

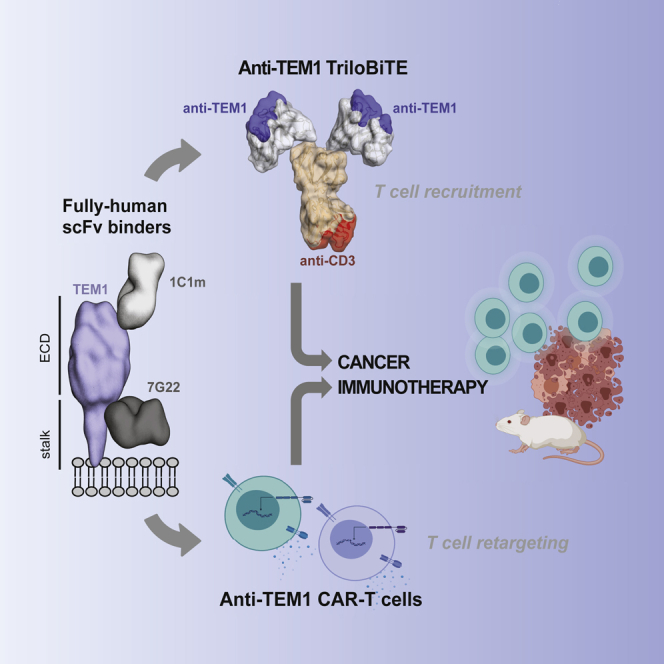
Summary
Tumor endothelial marker 1 (TEM1) is an emerging cancer target with a unique dual expression profile. First, TEM1 is expressed in the stroma and neo-vasculature of many human carcinomas but is largely absent from healthy adult tissues. Second, TEM1 is expressed by tumor cells of mesenchymal origin, notably sarcoma. Here, we present two fully human anti-TEM1 single-chain variable fragment (scFv) reagents, namely, 1C1m and 7G22, that recognize distinct regions of the extracellular domain and possess substantially different affinities. In contrast to other, well-described anti-TEM1 binders, these fragments confer cytolytic activity when expressed as 2nd generation chimeric antigen receptors (CARs). Moreover, both molecules selectively redirect human T cell effector functions toward TEM1+ tumor cells when incorporated into experimental soluble bispecific trivalent engagers that we term TriloBiTEs (tBs). Furthermore, systemic delivery of 1C1m-tB prevents the establishment of Ewing sarcoma tumors in a xenograft model. Our observations confirm TEM1 as a promising target for cancer immunotherapy and illustrate the prospective translational potential of certain scFv-based reagents.
Keywords: TEM1, endosialin/CD248, cancer immunotherapy, T cell engager, sarcoma, CAR, BiTE
Graphical abstract
Highlights
Discovery and characterization of two fully human scFv binders targeting human TEM1
Specific killing of TEM1+ cancer cell lines
Demonstration of a versatile soluble heterodimeric trivalent T cell engager format
Fierle et al. describe the targeting of the human TEM1 (CD248/endosialin) tumor marker by redirecting the lytic activity of T cells. This study indicates that both cell-based CAR and soluble engager paradigms may have utility in this regard and suggests a potential immunotherapeutic approach for the treatment of soft-tissue sarcomas.
Introduction
Over the last two decades, the options for treating cancer have been revolutionized by the rapidly expanding field of human immunotherapy. Natural and engineered cytotoxic T lymphocytes represent a potentially powerful weapon in the arsenal of current anti-cancer therapies. Strategies aimed at efficiently mobilizing patient-autologous effector T cells have achieved significant responses in several clinical settings.1, 2, 3, 4, 5 Beyond canonical peptide-major histocompatibility complex (MHC) restriction, recent approaches have exploited the redirection of bulk T cells toward diverse cell surface tumor antigens recognized by antibodies or other synthetic affinity reagents. The promise of such immunotherapeutic strategies in overcoming the limitations of the natural anti-tumor immune response has led to the development of two dominant paradigms: (1) the adoptive transfer of T cells engineered with a tumor-targeting chimeric antigen receptor (CAR); and (2) the non-selective recruitment and “engagement” of native T cells by systemically delivered, soluble bispecific “bridging” proteins. Both strategies seek to activate a polyclonal population of T cells in the absence of natural cognate T cell receptor (TCR)-MHC signaling.6, 7, 8
CARs are synthetic immune receptors that combine an extracellular ligand-binding domain, typically a single-chain variable fragment (scFv), with an intracellular signaling module containing immunoreceptor tyrosine-based activation motifs (ITAMs), commonly those found in CD3ζ, together with additional co-stimulatory endodomains.9,10 Several pioneering studies have revealed the clinical potential of CAR-T cells directed against CD19 expressed on (malignant) B cells, resulting in the approval of CD19-CARs by the US and European regulatory authorities.1, 2, 3
T-cell-engaging bispecifics represent an attractive off-the-shelf alternative to CAR-T cell therapy. Such molecules generally comprise a tumor-targeting moiety fused to an anti-CD3 recognition domain for the engagement of T cells.11,12 In this way, T-cell-engaging bispecifics bridge bystander T cells to tumor cells, facilitating the creation of an immunological synapse and triggering the effector-driven lysis of target cells.13 The most advanced molecules of this class are the so-called bispecific T cell engagers (BiTEs), which are based on two distinct scFvs separated by a flexible linker.11,12 Validating the concept of T cell retargeting with a bispecific mediator, the CD19xCD3 BiTE blinatumomab (Blincyto) has been approved by the US Food and Drug Administration (USFDA) for the treatment of severe relapsed B cell malignancies following impressive results in clinical trials.4,5,14 In addition, a plethora of potential targets and molecular formats are currently in development, covering a wide range of indications and conferring different binding geometries and molecular properties.15
Because both strategies unleash the considerable cytotoxic potential of T cells, therapy with CAR-T cells and BiTEs can be accompanied by severe adverse events, notably cytokine release syndrome (CRS) and neurotoxicities.11,16 Such outcomes are at least in part dependent on the chosen tumor-associated antigen since many antigens are also expressed on healthy cells, albeit often to a lesser extent. This is especially the case for many epithelial tumor targets, such as Her2, EpCAM, and CEACAM5, and such “on-target off-tumor” activity can represent a major obstacle to the treatment of solid tumors.17,18 There exists, therefore, substantial interest in identifying additional tumor-associated or tumor-specific antigens (TAAs) that may facilitate the improved selective targeting of tumor tissue.
Tumor endothelial marker 1 (TEM1; endosialin, CD248) is a cell surface antigen shown to be conserved between mice and humans that is frequently expressed in the stroma and neo-vasculature of many solid tumors.19, 20, 21, 22 In this context, TEM1 has been shown to localize to several cell types including pericytes,19 endothelial precursor cells,23 tumor stroma fibroblasts, and smooth muscle cells.24 Furthermore, TEM1 is expressed directly on tumor cells in a large proportion of high-grade human sarcomas25. Although its molecular function remains elusive, TEM1 is implicated in vascular cell adhesion and migration, angiogenesis, and tumor progression.26,27 Outside of its tumor association, the TEM1 protein has been shown to be absent from most mature adult healthy tissues, with significantly restricted expression profiles when compared to several current tumor therapeutic targets such as Her2, EGFR, and VEGF (Human Protein Atlas, https://www.proteinatlas.org/). Safety and validation studies have revealed that homozygous knockout (KO) mice appear fully viable and display apparently normal wound healing and reproductive function.27 Interestingly however, such mice show impaired tumor growth and invasiveness, suggesting that TEM1 has pro-tumorigenic activity. Furthermore, a vaccination study using a mouse TEM1-TetTox cDNA fusion vaccine was shown to break self-tolerance to TEM1, inducing a strong cellular CD3+ T cell response that led to the control of TEM1+ tumors without the induction of visible auto-pathology or impacts on wound healing and reproduction.28 These and other in vivo validation data have, in recent years, stimulated significant interest in exploiting TEM1 as a tumor target.19,20,22,29 For example, ontuxizumab (MORAb-004), a humanized monoclonal antibody, has shown favorable safety characteristics in discrete phase II clinical trials.30, 31, 32, 33, 34 However, no objective efficacy with regard to progression-free survival was observed despite the molecule being formatted as an ADCC-competent immunoglobulin G1 (IgG1) isotype.30, 31, 32, 33, 34 More recently, pre-clinical proof-of-concept studies have demonstrated the potential utility of fully human scFv-based radio- and NR-conjugates for theranostic TEM1+ tumor targeting.35, 36, 37, 38 Additionally, anti-TEM1 moieties conjugated to both small molecule and proteinaceous cytotoxins have also been described for the treatment of human sarcomas.29,39,40 However, therapeutic interventions that redirect and/or recruit T cells toward TEM1+ tumors have, to the best of our knowledge, not been explored. Such an approach is compelling, as TEM1-activated T cells offer the potential to disrupt global tumor architecture by targeting tumor islets, stromal compartments, and neo-vasculature and by driving epitope spreading and tumor regression28.
Here, we report that anti-TEM1 recognition by fully human scFvs can be leveraged for effective T cell redirection. Surprisingly, we observe that not all existing anti-TEM1 scFvs function in this regard, with activity appearing highly dependent on the discrete properties of the scFv molecule. Additionally, we evaluate a prototype soluble engager with a stabilized heterodimeric tri-valent structure that may offer advantages over classical BiTE and bispecific antibody formats.
Results
Previously described anti-TEM1 binders show functional impairment in a CAR format
Initially, our investigations centered on whether an anti-TEM1 scFv could show functionality in a second-generation CAR format. To this end, the previously described anti-TEM1 scFv sc78 and a scFv derived from MORAb-004 (ontuxizumab) were incorporated into a modular second-generation CAR construct comprising the spacer, TM, and cytosolic domains of CD28 fused to CD3ζ ITAM signaling elements and an in-frame monomeric GFP reporter.37,41
Surprisingly, neither sc78 nor MORAb-004 scFv showed activity in this generic CAR context. Specifically, HEK293-6E cells engineered to express these CAR constructs did not recognize the recombinant cognate TEM1 target antigen (Figure S1A). Furthermore, the sc78-CAR did not induce ITAM-driven NFAT signaling nor specific T cell activation or killing in the presence of TEM1+ target cells (Figures S1B–S1D). In the absence of a validated benchmark CAR clone, TEM1 therefore lacks a proof-of-concept for the therapeutic paradigm of T cell retargeting.
Identification and characterization of new scFvs recognizing distinct TEM1 extracellular epitopes
To explore the generality of these unexpected observations, we therefore decided to isolate, de novo, additional fully human anti-TEM1 scFvs. MORAb-004 and sc78, respectively, arose from rodents inoculated with TEM1+ human fibroblasts and from the recombinant hTEM1 extracellular domain (ECD) expressed and purified from Escherichia coli. In contrast, we expressed a truncated secreted ECD fragment of hTEM1 from a human host cell line to ensure correct folding and the presence of native post-translational modifications.42 From the resulting phagedisplay-derived scFv panel, one clone, namely, 1C1m (previously described as HS06mut)42, was successfully affinity matured, resulting in a high-affinity human/murine cross-reactive binder with a monovalent affinity of 1 nM toward hTEM1 and 6 nM toward mTEM1 (Figure S2A). Although the epitope of 1C1m resides in the relatively membrane-distal, tertiary-ordered region of the hTEM1 ECD,42 another promising binder, 7G22, was raised against the isolated membrane-proximal sialomucin stalk domain.43 This clone is specific for hTEM1 and has a modest monovalent binding affinity of approximately 360 nM (Figure S2B). Several studies have shown that targeting a more membrane-proximal epitope and thus decreasing the bridging distance between T cell and target cell can improve T cell activation both by CARs and soluble engager molecules.44, 45, 46, 47, 48 Thus, we decided to investigate clone 7G22 alongside the better characterized candidate 1C1m.
To assess the selectivity of these binders, the clones were reformatted as scFv-Fc fusions and tested for binding to a large human proteome array. Clone 1C1m appeared to be highly selective for TEM1, recognizing no other predicted cell surface protein and only a handful of other intracellular/nuclear proteins (Figures 1A and S3A). Although the binding profile of the membrane-proximal binder 7G22 was less restricted, TEM1 was still among the 30 strongest hits and few other membrane proteins were recognized (Figures 1B and S3B).
Figure 1.

Two fully human scFv-Fc clones with high selectivity for human TEM1
(A and B) Protein array probing the binding of the scFv-Fc clones 1C1m (A) and 7G22 (B) to 23,100 expressed human open reading frames (ORFs; degeneracy on the array results in ~16,000 unique proteins). Top panels represent the top 1,000 binding signals, and bottom panels show the strongest hits for each clone. Note that full-length TEM1 is spotted in triplicate on the array. Corrected protein-specific fluorescence signals (Prot s-c), obtained by subtracting the average signal from the corresponding protein on the negative control array, are plotted. See Figure S3 for the corresponding Z and i scores from this analysis.
(C) Whole-mount staining of the developing murine retina (P5). 1C1m staining (white) co-localizes with a subset of pericytes (stained against NG2; red).
(D) Cryo-sections of Lewis lung carcinoma (left) and MC38 colon carcinoma (right) xenografts were stained with 1C1m-Fc (red) or anti-NG2 (white). Endothelial cells are stained in green (pecam), and nuclei are visualized with DAPI. Scale bars in (C) and (D) represent 50 μm.
(E) Binding of scFv-Fc clones (1 μg/ml) to endogenous TEM1-expressing A673 cells (left) or a genetically engineered A673 TEM1KO line. Of note, this line is polyclonal and contains a proportion of residual TEM1+ cells. Representative histograms of n = 2 experiments shown in (E). See also Figures S2 and S3.
We next sought to further evaluate the specificity of 1C1m by staining TEM1-expressing cells in tissues with prominent neo-vascularization. Thus, the developing murine retina was stained using 1C1m-Fc as the detection antibody. In agreement with previous studies that reported TEM1 expression on a subset of pericytes in developing tissues,19,49 the staining signal originating from 1C1m-Fc was seen to co-localize to neonatal pericytes, which also expressed the pericyte marker NG2 (Figure 1C). In the tumor setting, multiple cell types have been shown to express TEM1, such as pericytes and fibroblasts.19, 20, 21,49,50 In line with this more diffuse expression pattern, 1C1m-Fc predominantly stained perivascular cells and also stromal cells in sections of Lewis lung carcinoma and MC38 colon carcinoma tissue (Figure 1D). As was the case in the developing retina, the 1C1m staining signal co-localized with the pericyte marker NG2.
Finally, we evaluated the specificity of 1C1m-Fc and 7G22-Fc using A673 Ewing sarcoma cells depleted for endogenous TEM1 through CRISPR-Cas9 genetic deletion. The resulting cell pool was maintained as a polyclonal population with approximately 90% of cells edited for CD248/TEM1, as determined from gene sequencing (data not shown). Comparative cell binding data for wild-type (WT) versus the KO cell pool further confirmed the TEM1 specificity of these two anti-TEM1 clones (Figure 1E). In contrast, the previously described sc78-Fc showed substantial binding to the TEM1-depleted cell pool. Additionally, we observed that the 1C1m-Fc staining ratio for A673 WT versus the TEM1-depleted KO line is indistinct from that of the clinically investigated MORAb-004 (Figure S2F). Our data would therefore indicate that the 1C1m and 7G22 scFvs are suitable reagents for the selective recognition of native cell surface TEM1.
CAR-T cells equipped with anti-TEM1 scFv clones specifically redirect human T cells to cognate target cells
To assess the potential of 1C1m and 7G22 as CARs, we incorporated these scFvs into the same CAR cassette described above and transduced them into human T cells. The transduction efficiency was typically between 60% and 80% for all CARs (data not shown). The resulting 1C1m-CAR-T cells clearly recognized the recombinant soluble antigen, in contrast to the previously tested sc78-CAR (Figure 2A).
Figure 2.

CAR-T cells equipped with anti-TEM1 scFv domains specifically recognize TEM1+ target cells
(A) CAR expression and antigen binding by primary human T cells transduced with 2nd generation anti-TEM1-CAR constructs. Binding of cognate biotinylated recombinant antigen (sc78: mTEM1(ECD); 1C1m: hTEM1(ECD); 7G22: TEM1(Δn)-SpyC-bioSpyT) is shown.
(B) Jurkat NFAT-reporter cells transduced with anti-TEM1 CARs express mCherry in response to stimulation with cognate target cells A673 (TEM1+) and Raji (CD19+) for the anti-CD19 CAR FMC63, which served as a positive control. Irrelevant HEK293-6E cells served as a negative control.
(C) Expression of early activation markers CD69 and CD25 by anti-TEM1 CAR-T cells after 16 h of co-culture with TEM1+ A673 cells or TEM1− AsPC-1 cells. Anti-CD19 CAR-T cells (FMC63) served as a positive control and were co-cultured with CD19+ Raji cells.
(D) Real-time kinetics of target cell killing (TEM1+ A673 cells) by anti-TEM1-CAR-T cells over the course of 30 h (E:T ratio 5:1).
(E) Cytokine production by anti-TEM1-CAR-T cells after 24 h of co-culture with TEM1+ target cells (E:T ratio 5:1) was measured by cytometric bead array. Cytokine concentrations in the culture media were determined from a standard curve. Data are represented as mean ± SD. Representative results of n = 2 independent experiments are shown. See also Figure S4.
Although 7G22-CAR-T cells showed only weak binding to the soluble recombinant sialomucin stalk domain of TEM1, both 1C1m- and 7G22-CARs induced NFAT signaling in fluorescent Jurkat reporter cells when stimulated with TEM1+ target cells (Figure 2B). Similarly, both anti-TEM1 CARs activated primary human T cells in the presence of cognate target cells, leading to the specific lysis of TEM1+ tumor cells (Figures 2C and 2D). Importantly, TEM1-negative control cells were spared in both cases. In line with our previous observations, the sc78-CAR did not activate T cells nor did it mediate specific cytotoxicity. In addition, 1C1m-CAR-T cells and, to a lesser extent, 7G22-CAR-T cells secreted Th1 effector cytokines interferon gamma (IFN-γ), interleukin-2 (IL-2), and transforming growth factor α (TNF-α) when co-cultured with TEM1+ target cells (Figure 2E).
Collectively, these data are consistent with the selective immunotherapeutic targeting of TEM1 by CAR-T cells. However, although 1C1m- and 7G22-CARs clearly mediated target-cell-specific cytotoxicity, the amplitudes of the respective T cell signaling and cytokine productivity responses appeared modest compared with those observed for the archetypal anti-CD19 CAR reference (Figures 2B, 2C, and S4). For instance, the level of IFN-γ secreted in response to cognate target cells was greater than 10-fold reduced in 1C1m TEM1-CAR T cells when compared alongside the FMC63 anti-CD19 control CAR (Figure S4C). A possible explanation for this finding is that the common CD28 extra-membrane spacer used in this construct results in a sub-optimal CAR bridging geometry for 1C1m. To investigate this idea further, we constructed a panel of 1C1m CARs with 12 distinct spacer moieties of varying lengths and complexities (ranging from 13 amino acid [aa]/1.3 kDa to 246 aa/27.6 kDa). However, none of these linker variants conferred a significant improvement in either NFAT activation or effector cytokine production when compared alongside the CD28-derived spacer (linker f; Figures S4A and S4C).
A soluble heterodimeric anti-TEM1 engager format specifically redirects T cell effector functions
We next asked whether uncoupling the anti-TEM1 targeting moiety from the T cell surface in the form of a soluble T cell engager could improve the apparent efficacy of 1C1m and 7G22, overcoming any possible mechanistic constraints of the CAR format. We thus devised a T cell engager, intermediate in size between a classical BiTE and a bispecific IgG, that would allow bivalent avidity-driven target binding. Adapting a previously reported scaffold based around the (murine) Fab domain, we constructed a human Fab constant domain CH1/Cκ dimerization core that we further stabilized by introducing buried interface mutations.51,52 To this we fused the variable domains of a humanized anti-hCD3 antibody (UCHT1) to generate a monomeric anti-CD3 Fab targeting moiety. Flexible linker sequences were included at the C terminus of both CH1 and Cκ, to which scFv domains directed against a tumor antigen of interest could be fused by modular cloning. The result was a heterodimeric bispecific trivalent molecule, which we term a “TriloBiTE” (Tri-lobed Bi-specific T cell Engager [tB]; Figure 3A).
Figure 3.

The soluble TriloBiTE (tB) heterodimer can redirect T cell effector functions
(A) Schematic representation of the TriloBiTE (tB) format. The structural composite was generated in PyMOL (version 2.0, Schrödinger, LLC) using domains from PDB: 1HZH.
(B) Reducing and non-reducing SDS-PAGE of various tBs (1 μg). Expected molecular weight is ~55 kDa for reduced tB and ~110 kDa for non-reduced protein.
(C) Analytical size-exclusion chromatography of purified TriloBiTE variants in PBS (pH 7.4) following a single freeze-thaw cycle.
(D) Thermostability of purified tBs in PBS was determined by differential scanning fluorimetry. Melting temperatures were calculated based on the minima of the derivative of fluorescence as a function of temperature for the first unfolding transition.
(E) Jurkat-NFAT reporter assay showing target- and concentration-dependent induction of luciferase activity by the anti-CD19 tB (0.01 pM–0.5 nM) at an E:T ratio of 5:1.
(F) Specific killing of CD19+ target cells by purified human CD8+ T cells in response to stimulation with FMC63-tB (0.01 pM–0.5 nM). Cell death was assessed using a caspase 3/7-cleavable fluorescent dye after 4 h of co-culture at an E:T ratio of 5:1.
(G) Quantification of effector cytokines secreted by human CD8+ T cells activated with FMC63-tB (0.01 pM–0.5 nM) in the presence of CD19+ Raji cells (E:T ratio 5:1). (E–G) Data are represented as mean ± SD. Representative curves of n = 2 independent experiments are shown. HEK293-6E data represent a CD19− control. See also Figure S5.
As an investigational molecule, we first engineered a bivalent CD19-specific TriloBiTE by fusing the clinically validated murine anti-CD19 scFv clone FMC63 to both chains of the anti-CD3 Fab arm.53 Similarly, anti-TEM1 tBs were engineered using the 1C1m and 7G22 scFv binders and the “benchmark” clone sc78. Transient expression from co-transfected HEK293-6E cells followed by affinity chromatography yielded good protein quantities (≈20 mg/L) with apparent homogeneity of up to 95%, depending on the clone (Figures 3B and 3C). Interestingly, the purified sc78-tB comprised a significant oligomeric or aggregated component in PBS buffer as revealed by SEC. Encouragingly, the absence of a faster eluting leading peak or shoulder in the analytical SEC profiles indicates a quantitative association of the two tB heterodimeric chains. We speculate that the engineered CH1/Ck interface is sufficiently stable so that it is not an obligate requirement for the two native cysteines, retained at the Fab C-termini to covalently associate. Hence, a proportion of tB contains heterodimer that can be dissociated by SDS under non-reducing conditions, resulting in the 58-kDa component visible in Figure 3B. Finally, differential scanning fluorimetry (DSF) of these anti-TEM1 tBs indicated encouraging thermostability of the tB tertiary structure, with transitional melting temperatures comparable to those obtained with the corresponding scFv-Fc clones (Figure 3D; data not shown).
We next investigated whether these anti-TEM1 tBs were able to recognize their cognate target cells. As expected, all tB variants recognized CD3+ Jurkat cells and specifically bound to endogenous hTEM1-expressing A673 cells (Figure S5A). Consistent with its behavior in the scFv-Fc format, the sc78-tB also bound significantly to A673 TEMKO cells, whereas 1C1m-tB stained only the residual fraction of TEM1-expressing cells within this polyclonal cell pool. The lower affinity 7G22-tB only stained the WT A673 cells. No binding was observed to two additional TEM1-negative cell lines (AsPC-1 and MS1; Figure S5A).
The potential of the tB format to redirect T cells specifically to a tumor antigen of choice was investigated using CD19 as a proof-of-concept target. To assess the efficacy of T cell engagement and activation by the TCR/CD3 complex, we used a Jurkat cell line engineered to correlate the strength and duration of CD3 ITAM signaling to the level of luciferase expression driven by an NFAT-responsive promotor. Using this system, we found that FMC63-tB induced NFAT signaling in a concentration-dependent manner and only in the presence of CD19+ Raji cells (Figure 3E). We next sought to assess the potential of the FMC63-tB to redirect the effector functions of primary human T cells. In co-culture, the FMC63-tB specifically activated a polyclonal population of primary human CD8+ T cells, which then lysed CD19+ Raji cells but spared CD19-negative HEK293 cells (Figure 3F). Moreover, T cells activated with the FMC63-tB secreted relevant effector cytokines with the magnitude of the cytokine response being dependent on the tB concentration (Figure 3G).
Taken together, these data validate the tB as an attractive structural format for soluble, productive T cell engagement. Moreover, the anti-TEM1 tBs appear to possess not only promising expression and biophysical properties but also the functionality required to recognize and cross-link TEM1+ tumor and human T cells.
Anti-TEM1 tBs redirect primary human T cell effector functions toward cognate TEM1+ target cells
We next asked whether the anti-TEM1 tBs could redirect human T cell effector activity toward TEM1+ cell lines and ultimately drive target-specific killing. Encouragingly, in co-culture, all anti-TEM1 tBs activated primary human T cells in a TEM1+target-selective manner (Figure 4A). This activation was accompanied by only a modest upregulation of the exhaustion marker PD-1 (Figure 4B). As expected, the TEM1-depleted KO cell pool triggered substantially lower levels of activation (Figure 4C). Furthermore, all three anti-TEM1 tBs redirected T cells to kill relevant endogenous TEM1-expressing target cells (Figure 4D) and induced the expression of granzyme B and perforin (Figure S5C), as well as effector cytokines (Figure 4E). Interestingly, stimulation with 7G22-tB led to a much stronger cytokine response than exposure to the other anti-TEM1 tBs, despite the otherwise comparable T cell activation and fixed time point cytotoxicity response (Figures 4A, 4C, and 4D). This effect was particularly pronounced for TNF-α and IL-2, the release of which commonly requires a higher TCR signaling threshold than that for IFN-γ.54 Because 7G22 recognizes its epitope with substantially weaker affinity than sc78 and 1C1m (360 nM versus 5.6 or 1 nM, respectively), such observations suggest that epitope location, rather than raw affinity of the scFv, is likely to prove the key determinant for optimal tB-mediated engagement of T effectors with TEM1+ target cells.
Figure 4.

Anti-TEM1 tBs redirect primary human T cell effector functions toward TEM1+ target cells
(A and B) Anti-TEM1-tBs specifically activate primary human T cells in the presence of TEM1-expressing A673 and SK-N-AS cells. Upregulation of early activation markers CD69 and CD25 (A) and the inhibitory co-receptor PD1 (B) was measured by flow cytometry after 16 h of co-culture (E:T ratio 1:1, 1 nM tB).
(C) Upregulation of CD69 and CD25 by CD8+ T cells co-cultured with TEM1+ A673 WT cells or TEM1-depleted A673 TEM1KO cells in the presence of anti-TEM1-tBs.
(D) Anti-TEM1-tBs redirect primary human T cells to kill endogenous TEM1-expressing cells (A673 or SK-N-AS), but spare TEM1-negative AsPC-1 cells. tBs were added at 1 nM, and specific cell killing was calculated as a percentage of complete lysis achieved with 1% Triton X-100. Statistical test was a 2-way ANOVA with multiple comparisons. The significance rating is based on the comparison of the test sample to the “no tB”control for each cell line.
(E) Quantification of effector cytokines secreted by polyclonal human T cells stimulated with anti-TEM1 tBs in the presence of TEM1-expressing A673 or SK-N-AS cells or irrelevant AsPC-1 cells for 24 h (E:T ratio 5:1). IFN-γ and TNF-α secretion were quantified after stimulation with 1 nM tB, and IL-2 was measured after activation with 10 nM tB.
(F ang G) Cytotoxicity exerted by human T cells that were redirected with anti-TEM1 tBs is dependent on TEM1. Filled symbols, TEM1+ A673 WT cells; open symbols, A673 TEM1KO cells. (F) Real-time kinetics of cell killing by pan-T cells redirected by different anti-TEM1 tBs (0.1 nM) over the course of 30 h (E:T ratio 5:1). (G) Cell killing was assessed after 24 h of co-culture (E:T ratio 5:1) as a function of tB concentration. Data are represented as mean ± SD. Representative results of n = 3 experiments (A–C, F, and G) or n = 2 experiments (D and E) using T cells from different donors are shown. See also Figure S5.
In addition to fixed time point cell killing, we also characterized tB-mediated kinetic killing profiles by using a time-lapse microscopy-based assay. Despite the above observed differences in cytokine production, all three anti-TEM1 tBs were shown to mediate the rapid and complete lysis of TEM1+ A673 cells (Figure 4F). However, in this kinetic assay format, the 1C1m-tB stimulated significant redirected T cell killing at picomolar concentrations, whereas approximately 100-times higher doses were required with 7G22- and sc78-tB (Figure 4G). Again, Tcell-mediated cytotoxicity was dependent on the presence of TEM1. In addition, a null-tB using an irrelevant scFv as the targeting moiety did not induce any cell killing, excluding a non-specific Tcellactivating effect of the anti-CD3 arm (Figure S5D).
The modular design of the tB enables the incorporation of either one or two tumor-targeting scFvs, in principle facilitating the optimization of functional activity and selectivity through the manipulation of affinity and/or avidity. To illustrate this principle, we expressed mono- and bivalent tB variants of both the low-affinity parental 1C1 and the single-amino-acid-substituted high-affinity 1C1m and compared their cytotoxic engager activity in co-culture assays (Figure S5E). Against A673 Ewing sarcoma cells expressing endogenous levels of TEM1, the bivalent 1C1m-tB triggered potent cytotoxicity at the lowest concentrations and mediated rapid cell killing. However, although the low-affinity monovalent variant was largely ineffective at concentrations below 1 nM, the bivalent lower-affinity tB (1C1-tB) showed killing kinetics comparable to that of the high-affinity monovalent variant.
A further comparison of the two high affinity clones 1C1m-tB and sc78-tB at two discrete concentrations revealed the former to be significantly more potent. At a concentration of 0.6 nM, both clones, as expected, rapidly redirected primary human T cells to specifically lyse relevant target cells (Figures 5A and 5B). However, at a 10-fold lower tB concentration, only the 1C1m-tB could maintain an appreciable and rapid killing kinetic, consistent with the titration data shown in Figure 4G. As both clones possess comparable single-digit nanomolar dissociation constants (KDs), this differential potency likely relates to the intrinsic properties of their respective target epitopes on the TEM1 ECD. Interestingly, although stimulation with sc78-tB achieved higher levels of cytokine production overall, 1C1m-tB induced measurable dose-dependent IFN-γ secretion at far lower concentrations (Figure 5C).
Figure 5.

1C1m-tB is highly potent in vitro
(A) Real-time kinetics of cell killing by primary T cells activated with 0.6 nM or 60 pM tB. An increase in Cytotox red signal indicates lysis of TEM1+ A673 cells (closed symbols), whereas TEM1-negative AsPC-1 cells (open symbols) are spared.
(B) Time-dependent killing of TEM1-positive A673 cells by primary human T cells in the presence of 0.6 nM 1C1m-tB. Clusters of dead cells emit red fluorescence.
(C) Induction of effector cytokine production in human pan-T cells with increasing concentrations of anti-TEM1 tB. Cytokine levels in the supernatant were measured after 24 h of co-culture with TEM1+ A673 cells. Data are represented as mean ± SD. Representative results of n = 3 experiments using T cells from different donors are shown.
1C1m-tB prevents the establishment of A673 tumors in vivo
In light of the observed in vitro potency achieved with 1C1m-tB, we next investigated its potential to redirect T cell activity in vivo. A preliminary serum clearance study (intravenous [i.v.]) was conducted using 1mg/kg 1C1-tB (see STAR Methods). A peak serum concentration of 4.6 μg/ml was reached after 60 min with levels achieving near-clearance after 48 h and a practical half-life of 11 h (Figures 6A and 6B).
Figure 6.

1C1m-tB prevents A673 tumor formation in an A673 xenograft study
(A and B) Serum clearance of 1C1-tB in female NSG mice was analyzed after administration of 1 mg/kg 1C1-tB i.v.
(B) Serum concentrations were calculated using a standard curve (A). Serum samples from two animals were pooled for each time point. The following parameters were calculated: cmax = 4.604 μg/ml at tmax = 1 h; AUC = 8859; t1/2 = 11 h.
(C–H) The potential of 1C1m-tB to induce the lysis of A673 cells by primary human T cells was assessed in a s.c. xenograft model. A total of 106 A673 Ewing’s sarcoma cells were implanted s.c. into the right flank of 10-week-old female NSG mice, either alone or admixed with 107 purified human pan-T cells. At 1 h, 24 h, and 48 h after tumor implantation, mice received 1 mg/kg 1C1m-tB in 100 μl PBS or PBS only administered i.v. (C) No apparent toxicities were observed upon tB administration, and body weight remained comparable between the groups throughout the study. (D–F) Tumor volume measurements for individual animals are shown as lines. Arrows indicate the time of tB administration. (G) Mean tumor growth in the three experimental groups. Data are represented as mean ± SD. Statistical significance (multiple paired t test) between the “T cells only” and “1C1m-tB” treatment groups was reached at day 15 (p = 0.016916) and maintained until the end of the study (p = 0.000045 on day 45). (H) Tumor volume was compared at end point day 45 for “T cells only” and “1C1m-tB” treatment groups and day 22–29 for the untreated group. Statistical significance was assessed with a paired two-tailed t test. p value (untreated versus 1C1m-tB) < 0.0001; p value (T cells only versus 1C1m-tB) = 0.0002. n = 10 animals per group.
We next implanted 106 human sarcoma A673 cells subcutaneously (s.c.) into the right flank of female NOD/SCID/IL-2Rγ KO (NSG) mice, either alone or mixed with 107 purified and expanded primary human donor T cells. One hour after tumor implantation, the mice received 1mg/kg 1C1m-tB or PBS vehicle control administered i.v. into the tail vein. The tB dose was repeated twice, at 24 h and 48 h after tumor implantation. Subsequently, the outgrowth of s.c. tumors was followed for 45 days or until animals had to be euthanized for ethical reasons. Neither the injection of human T cells nor the administration of 1C1m-tB had an impact on body weight, and no apparent toxicities were observed during the experiment (Figure 6C). As expected, the A673 cells rapidly formed tumors in the untreated animals (Figure 6D). The co-administration of human T cells together with the tumor cells substantially delayed tumor outgrowth, but all animals eventually developed tumors (Figure 6E). In contrast, the treatment with systemically delivered 1C1m-tB prevented tumor establishment or significantly suppressed tumor formation compared to the Tcellonly group (Figures 6F–6H). In this group, 5 out of the 10 mice did not develop tumors, indicating that all tumor cells were eradicated (Figures 6G and 6H). Other animals only developed flat, scar-like lesions, which never developed into a three-dimensional tumor mass. Hence, this study suggests that 1C1m-tB is active systemically in vivo, redirecting human T cells to kill TEM1-expressing target cells and therefore preventing the establishment of A673 tumors.
Discussion
The focus of the current study was to explore the potential of the TEM1tumor-associated antigen as a target for Tcellredirected immunotherapy. Although we observe that previously reported TEM1 binders appear to have deficiencies in this regard, two fully human anti-TEM1 scFv clones that we have recently isolated show encouraging T cell recruitment behavior. Using these clones as tumor recognition domains, we demonstrate their efficacy both in a generic CAR context and when incorporated into humanized heterodimeric soluble T cell engagers, which we term TriloBiTEs (tBs). The resulting anti-TEM1 tBs selectively redirect human T cells toward TEM1-expressing cells in vitro and in vivo, stimulating T cell effector functions and mediating tumor cell lysis.
Given its exclusive expression within the tumor stroma and neo-vasculature, TEM1 represents an ideal biomarker for the detection and monitoring of cancerous lesions. In this context, the previously reported MORAb-004 and sc78 molecules have been evaluated as tools for both PET and near-infrared (NIR) optical imaging and show strong localization to TEM1+ tumors, supporting the validation of TEM1 as a tumor target.36,55 Besides tumor stromal and vasculature cells, cancer cells of mesenchymal origin, notably sarcoma, have been found to express TEM1 directly.25,40 Although a heterogeneous group of tumors, sarcomas often share an aggressive pathological behavior and resistance to chemotherapy. This, coupled with the observed high prevalence of TEM1 expression across different sarcoma subtypes, has encouraged the development of several exploratory antibody-drug conjugates (ADCs).29,35,39,40
Engineered immunotherapy, targeting principally lineage-restricted cell-surface markers such as CD19, has shown great promise for the treatment of hematological malignancies.1,2,4,11,53,56 However, with a few exceptions, the clinical impact against many solid tumors has proven less encouraging. This issue is generally attributed to the cumulative impact of multiple confounding factors that include (1) the relative paucity of truly tumor-selective antigens, (2) the (often) broad heterogeneity of target antigen distribution and abundance, and (3) the physical and powerful immunosuppressive barriers resident within the TME6. Conceptually, targeting TEM1 could address some of these aspects. First, TEM1 has a very restricted expression profile in normal somatic tissues but is expressed with high incidence on the surface of various sarcoma cells.22,25,40,50 Second, the widespread expression of TEM1 within the neo-vasculature of many human carcinomas provides a conceptual mechanism for physically disrupting global tumor integrity in the relative absence of stroma-mediated immunosuppression.19,21,57 Indeed, a vaccine-based strategy targeting vascular TEM1 demonstrated that vascular disruption and elimination of TEM1-expressing stromal elements also led to in situ vaccination by facilitating the epitope spreading of tumor cell antigens.28 Although beyond the scope of our study, this consideration offers the opportunity of widespread tumor targeting irrespective of the tissue of origin.
The efficiency of T cell retargeting with both CARs and soluble engager molecules is critically impacted by a number of design parameters, such as binding strength (affinity/avidity), epitope location, and format architecture. Previous reports suggest that, for a given epitope, the potency of bispecific engagers is correlated with targeting affinity, especially for targets with very low cell-surface abundance.58 However, our data also confirm that a bivalent targeting scaffold such as the tB described here can, to some extent, both compensate for modest clone affinity and shed light on the relative functional “quality” of distinct TEM1 epitopes. Clone 7G22 was uniquely selected against the membrane-proximal sialomucin stalk domain of human TEM1 and, as a clone isolated directly from an IgM/D-derived naive library, its affinity was found to be rather low (Figure S2). Nevertheless, in tB format, this clone demonstrated cell killing activities comparable to those seen with the higher affinity clones 1C1m and sc78 and notably mediated a strong cytokine response (Figure 4). Targeting a more membrane-proximal epitope has been shown to improve the potency of both soluble T cell engagers and CARs directed against several large protein ECDs.44, 45, 46, 47, 48 Underlying these observations is the architecture of the immune synapse, where the membranes of T cell and target cell/APC have to be brought into close proximity to exclude large phosphatases such as CD45.59 Given the relatively large size of the multidomain TEM1 ECD, the presumed reduction in bridging distance may explain the surprising efficacy of the unoptimized 7G22 scFv.
As a class, bispecific engagers acting by CD3 ligation are extraordinarily potent, typically with narrow therapeutic windows. Hence, minimizing systemic toxicities is critical for advancing new molecules toward the clinic. Here, the design and molecular features of the T cell engager can play a crucial role. Depending on the antigen of interest, the modularity of the tB format offers the flexibility of monovalent affinity-driven recognition of single antigens or epitopes or of bivalent combinations that exploit avidity to stabilize target interactions. In principle, by using lower-affinity targeting scFvs, tBs could be configured or “tuned” to only trigger T cell activation in the presence of high(er) antigen expression densities encountered in the tumor and not the low(er) constitutive levels intrinsic to healthy tissues. Classical high-affinity monovalent engagers are arguably less discriminatory in this regard and can render recruited T cells sensitive to low antigen levels.60
Moreover, the therapeutic potential and safety of soluble T cell engagers may be determined by the persistence and duration of the T cell response.2 To achieve sustained T cell activation, the dosing regimen has to be adapted according to the pharmacokinetic properties of the molecular format. Formats with molecular weights at or below the glomerular filtration threshold, such as the classical tandem scFv BiTEs, are rapidly cleared from the circulation and thus require administration by continuous infusion.61 The increased mass and Stokes radius of the tB structure potentially balance the advantages of an increased half-life with clearance rates still sufficiently rapid to limit the duration of any unexpected severe adverse events.
Although the 1C1m scFv clearly redirects cytotoxic effector functions in vitro and in vivo, we observe only modest activation-induced cytokine levels, particularly when compared alongside a classical anti-CD19 clone such as the FMC63 used as a control in our study. However, although lower levels of cytokines may limit the expansion and persistence of CAR-T cells,54,62 a repeatedly administered soluble T cell engager would be expected to constantly (re)trigger a localized polyclonal T cell response in lymphocyte-infiltrated tumors without the requirement for long-term persistence of individual T cell clones. In this case, the required levels, thresholds, and functional roles played by individual cytokines are less clear. It should be noted, however, that CRS is a frequently reported adverse event observed in response to T cell redirection therapy.14,63 Interestingly, a recent study found that TNF-α, which is elevated in response to BiTE therapy and responsible for driving high IL-6 production and triggering CRS, is dispensable for efficient cytotoxic activity mediated by T cell engaging bispecifics.64 Indeed, the authors propose to block TNF-α signaling prior to the administration of an engager molecule to prevent CRS. Our 1C1m-tB observations likewise would indicate that effective redirected cytotoxicity is not conditional on a strong accompanying cytokine response. The in vivo/clinical significance of engager molecules possessing properties that intrinsically limit or attenuate cytokine cascades remains to be established.
In conclusion, our work suggests that TEM1 may represent an amenable target for immunotherapeutic T cell redirection strategies. Moreover, we present the soluble heterodimeric TriloBiTE as a validated experimental T cell engager format with convenient and flexible tumor-targeting utility.
Limitations of study
Although we have shown that the specific and potent killing potential of primary human T cells can be redirected toward human tumor cell lines expressing endogenous levels of TEM1, unanswered questions remain concerning the clinical safety of this approach. For example, despite our 1C1m-tB candidate possessing strong cross-reactivity with murine TEM1, the anti-CD3 effector arm recognizes only the human CD3 complex, thus limiting its in vivo applicability to either human hybrid CD3-engineered or immune-reconstituted and limited-duration xenograft models such as the NSG mice we used in the current study. In the absence of an equivalent murine surrogate of 1C1m-tB (compatible with true syngeneic immune-competent models), we are thus unable to evaluate the potential for “on-target off-tumor” or off-target toxicity attributed to 1C1m-tB. Additionally, published evidence suggests that TEM1 tissue expression profiles may vary across species. For example, ∼8% of naive human CD8+ T cells have been shown to express detectable levels of TEM1, which is not the case in mice.65 The implications of ablating this specific subset in a primate or human setting have not been explored but should be considered in the context of future in vivo toxicological studies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 1C1m anti-hu/muTEM1 scFv | Fierle et al.42 | N/A |
| 7G22 anti-huTEM1 scFv | This paper | N/A |
| sc78 scFv | Zhao et al.37 | N/A |
| MORAb-004 scFv | US07807382B2 | N/A |
| FMC63 anti-CD19 scFv | US7446179 | N/A |
| TriloBiTE anti-hCD3 core | This paper | N/A |
| Rat anti-mouse CD31 | Biolegend | RRID: AB_312908 |
| Rabbit anti-mouse NG2 | Merck Millipore | RRID: AB_11213678 |
| Alexa Fluor 555-conjugated goat anti-human IgG (H+L) | Life Technologies | RRID:AB_2535854 |
| Alexa Fluor 488-conjugated donkey anti-rat IgG (H+L) | Life Technologies | RRID:AB_2535794 |
| Alexa Fluor 647-conjugated donkey anti-rabbit IgG (H+L) | Life Technologies | RRID:AB_25361833 |
| Alexa Fluor 647-conjugated goat anti-human IgG, F(ab)2 fragment specific | Jackson Immunoresearch | RRID:AB_2337888 |
| Alexa Fluor 647-conjugated goat anti-human IgG, Fc fragment-specific | Jackson Immunoresearch | RRID:AB_2337889 |
| APC-conjugated mouse anti-human CD8 (clone SK1) | Biolegend | RRID:AB_2075388 |
| BV785-conjugated mouse anti-human CD4 (clone OKT4) | Biolegend | RRID:AB_2561365 |
| Alexa Fluor 700-conjugated mouse anti-human CD69 (clone FN50) | Biolegend | RRID:AB_493775 |
| PE-conjugated mouse anti-human CD25 (clone BC96) | Biolegend | RRID:AB_314276 |
| BV605-conjugated mouse anti-human PD1 (clone EH12.2H7) | Biolegend | RRID:AB_11124107 |
| FITC-conjugated mouse anti-human CD3 (clone OKT3) | Biolegend | RRID:AB_571906 |
| BV421-conjugated mouse anti-human Perforin (clone B-D48) | Biolegend | RRID:AB_11149688 |
| PE-conjugated mouse anti-human Granzyme B (clone GB11) | BD Biosciences | RRID:AB_10561690 |
| APC-conjugated Streptavidin | Biolegend | CAT#405207 |
| HRP-conjugated mouse anti-human IgG, F(ab)2-fragment specific | Jackson Immunoresearch | RRID:AB_2339089 |
| PE-conjugated goat anti-mouse IgG (H+L) | ThermoFisher Scientific | RRID:AB_2534043 |
| Biological samples | ||
| Buffy coats | Service de Transfusion, Epalinges, Switzerland | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human IL-2 | Peprotech | CAT#200-02-50UG |
| Recombinant human IL-7 | Miltenyi Biotec | CAT#130-095-367 |
| Recombinant human IL-15 | Miltenyi Biotec | CAT#130-095-765 |
| hTEM1(Δn)-SpyC | Fierle et al. (2019) | N/A |
| h/mTEM1 ECD (biotinylated) | Fierle et al. (2019) | N/A |
| CD19 (biotinylated) | BioCat | CAT# CD9-H82E9 |
| Critical commercial kits and assays | ||
| Caspase 3/7 apoptosis assay | Life Technologies | CAT#C10427 |
| Human CD8 T cell isolation kit | Miltenyi Biotec | CAT#130-096-495 |
| Human pan-T cell isolation kit | Miltenyi Biotec | CAT#130-096-535 |
| CD3/CD28 T cell activator kit | Life Technologies | CAT#11161D |
| CytoTox 96 kit (LDH assay) | Promega | CAT#G1780 |
| Human IFN-γ ELISA MAX Standard kit | Biolegend | CAT#430102 |
| Human IL-2 ELISA MAX Standard kit | Biolegend | CAT#431801 |
| Cytometric bead array, MultiCyt Qbeads customized | Bucher Biotec | CAT#90603 |
| FAM-MGB-TaqMan probe (hCD248; Hs00535586_s1) | ThermoFisher Sci | CAT#4331182 |
| FAM-MGB-TaqMan probe (hGAPDH; Hs02786624_g1) | ThermoFisher Sci | CAT#4331182 |
| Experimental models: Cell lines | ||
| A673 | ATCC | RRID:CVCL_0080 |
| A673-KO | Synthego | https://www.synthego.com |
| 2H11 | ATCC | RRID:CVCL_6762 |
| MS1 | ATCC | RRID:CVCL_6502 |
| SK-N-AS | ATCC | RRID:CVCL_1700 |
| HEK293T | ATCC | RRID:CVCL_0063 |
| AsPC-1 | ATCC | RRID:CVCL_0152 |
| Raji | ATCC | RRID:CVCL_0511 |
| Jurkat-NFAT-Lucia | Invivogen | RRID:CVCL_KS47 |
| Jurkat-NFAT-mCherry | Melita Irving, UNIL | N/A |
| Lewis lung carcinoma | ATCC | RRID:CVCL_4358 |
| MC38 | ATCC | RRID:CVCL_0A68 |
| HEK293-6E | NRC Canada | RRID:CVCL_HF20 |
| Experimental models: Organisms/strains | ||
| NOD/SCID/IL-2Rγ KO (NSG) mice | In-house colony | RRID:IMSR_NM-NSG-001 |
| C57BL/6 | In-house colony | RRID:IMSR_JAX:000664 |
| Software and algorithms | ||
| GraphPad Prism (9.0) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo v10 | BD Lifesciences | https://www.flowjo.com/ |
| Incucyte Base Software | Essen Bioscience | https://www.essenbioscience.com/en/products/software/incucyte-base-software/ |
| Adobe Illustrator (CC) | Adobe | https://www.adobe.com/products/illustrator.html |
| Microsoft Excel (2019 MSO) | Microsoft | Product ID: 00414-50000-00000-AA651 |
| Other | ||
| FectoPRO transfection reagent | Polyplus | CAT#116-010 |
| Turbofect transfection reagent | Life Technologies | CAT#R0532 |
| Cytotox Red reagent | Essen Bioscience | CAT#4632 |
| QUANTI-Luc luciferase substrate | Invivogen | CAT#rep-qlc-1 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Steven M. Dunn (steven.dunn@chuv.ch).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell lines
A-673 (Homo sapiens, female, ATCC CRL-1598; RRID:CVCL_0080), 2H11 (Mus musculus, male, ATCC CRL-2163; RRID:CVCL_6762) and MS1 (Mus musculus, sex unknown, ATCC CRL-2279; RRID:CVCL_6502) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with GlutaMAX and 10% fetal bovine serum (FBS). A673 TEM1KO cells were purchased from Synthego and cultured in DMEM/10% FBS. TEM1 was deleted genetically using CRISPR-Cas9 and the absence of TEM1 mRNA/protein in ∼95% of the line was confirmed by RT-PCR/flow cytometry. SK-N-AS (Homo sapiens, female, ATCC CRL-2137; RRID:CVCL_1700) cells were additionally supplemented with 0.1 mM non-essential amino acids (GIBCO, Life Technologies, Cat #11140050). HEK293T (Homo sapiens, fetus, ATCC CRL-11268; RRID:CVCL_0063) cells were cultured in DMEM, 10% FBS and 100 U/ml penicillin/streptomycin (GIBCO, Life Technologies, Cat#15140122). AsPC-1 (Homo sapiens, female, ATCC CRL-1682; RRID:CVCL_0152), Raji (Homo sapiens, male, ATCC CCL-86; RRID:CVCL_0511) and Jurkat NFAT reporter cells (Homo sapiens, male, NFAT: Invivogen, Cat#jktl-nfat; RRID:CVCL_KS47, mCherry: kindly provided by Melita Irving, Ludwig Center for Cancer Research Lausanne) were cultured in RPMI-1640 Glutamax (Life Technologies, Cat#61870010) containing 10% FBS. Jurkat Lucia cells were maintained under selective pressure using 100 μg/ml zeocin (Invivogen, Cat#ant-zn-1). All cells were maintained at 37°C, 5% CO2 in a humidified incubator and the absence of mycoplasma from all cell lines was confirmed by regular testing (GATC service). Cell line authentication has not been performed.
Primary T cell cultures
For the isolation of primary T cells, peripheral blood mono-nucleated cells (PBMCs) were isolated from fresh buffy coats obtained from healthy volunteer donors (Service de transfusion, Epalinges, Switzerland). PBMCs were separated by density centrifugation using Lymphoprep (Axonlab, Cat#1114545). Pan-T cells were subsequently extracted by magnetic separation using a human pan-T cell isolation kit (Miltenyi Biotec, Cat#130-096-535) and stimulated with human T cell activator CD3/CD28 beads (Life Technologies, Cat#11161D) and 50 RU IL-2 (Peprotech, Cat#200-02-50UG) for 5 days. After the removal of the beads, primary T cells were further expanded with IL-7 and IL-15 (Miltenyi Biotec, Cat#130-095-367 and Cat#130-095-765) for a further 5–10 days.
In vivo studies
All animal experimentation was performed in accordance with the guidelines of the Swiss Federal Veterinary Office and approved by the Cantonal Veterinary Office under the license number 2797.1. Female NOD/SCID/IL-2Rγ KO (NSG) mice were bred and housed under specific- and opportunistic pathogen-free (SOPF) conditions.
To gain insights into the pharmacokinetics (PK) of TriloBiTE (tB) molecules, 1 mg/kg 1C1-tB was injected into the tail vein of ten 8-week-old female NOD/SCID/IL-2Rγ KO (NSG) mice (bred in house, RRID:IMSR_NM-NSG-001). The tB was purified and prepared as described above and injected in a total volume of 100 μl, in PBS. Blood was collected by tail vein puncture at two time points per animal, resulting in 10 different duplicate time points (10 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 24 h, 48 h and 1 week after tB injection). In addition, prior to tB administration, a sample was taken from the facial vein of each animal. Serum was obtained by centrifugation and stored at −80°C.
For the A673 xenograft experiment, 30 female NSG mice (10-weeks old, RRID:IMSR_NM-NSG-001) were implanted with 106 A673 cells (ATCC CRL-1598; RRID:CVCL_0080, cultured and expanded as described above) s.c. on the right flank. 10 out of the 30 animals received only A673 cells and 20 mice received the tumor cells mixed with 107 primary human T cells. Human pan-T cells were isolated from a fresh buffy coat and expanded using CD3/CD28 beads as described previously. One hour after tumor implantation, 10 of the T cell-implanted mice received 1 mg/kg 1C1m-tB in 100 μl PBS into the tail vein. Control groups (n = 10) received 100 μl PBS. The i.v. dosing of 1C1m-tB or PBS vehicle control was repeated 24 h and 48 h after tumor implantation. Subsequently, mice were monitored thrice weekly and tumors were measured using calipers for a total of 45 days, or until the tumor volume approached ∼1000 mm3. Littermates of the same sex were allocated randomly to the different experimental groups. Tumor volume measurements were collected in a blinded manner.
Method details
Recombinant protein expression and purification
ScFv candidates were reformatted into a pTT expression vector containing the constant region of human IgG1 to produce scFv-Fc fusion proteins. To generate the TriloBiTE (tB) construct, heavy and light chain of the humanized and chimeric anti-CD3 clone UCHT1 were synthesized in Fab format (GeneArt, Thermo Fisher Scientific). Stabilizing mutations have been introduced into the human CH1-CK interface51. Both heavy and light chain of the resulting chimeric molecule were separately cloned into a pTT-based mammalian episomal expression vector. Both constructs contained modular cloning sites (NcoI/SalI) to accommodate scFv clones, which were C-terminally fused to the CH1 or CK domain of UCHT1 via a flexible (GS)-linker: GGGGSGGGSGGGS for CK and DKTHTGGGGSGGGGS for CH1. The sequence of the anti-CD19 scFv (FMC63) was extracted from Sequence 2, patent US7446179. Recombinant protein was produced using the HEK293-6E/pTT expression system and FectoPRO transfection protocol (Polyplus, #116-010), as described previously42. ScFv-Fc fusions were purified from clarified expression media using a HiTrap MabSelect column (GE Healthcare, Cat#11003494), followed by extensive dialysis against phosphate-buffered saline (PBS; Slide-A-Lyzer G2 dialysis cassettes; Life Technologies, Cat#87731). tBs were purified from clarified expression media by immobilized metal ion affinity chromatography (IMAC) using a HisTrap excel column (GE Healthcase, Cat#17371205) at a flow-rate of 1 ml/min. The column was equilibrated with 50 mM Tris, 0.5 M NaCl, 10 mM imidazole, pH 7.5 and protein was eluted with 50 mM Tris, 0.5 M NaCl, 300 mM imidazole, pH 7.4 in 1 mL fractions. Monomeric peak fractions were immediately separated by preparative size-exclusion chromatography using a Superdex 200 Increase 10/300 GL column (GE Healthcare, Cat#28990944) at a flow-rate of 0.75 ml/min. PBS (0.01 M phosphate, 0.14 M NaCl, pH 7.4) was used as sample diluent and eluent. All chromatography experiments were run on an ÄKTApure chromatography system (GE Healthcare).
The mouse IgG2a parental clone of the anti-TEM1 benchmark MORAb-004 was produced from the hybridoma cell line FB5, obtained from the MSKCC, Antibody & Bioresource Core facility, (417 E. 68th Street, NY 10065). Hybridoma cell culture and antibody production in serum-free medium (ThermoFisher Scientific, Cat#12045-076) were performed as recommended by the supplier. The IgG was purified from clarified expression media using a HiTrap MabSelect column (Cytiva, Cat#11003494), followed by dialysis against PBS (PBS; Slide-A-Lyzer G2 dialysis cassette; Life Technologies, Cat#87731).
Biophysical protein characterization
Purified protein samples were typically quality controlled by SDS-PAGE, loading 2 μg per sample on a Novex 4%–12% Bis-Tris gel (Life Technologies, Cat#NP0321) under reducing/non-reducing conditions. Samples were heated at 70°C for 10 min, loaded onto the gel and separated for 38 min at 200 V. Separated protein bands were visualized by Coomassie Blue staining (InstantBlue; Expedeon, Cat#ISB1L).
Additionally, the homogeneity of purified tBs was assessed and controlled by analytical size-exclusion chromatography. To this end, 100 μl concentrated (≈1 mg/ml) protein sample was injected and separated over a Superdex 200 Increase 5/150 GL analytical grade column (GE Healthcare, Cat#28990945) at a flow rate of 0.45 ml/min. PBS (0.01 M phosphate, 0.14 M NaCl, pH 7.4) was used as sample diluent and eluent.
ScFv stability in the tB format was confirmed by differential scanning fluorimetry following the Protein Thermal Shift Assay protocol from Applied Biosystems (Cat#4461146), following the manufacturer’s instructions. Melting curves were generated with a 7500 Fast RT-PCR machine (Applied Biosystems), starting at 25°C and gradually increasing the temperature by 0.05°C/s until reaching 99°C and data analysis was carried out using Applied Biosystems 7500 Fast RT-PCR software. Samples were formulated in PBS.
Surface plasmon resonance (SPR) analysis was performed on a Biacore T200 instrument (GE Healthcare), as described previously42. Briefly, mTEM1-SpyC:bio-SpyT or hTEM1(Δn)-SpyC:bio-SpyT antigen complexes were immobilized on a Series S SA sensor chip (GE Healthcare, Cat#BR-1005-31) at a density of 150 RU and monovalent 1C1m or 7G22 analyte was injected/dissociated at 30 μl/min in the following concentrations: 0 nM, 1.25 nM, 2.5 nM, 5 nM and 10 nM for 1C1m and 0 nM, 1 nM, 2 nM, 4 nM and 8 nM for 7G22. Surfaces were regenerated between cycles/experiments by injecting 10 mM glycine-HCl, pH 1.5 for 30 s.
Protein array
The analysis of antibody specificity was performed by Cambridge Protein Arrays Ltd. (UK), using the HuProtTM v4.0 Human Proteome Microarray which contains the expression products (as GST-fusions) of over 23,000 clones encoding human recombinant proteins, representing more than 16,000 different genes that cover ∼80% of the annotated human protein coding genome. Briefly, the scFv-Fc samples were incubated with separate, pre-blocked arrays at a concentration of 33 nM (3.5 μg/ml), diluted in blocking buffer (PBS/0.05% Tween/2% BSA) and binding was detected using an anti-human Fc-550 fluorescent probe (2.5 μg/ml in blocking buffer). Wash steps were performed after incubation with samples and secondary antibodies, using 3x PBS/0.05% Tween followed by 1x 5 min wash with PBS/0.05% Tween. The dried array slides were scanned on a Tecan LS400 microarray scanner at 532 nm and 633 nm excitation. For data analysis, fluorescence signals obtained from the corresponding protein on a negative control array were subtracted and signal strength was analyzed in relation to the amount of immobilized protein, assessed by staining for the GST-tag of the immobilized proteins (anti-GST-650; 0.5 μg/ml in blocking buffer).
Quantifying TEM1 mRNA expression by RT-qPCR
For mRNA isolation, 107 cells were seeded into a T75 tissue culture flask and harvested after 24 h with trypsin-EDTA. Detached cells were collected by centrifugation (5 min at 250x g) and washed once in ice-cold PBS. The cell pellets were either snap-frozen on dry ice and stored at −80°C or immediately processed for mRNA isolation. Cells were lysed by adding 600 μl buffer RLT and samples were homogenized by passing the lysate through an RNase-free syringe with 20-gauge needle (Braun). Subsequently, mRNA was isolated by absorption of RNA longer than 200 bases to the silica-based membrane of microspin columns using the QIAGEN RNeasy Mini kit (QIAGEN, Cat #74106), following the manufacturer’s instructions. Reverse transcription of mRNA into cDNA was performed using the Takara Primescript First Strand cDNA synthesis kit (Takara, Cat #6110B). Briefly, 5 μM oligo dT primer were annealed with 1 μg total mRNA in a total volume of 10 μl containing dNTPs (1 mM each) at 65°C for 5 min. 20 units of RNase inhibitor and 200 U of PrimeScript reverse transcriptase were then added to the annealed RNA/primer mixture and the reaction incubated for 1 h at 42°C. The reaction was stopped by heating to 70°C for 15 min and the samples were immediately cooled on ice before snap-freezing on dry ice for storage (at −80°C) or analysis by qPCR. For TaqMan qPCR, approximately 50 ng cDNA were mixed with 10 μl TaqMan Fast Universal Master Mix (Applied Biosystems, Cat #4352042) and 1 μl TaqMan probe in a total reaction volume of 20 μl. FAM-MGB-TaqMan probes (ThermoFisher Scientific) were used to detect target and reference genes: hCD248 (Cat #4331182_Hs00535586_s1), hGAPDH (Cat #4331182_Hs02786624_g1). Real-time PCR amplification of target and housekeeping genes was performed in a 7500 Fast RT-PCR machine (Applied Biosystems) and relative gene expression levels in different cell lines were analyzed by comparative threshold cycle (Ct) quantification. Expression levels are presented relative to GAPDH.
Immunohistochemistry
Retinas were collected from 5-day old pups, fixed in 4% paraformaldehyde (PFA) for 4 h at 4°C, washed with PBS and blocked overnight at 4°C with 5% donkey serum, 0.5% BSA, 0.3% Triton X-100, 0.1% sodium azide/PBS. For tumor staining experiments, 5x 105 Lewis lung carcinoma (ATCC, Cat# CRL-1642) or MC38 colon carcinoma (kindly gifted by Jeffrey Schlom, NIH) cells were resuspended in 100 μl PBS and injected subcutaneously into the right flank of adult C57BL/6 mice. Mice were sacrificed when tumors reached 1 cm3. Mice were sacrificed with 15 μl/g of 10% Ketasol, 8% Xylasol and were perfused intracardially with PBS and 4% PFA. Tumors were collected and further fixed overnight in 4% PFA. Washed tumors were then incubated in 30% sucrose for 12 h before embedding in optimal cutting temperature (OCT) compound (Tissue-Tek, Cat# 4583) for frozen sectioning. 8 μm cryosections were prepared at the histology facility of UNIL and blocked as described above. For the staining, both retinas and tumor sections were incubated overnight at 4°C with 5 μg/ml 1C1m-Fc, rat anti-mouse CD31 antibody (Biolegend, Cat# 338002, 1:500) and rabbit anti-mouse NG2 antibody (Merck Millipore, Cat# AB5320, 1:1000), all diluted in blocking buffer. The following day, the retinas were washed 5x 10 min and the tumor sections 3x 10 min with blocking buffer and then incubated overnight at 4°C with the following secondary antibodies: Alexa Fluor 555-conjugated goat anti-human antibody (Invitrogen, Cat# A21433), Alexa Fluor 488-conjugated donkey anti-rat antibody (Life Technologies, Cat# A21208) and Alexa Fluor 647-conjugated donkey anti-rabbit antibody (Life Technologies, Cat# A31573), all diluted 1:500 in blocking buffer. Wash steps were repeated and imaged were acquired using an upright Zeiss Axio Imager Z1 microscope.
Manufacturing of CAR-T cells
For the manufacturing of CAR-T cells, scFv sequences encoding anti-TEM1 molecules were constructed for clones sc78 (extracted from Zhao et al.37) and MORAb-004 (extracted from US07807382B2), and anti-CD19 (FMC63; sequence extracted from patent US7446179). These scFvs were fused to a spacer/hinge transmembrane region and intracellular costimulatory domain derived from hCD28, followed by an intracellular hCD3ζ signaling domain. The resulting 2nd generation CAR cassettes were cloned in-frame to a monomeric green fluorescent protein ORF (TagGFP2, Evrogen) into a modified pRRL lentiviral vector (originally developed by Didier Trono, EPFL). Lentivirus was produced by transient transfection of HEK293T cells using pCMVR8.74 and pMD2.G plasmids for packaging (origin: Didier Trono lab, EPFL) and Turbofect transfection reagent (Life Technologies, Cat# R0532). Virus-containing supernatant was harvested after 48 h, concentrated by ultracentrifugation and 100 μl were added directly to 5 × 106 Jurkat-NFAT reporter cells or primary human T cells pre-plated in 48-well plates on the previous day. Primary T cells were transduced the day after isolation. All transduced cells were expanded for 10–14 days before performing functional assays.
Flow cytometry assays
For typical binding experiments, adherent cells were detached using 10 mM EDTA and 0.5x106 pre-blocked cells were resuspended in 100 μl purified TriloBiTE or scFv-Fc (diluted to 1 μg/ml in 5% FBS, PBS). After 45 min of incubation on ice, the wells were washed 3x with FACS buffer. 50 μl Alexa Fluor 647-conjugated goat anti-human Fab antibody (Jackson ImmunoResearch, Cat# 109-605-097) was added for 30 min on ice to detect binding TriloBiTEs. Binding of scFv-Fc molecules was detected using an Alexa Fluor 647 AffiniPure Goat Anti-Human IgG probe (Jackson Immunoresearch, Cat# 109-605-098, 1:200 dilution). Then, the samples were washed again 3x with FACS buffer. Binding of the murine parent of MORAb-004 (FB5) to A673 wt and TEM1KO cells was detected using a PE-conjugated goat anti-mouse IgG (H+L) antibody (ThermoFisher Scientific, Cat# A10543; diluted 1/50in FACS buffer.
For measuring early T cell activation, 0.5x106 target cells were seeded into 24-well plates. Subsequently, 0.5x106 purified and expanded (CAR-)T cells were added the wells. When measuring TriloBiTE-mediated T cell activation, purified tB protein was added to a final concentration of 1 nM. After 16-18 h of co-culture, the stimulated T cells were recovered and washed once in FACS buffer. Cells were blocked with FACS buffer for 20 min on ice and incubated with the following staining mix: APC anti-hCD8 (Biolegend Cat# 344722), BV785 anti-hCD4 (Biolegend #317441), Alexa Fluor 700 anti-hCD69 (Biolegend Cat# 310922), PE anti-hCD25 (Biolegend Cat# 302606), BV605 anti-hPD1 (Biolegend Cat# 329923). After 30 min of incubation on ice, the cells were washed again three times. For perforin/granzyme B staining, the cells were stained with FITC anti-hCD3 (Biolegend Cat# 317305), BV421 anti-human perforin (Biolegend Cat# 353307) and PE anti-human granzyme B (BD Biosciences Cat# 561142).
Antigen binding by CAR-T cells was assessed by incubating 0.5x106 CAR-T cells with 1 μg/ml bio-h/mTEM1 (produced in-house) and APC-conjugated streptavidin (1:2000; Biolegend Cat# 405207), both diluted and mixed in FACS buffer. Mammalian expression supernatant containing TEM1(Δn)-SpyC fusion protein was mixed with 1 μM of bio-SpyT at RT for 2 h with gentle rotation. The resulting covalent TEM1-SpyC:bSpyT complex was separated from free bSpyT by buffer-exchange into PBS using a spin column with a 10 KDa cut-off (Vivaspin 6; GE Healthcare, Cat# 28932296).The resulting biotinylayed SpyT-SpyC-antigen complex was used as the staining antigen for 7G22-CAR-T cells. The cells were incubated with the staining mix for 30 min on ice and subsequently washed three times with FACS buffer. Immediately before data acquisition, dead cells were stained with 4′,6-Diamidino-2-phenylindole (DAPI, 1:2000 dilution). Data was acquired using an LSR-II flow cytometer equipped with FACSDIVA software (BD Biosciences). Data analysis and plotting were carried out using FlowJo v10 (FlowJo LLC).
Jurkat NFAT activation reporter cell assays
In order to measure anti-CD19 TriloBiTE-mediated NFAT activation responses, 105 Raji target cells were seeded in 96-well U-bottom plates. Subsequently, 105 Jurkat NFAT-Lucia reporter cells (Invivogen), were added to each well. Purified TriloBiTEs (diluted in PBS) were added in 3-fold serial dilution, starting from 1 nM. Phorbol myristate acetate (PMA)/ionomycin was included as a positive response control. After 24 h of co-culture, the supernatants were collected and mixed with an equal volume of QUANTI-Luc luciferase substrate (Invivogen, Cat# rep-qlc-1). Luminescence was measured immediately using a BioTec H1MFG Synergy plate reader.
For the assessment of CAR-induced ITAM-signaling, Jurkat-NFAT-mCherry reporter cells (kindly provided by Melita Irving, Ludwig Cancer Research Lausanne) were transduced with CAR-GFP constructs as described above. 10-14 days after transduction, 106 CAR-GFP transduced Jurkat reporters were seeded in 24-well plates together with 106 target cells. After 24 h of co-culture, Jurkat-NFAT-mCherry cells were harvested by pipetting, washed in FACS buffer (5% FBS, PBS) and analyzed for GFP and mCherry expression by flow cytometry.
Primary T cell cytotoxicity assays
Target cell killing mediated by the FMC63-tB was assessed using a Caspase 3/7 flow cytometry assay (Life technologies, Cat# C10427). Specifically, 105 target cells and 0.5x106 primary human CD8+ T cells (isolated using a human CD8 T cell isolation kit; Miltenyi Biotec, Cat# 130-096-495) were mixed in 96-well assay plates. Anti-CD19 tBs were added in 3-fold serial dilution starting at 5 nM. Additionally, cells in control wells were lysed with 20% ethanol. After 4 h of incubation, the supernatant was removed and the cells were resuspended in 20 μl Caspase 3/7 solution (previously diluted 1:1000 in FACS buffer, as recommended by the manufacturer). Cells were incubated with the caspase dye for 30 min at 37°C and the sample plate was read directly on an Intellicyt iQue TM Screener PLUS instrument (10 s sampling; 1 μl/s).
For the assessment of specific target cell killing using real-time kinetic cell imaging, 2x104 adherent target cells were seeded in 96-well flat-bottom plates and allowed to attach for ∼20 h. When approximately 30% confluency was observed, soluble tBs were added as 3-fold serial dilutions, typically starting from 5 nM. Positive control wells were lysed using 1% Triton X-100. 1.25x106 purified and expanded primary human T cells were added to the plate to reach an E:T ratio of around 5:1. Transduced and expanded CAR-T cells (prepared as described above) were equally added at 1.25x106 cells/well, in this case omitting the soluble engager molecules. Cytotox Red reagent (Essen Bioscience, Cat# 4632) was added to a final dilution of 1:4000, and resultant cell death was monitored as an increase in fluorescence over time.
Alternatively, specific target cell killing was assessed by measuring LDH release with the CytoTox 96 kit from Promega (Cat# G1780), following the manufacturer’s instructions. Target cells, (CAR-)T cells and tBs were prepared as described above and the assay was incubated for 24 h at 37°C. Control wells were lysed using 10% Triton X-100. Subsequently, 50 μl clarified culture supernatant was mixed with 50 μl CytoTox 96 Reagent and incubated at RT for 30 min (protected from light). The reaction was stopped by adding 50 μl stop solution and LDH activity was quantified colorimetrically, measuring absorbance at 490 nm on a BioTek H1MFG Synergy plate reader. Background signal was subtracted from all samples and corrected cell killing (spontaneous release by targets and effectors subtracted) were calculated as a percentage of maximum lysis.
Quantification of effector cytokines
Supernatants of co-cultures set up as described above were tested for the presence of T cell-secreted effector cytokines. Cytokines were either quantified by sandwich ELISA (human IFN-γ or IL-2 ELISA MAX Standard kits, Biolegend Cat# 430102 and Cat# 431801) or by cytokine bead array (MultiCyt QBeads customized Human 3-Plex [IFN-γ, IL-2, TNF-α], Bucher Biotec/Sartorius Cat# 90603). Both assays were performed according to the manufacturer’s instructions. In both cases, standard curves were prepared to quantify secreted cytokines. ELISA-based assays were read out on a BioTek H1MFG Synergy plate reader using TMB as a substrate for the HRP-conjugated secondary antibody. Plates for the MultiCyt bead array were analyzed on an Intellicyt iQue TM Screener PLUS instrument (10 s sampling; 1 μl/s).
PK elisa
Nunc maxisorb plates were pre-coated with neutravidin, blocked, and coated with 0.5 μg/ml biotinylated hTEM1-ECD protein, according to the standard ELISA protocol described in chapter 2. A tB standard curve was prepared by 2-fold dilution of purified 1C1-tB in 2% pooled murine serum (baseline)/ PBST/ 1% BSA, starting at 10 μg/ml. Serum samples from duplicate animals were pooled and diluted 1/50 in 1% BSA/ PBST. Both samples and standard curve were added to the coated ELISA plate and incubated for 1 h at RT. tBs were detected using an HRP-conjugated anti-human Fab antibody (Jackson ImmunoResearch, Cat# 209-035-097; 1:10000). ELISA plates were read on a BioTek H1MFG Synergy plate reader using TMB as a substrate. 1C1-tB serum concentrations were calculated based on the obtained standard curve.
Quantification and statistical analysis
For statistical analyses, the Student’s t test was used for comparison between two datasets. Excel or Prism software was used for statistical analysis. Data are presented as mean ± standard deviation as indicated in figure legends. A p value of < 0.05 is considered statistically significant and is denoted by ∗. Images shown were randomly chosen within a well of interest. Other statistical details can be found in figure legends. No data were excluded.
Acknowledgments
We would like to thank the Mouse Pathology, Animal and Cellular Imaging facilities of the University of Lausanne. In particular, we would like to thank Anne Wilson for excellent FACS support; Maria Fastré, Francis Derouet, and Wilson Castro for their support and guidance with the in vivo study; and Gerd Ritter for critical reading of the manuscript. Funding for this study was provided by Ludwig Cancer Research.
Author contributions
Conceptualization, S.M.D., G.C., and T.V.P.; methodology, J.K.F. and S.M.D.; investigation, J.K.F., M.B., M.d., V.A., L.W., M.B., and J.A.-S.; writing – original draft, J.K.F., and S.M.D.; writing – review and editing, J.K.F., S.M.D., G.C., and T.V.P.; supervision, S.M.D., G.C., and T.V.P.; funding acquisition, S.M.D., and G.C.
Declaration of interests
J.K.F., S.M.D., and G.C. are named inventors on WO2020243455 disclosing 1C1m. All other authors declare no competing interests.
Published: July 29, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100362.
Contributor Information
George Coukos, Email: george.coukos@chuv.ch.
Steven M. Dunn, Email: steven.dunn@chuv.ch.
Supplemental information
Data and code availability
This study did not generate or analyze datasets or code.
References
- 1.Grupp S.A., Kalos M., Barrett D., Aplenc R., Porter D.L., Rheingold S.R., Teachey D.T., Chew A., Hauck B., Wright J.F. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bargou R., Leo E., Zugmaier G., Klinger M., Goebeler M., Knop S., Noppeney R., Viardot A., Hess G., Schuler M. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–977. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 5.Topp M.S., Kufer P., Gökbuget N., Goebeler M., Klinger M., Neumann S., Horst H.A., Raff T., Viardot A., Schmid M. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011;29:2493–2498. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- 6.June C.H., O’Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 7.Dreier T., Lorenczewski G., Brandl C., Hoffmann P., Syring U., Hanakam F., Kufer P., Riethmuller G., Bargou R., Baeuerle P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer. 2002;100:690–697. doi: 10.1002/ijc.10557. [DOI] [PubMed] [Google Scholar]
- 8.Dreier T., Baeuerle P.A., Fichtner I., Grün M., Schlereth B., Lorenczewski G., Kufer P., Lutterbüse R., Riethmüller G., Gjorstrup P., Bargou R.C. T cell costimulus-independent and very efficacious inhibition of tumor growth in mice bearing subcutaneous or leukemic human B cell lymphoma xenografts by a CD19-/CD3- bispecific single-chain antibody construct. J. Immunol. 2003;170:4397–4402. doi: 10.4049/jimmunol.170.8.4397. [DOI] [PubMed] [Google Scholar]
- 9.Imai C., Mihara K., Andreansky M., Nicholson I.C., Pui C.H., Geiger T.L., Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 10.Maher J., Brentjens R.J., Gunset G., Rivière I., Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 11.Löffler A., Kufer P., Lutterbüse R., Zettl F., Daniel P.T., Schwenkenbecher J.M., Riethmüller G., Dörken B., Bargou R.C. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood. 2000;95:2098–2103. [PubMed] [Google Scholar]
- 12.Mack M., Riethmüller G., Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. USA. 1995;92:7021–7025. doi: 10.1073/pnas.92.15.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Offner S., Hofmeister R., Romaniuk A., Kufer P., Baeuerle P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006;43:763–771. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 14.Topp M.S., Gökbuget N., Stein A.S., Zugmaier G., O’Brien S., Bargou R.C., Dombret H., Fielding A.K., Heffner L., Larson R.A. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 15.Labrijn A.F., Janmaat M.L., Reichert J.M., Parren P.W.H.I. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019;18:585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 16.Gust J., Hay K.A., Hanafi L.A., Li D., Myerson D., Gonzalez-Cuyar L.F., Yeung C., Liles W.C., Wurfel M., Lopez J.A. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017;7:1404–1419. doi: 10.1158/2159-8290.CD-17-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thistlethwaite F.C., Gilham D.E., Guest R.D., Rothwell D.G., Pillai M., Burt D.J., Byatte A.J., Kirillova N., Valle J.W., Sharma S.K. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017;66:1425–1436. doi: 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bagley R.G., Honma N., Weber W., Boutin P., Rouleau C., Shankara S., Kataoka S., Ishida I., Roberts B.L., Teicher B.A. Endosialin/TEM 1/CD248 is a pericyte marker of embryonic and tumor neovascularization. Microvasc. Res. 2008;76:180–188. doi: 10.1016/j.mvr.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Brady J., Neal J., Sadakar N., Gasque P. Human endosialin (tumor endothelial marker 1) is abundantly expressed in highly malignant and invasive brain tumors. J. Neuropathol. Exp. Neurol. 2004;63:1274–1283. doi: 10.1093/jnen/63.12.1274. [DOI] [PubMed] [Google Scholar]
- 21.Christian S., Winkler R., Helfrich I., Boos A.M., Besemfelder E., Schadendorf D., Augustin H.G. Endosialin (Tem1) is a marker of tumor-associated myofibroblasts and tumor vessel-associated mural cells. Am. J. Pathol. 2008;172:486–494. doi: 10.2353/ajpath.2008.070623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rouleau C., Curiel M., Weber W., Smale R., Kurtzberg L., Mascarello J., Berger C., Wallar G., Bagley R., Honma N. Endosialin protein expression and therapeutic target potential in human solid tumors: sarcoma versus carcinoma. Clin. Cancer Res. 2008;14:7223–7236. doi: 10.1158/1078-0432.CCR-08-0499. [DOI] [PubMed] [Google Scholar]
- 23.Bagley R.G., Rouleau C., St Martin T., Boutin P., Weber W., Ruzek M., Honma N., Nacht M., Shankara S., Kataoka S. Human endothelial precursor cells express tumor endothelial marker 1/endosialin/CD248. Mol. Cancer Ther. 2008;7:2536–2546. doi: 10.1158/1535-7163.MCT-08-0050. [DOI] [PubMed] [Google Scholar]
- 24.MacFadyen J.R., Haworth O., Roberston D., Hardie D., Webster M.T., Morris H.R., Panico M., Sutton-Smith M., Dell A., van der Geer P. Endosialin (TEM1, CD248) is a marker of stromal fibroblasts and is not selectively expressed on tumour endothelium. FEBS Lett. 2005;579:2569–2575. doi: 10.1016/j.febslet.2005.03.071. [DOI] [PubMed] [Google Scholar]
- 25.Rouleau C., Smale R., Fu Y.S., Hui G., Wang F., Hutto E., Fogle R., Jones C.M., Krumbholz R., Roth S. Endosialin is expressed in high grade and advanced sarcomas: evidence from clinical specimens and preclinical modeling. Int. J. Oncol. 2011;39:73–89. doi: 10.3892/ijo.2011.1020. [DOI] [PubMed] [Google Scholar]
- 26.Tomkowicz B., Rybinski K., Foley B., Ebel W., Kline B., Routhier E., Sass P., Nicolaides N.C., Grasso L., Zhou Y. Interaction of endosialin/TEM1 with extracellular matrix proteins mediates cell adhesion and migration. Proc. Natl. Acad. Sci. USA. 2007;104:17965–17970. doi: 10.1073/pnas.0705647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nanda A., Karim B., Peng Z., Liu G., Qiu W., Gan C., Vogelstein B., St Croix B., Kinzler K.W., Huso D.L. Tumor endothelial marker 1 (Tem1) functions in the growth and progression of abdominal tumors. Proc. Natl. Acad. Sci. USA. 2006;103:3351–3356. doi: 10.1073/pnas.0511306103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Facciponte J.G., Ugel S., De Sanctis F., Li C., Wang L., Nair G., Sehgal S., Raj A., Matthaiou E., Coukos G., Facciabene A. Tumor endothelial marker 1-specific DNA vaccination targets tumor vasculature. J. Clin. Invest. 2014;124:1497–1511. doi: 10.1172/JCI67382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouleau C., Gianolio D.A., Smale R., Roth S.D., Krumbholz R., Harper J., Munroe K.J., Green T.L., Horten B.C., Schmid S.M., Teicher B.A. Anti-Endosialin Antibody-Drug Conjugate: Potential in Sarcoma and Other Malignancies. Mol. Cancer Ther. 2015;14:2081–2089. doi: 10.1158/1535-7163.MCT-15-0312. [DOI] [PubMed] [Google Scholar]
- 30.D’Angelo S.P., Hamid O.A., Tarhini A., Schadendorf D., Chmielowski B., Collichio F.A., Pavlick A.C., Lewis K.D., Weil S.C., Heyburn J. A phase 2 study of ontuxizumab, a monoclonal antibody targeting endosialin, in metastatic melanoma. Invest. New Drugs. 2018;36:103–113. doi: 10.1007/s10637-017-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grothey A., Strosberg J.R., Renfro L.A., Hurwitz H.I., Marshall J.L., Safran H., Guarino M.J., Kim G.P., Hecht J.R., Weil S.C. A Randomized, Double-Blind, Placebo-Controlled Phase II Study of the Efficacy and Safety of Monotherapy Ontuxizumab (MORAb-004) Plus Best Supportive Care in Patients with Chemorefractory Metastatic Colorectal Cancer. Clin. Cancer Res. 2018;24:316–325. doi: 10.1158/1078-0432.CCR-17-1558. [DOI] [PubMed] [Google Scholar]
- 32.Jones R.L., Chawla S.P., Attia S., Schöffski P., Gelderblom H., Chmielowski B., Le Cesne A., Van Tine B.A., Trent J.C., Patel S. A phase 1 and randomized controlled phase 2 trial of the safety and efficacy of the combination of gemcitabine and docetaxel with ontuxizumab (MORAb-004) in metastatic soft-tissue sarcomas. Cancer. 2019;125:2445–2454. doi: 10.1002/cncr.32084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norris R.E., Fox E., Reid J.M., Ralya A., Liu X.W., Minard C., Weigel B.J. Phase 1 trial of ontuxizumab (MORAb-004) in children with relapsed or refractory solid tumors: A report from the Children’s Oncology Group Phase 1 Pilot Consortium (ADVL1213) Pediatr. Blood Cancer. 2018;65:e26944. doi: 10.1002/pbc.26944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rybinski K., Imtiyaz H.Z., Mittica B., Drozdowski B., Fulmer J., Furuuchi K., Fernando S., Henry M., Chao Q., Kline B. Targeting endosialin/CD248 through antibody-mediated internalization results in impaired pericyte maturation and dysfunctional tumor microvasculature. Oncotarget. 2015;6:25429–25440. doi: 10.18632/oncotarget.4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C., Chacko A.M., Hu J., Hasegawa K., Swails J., Grasso L., El-Deiry W.S., Nicolaides N., Muzykantov V.R., Divgi C.R., Coukos G. Antibody-based tumor vascular theranostics targeting endosialin/TEM1 in a new mouse tumor vascular model. Cancer Biol. Ther. 2014;15:443–451. doi: 10.4161/cbt.27825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li C., Wang J., Hu J., Feng Y., Hasegawa K., Peng X., Duan X., Zhao A., Mikitsh J.L., Muzykantov V.R. Development, optimization, and validation of novel anti-TEM1/CD248 affinity agent for optical imaging in cancer. Oncotarget. 2014;5:6994–7012. doi: 10.18632/oncotarget.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao A., Nunez-Cruz S., Li C., Coukos G., Siegel D.L., Scholler N. Rapid isolation of high-affinity human antibodies against the tumor vascular marker Endosialin/TEM1, using a paired yeast-display/secretory scFv library platform. J. Immunol. Methods. 2011;363:221–232. doi: 10.1016/j.jim.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delage J.A., Faivre-Chauvet A., Fierle J.K., Gnesin S., Schaefer N., Coukos G., Dunn S.M., Viertl D., Prior J.O. 177Lu radiolabeling and preclinical theranostic study of 1C1m-Fc: an anti-TEM-1 scFv-Fc fusion protein in soft tissue sarcoma. EJNMMI Res. 2020;10:98. doi: 10.1186/s13550-020-00685-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capone E., Piccolo E., Fichera I., Ciufici P., Barcaroli D., Sala A., De Laurenzi V., Iacobelli V., Iacobelli S., Sala G. Generation of a novel Antibody-Drug Conjugate targeting endosialin: potent and durable antitumor response in sarcoma. Oncotarget. 2017;8:60368–60377. doi: 10.18632/oncotarget.19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo Y., Hu J., Wang Y., Peng X., Min J., Wang J., Matthaiou E., Cheng Y., Sun K., Tong X. Tumour endothelial marker 1/endosialin-mediated targeting of human sarcoma. Eur. J. Cancer. 2018;90:111–121. doi: 10.1016/j.ejca.2017.10.035. [DOI] [PubMed] [Google Scholar]
- 41.Diaz L.A., Jr., Coughlin C.M., Weil S.C., Fishel J., Gounder M.M., Lawrence S., Azad N., O’Shannessy D.J., Grasso L., Wustner J. A first-in-human phase I study of MORAb-004, a monoclonal antibody to endosialin in patients with advanced solid tumors. Clin. Cancer Res. 2015;21:1281–1288. doi: 10.1158/1078-0432.CCR-14-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fierle J.K., Abram-Saliba J., Brioschi M., deTiani M., Coukos G., Dunn S.M. Integrating SpyCatcher/SpyTag covalent fusion technology into phage display workflows for rapid antibody discovery. Sci. Rep. 2019;9:12815. doi: 10.1038/s41598-019-49233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Christian S., Ahorn H., Koehler A., Eisenhaber F., Rodi H.P., Garin-Chesa P., Park J.E., Rettig W.J., Lenter M.C. Molecular cloning and characterization of endosialin, a C-type lectin-like cell surface receptor of tumor endothelium. J. Biol. Chem. 2001;276:7408–7414. doi: 10.1074/jbc.M009604200. [DOI] [PubMed] [Google Scholar]
- 44.Hombach A.A., Schildgen V., Heuser C., Finnern R., Gilham D.E., Abken H. T cell activation by antibody-like immunoreceptors: the position of the binding epitope within the target molecule determines the efficiency of activation of redirected T cells. J. Immunol. 2007;178:4650–4657. doi: 10.4049/jimmunol.178.7.4650. [DOI] [PubMed] [Google Scholar]
- 45.James S.E., Greenberg P.D., Jensen M.C., Lin Y., Wang J., Till B.G., Raubitschek A.A., Forman S.J., Press O.W. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 2008;180:7028–7038. doi: 10.4049/jimmunol.180.10.7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bluemel C., Hausmann S., Fluhr P., Sriskandarajah M., Stallcup W.B., Baeuerle P.A., Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 2010;59:1197–1209. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J., Stagg N.J., Johnston J., Harris M.J., Menzies S.A., DiCara D., Clark V., Hristopoulos M., Cook R., Slaga D. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell. 2017;31:383–395. doi: 10.1016/j.ccell.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi J., Li X., Peng H., Cook E.M., Dadashian E.L., Wiestner A., Park H., Rader C. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc. Natl. Acad. Sci. USA. 2018;115:E5467–E5476. doi: 10.1073/pnas.1719905115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacFadyen J., Savage K., Wienke D., Isacke C.M. Endosialin is expressed on stromal fibroblasts and CNS pericytes in mouse embryos and is downregulated during development. Gene Expr. Patterns. 2007;7:363–369. doi: 10.1016/j.modgep.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 50.Dolznig H., Schweifer N., Puri C., Kraut N., Rettig W.J., Kerjaschki D., Garin-Chesa P. Characterization of cancer stroma markers: in silico analysis of an mRNA expression database for fibroblast activation protein and endosialin. Cancer Immun. 2005;5:10. [PubMed] [Google Scholar]
- 51.Chen W., Bardhi A., Feng Y., Wang Y., Qi Q., Li W., Zhu Z., Dyba M.A., Ying T., Jiang S. Improving the CH1-CK heterodimerization and pharmacokinetics of 4Dm2m, a novel potent CD4-antibody fusion protein against HIV-1. MAbs. 2016;8:761–774. doi: 10.1080/19420862.2016.1160180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schoonjans R., Willems A., Schoonooghe S., Fiers W., Grooten J., Mertens N. Fab chains as an efficient heterodimerization scaffold for the production of recombinant bispecific and trispecific antibody derivatives. J. Immunol. 2000;165:7050–7057. doi: 10.4049/jimmunol.165.12.7050. [DOI] [PubMed] [Google Scholar]
- 53.Kochenderfer J.N., Feldman S.A., Zhao Y., Xu H., Black M.A., Morgan R.A., Wilson W.H., Rosenberg S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itoh Y., Germain R.N. Single cell analysis reveals regulated hierarchical T cell antigen receptor signaling thresholds and intraclonal heterogeneity for individual cytokine responses of CD4+ T cells. J. Exp. Med. 1997;186:757–766. doi: 10.1084/jem.186.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chacko A.M., Li C., Nayak M., Mikitsh J.L., Hu J., Hou C., Grasso L., Nicolaides N.C., Muzykantov V.R., Divgi C.R., Coukos G. Development of 124I immuno-PET targeting tumor vascular TEM1/endosialin. J. Nucl. Med. 2014;55:500–507. doi: 10.2967/jnumed.113.121905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.A., Lanier B.J. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kiyohara E., Donovan N., Takeshima L., Huang S., Wilmott J.S., Scolyer R.A., Jones P., Somers E.B., O’Shannessy D.J., Hoon D.S. Endosialin Expression in Metastatic Melanoma Tumor Microenvironment Vasculature: Potential Therapeutic Implications. Cancer Microenviron. 2015;8:111–118. doi: 10.1007/s12307-015-0168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slaga D., Ellerman D., Lombana T.N., Vij R., Li J., Hristopoulos M., Clark R., Johnston J., Shelton A., Mai E. Avidity-based binding to HER2 results in selective killing of HER2-overexpressing cells by anti-HER2/CD3. Sci. Transl. Med. 2018;10:eaat5775. doi: 10.1126/scitranslmed.aat5775. [DOI] [PubMed] [Google Scholar]
- 59.Cordoba S.P., Choudhuri K., Zhang H., Bridge M., Basat A.B., Dustin M.L., van der Merwe P.A. The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood. 2013;121:4295–4302. doi: 10.1182/blood-2012-07-442251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Root A.R., Cao W., Li B., LaPan P., Meade C., Sanford J., Jin M., O’Sullivan C., Cummins E., Lambert M. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies (Basel) 2016;5:6. doi: 10.3390/antib5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu M., Wu B., Brandl C., Johnson J., Wolf A., Chow A., Doshi S. Blinatumomab, a Bispecific T-cell Engager (BiTE(®)) for CD-19 Targeted Cancer Immunotherapy: Clinical Pharmacology and Its Implications. Clin. Pharmacokinet. 2016;55:1271–1288. doi: 10.1007/s40262-016-0405-4. [DOI] [PubMed] [Google Scholar]
- 62.Au-Yeung B.B., Zikherman J., Mueller J.L., Ashouri J.F., Matloubian M., Cheng D.A., Chen Y., Shokat K.M., Weiss A. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc. Natl. Acad. Sci. USA. 2014;111:E3679–E3688. doi: 10.1073/pnas.1413726111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Przepiorka D., Ko C.W., Deisseroth A., Yancey C.L., Candau-Chacon R., Chiu H.J., Gehrke B.J., Gomez-Broughton C., Kane R.C., Kirshner S. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015;21:4035–4039. doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- 64.Li J., Piskol R., Ybarra R., Chen Y.J., Li J., Slaga D., Hristopoulos M., Clark R., Modrusan Z., Totpal K. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019;11:eaax8861. doi: 10.1126/scitranslmed.aax8861. [DOI] [PubMed] [Google Scholar]
- 65.Hardie D.L., Baldwin M.J., Naylor A., Haworth O.J., Hou T.Z., Lax S., Curnow S.J., Willcox N., MacFadyen J., Isacke C.M., Buckley C.D. The stromal cell antigen CD248 (endosialin) is expressed on naive CD8+ human T cells and regulates proliferation. Immunology. 2011;133:288–295. doi: 10.1111/j.1365-2567.2011.03437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or code.