT. After 3 washes with 0.1 M PB the slices were stained with DAPI as mentioned and mounted on glass with Mowiol. For the migration analysis, confocal images (10 × objective, 0.5zoom, 2048 × 2048 pixels) were taken from 2 to 3 non-consecutive sections from at least three successfully targeted animals per plasmid. Images were rotated to correctly position the cortical layers, and the number of cells in different layers were counted using ImageJ (Analyze particles option). The results were exported to a spreadsheet for further analysis. Cortical areas from the pia to the ventricle were divided in 10 equal-sized bins and the percentage of tdTomato-positive cells per bin was calculated.
Quantification and statistical analysis
RNA-sequencing data analysis
RNaseq reads were quality controlled using the program FastQC (cite @misc, version 0.11.5) and mapped to the human genome (hg38) using the program STAR (version 2.5.2a84). Gene counts were calculated based on the Ensembl transcript models (GRCh38.92) and the analysis, including normalization and differential gene expression, was performed in R.73,85, 86, 87
Data analysis of DIA proteomics
Raw data were annotated and normalized with local normalization in Spectronaut (Biognosys). Peptide quantity was determined as the sum of precursor intensities, and protein quantity as the sum of the top 3 peptide quantities. Differential abundances of proteins were calculated by fitting linear models for each protein with the limma R package,73 and applying an Empirical Bayes method to moderate the variances.74 Different conditions were compared by calculating contrasts with multcomp75 and emmeans76 packages. The computed p values were adjusted for multiple testing by controlling the false discovery rate.77 All calculations were performed in R.78
Statistical analysis of migration assay
For the IUE experiments on neuronal migration, the analysis was performed on the number of targeted cells corresponding to the first fours bins, considered to correspond to the cortical plate of the somatosensory cortex of the P1 mice. Statistical analysis was performed using one-way ANOVA, followed by Bonferroni’s multiple comparison test.
Acknowledgments
We would like to thank Children’s Hospital in Boston, under the leadership of Prof. Christopher Walsh, for recruitment of patients into the study; Harvard iPSC core facility and the team of Laurence Daheron for the reprogramming and quality control of hiPSC lines; Prof. Michael Greenberg for expertise and brainstorming on modeling AS in hiPSC; R. Schmucki and F. Koechl for help with RNA-seq analysis; D. Avilla, P. Jakob, and S. Golling for help with LC-MS data acquisition; the Roche postdoctoral fellowship program for funding N.J.P., C.W., and P.G. and the Roche RiSE internship program for funding S.M.
Author contributions
V.C., N.J.P., Y.M., and P.L. performed the neuronal differentiations. N.J.P. and M.T. planned and performed the proteomics experiments. N.J.P., P.L., and S.M. planned and performed the immunostaining experiments. N.J.P., C.W., and T.D. planned and performed the RNA experiments. P.G. and N.J.P. performed the EV work. M.E., M.B., and B.B. performed the data analysis. A.M.P. performed the bacterial ubiquitination assays. F.I.Z. performed the Y2H and migration assays. E.M. performed the brain analysis. T.D., Y.E., M.H., M.E., B.D., M.T., V.C., C.P., and R.J. supervised the research. N.J.P., B.D., Y.E., and R.J. wrote the manuscript. All of the authors reviewed the manuscript.
Declaration of interests
N.J.P., V.C., C.W., P.L., S.M., P.G., T.D., M.T., Y.M., B.B., C.P., S.R., M.H., M.B., T.K., T.D., M.E., and R.J. are employed by F. Hoffmann-La Roche. Parts of the work in this study have been filed in the patent WO2020/148310. The remaining authors declare no competing financial interests.
Published: August 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100360.
Contributor Information
Nikhil J. Pandya, Email: pandya.nikhil_janak@gene.com.
Ravi Jagasia, Email: ravi.jagasia@roche.com.
Supplemental information
Data and code availability
All RNA-Seq and proteomics data generated in this study are available in SRA (https://www.ncbi.nlm.nih.gov/sra) and Massive (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Matsuura T., Sutcliffe J.S., Fang P., Galjaard R.J., Jiang Y.H., Benton C.S., Rommens J.M., Beaudet A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 2.Fang P., Lev-Lehman E., Tsai T.F., Matsuura T., Benton C.S., Sutcliffe J.S., Christian S.L., Kubota T., Halley D.J., Meijers-Heijboer H. The spectrum of mutations in UBE3A causing Angelman syndrome. Hum. Mol. Genet. 1999;8:129–135. doi: 10.1093/hmg/8.1.129. [DOI] [PubMed] [Google Scholar]
- 3.Cook E.H., Jr., Lindgren V., Leventhal B.L., Courchesne R., Lincoln A., Shulman C., Lord C., Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am. J. Hum. Genet. 1997;60:928–934. [PMC free article] [PubMed] [Google Scholar]
- 4.Clayton-Smith J., Laan L. Angelman syndrome: a review of the clinical and genetic aspects. J. Med. Genet. 2003;40:87–95. doi: 10.1136/jmg.40.2.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buiting K., Williams C., Horsthemke B. Angelman syndrome - insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016;12:584–593. doi: 10.1038/nrneurol.2016.133. [DOI] [PubMed] [Google Scholar]
- 6.Chamberlain S.J., Lalande M. Angelman syndrome, a genomic imprinting disorder of the brain. J. Neurosci. 2010;30:9958–9963. doi: 10.1523/JNEUROSCI.1728-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang H.S., Allen J.A., Mabb A.M., King I.F., Miriyala J., Taylor-Blake B., Sciaky N., Dutton J.W., Jr., Lee H.M., Chen X. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481:185–189. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meng L., Ward A.J., Chun S., Bennett C.F., Beaudet A.L., Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–412. doi: 10.1038/nature13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonzogni M., Wallaard I., Santos S.S., Kingma J., du Mee D., van Woerden G.M., Elgersma Y. A behavioral test battery for mouse models of Angelman syndrome: a powerful tool for testing drugs and novel Ube3a mutants. Mol. Autism. 2018;9:47. doi: 10.1186/s13229-018-0231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rotaru D.C., Mientjes E.J., Elgersma Y. Angelman Syndrome: From Mouse Models to Therapy. Neuroscience. 2020;445:172–189. doi: 10.1016/j.neuroscience.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 11.Chamberlain S.J., Chen P.F., Ng K.Y., Bourgois-Rocha F., Lemtiri-Chlieh F., Levine E.S., Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. Proc. Natl. Acad. Sci. USA. 2010;107:17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fink J.J., Robinson T.M., Germain N.D., Sirois C.L., Bolduc K.A., Ward A.J., Rigo F., Chamberlain S.J., Levine E.S. Disrupted neuronal maturation in Angelman syndrome-derived induced pluripotent stem cells. Nat. Commun. 2017;8:15038. doi: 10.1038/ncomms15038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frohlich J., Miller M.T., Bird L.M., Garces P., Purtell H., Hoener M.C., Philpot B.D., Sidorov M.S., Tan W.H., Hernandez M.C. Electrophysiological phenotype in Angelman syndrome differs between genotypes. Biol. Psychiatry. 2019;85:752–759. doi: 10.1016/j.biopsych.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang H.S., Burns A.J., Nonneman R.J., Baker L.K., Riddick N.V., Nikolova V.D., Riday T.T., Yashiro K., Philpot B.D., Moy S.S. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav. Brain Res. 2013;243:79–90. doi: 10.1016/j.bbr.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pandya N.J., Avila D., Dunkley T., Jagasia R., Tzouros M. In: Li K.W., editor. Springer; 2019. TMT-MS3-Enabled proteomic quantification of human IPSC-Derived neurons; pp. 103–117. (Neuroproteomics). [Google Scholar]
- 16.Oughtred R., Stark C., Breitkreutz B.J., Rust J., Boucher L., Chang C., Kolas N., O’Donnell L., Leung G., McAdam R. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019;47(D1):D529–D541. doi: 10.1093/nar/gky1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunkley T., Costa V., Friedlein A., Lugert S., Aigner S., Ebeling M., Miller M.T., Patsch C., Piraino P., Cutler P., Jagasia R. Characterization of a human pluripotent stem cell-derived model of neuronal development using multiplexed targeted proteomics. Proteomics Clin. Appl. 2015;9:684–694. doi: 10.1002/prca.201400150. [DOI] [PubMed] [Google Scholar]
- 18.Costa V., Aigner S., Vukcevic M., Sauter E., Behr K., Ebeling M., Dunkley T., Friedlein A., Zoffmann S., Meyer C.A. MTORC1 inhibition corrects neurodevelopmental and synaptic alterations in a human stem cell model of tuberous sclerosis. Cell Rep. 2016;15:86–95. doi: 10.1016/j.celrep.2016.02.090. [DOI] [PubMed] [Google Scholar]
- 19.Kruse R., Krantz J., Barker N., Coletta R.L., Rafikov R., Luo M., Højlund K., Mandarino L.J., Langlais P.R. Characterization of the CLASP2 protein interaction network identifies SOGA1 as a microtubule-associated protein. Mol. Cell. Proteomics. 2017;16:1718–1735. doi: 10.1074/mcp.RA117.000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burette A.C., Judson M.C., Burette S., Phend K.D., Philpot B.D., Weinberg R.J. Subcellular organization of UBE3A in neurons. J. Comp. Neurol. 2017;525:233–251. doi: 10.1002/cne.24063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su H., Fan W., Coskun P.E., Vesa J., Gold J.A., Jiang Y.H., Potluri P., Procaccio V., Acab A., Weiss J.H. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci. Lett. 2011;487:129–133. doi: 10.1016/j.neulet.2009.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santini E., Turner K.L., Ramaraj A.B., Murphy M.P., Klann E., Kaphzan H. Mitochondrial superoxide contributes to hippocampal synaptic dysfunction and memory deficits in Angelman syndrome model mice. J. Neurosci. 2015;35:16213–16220. doi: 10.1523/JNEUROSCI.2246-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llewellyn K.J., Nalbandian A., Gomez A., Wei D., Walker N., Kimonis V.E. Administration of CoQ10 analogue ameliorates dysfunction of the mitochondrial respiratory chain in a mouse model of Angelman syndrome. Neurobiol. Dis. 2015;76:77–86. doi: 10.1016/j.nbd.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Avagliano Trezza R., Sonzogni M., Bossuyt S.N.V., Zampeta F.I., Punt A.M., van den Berg M., Rotaru D.C., Koene L.M.C., Munshi S.T., Stedehouder J. Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat. Neurosci. 2019;22:1235–1247. doi: 10.1038/s41593-019-0425-0. [DOI] [PubMed] [Google Scholar]
- 25.Zampeta F.I., Sonzogni M., Niggl E., Lendemeijer B., Smeenk H., de Vrij F.M.S., Kushner S.A., Distel B., Elgersma Y. Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms. Hum. Mol. Genet. 2020;29:3032–3043. doi: 10.1093/hmg/ddaa194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobson A.D., MacFadden A., Wu Z., Peng J., Liu C.W. Autoregulation of the 26S proteasome by in situ ubiquitination. Mol. Biol. Cell. 2014;25:1824–1835. doi: 10.1091/mbc.E13-10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Utine G.E., Taşkıran E.Z., Koşukcu C., Karaosmanoğlu B., Güleray N., Doğan Ö.A., Kiper P.Ö., Boduroğlu K., Alikaşifoğlu M. HERC1 mutations in idiopathic intellectual disability. Eur. J. Med. Genet. 2017;60:279–283. doi: 10.1016/j.ejmg.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 28.Morice-Picard F., Benard G., Rezvani H.R., Lasseaux E., Simon D., Moutton S., Rooryck C., Lacombe D., Baumann C., Arveiler B. Complete loss of function of the ubiquitin ligase HERC2 causes a severe neurodevelopmental phenotype. Eur. J. Hum. Genet. 2016;25:52–58. doi: 10.1038/ejhg.2016.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harlalka G.V., Baple E.L., Cross H., Kühnle S., Cubillos-Rojas M., Matentzoglu K., Patton M.A., Wagner K., Coblentz R., Ford D.L. Mutation of HERC2 causes developmental delay with Angelman-like features. J. Med. Genet. 2013;50:65–73. doi: 10.1136/jmedgenet-2012-101367. [DOI] [PubMed] [Google Scholar]
- 30.Martínez-Noël G., Galligan J.T., Sowa M.E., Arndt V., Overton T.M., Harper J.W., Howley P.M. Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol. Cell. Biol. 2012;32:3095–3106. doi: 10.1128/MCB.00201-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kühnle S., Kogel U., Glockzin S., Marquardt A., Ciechanover A., Matentzoglu K., Scheffner M. Physical and functional interaction of the HECT ubiquitin-protein ligases E6AP and HERC2. J. Biol. Chem. 2011;286:19410–19416. doi: 10.1074/jbc.M110.205211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horie M., Watanabe K., Bepari A.K., Nashimoto J., Araki K., Sano H., Chiken S., Nambu A., Ono K., Ikenaka K. Disruption of actin-binding domain-containing Dystonin protein causes dystonia musculorum in mice. Eur. J. Neurosci. 2014;40:3458–3471. doi: 10.1111/ejn.12711. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y., Kaneko N., Asai N., Enomoto A., Isotani-Sakakibara M., Kato T., Asai M., Murakumo Y., Ota H., Hikita T. Girdin is an intrinsic regulator of neuroblast chain migration in the rostral migratory stream of the postnatal brain. J. Neurosci. 2011;31:8109–8122. doi: 10.1523/JNEUROSCI.1130-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker J.D., Shelton L.B., Zheng D., Favretto F., Nordhues B.A., Darling A., Sullivan L.E., Sun Z., Solanki P.K., Martin M.D. Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 2017;15:e2001336. doi: 10.1371/journal.pbio.2001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorenzi M.V., Castagnino P., Chen Q., Hori Y., Miki T. Distinct expression patterns and transforming properties of multiple isoforms of Ost, an exchange factor for RhoA and Cdc42. Oncogene. 1999;18:4742–4755. doi: 10.1038/sj.onc.1202851. [DOI] [PubMed] [Google Scholar]
- 36.Abed M., Verschueren E., Budayeva H., Liu P., Kirkpatrick D.S., Reja R., Kummerfeld S.K., Webster J.D., Gierke S., Reichelt M. The Gag protein PEG10 binds to RNA and regulates trophoblast stem cell lineage specification. PLoS ONE. 2019;14:e0214110. doi: 10.1371/journal.pone.0214110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campillos M., Doerks T., Shah P.K., Bork P. Computational characterization of multiple Gag-like human proteins. Trends Genet. 2006;22:585–589. doi: 10.1016/j.tig.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Clark M.B., Jänicke M., Gottesbühren U., Kleffmann T., Legge M., Poole E.S., Tate W.P. Mammalian gene PEG10 expresses two reading frames by high efficiency -1 frameshifting in embryonic-associated tissues. J. Biol. Chem. 2007;282:37359–37369. doi: 10.1074/jbc.M705676200. [DOI] [PubMed] [Google Scholar]
- 39.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 40.Huttlin E.L., Bruckner R.J., Paulo J.A., Cannon J.R., Ting L., Baltier K., Colby G., Gebreab F., Gygi M.P., Parzen H. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017;545:505–509. doi: 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avagliano Trezza R., Punt A.M., Mientjes E., van den Berg M., Zampeta F.I., de Graaf I.J., van der Weegen Y., Demmers J.A.A., Elgersma Y., Distel B. Mono-ubiquitination of Rabphilin 3A by UBE3A serves a non-degradative function. Sci. Rep. 2021;11:3007. doi: 10.1038/s41598-021-82319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaaroor-Regev D., de Bie P., Scheffner M., Noy T., Shemer R., Heled M., Stein I., Pikarsky E., Ciechanover A. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:6788–6793. doi: 10.1073/pnas.1003108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skogberg G., Gudmundsdottir J., van der Post S., Sandström K., Bruhn S., Benson M., Mincheva-Nilsson L., Baranov V., Telemo E., Ekwall O. Characterization of human thymic exosomes. PLoS ONE. 2013;8:e67554. doi: 10.1371/journal.pone.0067554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hurwitz S.N., Rider M.A., Bundy J.L., Liu X., Singh R.K., Meckes D.G., Jr. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget. 2016;7:86999–87015. doi: 10.18632/oncotarget.13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nonhoff U., Ralser M., Welzel F., Piccini I., Balzereit D., Yaspo M.L., Lehrach H., Krobitsch S. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol. Biol. Cell. 2007;18:1385–1396. doi: 10.1091/mbc.E06-12-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paul S., Dansithong W., Figueroa K.P., Scoles D.R., Pulst S.M. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 2018;9:3648. doi: 10.1038/s41467-018-06041-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Youn J.Y., Dunham W.H., Hong S.J., Knight J.D.R., Bashkurov M., Chen G.I., Bagci H., Rathod B., MacLeod G., Eng S.W.M. High-density proximity mapping reveals the subcellular organization of mRNA-associated granules and bodies. Mol. Cell. 2018;69:517–532.e11. doi: 10.1016/j.molcel.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 48.Cinti A., Le Sage V., Ghanem M., Mouland A.J. HIV-1 gag blocks selenite-induced stress granule assembly by altering the mRNA cap-binding complex. MBio. 2016;7:e00329. doi: 10.1128/mBio.00329-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reineke L.C., Lloyd R.E. Diversion of stress granules and P-bodies during viral infection. Virology. 2013;436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wheeler J.R., Matheny T., Jain S., Abrisch R., Parker R. Distinct stages in stress granule assembly and disassembly. eLife. 2016;5:e18413. doi: 10.7554/eLife.18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y.B., Tewari A., Salameh J., Arystarkhova E., Hampton T.G., Brashear A., Ozelius L.J., Khodakhah K., Sweadner K.J. A dystonia-like movement disorder with brain and spinal neuronal defects is caused by mutation of the mouse laminin β1 subunit, Lamb1. eLife. 2015;4:e11102. doi: 10.7554/eLife.11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmed M., Marziali L.N., Arenas E., Feltri M.L., Ffrench-Constant C. Laminin α2 controls mouse and human stem cell behaviour during midbrain dopaminergic neuron development. Development. 2019;146:dev172668. doi: 10.1242/dev.172668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen L., Chu C., Kong X., Huang T., Cai Y.D. Discovery of new candidate genes related to brain development using protein interaction information. PLoS ONE. 2015;10:e0118003. doi: 10.1371/journal.pone.0118003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saunders A., Macosko E.Z., Wysoker A., Goldman M., Krienen F.M., de Rivera H., Bien E., Baum M., Bortolin L., Wang S. Molecular diversity and specializations among the cells of the adult mouse brain. Cell. 2018;174:1015–1030.e16. doi: 10.1016/j.cell.2018.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jäkel S., Agirre E., Mendanha Falcão A., van Bruggen D., Lee K.W., Knuesel I., Malhotra D., Ffrench-Constant C., Williams A., Castelo-Branco G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature. 2019;566:543–547. doi: 10.1038/s41586-019-0903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y., Sloan S.A., Clarke L.E., Caneda C., Plaza C.A., Blumenthal P.D., Vogel H., Steinberg G.K., Edwards M.S.B., Li G. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y., Chen K., Sloan S.A., Bennett M.L., Scholze A.R., O’Keeffe S., Phatnani H.P., Guarnieri P., Caneda C., Ruderisch N. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller J.A., Ding S.L., Sunkin S.M., Smith K.A., Ng L., Szafer A., Ebbert A., Riley Z.L., Royall J.J., Aiona K. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. doi: 10.1038/nature13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Küry S., van Woerden G.M., Besnard T., Proietti Onori M., Latypova X., Towne M.C., Cho M.T., Prescott T.E., Ploeg M.A., Sanders S., Undiagnosed Diseases Network. GEM HUGO. Deciphering Developmental Disorders Study De novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. Am. J. Hum. Genet. 2017;101:768–788. doi: 10.1016/j.ajhg.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yi J.J., Paranjape S.R., Walker M.P., Choudhury R., Wolter J.M., Fragola G., Emanuele M.J., Major M.B., Zylka M.J. The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J. Biol. Chem. 2017;292:12503–12515. doi: 10.1074/jbc.M117.788448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaneko-Ishino T., Ishino F. The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front. Microbiol. 2012;3:262. doi: 10.3389/fmicb.2012.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ono R., Nakamura K., Inoue K., Naruse M., Usami T., Wakisaka-Saito N., Hino T., Suzuki-Migishima R., Ogonuki N., Miki H. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat. Genet. 2006;38:101–106. doi: 10.1038/ng1699. [DOI] [PubMed] [Google Scholar]
- 63.Golda M., Mótyán J.A., Mahdi M., Tőzsér J. Functional study of the retrotransposon-derived human PEG10 protease. Int. J. Mol. Sci. 2020;21:E2424. doi: 10.3390/ijms21072424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whiteley A.M., Prado M.A., de Poot S.A.H., Paulo J.A., Ashton M., Dominguez S., Weber M., Ngu H., Szpyt J., Jedrychowski M.P. Global proteomics of Ubqln2-based murine models of ALS. J. Biol. Chem. 2021;296:100153. doi: 10.1074/jbc.RA120.015960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blokhuis A.M., Koppers M., Groen E.J.N., van den Heuvel D.M.A., Dini Modigliani S., Anink J.J., Fumoto K., van Diggelen F., Snelting A., Sodaar P. Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta Neuropathol. 2016;132:175–196. doi: 10.1007/s00401-016-1575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pastuzyn E.D., Day C.E., Kearns R.B., Kyrke-Smith M., Taibi A.V., McCormick J., Yoder N., Belnap D.M., Erlendsson S., Morado D.R. The neuronal gene arc encodes a repurposed retrotransposon gag protein that mediates intercellular RNA transfer. Cell. 2018;172:275–288.e18. doi: 10.1016/j.cell.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ashley J., Cordy B., Lucia D., Fradkin L.G., Budnik V., Thomson T. Retrovirus-like gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell. 2018;172:262–274.e11. doi: 10.1016/j.cell.2017.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kühnle S., Mothes B., Matentzoglu K., Scheffner M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc. Natl. Acad. Sci. USA. 2013;110:8888–8893. doi: 10.1073/pnas.1302792110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koopmans F., Pandya N.J., Franke S.K., Phillippens I.H.C.M.H., Paliukhovich I., Li K.W., Smit A.B. Comparative hippocampal synaptic proteomes of rodents and primates: differences in neuroplasticity-related proteins. Front. Mol. Neurosci. 2018;11:364. doi: 10.3389/fnmol.2018.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pandya N.J., Seeger C., Babai N., Gonzalez-Lozano M.A., Mack V., Lodder J.C., Gouwenberg Y., Mansvelder H.D., Danielson U.H., Li K.W. Noelin1 affects lateral mobility of synaptic AMPA receptors. Cell Rep. 2018;24:1218–1230. doi: 10.1016/j.celrep.2018.06.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Plubell D.L., Wilmarth P.A., Zhao Y., Fenton A.M., Minnier J., Reddy A.P., Klimek J., Yang X., David L.L., Pamir N. Extended multiplexing of tandem mass tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell. Proteomics. 2017;16:873–890. doi: 10.1074/mcp.M116.065524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phipson B., Lee S., Majewski I.J., Alexander W.S., Smyth G.K. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat. 2016;10:946–963. doi: 10.1214/16-AOAS920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hothorn T., Bretz F., Westfall P. Simultaneous inference in general parametric models. Biom. J. 2008;50:346–363. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 76.Lenth R., Singmann H., Love J., Buerkner P., Herve M. 2019. Estimated Marginal Means, aka Least-Squares Means.https://cran.r-project.org/web/packages/emmeans/emmeans.pdf [Google Scholar]
- 77.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 78.R Development Core Team . R Foundation for Statistical Computing; 2019. R: A language and environment for statistical computing. [Google Scholar]
- 79.Gkourtsa A., van den Burg J., Avula T., Hochstenbach F., Distel B. Binding of a proline-independent hydrophobic motif by the Candida albicans Rvs167-3 SH3 domain. Microbiol. Res. 2016;190:27–36. doi: 10.1016/j.micres.2016.04.018. [DOI] [PubMed] [Google Scholar]
- 80.Nelson J.K., Sorrentino V., Avagliano Trezza R., Heride C., Urbe S., Distel B., Zelcer N. The deubiquitylase USP2 regulates the ldlr pathway by counteracting the E3-ubiquitin Ligase IDOL. Circ. Res. 2016;118:410–419. doi: 10.1161/CIRCRESAHA.115.307298. [DOI] [PubMed] [Google Scholar]
- 81.Witwer K.W., Buzás E.I., Bemis L.T., Bora A., Lässer C., Lötvall J., Nolte-’t Hoen E.N., Piper M.G., Sivaraman S., Skog J. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles. 2013;2 doi: 10.3402/jev.v2i0.20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat. Protoc. 2006;1:1552–1558. doi: 10.1038/nprot.2006.276. [DOI] [PubMed] [Google Scholar]
- 83.Taniguchi Y., Young-Pearse T., Sawa A., Kamiya A. In utero electroporation as a tool for genetic manipulation in vivo to study psychiatric disorders: from genes to circuits and behaviors. Neuroscientist. 2012;18:169–179. doi: 10.1177/1073858411399925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-Seq and proteomics data generated in this study are available in SRA (https://www.ncbi.nlm.nih.gov/sra) and Massive (https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.