

Abstract
Infectious diseases are potential drivers for human evolution, through a complex, continuous and dynamic interaction between the host and the pathogen/s. It is this dynamic interaction that contributes toward the clinical outcome of a pathogenic disease. These are modulated by contributions from the human genetic variants, transcriptional response (including noncoding RNA) and the pathogen’s genome architecture. Modern genomic tools and techniques have been crucial for the detection and genomic characterization of pathogens with respect to the emerging infectious diseases. Aided by next-generation sequencing (NGS), risk stratification of host population/s allows for the identification of susceptible subgroups and better disease management. Nevertheless, many challenges to a general understanding of host–pathogen interactions remain. In this review, we elucidate how a better understanding of the human host-pathogen interplay can substantially enhance, and in turn benefit from, current and future applications of multi-omics based approaches in infectious and rare diseases. This includes the RNA-level response, which modulates the disease severity and outcome. The need to understand the role of human genetic variants in disease severity and clinical outcome has been further highlighted during the Coronavirus disease 2019 (COVID-19) pandemic. This would enhance and contribute toward our future pandemic preparedness.
Keywords: human variants, genetic diversity, pathogens, gene expression, noncoding RNA, disease severity
Introduction
Even today, one of the leading causes of morbidity and mortality globally is infectious diseases [1–5]. This highlights the functional role of widespread infectivity of the pathogen/s that is leading to mortality among human population/s. The rapid emergence of infectious diseases within the last two decades and their exacerbation to a pandemic scale with possible role of globalization is increasing the risk of life-threatening infections, both acute and chronic. This also has challenged the healthcare infrastructure globally with economic consequences. The outcome of the disease is determined by the dynamic interplay between the host and the pathogen. We summarize here the modern tools, techniques and scientific discoveries, which have been important to understand this relationship and the associations thus observed. The need for multidimensional studies combining genomic data of both host and pathogen, overlaid with clinical and demographic status of patients, would be pivotal in the modern era. In the current ongoing pandemic, initiatives such as COVID-19 Host Genetics Initiative serve as an important advancement to understand and highlight the milieu of host–pathogen interactome. The initiative paves the way for comprehensive meta-analysis projects bringing worldwide researchers together to identify determinants of COVID-19 susceptibility, severity and outcomes. Simultaneously, it has laid the groundwork for diagnostic markers and therapeutic target discovery [6].
Human genetic diversity
The genetic basis of diseases is a reflection of the evolution of the human genome. One of the fundamental characteristics that portray the host component in pathogenic diseases is the fact that life-threatening clinical disease is manifested only in a small percentage of infected individuals. This variation highlights the importance of studying human genetic diversity in the context of pathogenic diseases. To understand these relevant genetic components, an understanding of differences in the human genome is important. Approximately 90% of human allelic variations are polymorphisms that date back to our African origin [7]. New mutations arise in the human population naturally at the rate of 175 mutations per diploid human genome per generation [8].
Importance of studying genetic diversity in the background of pathogenic diseases
During the current COVID-19 global pandemic, identifying a susceptible group of population can significantly modulate the outcome of the pandemic vis-a-vis human population. This includes priority healthcare access and close monitoring. Thus, any leads in that direction are too important to be undermined. The history of human genetic susceptibility in pathogenic disease outbreaks dates back to the identification of resistance factors for the disease, inclusive of the individual’s heterozygous allele state for sickle variants of erythrocytes having resistance to malaria in the 1950s [9, 10]. Another factor is the lack of expression of the Duffy antigen receptor for chemokines (DARC) on red cells due to single nucleotide polymorphism (SNP) rs2814778 leading to a negligible infection in Western and Central Africa [10, 11].
Similarly, the Delta 32 mutation at rs333 in the entry receptor C-C chemokine receptor type 5 (CCR5) for Human Immunodeficiency Virus-Acquired Immunodeficiency Syndrome (HIV-AIDS) [12] confers resistance to the individual [13]. This mutation is understood to have evolved 700 years ago under the pathogenic evolutionary pressure of bubonic plague [14]. The survival of these alleles from the pandemic in the current human populations underscores the need to identify associations of host genetic elements with respect to infectious diseases. Extending the implications of the above example, studies can be used to classify a population according to the risk of acquiring infection and severe clinical manifestations of the disease. A well-known example is the O blood group in ABO blood grouping. The O blood group confers a protective effect to host in COVID-19 [15], increasing the susceptibility to cholera [16–18]. Furthermore, along with the O blood group, Rh-negative type compared to other blood groups was less susceptible to SARS-CoV-2 infection and had better clinical outcome. In addition, the AB group is known to require increased respiratory support (invasive), and is at higher risk of death due to COVID-19 [19]. Genome-wide association study (GWAS) by Genetics Of Mortality In Critical Care (GenOMICC) has studied the critically ill COVID-19 patients in UK to discover host genetic variants associated with critical illness. The study has highlighted that although ABO locus was previously associated with COVID-19, it did not show the same in their study. But the presence of signal close to genome-wide significance at the ABO locus potentially indicates its role in COVID-19. Whether ABO locus is associated with critical illness or not - is a matter of future research and more studies in this direction would be essential for in-depth understanding.
This is possibly relevant for large number of pathogenic diseases that have no/asymptomatic effect on a subset of the population. An example of such a disease previously shown to have a major impact on global food supply is the Creutzfeldt–Jakob disease caused by the prion protein. An SNP (rs1799990) leading to heterozygosity of methionine/valine at 129-codon of their prion protein gene confers immunity to the disease [20]. In the modern age endeavor toward personalized medicine and continual decrease in the cost of next-generation sequencing (NGS) enabled human genome sequencing, it is possible to understand and elucidate the genomic architecture of any given population. This information when overlaid with the rate at which diseases are spreading through a population can give us insights into population susceptibility for infection/s. The targeted administration of therapeutic interventions on susceptible groups of populations could reduce the load on medical infrastructure.
Diseases and its associated human host genetic variation
Statistical and genomics based measures are used to understand the host component involved in disease susceptibility. Human genetic components driving disease infection and prognosis were identified by a great variety of approaches. This includes twin studies, linkage analysis, complex segregation analysis and whole-genome sequencing approaches such as GWAS.
Assessment of human genetic components in twin studies identified 86% of hereditary components in Measles [21]. Even tuberculosis has a genetic component as highlighted by twin studies [22] which were later proven to be the result of defects in IL12/IFNγ dependent signaling pathway leading to a condition termed Mendelian susceptibility to mycobacterial disease (MSMD). In addition, Hepatitis B has also shown different associations in twin and population studies [23–25]. Furthermore, Hepatitis B viral clearance associated with hepatocellular carcinoma has host genetic elements modulating disease severity, such as human leukocyte antigen HLA-DP and HLA-DQ loci [26].
Moreover, an unbiased view of genomes of affected individuals is provided by GWAS, which has revolutionized the area of disease genetics allowing the field to move out of the candidate gene approach [27]. It has enabled the identification of many lead SNPs associated with diseases. The GWAS catalog is maintained by EMBL at https://www.ebi.ac.uk/gwas [28]. The GWAS Catalog contains 5037 publications and 257 351 variant-trait association (as on 5 May 2021). GWAS studies require large sample sizes as millions of genomes and variants are analyzed together. It is ideal to have a population set with a homogeneous ethnic background to avoid spurious associations [29]. Mycobacterium leprae or M. lepromatosis causes leprosy by long-term infection damaging the nervous system, eyes and respiratory tracts. Using GWAS, several candidate genes have been observed as host genetic factors modulating disease severity, including TNFA, IL10, PARK2 in the past and more recently LACC1 [30]. Genetic components and mutations in the interleukin-coding genes have been observed to be involved in many diseases as shown in Table 1.
Table 1.
List of infections with associated human genetic variation
| Disease | Associated genetic element | Study methodology | Disease | Reference |
|---|---|---|---|---|
| COVID-19 | SLC6A20, LZTFL1, CCR9, FYCO1, CXCR6, XCR1, ABO, OAS, TYK2, DPP9, IFNAR2, CCR2 | GWAS | COVID-19 | [15, 31, 32] |
| AIDS | CCR5 | GWAS, PCR, PCR-RFLP | AIDS | [33, 34] |
| Hepatitis | IL28B | GWAS | Hepatitis | [35–37] |
| Hepatitis | Different HLA types, TLR-3, TLR-9, NTCP | GWAS | Hepatitis | [25, 38–43] |
| Dengue | MICB, TNF, CD209, FcγRIIA, TPSAB1, CLEC5A, IL10, PLCE1 | GWAS | Dengue | [44–46] |
| Malaria | TLRs, TNFs, HBB, ABO, ATP2B4 | PCR-RFLP and sequencing, PCR, snp directed seq, GWAS | Malaria | [47–52] |
| Tuberculosis | TLRs, IFN-γ, AGMO, FOXP1, UBLCP1 | GWAS, Candidate gene approach | Tuberculosis | [53–58] |
| Leprosy | IL10, PACRG, NOD2, HLA-DRB1/DQA1, LTA, GATA3, IFNG, TLR1 | Case control, GWAS | Leprosy | [30, 59–64] |
| Meningococcal disease | CEACAM, SPLUNC1, CFH/CFHR3, IL-1, Compliment factors | Candidate gene approach, GWAS | Meningococcal disease | [65–69] |
| Creutzfeldtjakob disease | PRNP | GWAS | Creutzfeldt Jakob disease | [70] |
| Pneumonia | MBL2, CD14, IRAK-4, MyD88, TIRAP | Candidate gene approach, GWAS | Pneumonia | [71–76] |
| MSMD | IFNGR1, IFNGR2, STAT1, IL12B, IL12RB1, ISG15, IRF8, NEMO, CYBB | Candidate gene approach, GWAS | MSMD | [77–80] |
| Cold Sores | TLR3, TRAF3, UNC93B1, KIF1B | Candidate gene approach, GWAS | Cold Sores | [81–84] |
| Warts, Cervical cancer | CXCL12, KLF12, NR5A2, MIR365, ARRDC3 | Case control, GWAS | Warts, Cervical cancer | [85–87] |
| Gastroenteritis | FUT2, FUT3, ABH | Candidate gene approach, GWAS | Gastroenteritis | [88–90] |
| Candidiasis | TLR1, TLR3, Dectin-1, CARD9, STAT1 | GWAS, Candidate gene approach, mice model | Candidiasis | [91–94] |
| Skin/respiratory Infections | DAPK3, XRN1, IL4, DEFB1, CRP, VDR | GWAS, Mice Models | Skin/respiratory Infections | [95–100] |
| Whipple’s disease | HLA-B27, IRF4 | GWAS | Whipple’s disease | [101–103] |
| Infectious mononucleosis, cancers | MDC1, RAD54L, TP53BP1, RPA1, LIG3 | Candidate gene approach, Genotyping | Infectious mononucleosis, cancers | [104, 105] |
| Influenza | IFITM3, IRF7, TMPRSS2, TLR3 | PCR amplification and sequencing, GWAS | Influenza | [106–110] |
| Mononucleosis, pneumonia | TLRs, MBL | PCR-RFLP and sequencing, | Mononucleosis, pneumonia | [111–115] |
| Respiratory infections | IFN-γ, IL-4, SLC39A1 | GWAS, Case control studies | Respiratory infections | [116–119] |
Host components and COVID-19
The current COVID19 pandemic is characterized by complexity of clinical phenotypes, with the majority of severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) infections being asymptomatic or mild (Figure 1). In brief, we are making an effort toward threading the available literature on host factors and clinical characteristics leading to variability in disease outcome of COVID-19.
Figure 1 .
COVID-19 and host association. The wealth of literature and the discoveries highlight the multicomponent role of the differential factors and their role in the current COVID-19 pandemic. The subcomponents of the major factors have been captured in the figure.
Ethnic diversity, one of the fundamental host population characteristics, has a role toward susceptibility in pathogenic diseases, even in the case of COVID-19. Blacks, South Asian populations such as Pakistanis have an elevated risk of contracting COVID-19 and are highly probable toward SARS-CoV-2 infection, as highlighted in the UK-based population study. It is important to note that Niedzwiedz et al. [120] also observed increased association of higher risk of infection (without hospitalization) in those who are socioeconomically disadvantaged and with no qualifications. At the same time, adjustment for the above factors only led to modest attenuation for the hospital cases. The meta-study from 49 562 COVID-19 patients from 46 studies across 19 countries worldwide associated and categorized the mutations according to their prevalence in COVID-19 diagnosed, hospitalized and critical cases. The study also reported 13 SNPs with significant associations to the genes: SLC6A20, LZTFL1, RPL24, FOXP4, TMEM65, ABO, OAS1, KANSL1, TAC4, DPP9, RAVER1, PLEKHA4 and IFNAR2 [121]. Moreover, the 3p21.31 gene cluster is associated in COVID-19 and it increases susceptibility to severe disease manifestation [15].
Other determinants such as age, gender and comorbidities are also shown to modulate the clinical variability of COVID-19. These factors are looked primarily from the standpoint of human SARS-CoV-2 receptors angiotensin-converting enzyme 2 (ACE2), entry point for SARS-CoV-2 and a directly associated serine protease involved in SARS spike protein cleavage-Transmembrane protease, serine (TMPRSS). In older patients, higher severity and mortality of the disease was reported and explained by age-related dynamics of host factor expression of ACE2 [122]. A 10-year increase in age showed a 1.2-fold increase in ACE2 expression. A lower nasal epithelial ACE2 expression and COVID-19 prevalence is reported in children [123, 124], although no decrease in ACE2 protein levels is reported in children. Moreover, this observed correlation needs further investigation, as some studies have found no correlation between ACE2 expression and COVID-19 pulmonary risk factors among children [125].
In contrast to the above cited examples, large population studies with severe COVID-19 evaluated independent risk factors such as male gender, asthma, cardiovascular disease, chronic obstructive pulmonary disorder (COPD), diabetes and smoking, compared to age-matched healthy controls. The findings indicate no significant difference in ACE2 localization [125–127]. However, the role of ACE2 variation in susceptibility to SARS infection needs further studies and investigations as similar associations with entry receptors have been reported during other viral diseases such as MERS [128] and HIV [129]. Additionally, SNPs in the entry receptor (ACE2) and TMPRSS2 may also affect the contrasting reports.
Studies showed that there is no correlation between ACE2 polymorphisms and COVID-19 susceptibility [130, 131]. Although, the increased frequencies of ACE2 SNPs (rs4830542, rs4240157, rs2074192, rs233575 and rs879922) in the European and the admixed American population have been associated with severe illness, while lower frequencies of the same confers protection in East and South Asian populations [132]. The SNP rs2285666 has been associated with different disease outcomes in the same population [133]. The G allele is associated with increased infection and fatality risk, whereas the A allele has been reported in milder cases of infection [134].
Few SNPs, rs73635825 (S19P) and rs143936283 (E329G), were observed in an in silico study [135]. Although numerous studies have reported the association of SNP’s in the ACE2 gene and COVID outcome, the functional role of these SNPs has been studied by Hashizume et al. [136]. The study reported that seven globally identified SNPs had no effect on gene expression levels of ACE2 or virus infectivity. The SARS-CoV-2 spike protein is activated by cathepsin-mediated or II transmembrane serine proteases (TTSPs) mediated cleavage, thus leading to spike protein binding and viral entry into the host cell. A study has identified two Expression quantitative trait loci (eQTL) (rs12329760 and rs75603675) that may confer COVID-19 susceptibility differences using the QTLbase database [137].
Seeing the non-reproducibility of SNP studies among different ethnic groups, screening of ACE2 and TMPRSS2 SNPs in specific populations could be something important to explore in the future. These variabilities indicate and necessitate further studies to identify the involvement of other host factors modulating COVID-19 susceptibility and severity.
Gene expression differences driven by noncoding genetic diversity lead to inter-individual variability in pathogenic diseases
Other than genetic diversity in the coding region, a major factor contributing to phenotypic diversity among the human populations is the difference in gene expression levels [138]. Further, the variation in gene expression levels is also attributed to the genetic diversity in the noncoding region of the genome. In this section, we discuss in detail the functional role played by the genetic variations in the noncoding region of the genome which has functional role in modulating the downstream gene expression. This is corroborated by the fact that most of the GWAS studies report that mutations are present in noncoding regions [139]. A 17% difference in gene expression was observed between African and European populations [140], which are also subsequently validated in larger studies as well [141]. The highly conserved genomic regions inclusive of enhancer, promoters and transcription factor binding sites, among others, are genetic factors that modulate gene expression. Transcriptome-wide association studies (TWAS) functionally annotate the effect of SNPs at the transcriptional level and identify regulatory SNPs called eQTLs [142, 143]. For COVID-19, the study [32] identified SNPs rs10735079, rs74956615, rs2109069 and rs2236757 by GWAS. Then, using TWAS the same group associated disease severity with increased expression of oligoadenylatesynthetase 3 (OAS3) and with C-C chemokine receptor type 2 (CCR2) around rs10735079 and rs1138594, respectively, in lung tissue [15]. Similarly, in latent tuberculosis, SNP rs62292160 was shown to increase the expression of IL4 [144]. In Sporadic Creutzfeldt–Jakob disease (CJD), increased expression of STX6 in multiple brain regions was associated with the risk of disease contraction. The SNPs rs12754041, rs10797664 and rs6425657, each in strong linkage disequilibrium with the SNP rs3747957, showed a high probability of being causal [145]. The advancement in genomic tools has made it possible to identify and find associated elements with gene expression levels. This paves the way to discover the functional mechanisms causing the underlying alterations and thereby, aid in targeting and developing therapeutics.
Beyond gene expression: RNA maturation and transposable elements in infectious disease outcome
Various studies have reported the differential expression of cytokine, chemokine and interferon genes and their possible association with disease severity, in case of sepsis, and MTB infection [146–148]. This leads us to think whether the differential gene expression is the sole determinant of disease severity? Of the plausible other factors with functional role in disease severity, alternate transcripts and noncoding RNA seems to be important modulators.
One of the crucial steps in RNA maturation is alternative splicing (AS) that allows the retention of exons, or parts of exons, and introns in mature transcripts and causes the proteome diversity expansion. Infections can cause global changes in the alternate splicing pattern, which could be due to intrinsic factors such as polymorphism at the splice sites, signaling events or due to direct intervention by virulence factors. Recent studies have shown a change in AS landscape in host cells during a viral infection [149]. In vitro studies of HIV infection identified alternative conformations for the HIV-1 Rev-responsive element (RRE) and 5′ untranslated region (UTR), increasing the possibility of alternative structures playing an important part in the transport of viral RNA from nuclease and in its subsequent packaging in virions [150]. For better understanding of the fundamental question, if the RNA structure affects splicing, it is possibly pertinent to distinguish multiple conformations for the same sequence in cells. The expression of genes in HIV-1 from the same primary transcript is aided by the ability of RNA to form alternative conformations at critical splice sites [151].
Transposable elements, particularly L1 and Alu elements, are capable of introducing novel splice sites [152]. Indeed, Alu insertions into a gene introduces both splice acceptor and donor sites, and thereby holds potential of creating new exons [153]. Most Alu-derived exons undergo AS, contributing to transcript diversity. Moreover, mRNA translation is regulated by the enriched presence of Alu in the 5′UTR of human genes. Additionally, numerous AS events of Alu-derived exons are tissue-specific, possibly suggesting TE contribution to cell type, defined by transcriptome differences [154–156]. Human genes make use of alternative polyadenylation (polyA) sites, and TEs render the cues for some of these events, suggesting the role of TEs in regulating the 3′ end processing of host transcripts. Transcript diversity is further promoted by TEs, by providing alternative promoters for host genes. High-throughput techniques have manifested the all-round role of TEs as alternative gene promoters, and their contribution to tissue-specific expression profiles in normal tissues.
Several mechanisms, including genetic and epigenetic pathways, are known for intronic Alu-mediated gene expression. An intronic Alu polymorphism within the ACE gene was shown to be associated with the SARS-CoV-2 infection severity and morbidity [157, 158]. Many studies have highlighted Alus as a key modulator of gene expression with involvement in diverse physiological processes [159, 160]. Knowing this, along with the knowledge of human demographics, the role Alu polymorphisms in the host response to SARS-CoV-2 infection becomes worthy to consider, especially the Alu polymorphism in the key genes for immune response. The retrotransposition of Line1into chromosomal DNA may result in genomic instability, whereas reverse transcription in the cytosol may activate innate immune sensors [161]. Jones et al. [161] proved that HIV-1 infection enhances L1 retrotransposition in Jurkat cells in a Vif- and Vpr-dependent manner. They also reported extrachromosomal L1 DNA buildup in primary CD4+ T cells as an outcome of HIV-1 infection. These data indicate an unexplored interaction between HIV-1 and endogenous retrotransposable elements, with possible role in the regulation of innate immune response to HIV-1 infection, genomic instability and cytopathicity associated with the infection. Alternate transcripts and transposable elements have been extensively studied with respect to metabolic disorders and in response to stress conditions. Studies focused to elucidate the role of these elements and of noncoding RNA in the context of infectious diseases may be an important area of research to explain and understand the observed diversity of clinical outcome.
Disease severity from the aspect of pathogen
In the previous sections we have discussed the host components modulating the disease outcome. However, the outcome of an infection is determined by the complex interplay of the pathogen and the host immune system. Pathogen virulence and their role in the mortality of the host are also defined by the nature of the pathogen. Hence, the need to understand and factor in the pathogen’s role in disease outcome is highlighted in this section.
M. tuberculosis, the leading cause of infectious disease mortality, is classified into eight lineages (Lineage 1–8) having diverse geographical host associations. The L1–3, L4, and L5-6, and L7 are restricted to Asia, Europe-American, West Africa and Ethiopia-specific populations, respectively [162]. Recent studies have shown the sub-lineages of the L2 and L4 to be associated with higher virulence and are more geographically widespread [163]. This increased virulence is attributed to the delay or decreased host immune response causing enhanced transmission of the infection [164, 165]. Additionally, the severity of the L2 lineage infection when associated with host CD209_336 A/G SNP in patients leads to poorer outcomes and are at increased risk of mortality [166]. Apart from pathogens, virulence factors (VFs) are the critical elements essential in determining both the basis of cellular and molecular pathogenesis. VFs aid in the establishment of colonies [167–169], modulating host mechanisms for replication and survival [170–173] and evasion of immune response [174, 175].
Interestingly, while known to cause asymptomatic infections, H. pylori virulence factors cytotoxin-associated gene A (CagA), vacuolating cytotoxin A (vacA) and blood group antigen binding adhesin (BabA) are associated with the development and severity of diseases including peptic ulcer disease, gastric adenocarcinoma and gastric high-grade B cell lymphoma [176–179]. The chromosomal integrity of the cag-pathogencity (cag-PAI) island or the lack thereof has been associated with pathological progression. Severe pathology is linked with the clinical isolates of H. pylori having deletion or rearrangement in the cagA promoter. Intact cag-PAI was reported to be in the strains from East Asian ancestry than in the European and African strains [180]. This variation may be indicative of the certain subset of populations being susceptible to severe disease outcome, while the remaining population could be predisposed to benign disease condition only, even if infection occurs.
Finally, clinical isolates and/or variants also characterize the infection and the development of disease. Possibly, the most relevant example of the role of variants in the diverse clinical presentation and disease development is that of SARS-CoV-2 and its numerous variant of concern (VOC) induced infection [181, 182]. SARS-CoV-2 virus being a positive sense RNA genome is more prone to genomic modifications, including deletion mutations and SNPs leading to the selection of the virus either toward increased or decreased virulence. The D614G mutation in the surface glycoprotein region along with P323L in the RNA-dependent RNA polymerase (RdRp) is among the few mutations that have become globally predominant. The predominance could be explained by the higher viral load in the host [183] causing increased infectivity by D614G. Mutations have also been associated with the clinical outcome of the disease. A study by Nagy et al. [184] have shown association of five mutations, L84S in the ORF8 protein, L37F in the NSP6 protein, G196V in the ORF3a protein, F308Y in the NSP4 protein and the S197L mutation, in the nucleocapsid phosphoprotein with mild cases. Additionally, they also reported 15 mutations within seven genes: L54F, D614G and V1176F in the surface (S) glycoprotein, A97V and P323L in the RdRp, Q57H and G251V in the ORF3a protein, P13L, S194L, R203K, G204R and I292T in the nucleocapsid phosphoprotein, I33T in the ORF6 protein, S1197R and T1198K mutations in the NSP3 protein are associated with the severe cases of COVID-19.
Continuous evolution of SARS-CoV-2, new mutation and their selection and subsequent emergence of lineages may confer it an evolutionary advantage. Constant and continuous genomic surveillance of mutations will not only be extremely useful in keeping track of viral evolution but also would be resourceful in the development of the vaccines.
Future perspectives
NGS technologies today have accelerated the identifications and characterization of pathogenic organisms, especially detection of emerging variants [185]. The expansion of current techniques to identify and stratify host populations on the basis of infectious disease risk will improve our understanding of host–pathogen interaction and the role of host factors in modulating disease severity.
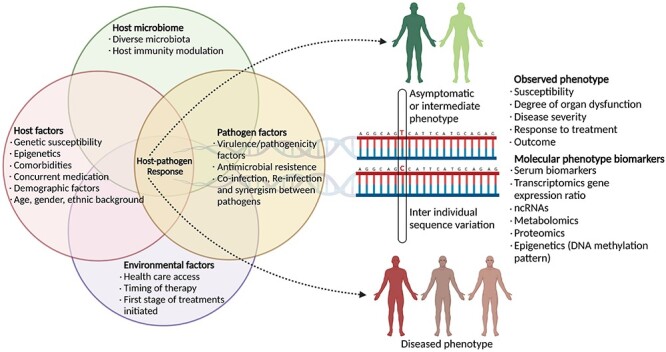
More studies are required to thread together the information across hierarchies and integrate genomic studies of both the pathogen and host along with respective epidemiological, transcriptomic, and clinical information. The threading of all facets including human genetic diversity, genetic elements, genes, noncoding RNA, transposable elements of the host as well as the pathogen, in the bottom-top approach, may link and piece together valuable information (Figure 2). The information thus gathered would aid in the identification of targets for drugs and therapeutics. Furthermore, the formulation of specific hypotheses based on population-wide studies would provide us with anticipated knowledge for experimental testing.
Figure 2.

Key players in delineating clinical outcome of pathogenic diseases. The outcome of the epidemic/pandemic at the local and global level has been modulated by many factors, inclusive of human genetic diversity, transcriptome, noncoding RNA and the pathogen/s genomic architecture. The multicomponent aspect highlights the role of integrative genomics for better understanding of disease severity and mortality.
Key Points
Human genetic variants are an important modulator of the host response to pathogen infection.
Among other factors, the host response at RNA level is shaped by differential expression of genes, alternate transcripts and the noncoding RNA.
The pathogen genome architecture and its ability to elicit or evade immune response is also an important factor.
Integrative Genomics of host-pathogen is integral to understand and elucidate the INTERACTOME shaping disease severity and outcome.
Identification of population subgroups toward susceptibility/protection against infection would help public health decision making.
Acknowledgement
PC acknowledges the CSIR for his Research Fellowship.
Ranjeet Maurya: My research aims to elucidate and understand the role of host response in modulating pathogenicity through application of integrative genomics approaches.
Akshay Kanakan: I am interested in expanding the applications of genomics toward development of Point-of-Care diagnostics.
Janani Srinivasa Vasudevan: I am interested to understand the host–pathogen interaction from the Genomics perspective.
Partha Chattopadhyay: My research is toward elucidating the role of noncoding RNA in modulating responses to pathogen challenge.
Rajesh Pandey: My lab is working toward INtegrative GENomics of HOst-PathogEn (INGEN-HOPE).
Contributor Information
Ranjeet Maurya, INtegrative GENomics of HOst-PathogEn (INGEN-HOPE) laboratory, CSIR-Institute of Genomics and Integrative Biology (CSIR-IGIB), Mall Road, Delhi-110007, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India.
Akshay Kanakan, INtegrative GENomics of HOst-PathogEn (INGEN-HOPE) laboratory, CSIR-Institute of Genomics and Integrative Biology (CSIR-IGIB), Mall Road, Delhi-110007, India.
Janani Srinivasa Vasudevan, INtegrative GENomics of HOst-PathogEn (INGEN-HOPE) laboratory, CSIR-Institute of Genomics and Integrative Biology (CSIR-IGIB), Mall Road, Delhi-110007, India.
Partha Chattopadhyay, INtegrative GENomics of HOst-PathogEn (INGEN-HOPE) laboratory, CSIR-Institute of Genomics and Integrative Biology (CSIR-IGIB), Mall Road, Delhi-110007, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India.
Rajesh Pandey, INtegrative GENomics of HOst-PathogEn (INGEN-HOPE) laboratory, CSIR-Institute of Genomics and Integrative Biology (CSIR-IGIB), Mall Road, Delhi-110007, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India.
Funding support
This research was funded by the Council of Scientific & Industrial Research (CSIR) [Project code: MLP-2005]; Fondation Botnar [Project code: CLP-0031]; Indo-US Science and Technology Forum (IUSSTF) [Project code: CLP-0033]; and Intel Corporation [Project code: CLP-0034]; Bill and Melinda Gates Foundation [Project code: CLP-0036].
Conflict of interest
Authors wish to declare to have no conflict of interest.
References
- 1.The top 10 causes of death . https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death(14 Jun 2021, date last accessed).
- 2.GBD 2016 Diarrhoeal Disease Collaborators . Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect Dis 2018;18:1211–28. 10.1016/S1473-3099(18)30362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michaud CM. Global burden of infectious diseases. In: Encyclopedia of Microbiology. Elsevier; 2009. 444–54. 10.1016/B978-012373944-5.00185-1 [DOI] [Google Scholar]
- 4.GBD 2015 LRI Collaborators . Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory tract infections in 195 countries: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect Dis 2017;17:1133–61. 10.1016/S1473-3099(17)30396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morens DM, Folkers GK, Fauci AS. The challenge of emerging and re-emerging infectious diseases. Nature 2004;430:242–9. 10.1038/nature02759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.COVID-19 Host Genetics Initiative . The COVID-19 host genetics initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet 2020;28:715–8. 10.1038/s41431-020-0636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbujani G, Magagni A, Minch E, et al. An apportionment of human DNA diversity. Proc Natl Acad Sci U S A 1997;94:4516–9. 10.1073/pnas.94.9.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics 2000;156:297–304. 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allison AC. Protection afforded by sickle-cell trait against subtertian malareal infection. Br Med J 1954;1:290–4. 10.1136/bmj.1.4857.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carter R, Mendis KN. Evolutionary and historical aspects of the burden of malaria. Clin Microbiol Rev 2002;15:564–94. 10.1128/CMR.15.4.564-594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller LH, Mason SJ, Clyde DF, et al. The resistance factor to plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N Engl J Med 1976;295:302–4. 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 12.Murphy PM. Viral exploitation and subversion of the immune system through chemokine mimicry. Nat Immunol 2001;2:116–22. 10.1038/84214. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y, Paxton WA, Wolinsky SM, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med 1996;2:1240–3. 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- 14.Stephens JC, Reich DE, Goldstein DB, et al. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet 1998;62:1507–15. 10.1086/301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Severe Covid-19 GWAS Group, Ellinghaus D, Degenhardt F, et al. Genomewide association study of severe Covid-19 with respiratory failure. N Engl J Med 2020;383:1522–34. 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhuri A, DasAdhikary CR. Possible role of blood-group secretory substances in the aetiology of cholera. Trans R Soc Trop Med Hyg 1978;72:664–5. 10.1016/0035-9203(78)90031-7. [DOI] [PubMed] [Google Scholar]
- 17.Levine MM, Nalin DR, Rennels MB, et al. Genetic susceptibility to cholera. Ann Hum Biol 1979;6:369–74. 10.1080/03014467900003751. [DOI] [PubMed] [Google Scholar]
- 18.Black RE, Levine MM, Clements ML, et al. Association between O blood group and occurrence and severity of diarrhoea due to Escherichia coli. Trans R Soc Trop Med Hyg 1987;81:120–3. 10.1016/0035-9203(87)90302-6. [DOI] [PubMed] [Google Scholar]
- 19.Zietz M, Zucker J, Tatonetti NP. Associations between blood type and COVID-19 infection, intubation, and death. Nat Commun 2020;11:5761. 10.1038/s41467-020-19623-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saba R, Booth SA. The genetics of susceptibility to variant Creutzfeldt-Jakob disease. Public Health Genomics 2013;16:17–24. 10.1159/000345203. [DOI] [PubMed] [Google Scholar]
- 21.Gedda L, Rajani G, Brenci G, et al. Heredity and infectious diseases: a twin study. Acta Genet Med Gemellol (Roma) 1984;33:497–500. 10.1017/s000156600000595x. [DOI] [PubMed] [Google Scholar]
- 22.Comstock GW. Tuberculosis in twins: a re-analysis of the Prophit survey. Am Rev Respir Dis 1978;117:621–4. 10.1164/arrd.1978.117.4.621. [DOI] [PubMed] [Google Scholar]
- 23.Lin TM, Chen CJ, Wu MM, et al. Hepatitis B virus markers in Chinese twins. Anticancer Res 1989;9:737–41. [PubMed] [Google Scholar]
- 24.Frodsham AJ, Zhang L, Dumpis U, et al. Class II cytokine receptor gene cluster is a major locus for hepatitis B persistence. Proc Natl Acad Sci U S A 2006;103:9148–53. 10.1073/pnas.0602800103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z, Wang C, Liu Z, et al. Host genetic determinants of hepatitis B virus infection. Front Genet 2019;10:696. 10.3389/fgene.2019.00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu L, Zhai X, Liu J, et al. Genetic variants in human leukocyte antigen/DP-DQ influence both hepatitis B virus clearance and hepatocellular carcinoma development. Hepatology 2012;55:1426–31. 10.1002/hep.24799. [DOI] [PubMed] [Google Scholar]
- 27.Ozaki K, Ohnishi Y, Iida A, et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet 2002;32:650–4. 10.1038/ng1047. [DOI] [PubMed] [Google Scholar]
- 28.MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017;45:D896–901. 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett SN, Caporaso N, Fitzpatrick AL, et al. Phenotype harmonization and cross-study collaboration in GWAS consortia: the GENEVA experience. Genet Epidemiol 2011;35:159–73. 10.1002/gepi.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cambri G, Mira MT. Genetic susceptibility to leprosy-from classic immune-related candidate genes to hypothesis-free. Whole Genome Approaches Front Immunol 2018;9:1674. 10.3389/fimmu.2018.01674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020;370:eabd4570. 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pairo-Castineira E, Clohisey S, Klaric L, et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021;591:92–8. 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 33.Ni J, Wang D, Wang S. The CCR5-Delta32 genetic polymorphism and HIV-1 infection susceptibility: a meta-analysis. Open Med (Wars) 2018;13:467–74. 10.1515/med-2018-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLaren PJ, Coulonges C, Bartha I, et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc Natl Acad Sci U S A 2015;112:14658–63. 10.1073/pnas.1514867112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coppola N, Pisaturo M, Sagnelli C, et al. Role of genetic polymorphisms in hepatitis C virus chronic infection. World J Clin Cases 2015;3:807–22. 10.12998/wjcc.v3.i9.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu AH, Khalili M. The birth and potential death of IL28B genotyping for hepatitis C therapy. Pers Med 2015;12:55–7. 10.2217/pme.14.60. [DOI] [PubMed] [Google Scholar]
- 37.Rauch A, Kutalik Z, Descombes P, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology 2010;138:1338–451345.e1. 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 38.Miao F, Sun H, Pan N, et al. Association of human leukocyte antigen class I polymorphism with spontaneous clearance of hepatitis B surface antigen in Qidong Han population. Clin Dev Immunol 2013;2013:145725. 10.1155/2013/145725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seshasubramanian V, Soundararajan G, Ramasamy P. Human leukocyte antigen A, B and hepatitis B infection outcome: a meta-analysis. Infect Genet Evol 2018;66:392–8. 10.1016/j.meegid.2017.07.027. [DOI] [PubMed] [Google Scholar]
- 40.Kamatani Y, Wattanapokayakit S, Ochi H, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet 2009;41:591–5. 10.1038/ng.348. [DOI] [PubMed] [Google Scholar]
- 41.Wang P, Mo R, Lai R, et al. Genetic variations of NTCP are associated with susceptibility to HBV infection and related hepatocellular carcinoma. Oncotarget 2017;8:105407–24. 10.18632/oncotarget.22211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Qahtani A, Al-Ahdal M, Abdo A, et al. Toll-like receptor 3 polymorphism and its association with hepatitis B virus infection in Saudi Arabian patients. J Med Virol 2012;84:1353–9. 10.1002/jmv.23271. [DOI] [PubMed] [Google Scholar]
- 43.He D, Tao S, Guo S, et al. Interaction of TLR-IFN and HLA polymorphisms on susceptibility of chronic HBV infection in southwest Han Chinese. Liver Int 2015;35:1941–9. 10.1111/liv.12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xavier-Carvalho C, Cardoso CC, Souza KF, et al. Host genetics and dengue fever. Infect Genet Evol 2017;56:99–110. 10.1016/j.meegid.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 45.Khor CC, Chau TNB, Pang J, et al. Genome-wide association study identifies susceptibility loci for dengue shock syndrome at MICB and PLCE1. Nat Genet 2011;43:1139–41. 10.1038/ng.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pare G, Neupane B, Eskandarian S, et al. Genetic risk for dengue hemorrhagic fever and dengue fever in multiple ancestries. EBioMedicine 2020;51:102584. 10.1016/j.ebiom.2019.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mendonça VRR, Goncalves MS, Barral-Netto M. The host genetic diversity in malaria infection. J Trop Med 2012;2012:1. 10.1155/2012/940616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basu M, Maji AK, Chakraborty A, et al. Genetic association of toll-like-receptor 4 and tumor necrosis factor-alpha polymorphisms with plasmodium falciparum blood infection levels. Infect Genet Evol 2010;10:686–96. 10.1016/j.meegid.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 49.Leoratti FMS, Farias L, Alves FP, et al. Variants in the toll-like receptor signaling pathway and clinical outcomes of malaria. J Infect Dis 2008;198:772–80. 10.1086/590440. [DOI] [PubMed] [Google Scholar]
- 50.Clark TG, Diakite M, Auburn S, et al. Tumor necrosis factor and lymphotoxin-alpha polymorphisms and severe malaria in African populations. J Infect Dis 2009;199:569–75. 10.1086/596320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohashi J, Naka I, Patarapotikul J, et al. A single-nucleotide substitution from C to T at position −1055 in the IL-13 promoter is associated with protection from severe malaria in Thailand. Genes Immun 2003;4:528–31. 10.1038/sj.gene.6364010. [DOI] [PubMed] [Google Scholar]
- 52.Malaria Genomic Epidemiology Network . Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and Oceania. Nat Commun 2019;10:5732. 10.1038/s41467-019-13480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tong H, Velavan TP, Thye T, et al. Human genetic factors in tuberculosis: an update. Trop Med Int Health 2017;22:1063–71. 10.1111/tmi.12923. [DOI] [PubMed] [Google Scholar]
- 54.Thye T, Owusu-Dabo E, Vannberg FO, et al. Common variants at 11p13 are associated with susceptibility to tuberculosis. Nat Genet 2012;44:257–9. 10.1038/ng.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thye T, Vannberg FO, Wong SH, et al. Genome-wide association analyses identifies a susceptibility locus for tuberculosis on chromosome 18q11.2. Nat Genet 2010;42:739–41. 10.1038/ng.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grant AV, Sabri A, Abid A, et al. A genome-wide association study of pulmonary tuberculosis in Morocco. Hum Genet 2016;135:299–307. 10.1007/s00439-016-1633-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sobota RS, Stein CM, Kodaman N, et al. A locus at 5q33.3 confers resistance to tuberculosis in highly susceptible individuals. Am J Hum Genet 2016;98:514–24. 10.1016/j.ajhg.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang D, Xia M, Cui Z. New triterpenoids isolated from the root bark of Ulmus pumila L. Chem Pharm Bull 2006;54:775–8. 10.1248/cpb.54.775. [DOI] [PubMed] [Google Scholar]
- 59.Uaska Sartori PV, Penna GO, Bührer-Sékula S, et al. Human genetic susceptibility of leprosy recurrence. Sci Rep 2020;10:1284. 10.1038/s41598-020-58079-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fitness J, Tosh K, Hill AVS. Genetics of susceptibility to leprosy. Genes Immun 2002;3:441–53. 10.1038/sj.gene.6363926. [DOI] [PubMed] [Google Scholar]
- 61.Santos AR, Suffys PN, Vanderborght PR, et al. Role of tumor necrosis factor-alpha and interleukin-10 promoter gene polymorphisms in leprosy. J Infect Dis 2002;186:1687–91. 10.1086/345366. [DOI] [PubMed] [Google Scholar]
- 62.Chopra R, Ali S, Srivastava AK, et al. Mapping of PARK2 and PACRG overlapping regulatory region reveals LD structure and functional variants in association with leprosy in unrelated indian population groups. PLoS Genet 2013;9:e1003578. 10.1371/journal.pgen.1003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grant AV, Alter A, Huong NT, et al. Crohn’s disease susceptibility genes are associated with leprosy in the Vietnamese population. J Infect Dis 2012;206:1763–7. 10.1093/infdis/jies588. [DOI] [PubMed] [Google Scholar]
- 64.Sales-Marques C, Salomão H, Fava VM, et al. NOD2 and CCDC122-LACC1 genes are associated with leprosy susceptibility in Brazilians. Hum Genet 2014;133:1525–32. 10.1007/s00439-014-1502-9. [DOI] [PubMed] [Google Scholar]
- 65.Hodeib S, Herberg JA, Levin M, et al. Human genetics of meningococcal infections. Hum Genet 2020;139:961–80. 10.1007/s00439-020-02128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davila S, Wright VJ, Khor CC, et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet 2010;42:772–6. 10.1038/ng.640. [DOI] [PubMed] [Google Scholar]
- 67.Callaghan MJ, Rockett K, Banner C, et al. Haplotypic diversity in human CEACAM genes: effects on susceptibility to meningococcal disease. Genes Immun 2008;9:30–7. 10.1038/sj.gene.6364442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Titmarsh CJ, Moscovis SM, Hall S, et al. Comparison of cytokine gene polymorphisms among Greek patients with invasive meningococcal disease or viral meningitis. J Med Microbiol 2013;62:694–700. 10.1099/jmm.0.058073-0. [DOI] [PubMed] [Google Scholar]
- 69.Sanders MS, Well GTJ, Ouburg S, et al. Single nucleotide polymorphisms in TLR9 are highly associated with susceptibility to bacterial meningitis in children. Clin Infect Dis 2011;52:475–80. 10.1093/cid/ciq155. [DOI] [PubMed] [Google Scholar]
- 70.Mead S, Poulter M, Uphill J, et al. Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol 2009;8:57–66. 10.1016/S1474-4422(08)70265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Picard C, Bernuth H, Ghandil P, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine 2010;89:403–25. 10.1097/MD.0b013e3181fd8ec3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khor CC, Chapman SJ, Vannberg FO, et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet 2007;39:523–8. 10.1038/ng1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kloek AT, Brouwer MC, Beek D. Host genetic variability and pneumococcal disease: a systematic review and meta-analysis. BMC Med Genomics 2019;12:130. 10.1186/s12920-019-0572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Endeman H, Herpers BL, Jong BAW, et al. Mannose-binding lectin genotypes in susceptibility to community-acquired pneumonia. Chest 2008;134:1135–40. 10.1378/chest.08-0642. [DOI] [PubMed] [Google Scholar]
- 75.Yuan FF, Marks K, Wong M, et al. Clinical relevance of TLR2, TLR4, CD14 and FcgammaRIIA gene polymorphisms in Streptococcus pneumoniae infection. Immunol Cell Biol 2008;86:268–70. 10.1038/sj.icb.7100155. [DOI] [PubMed] [Google Scholar]
- 76.Kenyan Bacteraemia Study Group, Wellcome Trust Case Control Consortium 2 (WTCCC2), Rautanen A, et al. Polymorphism in a lincRNA associates with a doubled risk of pneumococcal bacteremia in Kenyan children. Am J Hum Genet 2016;98:1092–100. 10.1016/j.ajhg.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol 2014;26:454–70. 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dorman SE, Picard C, Lammas D, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet 2004;364:2113–21. 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 79.Bustamante J, Arias AA, Vogt G, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 2011;12:213–21. 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Newport MJ, Huxley CM, Huston S, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med 1996;335:1941–9. 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- 81.Casrouge A, Zhang S-Y, Eidenschenk C, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006;314:308–12. 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 82.Zhang S-Y, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007;317:1522–7. 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 83.Kleinstein SE, Shea PR, Allen AS, et al. Genome-wide association study (GWAS) of human host factors influencing viral severity of herpes simplex virus type 2 (HSV-2). Genes Immun 2018;20:112–20. 10.1038/s41435-018-0013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang S-Y, Abel L, Casanova J-L. Mendelian predisposition to herpes simplex encephalitis. Handb Clin Neurol 2013;112:1091–7. 10.1016/B978-0-444-52910-7.00027-1. [DOI] [PubMed] [Google Scholar]
- 85.Okuyama NCM, Cezar-Dos-Santos F, Pereira ÉR, et al. Genetic variant in CXCL12 gene raises susceptibility to HPV infection and squamous intraepithelial lesions development: a case-control study. J Biomed Sci 2018;25:69. 10.1186/s12929-018-0472-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Adebamowo SN, Adeyemo AA, Rotimi CN, et al. Genome-wide association study of prevalent and persistent cervical high-risk human papillomavirus (HPV) infection. BMC Med Genet 2020;21:231. 10.1186/s12881-020-01156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takeuchi F, Kukimoto I, Li Z, et al. Genome-wide association study of cervical cancer suggests a role for ARRDC3 gene in human papillomavirus infection. Hum Mol Genet 2019;28:341–8. 10.1093/hmg/ddy390. [DOI] [PubMed] [Google Scholar]
- 88.Nordgren J, Sharma S, Kambhampati A, et al. Innate resistance and susceptibility to norovirus infection. PLoS Pathog 2016;12:e1005385. 10.1371/journal.ppat.1005385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Currier RL, Payne DC, Staat MA, et al. Innate susceptibility to norovirus infections influenced by FUT2 genotype in a United States pediatric population. Clin Infect Dis 2015;60:1631–8. 10.1093/cid/civ165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bustamante M, Standl M, Bassat Q, et al. A genome-wide association meta-analysis of diarrhoeal disease in young children identifies FUT2 locus and provides plausible biological pathways. Hum Mol Genet 2016;25:4127–42. 10.1093/hmg/eddw264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smeekens SP, Veerdonk FL, Kullberg BJ, et al. Genetic susceptibility to Candida infections. EMBO Mol Med 2013;5:805–13. 10.1002/emmm.201201678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Glocker E-O, Hennigs A, Nabavi M, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 2009;361:1727–35. 10.1056/NEJMoa0810719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Veerdonk FL, Plantinga TS, Hoischen A, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 2011;365:54–61. 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- 94.Thompson A, Griffiths JS, Walker L, et al. Dependence on Dectin-1 varies with multiple Candida species. Front Microbiol 2019;10:1800. 10.3389/fmicb.2019.01800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thomson W, Harrison B, Ollier B, et al. Quantifying the exact role of HLA-DRB1 alleles in susceptibility to inflammatory polyarthritis: results from a large, population-based study. Arthritis Rheum 1999;42:757–62. 10.1002/1529-0131(199904)42:4<757::AID-ANR20>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 96.Shukla SK, Rose W, Schrodi SJ. Complex host genetic susceptibility to Staphylococcus aureus infections. Trends Microbiol 2015;23:529–36. 10.1016/j.tim.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 97.Ye Z, Vasco DA, Carter TC, et al. Genome wide association study of SNP-, gene-, and pathway-based approaches to identify genes influencing susceptibility to Staphylococcus aureus infections. Front Genet 2014;5:125. 10.3389/fgene.2014.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ruimy R, Angebault C, Djossou F, et al. Are host genetics the predominant determinant of persistent nasal Staphylococcus aureus carriage in humans? J Infect Dis 2010;202:924–34. 10.1086/655901. [DOI] [PubMed] [Google Scholar]
- 99.Nurjadi D, Herrmann E, Hinderberger I, et al. Impaired β-defensin expression in human skin links DEFB1 promoter polymorphisms with persistent Staphylococcus aureus nasal carriage. J Infect Dis 2013;207:666–74. 10.1093/infdis/jis735. [DOI] [PubMed] [Google Scholar]
- 100.Messaritakis I, Samonis G, Dimopoulou D, et al. Staphylococcus aureus nasal carriage might be associated with vitamin D receptor polymorphisms in type 2 diabetes. Clin Microbiol Infect 2014;20:920–5. 10.1111/1469-0691.12587. [DOI] [PubMed] [Google Scholar]
- 101.Dobbins WO. HLA antigens in Whipple’s disease. Arthritis Rheum 1987;30:102–5. 10.1002/art.1780300115. [DOI] [PubMed] [Google Scholar]
- 102.McKinley R, Grace CS. Whipple’s disease in an HLA-B27 positive female. Aust N Z J Med 1985;15:758–60. [PubMed] [Google Scholar]
- 103.Guérin A, Kerner G, Marr N, et al. IRF4 haploinsufficiency in a family with Whipple’s disease. Elife 2018;7:e32340. 10.7554/eLife.32340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shen G-P, Pan Q-H, Hong M-H, et al. Human genetic variants of homologous recombination repair genes first found to be associated with Epstein-Barr virus antibody titers in healthy Cantonese. Int J Cancer 2011;129:1459–66. 10.1002/ijc.25759. [DOI] [PubMed] [Google Scholar]
- 105.Houldcroft CJ, Kellam P. Host genetics of Epstein-Barr virus infection, latency and disease. Rev Med Virol 2015;25:71–84. 10.1002/rmv.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gounder AP, Boon ACM. Influenza pathogenesis: the effect of host factors on severity of disease. J Immunol 2019; 202:341–50. 10.4049/jimmunol.1801010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Everitt AR, Clare S, Pertel T, et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012;484:519–23. 10.1038/nature10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang Y-H, Zhao Y, Li N, et al. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat Commun 2013;4:1418. 10.1038/ncomms2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cheng Z, Zhou J, To KK-W, et al. Identification of TMPRSS2 as a susceptibility gene for severe 2009 pandemic A(H1N1) influenza and A(H7N9) influenza. J Infect Dis 2015;212:1214–21. 10.1093/infdis/jiv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee N, Cao B, Ke C, et al. IFITM3, TLR3, and CD55 gene snps and cumulative genetic risks for severe outcomes in chinese patients with h7n9/h1n1pdm09 influenza. J Infect Dis 2017;216:97–104. 10.1093/infdis/jix235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hu Y, Wu D, Tao R, et al. Association between mannose-binding lectin gene polymorphism and pediatric cytomegalovirus infection. Viral Immunol 2010;23:443–7. 10.1089/vim.2009.0109. [DOI] [PubMed] [Google Scholar]
- 112.Wujcicka W, Paradowska E, Studzińska M, et al. TLR9 2848 GA heterozygotic status possibly predisposes fetuses and newborns to congenital infection with human cytomegalovirus. PLoS One 2015;10:e0122831. 10.1371/journal.pone.0122831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Taniguchi R, Koyano S, Suzutani T, et al. Polymorphisms in TLR-2 are associated with congenital cytomegalovirus (CMV) infection but not with congenital CMV disease. Int J Infect Dis 2013;17:e1092–7. 10.1016/j.ijid.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 114.Jabłońska A, Paradowska E, Studzińska M, et al. Relationship between toll-like receptor 2 Arg677Trp and Arg753Gln and toll-like receptor 4 Asp299Gly polymorphisms and cytomegalovirus infection. Int J Infect Dis 2014;25:11–5. 10.1016/j.ijid.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 115.Gelemanović A, Dobberpuhl K, Krakar G, et al. Host genetics and susceptibility to congenital and childhood cytomegalovirus infection: a systematic review. Croat Med J 2016;57:321–30. 10.3325/cmj.2016.57.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tahamtan A, Askari FS, Bont L, et al. Disease severity in respiratory syncytial virus infection: role of host genetic variation. Rev Med Virol 2019;29:e2026. 10.1002/rmv.2026. [DOI] [PubMed] [Google Scholar]
- 117.Pasanen A, Karjalainen MK, Bont L, et al. Genome-wide association study of polymorphisms predisposing to bronchiolitis. Sci Rep 2017;7:41653. 10.1038/srep41653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Larkin EK, Hartert TV. Genes associated with RSV lower respiratory tract infection and asthma: the application of genetic epidemiological methods to understand causality. Future Virol 2015;10:883–97. 10.2217/fvl.15.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gentile DA, Doyle WJ, Zeevi A, et al. Cytokine gene polymorphisms moderate illness severity in infants with respiratory syncytial virus infection. Hum Immunol 2003;64:338–44. 10.1016/S0198-8859(02)00827-3. [DOI] [PubMed] [Google Scholar]
- 120.Niedzwiedz CL, O’Donnell CA, Jani BD, et al. Ethnic and socioeconomic differences in SARS-CoV-2 infection: prospective cohort study using UK Biobank. BMC Med 2020;18:160. 10.1186/s12916-020-01640-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.The COVID-19 Host Genetics Initiative, ganna andrea . Mapping the human genetic architecture of COVID-19 by worldwide meta-analysis. medRxiv Published Online First12 March 2021. 10.1101/2021.03.10.21252820preprint: not peer reviewed. [DOI] [Google Scholar]
- 122.Chen J, Jiang Q, Xia X, et al. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell 2020;19:e13168. 10.1111/acel.13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bunyavanich S, Do A, Vicencio A. Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA 2020;323:2427–9. 10.1001/jama.2020.8707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dong Y, Mo X, Hu Y, et al. Epidemiological characteristics of 2143 pediatric patients with 2019 coronavirus disease in China. Pediatrics 2020;145(6):e20200702. Published Online First:16 March 2020. 10.1542/peds.2020-0702. [DOI] [Google Scholar]
- 125.Ortiz ME, Thurman A, Pezzulo AA, et al. Heterogeneous expression of the SARS-Coronavirus-2 receptor ACE2 in the human respiratory tract. EBioMedicine 2020;60:102976. 10.1016/j.ebiom.2020.102976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy region, Italy. JAMA 2020;323:1574–81. 10.1001/jama.2020.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study. Lancet 2020;395:1763–70. 10.1016/S0140-6736(20)31189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kleine-Weber H, Schroeder S, Krüger N, et al. Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus. Emerg Microbes Infect 2020;9:155–68. 10.1080/22221751.2020.1713705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Samson M, Libert F, Doranz BJ, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996;382:722–5. 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 130.Chiu RWK, Tang NLS, Hui DSC, et al. ACE2 gene polymorphisms do not affect outcome of severe acute respiratory syndrome. Clin Chem 2004;50:1683–6. 10.1373/clinchem.2004.035436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Itoyama S, Keicho N, Hijikata M, et al. Identification of an alternative 5′-untranslated exon and new polymorphisms of angiotensin-converting enzyme 2 gene: lack of association with SARS in the Vietnamese population. Am J Med Genet A 2005;136:52–7. 10.1002/ajmg.a.30779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shoily SS, Ahsan T, Fatema K, et al. Disparities in COVID-19 severities and casualties across ethnic groups around the globe and patterns of ACE2 and PIR variants. Infect Genet Evol 2021;92:104888. 10.1016/j.meegid.2021.104888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Möhlendick B, Schönfelder K, Breuckmann K, et al. ACE2 polymorphism and susceptibility for SARS-CoV-2 infection and severity of COVID-19. Pharmacogenet Genomics Published Online First:14 May 2021. 10.1097/FPC.0000000000000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Srivastava A, Bandopadhyay A, Das D, et al. Genetic association of ACE2 rs2285666 polymorphism with COVID-19 spatial distribution in India. Front Genet 2020;11:564741. 10.3389/fgene.2020.564741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hussain M, Jabeen N, Raza F, et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol 2020;92:1580–6. 10.1002/jmv.25832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hashizume M, Gonzalez G, Ono C, et al. Population-specific ACE2 single-nucleotide polymorphisms have limited impact on SARS-CoV-2 infectivity in vitro. Viruses 2021;13:67. 10.3390/v13010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hou Y, Zhao J, Martin W, et al. New insights into genetic susceptibility of COVID-19: an ACE2 and TMPRSS2 polymorphism analysis. BMC Med 2020;18:216. 10.1186/s12916-020-01673-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Idaghdour Y, Storey JD, Jadallah SJ, et al. A genome-wide gene expression signature of environmental geography in leukocytes of Moroccan Amazighs. PLoS Genet 2008;4:e1000052. 10.1371/journal.pgen.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Farh KK-H, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518:337–43. 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Barash A, Machluf Y, Ariel I, et al. The pursuit of COVID-19 biomarkers: putting the spotlight on ACE2 and TMPRSS2 regulatory sequences. Front Med (Lausanne) 2020;7:582793. 10.3389/fmed.2020.582793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stranger BE, Nica AC, Forrest MS, et al. Population genomics of human gene expression. Nat Genet 2007;39:1217–24. 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jin H-J, Jung S, DebRoy AR, et al. Identification and validation of regulatory SNPs that modulate transcription factor chromatin binding and gene expression in prostate cancer. Oncotarget 2016;7:54616–26. 10.18632/oncotarget.10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wainberg M, Sinnott-Armstrong N, Mancuso N, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet 2019;51:592–9. 10.1038/s41588-019-0385-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ai J-W, Zhang H, Zhou Z, et al. Gene expression pattern analysis using dual-color RT-MLPA and integrative genome-wide association studies of eQTL for tuberculosis suscepitibility. Respir Res 2021;22:23. 10.1186/s12931-020-01612-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jones E, Hummerich H, Viré E, et al. Identification of novel risk loci and causal insights for sporadic Creutzfeldt-Jakob disease: a genome-wide association study. Lancet Neurol 2020;19:840–8. 10.1016/S1474-4422(20)30273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sutherland JS, Loxton AG, Haks MC, et al. Differential gene expression of activating Fcγ receptor classifies active tuberculosis regardless of human immunodeficiency virus status or ethnicity. Clin Microbiol Infect 2014;20:O230–8. 10.1111/1469-0691.12383. [DOI] [PubMed] [Google Scholar]
- 147.Giacomini E, Iona E, Ferroni L, et al. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol 2001;166:7033–41. 10.4049/jimmunol.166.12.7033. [DOI] [PubMed] [Google Scholar]
- 148.Liepelt A, Hohlstein P, Gussen H, et al. Differential gene expression in circulating CD14+ monocytes indicates the prognosis of critically ill patients with sepsis. J Clin Med 2020;9:127. 10.3390/jcm9010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Boudreault S, Roy P, Lemay G, et al. Viral modulation of cellular RNA alternative splicing: a new key player in virus-host interactions? Wiley Interdiscip Rev RNA 2019;10:e1543. 10.1002/wrna.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sherpa C, Rausch JW, Le Grice SFJ, et al. The HIV-1 rev response element (RRE) adopts alternative conformations that promote different rates of virus replication. Nucleic Acids Res 2015;43:4676–86. 10.1093/nar/gkv313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tomezsko PJ, Corbin VDA, Gupta P, et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 2020;582:438–42. 10.1038/s41586-020-2253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li WH, Gu Z, Wang H, et al. Evolutionary analyses of the human genome. Nature 2001;409:847–9. 10.1038/35057039. [DOI] [PubMed] [Google Scholar]
- 153.Lev-Maor G, Sorek R, Shomron N, et al. The birth of an alternatively spliced exon: 3′ splice-site selection in Alu exons. Science 2003;300:1288–91. 10.1126/science.1082588. [DOI] [PubMed] [Google Scholar]
- 154.Sorek R, Ast G, Graur D. Alu-containing exons are alternatively spliced. Genome Res 2002;12:1060–7. 10.1101/gr.229302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lin L, Shen S, Tye A, et al. Diverse splicing patterns of exonized Alu elements in human tissues. PLoS Genet 2008;4:e1000225. 10.1371/journal.pgen.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature 2012;489:101–8. 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Delanghe JR, Speeckaert MM, De Buyzere ML. The host’s angiotensin-converting enzyme polymorphism may explain epidemiological findings in COVID-19 infections. Clin Chim Acta 2020;505:192–3. 10.1016/j.cca.2020.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hatami N, Ahi S, Sadeghinikoo A, et al. Worldwide ACE (I/D) polymorphism may affect COVID-19 recovery rate: an ecological meta-regression. Endocrine 2020;68:479–84. 10.1007/s12020-020-02381-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Deininger P. Alu elements: know the SINEs. Genome Biol 2011;12:236. 10.1186/gb-2011-12-12-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pandey R, Mukerji M. From “JUNK” to just unexplored noncoding knowledge: the case of transcribed Alus. Brief Funct Genomics 2011;10:294–311. 10.1093/bfgp/elr029. [DOI] [PubMed] [Google Scholar]
- 161.Jones RB, Song H, Xu Y, et al. LINE-1 retrotransposable element DNA accumulates in HIV-1-infected cells. J Virol 2013;87:13307–20. 10.1128/JVI.02257-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Galagan JE. Genomic insights into tuberculosis. Nat Rev Genet 2014;15:307–20. 10.1038/nrg3664. [DOI] [PubMed] [Google Scholar]
- 163.Gagneux S. Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci 2012;367:850–9. 10.1098/rstb.2011.0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Krishnan N, Malaga W, Constant P, et al. Mycobacterium tuberculosis lineage influences innate immune response and virulence and is associated with distinct cell envelope lipid profiles. PLoS One 2011;6:e23870. 10.1371/journal.pone.0023870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sarkar R, Lenders L, Wilkinson KA, et al. Modern lineages of Mycobacterium tuberculosis exhibit lineage-specific patterns of growth and cytokine induction in human monocyte-derived macrophages. PLoS One 2012;7:e43170. 10.1371/journal.pone.0043170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ogarkov O, Mokrousov I, Sinkov V, et al. Lethal ’ combination of Mycobacterium tuberculosis Beijing genotype and human CD209-336G allele in Russian male population. Infect Genet Evol 2012;12:732–6. 10.1016/j.meegid.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 167.Tram TTB, Nhung HN, Vijay S, et al. Virulence of Mycobacterium tuberculosis clinical isolates is associated with sputum pre-treatment bacterial load, lineage, survival in macrophages, and cytokine response. Front Cell Infect Microbiol 2018;8:417. 10.3389/fcimb.2018.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Song Y, Yang C, Chen G, et al. Molecular insights into the master regulator CysB-mediated bacterial virulence in Pseudomonas aeruginosa. Mol Microbiol 2019;111:1195–210. 10.1111/mmi.14200. [DOI] [PubMed] [Google Scholar]
- 169.Hotomi M, Yuasa J, Briles DE, et al. Pneumolysin plays a key role at the initial step of establishing pneumococcal nasal colonization. Folia Microbiol (Praha) 2016;61:375–83. 10.1007/s12223-016-0445-z. [DOI] [PubMed] [Google Scholar]
- 170.Crofts AA, Giovanetti SM, Rubin EJ, et al. Enterotoxigenic E. coli virulence gene regulation in human infections. Proc Natl Acad Sci U S A 2018;115:E8968–76. 10.1073/pnas.1808982115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bachert C, Holtappels G, Merabishvili M, et al. Staphylococcus aureus controls interleukin-5 release in upper airway inflammation. J Proteomics 2018;180:53–60. 10.1016/j.jprot.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 172.Singh N, Kumar A. Virulence factor SenX3 is the oxygen-controlled replication switch of Mycobacterium tuberculosis. Antioxid Redox Signal 2015;22:603–13. 10.1089/ars.2014.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bomjan R, Zhang M, Zhou D. Yshb promotes intracellular replication and is required for salmonella virulence. J Bacteriol 2019;201:e00314-19. 10.1128/JB.00314-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bhattacharya M, Berends ETM, Chan R, et al. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U S A 2018;115:7416–21. 10.1073/pnas.1721949115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dieudonné-Vatran A, Krentz S, Blom AM, et al. Clinical isolates of Streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a PspC allele-dependent fashion. J Immunol 2009;182:7865–77. 10.4049/jimmunol.0802376. [DOI] [PubMed] [Google Scholar]
- 176.Loh JT, Shaffer CL, Piazuelo MB, et al. Analysis of cagA in Helicobacter pylori strains from Colombian populations with contrasting gastric cancer risk reveals a biomarker for disease severity. Cancer Epidemiol Biomarkers Prev 2011;20:2237–49. 10.1158/1055-9965.EPI-11-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rhead JL, Letley DP, Mohammadi M, et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007;133:926–36. 10.1053/j.gastro.2007.06.056. [DOI] [PubMed] [Google Scholar]
- 178.Chen M-Y, He C-Y, Meng X, et al. Association of Helicobacter pylori babA2 with peptic ulcer disease and gastric cancer. World J Gastroenterol 2013;19:4242–51. 10.3748/wjg.v19.i26.4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sheu S-M, Sheu B-S, Chiang W-C, et al. H. pylori clinical isolates have diverse babAB genotype distributions over different topographic sites of stomach with correlation to clinical disease outcomes. BMC Microbiol 2012;12:89. 10.1186/1471-2180-12-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Kauser F, Khan AA, Hussain MA, et al. The cag pathogenicity island of Helicobacter pylori is disrupted in the majority of patient isolates from different human populations. J Clin Microbiol 2004;42:5302–8. 10.1128/JCM.42.11.5302-5308.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Challen R, Brooks-Pollock E, Read JM, et al. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ 2021;372:n579. 10.1136/bmj.n579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Toyoshima Y, Nemoto K, Matsumoto S, et al. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J Hum Genet 2020;65:1075–82. 10.1038/s10038-020-0808-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Volz E, Hill V, McCrone JT, et al. Evaluating the effects of SARS-CoV-2 spike mutation D614G on transmissibility and pathogenicity. Cell 2021;184:64–75.e11. 10.1016/j.cell.2020.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Nagy Á, Pongor S, Győrffy B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int J Antimicrob Agents 2021;57:106272. 10.1016/j.ijantimicag.2020.106272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Frey KG, Bishop-Lilly KA. Current and Emerging Technologies for the Diagnosis of Microbial Infections. Elsevier; 2015. 525–54. 10.1016/bs.mim.2015.06.00442Methods in MicrobiologyNext-Generation Sequencing for Pathogen Detection and Identification [DOI] [Google Scholar]