

Abstract
The identification of protein–ligand interaction plays a key role in biochemical research and drug discovery. Although deep learning has recently shown great promise in discovering new drugs, there remains a gap between deep learning-based and experimental approaches. Here, we propose a novel framework, named AIMEE, integrating AI model and enzymological experiments, to identify inhibitors against 3CL protease of SARS-CoV-2 (Severe acute respiratory syndrome coronavirus 2), which has taken a significant toll on people across the globe. From a bioactive chemical library, we have conducted two rounds of experiments and identified six novel inhibitors with a hit rate of 29.41%, and four of them showed an IC50 value <3 μM. Moreover, we explored the interpretability of the central model in AIMEE, mapping the deep learning extracted features to the domain knowledge of chemical properties. Based on this knowledge, a commercially available compound was selected and was proven to be an activity-based probe of 3CLpro. This work highlights the great potential of combining deep learning models and biochemical experiments for intelligent iteration and for expanding the boundaries of drug discovery. The code and data are available at https://github.com/SIAT-code/AIMEE.
Keywords: deep learning, drug discovery, SARS-CoV-2 3CL inhibitors, model interpretation
Introduction
Drug discovery is one of the most powerful weapons in the fight against diseases. Due to recent changes in human behavior, such as globalization and an increasing stress placed on the natural environment, the arms race between humanity and disease has intensified. The ongoing COVID-19 pandemic has so far sickened more than 100 million and killed over 2 million people across the globe as of January 2021 [1], and it has been just over a year since the SARS-CoV-2 was reported [2–4]. Therefore, it is imperative to be constantly updating our technology to address the challenges posed by existing and possible new emerging diseases. For SARS-CoV-2, the viral main protease (Mpro or 3CLpro) is an attractive drug target for COVID-19 drug discovery, given its essential role in viral infection and there being no human homolog [5–9]. However, so far, no specific drugs against SARS-CoV-2 have been approved. Considering high-throughput screening is not available in most biological safety level 3 laboratories (where SARS-CoV-2 cell-based assays can be performed), a feasible and cost-effective way is to combine in vitro experiments with in silico screening.
Deep learning has recently been applied successfully in many fields, including drug discovery [10–13]. The deep learning-based method may combine the advantages of structure-based and ligand-based drug design methods and may lead to superior performance [14–18]. More importantly, the rapidly increasing amount of data about SARS-CoV-2 enables deep learning to efficiently extract useful features and thus significantly improve the prediction accuracy. There have been several applications of deep learning models for screening inhibitors targeting important viral proteins, such as 3CLpro and the spike protein, since the outbreak of COVID-19 [19–23], although most of these models did not verify their predictions in vitro. Presently, it is hard to judge whether traditional methods or AI-aided methods are better suited to practical applications of drug discovery. Besides the quantity and quality requirements of the data, deep learning-based methods rely highly on appropriate design of experiments and might suffer from the generalizability issue. Thus, the gap between deep learning-based methods and experimental approaches remains. Ideally, one of the most promising ways is to combine deep learning models and biochemical experiments to build a closed and iterative loop, enabling the model to constantly learn and boosting its accuracy for the target task [24].
In the present study, a novel framework was proposed for integrating an AI model and enzymological experiments (AIMEE) to identify inhibitors against the SARS-CoV-2 3CL protease. Our framework involves three stages. First, our model was pretrained with the necessary information, including protein sequences, protein structures and protein–ligand interactions. The model has achieved the top performance on a benchmark dataset over all existing scoring functions. Second, the model was utilized on a carefully curated imbalanced 3CLpro inhibitor dataset and then applied the resulting model to screen a chemical library, including approved, clinical-stage drugs and bioactive compounds. Next, compounds with high predicted probability were selected for verifying their binding with 3CLpro in vitro. Third, the new experimental data was integrated and the model was updated according to the experimental results. Finally, we iterated steps two and three.
Through this approach, we identified several 3CLpro inhibitors, including six inhibitors with the IC50 value (half-maximal inhibitory concentration) <20 μM. Importantly, some inhibitors showed a potential clinical value when treating COVID-19. For example, bacitracin, which shows the inhibition of 3CLpro with an IC50 of 1.353 μM, was approved for intramuscular injection in the treatment of staphylococcal pneumonia and empyema in infants. Remarkably, the 100 μM inhibitor primary screening hit rate rose from 4.46% in the first round to 58.82% in the second round, and the strong binding (IC50 value <20 μM) hit rate rose from 0.13% to 29.41% (Figure 1), which suggests a significantly increased accuracy of the model during the process. Computationally, a highly imbalanced dataset with numerous inactive compounds against a limited number of active compounds could lead to a negative impact on the model performance. We demonstrated that our method reduces this negative effect by leveraging various techniques, which could be introduced into many drug discovery applications. Furthermore, we demonstrated the binding positions of the identified inhibitors, explored the logic behind the model and evaluated how the model discerns the key sites of the identified compounds. Based on this biological interpretation, we showed a commercially available compound to be activity-based probe (ABP) [25], which could be used for the activity-based protein profiling (ABPP) of the target protein to study its enzymological properties and functions during infection [26]. This work highlights a promising prospect of uniting deep learning models and biochemical experiments for intelligent iteration and for expanding the boundaries of drug discovery.
Figure 1 .
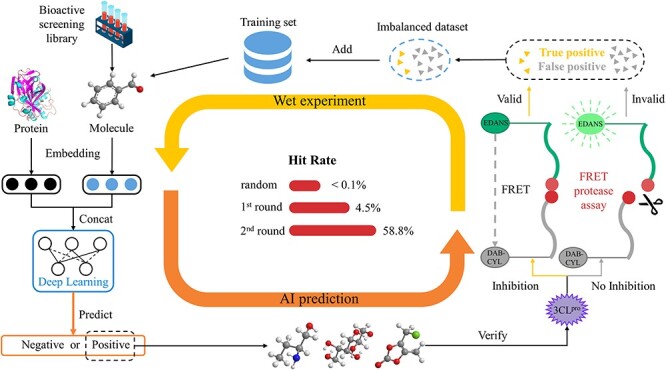
Schematic of the proposed framework and the hit rates in the closed-loop. The framework consists of two parts: model training and enzymological experiments. After several rounds of iteration, the screening hit rate increased.
Results and discussion
Model pretraining and performance
Among recently proposed deep learning-based methods, structure-based methods, utilizing the strong feature extraction capability of deep learning to capture information from protein–ligand complexes, showed a huge improvement compared to docking methods. However, these methods are limited by the lack of data and cannot extract the same information from sequence-only data, which are far more abundant than structural data. On the other hand, sequence-based methods may be disadvantaged by incomplete sample information. Here, we designed a graph-enhanced Transformer model to predict protein–ligand interactions, which combines the advantages of both structure-based and sequence-based methods. Figure 2A exhibits the architecture of proposed model and how our model selects 3CLpro inhibitors.
Figure 2 .
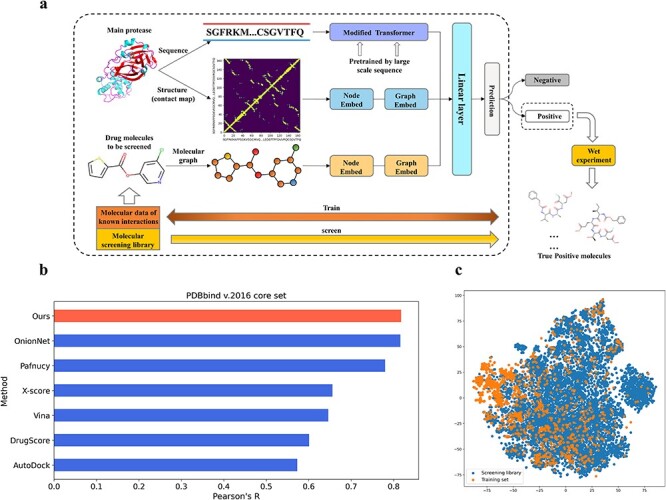
Model training and performance evaluation. (A) The architecture of the proposed model. (B) Our model has achieved the top performance on the benchmark PDBbind v2016 set. Pearson’s correlation coefficient (R), which is used to evaluate the linear correlation between the predicted and real values, is the most important measure for evaluating this benchmark dataset. (C) Dimensionality reduction by t-SNE of the molecule representations of screening library (blue) and training set (orange).
We explored multimodal inputs of proteins to capture information from different dimensions. At first, the modified Transformer [27] was pretrained using large-scale unlabeled protein sequences, which extracted protein evolutionary relationships using self-supervised learning. Then, for a protein with solved structural data, we utilized the pretrained Transformer and a graph attention network (GAT) to process its sequence and structure, respectively, to get the protein embedding vector. The corresponding drug molecule was processed by GAT to get the drug embedding vector. Next, these two representation vectors were concatenated and were fed into a linear regression layer to predict their binding.
We evaluated the model on the diverse 290 complexes within the PDBbind v.2016 core set and split the training and validation sets in the same manner as Pafnucy [16]. Pearson’s correlation coefficient R and root mean square error (RMSE), which refer to the linear correlation and the differences between the predicted and real values, respectively, were used as the evaluation metrics. Our model achieved RMSE = 1.274 and R = 0.818 on the PDBbind v.2016 core set. As shown in Figure 2B, our model has achieved the top performance on this structural benchmark dataset over existing methods, including docking and deep learning-based methods.
Deep learning-based screening
One of the greatest advantages of deep learning is its ability to constantly learn and improve its accuracy for a specific task. Here, to obtain novel 3CLpro inhibitors in an efficient and inexpensive way, we have built a closed-loop combining our deep learning model and wet lab experiments and have conducted two rounds of experiments.
In the first round of experiments, a curated dataset (dataset v1) containing 304 3CLpro inhibitors with measured IC50 values was used to train our model. Among these, 160 compounds with IC50 < 20 μM were regarded as positive samples, whereas another 144 compounds were regarded as negative samples. After training, our model selected 740 candidate compounds from a bioactive chemical library of 10 924 compounds consisting of approved, clinical-stage and bioactive compounds. Among these candidates, we verified 33 compounds which showed an inhibitory effect at 100 μM in primary screening, and then we calculated their IC50 values. Using IC50 of 20 μM as a hit cutoff, we identified one positive compound (IC50 = 1.353 μM) and 739 negative compounds for 3CLpro. Then, we incorporated these results and data carefully curated from other sources to build the 3CL inhibitor refined set (dataset v2). We have also expanded the negative set, which contains almost 300 000 negative compounds for SARS-CoV/SARS-CoV-2 3CLpro, for negative sample sampling, as described in the Material and Methods section. The lower the IC50 value, the better the inhibiting capability of the compound on the target enzyme. However, normally only a limited portion of molecules have a sufficient inhibiting capability against a specific target. From a biochemical point of view, most of the approved drugs function at therapeutic plasma concentrations <10 μM. The use of compounds with higher IC50 values might lead to a higher chance of side effects, such as off-target toxicity. In screening, if the IC50 cutoff is set to a much higher value than 10 μM, there would be many false positive hits. This, obviously, would have a large negative effect on our deep learning model. Moreover, an IC50 of 20 μM is widely used in in vitro screening as a cutoff to classify active and inactive compounds [8].
In the second round of experiments, the refined set consisting of 408 positive and 1859 negative compounds was used to retrain our model. At first, we tried to retrain our model directly on the merged set consisting of both the refined set and the negative set (i.e. 408 positive and over 300 000 negative compounds) to fully extract features from lots of negative compounds. However, the performance metrics were very low, indicating a negative impact on the model training. This high data imbalance seems to be a very common problem in bioassay datasets. A recent study showed this extreme imbalance issue results in very poor performance, especially for machine learning algorithms [e.g. average precision, recall and Matthews correlation coefficient (Mcc) of 0.012, 0.53 and 0.056, respectively, for a set consisting of 1181 positive and 256 343 negative compounds], although several strategies have been employed to try to mitigate this imbalance [28].
To ensure the reliability of the screening results, we explored a strategy to improve model performance. First, we randomly selected 80 000 samples from the negative set and merged them with the refined set. Then, the model was trained on the merged set using 3-fold cross validation, which means each fold contains a similar number of positive and negative samples. To test the model sensitivity for different negative samples, we repeated the experiment three times by randomly choosing 80 000 negative samples from the negative set. During training, a focal loss (FL) [29], which focuses more on the minority class and hard samples, was used instead of binary cross-entropy (CE) loss. At last, our model achieved average precision, recall and Mcc of 0.716, 0.515 and 0.605, respectively, on this highly imbalanced dataset (Table 1). These evaluation metrics indicated that for each fold consisting of 136 positive and 27 286 negative samples, our model had predicted 98 positive samples in which 70 are true positive. This was a great improvement on such an imbalanced dataset, and it is of important significance for drug discovery, especially for in silico screening, because biochemical experiments usually produce large numbers of negative but a small fraction of positive samples (e.g. 0.01–0.14% typical hit rate for a high-throughput screening). We also tested different numbers of negative sampling, and the results indicated that the 80 000 samples were appropriate considering various parameters, including accuracy, recall, Mcc and computational cost. Furthermore, the sensitivity analysis by three rounds of random down-sampling and 3-fold cross validation verified the stability of the model. Lastly, we used these models to screen the bioactive chemical library after excluding the refined set. For each down-sampling group, the compounds with predicted probability higher than 0.5 in at least 2-folds were curated. A total of 17 compounds were selected as the final candidates.
Table 1.
Model performance on the highly imbalanced 3CLpro inhibitor dataset
| Random sampling of 80 000 negative samples | 3-fold cross validation | Precision | Recall | Mcc |
|---|---|---|---|---|
| 1 | 1 | 0.660 | 0.515 | 0.581 |
| 2 | 0.742 | 0.529 | 0.625 | |
| 3 | 0.753 | 0.537 | 0.634 | |
| 2 | 1 | 0.682 | 0.537 | 0.603 |
| 2 | 0.740 | 0.544 | 0.633 | |
| 3 | 0.713 | 0.529 | 0.613 | |
| 3 | 1 | 0.627 | 0.507 | 0.562 |
| 2 | 0.805 | 0.456 | 0.604 | |
| 3 | 0.725 | 0.485 | 0.592 |
In vitro screening for 3CLpro inhibitors
After protein purification, we evaluated two well-known inhibitors of SARS-COV-2 3CLpro, namely GC-376 and boceprevir. The calculated IC50 values of GC376 and boceprevir for 3CLpro were 0.5508 μM and 20.62 μM (Figure 3B), which were similar to their previously reported IC50 values [7]. This result indicates that our enzymatic assay is reliable.
Figure 3 .
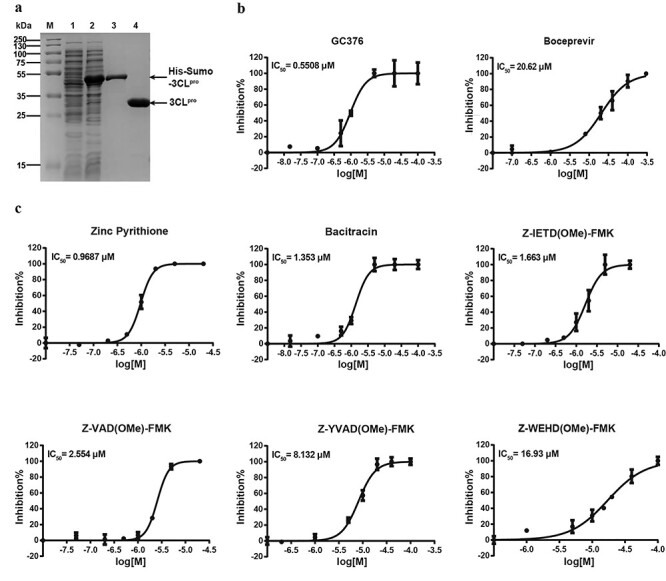
Purification of 3CLpro and enzyme inhibitory activity of the inhibitors. (A) Purification of 3CLpro. (B) The inhibitory effect of GC376 and boceprevir for SARS-CoV-2 3CLpro. (C) The inhibitory effect of identified inhibitors for SARS-CoV-2 3CLpro.
With the established FRET-based assay, the first-round screening was done with the predicted 740 compounds. Encouragingly, there were multiple compounds that showed inhibition against SARS-COV-2 3CLpro at 100 μM concentration (Supplementary Figure S2, available online at http://bib.oxfordjournals.org). Among these compounds, bacitracin, which showed an IC50 value of 1.353 μM, was regarded as showing inhibition and was included in the positive dataset, whereas another 739 compounds with IC50 values >20 μM were regarded as negative and were included in the negative dataset. As described above, we retrained our model and then predicted 17 candidates for the second-round screening. Among them, 10 compounds showed inhibition at 100 μM concentration (Supplementary Figure S2, available online at http://bib.oxfordjournals.org), and we further tested their IC50 values. The dose–response curves of these compounds were determined under conditions containing 40 μM substrate, 2 μM 3CLpro and different concentrations of tested compounds. There were five compounds exhibiting desirable inhibitory effects in this experiment.
Combining the results from the first and second rounds of screening, we have identified six strong inhibitors for SARS-CoV-2 3CLpro (Figure 3C). Among these molecules, zinc pyrithione inhibited the target enzyme at the lowest concentration in our screening, with an IC50 value of 0.9687 μM. It is commonly tested for fungistatic and bacteriostatic use in preclinical research. Zinc pyrithione has also been reported to inhibit the replication of various ribonucleic acid (RNA) viruses, including SARS-coronavirus and equine arteritis virus. The underlying mechanism is due to direct inhibition of the RNA-dependent RNA polymerase (RdRp), thus impairing the RNA synthesis activity of viruses [30]. Our study reveals that zinc pyrithione can also bind to 3CLpro. Therefore, it is plausible that zinc pyrithione can be regarded as a potential candidate against SARS-CoV-2. Similarly, bacitracin, which has been an FDA-approved drug for years due to its known antibiotic activity, also showed strong inhibition of 3CLpro with an IC50 of 1.353 μM. More promisingly, bacitracin can be used as a pediatric medication via intramuscular injection for the systemic treatment of infantile streptococcal pneumonia and empyema. Bacitracin can also be administered as a topical ophthalmic ointment to treat superficial eye infections involving the conjunctiva and cornea. The ability to inhibit the SARS-CoV-2 3CLpro, combined with the fact that both these drugs have already been approved for clinical treatments, demonstrates that both compounds show great promise to work as anti-SARS-CoV-2 medications in clinical treatments.
As a second type of compound, four peptidyl fluorine-methyl-ketones (pFMK), showed potent 3CLpro inhibitory efficiency (IC50 < 20 μM), although by varying the peptide sequences, there was a nearly 10-fold difference. This is not surprising, since peptide sequences normally provide the enzyme recognition sites for the molecules. pFMKs are well known covalent caspase inhibitors in biomedical research. While caspases play key roles in apoptosis, pyroptosis and various immune responses, they all share the same active site residue of cysteine with our target enzyme 3CLpro. This indicates FMK could be a powerful warhead to react with the active site cysteine of 3CLpro and to block its proteolytic activity in a covalent and irreversible manner. We further predicted ADME properties of these inhibitors using SwissADME [31] (Supplementary File S2, available online at http://bib.oxfordjournals.org).
Among the pFMKs, Z-VAD (OMe)-FMK is a well-characterized pan-caspase inhibitor that irreversibly binds the catalytic site of various caspases. It prevents cell shrinkage and deoxyribonucleic acid (DNA) fragmentation by inhibiting caspase-2, -3, -6 and -8 in flounder immune cells [32] and prevents an increase of p53, PARP-1 and caspase-3 levels in retinal ganglion cells [33]. Through an extension of the peptide chain of Z-VAD(OMe)-FMK with a tyrosine residue, the compound Z-YVAD(OMe)-FMK can block IL-1β secretion, which often initiates hyperinflammation in disease by inhibiting caspase-1 activity [34]. More recently, Z-WEHD(OMe)-FMK was proven to be a potent, cell-permeable and irreversible caspase-1/5 inhibitor and was identified as a robust inhibitor of cathepsin B activity, which is a member of a different cysteine protease family. The known off-target effect of Z-WEHD(OMe)-FMK on cathepsin B might also explain its lower inhibition efficiency for 3CLpro compared to the two former compounds.
The last but the most potent pFMK molecule in our assay is Z-IETD(OMe)-FMK, with a IC50 of 1.663 μM. It is a specific caspase-8 inhibitor that disrupts the extrinsic caspase pathway [35] and only partially inhibits the cleavage of caspase-3 and PARP. At non-toxic doses, Z-IETD(OMe)-FMK was found to be immunosuppressive. It was shown to block NF-κB in activated primary T cells but has little inhibitory effect on the secretion of IL-2 and IFN-γ during T cell activation [36]. In the case of SARS-CoV-2, hyperinflammation and the cytokine storm are severe problems after the early stage of the disease. The immunosuppressive function of Z-IETD(OMe)-FMK might help it treat the disease by preventing the damage caused by these more severe effects of the immune response, such as hyperinflammation and the cytokine storm.
Model interpretation
As previously mentioned, the mono-fluorinated derivatives are generally irreversible covalent inhibitors of many proteases, which form a covalent thioether adduct with various biological targets, including cysteine protease [37–39]. Based on this mechanism and some released peptidic covalent reversible/irreversible inhibitors in complex with 3CLpro (e.g. GC376, boceprevir and N3), we assumed that these pFMK inhibitors identified in this study first near to the active site of 3CLpro, then their C-terminal warhead [i.e. fluoromethyl ketone (FMK)] could stable covalently bind to the residue Cys145 of 3CLpro by a nucleophilic attack. To confirm this prediction, we performed a covalent docking following a two-point attractor and flexible side chain methods by AutoDock [40, 41]. All of these pFMK inhibitors formed a covalent bond by displacing the fluoride group with the thiolate group of Cys145 (Supplementary Figure S3, available online at http://bib.oxfordjournals.org). Although covalent inhibitors have become more popular in recent years and have shown exciting promise in SARS-CoV-2 therapy [39, 42], it is not easy to screen them in silico, especially by using docking methods. The process is complicated and time-consuming and has various limitations. To further complicate matters, large compounds, such as bacitracin, cannot be docked correctly by AutoDock. Instead, we performed bacitracin docking by DINC [43] (a protocol improved docking of large ligands) (Supplementary Figure S4, available online at http://bib.oxfordjournals.org).
On the contrary, our deep learning model can select most compounds simultaneously regardless of their length and regardless of whether they form covalent or non-covalent interactions with the target. However, the results from black-box machine learning model, where humans are not able to understand the process, may confuse people and increase the risk of following false leads, especially in biology and chemistry. Moreover, it is hard to optimize the model and the results without a clear interpretation of the model. Therefore, it is important to map the features extracted by the deep learning model to domain knowledge in order to update our understanding. To explain our model, we explored why our model selects the inhibitors that it does and understand how our model discerns the important sites for binding.
At first, we assumed that our model predicts positive samples due to similar chemical structures in the training set. To test this assumption, we compared the chemical similarity between identified inhibitors and positive compounds in the training set using the Tanimoto similarity score [44]. The Tanimoto nearest neighbor in the training set of Zinc Pyrithione was 1-oxidopyridine-2-thione, a monomer of Zinc Pyrithione. The Tanimoto nearest neighbors of four pFMK inhibitors were Z-FA-FMK and Z-DEVD(OMe)-FMK, which had been reported recently to inhibit 3CLpro with IC50 values of 11.39 μM and 6.81 μM, respectively [9]. In that study, Z-FA-FMK inhibited SARS-CoV-2 in vitro at the nanomolar level (EC50 = 0.13 μM) without apparent cytotoxicity, while Z-DEVD(OMe)-FMK did not show potent antiviral activity (EC50 > 20 μM). To investigate the influence of nearest neighbors on the predicted results, we removed Z-DEVD(OMe)-FMK and Z-FA-FMK from the training set and retrained the model. The results show that Z-VAD(OMe)-FMK and Z-IETD(OMe)-FMK were then predicted as negative, whereas Z-WEHD(OMe)-FMK and Z-YVAD(OMe)-FMK were still predicted as positive. Presumably, the model does not only rely on the structural similarity but also relies on hidden features of compounds. To test this hypothesis, we compared the final embedding of compounds from the screening library after dimensionality reduction by t-SNE. As shown in Figure 4A, Z-FA-FMK, Z-DEVD(OMe)-FMK and the four identified FMK inhibitors were very close in space after model processing (right, indicated by black arrow), whereas they distributed relatively far before processing (left). These results suggest that the model selects compounds based on their spatial distance to positive compounds in high-dimensional space.
Figure 4 .
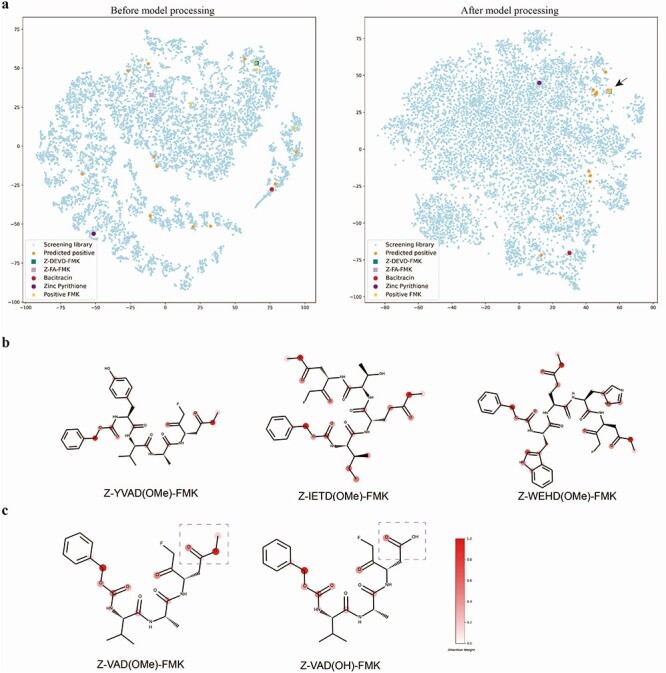
Visualization and model interpretation. (A) Dimensionality reduction by t-SNE of the molecule representation before model processing (left) and after model processing (right). (B) The attention weights learned by the model indicate important atoms. (C) The difference between Z-VAD(OMe)-FMK (left, predicted positive) and Z-VAD(OH)-FMK (right, predicted negative) is the methylated modification of aspartic acid side chain (indicated by purple dotted lines), and our model thinks this modification is critical for binding.
As mentioned previously, the interactions of other functional groups of these derivatives facilitate the covalent bond between the FMK warhead and Cys145. It raises the question of whether all pFMK derivatives can bind to 3CLpro or to only those with particular features have this binding capability. For our model, the embedding vector of each molecule has its own chemical implications, which has converged information from its atom and bond embeddings through the attention mechanism. To understand this question, we explored why the model considers a compound to be important at the atomic level. We visualized the important atoms ranked by attention weight. As indicated by the color bar (Figure 4B), the redder in color the atom, the more important it is based on attention weight. We can clearly see some common features across these inhibitors. The carbon near the N-terminal benzene ring (Z) and the carbonyl near the FMK have high attention weights. More importantly, the methylated modification of the last amino acid (D) is considered essential. Interestingly, Z-VAD(OH)-FMK which does not have that methylated modification is predicted as negative. As shown in Figure 4C, the difference between Z-VAD(OMe)-FMK (predicted positive) and Z-VAD(OH)-FMK (predicted negative) is the methylated modification of the aspartic acid side chain, and our model thinks this modification is critical for binding. To prove this hypothesis, we have tested the binding between Z-VAD(OH)-FMK and 3CLpro in vitro and calculated the IC50 value to be 116.3 μM (Supplementary Figure S5a, available online at http://bib.oxfordjournals.org). This result suggests that a methylated modification strongly improves the binding ability of these derivatives to 3CLpro. Comparing to the IC50 value of 2.554 μM for Z-VAD(OMe)-FMK (Figure 3C), we considered that a methylated modification strongly improves the binding ability of these derivatives to 3CLpro. One possible explanation is that the methylated modification could significantly reduce the molecular polarity and disrupt the molecular interaction force between the carboxylic group and the cysteine sulfhydryl group of 3CLpro, thus leading to a biologically inactive structure. This is very impressive because our model has successfully captured this important domain knowledge even without any relevant negative samples in the training set. We have also performed docking of Z-VAD(OH)-FMK to 3CLpro and the simulated binding energy is lower than Z-VAD(OMe)-FMK, indicating a stronger binding affinity of Z-VAD(OH)-FMK, as predicted by the docking method (Supplementary Figure S5b, available online at http://bib.oxfordjournals.org). This result suggests that molecular docking cannot capture this critical feature accurately.
Moreover, we have collected and tested all pFMK derivatives from PubChem using our model. From all these 57 derivatives, except 6 known inhibitors identified in this study and previous studies, we have predicted another 10 derivatives that are selected as positive for 3CLpro (Supplementary Figure S6, Supplementary Table S1, available online at http://bib.oxfordjournals.org). All compounds without the methylated modification of the last amino acid side chain are predicted as negative. These observations indicate that the attention weight of our model has indeed captured the domain knowledge at the atomic level. This model interpretation can be used in many applications for mapping to domain knowledge and for even discovering new knowledge, which may guide drug optimization.
Biotin-VAD(OMe)-FMK is an ABP of 3CLpro
ABPs could be used as a chemical antibody to report on the expression of a target protein and have been shown to be remarkable tools due to their ability to label and enrich variable enzymatic activities [45]. An ABP typically consists of three elements: a reactive group (sometimes called a ‘warhead’), a reporter group (biotin group or fluorophore) and a linker group which could be a peptide sequence used for connecting the two other parts, while functioning as the recognition site for the target enzyme (Supplementary Figure S7, available online at http://bib.oxfordjournals.org). As mentioned, the covalent bond between the FMK and Cys145 and the methylated modification of the P1 amino acid (D) were considered important modifications for binding of 3CLpro. More interestingly, biotin-VAD(OMe)-FMK, which was predicted as positive for binding 3CLpro, is commercially available. Therefore, we tested whether biotin-VAD(OMe)-FMK would be a potential ABP for 3CLpro. As shown in Supplementary Figure S8, available online at http://bib.oxfordjournals.org, biotin-VAD(OMe)-FMK indeed inhibits3CLpro with an IC50 of 6.675 μM, suggesting it is a potent inhibitor of 3CLpro. Next, we performed comparative ABPP experiments using different concentrations of biotin-VAD(OMe)-FMK to label 3CLpro. It was found that the labeling of 3CLpro by biotin-VAD(OMe)-FMK is remarkably concentration-dependent (Figure 5A). Subsequently, we used the same concentration of biotin-VAD(OMe)-FMK to incubate with 3CLpro for different periods of time. The results showed that the labeling was also time-dependent and a clear band could be visualized even within 5 s labeling (Figure 5B). These experiments validated biotin-VAD(OMe)-FMK as an ABP for 3CLpro3CL protease.
Figure 5 .
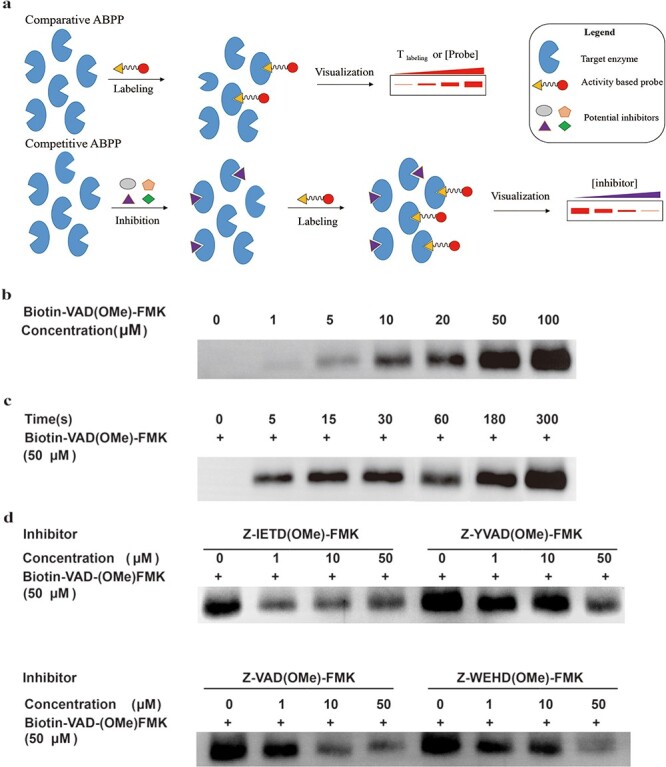
Comparative and competitive ABPP of 3CLpro by biotin-VAD(OMe)-FMK using a western blot assay. (A) Schematic of comparative and competitive ABPP. (B) 100 ng 3CLpro was exposed to increasing concentrations of biotin-VAD(OMe)-FMK for 3 min. (C) 100 ng 3CLprowas exposed to 50 μM of biotin-VAD(OMe)-FMK for different times. (D) 100 ng 3CLpro was pre-incubated with increasing concentrations of four inhibitors for 15 min, followed by incubation of 50 μM biotin-VAD(OMe)-FMK for 3 min.
Next, we ran competitive ABPP experiment, with biotin-VAD(OMe)-FMK and four covalent 3CL protease inhibitors we identified, including Z-IETD(OMe)-FMK, Z-YVAD(OMe)-FMK, Z-VAD(OMe)-FMK and Z-WEHD(OMe)-FMK. First, 3CLpro was pre-incubated with different concentrations of inhibitors, then biotin-VAD(OMe)-FMK was used to label the residual protease [46]. As the concentration of inhibitors increased, the bands gradually became weaker. The results of competitive ABPP are consistent with the fluorescent substrate assay to detect 3CLpro activity. These results suggest that biotin-VAD(OMe)-FMK could be a powerful ABP for 3CLpro and has a potential application for the labeling of 3CLpro in cell lysate or in vitro, or possibly even in vivo. There are two main advantages of this ABP. First, biotin-VAD(OMe)-FMK is commercially available. Second, the labeling speed of this probe is very fast. Combined with previous results where Z-VAD(OH)-FMK showed weak inhibition for 3CLpro, these competitive ABPP results prove that the methylated modification of P1 amino acid (D) is truly important for the binding of 3CLpro. This finding suggests that model interpretability is absolutely necessary, especially in the application of biology and chemistry, which could help us to discover new research paths and to gain new insights.
Conclusion
In this work, we propose a framework, namely AIMEE, combining a deep learning model and an enzymatic assay to identify inhibitors against SARS-CoV-2 3CLpro. Through this closed-loop and experimental design, our method has achieved excellent performance on a 3CLpro-specific highly imbalanced dataset and has identified and verified six novel potent SARS-CoV-2 3CLpro inhibitors. Among them, there are four outstanding compounds with an IC50 value <3 μM. More importantly, some of these inhibitors have potential clinical value as COVID-19 therapies. It should be noted that the hit rate of primary screening (100 μM inhibitor) and strong binding (IC50 < 20 μM) in the second round is 58.82% and 29.41%, respectively. This is significant not only because of low cost but also because high-throughput screening is not readily available due to the BSL3 level of the virus. It proves that building a closed-loop deep learning model combined with wet lab experiments is one of the most effective and promising ways to identify novel inhibitors for a specific target. In addition, it is also possible to easily transfer this 3CLpro model to a homolog target or a protein target which resembles 3CLpro in high-dimensional space.
Furthermore, we explored the logic behind the model and evaluated how the model discerns the key sites of the identified compounds. Based on the model interpretation, we have proven that the methylated modification of the FMK-nearest amino acid side chain is particularly critical for the binding between the pFMK derivatives and 3CLpro. As demonstrated in a previous study, this modification facilitates the binding by avoiding compound cyclization. The mapping of the features captured by a deep learning model to domain knowledge is particularly enlightening, especially for a field relying heavily on wet lab experiments. Since knowledge in such fields has been accumulated over a long period of time and the low-hanging fruit has become so rare, it is now more difficult to get new scientific breakthroughs. Artificial intelligence, particularly deep learning models, may elucidate new paths of research and help us to gain new insights. In this study, we selected a commercially available compound, biotin-VAD(OMe)-FMK, and proved it could act as an ABP for SARS-CoV-2 3CLpro. This opens an avenue for new applications for existing drugs. Taken together, this work highlights the utility of a novel approach incorporating deep learning models and wet lab experiments to expand the boundaries of biological and chemical studies and drug discovery.
Materials and methods
Data and tools
The PDBbind dataset (http://www.pdbbind.org.cn/), which provides protein–drug complex structures with experimentally measured binding values, is commonly used as a benchmark for validating scoring functions [47]. Here, PDBbind v2016 core set with diverse 290 complexes covering all protein classes in the PDBbind refined set was used as a test set for comparisons with other methods. The PDBbind dataset splitting is briefly described as follows. First, the core set containing 290 protein–ligand complexes is utilized as test set. Second, 1000 complexes within the refined set were separated to be used as a validation set (same as Pafnucy [16]). Third, the other complexes from the refined and general sets are combined to be used as the training set. Thus, a total of 13 196 complexes are used for model evaluation. Additionally, CASF-2013 with 195 samples and Astex Diverse Set with 73 samples are also used as independent test sets.
We have collected SARS-CoV/SARS-CoV-2 3CLpro inhibitors (including positive and negative) from various papers [6–9, 48] and public datasets such as PubChem (https://pubchem.ncbi.nlm.nih.gov/bioassay/1706), GHDDI (https://ghddi-ailab.github.io/Targeting2019-nCoV/) and Diamond (https://www.diamond.ac.uk/covid-19/for-scientists.html) as well as validated results in our wet lab experiment. This refined dataset contains carefully selected 3CLpro inhibitors with measured IC50 value and negative compounds which were verified in our experiment. We chose an IC50 of 20 μM as the threshold to determine the positive and negative samples. The refined set then consisted of 408 positive and 1828 negative samples. The negative set, which contained almost 300 000 negative compounds for SARS-CoV/SARS-CoV-2 3CLpro, was used as a negative sample sampling source for training. After removing overlaps with the training set, a collection of 10 924 compounds consisting of approved and clinical-stage drugs and bioactive compounds was used for in silico screening.
The compound data were processed by RDKit (https://www.rdkit.org). The molecular docking was performed using AutoDock [40] and DINC [43]. The visualization of docking result was performed using open source PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.). The ADME prediction was conducted using SwissADME which integrates some open-source algorithms and their own models [31]. This tool is freely available at http://www.swissadme.ch/.
All chemicals were analytical grade and used without further purification. All stock solutions of compounds were prepared in dimethyl sulfoxide (DMSO), and the compounds were purchased from MedChemExpress.
Model
To fully utilize available information from biological evolutionary relationships and structural information, we explored multimodal inputs of proteins from different dimensions. Basically, our model consisted of four parts: (i) protein sequence which were pretrained by large scale of sequences using masked language model [49]; (ii) protein and structure (contact map) data which were processed by the Transformer [27] and GAT [50], respectively, and then concatenated to get protein embedding vector; (iii) drug smiles that were processed by GAT (attentive FP model [51]) to get drug embedding vector and (iv) protein and drug embedding vectors which were concatenated and were fed into fully connected layers to predict interaction.
Transformer
The most critical part within the Transformer is multihead attention [27]. Multihead attention stacks several modules, namely ‘scaled dot-product attention’, and allows the model to perform parallel computing.
The input of each scaled dot-product attention layer consists of queries (q), keys (k) and values (v). Practically, the q, k and v are packed into matrices Q, K and V. Then, the matrix of outputs is calculated as
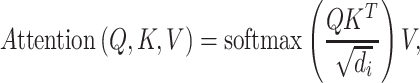 |
(1) |
where 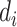 is the dimension of q and k. Multihead attention stacks h attention layers, and the output matrix is
is the dimension of q and k. Multihead attention stacks h attention layers, and the output matrix is
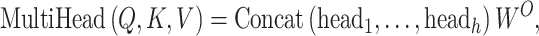 |
(2) |
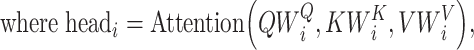 |
(3) |
where the projections are parameter matrices 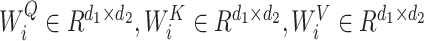 and
and 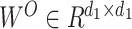 , where
, where 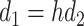 , and h is the number of heads. We tried different amounts of protein sequence for the Transformer pretraining and finally selected TAPE [52], which was pretrained on 31 million protein domains from Pfam dataset [53]. During training, the first eight layers of this module were frozen.
, and h is the number of heads. We tried different amounts of protein sequence for the Transformer pretraining and finally selected TAPE [52], which was pretrained on 31 million protein domains from Pfam dataset [53]. During training, the first eight layers of this module were frozen.
Graph attention network
The key point of GAT is leveraging a self-attention strategy, which was briefly described above, to compute a hidden context of each node by converging its neighbors in the graph [50]. More specifically, for a single graph attentional layer, the representation vector of node i is calculated as
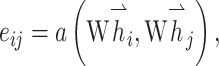 |
(4) |
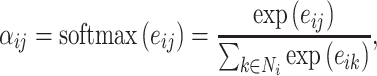 |
(5) |
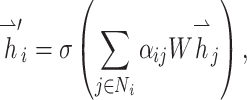 |
(6) |
where 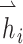 ,
, 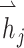 are the feature vectors of target node i and neighbor node j.
are the feature vectors of target node i and neighbor node j. 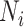 represents all neighbors of node i. W is the trainable weights, while
represents all neighbors of node i. W is the trainable weights, while 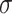 is activate function. Here, we use exponential linear unit (ELU) activate function [54]. By aggregating information of every neighbors in the graph through attention, we obtained the representation vector of node i,
is activate function. Here, we use exponential linear unit (ELU) activate function [54]. By aggregating information of every neighbors in the graph through attention, we obtained the representation vector of node i, 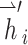 .
.
Then, for l iterations, the GAT was processed in a similar manner to a graph neural network, which involved message passing and readout phases:
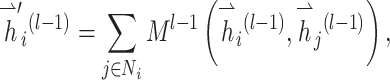 |
(7) |
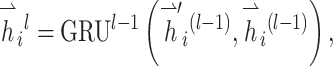 |
(8) |
where 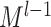 and GRU represent graph attention mechanism and activate function gated recurrent unit [55], respectively;
and GRU represent graph attention mechanism and activate function gated recurrent unit [55], respectively; 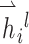 is the representation vector of node i after l iterations, which aggregates the information from node i and its neighbors of iteration l − 1.
is the representation vector of node i after l iterations, which aggregates the information from node i and its neighbors of iteration l − 1.
Focal loss
We used FL to address the negative impact on the model caused by an imbalanced dataset. The FL was originally designed to address the one-stage object detection scenario in which foreground is much less than background [29]. Typically, the CE loss for a binary classification model (where label is 0 or 1) is defined as
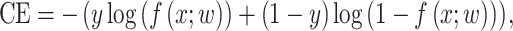 |
(9) |
where f() and 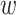 correspond to the classifier and trainable weights;
correspond to the classifier and trainable weights; 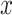 and
and 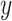 correspond to the input and label, respectively. For simplification, the predicted probability
correspond to the input and label, respectively. For simplification, the predicted probability 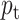 by the classifier is defined as
by the classifier is defined as
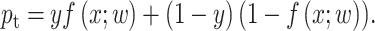 |
(10) |
Thus, CE could be simplified as
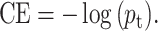 |
(11) |
For imbalanced dataset, to balance the importance of positive/negative samples and to reduce the impact to gradient by the majority of easily classified negative samples, a weighting factor 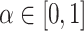 (
(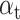 is defined similar to
is defined similar to 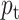 ) and a modulating factor
) and a modulating factor 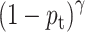 (tunable focusing parameter
(tunable focusing parameter 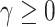 ) were added in CE, thus the FL is defined as
) were added in CE, thus the FL is defined as
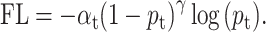 |
(12) |
Evaluation metrics
Pearson’s correlation coefficient R and RMSE, which refer to the linear correlation and the differences between the predicted and real values, respectively, were used as evaluation metrics:
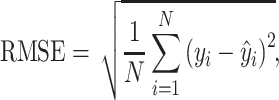 |
(13) |
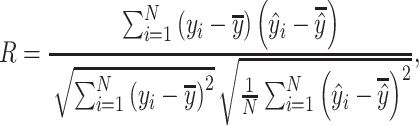 |
(14) |
where N is the size of a dataset, yi is the real value, whereas ŷi is the predicted value; 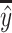 is the average of real values, whereas
is the average of real values, whereas 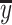 is the average of the predicted value.
is the average of the predicted value.
We used three metrics including precision, recall and Mcc to evaluate the model performance on the imbalanced dataset:
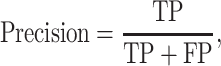 |
(15) |
 |
(16) |
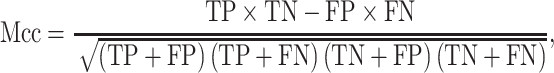 |
(17) |
where TP is the number of true positives, TN is the number of true negatives, FP is the number of false positives, FN is the number of false negatives, P indicates positive and N indicates negative.
Purification of 3CLpro
A previous study demonstrated that the first serine residue on the N-terminus of SARS-COV 3CLpro is important for its activity and substrate binding, and additional residues at the N- and C-terminus of 3CLpro would significantly decrease the enzyme activity [56]. Thus, it is important to produce a native 3CLpro for in vitro inhibitor screening with no additional residues on either end. Here, we chose a novel strategy to obtain a native 3CLpro of SARS-CoV-2. The full gene of SARS-CoV-2 3CLpro was cloned and inserted into a pET28b-small ubiquitin-like modifier protein (SUMO) vector, generating a His6-SUMO-tagged fusion protein. Recombinant 3CLpro was expressed in Escherichia coli (DE3) and subsequently purified. After digesting with SUMO protease (ULP1), we obtained the native SARS-CoV-2 3CLpro without any redundant residues at either the N- or C-terminus (Figure 3A). In addition, the yield of SARS-CoV-2 3CLpro was significantly improved (115 mg/l).
Construction of recombinant plasmid
The full-length gene encoding SARS-CoV-2 3CLpro (ORF1ab polyprotein residues 3264-3569, GenBank accession number MN908947.3) was optimized and synthesized for E. coli expression (Genscript). All primers and fluorescence substrates were synthesized by Genscript.
The synthesized gene was amplified using PCR with the forward primer 5′-CACAGAGAACAGATTGGT GGAAGCGGTTTCCGTAAGATGGCG-3′ and the reverse primer 5′-CTCAGCTTCCTT TCGGGCTTTGTTATTGAAAGGTCACACCGCTGC-3′. This PCR product was employed as the target gene. Then, we amplified the vector pET-28b-SUMO with the forward primer 5′-CGCCATCTTACGGAAACCGCTTCCACCAATCTGTTCTCTGTGAG-3′ and the reverse primer 5′-GTGCAGCGGTGTGACCTTTCAATAACAAAGCCCGAAAGGAAGCTG-3′. Through homologous recombination, we connected the target gene with the vector skeleton (ClonExpress II One Step Cloning Kit). At the N-terminus, the construct designed for SARS-CoV-2 3CLpro contains a six-histidine tag (His6-tag) and a SUMO gene. The C-terminus does not contain any additional amino acids. The native N-terminus was generated after the treatment with SUMO protease, also known as Ulp1, which is highly specific for the SUMO protein fusion, recognizing the tertiary structure of SUMO rather than an amino acid sequence. After the digest with Ulp1, we gained a natural SARS-COV-2 3CLpro. The gene sequence of the 3CLpro was verified by sequencing at Sangon Biotech (Shanghai).
Protein expression and purification
The sequence-verified SARS-CoV-2 3CLpro construct was transformed into E. coli strain BL21-DE3 (Transgene). Transformed clones were pre-cultured at 37°C in 100 ml Luria broth medium with kanamycin (100 μg/ml) overnight, and the incubated culture was inoculated into 1 l Luria broth medium supplied with kanamycin (100 μg/ml) at 37°C. When the cells were grown to OD600 of 0.6–0.8, 0.2 mM isopropyl-D-thiogalactoside (IPTG) was added to the culture to induce the expression of the 3CLpro gene at 16°C. After 20 h, cells were harvested by centrifugation at 4500 × g, 4°C for 10 min. The cell pellets were washed with phosphate-buffered saline twice and then were resuspended in 40 ml lysis buffer (20 mM Tris, 300 mM NaCl, pH 8.0; pH of all buffers was adjusted at room temperature) and then lysed by high-pressure homogenization. The lysate was clarified by ultracentrifugation at 30 000 × g at 4°C for 30 min. The supernatant was loaded onto a 5 ml HisTrap HP column (GE Healthcare) equilibrated with lysis buffer containing 20 mM imidazole. The HisTrap FF column was washed with 150 ml lysis buffer containing 20 mM imidazole to remove nonspecific binding proteins and eluted with elution buffer (20 mM Tris, 150 mM NaCl, 500 mM imidazole, pH 7.8) with a linear gradient of imidazole ranging from 0 mM to 500 mM, 20 column volumes using an AKTA Pure fast protein liquid chromatography (FPLC). Next, the fractions containing target protein were pooled using Amicon Ultra 15 centrifugal filters (10 kDa, Merck Millipore) at 4500 × g, 4°C to concentrate the protein. The protein was then mixed with SUMO protease at a molar ratio of 1:100 and was dialyzed into reaction buffer (20 mM Tris, 100 mM NaCl, 1 mM DTT, 1 mM EDTA; pH 7.8) at 30°C, 1100 rpm, for 1 h. The digested products were loaded onto a Superdex 16/600 size exclusion column equilibrated with elution buffer (330 mM Na2HPO4, 167 mM NaH2PO4, 150 mM NaCl; pH 7.3) and then the eluted fractions were pooled and concentrated.
SARS-CoV-2 3CLpro kinetic assay
The standard curve was generated as described below: 2 μM SARS-CoV-2 3CLpro was incubated with varying concentrations of FRET substrate (0.5–40 μM), and the reaction progress was monitored until the fluorescence signals reached plateau, at which point, we deemed all the FRET substrate was digested by 3CLpro.
For the measurements of Km/Vmax, screening of the protease inhibitor library, as well as IC50 measurements, proteolytic reaction with 2 μM SARS-CoV-2 in 50 μl of reaction buffer was carried out at 30°C in a fluorescence microplate reader with filters for excitation at 360 nm and emission at 460 nm. Reactions were monitored every 40 s. For Km/Vmax measurements, a FRET substrate concentration ranging from 0 to 200 μM was applied. The initial velocity of the proteolytic activity was calculated by linear regression for the first 8 min of the kinetic progress curves. The initial velocity was plotted against the FRET concentration with the classic Michaelis–Menten equation.
FRET protease assays
The fluorescent substrate harbors the cleavage site of SARS-CoV-2 3CLpro and uses Edans and Dabcyl, respectively, as a donor and quencher pair (Dabcyl-KTSAVLQ↓SGFRKM-E(Edans)-NH2; GenScript). The peptide substrate contains a 14 amino sequence with Dabcyl and Edans attached to its N- and C-terminals, respectively. The fluorescent substrate in a buffer composed of 20 mM Tris, 100 mM NaCl, 1 mM DTT; pH 7.3 was used for the fluorescence resonance energy transfer (FRET) protease assay. Fluorescence readings were obtained by using an excitation wavelength of 360 nm and an emission wavelength of 460 nm in a fluorescence microplate reader (BioTek Synergy4) 30 min after the addition of substrate. Initially, we mixed the SARS-CoV-2 3CLpro with each compound at a final concentration of 2.0 μM in assay buffer or DMSO in assay buffer as a negative control and incubated the reaction mixture at 30°C for 5 min. Next, we added the substrate dissolved in the reaction buffer for a final reaction volume of 50 μl. The final substrate concentrations varied from 5 μM to 640 μM (5 μM, 10 μM, 20 μM, 40 μM, 80 μM, 160 μM, 320 μM and 640 μM). A calibration curve was generated by measuring different concentrations (from 0.15 μM to 20 μM) of free Edans in a final volume of 50 μl reaction buffer. Initial velocities were determined from the linear section of the curve, and the relative fluorescence units (RFUs) were determined by subtracting background values (substrate-containing well without protease) from the raw fluorescence values. For the determination of the IC50, we incubated 2 μM of SARS-CoV-2 3CLpro with compounds at different final concentrations in reaction buffer at 30°C for 5 min. Afterward, we added the FRET substrate to the reaction mixture for a final concentration of 40 μM and a final total volume of 50 μl to initiate the reaction. Measurements of inhibitory activities of the compounds were performed in triplicate and are presented as the mean ± SD.
ABPP assay
Comparative ABPP assay
For the comparative ABPP assay, we prepared 20 μg of total protein for each sample and made the total volume up to 9 μl by the addition of assay buffer. Next, 1 μl of 10× working stock of biotin-VAD(OMe)-FMK was added, generating final ABP concentrations of 10 μM, 25 μM and 50 μM. We then incubated the mixture at 37°C for different lengths of time (30 s, 1 min and 3 min). We added 2.5 μl 5× loading buffer and boiled sample at 100°C for 5 min. In this assay, we used deactivated lysate by boiling it with 1% (wt/vol) sodium dodecyl sulfate (SDS) as negative control. The proteins were then separated based on size using a 12% (wt/vol) SDS-polyacrylamide gel electrophoresis (PAGE).
Concentration-dependent experiments
We prepared 100 ng of purified 3CL protease for each sample. The protein solution was premixed with assay buffer, then we aliquoted the premixed solution into different tubes. Next, we added 1 μl of 10× working stock of biotin-VAD(OMe)-FMK, generating final ABP concentrations of 0 μM, 1 μM, 5 μM, 10 μM, 20 μM, 50 μM and 100 μM. The mixture was incubated at 37°C for 15 min. Afterward, the tubes were placed on ice to stop the reaction. Next, we placed the tubes on ice, added 2.5 μl 5× loading buffer to each sample and boiled the samples for 5 min at 100°C. The samples were then separated based on size using a 12% (wt/vol) SDS-PAGE.
Time-dependent experiments
We prepared 100 ng of purified 3CL protease for each sample. The protein solution was premixed with assay buffer, then aliquoted into different tubes. Next, we added 1 μl of 10× working stock of biotin-VAD(OMe)-FMK, generating final ABP concentrations of 50 μM. The reaction mixture was incubated at 37°C for 0 s, 5 s, 15 s, 30 s, 60 s, 180 s and 300 s. Afterward, we placed the tubes on ice to stop the reaction, added 2.5 μl 5× loading buffer to each sample and boiled the samples for 5 min at 100°C. The samples were then run separated based on size using a 12% (wt/vol) SDS-PAGE.
Competitive ABPP assay
We prepared 100 ng of purified 3CL protease for each sample by premixing the concentrated protein solution with assay buffer and then aliquoting it into different tubes. Next, we added different 3CLpro inhibitors Z-IETD(OMe)-FMK, Z-VAD (OMe)-FMK, Z-YVAD(OMe)-FMK and Z-WEHD(OMe)-FMK in the following concentrations: 0μM, 1 μM, 10 μM and 50 μM. We then incubated the mixture at 37°C for 15 min. Afterward, we placed the tubes on ice to stop the reaction. Lastly, in order to label the residual active site after inhibition with the 3CLpro inhibitors, we added 1 μl of 500 μM biotin-VAD(OMe)-FMK, the ABP, to a final ABP concentration of 50 μM. The samples were then incubated at 37°C for 3 min.
Key Points
We propose a novel framework integrating deep learning model and enzymological experiments to screen inhibitors. Based on this framework, we identified six novel strong inhibitors against 3CL protease of SARS-CoV-2.
We leveraged multimodal inputs of proteins to predict protein–ligand interactions, which combines the advantages of both structure-based and sequence-based methods, achieving the top performance on the benchmark dataset PDBbind over existing methods.
We demonstrated that our method reduces the negative effect on model performance caused by highly imbalanced dataset during training, which could be introduced into many drug discovery applications.
We explored the model interpretability, mapping the deep learning extracted features to the domain knowledge of chemical properties and finding a methylated modification which strongly improves the binding ability of FMK derivatives to 3CLpro.
Based on the knowledge extracted by our model, a commercially available compound was selected and proven to be an ABP of 3CLpro. This finding suggests that this model interpretation could help discover new research paths and help us to gain new insights
Supplementary Material
Acknowledgements
We would like to thank Diana Czuchry for her helpful feedback on a draft of the paper.
Fan Hu is an assistant professor at the Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research focuses on computational biology, drug discovery and machine learning.
Lei Wang is a graduate student at the CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. Her research focuses on chemical biology, biological chemistry and proteomics.
Yishen Hu is a graduate student at the Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research focuses on machine learning and drug discovery.
Dongqi Wang is a graduate student at the Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research focuses on machine learning and drug discovery.
Weijie Wang is a technician at the CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research focuses on chemical biology, biological chemistry and proteomics.
Jianbing Jiang is an assistant professor at the School of Pharmaceutical Sciences, Shenzhen University Health Science Center. His research focuses on natural product chemical biology and activity-based probes.
Nan Li is an associate professor at the CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research focuses on chemical biology, biological chemistry and proteomics.
Peng Yin is an associate professor at the Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. His research focuses on computational biology and machine learning.
Contributor Information
Fan Hu, Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China.
Lei Wang, CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China.
Yishen Hu, Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China.
Dongqi Wang, Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China.
Weijie Wang, CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China.
Jianbing Jiang, School of Pharmaceutical Sciences, Shenzhen University Health Science Center, Shenzhen, 518055, China.
Nan Li, CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China.
Peng Yin, Guangdong-Hong Kong-Macao Joint Laboratory of Human-Machine Intelligence-Synergy Systems, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China.
Data and code availability
The code and data are available at https://github.com/SIAT-code/AIMEE.
Funding
National Key Research and Development Program of China (2018YFA0902703); Strategic Priority Research Program of Chinese Academy of Sciences (No XDB 38040200); National Natural Science Foundation of China (Nos 11801542, 31800694 and 31971354); Shenzhen Science and Technology Innovation Committee (Nos JCYJ20170818164014753, JCYJ20170818163445670, JCYJ20180703145002040).
Reference
- 1.WHO . World Health Organization: Coronavirus Disease (COVID-2019) Situation Reports. 2021. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports.
- 2.Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020; 382: 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020; 27: 325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020; 395: 565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L, Lin D, Sun X, et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (80-) 2020; 368: 409–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin Z, Du X, Xu Y, et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020; 582: 289–93. [DOI] [PubMed] [Google Scholar]
- 7.Fu L, Ye F, Feng Y, et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat Commun 2020; 11: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma C, Sacco MD, Hurst B, et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 2020; 30: 678–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu W, Xu M, Chen CZ, et al. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol Transl Sci 2020; 3(5): 1008–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krizhevsky A, Sutskever I, Hinton GE. ImageNet classification with deep convolutional neural networks. Commun ACM 2017; 60: 84–90. [Google Scholar]
- 11.Voulodimos A, Doulamis N, Doulamis A, et al. Deep learning for computer vision: a brief review. Comput Intell Neurosci 2018; 2018: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young T, Hazarika D, Poria S, et al. Recent trends in deep learning based natural language processing. IEEE Comput Intell Mag 2018; 13: 55–75. [Google Scholar]
- 13.Schneider P, Walters WP, Plowright AT, et al. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov 2020; 19: 353–64. [DOI] [PubMed] [Google Scholar]
- 14.Wallach I, Dzamba M, Heifets A. AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. Data Min Knowl Discov 2015; 22: 31–72. [Google Scholar]
- 15.Ragoza M, Hochuli J, Idrobo E, et al. Protein-ligand scoring with convolutional neural networks. J Chem Inf Model 2017; 57: 942–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stepniewska-Dziubinska MM, Zielenkiewicz P, Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018; 34: 3666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Öztürk H, Özgür A, Ozkirimli E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics 2018; 34:i821–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Tan X, Wang D, et al. TransformerCPI: improving compound–protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 2020; 36(16): 4406–14. [DOI] [PubMed] [Google Scholar]
- 19.Vellingiri B, Jayaramayya K, Iyer M, et al. COVID-19: a promising cure for the global panic. Sci Total Environ 2020; 725:138277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beck BR, Shin B, Choi Y, et al. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J 2020; 18: 784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ton AT, Gentile F, Hsing M, et al. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol Inform 2020; 2000028: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang B, Hu Y, Chen L, et al. Mining of epitopes on spike protein of SARS-CoV-2 from COVID-19 patients. Cell Res 2020; 30: 702–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu F, Jiang J, Yin P. Prediction of potential commercially inhibitors against SARS-CoV-2 by multi-task deep model. arXiv preprint, arXiv: 2003.00728, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stokes JM, Yang K, Swanson K, et al. A deep learning approach to antibiotic discovery. Cell 2020; 180:688–702.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanman LE, Bogyo M. Activity-based profiling of proteases. Annu Rev Biochem 2014; 83: 249–73. [DOI] [PubMed] [Google Scholar]
- 26.Whidbey C, Wright AT. Activity-based protein profiling—enabling multimodal functional studies of microbial communities. Annu Rev Biochem 2018; 420: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaswani A, Shazeer N, Parmar N, et al. Attention is all you need. arXiv preprint, arXiv: 1706.03762, 2017. [Google Scholar]
- 28.Korkmaz S. Deep learning-based imbalanced data classification for drug discovery. J Chem Inf Model 2020; 60(9): 4180–90. [DOI] [PubMed] [Google Scholar]
- 29.Lin T-Y, Goyal P, Girshick R, et al. Focal loss for dense object detection. In: Proceedings of the IEEE International Conference on Computer Vision, 2017 . p. 2980–88. Oct. 22 2017 to Oct. 29 2017. Venice, Italy. IEEE, USA.
- 30.te Velthuis AJW, van den Worm SHE, Sims AC, et al. Zn2+ inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog 2010; 6:e1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 2017; 7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S, Li J, Peng W, et al. Characterization of the responses of the caspase 2, 3, 6 and 8 genes to immune challenges and extracellular ATP stimulation in the Japanese flounder (Paralichthys olivaceus). BMC Vet Res 2019; 15:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G-Y, Fan B, Su G-F. Acute energy reduction induces caspase-dependent apoptosis and activates p53 in retinal ganglion cells (RGC-5). Exp Eye Res 2009; 89: 581–9. [DOI] [PubMed] [Google Scholar]
- 34.Zheng R, Tao L, Jian H, et al. NLRP3 inflammasome activation and lung fibrosis caused by airborne fine particulate matter. Ecotoxicol Environ Saf 2018; 163: 612–9. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Pemberton A, Morrison WI, et al. Granzyme B is an essential mediator in CD8 + T cell killing of Theileria parva -infected cells. Infect Immun 2018; 87: e00386–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence CP, Chow SC. Suppression of human T cell proliferation by the caspase inhibitors, z-VAD-FMK and z-IETD-FMK is independent of their caspase inhibition properties. Toxicol Appl Pharmacol 2012; 265: 103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powers JC, Asgian JL, Ekici ÖD, et al. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem Rev 2002; 102: 4639–750. [DOI] [PubMed] [Google Scholar]
- 38.Citarella A, Micale N. Peptidyl fluoromethyl ketones and their applications in medicinal chemistry. Molecules 2020; 25:4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cannalire R, Cerchia C, Beccari AR, et al. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J Med Chem 2020. doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 2009; 30: 2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bianco G, Forli S, Goodsell DS, et al. Covalent docking using autodock: two-point attractor and flexible side chain methods. Protein Sci 2016; 25: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ghosh AK, Samanta I, Mondal A, et al. Covalent inhibition in drug discovery. ChemMedChem 2019; 14: 889–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dhanik A, McMurray JS, Kavraki LE. DINC: a new AutoDock-based protocol for docking large ligands. BMC Struct Biol 2013; 13:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bajusz D, Rácz A, Héberger K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Chem 2015; 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li N, Overkleeft HS, Florea BI. Activity-based protein profiling: an enabling technology in chemical biology research. Curr Opin Chem Biol 2012; 16: 227–33. [DOI] [PubMed] [Google Scholar]
- 46.Li N, Kuo C-L, Paniagua G, et al. Relative quantification of proteasome activity by activity-based protein profiling and LC-MS/MS. Nat Protoc 2013; 8: 1155–68. [DOI] [PubMed] [Google Scholar]
- 47.Wang R, Fang X, Lu Y, et al. The PDBbind database: collection of binding affinities for protein−ligand complexes with known three-dimensional structures. J Med Chem 2004; 47: 2977–80. [DOI] [PubMed] [Google Scholar]
- 48.Riva L, Yuan S, Yin X, et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020; 586(7827): 113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Devlin J, Chang M-W, Lee K, et al. BERT: pre-training of deep bidirectional transformers for language understanding. arXiv preprint, arXiv: 1810.04805, 2018. [Google Scholar]
- 50.Veličković P, Cucurull G, Casanova A, et al. Graph attention networks. arXiv preprint, arXiv: 1710.10903, 2017. [Google Scholar]
- 51.Xiong Z, Wang D, Liu X, et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J Med Chem 2019; 63: 8749–60. [DOI] [PubMed] [Google Scholar]
- 52.Rao R, Bhattacharya N, Thomas N, et al. Evaluating protein transfer learning with TAPE. NIPS 2019; 32: 9689. [PMC free article] [PubMed] [Google Scholar]
- 53.El-Gebali S, Mistry J, Bateman A, et al. The Pfam protein families database in 2019. Nucleic Acids Res 2019; 47: D427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clevert D-A, Unterthiner T, Hochreiter S. Fast and accurate deep network learning by exponential linear units (ELUs). arXiv preprint, arXiv:1511.07289v5, 2015. [Google Scholar]
- 55.Cho K, van Merrienboer B, Gulcehre C, et al. Learning phrase representations using RNN encoder-decoder for statistical machine translation. arXiv preprint, arXiv: 1406.1078, 2014. [Google Scholar]
- 56.Xue X, Yang H, Shen W, et al. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J Mol Biol 2007; 366: 965–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and data are available at https://github.com/SIAT-code/AIMEE.