

Abstract
Background
Macrophages are pivotal cells in sarcoidosis. Monocytes-derived (MD) macrophages have recently been demonstrated to play a major role especially in pulmonary sarcoidosis. From inflammatory tissues to granulomas, they may be exposed to low oxygen tension environments. As hypoxia impact on sarcoidosis immune cells has never been addressed, we designed the present study to investigate MD-macrophages from sarcoidosis patients in this context. We hypothesized that hypoxia may induce functional changes on MD-macrophages which could have a potential impact on the course of sarcoidosis.
Methods
We studied MD-macrophages, from high active sarcoidosis (AS) (n=26), low active or inactive sarcoidosis (IS) (n=24) and healthy controls (n=34) exposed 24 hours to normoxia (21% O2) or hypoxia (1.5% O2). Different macrophage functions were explored: hypoxia-inducible factor-1α (HIF-1α) and nuclear factor-kappa B (NF-κB) activation, cytokines secretion, phagocytosis, CD80/CD86/HLA-DR expression, profibrotic response.
Results
We observed that hypoxia, with a significantly more pronounced effect in AS compared with controls and IS, increased the HIF-1α trans-activity, promoted a proinflammatory response (TNFα, IL1ß) without activating NF-κB pathway and a profibrotic response (TGFß1, PDGF-BB) with PAI-1 secretion associated with human lung fibroblast migration inhibition. These results were confirmed by immunodetection of HIF-1α and PAI-1 in granulomas observed in pulmonary biopsies from patients with sarcoidosis. Hypoxia also decreased the expression of CD80/CD86 and HLA-DR on MD-macrophages in the three groups while it did not impair phagocytosis and the expression of CD36 expression on cells in AS and IS at variance with controls.
Conclusions
Hypoxia had a significant impact on MD-macrophages from sarcoidosis patients, with the strongest effect seen in patients with high active disease. Therefore, hypoxia could play a significant role in sarcoidosis pathogenesis by increasing the macrophage proinflammatory response, maintaining phagocytosis and reducing antigen presentation, leading to a deficient T cell response. In addition, hypoxia could favor fibrosis by promoting profibrotic cytokines response and by sequestering fibroblasts in the vicinity of granulomas.
Keywords: pulmonary sarcoidosis, hypoxia-inducible factor 1, plasminogen activator inhibitor-1, macrophages, fibrosis, monocytes
Introduction
Sarcoidosis is a systemic granulomatous disease of unknown cause affecting the lung with a prevalence close to 90% (1, 2). Granulomas, the key lesion in sarcoidosis, are well circumscribed and coalescent clusters of macrophage-derived epithelioid and giant cells, associated with lymphocytes, subsequently wrapped by lamellar fibrosis (3). The prevailing hypothesis explaining granuloma formation is that exposure to unknown antigens triggers a disproportionate inflammatory response in genetically predisposed individuals (4). Sarcoidosis is particularly challenging in regards of identifying factors involved in initiation of inflammation, granuloma perpetuation and evolution towards fibrosis as observed mostly in pulmonary, but also in hepatic, renal or cardiac involvement (3, 5–7). These events are determinant for sarcoidosis outcomes which vary from benign to life-threatening especially due to pulmonary fibrosis and/or pulmonary hypertension (8).
Macrophages are pivotal cells in the sequence of events associated with sarcoidosis pathogenesis, granulomas having been clearly demonstrated constituted of macrophages-derived cells (9–13). Different types of macrophage populations (alveolar and interstitial) derivating from distinct precursors of embryonic/fetal and myeloid origin, reside in the lung (14, 15). Notably, upon injury, the myeloid precursors generate monocyte-derived (MD) macrophages, though to play a major role in fibrosis (16) and sarcoidosis (11, 17–20).
In the lung of patients, these cells are an important source of TNFα, a major cytokine in the pathogenesis of sarcoidosis (11). Circulating mononuclear cells from sarcoidosis patients are also able to reconstitute granulomas in vitro, and present a specific transcriptional profile compared to other granulomatosis such as tuberculosis (17, 18). In addition, analysis of granulomas from sarcoidosis recurrence after lung transplantation demonstrated that constitutive cells derived from recipient blood cells (19, 20), indicating that peripheral blood cells are major contributors in granuloma formation.
Hypoxia, defined as an imbalance between impaired tissue oxygen supply and cell demand, is a micro-environmental condition known to modulate innate and adaptive immunity (21) by promoting pro-inflammatory response (22) and to contribute to the pathophysiology of fibrotic diseases (23). Hypoxia impact on macrophages is particularly relevant to study in sarcoidosis as a previous study showed that the absence of in-depth vascularization makes sarcoidosis granulomas hypoxic (24) as for tuberculous granulomas (25). Moreover, from inflammatory tissues to granulomas, MD-macrophages may be exposed to low oxygen tension environments mainly due to O2 consumption related to hypermetabolism of inflammatory cells (26). The cellular response to hypoxia is mostly controlled by the hypoxia-inducible factor (HIF) transcription factor and its target genes harboring the hypoxia-response element (27). Several studies in sarcoidosis reported the expression of HIF-target genes within lung and lymph node granulomas, while HIF-1α expression was inconsistently found (28, 29). Hypoxia and the HIF-1 signaling impact on the processes controlling granuloma evolution are still unclear and their effects on sarcoidosis immune cells have never been addressed.
We hypothesized that hypoxia may induce macrophage functional changes with a potential impact on the course of the disease. Therefore, we studied the consequences of hypoxia on a set of main functions of MD-macrophages from sarcoidosis patients and healthy controls. We show that hypoxia had a significant impact on MD-macrophages from sarcoidosis patients, with the strongest effect seen in patients with high active disease. Therefore, hypoxia could play a significant role in sarcoidosis pathogenesis and fibrosis outcome.
Materials and Methods
Additional details are provided in the Online Supplement.
Patients and Controls
This prospective monocentric study was conducted in the Pulmonary Department of the Avicenne University Hospital, France between 2017 and 2019 and received institutional review board approval (CPP Ile-de-France X 2016-10-02) according to French legislation. Written informed consent for all participants was obtained for biological investigations.
Patients were over 18 with a pulmonary sarcoidosis according to guidelines (30, 31). The diagnosis of sarcoidosis was validated in multidisciplinary meetings with histopathology confirmation in 46 of the 50 patients, and 4 patients had an initial Lofgren syndrome.
Individuals with corticosteroid/immunosuppressive therapy in the past 6 months were excluded. As the lung is the most affected organ in sarcoidosis and disease activity is difficult to define, we used a computed tomography (CT) score developed in pulmonary sarcoidosis: the abbreviated Computed-Tomography Activity Score (aCTAS) (32). Briefly, aCTAS comprised the sum of the presence (1) or absence (0) of nodularity, ground glass opacification, interlobular septal thickening and consolidation (see Figure E1). According to aCTAS, patients were divided in high Active Sarcoidosis (AS) (n=26) with an aCTAS≥2, and low active or Inactive Sarcoidosis (IS) groups (n=24) with an aCTAS<2. Bioclinical and imaging data were recorded at the time of blood draw. Healthy volunteers (n=34) over 18, with no history of sarcoidosis or any known current disease were evaluated as “controls” and matched for age (± 5 years), sex and smoking status with sarcoidosis patients.
In addition, paraffin embedded surgical pulmonary biopsies done before this study for diagnosis in three patients with active sarcoidosis were retrieved from the pathology department archives.
Peripheral Blood Mononuclear Cells (PBMCs) Isolation and Macrophage Differentiation
Twenty mL of total blood sampled in EDTA tubes for routine hematology analysis were collected. We isolated PBMCs using Ficoll density gradient separation and monocytes were purified using the Pan Monocyte Isolation Kit (Miltenyi-Biotec, France). A >90% purity of enriched monocytes was controlled by flow cytometry using APC-CyTM7 mouse anti-Human CD14 (BD Biosciences #557831). Monocytes were cultured in RPMI containing 10% fetal bovine serum (FBS) and differentiated into macrophages with M-CSF (33) (Bio-Techne, France) at physiologic circulating concentration of 5ng/ml (34) for 10 days at 21% O2. The yield of isolated monocytes from either controls or sarcoidosis patients (AS and IS) allowed to only perform two or three different types of assays on the same sample.
Normoxic and Hypoxic Culture Conditions
After these 10 days of differentiation, MD-macrophages were maintained under either hypoxic or normoxic conditions for 24hrs. Cell cultures under hypoxic conditions were performed in a hermetic chamber containing 1.5% O2 for 24hrs to reproduce deep hypoxia (pO2<10mmHg) already evidenced in vivo in sarcoid granulomas by fluoromisonidazole uptake (35). Normoxic cells were maintained at 21% O2 for 24hrs as used in sarcoidosis in vitro models (18, 36). After normoxic or hypoxic exposures, cells or cell lysates and/or supernatants were processed for experiments 2.4 to 2.9.
HIF-1α and Nuclear Factor-kappa B (NF-κB) Activation Assays
After hypoxia or normoxia for 24hrs, cell extracts from MD-macrophages were obtained by addition of 100µL lysis buffer (250 mM NaCl, 50 mM HEPES pH 7.0, 5 mM EDTA, 1 mM dithiothreitol (DTT), 0.1% Nonidet NP40, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 50 μg/ml phenylmethylsulfonyl fluoride, 2 mM sodium pyrophosphate, 1 mM sodium orthovanadate). After centrifugation (10 000×g, 30 min, 4°C), cell lysate protein content was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, France #23225). We evaluated the activation status of HIF-1α, NF-κB-p50 and -p65 using specific oligonucleotide DNA-binding enzyme-linked immunosorbent assays (ELISA) based kit (TransAM® HIF-1 (#47096), NF-kB p50 (#41096) and NF-kB p65 (#40096), Active Motif, Belgium) according to the manufacturer’s protocol.
Immunofluorescent Staining
We cultured MD-macrophages on Labtek 8 well-chambers slides (Ibidi, France) at a density of 0.2 106 cells/well and placed either under control atmosphere or hypoxia for 24hrs. Slides were incubated with an anti-HIF1α rabbit antibody (HIF1alpha (Novus) # NB100479). To detect an unsuspected hypoxic culture environment, reduction of pimonidazole was assayed. Intracellular hypoxia was assessed by adding 400µM pimonidazole (Hypoxyprobe™) in the medium for 2hrs, then cells were immunostained using an anti-pimonidazole mouse IgG1 monoclonal antibody (MAb1) (1/50 Hypoxyprobe ™ kit). Pimonidazole forms adducts with thiol containing proteins only in cells that have an oxygen concentration less than 14 micromolar – equivalent to a partial pressure pO2 = 10 mmHg at 37°C (according to Hypoxyprobe™).
Phagocytosis Functional Test
Phagocytosis was assayed in MD-macrophages using 1µm fluospheres™ yellow-green (505/515) (#F8823 Invitrogen) with an equivalent of 200 fluospheres per cell (37). MD-macrophages were incubated with fluospheres during 30 min in culture plates and then washed 4 times with PBS to remove free microspheres and fixed with 4% PFA. Phagocytosis was estimated by the number of cells with at least one particle compared to the total number of cells observed on 4 fields (objective x100, 24-well plates).
Flow Cytometry
Cell surface marker expression of CD14, and/or CD80, CD86, HLA-DR molecules known to be associated with classic macrophage activation (38) and/or CD36, CD163 markers of alternative macrophage activation (38) and/or isotypes-matched IgG were assayed on MD-macrophages using fluorescent labelled antibodies (see Table E2). At least 20 x 103 cells per sample were analyzed on a flow cytometer Canto II, BD Biosciences and data analysis was performed using BD FACS Diva™ software. Samples were gated on cells using FSC/SSC and doublet discrimination to identify singlets using SS-W vs SS-A, MD-macrophages were identified on the basis of CD14+ expression (see Figure E2).
RNA Extraction, Reverse Transcription and Quantitative-PCR (RT-qPCR)
The RNA was extracted from MD-macrophages using Trizol (Thermo Fisher Scientific, France #15596018) according to the manufacturer’s protocol. Reverse transcription was performed using reserve transcriptase system from Promega (M-MLV reverse transcriptase #M1708, dNTP mix #U151B, M-MLV RT 5X buffer #M531A Random, RNAsin #N2518).
Messenger RNA levels of cytokines associated with sarcoidosis and fibrosis were assayed by quantitative PCRs in presence of ABsolute QPCR Mix (Thermo Fisher Scientific #AB1162) with primer sets specific to IL1B, TNF-A, IL-18, IL-10, CXCL8, PDGFB, VEGF, TGFB1 (see Table E1 , in the supplement). A probe set for UBC was used as the normalization standard. The PCR and relative quantifications were performed in a real-time PCR system (StepOnePlus Real-Time PCR system, Applied BioSystems).
Cytokines Assay
“Conditioned media” (CM) from MD-macrophages (500µl of RPMI medium without FBS/1. 106 cells) were collected after 24hrs (under normoxia or hypoxia culture conditions). Presence of cytokines in CM (IL1-ß, CXCL8, TNFα, IL-10, PDGF-BB, PAI-1, VEGF-A, IL-18, IL-5) were assayed (undiluted, in duplicates) using multiplex-bead based assays (ProcartaPlex™ Multiplex Immunoassay (Invitrogen) #PPX-16-MXH497N) according to manufacturer’s instructions. Cytokines to be investigated were chosen according to RTqPCR results, and Human cytokine antibody array (proteome profiler™ Bio-techne #ARY022B) (data not shown; analysis performed on 1 control and 2 active sarcoidosis). Cytokine concentrations were automatically calculated or extrapolated if close to the lower limit of detection by the xPONENT software.
Concentrations of active (free) and total (free+latent) TGFß1 in MD-macrophages supernatants were determined by enzyme-linked immunosorbent assays (ELISA) using Duo Set kits (R&D systems #DY240) according to the manufacturer’s instructions.
Fibroblast Gap-Closure Assays
Migration of normal human lung fibroblasts (NHLFs) was assayed by placing 0.7mm inserts before cell seeding. The percentage of gap-closure was estimated by the difference between the initial and final gap areas at 24hrs of specific culture conditions, relatively to the initial area.
NHLFs were cultured for 24hrs with FBS10% (i) in CM of MD-macrophages in presence of either anti-human plasminogen activator inhibitor-1 (hPAI-1) antibody (Bio-Techne #AF1786) 20ng/ml, or vehicle, or (ii) in RPMI media in presence of either anti-hPAI-1 antibody, or goat IgG isotype control (Bio-Techne # AB-108-C) 20ng/ml, recombinant hPAI-1 (Bio-Techne #1786-PI-010) or vehicle.
Immunohistochemistry (IHC)
Tissue sections from the three archived pulmonary biopsies were submitted to a microwave antigen-retrieval technique for all antibodies or isotypes. Citrate buffer pH6 was used for PAI-1 and CD68 antibodies and Tris-EDTA pH9 for HIF-1α antibody. Antibodies against: HIF1α (2µg/ml rabbit polyclonal, bs0737, Bioss Inc), PAI-1 (1µg/ml, rabbit polyclonal, Bio-Techne; AF1786) and CD68 [1:1 (ready to use), mouse monoclonal, Dako; IS-61330-2] were used. After being rinsed, tissue sections were incubated with either biotinylated goat anti–rabbit IgG (7.5 μg/ml; Vector Laboratories) or biotinylated horse anti-mouse IgG (7.5 μg/ml; Vector Laboratories) for 30 minutes and detected with an avidin-biotin peroxidase complex detection kit (Vectastain Elite ABC kit; Vector Laboratories) using nickel-diaminobenzidine as a substrate. The precipitation reaction was enhanced with Tris-cobalt, and the sections were counterstained with 0.1% nuclear fast red.
Statistical Analysis
Demographic results were presented as means ± SD or proportions and compared using one-way Anova, student’s t-test or Chi2 test when appropriate. Biological results were represented using box and whiskers (showing 25th and 75th percentile and median), each point indicates a patient and/or control. Biological results were compared with two-way ANOVA-repeated measures with Sidak post-hoc test. PRISM software (v6, GraphPad, USA) was used. A p value<0.05 was considered as significant.
Results
Patients and Controls
Characteristics and comparison between groups, i.e., AS and IS and controls are shown in Table 1. Patients with AS compared to IS had higher angiotensin converting enzyme blood levels (p<0.0001), more impaired forced vital capacity (p=0.006) and diffusing capacity of the lung for carbon monoxide (p=0.003) and had to be more frequently treated for sarcoidosis during the 6 months following inclusion in the study (69% versus 21%). The proportion of patients with extra-pulmonary involvement was similar, but Scadding’s radiographic staging was different with a higher proportion of stage 1 in IS.
Table 1.
Subject characteristics.
| Characteristic | Controls (n=34) | AS (n=26) | IS (n=24) | p value |
|---|---|---|---|---|
| Age, years | 44.3 ± 12.5 | 46.7 ± 12.7 | 52.3 ± 13.6 | 0.08 |
| Female (n (%)) | 17 (50%) | 12 (46.6%) | 14 (58.3%) | 0.7 |
| Current and ex-smoker (n (%)) | 8 (22.8%) | 7 (26.9%) | 6 (25%) | 0.9 |
| ACE (UI/L) (mean ± SD) | NA | 119.3 ± 58 | 62 ± 30.5 | <0.0001 |
| Extra thoracic involvement (n (%)) | NA | 13 (50%) | 12 (50%) | 1 |
| Scadding CXR stage (n (%)) | ||||
| 0 | NA | 0 (0%) | 4 (16.6%) | <0.0001 |
| 1 | 1 (3.8%) | 12(50%) | ||
| 2 | 18 (69.2%) | 2 (8.3%) | ||
| 3 | 1 (3.8%) | 1 (4.1%) | ||
| 4 | 6 (23.1%) | 5 (20.8%) | ||
| Pulmonary function | ||||
| FVC % [predicted; mean ± SD] | NA | 77.6 ± 21.6 | 94.2 ± 19 | 0.006 |
| DLCO % [predicted; mean ± SD] | 58 ± 20.6 | 74.5 ± 14.2 | 0.003 | |
| Treatment initiation* (n (%)) | NA | 18 (69.2%) | 5 (20.8%) | 0.0009 |
| aCTAS [32] (n (%)) | NA | |||
| 0 | 0 (0%) | 11 (45.8%) | ||
| 1 | 0 (0%) | 13 (54.2%) | ||
| 2 | 18 (69.2%) | 0 (0%) | ||
| 3 | 8 (30.7%) | 0 (0%) | ||
| 4 | 0 (0%) | 0 (0%) |
Data are expressed in mean ± SD or N (%); groups were compared for continuous variables with one-way ANOVA test (3 groups) or t-student test (2 groups) and for categorical variables with Chi2 test; *Immunosuppressive or corticosteroid treatment initiation ≤ 6 months after inclusion. AS, high active sarcoidosis, IS, low active or inactive sarcoidosis ACE, angiotensin converting enzyme; CXR, chest x-ray; FVC, forced vital capacity; DLCO, diffusing capacity of the lung for carbon monoxide; aCTAS, abbreviated CT activity Score (32); NA, not applicable.
Bold characters highlight significant values.
HIF-1α Is Activated by Hypoxia in MD-Macrophages From High Active Sarcoidosis
The transcription factor HIF-1α was immunodetected in MD-macrophages both in normoxic and hypoxic conditions, similarly between sarcoidosis and control groups (Figure 1A) HIF-1α was immunolocalized in the cytoplasm and nucleus of MD-macrophages (Figure 1B). Pimonidazole staining was only present in response to hypoxia and absent in normoxia (Figure 1A), suggesting that HIF-1α stabilization in MD-macrophages in normoxia is probably induced by other stimuli than hypoxia. This also demonstrates that MD-macrophages exposed to 1.5% oxygen have a partial pressure in oxygen ≤ 10 mmHg. HIF-1α activation status in MD-macrophages was assayed using a specific oligonucleotide-binding test. In normoxia, HIF-1α was similarly activated at a very low level in controls and sarcoidosis groups (Figure 1C). In contrast, in response to hypoxia, the level of HIF-1α activation was markedly increased in AS compared to IS and controls (Figure 1C).
Figure 1.

HIF-1α is activated by hypoxia in MD-macrophages from high active sarcoidosis. (A, B) Representative immunofluorescence staining of MD-macrophages. (A) Similar presence of HIF-1α (in red) in controls and in high active pulmonary sarcoidosis (AS) and low active or inactive sarcoidosis (IS) patients in both normoxia and hypoxia. Pimonidazole staining (in green) of MD-macrophages from AS is absent in normoxia (magnification x200). (B) Nuclear staining with DAPI (in blue) and accumulation of HIF-1α (in red) in MD-macrophages. Overlay image showing HIF-1α nuclear localization (magnification x400) (same results were found in 3 patients and 3 controls). (C) HIF-1α activation (measured as OD 450nm) assessed by TransAM® HIF-1α in controls, AS and IS patients, after 24hrs of normoxia or hypoxia. Results are expressed as box plot showing 25th and 75th percentile and median, each point indicates a patient and/or control (n= 7/group). *p < 0.05; ****p < 0.0001 in two-way ANOVA-repeated measures with Sidak post-hoc test.
Hypoxia Impaired Phagocytosis in Controls but Not in Sarcoidosis MD-Macrophages
It has been previously reported that phagocytosis, a main function of macrophages, can be modulated by HIF-1 (39) and involved in sarcoidosis pathogenicity (17, 40, 41). Therefore, phagocytosis was assessed in MD-macrophages exposed to normoxia or 24hrs-hypoxia and challenged with fluospheres (Figures 2A–C). In response to hypoxia, fluospheres phagocytosis was maintained in MD-macrophages from AS and IS, whereas it was decreased in controls (Figure 2D). Likewise, expression of the CD36 scavenger surface marker was maintained under hypoxic condition in MD-macrophages from sarcoidosis in contrast to controls (Figure 2E).
Figure 2.

Hypoxia impaired phagocytosis in controls but not in sarcoidosis MD-macrophages. (A–C) Representative image of MD-macrophages exposed to normoxia from high active sarcoidosis after 30 min incubation with fluospheres (1µm) followed by four PBS washes (magnification x 200); (A) fluorescent 1µm fluospheres (green) (B), phase-contrast images of MD-macrophages (C), overlay images showing fluospheres within MD-macrophages. (D) Effect of hypoxia on phagocytosis in controls and sarcoidosis patients estimated by the ratio of cells with at least one particle and the total number of cells. Each point indicates a patient and/or control (n=6/group). In sarcoidosis group, black dots represent high active sarcoidosis (AS), and red dots low active or inactive sarcoidosis (IS); (E) Effect of hypoxia on the CD36 scavenger receptor expression in controls, AS, IS; analyzed by flow cytometry of CD14+ MD-macrophages. The results are expressed as the mean CD36 fluorescence intensity (MFI). Each point indicates a patient and/or control (n= 6-9/group). Results are expressed as box plot showing 25th and 75th percentile and median *p < 0.05; ***p < 0.001 in two-way ANOVA-repeated measures with Sidak post-hoc test.
Hypoxia Decreased CD80 and CD86 Co-Stimulation Molecules and Human Leucocyte Antigen-DR (HLA-DR)
Hypoxia effect on the expression of CD80 and CD86, co-stimulatory molecules interacting to activate T lymphocytes, and of the HLA-DR presenting antigen molecule were determined using flow cytometry. In normoxic condition, CD80 and CD86 were similarly expressed on MD-macrophages from all groups (Figures 3A, B). Hypoxia significantly decreased the expression of CD80 and CD86 on MD-macrophages from controls and AS (Figures 3A, B). A higher HLA-DR expression was observed in normoxic condition on MD-macrophages in IS compared with controls. Hypoxia significantly decreased HLA-DR expression on MD-macrophages only from sarcoidosis sub-groups (Figure 3C). By contrast, CD163 surface expression, did not vary on MD-macrophages from sarcoidosis and controls exposed to hypoxia (see Figure E3).
Figure 3.

Hypoxia decreased CD80 and CD86 co-stimulation molecules and HLA-DR. (A–C) Effect of hypoxia on CD80, CD86 and HLA-DR surface expression on CD14+ MD-macrophages from controls, high active sarcoidosis (AS), and low active or inactive sarcoidosis (IS). Results are expressed as the mean fluorescence intensity (MFI) of CD80 (A), CD86 (B) and HLA-DR (C) analyzed by flow cytometry. Results are expressed as box plot showing 25th and 75th percentile and median, each point indicates a patient and/or control (n = 6-11/group). *p < 0.05; **p < 0.01; ****p < 0.0001 in two-way ANOVA-repeated measures with Sidak post-hoc test.
Hypoxia Induced a Pro-Inflammatory Response Without Activation of NF-κB in High Active Sarcoidosis
Pro-inflammatory cytokine secretion in CM was compared in normoxia and hypoxia. Baseline concentrations of chemokine (C-X-C motif) ligand 8 (CXCL8), tumor necrosis factor-α (TNFα), interleukin-1ß (IL-1ß) (Figures 4A–C), IL-18 and IL-5 (see Figure E4) under normoxia were similar between groups. Hypoxia significantly increased pro-inflammatory cytokine secretion only in MD-macrophages from AS (Figures 4A–C). In normoxia, IL-10 secretion was increased in AS compared to IS, while hypoxia significantly reduced its secretion (Figure 4D). Increase in pro-inflammatory cytokine secretion induced by hypoxia in MD-macrophages from AS was associated with an increase in their mRNA levels (see Figure E4). The pro-inflammatory response to hypoxia was not associated with NF-κB (p65 and p50 subunits) activation which was decreased in AS (Figures 4E, F).
Figure 4.

Hypoxia induced a pro-inflammatory response without activation of NF-κB in high active sarcoidosis. (A–D) Concentrations of CXCL8 (A), TNFα (B), IL-1ß (C) and IL-10 (D) assessed by Luminex® in conditioned media of normoxic and hypoxic MD-macrophages from controls, high active sarcoidosis (AS), and low active or inactive sarcoidosis (IS); results are expressed in pg/ml (n= 5-8 controls or patients/group). (E, F) NF-κB-p65 and p50 activation (measured as OD450nm) assessed by TransAM ®. Each point indicates a patient and/or control (n= 7-8/group). Results are expressed as box plot showing 25th and 75th percentile and median. *p < 0.05; **p < 0.01; ***p < 0.001 in two-way ANOVA-repeated measures with Sidak post-hoc test.
Hypoxia Promoted a Profibrotic Response in High Active Sarcoidosis
Secretion of profibrotic factors in CM of MD-macrophages, TGF-ß1, platelet-derived growth factor-BB (PDGF-BB), PAI-1 (Figures 5A–D) and vascular endothelial growth factor-A (VEGF-A) (see Figure E5) was studied. In normoxia, concentration of total TGF-ß1 was higher in CM from AS, especially the latent form of TGF-ß1 (Figure 5A). Hypoxia induced its decrease in AS, albeit remaining higher than in controls (Figure 5B). Likewise, hypoxia increased the secretion of PDGF-BB, PAI-1 (Figures 5C, D) and VEGF-A (see Figure E5) in all groups. These results were supported by an increase in mRNA transcript levels of profibrotic cytokines in MD-macrophages, especially TGFB1 and VEGF in AS (see Figure E5).
Figure 5.

Hypoxia promoted a profibrotic response in high active sarcoidosis. (A) Active (free) and total (free+latent) TGß1 (in pg/ml) measured by ELISA in conditioned media from MD-macrophages after 24hrs of normoxia. (n= 7-14 independent experiments). (B–D) Concentrations of total TGFß1 (B), PDGF-BB (C) and PAI-1 (D) assessed by Luminex® in conditioned media of normoxic and hypoxic MD-macrophages from controls, high active sarcoidosis (AS), and low active or inactive sarcoidosis (IS). Each point indicates a patient and/or control (n= 5-8/group). Results are expressed in pg/ml with box plot showing 25th and 75th percentile and median. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 in two-way ANOVA-repeated measures and two-way ANOVA with Sidak post-hoc test.
Secretion of PAI-1 by Hypoxic MD-Macrophages From High Active Sarcoidosis Inhibited Lung Fibroblast Migration
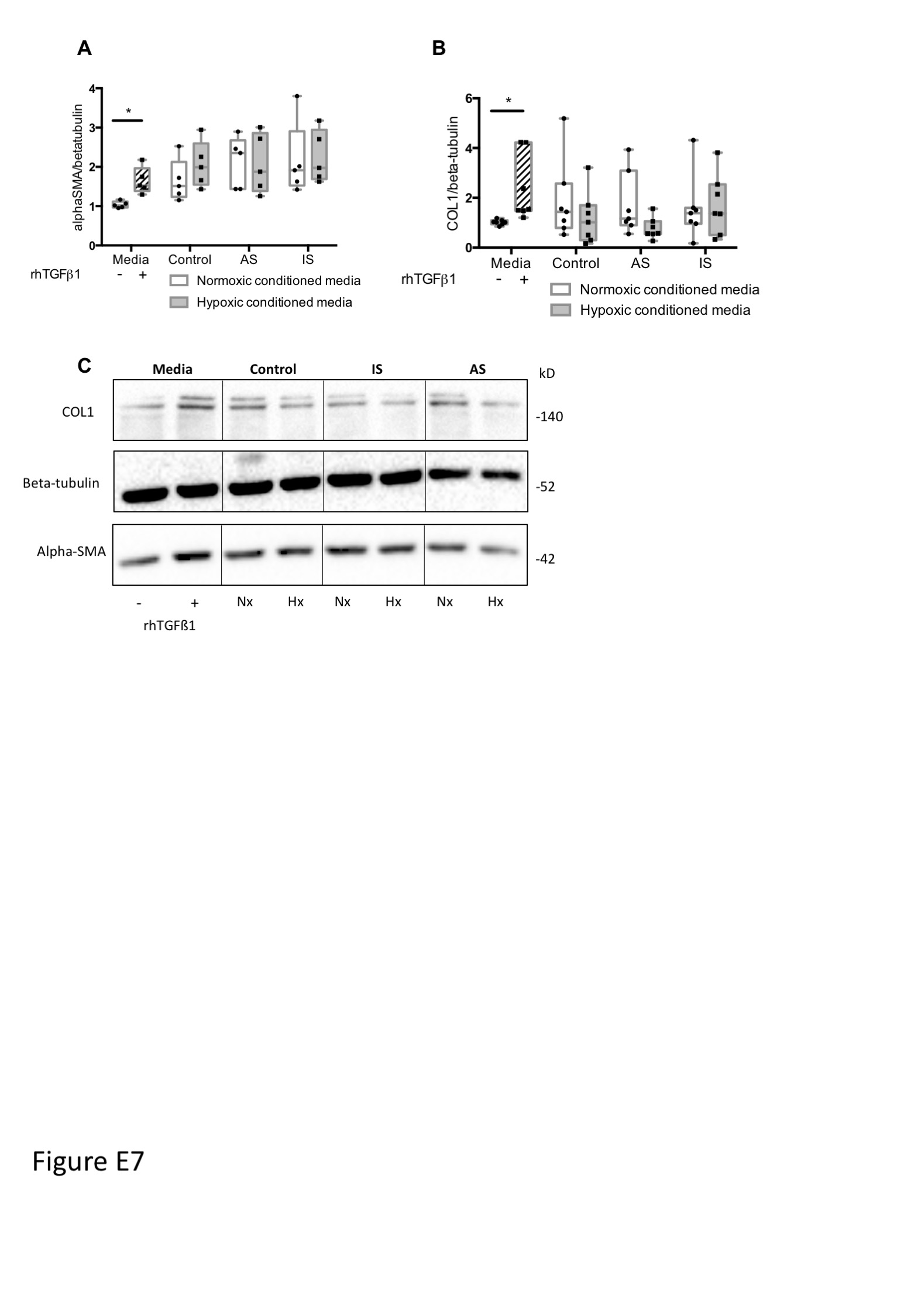
Among the profibrotic factors, PAI-1 secretion in hypoxia was two-fold higher in AS compared to IS and controls (Figure 5D). Accordingly, we hypothesized that PAI-1 may act on fibroblasts close to macrophages. The effect of CM from MD-macrophages exposed to either normoxia or hypoxia on gap closure in NHLF monolayers was studied. Exposure of NHLF to CM from hypoxia-exposed MD-macrophages from all groups significantly inhibited gap closure as compared with CM from normoxia-exposed MD-macrophages (Figures 6A, B). Hypoxic CM did not affect NHLF proliferation (see Figure E6) or fibroblast-myofibroblast differentiation as assessed by alpha-smooth actin and collagen type I quantification in Western blot (see Figure E7), indicating that inhibition of gap closure was related to a decrease in the ability of NHLF to migrate. The “immobilization” of NHLF was markedly observed with CM from hypoxic MD-macrophages from AS (Figure 6B).
Figure 6.

Secretion of PAI-1 by hypoxic MD-macrophages from high active sarcoidosis inhibited lung fibroblast migration. (A) Representative contrast-phase microscopy images of NHLF during gap closure assays at basal time and after 24hrs of incubation with conditioned media from controls or high active sarcoidosis (AS) or low active or inactive sarcoidosis (IS) patients MD-macrophages exposed to normoxia or hypoxia. (B) Quantitative analysis of NHLF gap closure assay comparing media alone or with 2500pg/ml recombinant human PAI-1 (rh-PAI-1) or normoxic or hypoxic conditioned media from MD-macrophages in controls, AS, and IS. Each point indicates a patient and/or control (n=5-7/group) (C) Correlation (Pearson Test) between PAI-1 level (pg/ml) in normoxic or hypoxic MD-macrophages conditioned media from sarcoidosis and controls and percentage of NHLF gap closure. Each point indicates a patient and/or control (n=3-6/group). (D) Dose effect of rh-PAI-1 on NHLF gap closure reversed by PAI-1 Ab (n=3 independent experiments); (E) Effect of PAI-1 antibody (PAI-1 Ab) added to the conditioned media on NHLF gap closure assay. Each point indicates a patient and/or control (n=3/group). Results are expressed as box plot showing 25th and 75th percentile and median (B, D) or mean with SD (E). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 in Anova two-way (D, E) and Anova two-way with repeated measures (B) with Sidak post-hoc test. #p < 0.05 between normoxic conditioned media from AS and IS.
Among the profibrotic factors secreted by MD-macrophages, PAI-1 levels negatively correlated with the percentage of NHLF gap closure (Figure 6C). Incubation of NHLF with increasing concentrations of rhPAI-1 reduced NHLF migration (Figure 6D). The inhibitory effect of rhPAI-1 on NHLF migration was prevented by addition of anti-PAI-1 antibody (Figure 6D). Inhibition of PAI-1 in the CM from hypoxic MD-macrophages with an anti-PAI-1 antibody restored NHLF migration, especially in AS (Figure 6E). These data indicate that PAI-1 secretion by hypoxic MD-macrophages from AS mostly contribute to inhibit human lung fibroblast migration.
Detection of HIF-1α and PAI-1 in Granulomas From Archived Pulmonary Biopsies
We immunodetected HIF-1α (Figures 7A, B) and PAI-1 (Figures 7D, E) in granulomas from the three archived pulmonary biopsies. Macrophages-derived mature epithelioid cells were identified on their characteristic large cytoplasm, eccentric reniform nuclei (42) and CD68+ labelling (Figures 7B, C). Nuclear localization of HIF-1α and PAI-1 was detected in the mature epithelioid cells constituting granulomas (Figures 7B, C, E). An isotype control is shown in Figure 7F.
Figure 7.

Detection of HIF-1α and PAI-1 in granulomas from pulmonary biopsies. (A, B) HIF-1α, (C) CD68 and (D, E) PAI-1 expression assayed by immunohistochemistry in archived lung biopsy from sarcoidosis patient. (A–F) are representative of the results obtained in lung biopsies from three sarcoidosis. (A, D) Granulomas are identified by a blue asterisks. (B, C) Serial sections of a granuloma wrapped with lamellar fibrosis (green triangle) showing HIF-1α expression in epithelioid cells characterized by their large cytoplasm, eccentric reniform nuclei (red arrow) (B) and CD68+ labelling (C). (A, D magnification x100 B, C, E, F magnification x200). Isotype control is shown in (F).
Discussion
Factors modulating the onset and progression of sarcoidosis are still poorly known. Here we showed that hypoxia, a microenvironmental factor most likely present within granulomas and inflammatory tissues, activated HIF-1 and induced a mixed proinflammatory-profibrotic MD-macrophage phenotype particularly marked in AS, as illustrated in Figure 8. These in vitro results were supported by immunohistochemistry data showing the expression of HIF-1α and its target PAI-1 in epithelioid cells constituting pulmonary sarcoidosis granulomas.
Figure 8.

Summary of hypoxia biological impact on MD-macrophages from controls versus high active sarcoidosis. Hypoxia induced in MD-macrophages a significantly more pronounced effect in high active sarcoidosis compared with controls. It increased the HIF-1α trans-activity, promoted a proinflammatory response without activating NF-κB pathway and a profibrotic response (TGFß1, PDGF-BB) with PAI-1 secretion associated with human lung fibroblast migration inhibition. Hypoxia also decreased the expression of CD80/CD86 and HLA-DR on MD-macrophages in the two groups while it did not impair phagocytosis and the surface expression of CD36 on cells from high active sarcoidosis at variance with controls. MØ, MD-macrophages.
One strength of our study was to prospectively include patients with either high active or low active or inactive sarcoidosis, not taking any therapy that could modify macrophage biology. The aCTAS score we used to evaluate the disease activity has already been validated by two independent studies (32, 43). This score was highly correlated with the range of FVC improvement under further sarcoidosis treatment, a good marker of the lesions reversibility. The choice to specifically study MD-macrophages in these patients was supported by recent works showing that in vitro models of granuloma using PBMCs can mimic sarcoidosis events such as phagosome-regulated mTOR or IL-13 signaling (17, 44). In addition, previous studies reported that immune cells constituting recurrent sarcoidosis granulomas in lung transplant allografts originated from the recipient monocyte-macrophage lineage (19, 20). Finally, the inflammatory phenotype of MD-macrophages in pulmonary sarcoidosis and their propensity to produce TNFα has been recently highlighted (11), supporting the role of these cells in sarcoidosis pathogenesis. However, the fact that experiments were almost exclusively performed in MD-macrophages constitutes a limitation of the present study. It would certainly be interesting to study the effects of hypoxia on other cell types involved in sarcoidosis such as T lymphocytes. A comparison of our data in MD-macrophages with data obtained in lung resident alveolar macrophages could also be informative but bronchoalveolar lavages are rarely done in the follow-up of sarcoidosis patients. Another limitation of our study is the fact that it was not possible to conduct all experiments for each patient for technical reasons, i.e. relatively to the low number of retrieved monocytes per blood sample.
To the best of our knowledge, the consequences of a hypoxic microenvironment on macrophages were never investigated in sarcoidosis, although granuloma hypoxia was inferred from morphometric analysis (24). By contrast, in tuberculosis pulmonary granulomas, hypoxia was directly demonstrated to play a major role in the disease course (25, 45). Recently, 18F-fluoromisonidazole uptake, a hypoxia-sensitive PET tracer, was observed in sarcoidosis lesions and associated with 18FFDG-PET uptake (35), suggesting that hypoxic lesions are metabolically active and associated with an active form of sarcoidosis. In addition, patients with high active sarcoidosis had more impaired lung function with lower DLCO, potentially resulting in local alveolar hypoxia.
Previous studies investigating HIF in sarcoidosis led to conflicting results (28, 29). Tzouvelekis et al. (29) reported increased expression of VEGF, a target of HIF, within sarcoidosis granulomas but failed to detect HIF-1α whereas Talreja et al. did (28). Such a difference in HIF-1α expression detection could be explained by different stages in the disease between the studies, the different antibodies used for HIF-1α detection and immunosuppressive treatments given to patients in the Tzouvelekis et al. study (28, 29). However, in line with Talreja et al. (28), we clearly immunodetected HIF-1α in pulmonary sarcoidosis granulomas. Such a finding prompted us to evaluate in vitro the effects of hypoxia on MD-macrophages from controls and untreated sarcoidosis patients.
In the present study HIF-1α nuclear expression was detected under normoxia in MD-macrophages from patients and controls. Talreja et al. also reported HIF-1α expression in freshly-isolated blood monocytes (28). The nuclear expression of HIF-1α in normoxic MD-macrophages is consistent with the fact that non-hypoxic stimuli such as TNF-α and IL-1β proinflammatory cytokines, NF-κB, reactive oxygen species, MAPK (46, 47) or mTOR pathway activation (48) are able to stabilize HIF-1α in immune cells. However, HIF-1α transcriptional activity was significantly more strongly induced by hypoxia in MD-macrophages from AS. Consistently, several HIF-target genes were induced by hypoxia in MD-macrophages from AS, such as VEGF and TGFB1 transcripts or PAI-1 protein. The TNFα and IL-1ß cytokines [involved in sarcoidosis pathogenesis (4, 49)] and CXCL8, induced by hypoxia only in AS, can also be directly upregulated by HIF in macrophages (50, 51). NF-κB activation, known to be also induced by hypoxia and leading to pro-inflammatory cytokine production (52) was decreased in AS, suggesting that it was not involved in this context. Moreover, HIF-1 can exert a negative feedback on NF-κB (53). The stronger response to hypoxia observed in MD-macrophages from AS is somewhat intriguing. It might be explained by still unexplored genetic polymorphisms, by epigenetic mechanisms and/or non-coding RNAs known to modulate HIF expression (54).
We investigated whether hypoxia could modulate phagocytosis and antigen processing/presentation, a key initial step of inflammatory lesions in sarcoidosis. We observed that phagocytosis was similar at baseline in control and sarcoidosis MD-macrophages. Interestingly, hypoxia markedly reduced phagocytosis in controls but not in sarcoidosis, suggesting that macrophage phagocytosis could be maintained under hypoxia in this disease. Previous papers reported that phagocytic activity of macrophages was increased in sarcoidosis and in an in vitro model of granuloma (17, 40, 41). An enhanced expression of phagocytosis-related genes, but a downregulation of genes involved in proteasome degradation were also observed, suggesting that the accumulation of intracellular phagocytic degradation products could participate in the chronicity of inflammation in sarcoidosis (40). We next evaluated the effect of hypoxia on the expression of CD80 and CD86 co-stimulatory molecules and on HLA-DR as these markers are associated with antigen presentation and expressed by epithelioid cells in sarcoidosis granuloma (55, 56). Hypoxia markedly decreased the expression of CD80 and CD86 on MD-macrophages in controls and AS while not significantly in IS. Hypoxia also downregulated the expression of HLA-DR in AS and IS MD-macrophages. These findings are in line with previous studies showing that hypoxia decreased the expression of CD80 in murine macrophages and CD80, CD86 and MHC class II in human dendritic cells, reducing their ability to initiate adaptive-immunity responses (57, 58). However, the role of antigenic presentation in sarcoidosis is still debated. Crouser et al. reported an enhanced presentation capacity in an in vitro model of sarcoidosis (17). Conversely, Grunewald et al. proposed that granuloma progression may occur when antigen recognition is not efficient enough, due to inadequate peptide presentation by modified HLA molecules or T cells incapacity to generate T cell clones, allowing granuloma persistence because of inefficient adaptive immune response (4). Here, we can speculate that hypoxia at the various steps towards granuloma formation may potentially exert a deleterious effect on antigen presentation processes, and therefore contribute to their persistence.
Mechanisms leading to sarcoidosis-associated pulmonary fibrosis are poorly understood. Granulomas are almost always surrounded by a concentric rim of collagen bundles (3). Macrophages are known to be involved in repair and fibrotic processes through different states of polarization (59). In sarcoidosis, macrophages may acquire a profibrotic M2-phenotype as shown in granulomas from patients’ biopsies or in vitro model (44, 60). In our study, hypoxia decreased the expression of M1 markers (CD80, CD86, HLA-DR), while M2 markers expression was maintained (CD163, CD36). However, hypoxia was also associated with a M1 pro-inflammatory cytokine response. In the literature, the effect of hypoxia on macrophage polarization remains unclear (61, 62). More than the over-simplified M1/M2 dichotomy, macrophages display remarkable plasticity and can change their phenotype in response to environmental factors.
Previous studies showed that under hypoxic conditions, macrophages can induce fibrosis in a HIF-dependent manner through the secretion of VEGF, PDGF-BB, or PAI-1 (63, 64). Here, we observed that MD-macrophages from AS secreted higher levels of PAI-1 in response to hypoxia as compared with controls or IS. The PAI-1 factor plays a key role in the development of pulmonary fibrosis by fibrotic matrix deposition (65). An increase in PAI-1 levels as well as a decrease in fibrinolytic activity has been reported in the bronchoalveolar lavage of patients with sarcoidosis (66). We clearly detected PAI-1 in granuloma epithelioid cells. Finally, we demonstrated that CM from hypoxic MD-macrophages inhibited pulmonary fibroblast migration, a phenomenon highly dependent on PAI-1. Consistently, PAI-1 binding to vitronectin has been previously shown to inhibit cell migration, leading to inefficient alveolar repair following injury and favoring the development of pulmonary fibrosis (67). Therefore, we hypothesize that PAI-1 secretion by granuloma cells could immobilize and sequester lung fibroblasts around granuloma, thus favoring the characteristic peripheral granuloma fibrosis.
In conclusion, this study shows that hypoxia exerts a significant and specific impact on MD-macrophages from sarcoidosis patients, with the strongest effect observed in patients with a high active disease. It may favor the development and persistence of granulomas in active sarcoidosis and fibrosis surrounding granulomas by promoting a mixed inflammatory/fibrosing response of macrophages, by reducing their antigen presentation capacities, leading to a deficient T cell response. Thus, the HIF pathway and PAI-1 could be involved in the pathogenesis of high active sarcoidosis, potentially representing new therapeutic targets. As a future direction, we plan to investigate interactions between HIF and other signaling pathways identified in the pathogenesis of sarcoidosis as mTOR, NLRP3 inflammasome, JAK/STAT, or heat shock proteins (49, 68, 69), already known to interfere with the HIF/hypoxia pathway (22, 70–72).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité de protection des personnes (CPP) Ile-de-France X 2016-10-02. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
FJ and VB designed research studies, contributed to the experiments, analyzed the data, and wrote the manuscript. HN and MK provided human samples and reviewed the manuscript. MP contributed to the experiments and reviewed the manuscript. CP, J-FB, DV, and HN wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by: Fonds de Recherche en Santé Respiratoire et Fondation du Souffle (2017), Financement de la Chancellerie des Universités de Paris-Legs Poix (2017), Bonus Qualité Recherche Université Sorbonne Paris- Nord (2016-2017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank Drs Diane Bouvry, Simon Chauveau, Morgane Didier, Olivia Freynet, Aurélie Hervé, Fatma Kort, Cécile Rotenberg, Lucile Sesé, MDs and Pr Yurdagül Uzunhan MD,PhD of the Pulmonology Department from the Avicenne University Hospital APHP (Bobigny, France), who contributed in the blood samples collection from sarcoidosis patients. We also acknowledge the technical help from the Histology Core (TisCel13) from the Sorbonne Paris-Nord University (Bobigny, France). We thank Elisabetta Dondi of Inserm U978 UFR SMBH, for technical assistance in flow cytometry analysis. We thank Nicolas Heddebaut of UMR1152 Faculté de Médecine, site Bichat, for the technical assistance for Luminex® analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.719009/full#supplementary-material
{kind=link}
References
- 1.Baughman RP, Teirstein AS, Judson MA, Rossman MD, Yeager H, Bresnitz EA, et al. Case Control Etiologic Study of Sarcoidosis (ACCESS) Research Group. Clinical Characteristics of Patients in a Case Control Study of Sarcoidosis. Am J Respir Crit Care Med (2001) 164:1885–9. 10.1164/ajrccm.164.10.2104046 [DOI] [PubMed] [Google Scholar]
- 2.Mañá J, Rubio-Rivas M, Villalba N, Marcoval J, Iriarte A, Molina-Molina M, et al. Multidisciplinary Approach and Long-Term Follow-Up in a Series of 640 Consecutive Patients With Sarcoidosis: Cohort Study of a 40-Year Clinical Experience at a Tertiary Referral Center in Barcelona, Spain. Med (Baltimore) (2017) 96:e7595. 10.1097/MD.0000000000007595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernaudin J-F, Moguelet P, Jeny F, Besnard V, Valeyre D, Kambouchner M. Chapter 6 - Pathology of Granuloma. In: Sarcoidosis [Internet] (2019). Philadelphia: Elsevier. Available at: http://www.sciencedirect.com/science/article/pii/B9780323544290000069 (Accessed 2020 Jul 9). [Google Scholar]
- 4.Grunewald J, Grutters JC, Arkema EV, Saketkoo LA, Moller DR, Müller-Quernheim J. Sarcoidosis. Nat Rev Dis Primer (2019) 5:45. 10.1038/s41572-019-0107-y [DOI] [PubMed] [Google Scholar]
- 5.Deutsch-Link S, Fortuna D, Weinberg EM. A Comprehensive Review of Hepatic Sarcoid. Semin Liver Dis (2018) 38:284–97. 10.1055/s-0038-1666853 [DOI] [PubMed] [Google Scholar]
- 6.Mahévas M, Lescure FX, Boffa J-J, Delastour V, Belenfant X, Chapelon C, et al. Renal Sarcoidosis: Clinical, Laboratory, and Histologic Presentation and Outcome in 47 Patients. Med (Baltimore) (2009) 88:98–106. 10.1097/MD.0b013e31819de50f [DOI] [PubMed] [Google Scholar]
- 7.Gajawada P, Cetinkaya A, von Gerlach S, Kubin N, Burger H, Näbauer M, et al. Myocardial Accumulations of Reg3A, Reg3γ and Oncostatin M are Associated With the Formation of Granulomata in Patients With Cardiac Sarcoidosis. Int J Mol Sci (2021) 22(8):22. 10.3390/ijms22084148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeny F, Uzunhan Y, Lacroix M, Gille T, Brillet P-Y, Nardi A, et al. Predictors of Mortality in Fibrosing Pulmonary Sarcoidosis. Respir Med (2020) 169:105997. 10.1016/j.rmed.2020.105997 [DOI] [PubMed] [Google Scholar]
- 9.Locke LW, Schlesinger LS, Crouser ED. Current Sarcoidosis Models and the Importance of Focusing on the Granuloma. Front Immunol (2020) 11:1719. 10.3389/fimmu.2020.01719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lepzien R, Rankin G, Pourazar J, Muala A, Eklund A, Grunewald J, et al. Mapping Mononuclear Phagocytes in Blood, Lungs, and Lymph Nodes of Sarcoidosis Patients. J Leukoc. Biol (2019) 105:797–807. 10.1002/JLB.5A0718-280RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lepzien R, Liu S, Czarnewski P, Nie M, Österberg B, Baharom F, et al. Monocytes in Sarcoidosis Are Potent TNF Producers and Predict Disease Outcome. Eur Respir J (2021) 58(1):2003468. 10.1183/13993003.03468-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Broos CE, van Nimwegen M, Hoogsteden HC, Hendriks RW, Kool M, van den Blink B. Granuloma Formation in Pulmonary Sarcoidosis. Front Immunol (2013) 4:437. 10.3389/fimmu.2013.00437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams DO. The Granulomatous Inflammatory Response. A Review. Am J Pathol (1976) 84:164–92. [PMC free article] [PubMed] [Google Scholar]
- 14.Evren E, Ringqvist E, Willinger T. Origin and Ontogeny of Lung Macrophages: From Mice to Humans. Immunology (2020) 160:126–38. 10.1111/imm.13154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop Into Functional Tissue-Resident Macrophages. Immunity (2016) 44:755–68. 10.1016/j.immuni.2016.02.017 [DOI] [PubMed] [Google Scholar]
- 16.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-Derived Alveolar Macrophages Drive Lung Fibrosis and Persist in the Lung Over the Life Span. J Exp Med (2017) 214:2387–404. 10.1084/jem.20162152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crouser ED, Locke LW, Julian MW, Bicer S, Sadee W, White P, et al. Phagosome-Regulated Mtor Signalling During Sarcoidosis Granuloma Biogenesis. Eur Respir J (2020) 57(3):2002695. 10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A3102 [DOI] [PubMed] [Google Scholar]
- 18.Crouser ED, White P, Caceres EG, Julian MW, Papp AC, Locke LW, et al. Schlesinger LS. A Novel in Vitro Human Granuloma Model of Sarcoidosis and Latent Tuberculosis Infection. Am J Respir Cell Mol Biol (2017) 57:487–98. 10.1165/rcmb.2016-0321OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ionescu DN, Hunt JL, Lomago D, Yousem SA. Recurrent Sarcoidosis in Lung Transplant Allografts: Granulomas are of Recipient Origin. Diagn. Mol Pathol Am J Surg Pathol Part B (2005) 14:140–5. 10.1097/01.pas.0000176765.26047.6f [DOI] [PubMed] [Google Scholar]
- 20.Milman N, Andersen CB, Burton CM, Iversen M. Recurrent Sarcoid Granulomas in a Transplanted Lung Derive From Recipient Immune Cells. Eur Respir J (2005) 26:549–52. 10.1183/09031936.05.00127404 [DOI] [PubMed] [Google Scholar]
- 21.Watts ER, Walmsley SR. Inflammation and Hypoxia: HIF and PHD Isoform Selectivity. Trends Mol Med (2019) 25:33–46. 10.1016/j.molmed.2018.10.006 [DOI] [PubMed] [Google Scholar]
- 22.McGettrick AF, O’Neill LAJ. The Role of HIF in Immunity and Inflammation. Cell Metab (2020) 32:524–36. 10.1016/j.cmet.2020.08.002 [DOI] [PubMed] [Google Scholar]
- 23.Delbrel E, Soumare A, Naguez A, Label R, Bernard O, Bruhat A, et al. HIF-1α Triggers ER Stress and CHOP-Mediated Apoptosis in Alveolar Epithelial Cells, a Key Event in Pulmonary Fibrosis(2018) (Accessed 2020 Jul 9). [DOI] [PMC free article] [PubMed]
- 24.Kambouchner M, Pirici D, Uhl J-F, Mogoanta L, Valeyre D, Bernaudin J-F. Lymphatic and Blood Microvasculature Organisation in Pulmonary Sarcoid Granulomas. Eur Respir J (2011) 37:835–40. 10.1183/09031936.00086410 [DOI] [PubMed] [Google Scholar]
- 25.Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, et al. Tuberculous Granulomas are Hypoxic in Guinea Pigs, Rabbits, and Nonhuman Primates. Infect Immun (2008) 76:2333–40. 10.1128/IAI.01515-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sitkovsky M, Lukashev D. Regulation of Immune Cells by Local-Tissue Oxygen Tension: HIF1 Alpha and Adenosine Receptors. Nat Rev Immunol (2005) 5:712–21. 10.1038/nri1685 [DOI] [PubMed] [Google Scholar]
- 27.Semenza GL. Oxygen Sensing, Homeostasis, and Disease. N Engl J Med Massachusetts Med Society (2011) 365:537–47. 10.1056/NEJMra1011165 [DOI] [PubMed] [Google Scholar]
- 28.Talreja J, Talwar H, Bauerfeld C, Grossman LI, Zhang K, Tranchida P, et al. HIF-1α Regulates IL-1β and IL-17 in Sarcoidosis. eLife (2019) 8:e44519. 10.7554/eLife.44519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tzouvelekis A, Ntolios P, Karameris A, Koutsopoulos A, Boglou P, Koulelidis A, et al. Expression of Hypoxia-Inducible Factor (HIF)-1a-Vascular Endothelial Growth Factor (VEGF)-Inhibitory Growth Factor (ING)-4- Axis in Sarcoidosis Patients. BMC Res Notes (2012) 5:654. 10.1186/1756-0500-5-654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ats/Ers/Wasog. Statement on Sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (Wasog) Adopted by the Ats Board of Directors and by the Ers Executive Committee, February 1999. Am J Respir Crit Care Med (1999) 160:736–55. 10.1164/ajrccm.160.2.ats4-99 [DOI] [PubMed] [Google Scholar]
- 31.Crouser ED, Maier LA, Wilson KC, Bonham CA, Morgenthau AS, Patterson KC, et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med (2020) 201:e26–51. 10.1164/rccm.202002-0251ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benamore R, Kendrick YR, Repapi E, Helm E, Cole SL, Taylor S, et al. CTAS: A CT Score to Quantify Disease Activity in Pulmonary Sarcoidosis. Thorax (2016) 71:1161–3. 10.1136/thoraxjnl-2016-208833 [DOI] [PubMed] [Google Scholar]
- 33.Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the Maintenance of Homeostasis. Cell Mol Immunol (2021) 18:579–87. 10.1038/s41423-020-00541-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morishita E, Sekiya A, Hayashi T, Kadohira Y, Maekawa M, Yamazaki M, et al. Increased Macrophage Colony-Stimulating Factor Levels in Patients With Graves’ Disease. Int J Hematol (2008) 88:272–7. 10.1007/s12185-008-0155-7 [DOI] [PubMed] [Google Scholar]
- 35.Furuya S, Naya M, Manabe O, Hirata K, Ohira H, Aikawa T, et al. 18F-FMISO PET/CT Detects Hypoxic Lesions of Cardiac and Extra-Cardiac Involvement in Patients With Sarcoidosis. J Nucl Cardiol Off Publ. Am Soc Nucl Cardiol (2019). 10.1007/s12350-019-01976-6 [DOI] [PubMed] [Google Scholar]
- 36.Zhang C, Chery S, Lazerson A, Altman NH, Jackson R, Holt G, et al. Anti-Inflammatory Effects of α-MSH Through P-CREB Expression in Sarcoidosis Like Granuloma Model. Sci Rep (2020) 10:7277. 10.1038/s41598-020-64305-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Enomoto R, Imamori M, Seon A, Yoshida K, Furue A, Tsuruda H, et al. Proposal for a New Evaluation of Phagocytosis Using Different Sizes of Fluorescent Polystyrene Microspheres. Adv Biol Chem Sci Res Publishing (2013) 03:556. 10.4236/abc.2013.36064 [DOI] [Google Scholar]
- 38.Nenasheva T, Gerasimova T, Serdyuk Y, Grigor’eva E, Kosmiadi G, Nikolaev A, et al. Macrophages Derived From Human Induced Pluripotent Stem Cells are Low-Activated “Naïve-Like” Cells Capable of Restricting Mycobacteria Growth. Front Immunol (2020) 11:1016. 10.3389/fimmu.2020.01016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anand RJ, Gribar SC, Li J, Kohler JW, Branca MF, Dubowski T, et al. Hypoxia Causes an Increase in Phagocytosis by Macrophages in a HIF-1α-Dependent Manner. J Leukoc Biol (2007) 82:1257–65. 10.1189/jlb.0307195 [DOI] [PubMed] [Google Scholar]
- 40.Talreja J, Farshi P, Alazizi A, Luca F, Pique-Regi R, Samavati L. RNA-Sequencing Identifies Novel Pathways in Sarcoidosis Monocytes. Sci Rep Nat Publishing Group (2017) 7:2720. 10.1038/s41598-017-02941-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pettersen HB, Johnson E, Osen SS. Phagocytosis of Agarose Beads by Receptors for C3b (CR1) and Ic3b (CR3) on Alveolar Macrophages From Patients With Sarcoidosis. Scand J Immunol (1990) 32:669–77. 10.1111/j.1365-3083.1990.tb03209.x [DOI] [PubMed] [Google Scholar]
- 42.Adams DO. The Structure of Mononuclear Phagocytes Differentiating In Vivo. II. The Effect of Mycobacterium Tuberculosis. Am J Pathol (1975) 80:101–16. [PMC free article] [PubMed] [Google Scholar]
- 43.Duan J, Xu Y, Zhu H, Zhang H, Sun S, Sun H, et al. Relationship Between CT Activity Score With Lung Function and the Serum Angiotensin Converting Enzyme in Pulmonary Sarcoidosis on Chest HRCT. Med (Baltimore) (2018) 97:e12205. 10.1097/MD.0000000000012205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Locke LW, Crouser ED, White P, Julian MW, Caceres EG, Papp AC, et al. IL-13-Regulated Macrophage Polarization During Granuloma Formation in an In Vitro Human Sarcoidosis Model. Am J Respir Cell Mol Biol (2019) 60:84–95. 10.1165/rcmb.2018-0053OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prosser G, Brandenburg J, Reiling N, Barry CE, Wilkinson RJ, Wilkinson KA. The Bacillary and Macrophage Response to Hypoxia in Tuberculosis and the Consequences for T Cell Antigen Recognition. Microbes Infect (2017) 19:177–92. 10.1016/j.micinf.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin N, Simon MC. Hypoxia-Inducible Factors: Key Regulators of Myeloid Cells During Inflammation. J Clin Invest (2016) 126:3661–71. 10.1172/JCI84426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF Transcription Factors, Inflammation, and Immunity. Immunity (2014) 41:518–28. 10.1016/j.immuni.2014.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weichhart T, Hengstschläger M, Linke M. Regulation of Innate Immune Cell Function by mTOR. Nat Rev Immunol (2015) 15:599–614. 10.1038/nri3901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huppertz C, Jäger B, Wieczorek G, Engelhard P, Oliver SJ, Bauernfeind F-G, et al. The NLRP3 Inflammasome Pathway is Activated in Sarcoidosis and Involved in Granuloma Formation. Eur Respir J (2020) 55:1900119. 10.1183/13993003.00119-2019 [DOI] [PubMed] [Google Scholar]
- 50.Lewis A, Elks PM. Hypoxia Induces Macrophage Tnfa Expression via Cyclooxygenase and Prostaglandin E2 In Vivo . (2019) (Accessed 2020 Aug 21). [DOI] [PMC free article] [PubMed]
- 51.Fang H-Y, Hughes R, Murdoch C, Coffelt SB, Biswas SK, Harris AL, et al. Hypoxia-Inducible Factors 1 and 2 Are Important Transcriptional Effectors in Primary Macrophages Experiencing Hypoxia. Blood (2009) 114:844–59. 10.1182/blood-2008-12-195941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor CT, Doherty G, Fallon PG, Cummins EP. Hypoxia-Dependent Regulation of Inflammatory Pathways in Immune Cells. J Clin Invest. (2016) 126:3716–24. 10.1172/JCI84433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.D’Ignazio L, Bandarra D, Rocha S. NF-κB and HIF Crosstalk in Immune Responses. FEBS J (2016) 283:413–24. 10.1111/febs.13578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choudhry H, Harris AL. Advances in Hypoxia-Inducible Factor Biology. Cell Metab (2018) 27:281–98. 10.1016/j.cmet.2017.10.005 [DOI] [PubMed] [Google Scholar]
- 55.Soler P, Boussaud V, Moreau J, Bergeron A, Bonnette P, Hance AJ, et al. In Situ Expression of B7 and CD40 Costimulatory Molecules by Normal Human Lung Macrophages and Epithelioid Cells in Tuberculoid Granulomas. Clin Exp Immunol (1999) 116:332–9. 10.1046/j.1365-2249.1999.00887.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.v Maarsseveen T, Mullink R, Rensen R, Stam J. HLA-DR Antigens in Epithelioid Cell Granulomas of Sarcoidosis, Using Semithin Frozen Sections. A Concept About Granuloma Formation. Sarcoidosis (1985) 2:148–53. [PubMed] [Google Scholar]
- 57.Lahat N, Rahat MA, Ballan M, Weiss-Cerem L, Engelmayer M, Bitterman H. Hypoxia Reduces CD80 Expression on Monocytes But Enhances Their LPS-Stimulated TNF-Alpha Secretion. J Leukoc Biol (2003) 74:197–205. 10.1189/jlb.0303105 [DOI] [PubMed] [Google Scholar]
- 58.Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen I-H, et al. Divergent Effects of Hypoxia on Dendritic Cell Functions. Blood Am Soc Hematology (2008) 112:3723–34. 10.1182/blood-2008-02-142091 [DOI] [PubMed] [Google Scholar]
- 59.Braga TT, Agudelo JSH, Camara NOS. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front Immunol (2015) 6:602. 10.3389/fimmu.2015.00602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shamaei M, Mortaz E, Pourabdollah M, Garssen J, Tabarsi P, Velayati A, et al. Evidence for M2 Macrophages in Granulomas From Pulmonary Sarcoidosis: A New Aspect of Macrophage Heterogeneity. Hum Immunol (2017) 41(9):898. 10.1016/j.humimm.2017.10.009 [DOI] [PubMed] [Google Scholar]
- 61.Raggi F, Pelassa S, Pierobon D, Penco F, Gattorno M, Novelli F, et al. Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front Immunol (2017) 8:1097. 10.3389/fimmu.2017.01097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X, et al. Hypoxia Promotes Glioma-Associated Macrophage Infiltration via Periostin and Subsequent M2 Polarization by Upregulating TGF-Beta and M-CSFR. Oncotarget (2016) 7:80521–42. 10.18632/oncotarget.11825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruthenborg RJ, Ban J-J, Wazir A, Takeda N, Kim J-W. Regulation of Wound Healing and Fibrosis by Hypoxia and Hypoxia-Inducible Factor-1. Mol Cells (2014) 37:637–43. 10.14348/molcells.2014.0150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, et al. Hypoxia-Inducible Factor-1α Mediates TGF-β-Induced PAI-1 Production in Alveolar Macrophages in Pulmonary Fibrosis. Am J Physiol Lung Cell Mol Physiol (2011) 300:L740–752. 10.1152/ajplung.00146.2010 [DOI] [PubMed] [Google Scholar]
- 65.Flevaris P, Vaughan D. The Role of Plasminogen Activator Inhibitor Type-1 in Fibrosis. Semin Thromb Hemost (2017) 43:169–77. 10.1055/s-0036-1586228 [DOI] [PubMed] [Google Scholar]
- 66.Lardot Y Sibille B Wallaert C Hermans D Lison null C. Depressed Urokinase Activity in Bronchoalveolar Lavage Fluid From Patients With Sarcoidosis, Silicosis or Idiopathic Pulmonary Rbrosis: Relationship to Disease Severity. Biomark Biochem Indic Expo Response Susceptibility Chem (1999) 4:361–72. 10.1080/135475099230741 [DOI] [PubMed] [Google Scholar]
- 67.Lazar MH, Christensen PJ, Du M, Yu B, Subbotina NM, Hanson KE, et al. Plasminogen Activator Inhibitor-1 Impairs Alveolar Epithelial Repair by Binding to Vitronectin. Am J Respir Cell Mol Biol (2004) 31:672–8. 10.1165/rcmb.2004-0025OC [DOI] [PubMed] [Google Scholar]
- 68.Linke M, Pham HTT, Katholnig K, Schnöller T, Miller A, Demel F, et al. Chronic Signaling via the Metabolic Checkpoint Kinase Mtorc1 Induces Macrophage Granuloma Formation and Marks Sarcoidosis Progression. Nat Immunol (2017) 18:293–302. 10.1038/ni.3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rotenberg C, Besnard V, Brillet P-Y, Giraudier S, Nunes H, Valeyre D. Dramatic Response of Refractory Sarcoidosis Under Ruxolitinib in a Patient With Associated JAK2-Mutated Polycythemia. Eur Respir J (2018) 52(6):52. 10.1183/13993003.01482-2018 [DOI] [PubMed] [Google Scholar]
- 70.Baird NA, Turnbull DW, Johnson EA. Induction of the Heat Shock Pathway During Hypoxia Requires Regulation of Heat Shock Factor by Hypoxia-Inducible Factor-1. J Biol Chem (2006) 281:38675–81. 10.1074/jbc.M608013200 [DOI] [PubMed] [Google Scholar]
- 71.Eickelberg O, Seebach F, Riordan M, Thulin G, Mann A, Reidy KH, et al. Functional Activation of Heat Shock Factor and Hypoxia-Inducible Factor in the Kidney. J Am Soc Nephrol. JASN (2002) 13:2094–101. 10.1097/01.ASN.0000022008.30175.5B [DOI] [PubMed] [Google Scholar]
- 72.Tong Z, Jiang B, Zhang L, Liu Y, Gao M, Jiang Y, et al. HSF-1 is Involved in Attenuating the Release of Inflammatory Cytokines Induced by LPS Through Regulating Autophagy. Shock Augusta Ga (2014) 41:449–53. 10.1097/SHK.0000000000000118 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.