

Abstract
Type 1 diabetes (T1D) has a multifactorial autoimmune etiology, involving environmental prompts and polygenic predisposition. We hypothesized that pancreata from individuals with and at risk for T1D would exhibit dysregulated expression of genes associated with monogenic forms of diabetes caused by nonredundant single-gene mutations. Using a “monogenetic transcriptomic strategy,” we measured the expression of these genes in human T1D, autoantibody-positive (autoantibody+), and control pancreas tissues with real-time quantitative PCR in accordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines. Gene and protein expression was visualized in situ with use of immunofluorescence, RNAscope, and confocal microscopy. Two dozen monogenic diabetes genes showed altered expression in human pancreata from individuals with T1D versus unaffected control subjects. Six of these genes also saw dysregulation in pancreata from autoantibody+ individuals at increased risk for T1D. As a subset of these genes are related to cellular stress responses, we measured integrated stress response (ISR) genes and identified 20 with altered expression in T1D pancreata, including three of the four eIF2α-dependent kinases. Equally intriguing, we observed significant repression of the three arms of the ISR in autoantibody+ pancreata. Collectively, these efforts suggest monogenic diabetes and ISR genes are dysregulated early in the T1D disease process and likely contribute to the disorder’s pathogenesis.
Introduction
Type 1 diabetes (T1D) is widely considered a multifactorial disorder, polygenic in etiology with environmental factors thought to contribute toward pathogenesis, resulting in autoimmune destruction of insulin-producing pancreatic β-cells (1,2). In contrast, monogenic diabetes comprises an expanding group of rare heterogeneous, single-gene disorders with a collective prevalence of ∼1–5% of all diabetes cases, depending on age of onset, geography, and ethnicity (3–6). Monogenic forms of diabetes distinguish critical proteins within human β-cell development and biology where no sufficient compensatory proteins or pathways exist in the presence of a sufficiently deleterious mutation, reflecting the critical nature of the protein and a lack of “redundancy” at that point within the affected pathway. Most forms of monogenic diabetes result, through a variety of mechanisms, in a reduced ability to process or secrete insulin, with some variants associated with insulin resistance (7).
We therefore studied genes associated with monogenic forms of diabetes with the rationale being a relevance to disease pathology. Traditionally, these nonredundant forms of diabetes have been classified based on age of onset—maturity-onset diabetes of the young (MODY) (8–10) and neonatal diabetes mellitus (NDM) (11,12), which includes transient NDM (TNDM1 and TNDM2) and permanent NDM (PNDM)—or as syndromic. However, to provide a physiological reference point, we have addressed the biological heterogeneity of monogenic diabetes genes by separating our studied genes into four physiological groups: immune, β-cell function, β-cell development, and endoplasmic reticulum (ER) function/stress.
We hypothesized that a phenotypic assessment of gene expression levels for the ever-expanding cohort of genes linked to monogenic diabetes could be enlightening in our understanding of multifactorial/polygenic T1D disease etiology and pathogenesis. In particular, we sought to address the question of the importance of genes causative in monogenic diabetes using real-time quantitative PCR (RTqPCR), immunofluorescence (IF), and in situ hybridization (ISH) studies on human pancreatic tissues from unaffected control organ donors and organ donors with T1D, autoantibody positivity (autoantibody+) (high risk for T1D), and type 2 diabetes from the Network for Pancreatic Organ donors with Diabetes (nPOD) repository.
Research Design and Methods
Donors
The JDRF nPOD program (www.jdrfnpod.com) recovers transplant-quality pancreata from organ donors as previously described (13). All procedures were approved by the University of Florida Institutional Review Board and the United Network for Organ Sharing (UNOS) according to federal guidelines, with informed consent obtained from each donor’s legal representative. For each donor, a medical chart review was performed in addition to assays for T1D-associated autoantibodies and C-peptide (14), with T1D diagnosed according to the guidelines established by the American Diabetes Association (15). Information for donors (patient number, autoantibody+ status, age, disease duration, sex, ethnicity, C-peptide, HbA1c, BMI, cause of death, and hiRES HLA) was obtained from nPOD records (Supplementary Table 1). Cause of death was validated via an independent medical chart review by a medical expert.
Sample Processing and RNA Extraction
Pancreata were recovered, placed in transport media on ice, and shipped via organ courier to the University of Florida where tissues were processed by a licensed Pathology Assistant as previously described (13). Tissue from pancreas was preserved as flash frozen or in RNAlater (QIAGEN, Valencia, CA) an average of 16 h from cross clamp. Total RNA was isolated following homogenization in QIAGEN RNeasy Plus Mini Kit isolation buffer as per the manufacturer’s instructions including treatment with DNase 1. RNA concentrations were determined with a Thermo Scientific NanoDrop 2000C (Thermo Fisher Scientific, Waltham, MA), and when necessary, integrity was verified by visualization of rRNA by gel electrophoresis and ethidium bromide staining.
RTqPCR, Gene Stability Ranking, and Quantitative PCR Minimum Guidelines
All samples were confirmed to be free of DNA contamination in control pancreata with an intron/exon primer pair and without reverse transcriptase. cDNA was produced with SuperScript II (Invitrogen, Carlsbad, CA) using oligo dT priming and subsequently used for RTqPCR with Thermo Scientific Luminaris Color HiGreen qPCR Master Mix, fluorescein (Thermo Fisher Scientific). Total RNA (0.5–1.0 μg) was used for each reaction with 20 µL cDNA, which was then diluted to 200 µL and then 2 µL diluted cDNA, was used for each 25 µL RT-qPCR reaction containing 600 nmol/L of each primer pair. Individual RT-qPCR reactions were carried out in duplicate in a Bio-Rad MyiQ. All samples were standardized for experimental design, nucleic acid isolation, measurement of total RNA concentration, reverse transcription, primer design/target specificity (16), and all RTqPCR parameters (17) in accordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines.
The MIQE guidelines (Supplementary Table 2) were followed to ensure the reliability/integrity of scientific data and to provide for experimental transparency/consistency among laboratories (17). Supplementary Table 2 illustrates a checklist for MIQE guidelines followed in this study. A fundamental axiom of MIQE is normalization of RTqPCR data by use of multiple reference genes (RGs) to address intra- and interkinetic variations in quantitative PCR studies (18,19). The following genes were identified as potential RGs based on the lowest standard deviation SD and coefficient of variation (CoV) of unnormalized quantification cycle (Cq) values across all unaffected and T1D pancreata: ASNS (asparagine synthetase), GLP1R (glucagon-like peptide 1 receptor), MAFB (V-maf musculoaponeurotic fibrosarcoma oncogene homolog B), NKX6-1 (NK6 homeobox 1), NRP1 (neuropilin 1), PFKM (phosphofructokinase, muscle), PPIA (cyclophilin A), and GCG (glucagon). For comparison and ranking of potential RGs with the lowest variation and highest stability across the control and T1D pancreata, four algorithms were evaluated simultaneously with the Web-based tool RefFinder (20) (https://github.com/fulxie/RefFinder), which incorporates four well-accepted algorithms: geNorm (21), Normfinder (22), BestKeeper (23), and the comparative Δ-Ct method (24) as well as its own comprehensive assessment (20). RefFinder requires an equal number of data points for all genes evaluated; therefore, only 45 donors were included in this analysis. RefFinder identified three human pancreas-specific RGs demonstrating the most stable expression in human pancreata, PPIA (SD 2.02, CoV 0.06), MAFB (SD 1.89, CoV 0.07), and ASNS (SD 2.15, CoV 0.07). We then used the geNorm algorithm (21) to generate separate normalization factors derived from the geometric mean of RGs PPIA for the unaffected and ASNS and MAFB for the T1D cohorts, respectively. The normalization factors were applied to the analysis of all monogenic diabetes and integrated stress response (ISR) genes to report quantitative changes in gene expression between unaffected and T1D pancreata, with the fold difference (FD) reported as the ratio of the means (T1D/unaffected). PPIA (cyclophilin A) also served as the interrun calibrator. With use of the interquartile range outlier test (25,26), only extreme outliers, defined as values below Q1 − 3(Q3 − Q1) or above Q3 + 3(Q3 − Q1) (where Q1 and Q3 are the first and third data quartile) were identified and excluded from statistical analyses (https://www.itl.nist.gov/div898/handbook/prc/section1/prc16.htm).
Primer Design
The respective monogenic diabetes genes analyzed in this study were selected based on review of the National Center for Monogenic Diabetes at the University of Chicago Monogenic Diabetes Registry for MODY Diabetes and Neonatal Diabetes, Kovler Diabetes Center (https://monogenicdiabetes.uchicago.edu); the Institute of Biomedical and Clinical Science, Medical School, University of Exeter, (https://medicine.exeter.ac.uk/research/biomedicalclinical/mole culargenetics-monogenic/); and OMIM (https://www.omim.org). We would like to point out, however, that this may not be an all-inclusive set of monogenic diabetes genes. Human gene symbols, based on HGNC (https://www.genenames.org) were used throughout this study, with gene names, chromosomal location, NCBI accession numbers, common protein names, OMIM no., chromosomal location, genomic coordinates, MODY number, NDM classification, and appropriate references provided in Supplementary Table 3. We used the public Primer-BLAST software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/ [16]), which incorporates the Primer3 program (27) for primer design and genome-wide BLAST analysis, along with the Needleman-Wunsch global alignment algorithm (28) to identify internal homology between primers and any unwanted targets in the human genome. This satisfies the requirements for primer specificity with comparison with both the human transcriptome and genome. Primers were designed, when possible, as exonic primers spanning an intron, with comparable GC content and an optimal Tm of 60°C (Supplementary Table 4).
IF
IF staining was completed using the PerkinElmer Opal 4-Color IHC Kit (NEL810001KT; PerkinElmer, Waltham, MA) as per the manufacturer’s instructions. After deparaffinization and rehydration, all slides were subjected to a microwaved treated antigen retrieval (AR) step with use of the kit’s appropriate AR buffer. Slides were then incubated in antibody diluent/blocking solution followed by incubation with primary antibody in antibody diluent/blocking solution. Slides were stained with the following antibodies: insulin (A0564; Dako [Research Resource Identifier (RRID):AB_10013624]), somatostatin (A0566; Dako [RRID:AB_2688022]), glucagon (ab10988; Abcam [RRID:AB_297642]), STAT5 (LS-B5540; LifeSpan BioSciences [RRID:AB_10915294]), GLIS3 (HPA056426; Sigma-Aldrich [RRID:AB_2683128]); GATA4 (sc-25310; Santa Cruz Biotechnology [RRID:AB_627667]), WFS1 (LS-B14378; LifeSpan BioSciences), and EIF2AK3/PERK (24390-1-AP; Proteintech). For detection and visualization, slides were subjected to the Polymer HRP Ms + Rb and the TSA Plus Fluorescent system (NEL703001KT, NEL741001KT, NEL744001KT, NEL745001KT; PerkinElmer). Coumarin (excitation maxima 402 nm; emission 443 nm) was assigned to insulin, fluorescein (excitation maxima 494 nm; emission 517 nm) to somatostatin, gene of interest (STAT5, WSF1, GATA4, GLIS3, and EIF2K3) to CY3 (excitation maxima 550 nm; emission 570 nm), and glucagon to CY5 (excitation maxima 648 nm; emission 667 nm). For each panel, negative and positive control slides were also stained to determine the exposure time and image processing necessary to provide optimal visualization of the antibody signal. All images were captured and processed with an Olympus IX73 florescence microscope with use of cellSense software.
RNAscope Coupled With IF
Fluorescent-labeled RNA probes (STAT5B, cat. no. PN 501151; GLIS3, PN 525701-C3; GATA4, PN 579821; WFS1, PN 436081; EI2AK3, custom reagent [targeting 330–1487 of NM_001313915.1]; PPP1R15A, PN 311141; PSEN1, PN 502001; COL6A2, PN 482611; ERN1/IRE1, PN 497331-C2; Hs-PPIB [positive control], 313901; and DapB [negative control], 310043) were purchased from Advanced Cell Diagnostics (Newark, CA). RNA staining was performed with an RNAscope Multiplex Fluorescent Reagent Kit v2 (PN 323100) according to the manufacturer’s instructions: slides were dewaxed 2 × 5 min in xylene and 2 × 2min in 100% EtOH and then air-dried. AR was performed for 15 min using RNAscope 1X Target Retrieval Reagents heated up to 98°C. Slides were dipped in double-distilled H2O and then 100% EtOH for 2 min and air dried. RNAscope Protease III was applied to the dry slides and incubated for 30 min at 40°C in the oven and then rinsed with RNAscope 1X Wash Buffer. After incubation with RNA probes for 30 min at 40°C, probes were amplified with RNAscope Multiplex FL v2 AMP 1 for 30 min and then RNAscope Multiplex FL v2 AMP 2 for 30 min, RNAscope Multiplex FL v2 AMP 3 for 15 min, and RNAscope Multiplex FL v2 HRP-C1 for 15 min. All amplification steps were performed at 40°C, and slides were washed between steps in RNAscope 1X Wash Buffer 2 × 2 min. Signal was developed with use of TSA Plus Cyanine 3 (NEL744001KT) 1:1,500 for 30 min at 40°C and blocked with HRP Blocker for 15 min at 40°C. Slides were washed in Tris-buffered saline and incubated overnight at 4°C with primary antibodies (mouse anti-CD99, cat. no. 318002, clone HCD99, 1:200, BioLegend [RRID:AB_604112], and rabbit anti–cytokeratin 19, cat. no. 13092, clone D7F7W, 1:100, Cell Signaling Technologies). On the next day, slides were washed in Tris-buffered saline and incubated for 2 h at room temperature with secondary antibodies (goat anti-rabbit Alexa Fluor 488, cat. no. A-11008, and goat anti-mouse Alexa Fluor 647, A-21235; Thermo Fisher Scientific). Images were acquired on a Leica DMI6000 widefield microscope.
Confocal Microscopy
Samples were imaged on a Nikon A1plus confocal microscope with a 20× (NA0.75) air objective (Nikon Instruments, Melville, NY). Excitation and emission for each target were as follows: insulin excitation 441/emission 450, somatostatin excitation 488/emission 525, WFS1/STAT5B excitation 561/emission 595, and glucagon excitation 647/emission 700. Three-dimensional (3D) Z-stacks were constructed with a 2.5-μm step size and compressed to a two-dimensional (2D) maximum intensity projection for display. All images for a given gene and condition (i.e., WFS1, healthy control subject) are displayed with identical lookup table values. Imaging parameters (laser power, pixel dwell, pinhole size, gain, offset) remained constant for all samples. All raw images of single islets were denoised in Nikon Elements (Nikon Instruments) for display.
Statistical Analysis
For each patient population, a two-tailed Wilcoxon rank-sum test comparing unaffected donors versus donors with disease was performed for each gene. The Storey method was used to control the false discovery rate; the adjusted P values (q values) were estimated with the qvalue R package (https://github.com/jdstorey/qvalue) with λ = 0. q values ≤0.024 were considered statistically significant. R (v3.6.1) was used for all calculations. GraphPad Prism v8.0 (GraphPad Software, La Jolla, CA) was used for all graphical presentations.
Data and Resource Availability
All of the data presented in this manuscript will be made available on request.
Results
Of relevance to the quality of human donor pancreatic RNA for gene expression analysis (29–32), normalized expression levels for ASNS, GCG, GLP1R, NRP1, and PFKM were not significantly different across donors grouped when correlated with cause of death (head trauma, anoxia, or cerebrovascular/stroke) or intensive care unit or organ transport times. A summary of metadata appears in Supplementary Table 1, where the subjects (unaffected control and T1D pancreata) are analyzed for experiments reported below.
Some Monogenic Diabetes Genes in Type 1 Diabetes, Autoantibody+, and Type 2 Diabetes Pancreata Are Differentially Expressed
Overall, 24 of the 45 monogenic diabetes genes showed an increased expression in T1D pancreas (Table 1). We then examined these 24 monogenic diabetes genes differentially expressed in the T1D pancreas in organs from a population of autoantibody+ donors (n = 20–24) considered at risk (33) for T1D, as well as in type 2 diabetes pancreata (n = 20). Of note, the vast majority of autoantibody+ donors were seropositive for only a single autoantibody (constituting pre–stage 1 T1D), with five seropositive for two autoantibodies (considered potentially stage 1–2 T1D [34], dysglycemia unknown). We identified five genes (BSCL2, DUT-N, EIF2AK3, ITCH, and MNX1; FD range = 1.58–3.41) displaying increased expression in autoantibody+ pancreata compared with unaffected controls and a single gene, HNF4A, that was highly repressed in autoantibody+ pancreata (FD = 0.08 vs. controls) but only marginally induced in the T1D group (FD = 1.78). In contrast, five monogenic diabetes genes (DUT-N, EIF2AK3, GLIS3, ITCH, and NR0B2) were significantly induced in type 2 diabetes pancreata compared with unaffected donor organs (Table 1).
Table 1.
Monogenic diabetes genes
| Gene | T1D (n = 25–30) | Autoantibody+ (n = 20–24) | Type 2 diabetes (n = 18–22) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| FD | P value | q value | FD | P value | q value | FD | P value | q value | |
| APPL1 | 1.97 | 0.056* | 0.029* | 1.08 | 0.223 | 0.078 | 1.01 | 0.491 | 0.141 |
| BSCL2 | 2.14 | 0.021 | 0.012 | 1.70 | 0.013 | 0.009 | 1.31 | 0.234 | 0.080 |
| CEL | 2.52 | 0.040 | 0.023 | 0.90 | 0.560 | 0.158 | 2.27 | 0.257 | 0.085 |
| DUT-M | 2.87 | 0.001 | 0.001 | 1.16 | 0.657 | 0.174 | 5.91 | 0.178 | 0.067 |
| DUT-N | 4.07 | 0.004 | 0.003 | 2.29 | <0.001 | 0.001 | 3.11 | <0.001 | <0.001 |
| EIF2AK3 | 8.32 | <0.001 | <0.001 | 3.41 | <0.001 | <0.001 | 4.03 | 0.004 | 0.004 |
| GATA4 | 4.35 | 0.004 | 0.004 | 0.71 | 0.431 | 0.127 | 1.37 | 0.242 | 0.081 |
| GATA6 | 2.99 | <0.001 | 0.001 | 0.91 | 0.243 | 0.081 | 1.84 | 0.208 | 0.076 |
| GLIS3 | 4.01 | <0.001 | 0.001 | 1.05 | 0.435 | 0.127 | 3.13 | 0.052* | 0.027* |
| HNF1A | 1.54 | 0.042 | 0.024 | 0.54 | 0.812 | 0.203 | 1.00 | 0.385 | 0.116 |
| HNF1B | 3.44 | <0.001 | 0.001 | 0.70 | 0.582 | 0.161 | 1.66 | 0.097 | 0.045 |
| HNF4A | 1.78 | 0.072* | 0.037* | 0.08 | 0.006 | 0.005 | 1.53 | 0.953 | 0.231 |
| ITCH | 2.81 | 0.003 | 0.003 | 2.54 | <0.001 | <0.001 | 3.41 | <0.001 | 0.001 |
| KLF11 | 2.12 | 0.021 | 0.012 | 0.42 | 0.243 | 0.081 | 1.25 | 0.670 | 0.174 |
| LRBA | 3.89 | 0.002 | 0.002 | 0.36 | 0.580 | 0.161 | 1.44 | 0.366 | 0.112 |
| MNX1 | 2.51 | 0.003 | 0.003 | 1.58 | 0.004 | 0.003 | 1.11 | 0.359 | 0.111 |
| NR0B2 | 1.88 | 0.037 | 0.022 | 1.04 | 0.497 | 0.141 | 2.41 | 0.002 | 0.002 |
| PLAGL1 | 2.87 | 0.001 | 0.001 | 1.14 | 0.624 | 0.168 | 1.56 | 0.155 | 0.062 |
| SIRT1 | 3.05 | 0.013 | 0.009 | 0.28 | 0.154 | 0.062 | 1.52 | 0.649 | 0.173 |
| STAT1 | 2.89 | 0.050* | 0.027* | 0.80 | 0.961 | 0.232 | 2.35 | 0.287 | 0.093 |
| STAT3 | 2.75 | 0.012 | 0.009 | 1.38 | 0.097 | 0.045* | 1.46 | 0.160 | 0.063 |
| STAT5B | 3.59 | 0.005 | 0.004 | 1.28 | 0.116 | 0.051 | 2.03 | 0.216 | 0.078 |
| TRMT10A | 2.25 | 0.018 | 0.011 | 0.65 | 0.442 | 0.128 | 0.69 | 0.219 | 0.078 |
| WFS1 | 4.41 | 0.002 | 0.002 | 0.89 | 0.767 | 0.193 | 1.35 | 0.207 | 0.076 |
Tabulated FDs, P values, and q values (estimation of false discovery rates) for the monogenic diabetes genes showing altered expression in T1D (left), autoantibody+ (middle), and type 2 diabetes (right) pancreata. Cq values from the unaffected (control) and T1D cohorts were independently normalized using the geometric mean of three pancreas-specific RGs, and the FD was calculated based on the ratio of the means (Research Design and Methods). See Research Design and Methods for statistical analysis (P values and q values). n refers to the number of independent pancreata in each cohort, and boldface type denotes genes showing altered expression relative to control donors: T1D, 24 altered genes out of a total of 45 monogenic diabetes genes; autoantibody+, 6 altered genes; and type 2 diabetes, 5 altered genes.
Genes for which either the P value (<0.05) or q value (≤0.024) approached significance.
The remaining 21 of the 45 monogenic diabetes genes were either not expressed differentially or not amplified in control or T1D pancreata, as shown in Supplementary Table 5, whereas four genes (ABCC8, GCK, NKX2.2, and RFX6) demonstrated a trend toward repression but did not achieve statistical significance. In addition, we have previously shown that INS, IAPP, and the INS-IGF2 read-through mRNA levels are dramatically inhibited in T1D pancreata (35). To address whether altered expression of these monogenic diabetes genes is indeed pancreas specific, we examined the expression of 19 genes altered in T1D pancreata by RTqPCR using total RNA isolated from 10 control and 10 T1D human spleens and found no genes differentially expressed in this organ (Supplementary Table 6).
Classification of Monogenic Diabetes Genes Into Physiological Groups Reveals Broad Differential Expression in Type 1 Diabetes Pancreata
The monogenic diabetes genes were separated into four physiological classifications: 1) immune, 2) β-cell function, 3) β-cell development, and 4) ER function/stress (Supplementary Table 7), with knowledge that certain genes associate with multiple categories (Supplementary Table 7).
Of the seven monogenic diabetes genes examined from the immune physiological group (Supplementary Table 7), normalized RTqPCR revealed significant differences in LRBA, SIRT1, STAT1, STAT3, and STAT5B expression in T1D versus unaffected pancreata, with FDs ranging from 2.75 to 3.89 (Table 1). For STAT5B a significant (3.59) FD was noted (Fig. 1A). Widefield IF for STAT5B, insulin (INS), glucagon (GCG), and somatostatin (SST), with IF for islet (CD99) and exocrine cell markers (KRT19) and ISH for STAT5 (Fig. 1B), along with STAT5B confocal ISH-IF (Fig. 1C) in unaffected and T1D human pancreata, revealed cytosolic expression of STAT5B most appreciably within the islets. These studies suggest that STAT5B colocalizes with glucagon and insulin in unaffected α- and β-cells, respectively (Fig. 1B and C). To demonstrate the uniformity of these results, we have included images of additional T1D and control pancreata (Supplementary Figs. 1 and 2). Most strikingly, we observed a number of islet cells that display only STAT5B expression with limited to no expression of INS, GCG, or SST in both unaffected and T1D islets (Fig. 1C and Supplementary Figs. 1 and 2).
Figure 1.
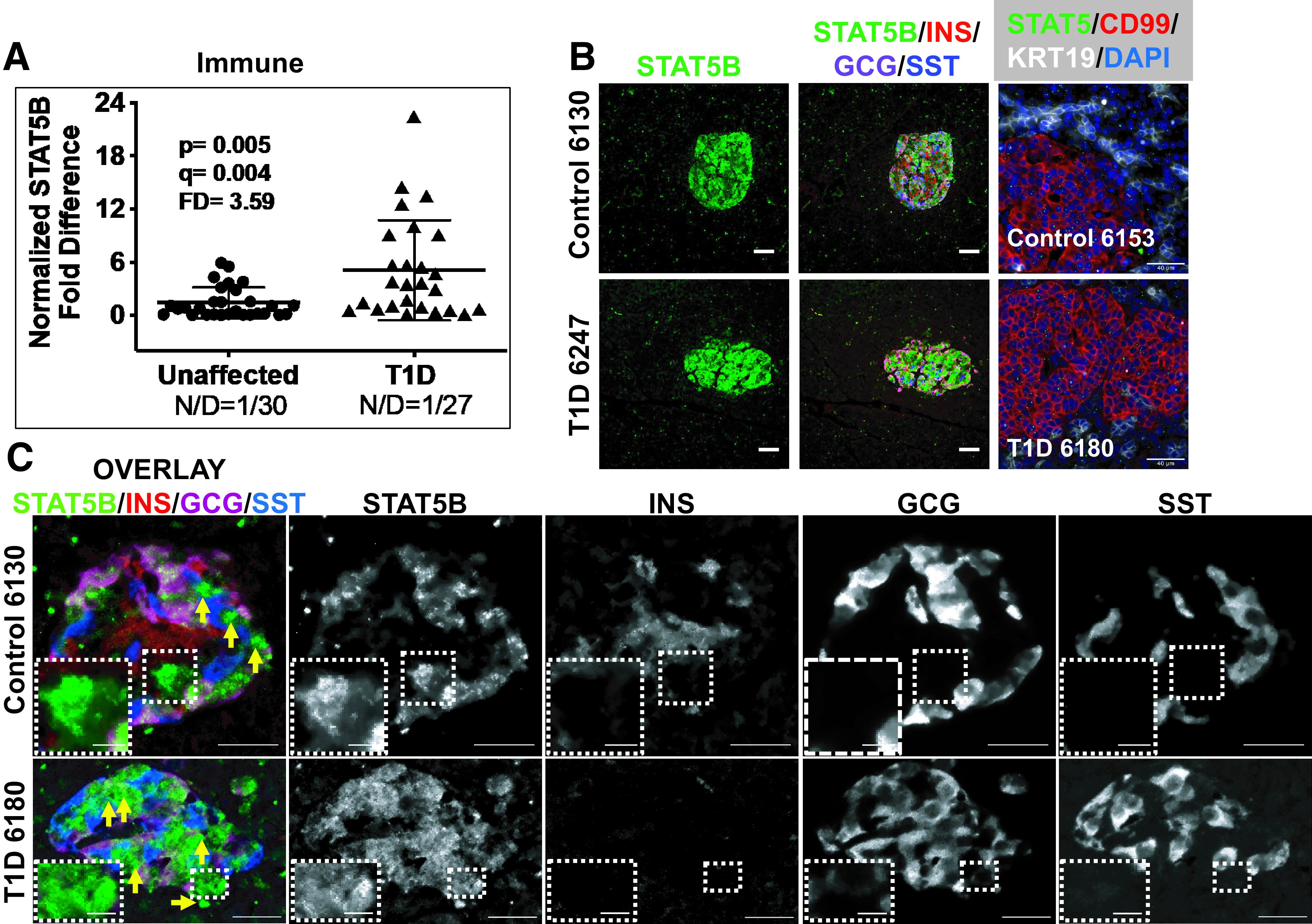
Monogenic diabetes genes were sorted into four physiological groups: immune, β-cell function, β-cell development, and ER function/stress (Supplementary Table 7). A: Scatter plot of gene expression data for STAT5B, a gene representing an immune monogenic diabetes gene, studied using RTqPCR in human organ donor pancreata. Cq values from the unaffected (control) and T1D cohorts were independently normalized using the geometric mean of three pancreas-specific RGs, and the FD is calculated based on the ratio of the means (Research Design and Methods). See Research Design and Methods for statistical analysis (P values and q values [estimation of false discovery rates]). N/D refers to number (N) of samples yielding no data out of the total (D). B: Widefield IF of STAT5B (green) and overlay with insulin (INS), glucagon (GCG), and somatostatin (SST) from a control and T1D pancreas. Final panel in each row shows combined STAT5B ISH (RNAscope [green dots]) coupled with IF for CD99 and KRT19. Magnification bars = 40 μm. C: Confocal imaging on a Nikon A1plus confocal microscope of STAT5B (green), insulin (INS), glucagon (GCG), and somatostatin (SST), showing an overlay of an islet from an unaffected and T1D pancreas (left panels). 3D Z-stacks were constructed with a 2.5-μm step size and compressed to a 2D maximum intensity projection for display. Individual channels in black and white identify cells positive only for STAT5B and negative for the other endocrine hormones, illustrated with yellow arrows (overlay) and with insets in the respective black and white channels.
We similarly observed significantly altered expression for numerous monogenic diabetes genes in the β-cell function group (Table 1), including DUT-N, DUT-M, GATA4, PLAGL1, and NR0B2, with the FDs ranging from 1.88 to 4.35 for T1D compared with unaffected control pancreata. GATA4 expression, FD 4.35 (Fig. 2A), was also examined by IF and ISH-IF, revealing primarily nuclear expression in both islets and the exocrine pancreas (Fig. 2B and Supplementary Figs. 3 and 4).
Figure 2.
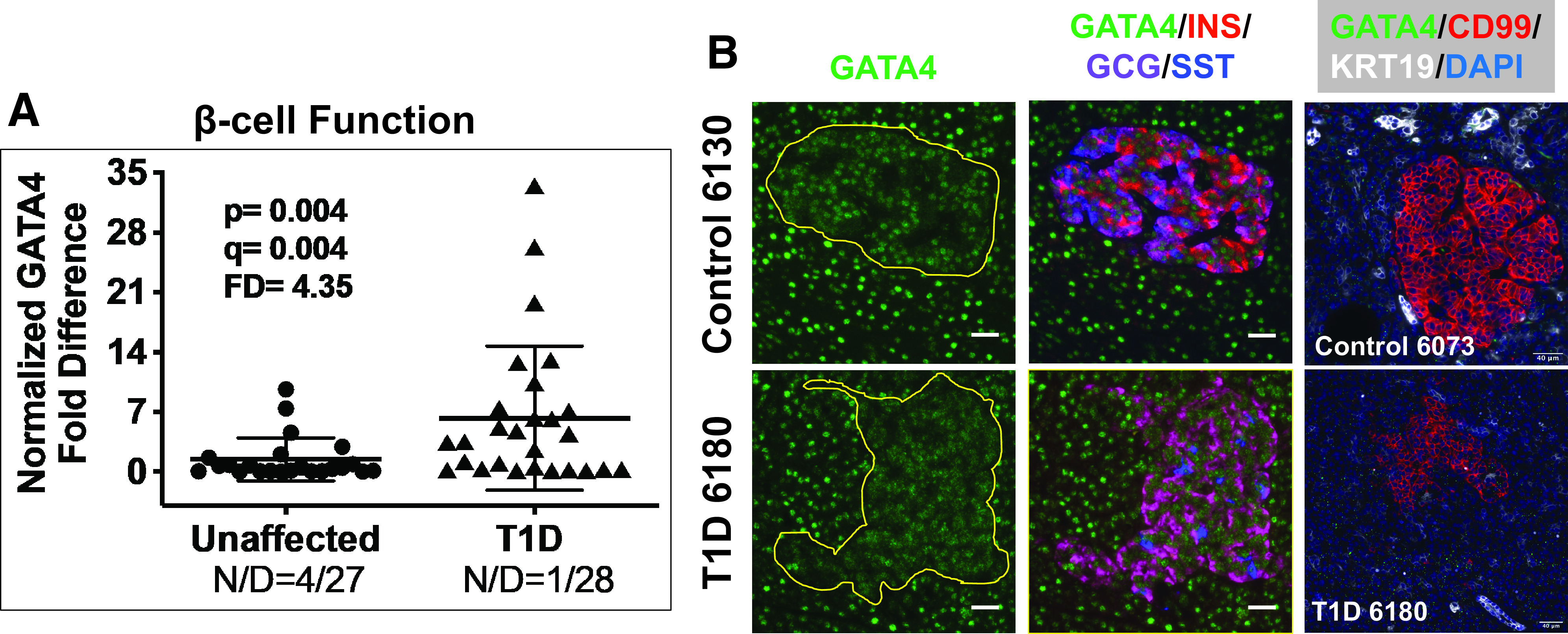
A: Scatter plot of RTqPCR data depicting expression levels for GATA4, a monogenic diabetes gene in the category β-cell function (Supplementary Table 7). The FD is calculated based on the ratio of the means (Research Design and Methods). See Research Design and Methods for statistical analysis (P values and q values [estimation of false discovery rates]). N/D refers to number (N) of samples yielding no data out of the total (D). B: Widefield IF of GATA4 (green) and overlay with insulin (INS), glucagon (GCG), and somatostatin (SST) from a control and T1D pancreas. Final panel in each row shows combined GATA4 ISH (RNAscope [green dots]) coupled with IF for CD99 and KRT19. Magnification bars = 40 μm.
RTqPCR analysis of genes belonging to the β-cell development group (Supplementary Table 7) identified altered expression in the T1D pancreas for GATA6, GLIS3, HNF1B, MNX1, and TRMT10A (Table 1), with FDs ranging from 2.25 to 4.01 relative to control donor pancreata. Examination of GLIS3, FD 4.01 (Fig. 3A), by IF and ISH-IF demonstrated expression in both islets and the exocrine pancreas in unaffected control donors (Fig. 3B). Close examination, however, reveals an obvious decrease in islet-specific expression of GLIS3 in the T1D pancreas with no obvious change in the exocrine region (Fig. 3B and Supplementary Figs. 5 and 6, yellow outlines). In addition, the combined ISH-IF (Fig. 3B, right) is consistent with decreased GLIS3 mRNA in the T1D islet and demonstrates that exocrine expression is most likely specific to ductal epithelial cells based on GLIS3 mRNA colocalization (ISH) with IF for KRT19, a type 1 keratin specific to ductal cells (36).
Figure 3.
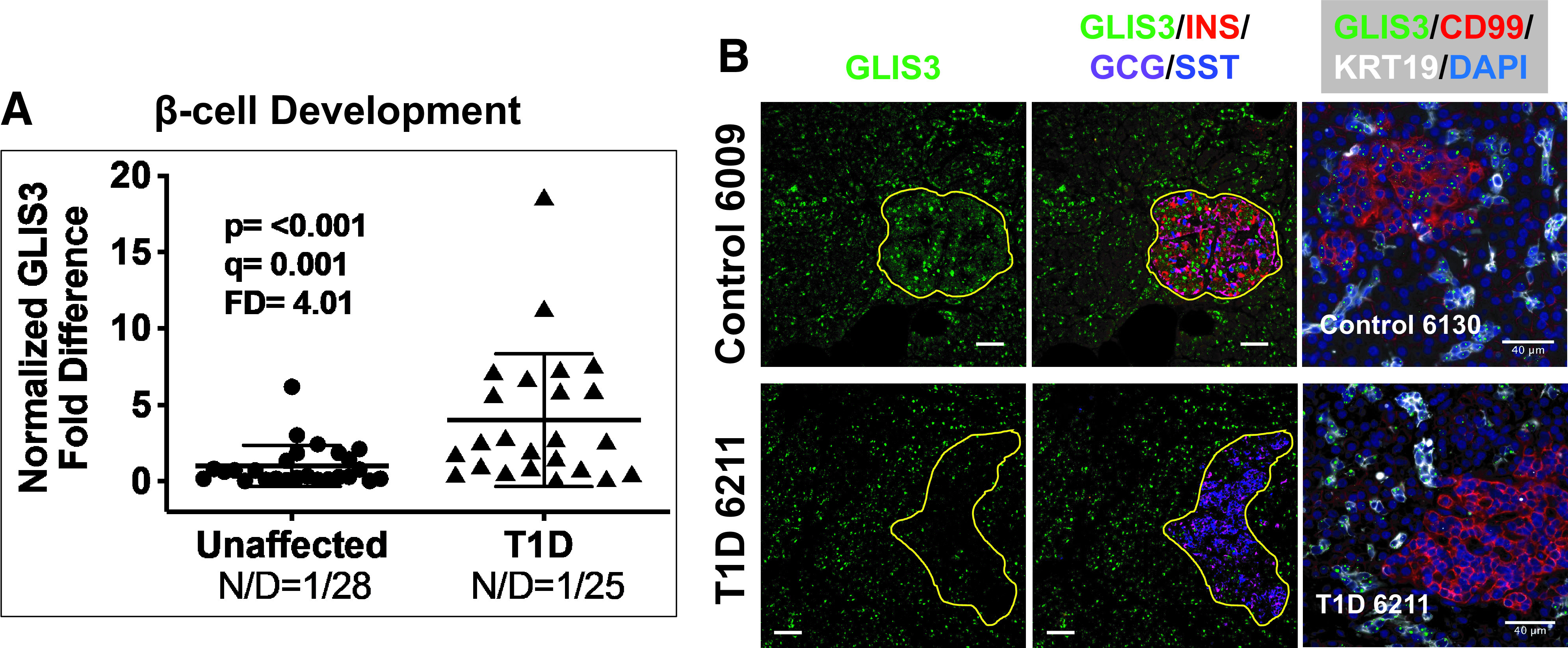
Scatter plot of RTqPCR data depicting expression levels for GLIS3, a monogenic diabetes gene in the category β-cell development (Supplementary Table 7). The FD is calculated based on the ratio of the means (Research Design and Methods). See Research Design and Methods for statistical analysis (P values and q values [estimation of false discovery rates]). N/D refers to number (N) of samples yielding no data out of the total (D). B: Widefield IF of GLIS3 (green) and overlay with insulin (INS), glucagon (GCG), and somatostatin (SST) from a control and T1D pancreas. Yellow outlines depict the islets to illustrate the significant reduction of GLIS3 in the T1D islet. Final panel in each row shows combined GLIS3 ISH (RNAscope [green dots]) coupled with IF for CD99 and KRT19. Magnification bars = 40 μm.
The final set of monogenic diabetes genes examined is grouped based on their association with ER function/stress (Supplementary Table 7), with altered expression in T1D pancreata observed for BSCL2, CEL, ITCH, and WFS1 (FD range from 2.2 to 4.41 [Table 1]). WFS1 (Wolframin, regulator of ER calcium homeostasis) expression FD of 4.41 (Fig. 4A) was also assessed by IF for evaluation of WFS1 (37) protein localization in the human pancreas (Fig. 4B and Supplementary Figs. 7 and 8). Consistent with the IF data, ISH-IF identified WFS1 mRNA in the islet along with scattered expression in exocrine regions of the pancreas from donors with T1D and control donors (Fig. 4B, right). With these data we noted islet-specific cytosolic expression with apparent colocalization involving both INS and GCG in unaffected control pancreas. Most strikingly, we consistently observed non-hormone-expressing WFS1+ cells in the vast majority of islets from both control and T1D pancreas. To confirm this observation, we used confocal microscopy on sections of unaffected control and T1D pancreata costained for WFS1 (Fig. 4C) in conjunction with INS, GCG, and SST. The combined z-stacks for the overlay and each separate channel illustrate individual cells (insets and yellow arrows) that only express WFS1, having no coregistration with endocrine hormones. The overlay panel (Fig. 4B) for the T1D pancreas 6180 also revealed a potential islet exclusively expressing cells positive only for WFS1 (green-only islet, single INS+ cell in the lower middle). The presence of islets/cells exclusively positive for WFS1 was reproduced in other islets from control and T1D pancreata (Supplementary Figs. 7 and 8). To be clear, cells only positive for WFS1 are also evident in islets that display hormone positivity as well (Fig. 4B and Supplementary Figs. 7 and 8).
Figure 4.
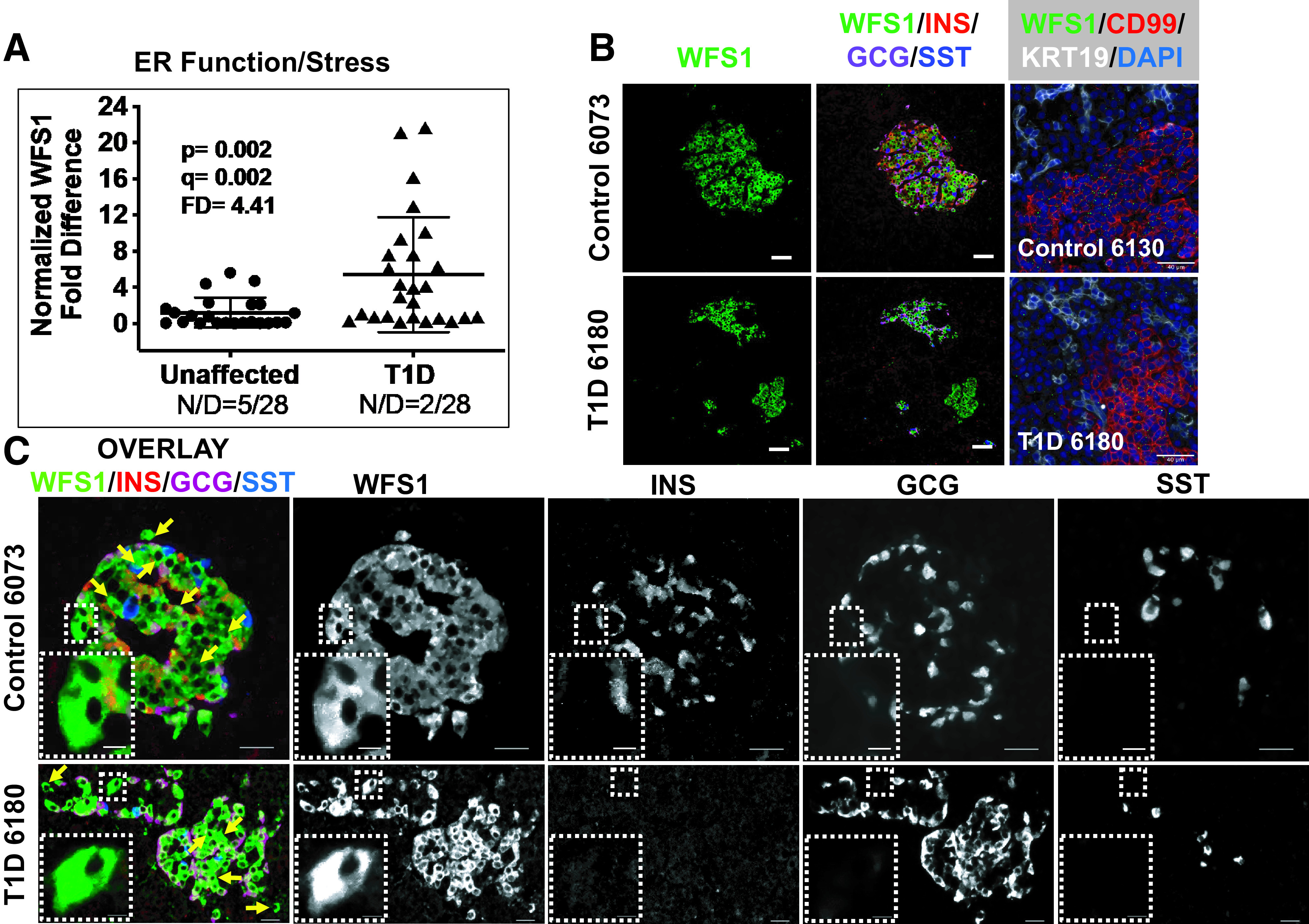
A: Scatter plot of RTqPCR data depicting expression levels for WFS1, a monogenic diabetes gene in the category ER function/stress (Supplementary Table 7). The FD is calculated based on the ratio of the means (Research Design and Methods). See Research Design and Methods for statistical analysis (P values and q values [estimation of false discovery rates]). N/D refers to number (N) of samples yielding no data out of the total (D). B: Widefield IF of WFS1 (green) and overlay with insulin (INS), glucagon (GCG), and somatostatin (SST) from a control and T1D pancreas. Final panel in each row shows combined WFS1 ISH (RNAscope [green dots]) coupled with IF for CD99 and KRT19. Magnification bars = 40 μm. C: Confocal imaging on a Nikon A1plus confocal microscope of WFS1 (green), insulin (INS), glucagon (GCG), and somatostatin (SST), showing an overlay of an islet from an unaffected and T1D pancreas (left panels). 3D Z-stacks were constructed with a 2.5-μm step size and compressed to a 2D maximum intensity projection for display. Individual channels in black and white identify cells positive only for WFS1 and negative for the other endocrine hormones, illustrated with yellow arrows (overlay) and with insets in the respective black and white channels.
Deeper Analysis of the ER Function and Stress Pathway Reveals That the Apex of the ISR Is Activated in Type 1 Diabetes Pancreata
EIF2AK3/PERK gene expression revealed a significantly increased FD of 8.3 (Fig. 5A). Using IF and ISH-IF in unaffected control and T1D pancreata (Fig. 5B and Supplementary Figs. 9 and 10), we noted EIF2AK3/PERK localization in both islets and the exocrine pancreas. The localization in control islets is consistently higher when compared with that in the exocrine tissue (Fig. 5B and Supplementary Fig. 9), whereas EIF2AK3/PERK localization in the T1D pancreas was more uniform in intensity in both the endocrine and exocrine tissue (Figs. 5B and Supplementary Fig. 10). We then examined 30 ISR-associated genes, identifying 20 that were significantly induced in T1D versus control pancreas (Table 2), with FDs ranging from 2.4 to 18.11. The ISR contains three arms where ATF6 (arm 1), IRE1α (ERN1) (arm 2), and each of the eIF2α kinases, PERK, HRI, PKR, and GCN2 (arm 3), constitutes the apex of each arm. RTqPCR data for genes at the apex of arms 1–3 of the ISR (Fig. 5C) demonstrate that all three arms are activated, including three of the four eIF2α kinases (EIF2AK3/PERK [Fig. 5A] and EIF2AK2/PKR and EIF2AK4/GCN2 [Fig. 5C]). These data therefore strongly implicate that the ISR is chronically activated in T1D pancreata, with disease duration for our T1D donor samples spanning 7 months–57 years (Supplementary Table 1).
Figure 5.
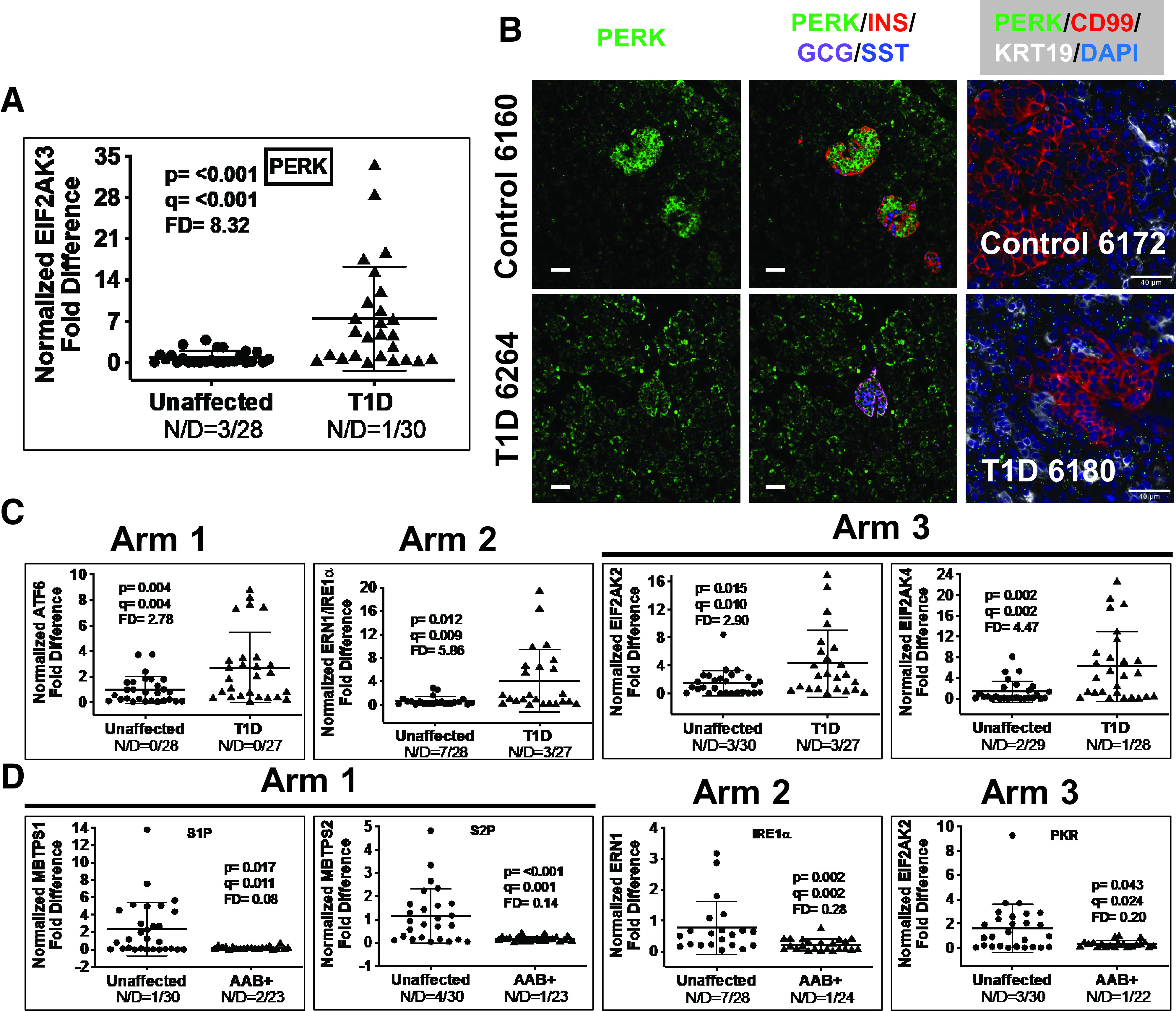
A: Scatter plot of RTqPCR data depicting expression levels for EIF2AK3/PERK, a representative gene in arm 3 of the ISR. The FD is calculated based on the ratio of the means (Research Design and Methods). See Research Design and Methods for statistical analysis (P values and q values [estimation of false discovery rates]). N/D refers to number (N) of samples yielding no data out of the total (D). B: Widefield IF of PERK (green) and overlay with insulin (INS), glucagon (GCG), and somatostatin (SST) from a control and T1D pancreas. Final panel in each row shows combined PERK ISH (RNAscope [green dots]) coupled with IF for CD99 and KRT19. Magnification bars = 40 μm. C: Scatter plots of RTqPCR data depicting expression levels for ISR genes from the apex of each ISR arm 1–3 in T1D vs. unaffected control pancreata: ATF6, ERN1/IRE1α, EIF2AK2/PKR, EIF2AK3/PERK, and EIF2AK4/GCN2. The FD is calculated based on the ratio of the means. See Research Design and Methods for statistical analysis (P values and q values [estimation of false discovery rates]). D: Scatter plot of RTqPCR data depicting expression levels for genes significantly repressed in autoantibody+ (AAB+) pancreata in each of the three arms of the ISR: MBTPS1/S1P, MBTPS2/S2P, ERN1/IRE1α, and EIF2AK2/PKR.
Table 2.
ISR genes
| Gene | T1D (n = 25–30) | AAB+ (n = 20–24) | ||||
|---|---|---|---|---|---|---|
| FD | P value | q value | FD | P value | q value | |
| ALS2 | 12.36 | <0.001 | <0.001 | 0.60 | 0.703 | 0.179 |
| ATF4 | 1.27 | 0.306 | 0.097 | ND | ||
| ATF6 | 2.78 | 0.004 | 0.004 | 0.85 | 0.869 | 0.216 |
| BLOC1S1 | 1.48 | 0.379 | 0.115 | ND | ||
| COL6A2 | 8.25 | <0.001 | <0.001 | 0.50 | 0.598 | 0.163 |
| DDIT3/CHOP | 1.83 | 0.073 | 0.037 | ND | ||
| DNAJB11 | 4.58 | <0.001 | <0.001 | 4.35 | <0.001 | <0.001 |
| DNAJB9 | 5.61 | <0.001 | <0.001 | 0.45 | 0.905 | 0.223 |
| DNAJC3 | 1.81 | 0.225 | 0.078 | ND | ||
| EDEM1 | 18.11 | <0.001 | <0.001 | 1.20 | 0.113 | 0.050 |
| EIF2AK1 | 2.12 | 0.142 | 0.058 | ND | ||
| EIF2AK2 | 2.90 | 0.015 | 0.010 | 0.20 | 0.043 | 0.024 |
| EIF2AK3 | 8.32 | <0.001 | <0.001 | 3.41 | <0.001 | <0.001 |
| EIF2AK4 | 4.47 | 0.002 | 0.002 | 0.79 | 0.170 | 0.065 |
| EIF2S1 | 2.14 | 0.082 | 0.039 | ND | ||
| ERN1/IRE1a | 5.86 | 0.012 | 0.009 | 0.28 | 0.002 | 0.002 |
| FEN1 | 8.37 | <0.001 | <0.001 | 2.47 | 0.001 | 0.001 |
| HSP90B1 | 4.07 | 0.002 | 0.002 | 0.56 | 0.358 | 0.111 |
| HSPA5 | 2.54 | 0.122 | 0.051 | ND | ||
| MBTPS1 | 3.12 | 0.026 | 0.015 | 0.08 | 0.017 | 0.011 |
| MBTPS2 | 2.77 | 0.159 | 0.063 | 0.14 | <0.001 | 0.001 |
| MSMO1 | 2.40 | 0.045 | 0.025 | 0.40 | 0.291 | 0.093 |
| MVK | 6.43 | <0.001 | <0.001 | 0.97 | 0.204 | 0.076 |
| NELFA | 7.48 | <0.001 | <0.001 | 1.65 | 0.017 | 0.011 |
| PDIA4 | 8.38 | <0.001 | <0.001 | 1.26 | 0.075 | 0.037 |
| PPP1R15A | 9.48 | <0.001 | <0.001 | 0.47 | 0.689 | 0.176 |
| TMBIM6 | 4.42 | <0.001 | <0.001 | 0.76 | 0.689 | 0.176 |
| VCP | 12.44 | <0.001 | <0.001 | 1.05 | 0.078 | 0.038 |
| XBP1s | 3.81 | 0.138 | 0.057 | ND | ||
| XBP1u | 1.48 | 0.361 | 0.111 | ND | ||
Tabulated FDs, P values, and q values for the ISR genes showing altered expression in T1D and autoantibody+ (AAB+) pancreata. n refers to the number of independent pancreata in each cohort, and boldface type denotes genes showing altered expression. Of 30 ISR genes tested, 20 showed differential expression in T1D pancreata. Of the 20 genes altered in T1D, 8 showed differential expression in the autoantibody+ cohort (4 genes were induced and 4 repressed). ND, not done.
To evaluate a potential role for the ISR in pre-T1D, we tested the expression levels of all 20 ISR genes showing altered expression in T1D pancreata within our autoantibody+ cohort (Table 2). We identified eight ISR-linked genes that showed significantly altered expression in pancreata from autoantibody+ donors, with four of these being induced (DNAJB11, EIF2AK3, FEN1, and NELFA [Table 2]). The remaining four dysregulated ISR genes showed a significant level of repression in autoantibody+ donor tissues associated with a remarkable inhibition at the apex of all three arms of the ISR (Table 2 and Fig. 5D).
Discussion
As a complement to traditional genetic linkage strategies, we used a transcriptomic analysis to test a novel hypothesis that genes classically associated with monogenic forms of diabetes (3–6) would be differentially expressed in human pancreas from individuals with T1D or those at increased risk for the disease. Studies were conducted on tissues from donors who did not carry monogenic diabetes–associated mutations, with features consistent with T1D or type 2 diabetes (HLA, age of onset, BMI, HbA1c, and C-peptide [Supplementary Table 1 and Supplementary Fig. 11]). In support of this strategy, our data demonstrate that 24 of the 45 genes associated with monogenic diabetes displayed altered expression in pancreata from T1D versus control donors, and 6 of these genes were also dysregulated in autoantibody+ organ donors, considered to have high risk (single autoantibody+) or pre-T1D (≥2 autoantibody+). The uniformity of the repressed HNF4A expression across the entire autoantibody+ cohort further advances the notion that even a single autoantibody may have relevant prognostic value.
The type 2 diabetes pancreas also demonstrated altered expression of five monogenic diabetes genes relative to unaffected control tissue. Not surprisingly, these genes fell primarily within the ER function/stress physiological group, while expression of monogenic diabetes genes associated with all four physiological groups (i.e., immune, β-cell function, β-cell development, and ER function/stress) were altered in T1D. Hence, genes exhibiting altered expression likely point toward mechanisms underlying T1D and type 2 diabetes pathogenesis, respectively.
Localization (IF) studies on select dysregulated monogenic diabetes and ISR genes from each physiological study group pinpointed genes that are expressed to a greater extent in the islets (e.g., STAT5B and WFS1), while others were expressed in both islet and exocrine regions of the pancreas (e.g., GATA4, GLIS3, and PERK). Hence, these data highlight a potential role for ISR activation within the exocrine pancreas in T1D pathogenesis, in line with our previous studies demonstrating reduced pancreas mass (38,39) and volume (40) as well as low serum levels of trypsinogen in recent-onset T1D patients and pre-T1D subjects (41). We also identified non-hormone-expressing cells in human islets that were positive for either the transcription factor STAT5B or an ER calcium channel/regulator, WFS1. Although both of these proteins were sometimes coregistered with GCG, INS, or SST, identifying α-, β-, and δ-cells, respectively, in control and T1D pancreata, there are cells scattered across the pancreas that express these proteins and are clearly hormone negative. We might speculate that these results represent a possible stem cell population, a state of dedifferentiation, cells exhausted of hormone at the time of death, or a previously undetected islet cell type. We are currently attempting to identify unique surface markers that could facilitate cell sorting and single-cell RNA-sequencing analysis to further characterize these cells
We believe the power of our monogenetic strategy has been borne out in identifying a plethora of noteworthy genes coupled with their unique pancreatic localization, as well as the potential functional implications of their dysregulated expression. In addition to monogenic diabetes genes, we extended our studies to ISR-linked genes, with 20 of 30 ISR genes studied displaying altered expression in T1D pancreata. These results included three of four eIF2α-dependent kinases (42), thus attendant with ISR (43) activation in T1D pathogenesis, strongly implicating an overarching, chronic stress response (44,45). Interestingly, we uncovered numerous additional loci with altered expression that are central to the regulation and function of each arm of the ISR pathway in T1D pancreata, suggesting a global activation of the ISR in the T1D pancreas, not simply extrapolated from the study of a single gene. The ISR can be induced by intrinsic stress associated, for example, with the accumulation of unfolded proteins in the ER mediated by PERK, or through extrinsic stressors including oxidative challenge (HRI), viral infection (PKR), or amino acid deprivation (GCN2) (42). While such stress responses may lead to activation of the unfolded protein response and/or ISR in type 2 diabetes (46–50), a similar argument for the ISR in T1D (47,51–54) has been hypothesized, but not directly demonstrated. Indeed, prior reports have implicated ER stress and the unfolded protein response as potentially contributing to inflammation and β-cell death in T1D, but to our knowledge, this represents a first report demonstrating comprehensive activation of all three arms of the ISR in human T1D pancreas tissue.
Of note, our studies are restricted due to the inherent limitations of our organ donor study group such that more mechanistic studies are not easily implemented in archival tissue. To this end, future studies in human islets or live human pancreas organ slices could more directly address pathway-specific and physiologically relevant mechanisms. From a diagnostic standpoint, our data are all derived from organ donor pancreata, which precludes any diagnostic assay in live patients; however, we are beginning studies to address some of these markers in peripheral blood. We thus believe our data afford the diabetes community a rich opportunity to investigate potential therapeutics targeting numerous metabolic and pancreas intrinsic signaling pathways. Our observations in the T1D pancreas taken in the context of a lifelong disease may be best described by the title of a review by Rutkowski and Kaufman entitled, “That Which Does Not Kill Me Makes Me Stronger: Adapting to Chronic ER Stress” (45).
Article Information
Acknowledgments. The authors thank the organ donors and their families for their precious contributions to nPOD for research, without which this work would not be possible. Organ procurement organizations partnering with nPOD to provide research resources are listed at https://www.jdrfnpod.org/for-partners/npod-partners/. The authors extend special thanks to Dr. Rhonda L. Bacher (Department of Biostatistics, University of Florida) regarding study design and statistical analyses.
Funding. This work was supported by nPOD (RRID:SCR_014541), a collaborative T1D research project sponsored by JDRF (5-SRA-2018-557-Q-R), and the Leona M. and Harry B. Helmsley Charitable Trust (grant 2018PG-T1D053). This research was also supported by the National Institutes of Health (DK108132, AI42288, and S10OD020026).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. C.H.W. researched the data and wrote the manuscript. H.H., D.E.B., J.J.L., and S.E. researched the data and reviewed and edited the manuscript. J.R.M. contributed to the study design, analyzed the data, and reviewed and edited the manuscript. D.R.M., I.K., and L.M.J. researched the data and reviewed and edited the manuscript. A.L.P., H.K., R.A.O., D.A.S., A.T.H., and B.B. contributed to discussion and reviewed and edited the manuscript. M.A.A. and H.S.N. conceived of the study and wrote the manuscript. C.H.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the nPOD 12th Annual Meeting, Tampa, FL, 23–26 February 2020.
Footnotes
H.S.N. and C.H.W. are co–senior authors.
This article contains supplementary material online at https://doi.org/10.2337/figshare.14612202.
References
- 1.Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet 2014;383:69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Diabetes Association . 2. Classification and diagnosis of diabetes: Standards of Medical Care in Diabetes—2017. Diabetes Care 2017(Suppl. 1):S11–S24 [DOI] [PubMed] [Google Scholar]
- 3.Hattersley AT, Patel KA. Precision diabetes: learning from monogenic diabetes. Diabetologia 2017;60:769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antosik K, Borowiec M. Genetic factors of diabetes. Arch Immunol Ther Exp (Warsz) 2016;64(Suppl. 1):157–160 [DOI] [PubMed] [Google Scholar]
- 5.Vaxillaire M, Bonnefond A, Froguel P. The lessons of early-onset monogenic diabetes for the understanding of diabetes pathogenesis. Best Pract Res Clin Endocrinol Metab 2012;26:171–187 [DOI] [PubMed] [Google Scholar]
- 6.Barbetti F, Ghizzoni L, Guaraldi F. Diabetes associated with single gene defects and chromosomal abnormalities. In Frontiers in Diabetes. Porta M, Matschinsky FM, Eds., Karger, 2017, p. 194 [Google Scholar]
- 7.Stekelenburg CM, Schwitzgebel VM. Genetic defects of the β-cell that cause diabetes. Endocr Dev 2016;31:179–202 [DOI] [PubMed] [Google Scholar]
- 8.Fajans SS, Bell GI. MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011;34:1878–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amed S, Oram R. Maturity-onset diabetes of the young (MODY): making the right diagnosis to optimize treatment. Can J Diabetes 2016;40:449–454 [DOI] [PubMed] [Google Scholar]
- 10.Bishay RH, Greenfield JR. A review of maturity onset diabetes of the young (MODY) and challenges in the management of glucokinase-MODY. Med J Aust 2017;207:223. [DOI] [PubMed] [Google Scholar]
- 11.Barbetti F, D’Annunzio G. Genetic causes and treatment of neonatal diabetes and early childhood diabetes. Best Pract Res Clin Endocrinol Metab 2018;32:575–591 [DOI] [PubMed] [Google Scholar]
- 12.Johnson MB, Hattersley AT, Flanagan SE. Monogenic autoimmune diseases of the endocrine system. Lancet Diabetes Endocrinol 2016;4:862–872 [DOI] [PubMed] [Google Scholar]
- 13.Campbell-Thompson ML, Montgomery EL, Foss RM, et al. Collection protocol for human pancreas. J Vis Exp 2012;63:4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell-Thompson M, Wasserfall C, Kaddis J, et al. Network for Pancreatic Organ Donors with Diabetes (nPOD): developing a tissue biobank for type 1 diabetes. Diabetes Metab Res Rev 2012;28:608–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.American Diabetes Association . Standards of Medical Care in Diabetes—2020 abridged for primary care providers. Clin Diabetes 2020;38:10–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 2012;13:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009;55:611–622 [DOI] [PubMed] [Google Scholar]
- 18.Kozera B, Rapacz M. Reference genes in real-time PCR. J Appl Genet 2013;54:391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dundas J, Ling M. Reference genes for measuring mRNA expression. Theory Biosci 2012;131:215–223 [DOI] [PubMed] [Google Scholar]
- 20.Xie F, Xiao P, Chen D, Xu L, Zhang B. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol. 2012;80:75–84 [DOI] [PubMed] [Google Scholar]
- 21.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:RESEARCH0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersen CL, Jensen JL, Ørntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004;64:5245–5250 [DOI] [PubMed] [Google Scholar]
- 23.Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol Lett 2004;26:509–515 [DOI] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001;25:402–408 [DOI] [PubMed] [Google Scholar]
- 25.Peck R, Olsen C, Devore JL. Introduction to Statistics and Data Analysis. Taylor M, Ed. Boston, MA, Brooks/Cole Cengage Learning, 2010 [Google Scholar]
- 26.Tampella G, Kerns HM, Niu D, et al. The Tec kinase-regulated phosphoproteome reveals a mechanism for the regulation of inhibitory signals in murine macrophages. J Immunol 2015;195:246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000;132:365–386 [DOI] [PubMed] [Google Scholar]
- 28.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol 1970;48:443–453 [DOI] [PubMed] [Google Scholar]
- 29.In’t Veld P, De Munck N, Van Belle K, et al. β-Cell replication is increased in donor organs from young patients after prolonged life support. Diabetes 2010;59:1702–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sullivan BA, Hollister-Lock J, Bonner-Weir S, Weir GC. Reduced Ki67 staining in the postmortem state calls into question past conclusions about the lack of turnover of adult human β-cells. Diabetes 2015;64:1698–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caballero F, Siniakowicz K, Hollister-Lock J, et al. Birth and death of human β-cells in pancreases from cadaver donors, autopsies, surgical specimens, and islets transplanted into mice. Cell Transplant 2014;23:139–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebrahimi A, Jung MH, Dreyfuss JM, et al. Evidence of stress in β cells obtained with laser capture microdissection from pancreases of brain dead donors. Islets 2017;9:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wasserfall C, Montgomery E, Yu L, et al. Validation of a rapid type 1 diabetes autoantibody screening assay for community-based screening of organ donors to identify subjects at increased risk for the disease. Clin Exp Immunol 2016;185:33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Insel RA, Dunne JL, Atkinson MA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015;38:1964–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wasserfall C, Nick HS, Campbell-Thompson M, et al. Persistence of pancreatic insulin mRNA expression and proinsulin protein in type 1 diabetes pancreata. Cell Metab 2017;26:568–575.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain R, Fischer S, Serra S, Chetty R. The use of cytokeratin 19 (CK19) immunohistochemistry in lesions of the pancreas, gastrointestinal tract, and liver. Appl Immunohistochem Mol Morphol 2010;18:9–15 [DOI] [PubMed] [Google Scholar]
- 37.Rigoli L, Bramanti P, Di Bella C, De Luca F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr Res 2018;83:921–929 [DOI] [PubMed] [Google Scholar]
- 38.Campbell-Thompson M, Fu A, Kaddis JS, et al. Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes 2016;65:719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campbell-Thompson ML, Kaddis JS, Wasserfall C, et al. The influence of type 1 diabetes on pancreatic weight. Diabetologia 2016;59:217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell-Thompson ML, Filipp SL, Grajo JR, et al. Relative pancreas volume is reduced in first-degree relatives of patients with type 1 diabetes. Diabetes Care 2019;42:281–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X, Campbell-Thompson M, Wasserfall CH, et al. Serum trypsinogen levels in type 1 diabetes. Diabetes Care 2017;40:577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donnelly N, Gorman AM, Gupta S, Samali A. The eIF2α kinases: their structures and functions. Cell Mol Life Sci 2013;70:3493–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep 2016;17:1374–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guan BJ, van Hoef V, Jobava R, et al. A unique ISR program determines cellular responses to chronic stress. Mol Cell 2017;68:885–900.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rutkowski DT, Kaufman RJ. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem Sci 2007;32:469–476 [DOI] [PubMed] [Google Scholar]
- 46.Rocha M, Diaz-Morales N, Rovira-Llopis S, et al. Mitochondrial dysfunction and endoplasmic reticulum stress in diabetes. Curr Pharm Des 2016;22:2640–2649 [DOI] [PubMed] [Google Scholar]
- 47.Meyerovich K, Ortis F, Allagnat F, Cardozo AK. Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. J Mol Endocrinol 2016;57:R1–R17 [DOI] [PubMed] [Google Scholar]
- 48.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 2012;81:767–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuchiya Y, Saito M, Kohno K. Pathogenic mechanism of diabetes development due to dysfunction of unfolded protein response. Yakugaku Zasshi 2016;136:817–825 [DOI] [PubMed] [Google Scholar]
- 50.Herbert TP, Laybutt DR. A reevaluation of the role of the unfolded protein response in islet dysfunction: maladaptation or a failure to adapt? Diabetes 2016;65:1472–1480 [DOI] [PubMed] [Google Scholar]
- 51.Engin F. ER stress and development of type 1 diabetes. J Investig Med 2016;64:2–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brozzi F, Eizirik DL. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J Med Sci 2016;121:133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhong J, Rao X, Xu JF, Yang P, Wang CY. The role of endoplasmic reticulum stress in autoimmune-mediated beta-cell destruction in type 1 diabetes. Exp Diabetes Res 2012;2012:238980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cnop M, Toivonen S, Igoillo-Esteve M, Salpea P. Endoplasmic reticulum stress and eIF2α phosphorylation: the Achilles heel of pancreatic β cells. Mol Metab 2017;6:1024–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]