

Summary
Functional profiling of a cancer patient’s tumor cells holds potential to tailor personalized cancer treatment. Here, we report the utility of fresh uncultured tumor-derived EpCAM+ epithelial cells (FUTCs) for ex vivo drug-response interrogation. Analysis of murine Kras mutant FUTCs demonstrates pharmacological and adaptive signaling profiles comparable to subtype-matched cultured cells. By applying FUTC profiling on non-small-cell lung cancer patient samples, we report robust drug-response data in 19 of 20 cases, with cells exhibiting targeted drug sensitivities corresponding to their oncogenic drivers. In one of these cases, an EGFR mutant lung adenocarcinoma patient refractory to osimertinib, FUTC profiling is used to guide compassionate treatment. FUTC profiling identifies selective sensitivity to disulfiram and the combination of carboplatin plus etoposide, and the patient receives substantial clinical benefit from treatment with these agents. We conclude that FUTC profiling provides a robust, rapid, and actionable assessment of personalized cancer treatment options.
Keywords: functional diagnostics, lung cancer, personalized medicine, KRAS, EGFR, drug sensitivity and resistance testing, ex vivo drug screening, targeted therapy, pharmacogenomics, ALK
Graphical abstract
Highlights
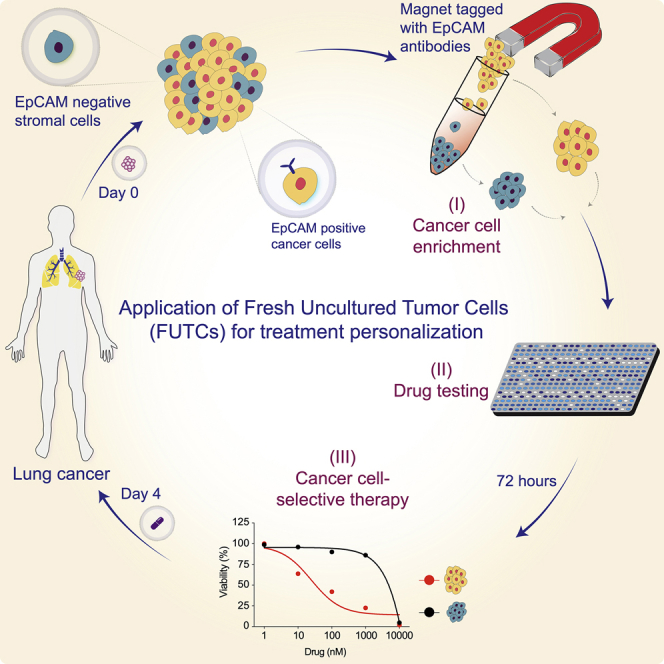
Fresh uncultured tumor-derived epithelial cells (FUTCs) are amenable for drug testing
Profiling of lung tumor and matched normal cells identifies cancer-selective treatments
NSCLC FUTCs with targetable drivers expose genotype-matched responses or non-responses
The FUTC approach can guide personalized therapy in compassionate patient care
Point-of-care functional diagnostic tests are unavailable for lung cancer. Talwelkar et al. present a pharmacological assay that uses fresh uncultured tumor cells (FUTCs) to guide personalized therapy for lung cancer patients. Besides identifying genotype-matched drug sensitivities, FUTC profiling also predicts clinical non-responses and can be used to expose resistance mechanisms.
Introduction
The concept of precision medicine means giving the right drug to the right individual at the right time. In this context, genotype-guided pairing of tumors with drugs that target tumor-selective driver mutations has been in clinical practice for >2 decades. For non-small-cell lung cancer (NSCLC) patients, inhibitors targeting EGFR, KRAS (G12C), ALK, BRAF, MET, and ROS are approved for genotype-guided indications that match for ∼40% of patients.1 However, even with the genetic matches, only 50%–70% of treated patients benefit from these treatments. When attempting to use genotype-guided therapies beyond currently approved indications, trials have shown that only 10% of patients can be paired with genomics-matched targeted treatments, and at best one-third of these receives clinical benefit.2,3 Hence, the prediction of successful precision anticancer treatments at the individual level remains challenging, largely because of extensive genomic and phenotypic heterogeneity of many cancer types, including NSCLC. Therefore, to identify precision medicines for the majority of NSCLC patients, we need additional tools beyond those that are currently used in the clinic.
Combining static genetic measurements with pharmacological interrogation of patient-derived cancer cells can provide a more comprehensive approach for predicting effective treatments4, 5, 6; however, proof of its clinical utility is lacking, particularly for solid tumors. For patients with hematological malignancies, we and others have successfully implemented individualized treatment strategies guided by ex vivo drug-response testing of patient biopsies.7,8 Based on these early successes of ex vivo testing to tailor patient treatments, multiple clinical trials were initiated for leukemia patients (e.g., NCT01620216, NCT04267081). For solid tumors, similar functional diagnostic methodologies can, however, not readily be adopted; the reasons include the fragile and short-lived nature of tissues (e.g., those observed in organotypic tumor slice cultures9,10) and a general lack of robust methods to isolate and propagate primary epithelial cells.
In promising recent developments, new culture and organoid approaches enabling the long-term expansion of primary epithelial cells have been introduced.4,11,12 Toward translational applications, the use of conditionally reprogrammed (CR) cultures derived from clinical lung tumors successfully identified novel actionable treatments and molecular mechanisms underlying drug resistance.4,13,14 Similarly, NSCLC organoids have been shown to recapitulate oncogenic addictions and the tissue architecture of the original tumors, offering an optional model for exploring inter- and intratumoral functional heterogeneity.15,16
While the implementation of CR or organoid cultures in principle permits drug-response profiling of patient-derived cells, these models take a long time to be established, and, more important, do not guarantee the expansion of malignant cells.4,15,17 Their functional interrogation will therefore unlikely lead to significant effects on personalized diagnostics and patient care.11 To circumvent the challenges associated with ex vivo cultures, we tested the utility of fresh uncultured tumor-derived cells (FUTCs) for drug-response assessment. Using this approach, we present a diagnostic assay for the rapid identification of actionable treatments using patient-derived tumor cells.
Results
Tumor-derived epithelial cultures often do not represent malignant cells
With the aim of identifying individualized treatment options, and to understand genetic driver-drug-response relationships, we established NSCLC patient-derived primary cultures to allow for pharmacogenomic screening. Patient-derived primary cultures were established using CR methodology, which allows ex vivo establishment and indefinite propagation of epithelial cells from both normal and malignant specimens.18 To confirm the identity of the tumor-derived cultures, we performed targeted next-generation sequencing for 578 cancer-related genes in 11 primary cultures, as well as matched tumor and adjacent normal lung tissues. Altogether, 71 nonsynonymous somatic mutations in 47 genes were detected in tumor tissues (Table 1). Of the 11 tested cultures, 9 lacked the oncogenic mutations detected in the reference tumor. Notably, the two cultures that showed a concordance of mutations with their respective source tissue were derived from different regions from the same tumor (Table 1). The success rate of establishing patient-derived malignant cultures was therefore limited to ∼10%. In accordance with previous findings,17,19,20 our results argue that CR culture establishment is not an effective approach for the generation of pharmacogenomic screening data from NSCLC patient samples.
Table 1.
Genomic analysis of NSCLC tumor tissue and tumor-derived CR cultures
| Sample no. | Sample ID | Gender/age | Tumor type | Tumor tissue (variant allele frequency) | Tumor-derived CR culturea (variant allele frequency) |
|---|---|---|---|---|---|
| 1 | PLT26; region-3 | F/55 | AC | TFG (0.10), CBFA2T3 (0.10), SMARCA4 (0.08), TP53 (0.07), TMPRSS2 (0.07), PPP2R1A (0.04), ALK+ | TFG (0.66), CBFA2T3 (0.73), SMARCA4 (0.45), TP53 (0.99), TMPRSS2 (0.54), PPP2R1A (0.50), ALK+ |
| 2 | PLT26; region-5 | F/55 | AC | TFG (0.15), CBFA2T3 (0.28), SMARCA4 (0.15), TP53 (0.28), TMPRSS2 (0.17), PPP2R1A (0.12), ALK+ | TFG (0.64), CBFA2T3 (0.66), SMARCA4 (0.49), TP53 (0.98), TMPRSS2 (0.50), PPP2R1A (0.58), ALK+ |
| 3 | PLT30 | M/75 | AC | NKX2-1 (0.33), ERB B4 (0.26), WASF 3 (0.25), KRAS (0.18), TP53 (0.15), HERPUD1 (0.14) | All wild type |
| 4 | PLT36 | F/62 | AC | GNAS (0.72), TP53 (0.40), LTBP2 (0.34), LCP1 (0.3), MAP2K1 (0.27), PDGFRB (0.25), GUCY1A2 (0.19), KMT2D (0.16), HECW1 (0.15), KMT2C (0.10) | All wild type |
| 5 | PLT41 | M/56 | AC | MYB (0.15), NSD3 (0.14), TP53 (0.14), KMT2C (0.11), ALK+ | All wild type |
| 6 | PLT42b | F/63 | AC | PDGFRB (0.05), TYK2 (0.04), ARID1A (0.03), IDH2 (0.03), TCL6 (0.03), ITK (0.03), NTRK3 (0.02), KRAS (0.02), WT1 (0.02) | All wild type |
| 7 | PLT62; region-1 | F/50 | AC | BCL6 (0.66), STK11 (0.43), KRAS (0.38), PRF1 (0.33), KIT (0.22), NEF 2L2 (0.20), CBFA2T3 (0.19), CLTCL1 (0.17) | All wild type |
| 8 | PLT62; region-2 | F/50 | AC | BCL6 (0.34), STK11 (0.26), KRAS (0.23), PRF1 (0.17), KIT (0.10), NEF2L2 (0.04), CBFA2T3 (0.07), CLTCL1 (0.10) | All wild type |
| 9 | PLT63 | F/68 | AC | CDKN2B (0.11), ALK+ | All wild type |
| 10 | PLT64 | F/25 | AC | ALK+ | All wild type |
| 11 | PLT66 | M/71 | AC | CIC (0.42), KRAS (0.36), CBF A2T3 (0.23), AKAP9 (0.22), NIN (0.22), AFF4 (0.21), PML (0.20), FGFR1OP (0.20), ABL1 (0.19), FLT3 (0.19), LTBP2 (0.19), PDGFRB (0.18), HEY1 (0.18), EPHA10 (0.17), UNC13D (0.16), CDH6 (0.16), ARID4B (0.14), BCL11B (0.14), UNC13D (0.11), CYP1B1 (0.10) | All wild type |
Passage #4.
All mutations identified with variant allele frequency <0.1.
Murine FUTCs capture drug responses of other ex vivo cell culture systems
To address the challenge of cell culture adaptation, we investigated whether freshly isolated epithelial cell adhesion molecule (EpCAM)-expressing epithelial cells can directly be used for drug-response assessment. We performed functional profiling of FUTCs and CR cultures derived from murine NSCLC tumors obtained from KrasG12D;Lkb1fl/fl (KL) and KrasG12D;p53fl/fl (KP) models. These models were selected because (1) they represent tumors with common genetic drivers and histopathological diversities detected in clinical specimens,21 (2) they allow CR culture establishment with relative ease, permitting comparisons of tumor-matched FUTCs and cultures (Figure 1A), and (3) we have gained understanding on their tumor subtype-selective drug sensitivities.22
Figure 1.

Murine FUTCs exhibit functional profiles comparable to conditionally reprogrammed cultures
(A) Schematic of comprehensive drug testing of KP and KL tumor-derived cultures to validate the ability of FUTCs to identify subtype-selective treatments.
(B) Viability assessment of FUTCs at 0 and 72 h.
(C–E) Dose-response curves of tumor-matched FUTCs and CR cultures treated for 72 h with (C) idasanutlin (MDM2 antagonist), or (D) tanespimycin (HSP90 inhibitor), or (E) gemcitabine (nucleoside analog).
(F) DSS calculated for (C)–(E) and compared between KL and KP subtypes for both FUTCs and CR cultures.
Data are represented as means ± SEMs. Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
First, we confirmed that FUTCs survive and grow during culture by measuring cellular ATP levels using CellTiter-Glo (CTG). FUTCs from the different tumor groups exhibited significantly higher CTG readouts after 3 days of culture (Figure 1B). Second, we assessed the utility of FUTCs to predict known genotype-selective drug sensitivities. We confirmed that only p53-expressing FUTCs responded to the Mdm2-p53 interaction inhibitor idasanutlin (Figures 1C and 1F). Moreover, KL-derived FUTCs exhibited selective responses to HSP90 inhibition (Figures 1D and 1F), corroborating findings from cultured lung cancer models.22, 23, 24 Lastly, we confirmed that gemcitabine, an approved chemotherapeutic agent for NSCLC treatment, potently inhibited the viability of both KL and KP cells (Figures 1E and 1F), matching previous findings in CR cultures.22 In further agreement with published data,25,26 a synergistic interaction was detected between MEK and ERBB receptor family inhibition in KL, but not in KP FUTCs (Figure 2A). We also could confirm adaptive reactivation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) signaling pathways, detected by the re-phosphorylation of ERK and AKT after extended MEK inhibition, described in many other NSCLC studies, including our murine CR cultures (Figures 2B–2D and S1).
Figure 2.

Murine FUTCs show treatment-adaptive signaling mechanisms
(A) Graphic model for subtype-selective adaptive activation of MAPK and PI3K-AKT pathways in murine Kras mutant NSCLC cultures.
(B) Dose-response curves of FUTCs and CR cultures treated trametinib (TR), afatinib (AF), and combination treatment. For the combination screen, 5 nM TR was used together with a dose series of AF.
(C) DSS calculated for (B) and compared between KL and KP subtypes for both FUTCs and CR cultures.
(D) Immunoblots of KL and KP FUTCs treated with vehicle (C; DMSO) and or treated with 50 nM TR for various time points (4, 24, 48, and 72 h), and probed with indicated antibodies.
Error bars represent ±SEMs. Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Patient-derived FUTCs are amenable for pharmacological profiling
To test a FUTC-based approach with patient-derived cells, we conducted a drug sensitivity and resistance testing (DSRT) study on 19 clinical NSCLC samples (Table S1). Tumor tissue analysis by immunohistochemistry (IHC) revealed inter- and intratumor heterogeneity for EpCAM expression levels, 13 of 19 tumor samples (68%) showed high EpCAM expression, while 11% and 21% samples showed moderate and low EpCAM expression, respectively (Table S2). To identify drug responses selective to tumor-derived EpCAM+ cells, responses were compared to tumor-derived non-epithelial EpCAM– cells, and also to healthy lung tissue-derived EpCAM+ cells as tissue-matched controls (Figure 3A). Similar to the murine FUTCs, the patient-derived FUTCs exhibited good viability in culture (Figures 3B, S2A, and S2B). Furthermore, analysis of KRAS mutations in 6 patient samples revealed an average 3.7-fold enrichment of cancer cells in tumor-derived EpCAM+ cell fractions compared to matched bulk tumor tissues (Figures 3C and S2C). Similarly, EpCAM+ fractions derived from samples carrying an ALK rearrangement exhibited enrichment of ALK fusion-carrying cells (Figure S2D).
Figure 3.

Application of patient-derived FUTCs for pharmacological screening
(A) Schematic of FUTC-based drug sensitivity and resistance testing from clinical specimens.
(B) Viability assessment of patient-derived cells at days 0 and 3 after seeding in 384-well plates. Heatmap showing percentages of total cancer cells and Ki-67+ cancer cells normalized to total cancer cells in the tumor tissue.
(C) Heatmap representing percentage of cancer cells and KRAS mutation variant allele frequencies in patient-matched tumor tissues versus tumor-derived EpCAM+ cells.
(D) DSS (66 compounds) of tumor-derived EpCAM+ cells were clustered by using a complete linkage method, coupled with Euclidean distance measurement.
(E) Heatmap of Pearson’s correlation coefficient values between technical replicates of the same sample, or samples from different patients (left). Representative correlation plots of DSS values, comparing technical replicate screens of PLT68 (right).
(F) Correlation plot comparing the association between the percentage of cancer cells in the tumor tissue and Z′prime factors obtained from respective DSRT screens.
Next, we performed screening with 66 lung cancer-selective drugs on 14 samples, or with smaller compound sets on 5 samples in which only limited EpCAM+ FUTCs could be isolated, and for 18/19 samples, we observed robust drug screening data (Z′ > 0.2). Unsupervised hierarchical clustering of the drug sensitivity scores revealed that technical replicates from the same patient sample cluster together, thus confirming robustness of the screening data generated using FUTCs (Figures 3D and 3E). Through multifactorial analyses, we showed that the robustness of the screening data correlated with the cancer cell fraction in the tumor tissue (Figures 3F and S2E). Moreover, samples with higher percentages of Ki-67+ cancer cells exhibited higher CTG activities ex vivo (Figures 3G and S2E). These results indicate that FUTCs are relevant ex vivo models of tumor tissue and that this single-cell population can be diagnostically profiled before culture establishment.
Responses to approved targeted therapies are exposed with patient-derived FUTCs
To further validate the diagnostic use of the FUTC assay, responses to well-established targeted receptor tyrosine kinase inhibitors were analyzed. Tumor-derived EpCAM+, but not EpCAM–, cells from 4 EGFR mutant patient samples (L858R and E746-A750del) responded to several classes of EGFR inhibitors. Third-generation mutant-selective osimertinib selectively and most effectively reduced the viability of EGFR mutant EpCAM+ tumor cells (drug sensitivity score [DSS] > 10), while the second-generation pan-ERBB inhibitor afatinib (AF) reduced the viability of both tumor and normal EpCAM+ cells (Figures 4A and 4B). These results are consistent with the known superior selectivity of osimertinib toward mutant EGFR.27, 28, 29, 30 Similarly, in FUTCs derived from a clinical sample carrying an activating exon 14 skipping mutation in the MET gene, the ALK/MET inhibitor crizotinib showed the strongest sensitivity (DSS > 10), and this was selective for EpCAM+ tumor cells (Figures 4C and 4D). Importantly, genotype-matched sensitivities were only observed in EpCAM+ cells, not in bulk tumor-derived cell populations, indicating that EpCAM enrichment is required for identification of cancer-specific vulnerabilities (Figures S3A and S3B).
Figure 4.

NSCLC FUTCs predict genotype-matched therapeutic responses
(A) Dose-response curves of gefitinib, osimertinib, and AF in patient-matched tumor-derived EpCAM+ and EpCAM– cells.
(B) DSS for gefitinib, osimertinib, and AF compared between EGFR mutant EpCAM+, EGFR wild-type EpCAM+, tumor EpCAM–, and normal EpCAM+ cells. Each dot represents an independent sample.
(C and D) Bar graph representing the DSS of crizotinib (C) and (D) ceritinib and crizotinib across all of the patient samples.
(E) Dose-response curves of crizotinib in patient-matched tumor-derived EpCAM+ or normal tissue-derived EpCAM+ cells.
(F) Dose-response curves of ceritinib in patient-matched tumor-derived EpCAM+ and EpCAM– cells.
(G) Representative IHC images of E-cadherin and vimentin staining in patient (PLT64)-derived EML4-ALK+ lung tumor tissues. Scale bars correspond to 200 or 20 μm for low or high magnifications, respectively. Boxes indicate areas shown in the higher magnification in a lower row.
Error bars represent ±SEMs. Two-tailed unpaired Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Contrasting with the above target-matched responses, EpCAM+ cells derived from three ALK fusion+ tumors showed low sensitivity to the ALK inhibitors ceritinib and crizotinib (Figures 4E and 4F). Since the epithelial-to-mesenchymal transition (EMT) can explain resistance to ALK inhibitors,31,32 primary tumor tissues were stained with epithelial E-cadherin and mesenchymal vimentin markers (Table S2; 6 of 18 patient samples showed vimentin staining-positive tumor cells). This confirmed EMT in all three ALK+ samples tested in the FUTC assay (Figures 4G and S2E), possibly explaining why these samples demonstrated a poor response to ALK inhibition. Thus, FUTC-based profiling can gauge targeted kinase inhibitor responses and non-responses in patient samples that carry the respective driver mutations.
Pharmacological profiling of KRAS mutant patient-derived FUTCs demonstrates inter- and intratumoral heterogeneity
We next asked how FUTC-based profiling captures sample-selective functional heterogeneity in KRAS mutant samples, which overall represent 25%–30% of NSCLC, yet stratify further due to differences in, among others, histopathology, metabolism, or co-occurring mutations. Comparison of drug responses of 7 KRAS mutant and 11 KRAS wild-type samples identified the MEK inhibitor trametinib (TR) as the most KRAS mutant-selective compound, but also other MAPK inhibitors showed KRAS-selective responses, albeit with a less stringent statistical difference (Figure 5A; Table S3). This aligned with the analysis of lung cancer cell lines in the Genomics of Drug Sensitivity in Cancer (GDSC) database,33 revealing MAPK inhibitors as the most KRAS mutant selective (Figure S3A). Moreover, TR selectively inhibited KRAS mutant cells without affecting tumor-derived EpCAM– and normal tissue-derived EpCAM+ control cells (Figure 5B). Correlation analysis of the drug sensitivities of individual KRAS mutant samples to average drug sensitivities of KRAS wild-type samples indicated intertumoral heterogeneity; each KRAS mutant sample exhibited a unique drug-response profile, and even for shared hits such as TR, the actual efficacies varied between samples. As anticipated, of a total of 27 hits, 14 compounds selective for KRAS mutant FUTCs either targeted the MAPK or the PI3K-AKT pathways (Figures 5C and S3B).
Figure 5.

Functional profiling of KRAS mutant FUTCs reveals inter- and intratumoral heterogeneity
(A) Volcano plot displaying an association between responses to drug candidates and KRAS mutation status. DSS comparisons were made between KRAS mutant cells (n = 7) and KRAS wild-type cells (n = 11) for 66 oncology drugs using a 2-sided Wilcoxon signed-rank test; DSS difference >±5 and p < 0.05 were considered as a hit and highlighted with red color.
(B) DSS for TR compared between different cell populations; each dot represents an independent sample.
(C) The number and target of unique hits identified for an individual patient sample. Boxes are color-coded based on target, and each box represents a single drug.
(D) Correlation plots of comparing DSS obtained from FUTCs derived from different tumor regions of the same patient specimen (PLT62). Drug candidates with DSS difference >±5 are color-coded with red and orange representing region-1 and region-2 selective hits, respectively.
(E) Dose-response curves following indicated MAPK inhibitor treatments in different regions of PLT62.
(F) Correlation plots comparing DSS of 66 oncology drugs as a single agent or in combination with 5 nM TR.
(G) Dot plot displaying DSS of single treatments of MAPK inhibitors (n = 4) and PI3K-AKT-mTOR (mammalian target of rapamycin) inhibitors (n = 6) as a single agent or in combination with 5 nM TR.
(H) Dose-response curves and DSS bar graph of TR, navitoclax (NA), and combination treatment. For the combination screen, 5 nM of TR was used together with the dose series of NA.
(I) Immunoblots of PLT87-derived KRAS mutant FUTCs treated with vehicle (C; DMSO) and or treated with 50 nM TR for various time points (4, 24, 48, and 72 h), and probed with indicated antibodies.
Error bars represent ±SEM. Two-tailed unpaired Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To assess intratumoral functional heterogeneity, drug responses in FUTCs derived from two different regions from the same patient sample (PLT62) were compared. Both tumor regions carried identical genetic alterations (Figure S4A), including STK11 and NFE2L2 mutations known to confer resistance to MEK inhibition in KRAS mutant lung cancer.24, 34 A combinatorial screen of 66 drugs in combination with TR showed that region-2 cells exhibited a relatively higher sensitivity to MAPK inhibitors (Figures 5D and 5E), while region-1 cells showed selective sensitivity to combinatorial treatment with TR plus PI3K-AKT pathway inhibitors (Figures 5F and 5G). Consistent with the drug sensitivity data, cancer cells from region-1 exhibited a higher percentage of pERK+ and p4EBP1+ cells, as well as cells dually stained for these markers, jointly indicating a potential tumor region-selective codependency on MAPK and PI3K-AKT pathways (Figures S4B and S4C). Interestingly, FUTCs from both tumor regions showed combinatorial response to TR plus the BCL-2/BCL-xL inhibitor navitoclax (NA) (Figure 5H), a combination described to convey synthetic lethality in KRAS mutant tumors.35
These data show that FUTC profiling can expose inter- and intratumoral functional heterogeneity in KRAS mutant NSCLC (Figures 5C–5G; Table S2), and tentatively indicates their use in identifying tissue-selective drug combinations. As in murine samples, feedback activation of the MAPK and PI3K-AKT pathways, measured by rebound ERK and AKT phosphorylation, was detected following TR treatment in KRAS mutant FUTCs from PLT87 (Figure 5I). Although analysis of more samples is warranted, these preliminary results suggests that deeper functional profiling of FUTCs may further help to identify drug combinations that target adaptive signaling mechanisms.
Compassionate implementation of a FUTC-based functional diagnostic assay
Finally, we report a case in which FUTC-based drug-response assessments were used to implement compassionate treatment for a patient with refractory metastatic lung cancer. This patient had been diagnosed with EGFR mutant stage IV lung adenocarcinoma (AC) 3.5 years earlier and had since been treated with both erlotinib and osimertinib, yet experienced disease recurrence, as evidenced by the emergence of multiple nodular lesions in the lungs and widespread metastatic lesions, concomitant with increases in the levels of carcinoembryonic antigen (CEA) and neuron-specific enolase (NSE) tumor markers. At this point, a metastatic lesion from the neck region was surgically resected, and FUTCs were used for replicate testing of 233 relevant drugs or drug combinations (Figures 6A–6C).
Figure 6.

FUTC-based functional testing guided treatment of a patient with refractory lung adenocarcinoma
(A) Treatment outline and measured CSE and NSA levels of a patient with EGFR mutant lung adenocarcinoma.
(B) A surgically removed tissue specimen was used for FUTC-based drug testing of 233 single drugs or drug combinations in duplicate.
(C) Correlation plot comparing the DSS from the 2 replicate tests.
(D) The drug sensitivity scores of the top 20 hits, with general toxicity based on activity on normal cells noted.
(E and F) Changes in the level of CEA and NSE (E) and (F) tumor size after carboplatin plus etoposide treatment.
(G) Representative images of hematoxylin and eosin (H&E) and IHC staining performed on surgically resected tumor tissue.
The drug sensitivity screen demonstrated poor response to multiple EGFR inhibitors (Figure S5A), in line with the acquired clinical resistance to erlotinib and osimertinib. To identify effective treatment options with minimal generic toxicities, we implemented a two-step process. In step 1, we identified the 20 treatments with the highest DSSs (Figure 6D). In step 2, we filtered the potent hits from step 1, based on whether the response was selective toward the patient’s cancer cells over normal lung CR cells (n = 6) derived from independent patients (Figure S5B). This allowed narrowing down of the list and eliminated 15 treatments with generic toxicities. From this analysis, we identified five treatments with selective potency toward the tumor-derived cells, including disulfiram and carboplatin combined with etoposide (Figure S5C). Conversely, the FUTC-based profiling results were in agreement with the clinical non-responses to therapies targeting PI3K and ALK, even though the tumor cells harbored activating mutations in PIK3CA and showed overexpression of ALK (Figures 6G, S5D, and S5E).
Based on these results, the scheduled carboplatin plus pemetrexed, which showed inferior sensitivity compared with carboplatin plus etoposide, was changed and the patient received disulfiram and five cycles of carboplatin plus etoposide with or without disulfiram. The patient received carboplatin/etoposide/disulfiram treatment every time an increase in the CEA/NSE level was observed. This resulted in a therapeutic benefit from the FUTC-defined chemotherapy during the first 3 treatment cycles (for the total span of 7 months), as detected by a decrease in tumor size analyzed by computed tomography imaging, as well as reduced levels of CEA and NSE (Figures 6E, 6F, and S6A–S6E). Interestingly, IHC analysis of the tumor tissue done a few months following treatment onset revealed two spatially intermixed subtypes of tumor populations: (1) an SCLC population with NSE expression and (2) a squamous cell carcinoma (SCC) population with CEA expression (Figure 6G). Histological transformation to SCLC or SCC are known acquired resistance mechanisms in EGFR mutant AC patients treated with EGFR inhibitors,36,37 and platinum plus etoposide is an effective treatment strategy for SCLC patients,38,39 which the FUTC-based drug testing had identified independently of the histological analyses. However, in line with the poor prognosis of SCLC, during the last two treatment cycles of carboplatin plus etoposide, NSE levels showed an initial decrease and then a steep increase, while CEA remained stable, indicating eventual chemoresistance and progression of the SCLC subpopulation, confirmed by pathology analysis of a new, recurrent metastatic lymph node biopsy (collected January 2020; Figures S6F and S6G). This compassionate case therefore demonstrates that therapeutic screening of FUTCs can be used to identify effective treatments, many of which cannot otherwise be predicted based on molecular diagnostics, in an individualized manner.
Discussion
Major advances have been made in the past decade in the area of precision cancer medicine and the development of diagnostic innovations, which includes solutions for the ex vivo propagation of carcinoma cells. However, unlike murine tumor cells, which generally adapt well to primary cultures, the majority of the patient-derived cultures we established represented non-malignant cells, corroborating conclusions from other recent studies.17,19,20 While the success rates of establishing cultures from pleural effusion are reportedly higher,4,13 their use is limited, as only ∼10% of all NSCLC patients present with malignant pleural effusions.36 Other preclinical models such as tumor organoids or xenografts also have suboptimal success rates of 20%–70% or 30%–40%, respectively,11,15,16,40,41 but these have an additional issue of long establishment times of 1–3 months for organoids or 2–10 months for xenografts.4,11,15,16,40 Prolonged ex vivo propagation in artificial conditions may cause a functional drift of cancer cells from their original identity, and also the patient’s tumor may evolve during this time, further compromising the compatibility of preclinical cultures with the diagnostic clinical decision-making process.
To overcome such limitations, and inspired by precision medicine approaches using liquid biopsy-sampled leukemic cells, we set out to assess whether uncultured tumor cell populations could be used for pharmacological profiling immediately following isolation, before their attachment to cell culture plates—the FUTC approach. We asked whether the study of epithelial cells in solution may be compromised (e.g., due to apoptotic priming related to anoikis). Encouragingly, both murine and human FUTCs demonstrated sustained or increased cell viability during the first 3 days of ex vivo culture. In addition, murine Kras mutant FUTCs were shown to mimic the MEKi-associated resistance bypass mechanisms and related drug sensitivities previously identified in primary cultures derived from these models. These validating findings supported further translational implementation of the approach, and target-matched drug responses were confirmed in FUTCs derived from surgical tissue samples carrying mutant EGFR, MET, or RAS, or rearrangement in ALK, with a positive correlation between cancer cell percentage and drug screen data quality. In KRAS-driven tumors, heterogeneity was observed at both inter- and intratumoral levels, which could be explained by underlying differences in biologies such as co-occurring mutations,26 metabolic dependencies explained (for example) by KRAS allelic copy gains,42 or phenotypes influenced by the tumor progenitor cells, such as tumor histopathology or oncogenic signaling activities.10,21 These data overall indicate that the FUTC assay holds promise for further diagnostic development, particularly for samples with high cancer cellularity. Nevertheless, for samples exhibiting lower cancer cellularity, a number of options can be explored to enhance the data reliability, for example, increasing the number of cells/well, increasing the number of concentration points per drug or technical replicates/compound, or testing multiple drugs for a particular target.
In previous studies, ex vivo drug-response testing on heterogeneous primary cancer and stromal cell mixtures failed to culminate in clinical benefits, possibly because these earlier assays did not involve the enrichment of tumor cells.43,44 In the FUTC assay, cancer cell-selective drug profiling is rather done on EpCAM+ epithelial cells isolated from tumor tissue, with parallel analysis of epithelial cells from adjacent normal tissue serving as a toxicity indicator. A recent study relatedly demonstrated that dynamic BH3 protein profiling reliably reveals drug-elicited apoptotic pathway signaling in freshly isolated breast and colorectal cancer tumor cells, further underscoring our conclusions.45 A caveat of the FUTC approach is that not all tumor epithelia express EpCAM, with 40%–85% of ACs and 85%–98% of SCCs reported to be EpCAM+.46, 47, 48, 49 To circumvent this, we believe that EpCAM– cancer cells can be enriched by the use of magnetic microbeads labeled with a cocktail of antibodies that will identify cells of epithelial origin (e.g., cytokeratin 8, cytokeratin 18).13 Nevertheless, in our dataset, 13 of 19 tumor tissue samples (68%) showed high EpCAM expression levels. Interestingly, genotype-matched drug sensitivities were observed in samples exhibiting moderate to low EpCAM expression levels. In addition, tested samples (8 of 8) showed enrichment of cancer cells following EpCAM selection, and while alternative purification methods are being investigated, EpCAM remains the gold standard for the capture of blood-circulating tumor cells.50, 51, 52 Implementing the EpCAM approach, reliable drug-response data, often matched to the targeted drivers, was obtained from 19 of 20 cases, highlighting the robustness of the assay. Even for drug screens exhibiting Z′ factor values <0.5, we observed reliable dose-response data and genotype-selective drug sensitivities in respective samples. While our preliminary results suggest that the FUTC approach can be adapted to smaller biopsy samples, rigorous investigation is necessary to further optimize its diagnostic utility.
The FUTC assay was implemented to guide the compassionate treatment of a stage IV EGFR mutant lung AC patient, with cells derived from an operable metastatic tumor in the neck area. While on EGFR inhibitor treatment, the patient had developed an aggressive disease with multiple metastatic lesions and genomic alterations in, among others, TP53, RB1, PIK3CA, and PTEN, commonly associated with EGFR inhibitor resistance. Even though the combined TP53 and RB1 mutations may have suggested a risk for histological transformation to SCLC,36,52 the official diagnosis was AC at the time of FUTC profiling. Nevertheless, in light of FUTC-based drug sensitivity data and the identified mutations, the patient received carboplatin plus etoposide treatment, generally recommended for the treatment of SCLC patients,19 leading to a substantial reduction in tumor burden and level of tumor markers. As far as we know, this represented a rare clinical case, with only one other report describing dual transformation of EGFR mutant lung ACs to NSE-marked SCLC and CEA-marked SCC.53 These cell populations were found intermixed in the biopsy sample, as if coexisting in symbiosis, perhaps implying that these were derived from a common progenitor cell. In addition to chemotherapy, the patient received another FUTC-guided treatment, the acetaldehyde dehydrogenase (ALDH2) inhibitor disulfiram, but its dosing was reduced as it is associated with delirium, possibly because the patient carried a variant ALDH2∗2 allele.54 The ex vivo approach also affirmed the previously known resistance to EGFR inhibitors and it prospectively predicted the non-responses to PI3K and ALK inhibitors, for which the patient was later treated based on molecular features. FUTC-based drug testing therefore both predicted the intrinsic resistance and sensitivity to clinically actionable regimens of the cancer.
This work demonstrates that tumor-derived FUTCs have promise for clinical application. The fast throughput nature of the assay, bypass of ex vivo culture, and ability to guide personalized treatment choices render the assay attractive for wider diagnostic use. However, to more broadly assess the clinical feasibility, more clinical profiling cases would need to be actioned, particularly for tumors that lack targetable drivers or metastatic-stage tumors that lack treatment options. It is important to note that since FUTCs allow for the identification of effective combinatorial treatments, the assay can be used to assess polytherapy options to circumvent the emergence of resistance to single treatments. Aligning with our findings, a Phase I clinical trial to test the safety of a triple combination of osimertinib, platinum, and etoposide to circumvent small cell conversion is under way for EGFR mutant patients (NCT03567642). In conclusion, FUTC-based functional profiling enables rapid assessment of tumor-selective drug responses and shows promise for further development as a personalized diagnostic assay to complement genomic and histopathological profiling.
Limitations of the study
While our study shows promising results for the FUTC drug screening approach, there are aspects that require further studies for broader application. First, the cancer cellularity of the profiled solid tumor tissue samples affect the drug screen data quality, and this can limit profiling success in some cases. Second, although our preliminary data using a smaller biopsy sample showed promise, this study aspect demands more extensive exploration to transform the FUTC approach into a routine diagnostic test. Finally, the translational functional diagnostic potential of the FUTC assay requires systematic validation in a clinical study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p63 | Abcam | Cat#ab124762; RRID: AB_10971840 |
| NKX2-1 | Abcam | Cat#ab133638; RRID: AB_2734144 |
| LKB1 | Cell Signaling Technology | Cat#13031; RRID: AB_2716796 |
| Ki-67 | Thermo Fisher Scientific | RM-9106-S0; RRID: AB_2341197 |
| pERK (Thr202/Tyr204) | Cell Signaling Technology | Cat#4370; RRID: AB_2315112 |
| pAKT (Ser473) | Cell Signaling Technology | Cat#4060; RRID: AB_2315049 |
| p4EBP1 (Thr37/46) | Cell Signaling Technology | Cat#2855; RRID :AB_560835 |
| E-cadherin | Cell Signaling Technology | Cat#3195; RRID: AB_2291471 |
| Vimentin | Abcam | Cat#ab92547; RRID: AB_10562134 |
| pEGFR (Tyr 1068) | Cell Signaling Technology | Cat#2234; RRID: AB_331701 |
| pERBB2 (Tyr1211/1222) | Cell Signaling Technology | Cat#2243; RRID: AB_490899 |
| pERBB3 (Tyr1289) | Cell Signaling Technology | Cat#4791; RRID: AB_2099709 |
| α-Tubulin | Cell Signaling Technology | Cat#2125; RRID: AB_2619646 |
| AKT | Cell Signaling Technology | Cat#2920; RRID: AB_1147620 |
| ERK | Cell Signaling Technology | Cat#9107; RRID: AB_10695739 |
| ALK | Cell Signaling Technology | Cat#3633; RRID: AB_11127207 |
| EpCAM | Abcam | Cat#71916; RRID: AB_1603782 |
| Biological samples | ||
| CR cultures derived from murine NSCLC tumors obtained from KrasG12D;Lkb1fl/fl (KL) mouse model | This Paper | N/A |
| FUTCs derived from murine NSCLC tumors obtained from KrasG12D;p53fl/fl (KP) mouse model | This Paper | N/A |
| CR cultures derived from murine NSCLC tumors obtained from KrasG12D;p53fl/fl (KP) mouse model | This Paper | N/A |
| Human normal lung CR cultures derived from tumor-adjacent normal lung tissue lung | This Paper | N/A |
| Human lung tumor-derived fresh uncultured tumor cells | This Paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase | Sigma | Cat#C2674-1G |
| Dispase | Invitrogen | Cat#17105041 |
| BSA | Sigma | Cat#A2153 |
| EpCAM (CD326) MicroBeads, mouse | Miltenyi Biotec | Cat#130-105-958 |
| DMEM | GIBCO | Cat#42430-025 |
| F12 | GIBCO | Cat#21765-029 |
| Y-27632, ROCK inhibitor | ENZO life sciences | Cat#ALX-270-333-M005 |
| Adenine | Sigma | Cat#A2786 |
| hrEGF | BD Bioscience | Cat#354052 |
| Insulin | Sigma | Cat#I2643 |
| Hydrocortisone | Sigma | Cat#H4001 |
| Cholera toxin | List Biological laboratory | Cat#100B |
| CellTiter-Glo | Promega | Cat#G9243 |
| EpCAM (CD326) MicroBeads, human | Miltenyi Biotec | Cat#130-061-101 |
| Polyacrylamide gels, 4–20% | Bio-Rad | Cat#4561096 |
| HBSS | Sigma | Cat#H6648 |
| Critical commercial assays | ||
| Tumor Dissociation Kit, human | Miltenyi Biotec | Cat#130-095-929 |
| BCA Protein Assay | G Biosciences | Cat#786-570 |
| gentleMACS Dissociator | Miltenyi Biotec | Cat#130-093-235 |
| Deposited data | ||
| Next-generation sequencing data for lung cancer patients | Mendeley Data | https://data.mendeley.com/datasets/bb4vfjnfkf/1 |
| Experimental models: Cell lines | ||
| J2 strain of Swiss-3T3 mouse fibroblasts | Kindly provided by Prof. Olli Kallioniemi at Sci Life Lab | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: KP: KrasLSL.G12D/+;p53fl/fl | Jackson laboratories | N/A |
| Mouse: KL: KrasLSL.G12D/+;Lkb1fl/fl | Jackson laboratories and Ron DePinho (MD Anderson). | N/A |
| Software and algorithms | ||
| BREEZE pipeline | Potdar et al. 55 | https://breeze.fimm.fi/28780_mc40mdewodkwmcaxnje4ode3nje0/index.php |
| Aiforia | N/A | https://www.aiforia.com/ |
| Spa-RQ | Bao et al. 56 | https://bitbucket.org/MagoBitbucket/spa-rq-tools/downloads/ |
| GDSC | Yang et al.33 | https://www.cancerrxgene.org/ |
| GraphPad Prism 9.1.0 | GraphPad | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Emmy Verschuren (emmy.verschuren@helsinki.fi).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell lines
The J2 strain of Swiss-3T3 mouse fibroblasts was kindly provided by Prof. Olli Kallioniemi at SciLifeLab. To culture 3T3 cells, we used DMEM supplemented with 10% FBS and cultures were maintained at 37°C and 5% CO2. 3T3 cells were passaged two to three times per week, and irradiated cells were used as a feeder layer for the propagation of CR cultures until the cells reached passage ten. Cultures were routinely checked for mycoplasma infection.
Animals
Mouse models harboring a conditional Kras mutant allele (KrasLSL-G12D/+) or loss-of-function TRp53 allele (p53fl/fl) were procured from the Jackson laboratories. Mouse models harboring loss-of-function Stk11/Lkb1 allele (Lkb1fl/fl) were received from Ron DePinho (MD Anderson). Around 10 week old KL and KP mice were intranasally administered with progenitor cell-directed Ad5-Cre viruses.21 Moribund mice were sacrificed by cervical dislocation, and tumor-bearing lungs were processed for FUTC-based pharmacological profiling. All mice used in this study were housed in the Laboratory Animal Center of the University of Helsinki. The facility provided animal care services including feeding, cage cleaning, health assessment, and room sanitation. Animals of both sexes were utilized for this work.
Preparation of murine lung-tumor derived FUTCs
Experiments involving KP and KL genetically engineered mouse models were conducted following the guidelines from the Finnish National Board of Animal Experimentation (permit number ESAVI/9752/04.10.07/2015) and all procedures were performed as described previously.21 For FUTC isolation, individual lung tumors were manually minced using a sterile scalpel and then enzymatically digested in HBSS containing 2 mg/ml collagenase, 0.3 mg/ml dispase and 10 mg/ml bovine serum albumin (BSA), using a Miltenyi gentleMACS dissociator, as described.22 Lung 2.01 program on MACS dissociator was used for cell homogenization and subsequently cells were filtered 70 μm cell strainers and centrifuged at 200xg for 10 min at 4°C. To separate EpCAM+ cells from tumor tissue dissociates, EpCAM (CD326) MicroBeads-dependent enrichment was implemented (130-105-958, MACS), as per the manufacturer’s instructions. Briefly, single cells obtained from tumor tissue were incubated for 15 minutes with EpCAM (CD326) microbeads in the refrigerator (2-8°C). These EpCAM microbead-labeled cells were subjected to magnetic separation using MACS LS Columns and MACS Magnetic Separator. LS Columns were placed on a Magnetic Separator and EpCAM microbead-labeled cells were applied onto the column. Unlabeled cells were collected in the flow-through while magnetically labeled cells were pushed out of the column using a plunger. These microbead-labeled EpCAM+ FUTCs were then either directly utilized for drug response assessment or for establishment of CR cultures. Detailed CR culture procedures are provided in the CR cultures method section.
Patient sample processing
Clinical samples used in this study were collected with patient’s informed consent at the Helsinki University Central Hospital (HUCH) and procedures were conducted in accordance with protocol approved by the Coordinating Ethics Committee of the University of Helsinki (License numbers: 85/13/03/00/2015 and HUS-1204-2019). Clinical features of cases used in this study are shown in Tables 1 and S1. All specimens used in this study were collected from adult patients; gender and age of the patients were not considered as significant factors in this study. To process clinical specimens for FUTC assay, within 30 mins after lobectomy, tumor and tumor-adjacent healthy lung tissue samples were transported from the hospital to the cell culture facility in cold HBSS. The tissue sample was divided into four parts, one each for DNA isolation, lysate preparation for western blot, histological analysis, and FUTC isolation. Subsequently, for single cell suspension, tumor tissue was manually minced using a sterile scalpel, and then enzymatically digested using a Tumor Dissociation Kit (Miltenyi, 130-095-929) and a gentleMACS Dissociator (130-093-235) following the manufacturer’s instructions. Single cell suspensions were then subjected to separation of EpCAM+ and EpCAM– cell fractions using Miltenyi’s human EpCAM (CD326) MicroBeads (130-061-101). EpCAM+/– cells derived from tumor tissue, and EpCAM+ cells derived from normal tissue were used for performing DSRT.
CR cultures
Both murine and human EpCAM+ cells were propagated using a Conditional Reprogramming (CR) culture protocol. In brief, EpCAM+ cells were plated on irradiated (30 gray) 3T3 cells in F-medium composed of 1:3 v/v DMEM: F-12 nutrient HAM supplemented with 5% FBS, 10 ng/ml EGF (BD Biosciences; 354052), 5 μg/ml insulin (Sigma; I2643), 24 μg/ml adenine (Sigma; A2786), 0.4 μg/ml hydrocortisone (Sigma; H4001), 10 ng/ml cholera toxin (List Biological laboratory; 100B), and 10 μM ROCK inhibitor (Y-27632; ENZO). All CR cultures were maintained at 37°C and 5% CO2. When CR cultures reached 80% confluence, they were differentially trypsinized using a two-step procedure the first to remove loosely attached feeder cells, and the second to trypsinize epithelial cells.
Method details
DSRT assay for murine NSCLC models
Tumor-derived fresh uncultured EpCAM+ cells and CR cultures at passage 4 were used for performing DSRT, as described previously.22 Briefly, 2500 FUTCs or 1500 CR cells per well were seeded in 384-well plates using a Biotek MultiFlo FX RAD dispenser in 20 μL F-medium. Following 24 h incubation, drugs were manually dispensed in 10 μL F-medium at eight concentrations, covering a 10,000-fold concentration range in triplicates. Additionally, multiple replicates of cells were treated with 0.1% dimethyl sulfoxide (DMSO) or 100 μM benzethonium chloride, serving as negative and positive controls, respectively. Following 72 h incubation at 37°C, 30 μL CellTiter-Glo reagent (Promega) was added in each well, and cell viability was measured using a PheraStar FS plate reader (BMG Labtech).
DSRT of patient-derived cells
To screen 66 lung cancer selective drugs, DSRT plates were prepared in advance by dispensing compounds into 384-well plates (3712, Corning) using an Echo 550 liquid handler (Labcyte), at five concentrations covering a 10,000-fold concentration range. As negative and positive controls, 0.1% DMSO and 100 μM benzethonium chloride were included in wells scattered across the plates. Pressurized Storage Pods (Roylan Developments Ltd.) were used to store pre-drugged DSRT plates and were used within one month. Tumor- and normal tissue-derived cells (2500 cells per well) were seeded in pre-drugged DSRT plates using a Biotek MultiFlo FX RAD dispenser, in 25 μL F-medium. Following 72 h incubation at 37°C, 25 μL CellTiter-Glo reagent (Promega) was added in each well, and cell viability was measured using a PheraStar FS plate reader (BMG Labtech). When the cell numbers were not sufficient for performing the 66-drug screen, drug screening for molecular target-selective drugs (5 - 10) were performed by seeding 2500 FUTCs/well in 384-well plates, and on the following day cells were exposed to drugs.
For the compassionate care case study, patient-derived tumor cells were directly utilized for DSRT without performing EpCAM-based immunomagnetic separation step as cytological and histological assessment of frozen tumor tissue sections prior to drug testing initiation revealed high (< 90%) cancer cellularity. In brief, tumor tissue within 60 minutes of surgical excision was embedded in Optimal Cutting Temperature (OCT) media and cryosections cut from the OCT block were stained with H&E. Stained sections were analyzed by a clinical pulmonary pathologist.
Western blotting
Reference tumor tissues, FUTC pellets, or CR cell pellets were lysed with RIPA buffer supplemented with fresh protease and phosphatase inhibitors (Roche). For protein quantification, BCA Protein Assay was used (G Biosciences; 786-570). Western blotting was performed using 20 μg of protein samples, using precast 4%–20% polyacrylamide gel (Bio-Rad, 4561096) for electrophoresis and PVDF membrane (Millipore, IPFL00010) for transfer. Western blot membranes were blocked using Odyssey Blocking Buffer (927-40000) at room temperature for 30 min, probed with primary antibodies at room temperature for 2 h or overnight at 4°C, and finally probed with IRDye secondary antibodies (LI-COR) diluted 1:10000 in Odyssey Blocking Buffer and scanned using an Odyssey infrared imager (LI-COR). We used Image Studio Lite software for western blot quantification.
Immunohistochemistry (IHC) analyses
Tissue processing and IHC procedures were performed as described previously22; all samples were processed using the equivalent experimental conditions. The Pannoramic 250 digital slide scanner (3DHISTECH, Budapest, Hungary) was used to acquire whole slide scans of stained tumor sections using a 20x objective, and the Pannoramic Viewer (3DHISTECH Ltd) was used at 1:4 magnification to export TIFF images.
To quantify the percentage of malignant epithelial region or cancer cellularity per tumor specimen, H&E stained whole tissue sections were uploaded to the deep learning-based image analysis cloud service, Aiforia (Fimmic Oy, Helsinki, Finland). Regional training sets were manually created by an imaging expert, by selecting regions-of-interest (206 regions) across all H&E-stained samples as two-layer sets, namely a parental layer for whole tumor tissue annotation and a layer for tumor epithelium-only annotation that was further guided by E-Cadherin staining on neighboring sections. The manually annotated tissue regions were used to train the Aiforia algorithm; multiple training-learning cycles were conducted until the predictions on all samples reached a satisfactory outcome. A total of 22 annotations for tissue regions and 184 annotations for tumor epithelial regions were included in the training. The results were validated by a clinical pulmonary pathologist, via the validation functions of Aiforia, and percentages of tumor epithelial regions per specimen were calculated. To quantify the percentage of cancer cells exhibiting EpCAM expression and to classify cancer cells based on the EpCAM intensity, we implemented a two-step process using the Aiforia algorithm. First, based on staining intensity and specificity cells are manually annotated as strongly or weakly positive. The manually annotated tissue regions exhibiting different patterns of EpCAM expression were used to train the Aiforia algorithm; multiple training-learning cycles were conducted until the predictions on all samples reached a satisfactory outcome. In the second step, trained Aiforia algorithm was used to calculate the percentage of cancer cells exhibiting strong or weak EpCAM positivity. To quantify the percentages of proliferating cancer cells, both region- and cell-based annotation tools provided by Aiforia were used. A two-layer training set was created, composed of a parental layer of tumor epithelial regions (based on H&E staining) and a cell-based layer to assign (i) proliferating (Ki-67 positive) and (ii) non-proliferating (Ki-67 negative) tumor cells. Multiple training-learning cycles were performed until the deep-learning prediction reached satisfactory outcome and then percentage of Ki-67 positivity were calculated using total number of proliferating and non-proliferating tumor cells.
To assess MAPK and PI3K-AKT-mTOR signaling pathway activities in tumor regions, images stained with phosphorylated ERK, and 4EBP1 were registered to directly adjacent E-cadherin-stained sections, and the overlap of staining was quantified using a spatial quantification and registration image analysis tool Spa-RQ, as described in Bao et al.55 To assess the co-activation between MAPK and PI3K-AKT-mTOR signaling pathways, images stained with phosphorylated ERK and 4EBP1 were registered to each other and the region of staining overlap was quantified by Spa-RQ. Spa-RQ employed a simple thresholding segmentation algorithm, a consistent threshold for each staining was applied to all the samples. The staining specificity and accuracy of the Spa-RQ was validated by a clinical pulmonary pathologist. To assess EMT, all tumor tissue sections were visually inspected for co-positivity of E-cadherin and vimentin in tumor epithelium-only regions. Tissue sections stained for phosphorylated EGFR, ERBB2, or ERBB3 proteins were visually inspected for positivity in tumor only regions.
Quantification and statistical analysis
DSRT data analysis
To determine screen-to-screen consistency of DSRT profiling, Z-prime factors were calculated by normalizing the raw luminescence values of drug-treated wells with positive and negative controls. Screens showing a Z-prime factor < 0.2 were not considered for further analysis (one of 20 cases). As we were analyzing dose response series with multiple assay points for each drug, we applied a slightly lower threshold for the Z-prime factor (> 0.2) than the cutoff that is most frequently used in standard high throughput single data point assays (> 0.5). Screens with Z-prime factor values from 0.2 to 0.5 still exhibited sigmoidal dose response curves and data consistency was observed across technical replicates, and with respect to the tumor genotype. Dose-response curves were plotted using a Marquardt-Levenberg algorithm via the in-house bioinformatic ‘Breeze’ pipeline.57 Next, dose–response curve parameters were employed to calculate the Drug Sensitivity Score (DSS), as described.58
Genomic sequencing and data analysis
A DNeasy Blood & Tissue kit (QIAGEN) was used to extract genomic DNA from healthy lung and tumor tissue samples and from CR cultures (Table 1). Genomic dsDNA (382-500 ng) was fragmented with a Covaris E220 evolution instrument (Covaris) to a mean fragment size of 200 base pairs (bp). For sample library preparation and enrichment, a KAPA Hyper library preparation kit was used, following the SeqCap EZ HyperCap Workflow User’s Guide Version 1.0 (Roche Nimblegen). In brief, pre-capture amplification was performed using 9 cycles, and captures were performed in multiplexes of 3 to 4 samples using 0.667-1 μg of each library. To identify somatic mutations, targeted next-generation sequencing was performed using the Illumina HiSeq2500 system in HiSeq high output mode using v4 chemistry or HiSeq Rapid run mode using v2 chemistry (Illumina), with the NimbleGen Cancer Panel to capture the exons of 578 cancer-related genes. NimbleGen probes used were 120522_HG19_Onco_R_EZ. Instead of SeqCap HE-Oligos, we used xGen Universal Blocking oligo TS mix (IDT). For post-capture amplification, ten cycles were used in two replicate reactions. The library was quantified for sequencing using the 2100 Bioanalyzer High sensitivity kit. Read length for the paired-end run was 2x101 bp. Pre-processing of short read data was done using the Trimmomatic software and included correction of the sequence data for adaptor sequences, bases with low quality, and reads less than 36 bp in length. The BWA-MEM algorithm was then used to map paired-end reads passing the pre-processing onto the human reference genome build 38 (EnsEMBL v82). Finally, variants were called according to the GATK best practice for somatic short variants (version 3.5.1), supplemented with cross-sample contamination and sequencing artifact filtering. In the process, tumor samples (tissue and CR culture) were paired with their patient-matched normal samples and variant calls were filtered against a panel of normals generated from the exome data of 24 healthy unrelated Finnish individuals sequenced in-house earlier.
Annotation for single nucleotide variants and short indels was performed using the Annovar tool against the RefGene database. In brief, variants called from samples were filtered for false-positives by removing variants not passing all GATK filters, residing in intronic and intergenic regions, and causing a synonymous or non-frameshift change as well as variant with a minor allele frequency ≥ 1% in the EPS, 1KG, general ExAC (ExAC), East Asian ExAC (ExAC_EAS), non-Finnish European ExAC (ExAC_NFE), Finnish ExAC (ExAC_FIN) databases, strand odd ratio for single nucleotide variants ≥ 3.00, and strand odd ratio for indels ≥ 11.00, coverage ≤ 10, and variant quality value ≤ 40. Finally, variants were removed if the variant allele frequency between the tumor and normal was < 2%. Tools used in the variant calling process and their versions have been outlined earlier.56 Surgical samples were used to generate information for 578 cancer-related genes and data generated using this methodology is presented in Table 1.
NGS analysis of lung cancer biopsies
Upfront NGS screenings were routinely performed for all the AC and ASC patients but not for SCC patients at Helsinki University Central Hospital (HUCH) before treatment initiation or surgery. DNA was extracted from paraffin samples after pre-treatments with the Maxwell RSC (Promega), according to the manufacturer’s instructions. The concentration of the DNA samples was measured using the NanoDrop ND 1000 (Thermo Fisher Scientific). Amplification-based NGS was performed to identify mutations in all exons of PIK3CA, EGFR, KIT, KRAS, MET, NRAS, and PDGFRA as well as exons 11-15 of BRAF-gene by using an in-house gene panel. Briefly, multiplex PCR was performed with 10 ng of genomic DNA, and then adapters were ligated to each PCR product. The amplicon libraries were constructed, and the quantity of amplicon libraries was determined using the Ion Library TaqMan Quantitation Kit (Thermo Fisher Scientific). Each library was diluted to a concentration of 10 pM, and pooled in equal volumes. Template preparation was performed with an Ion Chef Instrument, and sequencing was carried out on an Ion S5 System with PI Chip. Data was generated using the Torrent Suite Software version 5.8 (Thermo Fisher Scientific). The Ion Reporter Software version 4.6 (Thermo Fisher Scientific) was used to filter out non-coding and polymorphic variants. Mean sequencing depth ≥ 1000 was considered as successful sequencing and called variants were only accepted if allele frequency ≥ 1%. All variants listed after filtering were visualized in the Integrative Genomics Viewer (IGV) to manually discard alterations generated by incorrect calling. Core needle biopsy samples were used to generate information for 8 lung cancer-related genes and data generated using this methodology is presented in Table S1 and Figure S3.
KRAS mutation analysis
Snap-frozen cell pellets of tumor-derived EpCAM+ and EpCAM– cells were utilized to detect KRAS mutation variant allele frequencies using digital droplet PCR. DNA was extracted from cell pellets with the Maxwell RSC (Promega), according to the manufacturer’s instructions. The concentration of the DNA samples was measured using a NanoDrop ND 1000 (Thermo Fisher Scientific). Targeted WT and mutation probes for the KRAS mutation sites G12 and G13 were designed according to Bio-Rad specifications (https://www.bio-rad.com). For each reaction, 2 μl of the extracted DNA was used and performed in duplicate. The QX200 Droplet Generator partitioned the samples (20 μl into 20,000 droplets) for PCR amplification. Following amplification using a thermal cycler, droplets from each sample were analyzed individually on the QX200 Droplet Reader, where positive and negative droplets were counted to provide absolute quantification of the target DNA in digital form. The results were analyzed with the QuantaSoft Analysis Pro Software (v.1.0, Bio-Rad). Data generated for KRAS mutation variant allele frequencies is presented in Table S1 and Figure S3.
Statistical analyses
To assess statistical significance, a Student’s t test, nonparametric Mann-Whitney test, or a two-sided Wilcoxon signed-rank test (Figure 6A) were used. The results were considered statistically significant if a p value < 0.05 was observed. Error bars indicate standard deviation or standard error of the mean. Pearson’s correlation coefficients were used to assess the significance of correlations and displayed in the XY plots. All the graphs presented here, including dose-response curve fits were generated using GraphPad Prism 8 (GraphPad Software Inc). Statistical details and number of samples used for a particular result can be found in the figure legends.
Acknowledgments
We dedicate this work to the memory of Dyanne Søraas, whose courage and willpower continues to inspire us to advance lung cancer diagnostics and treatment. We acknowledge all of the patients who supported our research by consenting access to clinical specimens. We are thankful to Matti Kankainen and Soili Kytölä for advice on the analysis of next-generation sequencing (NGS) and droplet digital PCR (ddPCR) data, Merja Räsänen for processing patient consents, Ashwini Yadav for assistance on KRAS mutation-drug-response association analysis, Astrid Murumägi for reagents and guidance on the establishment of the CR culture protocol, and Vishal Sinha for assistance in drug-response correlation analysis. We thank the Sequencing Core Facility at FIMM, HiLIFE, at the University of Helsinki for NGS analysis, and the Laboratory of Genetics, HUS Diagnostic Center, HUSLAB, at the Helsinki University Hospital for NGS and ddPCR analysis. We thank the HiLIFE Laboratory Animal Centre Core Facility at the University of Helsinki for animal husbandry care and guidance. We thank the thoracic pathologists who helped with the clinical specimen selection, the Laboratory Animal Centre for husbandry support, the FIMM WebMicroscope facility for scanning histological slides, and the FIMM High-Throughput Biomedicine facility for drug screening resources. We thank members of the Verschuren and Wennerberg labs for support and guidance. The study received financial support from the University of Helsinki Integrative Life Science doctoral program (S.S.T.); the Ida Montinin Foundation (S.S.T.); the Väinö and Laina Kivi Foundation (S.S.T.); HUSLAB and the Finnish Medical Foundation (M.I.M.); the Academy of Finland (E.W.V.; grants 307111 and 328473); the Novo Nordisk Foundation (K.W.; the Novo Nordisk Foundation Center for Stem Cell Biology, DanStem; grant no. NNF17CC0027852); and a HiPOC 2020 grant from the University of Helsinki (E.W.V.). The Verschuren lab receives funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 859860 and the iCAN Digital Precision Cancer Medicine Flagship, Helsinki, Finland.
Author contributions
S.S.T., K.W., and E.W.V. conceived and designed the study; S.S.T. performed experiments for the development of the FUTC-based drug screening assay; J.B., A.H., and N.L. performed the experiments related to the IHC analysis; S.S.T., S.P., and J.B. performed the data analyses; L.S. advised on the compassionate case study design; S.S.T., E.W.V., and K.W. conducted the data interpretation; J.R., A.K., and M.I.M. assisted in collecting the clinical data and the primary tissue workflow, and received patients’ consents; M.I.M. and J.L. performed the clinical pathology analyses; and S.S.T., K.W., and E.W.V. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: August 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100373.
Contributor Information
Krister Wennerberg, Email: krister.wennerberg@bric.ku.dk.
Emmy W. Verschuren, Email: emmy.verschuren@helsinki.fi.
Supplemental information
Data and code availability
The authors declare that the main data supporting the findings of this study are available within the article. The data presented in Figure S3A of this study are available from the GDSC1 database portal (GDSC: https://www.cancerrxgene.org/). Processed NGS data is available online from the Mendeley Data (Mendeley Data: https://doi.org/10.17632/bb4vfjnfkf.1). Raw NGS data and IHC images generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
References
- 1.Francies H.E., McDermott U., Garnett M.J. Genomics-guided pre-clinical development of cancer therapies. Nat. Cancer. 2020;1:482–492. doi: 10.1038/s43018-020-0067-x. [DOI] [PubMed] [Google Scholar]
- 2.Tannock I.F., Hickman J.A. Molecular screening to select therapy for advanced cancer? Ann. Oncol. 2019;30:661–663. doi: 10.1093/annonc/mdz088. [DOI] [PubMed] [Google Scholar]
- 3.van der Velden D.L., Hoes L.R., van der Wijngaart H., van Berge Henegouwen J.M., van Werkhoven E., Roepman P., Schilsky R.L., de Leng W.W.J., Huitema A.D.R., Nuijen B. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature. 2019;574:127–131. doi: 10.1038/s41586-019-1600-x. [DOI] [PubMed] [Google Scholar]
- 4.Crystal A.S., Shaw A.T., Sequist L.V., Friboulet L., Niederst M.J., Lockerman E.L., Frias R.L., Gainor J.F., Amzallag A., Greninger P. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J.K., Liu Z., Sa J.K., Shin S., Wang J., Bordyuh M., Cho H.J., Elliott O., Chu T., Choi S.W. Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy. Nat. Genet. 2018;50:1399–1411. doi: 10.1038/s41588-018-0209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snijder B., Vladimer G.I., Krall N., Miura K., Schmolke A.S., Kornauth C., Lopez de la Fuente O., Choi H.S., van der Kouwe E., Gültekin S. Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: interim results from a single-arm, open-label, pilot study. Lancet Haematol. 2017;4:e595–e606. doi: 10.1016/S2352-3026(17)30208-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pemovska T., Kontro M., Yadav B., Edgren H., Eldfors S., Szwajda A., Almusa H., Bespalov M.M., Ellonen P., Elonen E. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013;3:1416–1429. doi: 10.1158/2159-8290.CD-13-0350. [DOI] [PubMed] [Google Scholar]
- 8.Tyner J.W., Yang W.F., Bankhead A., 3rd, Fan G., Fletcher L.B., Bryant J., Glover J.M., Chang B.H., Spurgeon S.E., Fleming W.H. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res. 2013;73:285–296. doi: 10.1158/0008-5472.CAN-12-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies E.J., Dong M., Gutekunst M., Närhi K., van Zoggel H.J., Blom S., Nagaraj A., Metsalu T., Oswald E., Erkens-Schulze S. Capturing complex tumour biology in vitro: histological and molecular characterisation of precision cut slices. Sci. Rep. 2015;5:17187. doi: 10.1038/srep17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Närhi K., Nagaraj A.S., Parri E., Turkki R., van Duijn P.W., Hemmes A., Lahtela J., Uotinen V., Mäyränpää M.I., Salmenkivi K. Spatial aspects of oncogenic signalling determine the response to combination therapy in slice explants from Kras-driven lung tumours. J. Pathol. 2018;245:101–113. doi: 10.1002/path.5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pauli C., Hopkins B.D., Prandi D., Shaw R., Fedrizzi T., Sboner A., Sailer V., Augello M., Puca L., Rosati R. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017;7:462–477. doi: 10.1158/2159-8290.CD-16-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman A.A., Letai A., Fisher D.E., Flaherty K.T. Precision medicine for cancer with next-generation functional diagnostics. Nat. Rev. Cancer. 2015;15:747–756. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kodack D.P., Farago A.F., Dastur A., Held M.A., Dardaei L., Friboulet L., von Flotow F., Damon L.J., Lee D., Parks M. Primary patient-derived cancer cells and their potential for personalized cancer patient care. Cell Rep. 2017;21:3298–3309. doi: 10.1016/j.celrep.2017.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuan H., Myers S., Wang J., Zhou D., Woo J.A., Kallakury B., Ju A., Bazylewicz M., Carter Y.M., Albanese C. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. N. Engl. J. Med. 2012;367:1220–1227. doi: 10.1056/NEJMoa1203055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim M., Mun H., Sung C.O., Cho E.J., Jeon H.J., Chun S.M., Jung D.J., Shin T.H., Jeong G.S., Kim D.K. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019;10:3991. doi: 10.1038/s41467-019-11867-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi R., Radulovich N., Ng C., Liu N., Notsuda H., Cabanero M., Martins-Filho S.N., Raghavan V., Li Q., Mer A.S. Organoid cultures as preclinical models of non-small cell lung cancer. Clin. Cancer Res. 2020;26:1162–1174. doi: 10.1158/1078-0432.CCR-19-1376. [DOI] [PubMed] [Google Scholar]
- 17.Gao B., Huang C., Kernstine K., Pelekanou V., Kluger Y., Jiang T., Peters-Hall J.R., Coquelin M., Girard L., Zhang W. Non-malignant respiratory epithelial cells preferentially proliferate from resected non-small cell lung cancer specimens cultured under conditionally reprogrammed conditions. Oncotarget. 2017;8:11114–11126. doi: 10.18632/oncotarget.14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Ory V., Chapman S., Yuan H., Albanese C., Kallakury B., Timofeeva O.A., Nealon C., Dakic A., Simic V. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu F., Lu Y., Tao L., Jiang Y.Y., Lin D.C., Wang L., Petersson F., Yoshiyama H., Koeffler P.H., Goh B.C., Loh K.S. Non-malignant epithelial cells preferentially proliferate from nasopharyngeal carcinoma biopsy cultured under conditionally reprogrammed conditions. Sci. Rep. 2017;7:17359. doi: 10.1038/s41598-017-17628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sette G., Salvati V., Giordani I., Pilozzi E., Quacquarini D., Duranti E., De Nicola F., Pallocca M., Fanciulli M., Falchi M. Conditionally reprogrammed cells (CRC) methodology does not allow the in vitro expansion of patient-derived primary and metastatic lung cancer cells. Int. J. Cancer. 2018;143:88–99. doi: 10.1002/ijc.31260. [DOI] [PubMed] [Google Scholar]
- 21.Nagaraj A.S., Lahtela J., Hemmes A., Pellinen T., Blom S., Devlin J.R., Salmenkivi K., Kallioniemi O., Mäyränpää M.I., Närhi K., Verschuren E.W. Cell of origin links histotype spectrum to immune microenvironment diversity in non-small-cell lung cancer driven by mutant KRAS and loss of Lkb1. Cell Rep. 2017;18:673–684. doi: 10.1016/j.celrep.2016.12.059. [DOI] [PubMed] [Google Scholar]
- 22.Talwelkar S.S., Nagaraj A.S., Devlin J.R., Hemmes A., Potdar S., Kiss E.A., Saharinen P., Salmenkivi K., Mäyränpää M.I., Wennerberg K., Verschuren E.W. Receptor tyrosine kinase signaling networks define sensitivity to erbb inhibition and stratify Kras-mutant lung cancers. Mol. Cancer Ther. 2019;18:1863–1874. doi: 10.1158/1535-7163.MCT-18-0573. [DOI] [PubMed] [Google Scholar]
- 23.Liu K., Guo J., Liu K., Fan P., Zeng Y., Xu C., Zhong J., Li Q., Zhou Y. Integrative analysis reveals distinct subtypes with therapeutic implications in KRAS-mutant lung adenocarcinoma. EBioMedicine. 2018;36:196–208. doi: 10.1016/j.ebiom.2018.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Skoulidis F., Byers L.A., Diao L., Papadimitrakopoulou V.A., Tong P., Izzo J., Behrens C., Kadara H., Parra E.R., Canales J.R. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitai H., Ebi H., Tomida S., Floros K.V., Kotani H., Adachi Y., Oizumi S., Nishimura M., Faber A.C., Yano S. Epithelial-to-mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS-mutant lung cancer. Cancer Discov. 2016;6:754–769. doi: 10.1158/2159-8290.CD-15-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kruspig B., Monteverde T., Neidler S., Hock A., Kerr E., Nixon C., Clark W., Hedley A., Laing S., Coffelt S.B. The ERBB network facilitates KRAS-driven lung tumorigenesis. Sci. Transl. Med. 2018;10:eaao2565. doi: 10.1126/scitranslmed.aao2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuzawa K., Yasuda H., Hamamoto J., Nukaga S., Hirano T., Kawada I., Naoki K., Soejima K., Betsuyaku T. Characterization of the efficacies of osimertinib and nazartinib against cells expressing clinically relevant epidermal growth factor receptor mutations. Oncotarget. 2017;8:105479–105491. doi: 10.18632/oncotarget.22297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soria J.C., Ohe Y., Vansteenkiste J., Reungwetwattana T., Chewaskulyong B., Lee K.H., Dechaphunkul A., Imamura F., Nogami N., Kurata T., FLAURA Investigators Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018;378:113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 29.Hirano T., Yasuda H., Tani T., Hamamoto J., Oashi A., Ishioka K., Arai D., Nukaga S., Miyawaki M., Kawada I. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget. 2015;6:38789–38803. doi: 10.18632/oncotarget.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee Y., Kim T.M., Kim D.W., Kim S., Kim M., Keam B., Ku J.L., Heo D.S. Preclinical Modeling of Osimertinib for NSCLC With EGFR Exon 20 Insertion Mutations. J. Thorac. Oncol. 2019;14:1556–1566. doi: 10.1016/j.jtho.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Fukuda K., Takeuchi S., Arai S., Katayama R., Nanjo S., Tanimoto A., Nishiyama A., Nakagawa T., Taniguchi H., Suzuki T. Epithelial-to-mesenchymal transition is a mechanism of ALK inhibitor resistance in lung cancer independent of ALK mutation status. Cancer Res. 2019;79:1658–1670. doi: 10.1158/0008-5472.CAN-18-2052. [DOI] [PubMed] [Google Scholar]
- 32.Kim H.R., Kim W.S., Choi Y.J., Choi C.M., Rho J.K., Lee J.C. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol. Oncol. 2013;7:1093–1102. doi: 10.1016/j.molonc.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang W., Soares J., Greninger P., Edelman E.J., Lightfoot H., Forbes S., Bindal N., Beare D., Smith J.A., Thompson I.R. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krall E.B., Wang B., Munoz D.M., Ilic N., Raghavan S., Niederst M.J. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. eLife. 2017;6:e18970. doi: 10.7554/eLife.18970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corcoran R.B., Cheng K.A., Hata A.N., Faber A.C., Ebi H., Coffee E.M., Greninger P., Brown R.D., Godfrey J.T., Cohoon T.J. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–128. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niederst M.J., Sequist L.V., Poirier J.T., Mermel C.H., Lockerman E.L., Garcia A.R., Katayama R., Costa C., Ross K.N., Moran T. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Y., Tang W.Y., Tong X., Ji H. Pathological transition as the arising mechanism for drug resistance in lung cancer. Cancer Commun. (Lond.) 2019;39:53. doi: 10.1186/s40880-019-0402-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan B.A., Coward J.I. Chemotherapy advances in small-cell lung cancer. J. Thorac. Dis. 2013;5(Suppl 5):S565–S578. doi: 10.3978/j.issn.2072-1439.2013.07.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marcoux N., Gettinger S.N., O’Kane G., Arbour K.C., Neal J.W., Husain H., Evans T.L., Brahmer J.R., Muzikansky A., Bonomi P.D. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2019;37:278–285. doi: 10.1200/JCO.18.01585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tentler J.J., Tan A.C., Weekes C.D., Jimeno A., Leong S., Pitts T.M., Arcaroli J.J., Messersmith W.A., Eckhardt S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dijkstra K.K., Monkhorst K., Schipper L.J., Hartemink K.J., Smit E.F., Kaing S., de Groot R., Wolkers M.C., Clevers H., Cuppen E., Voest E.E. Challenges in Establishing Pure Lung Cancer Organoids Limit Their Utility for Personalized Medicine. Cell Rep. 2020;31:107588. doi: 10.1016/j.celrep.2020.107588. [DOI] [PubMed] [Google Scholar]
- 42.Kerr E.M., Gaude E., Turrell F.K., Frezza C., Martins C.P. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burstein H.J., Mangu P.B., Somerfield M.R., Schrag D., Samson D., Holt L., Zelman D., Ajani J.A., American Society of Clinical Oncology American Society of Clinical Oncology clinical practice guideline update on the use of chemotherapy sensitivity and resistance assays. J. Clin. Oncol. 2011;29:3328–3330. doi: 10.1200/JCO.2011.36.0354. [DOI] [PubMed] [Google Scholar]
- 44.Schrag D., Garewal H.S., Burstein H.J., Samson D.J., Von Hoff D.D., Somerfield M.R., ASCO Working Group on Chemotherapy Sensitivity and Resistance Assays American Society of Clinical Oncology Technology Assessment: chemotherapy sensitivity and resistance assays. J. Clin. Oncol. 2004;22:3631–3638. doi: 10.1200/JCO.2004.05.065. [DOI] [PubMed] [Google Scholar]
- 45.Bhola P.D., Ahmed E., Guerriero J.L., Sicinska E., Su E., Lavrova E., Ni J., Chipashvili O., Hagan T., Pioso M.S. High-throughput dynamic BH3 profiling may quickly and accurately predict effective therapies in solid tumors. Sci. Signal. 2020;13:eaay1451. doi: 10.1126/scisignal.aay1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piyathilake C.J., Frost A.R., Weiss H., Manne U., Heimburger D.C., Grizzle W.E. The expression of Ep-CAM (17-1A) in squamous cell cancers of the lung. Hum. Pathol. 2000;31:482–487. doi: 10.1053/hp.2000.6711. [DOI] [PubMed] [Google Scholar]
- 47.Went P.T., Lugli A., Meier S., Bundi M., Mirlacher M., Sauter G., Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum. Pathol. 2004;35:122–128. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 48.Kim Y., Kim H.S., Cui Z.Y., Lee H.S., Ahn J.S., Park C.K., Park K., Ahn M.J. Clinicopathological implications of EpCAM expression in adenocarcinoma of the lung. Anticancer Res. 2009;29:1817–1822. [PubMed] [Google Scholar]
- 49.Pak M.G., Shin D.H., Lee C.H., Lee M.K. Significance of EpCAM and TROP2 expression in non-small cell lung cancer. World J. Surg. Oncol. 2012;10:53. doi: 10.1186/1477-7819-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krebs M.G., Hou J.M., Sloane R., Lancashire L., Priest L., Nonaka D., Ward T.H., Backen A., Clack G., Hughes A. Analysis of circulating tumor cells in patients with non-small cell lung cancer using epithelial marker-dependent and -independent approaches. J. Thorac. Oncol. 2012;7:306–315. doi: 10.1097/JTO.0b013e31823c5c16. [DOI] [PubMed] [Google Scholar]
- 51.Bidard F.C., Peeters D.J., Fehm T., Nolé F., Gisbert-Criado R., Mavroudis D., Grisanti S., Generali D., Garcia-Saenz J.A., Stebbing J. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15:406–414. doi: 10.1016/S1470-2045(14)70069-5. [DOI] [PubMed] [Google Scholar]
- 52.Offin M., Chan J.M., Tenet M., Rizvi H.A., Shen R., Riely G.J., Rekhtman N., Daneshbod Y., Quintanal-Villalonga A., Penson A. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019;14:1784–1793. doi: 10.1016/j.jtho.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yao Y., Zhu Z., Wu Y., Chai Y. Histologic transformation from adenocarcinoma to both small cell lung cancer and squamous cell carcinoma after treatment with gefitinib: a case report. Medicine (Baltimore) 2018;97:e0650. doi: 10.1097/MD.0000000000010650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin S., Chen J., Chen L., Histen G., Lin Z., Gross S., Hixon J., Chen Y., Kung C., Chen Y. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA. 2015;112:9088–9093. doi: 10.1073/pnas.1510757112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bao J., Walliander M., Kovács F., Nagaraj A.S., Hemmes A., Sarhadi V.K., Knuutila S., Lundin J., Horvath P., Verschuren E.W. Spa-RQ: An Image Analysis Tool to Visualise and Quantify Spatial Phenotypes Applied to Non-Small Cell Lung Cancer. Sci. Rep. 2019;9:17613. doi: 10.1038/s41598-019-54038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dufva O., Kankainen M., Kelkka T., Sekiguchi N., Awad S.A., Eldfors S., Yadav B., Kuusanmäki H., Malani D., Andersson E.I. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat. Commun. 2018;9:1567. doi: 10.1038/s41467-018-03987-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Potdar S., Ianevski A., Mpindi J.P., Bychkov D., Fiere C., Ianevski P., Yadav B., Wennerberg K., Aittokallio T., Kallioniemi O. Breeze: an integrated quality control and data analysis application for high-throughput drug screening. Bioinformatics. 2020;36:3602–3604. doi: 10.1093/bioinformatics/btaa138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yadav B., Pemovska T., Szwajda A., Kulesskiy E., Kontro M., Karjalainen R., Majumder M.M., Malani D., Murumägi A., Knowles J. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep. 2014;4:5193. doi: 10.1038/srep05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the main data supporting the findings of this study are available within the article. The data presented in Figure S3A of this study are available from the GDSC1 database portal (GDSC: https://www.cancerrxgene.org/). Processed NGS data is available online from the Mendeley Data (Mendeley Data: https://doi.org/10.17632/bb4vfjnfkf.1). Raw NGS data and IHC images generated in this study are available from the lead contact with a completed Materials Transfer Agreement.