

Diabetes, the “other pandemic,” has progressively increased in magnitude despite advances in knowledge about diabetes prevention over the last two decades (1). Therefore, diabetes will remain a major public health problem for the foreseeable future. More patients with diabetic complications inevitably accompany more people living with diabetes. Diabetic kidney disease (DKD) is one of the most serious, risky, and common, occurring in ∼30% of patients with type 1 diabetes and ∼40% of those with type 2 diabetes (2). Progressive DKD is now the foremost cause of kidney failure worldwide, accounting for half of all cases (3). However, in many regions, treatment for kidney failure by dialysis or transplantation is inaccessible and DKD becomes an unescapable death sentence. Indeed, DKD is now the most common cause of death in Mexico City (4). However, even such sobering observations fail to capture the true magnitude of the impact, as DKD independently increases risks of all-cause and cardiovascular mortality by more than fivefold even before patients develop kidney failure (5). Indeed, the mortality rate outpaces the rate of progression to kidney failure by more than 2:1 once macroalbuminuria develops (6). The need to find better ways to identify and treat DKD has never been more urgent. Recent therapeutic advances with the sodium–glucose cotransporter 2 inhibitors demonstrate clear benefits on top of the standard of care, yet substantial residual risk of kidney failure and death remains (7,8).
Uncovering the biological basis of disease is essential to further therapeutic advancement. While hyperglycemia is a well-recognized DKD risk factor, the traditional biomarker of glycated hemoglobin is an average measure that does not capture glycemic variability, an indicator of more disordered homeostasis. Glycemic variability per se is associated with increased levels of circulating markers of oxidative stress and inflammation, such as total antioxidant capacity and high-sensitivity C-reactive protein in young people with type 1 diabetes (9,10). Inflammation is increasingly recognized as a major driver of DKD (11). Innate immune responses generated by macrophages and T lymphocytes, along with a network of resident dendritic cells, are integral components of injury to the diabetic kidney. Kidney injury molecule-1 (KIM-1), also known as T-cell immunoglobulin and mucin domain-containing protein-1 (TIM-1) or KIM-1/TIM-1, is a cellular receptor located on T cells and dendritic cells that is involved in autoimmune responses and conditions (12,13). Although KIM-1 is nondetectable or expressed at low levels in the normal kidney, it is markedly upregulated in damaged proximal tubule cells in both acute and chronic kidney injuries (Fig. 1) (14–17). KIM-1 drives release of proinflammatory chemokines, such as monocyte chemoattractant protein-1 (also known as chemokine C-C motif ligand 2), and thereby recruits and activates leukocytes in the kidney (15). KIM-1 also promotes phagocytosis of injured and apoptotic cells by the tubuloepithelium as well as antigen presentation and dendritic cell activation of T lymphocytes (15,17). In human kidney diseases, including DKD, increased KIM-1 expression is associated with lower estimated glomerular filtration rate (eGFR) (14). Expression has also been observed in other kidney diseases, such as IgA nephropathy, where KIM-1–positive tubules are surrounded by an inflammatory infiltrate with a predominance of macrophages and T cells (18). Using cell lineage tracing in mice, distinct mucosa-associated invariant T cells that express an activation marker have been observed in fibrotic human kidneys, suggesting a mechanism for KIM-1 to arrive in the kidney on infiltrating T cells (19).
Figure 1.
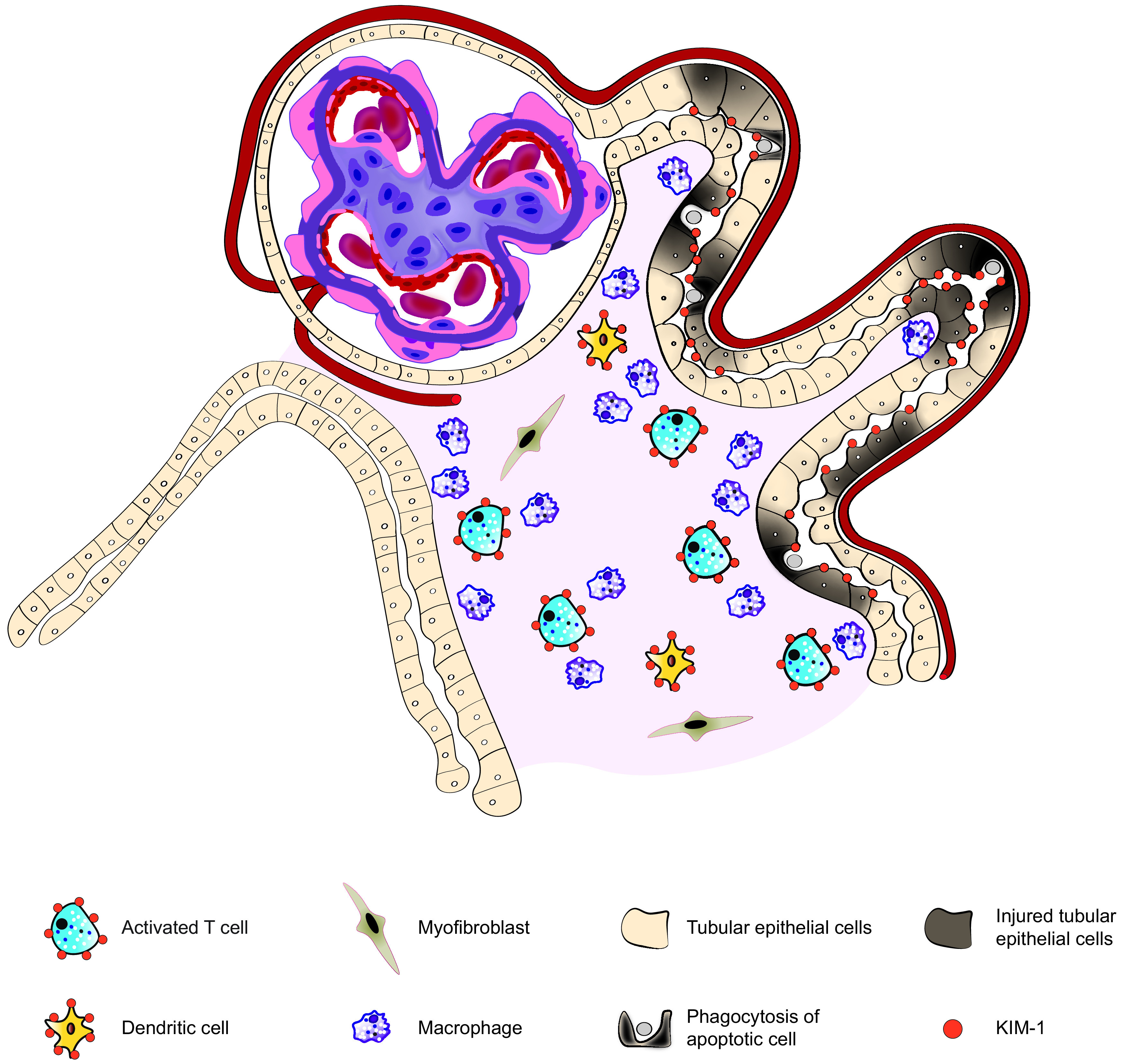
KIM-1 is a phosphatidylserine receptor that is upregulated on the apical surface of injured proximal tubular cells. KIM-1 expression is associated with transformation of tubular epithelial cells into phagocytes that consume apoptotic cells and cellular debris. KIM-1 also drives proximal tubular cells to release monocyte chemoattractant protein-1 (also known as chemokine C-C motif ligand 2) and promotes inflammation by leukocyte recruitment and activation in the diabetic kidney. Adapted with permission from Alicic RZ, Johnson E, Tuttle KR. Inflammatory mechanisms as new biomarkers and therapeutic targets for diabetic kidney disease. Adv Chronic Kidney Dis 2018;25:181–191.
In the study by Forbes et al. (20) in this issue of Diabetes, the role that KIM-1–induced inflammatory responses to glycemic variation may play in DKD was examined in a long-term observational study of youth with type 1 diabetes, a mouse model of atherosclerosis (apoE knockout) with and without streptozotocin-induced diabetes, human proximal tubular cell cultures, and human kidney tissue from patients with DKD and from control subjects without DKD. The main finding was that plasma KIM-1, but not urine KIM-1, was associated with higher levels of eGFR and increased albuminuria, predictors of progressive DKD. This observation suggested that KIM-1 was derived from the systemic circulation rather than the kidney. The investigators identified cytotoxic CD8+ T cells as a potential source. The mouse models revealed higher levels of T-cell–derived KIM-1 in response to either glycemic fluctuations without diabetes or overt diabetes. Plasma from high-risk youth with type 1 diabetes, identified by the highest tertile of albuminuria within the conventional normal range, induced human proximal tubular cell expression of collagen IV and sodium–glucose cotransporter 2 that was blocked by anti–KIM-1 antibodies. Human kidney tissue demonstrated that KIM-1–positive T cells were present in the tubulointerstitium of patients with DKD and focal segmental glomerulosclerosis but were greatest in those with DKD. These data implicate glycemic variability as a potential DKD risk factor through inflammatory mechanisms involving KIM-1 on circulating T cells that may infiltrate the kidney prior to local KIM-1 production by injured proximal tubular cells.
This provocative study suggests a primary role for systemic T-cell–derived KIM-1 as a driving force for progressive DKD. However, there are a number of limitations that require additional research to confirm this hypothesis (20). For example, the kidney tissue samples of DKD were from a separate cohort that was far older and likely comprised patients with type 2 diabetes. They had markedly reduced eGFR and severely increased albuminuria, representing advanced DKD, compared with the main study cohort with type 1 diabetes who had preserved eGFR and slightly increased levels of albuminuria. The inflammatory mechanisms and phenotypes of DKD may be quite different between early and late-stage disease, when KIM-1 levels in the proximal tubule are very high. The signal for T-cell recruitment to the diabetic kidney early in, or prior to, injury is unclear. Reverse causality must also be considered for the association of KIM-1 and DKD risk, as cause and effect cannot be discerned from observational or cross-sectional data. Indeed, higher circulating levels of KIM-1 could represent an adaptive response to mitigate cellular injury or may simply be a biomarker without direct involvement in DKD pathogenesis. Recognition of DKD as an inflammatory disease linked by metabolic perturbations and immune responses is a central tenet that requires broader and deeper exploration. The ultimate deliverable will be to unravel these mechanisms in a manner that translates to better care for patients with DKD at a stage when health can be maintained and devastating consequences prevented.
Article Information
Acknowledgments. The authors thank Emily J. Cox (Providence Medical Research Center, Providence Health Care, Spokane, WA) for technical assistance with generating Fig. 1.
Funding. K.R.T. is supported by four National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health (NIH) grants, a National Center for Advancing Translational Sciences/NIH grant, a National Institute on Minority Health and Health Disparities/NIH grant, and a Centers for Disease Control and Prevention contract.
Duality of Interest. K.R.T. is supported by research grants from Goldfinch Bio, Bayer, and Travere Therapeutics and has received consulting fees or speaking honoraria from Eli Lilly and Company, Boehringer Ingelheim, AstraZeneca, Gilead Sciences, Goldfinch Bio, Novo Nordisk, Bayer, Travere Therapeutics, and Janssen Global Services. No other potential conflicts of interest relevant to this article were reported.
Footnotes
See accompanying article, p. 1754.
References
- 1.International Diabetes Federation . IDF Diabetes Atlas, 9th edition, 2019. Accessed 3 April 2021. Available from https://www.diabetesatlas.org/en/sections/demographic-and-geographic-outline.html
- 2.Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol 2017;12:2032–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levin A, Tonelli M, Bonventre J, et al.; ISN Global Kidney Health Summit participants . Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 2017;390:1888–1917 [DOI] [PubMed] [Google Scholar]
- 4.Alegre-Díaz J, Herrington W, López-Cervantes M, et al. Diabetes and cause-specific mortality in Mexico City. N Engl J Med 2016;375:1961–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Afkarian M, Sachs MC, Kestenbaum B, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol 2013;24:302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA; UKPDS GROUP . Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int 2003;63:225–232 [DOI] [PubMed] [Google Scholar]
- 7.Perkovic V, Jardine MJ, Neal B, et al.; CREDENCE Trial Investigators . Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 2019;380:2295–2306 [DOI] [PubMed] [Google Scholar]
- 8.Heerspink HJL, Stefánsson BV, Correa-Rotter R, et al.; DAPA-CKD Trial Committees and Investigators . Dapagliflozin in patients with chronic kidney disease. N Engl J Med 2020;383:1436–1446 [DOI] [PubMed] [Google Scholar]
- 9.Hoffman RP, Dye AS, Huang H, Bauer JA. Glycemic variability predicts inflammation in adolescents with type 1 diabetes. J Pediatr Endocrinol Metab 2016;29:1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papachristoforou E, Lambadiari V, Maratou E, Makrilakis K. Association of glycemic indices (hyperglycemia, glucose variability, and hypoglycemia) with oxidative stress and diabetic complications. J Diabetes Res 2020;2020:7489795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pichler R, Afkarian M, Dieter BP, Tuttle KR. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol 2017;312:F716–F731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuchroo VK, Dardalhon V, Xiao S, Anderson AC. New roles for TIM family members in immune regulation. Nat Rev Immunol 2008;8:577–580 [DOI] [PubMed] [Google Scholar]
- 13.Xiao S, Zhu B, Jin H, et al. Tim-1 stimulation of dendritic cells regulates the balance between effector and regulatory T cells. Eur J Immunol 2011;41:1539–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Timmeren MM, van den Heuvel MC, Bailly V, Bakker SJ, van Goor H, Stegeman CA. Tubular kidney injury molecule-1 (KIM-1) in human renal disease. J Pathol 2007;212:209–217 [DOI] [PubMed] [Google Scholar]
- 15.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 2008;118:1657–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphreys BD, Xu F, Sabbisetti V, et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest 2013;123:4023–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brooks CR, Yeung MY, Brooks YS, et al. KIM-1-/TIM-1-mediated phagocytosis links ATG5-/ULK1-dependent clearance of apoptotic cells to antigen presentation. EMBO J 2015;34:2441–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin Q, Chen Y, Lv J, et al. Kidney injury molecule-1 expression in IgA nephropathy and its correlation with hypoxia and tubulointerstitial inflammation. Am J Physiol Renal Physiol 2014;306:F885–F895 [DOI] [PubMed] [Google Scholar]
- 19.Law BMP, Wilkinson R, Wang X, et al. Human tissue-resident mucosal-associated invariant T (MAIT) cells in renal fibrosis and CKD. J Am Soc Nephrol 2019;30:1322–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forbes JM, McCarthy DA, Kassianos AJ, et al. T-cell expression and release of kidney injury molecule-1 in response to glucose variations initiates kidney injury in early diabetes. Diabetes 2021;70:1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]