

Abstract
Infections are a major threat to human reproductive health, and infections in pregnancy can cause prematurity or stillbirth, or can be vertically transmitted to the fetus leading to congenital infection and severe disease. The acronym ‘TORCH’ (Toxoplasma gondii, other, rubella virus, cytomegalovirus, herpes simplex virus) refers to pathogens directly associated with the development of congenital disease and includes diverse bacteria, viruses and parasites. The placenta restricts vertical transmission during pregnancy and has evolved robust mechanisms of microbial defence. However, microorganisms that cause congenital disease have likely evolved diverse mechanisms to bypass these defences. In this Review, we discuss how TORCH pathogens access the intra-amniotic space and overcome the placental defences that protect against microbial vertical transmission.
Subject terms: Bacteria, Virology, Parasitology, Antimicrobial responses, Infectious diseases
Infections during pregnancy can be associated with devastating outcomes for the pregnant mother and developing fetus. In this Review, Megli and Coyne discuss placental defences and provide an overview of how various viral, bacterial and parasitic pathogens traverse the maternal–fetal interface and cause disease.
Introduction
Infections during pregnancy can be associated with devastating consequences to the pregnant mother and developing fetus. Vertical transmission, defined as infection of the fetus from the maternal host, is a major cause of morbidity and mortality in pregnancy. In some cases, bacterial, viral and parasitic infections induce dire outcomes in the fetus. The sequelae of infections in pregnancy include teratogenic effects, which cause congenital anomalies; growth restriction, stillbirth, miscarriage and neonatal death; prematurity; and maternal morbidity (Fig. 1). Vertical transmission of pathogens across the maternal–fetal interface can cause fetal infection, which can disrupt organogenesis and is associated with congenital anomalies of every major organ system (Table 1). Congenital anomalies are present in ~3% of live births and the proportion attributable to infection is poorly characterized1–3.
Fig. 1. Routes of transmission across the placenta and consequences of infection.

a | TORCH (Toxoplasma gondii, other, rubella virus, cytomegalovirus, herpes simplex virus) pathogens can access the intra-amniotic compartment through multiple mechanisms, including direct transplacental transmission, placental damage or disruption and/or fetal–maternal haemorrhage. In addition, pathogens can be transmitted by ascending the genital tract. b | Infections in pregnancy can affect the maternal host, fetus and/or the placenta itself. The results of infection and the inflammatory response have consequences at each site.
Table 1.
TORCH pathogens and the hallmarks of their infections
| Pathogen | Mode of transmission | Hallmarks of congenital infection |
|---|---|---|
| Bacteria | ||
| Listeria monocytogenes | Contaminated food | Stillbirth/pregnancy loss; preterm delivery; neonatal sepsis |
| Treponema pallidum | Sexual | Stillbirth/pregnancy loss; low birthweight; fetal hepatosplenomegaly; developmental delay; saddle nose deformity; rhinitis; dental deformities; chorioretinitis; anaemia; rash; dilated bowel; skin thickening; periostitis; bone fractures and demineralization |
| Streptococcus agalactiae | Commensal | Neonatal sepsis |
| Staphylococcus aureus | Commensal | Neonatal sepsis |
| Escherichia coli | Commensal | Neonatal sepsis |
| Viruses | ||
| Cytomegalovirus | Faecal–oral | Chorioretinitis; low birthweight; hearing loss; developmental delay; anaemia/thrombocytopenia; rash; stillbirth/pregnancy loss; ventriculomegaly; microcephaly; intracerebral calcification; echogenic bowel/abdominal calcifications; normal |
| Herpes simplex virus 1 and 2 | Sexual or oral contact | Neonatal meningitis; dermatological lesions; ventriculomegaly; microcephaly; intracerebral calcifications; chorioretinitis; optic atrophy; limb dysplasia |
| Varicella zoster virus | Respiratory droplets | IUGR; limb abnormalities |
| Lymphocytic choriomeningitis virus | Fomites (rodent based) | Ventriculomegaly/hydrocephalus; developmental delay; motor and sensory deficits; chorioretinitis; hearing loss |
| Zika virus | Arbovirus (Aedes species, sexual, blood borne) | Microcephaly; IUGR; hepatosplenomegaly; intrahepatic calcifications; ventriculomegaly; intracerebral calcifications; echogenic bowel; stillbirth/pregnancy loss |
| West Nile virus | Arbovirus (Culex species) | Chorioretinitis; meningitis/encephalitis; possible lissencephaly |
| Rift Valley fever virus | Arbovirus (Aedes species, Culex species, Anopheles and Mansonia species, contact with contaminated animal materials) | Stillbirth/pregnancy loss; preterm delivery |
| Human parvovirus B19 | Respiratory droplets | Anaemia; hydrops; stillbirth/pregnancy loss |
| Parasites | ||
|
Plasmodium falciparum Plasmodium vivax |
Arthropod vector (Anopheles species) | IUGR; preterm delivery; severe hypoglycaemia |
| Toxoplasma gondii | Ingestion of contaminated food or oocytes | Ventriculomegaly; hydrocephalus; intracerebral calcifications; choroid plexus cysts; hydrocephalus; ascites; IUGR; hepatosplenomegaly |
| Trypanosoma cruzi | Arthropod vector (Triatominae subfamily) | IUGR; respiratory failure; hepatosplenomegaly; meningitis; heart failure; hydrops; heart disease; megacolon |
IUGR, intrauterine growth restriction; TORCH, Toxoplasma gondii, other, rubella virus, cytomegalovirus, herpes simplex virus.
Pregnancy loss through miscarriage or stillbirth (defined as intrauterine fetal death after 20 weeks) can also be caused by infection. Approximately 10–30% of all stillbirths have an infectious aetiology4–7, although the low rates of diagnostic testing for infections in pregnancy might result in an underestimation of this value. The rate of pregnancy loss is variable with gestational age at infection and by specific pathogen. For example, infection with Treponema pallidum causes pregnancy loss or fetal death in up to 50% of cases8 whereas parvovirus B19 infection causes pregnancy loss or stillbirth in <3%9. The mechanisms of pregnancy loss in the setting of ‘TORCH’ (Toxoplasma gondii, other, rubella virus, cytomegalovirus, herpes simplex virus) infections can be pathogen mediated, placenta mediated and/or can be through inflammation-induced previable delivery. There are also outcomes of congenital infections that do not manifest until after delivery. These can include hearing loss, developmental delays and/or blindness as detailed below.
Inflammation initiated by an infection of the maternal host is also a known cause of preterm labour and can result in previable delivery or sequelae of prematurity10 with lifelong consequences to the neonate. Neonates can have sequelae from maternal infections even with full-term development11, demonstrating that the fetal response to infection is not restricted to effects of prematurity. Early onset sepsis, an important cause of neonatal morbidity and mortality particularly in premature or low birthweight infants, is strongly associated with maternal infection12–14.
In this Review, we highlight the molecular pathogenic mechanisms of select pathogens that have distinct effects during pregnancy. The acronym TORCH was coined to refer to pathogens known to traverse the maternal–fetal barrier and cause congenital disease in the fetus15. In addition to the traditional TORCH pathogens, we also describe the pathogenesis of emerging pathogens with important sequelae to the pregnant person, fetus and/or neonate.
Placental structure and defences
To understand the mechanisms of vertical transmission, it is crucial to understand the unique structure and function that exist at the maternal–fetal interface. The human placenta is a complex organ composed primarily of specialized fetus-derived cell types. As gestational age-specific differences in placental structure and cellular composition have been reviewed in detail elsewhere16,17, we provide a concise description of human placental structure and development below to provide a framework to address mechanisms of pathogenesis. An overview of the structure of the maternal–fetal interface is shown in Fig. 2a.
Fig. 2. Structure and cellular composition of the maternal–fetal interface.

a | The structure of the maternal–fetal interface includes the maternal decidua and the fetus-derived placenta. The maternal uterine microvasculature is remodelled to form spiral arteries, which deliver blood to chorionic villi in the intervillous space. b | The placenta undergoes a series of rapid morphological changes throughout gestation. In early pregnancy (left), the blastocyst differentiates into the embryo and the trophectoderm, the earliest cell type that will form the placenta. The invasive trophoblasts begin to invade the decidua, where the early syncytiotrophoblast forms and infiltrates into the endometrium. Throughout the first trimester (middle), chorionic villi form and remain immature until the later stages of this trimester. Immature villi are covered in the syncytiotrophoblast layer, with a contiguous layer of cytotrophoblasts lying below this layer. The stroma of the villi in the first trimester contains fetal vessels, which begin to form at ~6–8 weeks of gestation. The maternal microvasculature undergoes extensive remodelling during the first trimester, with the placenta transitioning to haemochorial at the end of this stage of gestation. In the second and third trimesters (right), chorionic villi mature and remain covered by the syncytiotrophoblast. However, unlike the immature villi of the first trimester, the cytotrophoblast layer becomes discontinuous in the later stages of gestation. At this stage, the fetal microvasculature is fully developed, and the villous stroma becomes enriched in fetus-derived Hofbauer cells, which reduce in number closer to full-term.
The human placenta develops when the fetus-derived trophoblasts, the main cell type that comprises the placenta, form the trophectoderm (Fig. 2b). The trophectoderm forms a cellular barrier early in development to prevent infection of the embryo. The process of placental development continues rapidly from that point onwards and is not fully complete until the end of the first trimester, undergoing a series of remarkable morphological changes (reviewed in ref.18). These changes result in the development of chorionic villi, which form the primary contact between the fetus-derived placenta and the maternal blood supply that will eventually bathe these structures.
Trophoblast stem cells give rise to cytotrophoblasts (CTBs), the proliferative mononuclear cells of the placenta, and the syncytiotrophoblast (STB), a multinucleated contiguous cell layer that covers the entire surface of placental chorionic villi. During the first trimester, the placenta undergoes substantial morphological changes that result in the villous structure it will have for the remainder of pregnancy (Fig. 2b). The end of the first trimester also marks the crucial transition to a haemochorial placenta, wherein maternal blood directly contacts the fetus-derived placenta. This process requires extravillous trophoblasts (EVTs) to remodel the maternal microvasculature, which will ultimately allow for delivery of maternal blood to the surface of the placental chorionic villi. This is a crucial point as we discuss mechanisms of microbial vertical transmission, which could differ markedly between the first trimester, when there is no direct contact between the placenta and maternal blood, and the later stages of pregnancy once the haemochorial placenta is established.
The STB is the foremost barrier against the haemochorial spread of infectious agents, particularly from the point of the establishment of a haemochorial placenta. As the proliferative CTBs lie subjacent to the STB, they also benefit from the protection of this fused cell layer. Chorionic villi contain additional barriers to infection that reside in the villous stroma, including fetus-derived macrophages (Hofbauer cells) and the fetal microvasculature, which must be breached for an infectious agent to reach fetal blood. Recent work also suggests that maternally derived immune cell types are in close proximity to the fetus-derived placenta and may thus impart an additional layer of immune protection19. Thus, when considering mechanisms of microbial vertical transmission, it is important to remember that transmission to the fetus requires infection of multiple cell types and/or breaching of cellular barriers, the most formidable of which is the STB, as is discussed below. Given its central role in protecting the fetus, it is perhaps not surprising that the placenta has evolved highly efficient mechanisms of antimicrobial defence. In the following sections, we review key physical and immunological mechanisms of placental defence from infection.
Physical defences
As described above, the STB covers the entirety of the chorionic villous surfaces. The STB contains upwards of 60 billion nuclei and has a surface area of 5 m2 in mid-gestation and 11–12 m2 at full-term20 — a single cell layer roughly the size of a small bedroom! The very nature of the fused STB provides a powerful barrier to microbial transmission, given that many pathogens bypass other cellular barriers by directly or indirectly weakening cell–cell junctions (reviewed in ref.21). In addition to a complete lack of cell junctions, the surface of the STB also poses a barrier to microbial attachment and invasion given the presence of a dense cortical actin network subjacent to the brush border (Fig. 3). Thus, the shared physical nature of the STB provides common structural mechanisms to resist diverse microorganisms. However, it should be noted that the STB is not the sole cellular barrier present in the human placenta. The stroma of the placenta also contains defensive barriers, including the microvasculature of fetal blood vessels, which would also need to be breached for microorganisms to access fetal blood. The physical defences of the villous surface restrict the attachment and/or invasion of non-viral pathogens, including Listeria monocytogenes and T. gondii22–24, but viruses appear to be capable of entering primary trophoblasts, suggesting that some viruses might overcome the physical barriers to microbial entry25–28.
Fig. 3. Placental defences against pathogens.

Given its role as a primary barrier to the haematogenous spread of infectious agents, the human placenta has evolved disparate and non-overlapping mechanisms of antimicrobial defence. These can be separated into at least three categories, physical defences (left), the constitutive release of antimicrobial effectors (middle) and/or robust innate immune response to infection (right). Physical defences include the lack of cell–cell junctions of the syncytiotrophoblast layer, preventing inflammation-mediated damage of intercellular junctions that could compromise the integrity of this barrier. Additional physical defences include the dense cortical actin network lying sub-apical to the dense brush border of the syncytiotrophoblast. Another form of defence involves the constitutive release of potent antimicrobial effectors such as antiviral microRNAs in extracellular vesicles, cytokines (for example, type III interferons), and antimicrobial peptides (middle). The placenta also responds to pathogens with potent innate immune signalling, which further enhances the release of antimicrobial defence substances (right).
Immunological defences
The decidua
The maternally facing surface of the placenta is in direct contact with the decidual lining of the uterus, and this leukocyte-rich layer is one of the first lines of immune defence at the maternal–fetal interface. The maternal decidua is in direct and/or indirect contact with fetal membranes, placental villi and the maternal circulation. The decidua can be divided based upon these contact sites — the decidua parietalis lines the uterine wall; the decidua basalis is the site of implantation and villous contact; and the decidua capsularis encases the fetal membrane. The decidual layers contain cytotoxic, helper and regulatory T cells, natural killer (NK) cells, innate lymphoid cells, macrophages and neutrophils29,30. Up to 40% of all decidual immune cells are leukocytes in the first trimester, and a leukocyte-rich infiltrate is also seen in parturition30–32. The composition of decidual cell populations displays gestational age-specific variation and is important for trophoblast invasion and for maintaining and establishing the placental vascular bed33,34. As such, particularly in the first trimester, decidual cells remain in close proximity to invading EVTs, and provide maternally derived immune defences at these sites (reviewed in ref.35). Despite its composition as an immune cell-enriched site, the decidua has been suggested to be a key reservoir for TORCH pathogens. Use of decidual explants and data from pathological examinations of clinical specimens suggest that the decidua may be a primary site of replication for several TORCH pathogens including cytomegalovirus (CMV)36, Zika virus (ZIKV)37 and L. monocytogenes38, and could thus form a reservoir for pathogens at the key site of the maternal–fetal interface.
The interaction of the decidua with various pathogens in vivo is complex, as decidual leukocytes have distinct phenotypic differences from those in the peripheral circulation. Maternally derived decidual macrophages exhibit unique properties compared with other macrophages resident in tissue or systemically circulating monocytes19,30,39,40. Studies suggest that the decidual composition of immune cells and their interaction with trophoblasts may alter their susceptibility to pathogens. For example, decidual NK cells possess the remarkable ability to transfer granulysins to trophoblasts to protect from L. monocytogenes infection without actively killing these cells41. These and other studies highlight the often unique and complex strategies of antimicrobial protection that exist at the maternal–fetal interface.
The immune cells in the decidua are in contact with decidual stromal cells, which can influence immune cell behaviour. In particular, the immune cell-rich decidua has altered activity in models of co-culture, reflecting that the complex architecture of the immune cell network within the decidual stroma modulates the immune inflammatory response. For example, decidual stromal cell function is altered with co-culture with macrophages42, dendritic cells and NK cells in a prostaglandin-dependent manner43. Moreover, the response of decidual stromal cells to pathogen stimulation is different from that of endometrial stromal cells44–46, suggesting that the microenvironment of the placenta has a role in innate immune defences. Decidual explants have been used to demonstrate that CD8+ effector memory T cells and NK cells can regulate decidual stromal CMV47 and herpes simplex virus (HSV)48 infections. These multicellular interactions are difficult to dissect and model but have important implications for understanding the immune landscape at the maternal–fetal interface as each individual component (for example, stromal cell regulation, hormonal microenvironment, composition of leukocytes) might have a role in immune defence against infection.
Chorionic villi
Fetal immune cell populations in placental development and in pregnancy-associated pathologies have been reviewed elsewhere49,50; therefore, we limit our discussion in this section to specific trophoblast-derived defensive strategies.
In addition to providing a physical defence from infection, it is also clear that villous trophoblasts form a chemical barrier to microbial vertical transmission. Trophoblasts have robust innate immune activity and secrete immunomodulators that are important for restricting infection (Fig. 3). In the case of viral infection, human trophoblasts constitutively release antiviral interferons that restrict infection in both an autocrine and a paracrine manner51,52, which appears to be specific for the STB. The release of interferons is a highly unique feature of trophoblasts as interferons are generally only induced in response to the detection of a viral infection. As the antiviral effectors of the interferon pathway, interferon-stimulated genes (ISGs), can exert potent cytotoxic and pro-inflammatory properties. Trophoblast-derived interferons are exclusively those of the type III interferon family: IFNλ1, IFNλ2 and IFNλ3. In mice, type III interferons also confer protection from viral infections, and deletion of the receptor for these interferons in the placenta sensitizes the fetus to ZIKV infection53. The type III interferon-mediated restriction of infection in mice is tightly associated with gestational age, with the greatest protection occurring after placentation53,54. Moreover, IFNλs delivered prophylactically or therapeutically in the setting of ZIKV infection in mice protect against vertical transmission53,54. In addition to interferons, human trophoblasts also secrete antiviral microRNAs that are packaged into placental exosomes, which confer broad antiviral protection in non-placental recipient cells and can be isolated from the serum of pregnant women25,51,55–57. Given these non-redundant and potent antiviral defences, it is perhaps not surprising that most viruses are unable to directly replicate in the STB. However, it should be noted that the effects of type I interferons (IFNα and IFNβ) may be distinct from those of the type III interferons. Induction of maternal type I interferon signalling has been shown to damage the placenta in mice in vivo and in human ex vivo tissue explants58. Although the mechanisms for these opposing effects have yet to be fully elucidated, at least one ISG, interferon-induced transmembrane protein (IFITM), inhibits fusion of CTBs with the STB59,60, which may indicate that the expression of this and perhaps other ISGs must be tightly regulated in the placenta to prevent interferon-mediated damage.
In the case of bacterial or parasitic infection, many studies highlight the resistant nature of the STB, which has largely been attributed to physical barrier properties22–24,61. However, similar to the properties of antiviral resistance, it is clear that the human placenta also uses additional molecular pathways to resist infection by non-viral pathogens. In addition to the constitutive release of antiviral interferons, the human placenta secretes other cytokines that likely function to limit infections in both an autocrine and a paracrine manner. For example, the constitutive activation of the inflammasome in placental trophoblasts results in the constitutive release of cytokines, including IL-1β, IL-18 and IL-1α, which can be found circulating systemically in pregnant women62. In addition, infection of human placental explants with L. monocytogenes robustly induces inflammasome signalling, the inhibition of which sensitizes trophoblasts to infection62. Like the effects of interferons, the paracrine effects of inflammasome-associated cytokines also protect from infection, as placenta-derived IL-1β primes circulating immune cells for subsequent inflammasome activation and protects from L. monocytogenes infection62. By contrast, inflammasome activation in the STB has also been implicated in increased severity of neonatal outcomes associated with placental malaria63, suggesting that similar to interferon signalling, the balance of this signalling is crucial.
There is no evidence to suggest that placenta-derived secreted products defend against parasite infections directly. In fact, the same placenta-derived factors that protect against viruses exert no effect on T. gondii infection56. Instead, in the case of T. gondii, the placenta releases diverse immunomodulatory factors including the regulatory T cell chemoattractants C-C motif chemokine 22 (CCL22) and CCL17 in response to infection23 (Fig. 3). However, the impact of this signalling on maternal and/or fetal consequences of infection has yet to be defined.
TORCH pathogens
As discussed above, TORCH pathogens make up a limited number of microorganisms with known teratogenic effects. In the following sections, we review key members of this group that fall into the categories of bacteria, viruses and parasites, and the consequences of these infections (Table 1).
Bacteria
Listeria monocytogenes
L. monocytogenes is a small Gram-positive bacterium that is transmitted through contaminated food. Listeriosis in pregnancy causes prematurity and stillbirth and enhances maternal morbidity with increased rates of meningitis and sepsis during pregnancy (Table 1). In a recent outbreak in South Africa, the mortality rate for infants was 28% and for pregnant women it was 8%64. Pregnancy-associated morbidity during this outbreak was responsible for 50% of cases presenting to health care64. Despite the immaturity of the fetal immune system in early pregnancy, treatment of L. monocytogenes in the first trimester after exposure has been shown to improve fetal outcomes65–67. By contrast, treatment after the first trimester has not been definitively shown to change the course of the disease for the fetus but is recommended to improve maternal outcomes68. This is likely due to enhanced susceptibility of the fetus to L. monocytogenes infection after the first trimester and suggests that there are gestational age-specific differences in vertical transmission.
After entry through the enteric mucosa, L. monocytogenes spreads cell to cell by manipulating actin polymerization. The mechanism of transplacental transmission of the bacterium remains unknown. In ex vivo cultures, the STB is highly resistant to infection24,62. The bacterial virulence factors required for entry into other cells (for example, listeriolysin O and internalin B) are not required for entry into human choriocarcinoma trophoblast cell lines69. The bacterial internalin protein InlA is important for binding to E-cadherin on primary trophoblasts70, but its role in crossing the placental barrier in vivo remains unclear, with differential effects in different animal models71,72. Together, these data suggest that transmission across the placental barrier may require additional, yet uncharacterized, bacterial virulence factors. Accordingly, there was minimal strain variation with respect to the ability to infect the placenta in studies using a competitive in vivo model in guinea pigs73, suggesting that the trophoblast-specific virulence factors are conserved in epidemic strains.
After placental colonization, the bacterium spreads to fetal tissues and can be reseeded back to maternal organs74. In mice, actin polymerization through the bacterial protein ActA has been shown to be important for cell-to-cell spread and transmission across the maternal–fetal interface75. The secreted bacterial colonization factor internalin P is important for distorting cells and allowing the bacteria to traverse the basement membrane76, but the mechanisms of transport across the STB to the basement membrane have yet to be characterized. Importantly, fetal infection is associated with placental abscess development and innate immune cell recruitment to the maternal–fetal interface77. Studies in non-human primate models demonstrate that the bacterium is not required to traverse the maternal–fetal barrier to cause placental inflammation, fetal loss and compromise78, suggesting that pregnancy loss may also occur in the absence of bacterial colonization of the fetus.
Treponema pallidum
T. pallidum is a well-characterized teratogen. This bacterium is 6–20 µm in length, has a small genome of only 1,041 open reading frames and has an outer membrane covering a layer of peptidoglycan but minimal surface-exposed proteins and no lipopolysaccharide79. The lack of surface proteins may facilitate evasion of the immune response and the remarkable longevity of the bacterium in the human host79. T. pallidum can colonize and cause disease in all fetal organ systems and is the causative agent of congenital syphilis (Table 1). More than 50% of women with syphilis have adverse pregnancy outcomes80–82, and fetal transmission causes a wide variety of neonatal pathologies including meningitis, osteochondritis, bone marrow suppression and hydrops83–85 (Table 1). Congenital syphilis is on the rise in the United States, with a reported 1,306 cases in 2018 (ref.86) and is a major cause of infection-mediated fetal loss and neonatal morbidity in the developing world, causing an estimated 150,000 stillbirths, 60,000 neonatal deaths and 100,000 infections of infants annually81,82.
The pathophysiology of T. pallidum congenital transmission remains largely unknown. The sequelae of the fetus with congenital syphilis depends both on the maternal stage of infection and the gestational age of the fetus84,85. Congenital transmission occurs less frequently with subsequent pregnancies, an observation known as Kassowitz’s law. This observation suggests that the maternal immune response to T. pallidum can limit the frequency of congenital syphilis, but the vertical transmission risk is never eliminated87–90. This has also been shown in a guinea pig model of congenital syphilis, which is the best-described model for this phenomenon90. Additionally, in the guinea pig model, the fetal IgM response corresponds more with vertical transmission than spirochaetal DNA91, suggesting that the fetal immune response is important in the pathogenesis of congenital syphilis.
Inflammation and maceration of placental tissue has been demonstrated in cases of pregnancy loss or intrauterine fetal demise associated with congenital syphilis in the absence of congenital anomalies, suggesting that the up to 50% fetal loss rate associated with maternal syphilis might be attributed to the inflammatory response at the placenta itself92,93. Along with this, the Jarisch–Herxheimer reaction (heightened inflammatory responses that occur with antibiotic treatment of syphilis in pregnancy) is associated with preterm contractions and evidence of uteroplacental insufficiency94, suggesting that placental inflammation has a role in the pathogenesis of preterm delivery associated with syphilis.
As discussed above, the placenta forms a barrier that effectively restricts many bacterial pathogens. Although there is evidence that systemic inflammation can cause preterm birth and fever itself can be teratogenic, there is no evidence that other bacterial pathogens (for example, Klebsiella species, methicillin-resistant Staphylococcus aureus (MRSA), Escherichia coli) routinely traverse the placental barrier and cause fetal infection. These pathogens are important causes of maternal morbidity in the setting of systemic maternal infection (for example, pyelonephritis), but are not major causes of fetal infection, suggesting that the maternal–fetal barrier to multiple bacterial pathogens remains uncompromised in the case of maternal systemic illness and bacteraemia. The mechanisms underlying the antibacterial defences of the placenta in the setting of bacteraemia remain largely uncharacterized.
By contrast, there is a subset of well-described bacterial pathogens that ascend the genital tract and cause chorioamnionitis (reviewed in refs95,96). Intra-amniotic infection classically occurs in the setting of ruptured membranes, which suggests that the bridge of the fetal membrane provides an important barrier to fetal infection. Pathogens endemic to the vaginal microflora such as group B Streptococcus (GBS), E. coli and Bacteroides species have been found in placental and neonatal cultures97. Both primate and murine models demonstrate that vaginal microorganisms can traverse the genital tract and cause disseminated disease in the fetal cavity. Typically, infections arise from genitourinary microbiota and can be polymicrobial97,98. To cause disease in the uterine cavity, these bacteria would have to ascend the vaginal microbial biofilm and the antimicrobial cervical mucous plug to contact the fetal membranes and cause an inflammatory response locally.
The best-studied microorganism associated with ascending infection and neonatal disease is GBS, which can cause neonatal sepsis with transmission. Screening for colonization and intrapartum prophylaxis with antibiotics decreases the incidence of early onset neonatal sepsis99. GBS biofilms on fetal membranes have been observed100 and it is one of the most commonly cultured bacteria in the setting of intra-amniotic infections97. GBS colonizes the genital tract in ~10–30% of women101. The predilection of some isolates to cause neonatal disease remains unclear, but GBS activates the innate immune neutrophil and macrophage response to initiate cytokine release, cell migration and the development of extracellular traps and bacteriolysis102,103. The mechanisms of pathogenic switching and specific virulence determinants for ascending the genital tract include pili, adhesins and altered regulatory systems (reviewed in refs104,105). E. coli can also adhere to fetal membranes, although to a lesser degree than GBS106. E. coli, like GBS, inhabits the genital and gastrointestinal tract and is commonly isolated in the setting of intra-amniotic infection. With implementation of universal screening and intrapartum prophylaxis with antibiotics to GBS, E. coli has become the most common aetiological agent of late-onset sepsis in neonates97,107. In obstetric isolates, phylogenetic subgroups are variable but genes on pathogenicity islands for type 1 fimbriae and iron acquisition are enriched108, suggesting that horizontal gene acquisition is important to cause disease in pregnancy. Similar to other bacterial pathogens, the specific virulence factors that allow E. coli to cause clinical disease and ascend the reproductive tract during pregnancy remain under-studied.
Viruses
Both DNA and RNA viruses can traverse the maternal–fetal interface and cause fetal disease. In the following sections, we discuss key teratogenic viruses and what is known regarding their routes of transplacental transmission and clinical outcomes. Although rubella virus was a major source of congenital disease before successful vaccination efforts, we have restricted our discussion to the most common viruses currently associated with contemporary congenital disease — CMV, HSV1 and HSV2, parvovirus B19, HIV and ZIKV (Table 1).
Cytomegalovirus
CMV is a member of the Herpesviridae family and is one of the most common causes of vertical infections globally. It is the single most common cause of congenital hearing loss in the United States, and the global burden of disease is also high, although likely underestimated109. Like other herpesviruses, CMV enters a latent state after acute infection and can be subsequently reactivated. In the setting of maternal primary infection, there is a ~40% risk of fetal transmission. By contrast, the risk of transmission to the fetus after reactivation is <0.05%, but causes most of the clinical disease110,111. Gestational age-specific variation in infectivity and fetal consequences have been described112, suggesting that alterations in the maternal–fetal interface during gestation regulates the mechanisms of CMV vertical infection. Placental histology in congenital CMV infection ranges from normal to acute and chronic intervillositis113. The classic, owl’s eye viral inclusion bodies occur more frequently in the first and second trimesters than in full-term placentae114.
Given that other recent reviews have provided thorough overviews of the mechanisms by which CMV might be vertically transmitted115–117, here we provide a more focused overview. CMV has been shown to infect trophoblasts, but with substantial gestational age-related differences in infectivity28. Given that human CMV does not readily infect rodents, in vivo studies of the mechanisms of CMV vertical transmission are limited to human placental primary cell cultures and/or explant models. Although guinea pig models of CMV infections in pregnancy have been described118, these do not fully recapitulate the phenotype of human CMV infections during pregnancy. Studies of CMV infection in human tissue demonstrate active replication in maternally derived decidua, which may generate a viral reservoir that could enhance the likelihood of CMV crossing the fetal barrier in the setting of primary infection36,119. Specifically, apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3A (APOBEC3A) has been implicated in control of CMV replication in the decidua, but not in placental villi37. Studies have also shown that CMV may preferentially infect placental pericytes to gain access to the fetus and that this might be a primary mode of infection in the setting of maternal viraemia120. The role that these cells have at the maternal–fetal interface remains largely uncharacterized, but they may represent an important site from which CMV can traverse the placenta and cause congenital infection particularly with advancing gestation.
Herpes simplex virus
HSV1 and HSV2 can be transmitted to the fetus via the transplacental route, but transmission through contact with a virus-shedding lesion in the genital tract is a far more common mode of vertical transmission. HSV1 and HSV2 are neurotropic and lie dormant in the dorsal root ganglion after primary infection. On reaching the fetus, these viruses are highly teratogenic and are associated with the classic triad of manifestations in skin (aplasia cutis, scarring, erosions), central nervous system (ventriculomegaly, microcephaly, intracranial calcifications) and eyes (chorioretinitis, atrophy), along with skeletal manifestations and fetal loss. A mouse model recapitulates vertical transmission, with fetal loss, congenital malformations and neurotropism, demonstrating that haematogenous dissemination (rather than ascending infection) is responsible for effects on the fetus121.
The mechanisms of transplacental transfer of HSV1 and HSV2 are largely uncharacterized. The STB is resistant to HSV1 and HSV2 infection, but EVTs are permissive25,122. Although the HSV entry mediators HveA, HveB and HveC are expressed in EVTs, viral transmission is not blocked by antibodies to HveA122. The maternal surface of the placenta is positive for HSV1 or HSV2 in 9–28% of women who are asymptomatic at the time of delivery without evidence of fetal transmission123,124, suggesting that the placenta and/or maternal immune response presents a barrier to transplacental transmission in women with non-primary infection.
Parvovirus B19
Parvovirus B19 is a non-enveloped single-stranded DNA virus in the Parvoviridae family. Infection with parvovirus B19 is common during childhood, causing low-grade fever, maculopapular rash and slapped cheek facial rash. Humoral immunity is protective against infection, and 70% of the adult population is immune125. The rate of transmission of maternal parvovirus B19 infection to the fetus is 17–33%, and most fetuses have spontaneous resolution with no sequelae, although ~3% can go on to develop non-immune hydrops from fetal anaemia9. Fetal loss and stillbirth are associated with infection even in the absence of hydrops. The sequelae of infection vary with gestational age, such that infection after 20 weeks confers a 0.5% risk of fetal loss, whereas before 20 weeks the rate is increased 30-fold9.
Parvovirus B19 exhibits strong tropism for erythroid precursors and immunohistochemistry demonstrates that after traversing the placenta the virus reaches the fetal endothelium126. The parvovirus B19 VP2 capsid protein has been shown to bind to globoside, which is present on the surfaces of the STB and CTBs127. The VP2 capsid protein has also been shown to bind to villous trophoblast cells through globoside glycolipids128. After viral entry, non-structural protein 1 (NS1) induces apoptosis in cells129. Consistent with this, apoptosis has also been demonstrated in the placenta in severely affected pregnancies, suggesting that fetal demise in the absence of hydrops may result from placental damage that directly affects function130. The rare but classic sequela of parvovirus B19 is non-immune fetal hydrops caused by transient, severe anaemia with loss of both erythrocytes and nucleated erythroid precursors129. Dichorionic twin discordance in infection and in clinical sequelae has been reported131,132, suggesting that the placental and fetal responses are important for limiting clinical disease, but the immune mechanisms by which this occurs are unclear.
HIV
Congenital transmission of HIV remains associated with global neonatal morbidity. HIV can be vertically transmitted via the transplacental route, during delivery and/or through breastfeeding postnatally. In the absence of antiretroviral therapy (ART), ~25% of infants born to HIV-positive women become infected compared with <2% of those whose mothers are on ART133. The relative contributions of the various routes of vertical transmission suggest that each has a role in congenital HIV infection, but the majority of congenital infections occur with intrapartum transmission and direct exposure to maternal secretions and blood. Ex vivo, chorionic villous explants isolated from both first trimester and full-term placentae support HIV infection, suggesting the possibility that the placenta could be infected during pregnancy134,135. However, although HIV proteins and/or nucleic acids can be detected in placental tissue collected from HIV-positive women, this occurs relatively infrequently, with studies demonstrating that the majority of placentae collected from HIV-positive women are negative for HIV136–138. These data concur with epidemiological data that suggest that, although transplacental transmission of HIV is possible, it is an infrequent (<1%) occurrence139,140. However, maternal co-infection with other pathogens such as CMV or malaria may further increase the risk of vertical transmission and/or adverse outcomes (reviewed in ref.141).
Zika virus
ZIKV is a positive-strand RNA virus in the Flaviviridae family that caused a large outbreak of >2,000 cases of congenital disease in 2015–2016, with an epicentre in Brazil142,143. ZIKV is transmitted through vector-dependent (Aedes aegypti mosquitoes) and vector-independent (for example, sexual, blood transfusion and vertical transmission) routes. Maternal ZIKV infections are associated with a range of fetal clinical outcomes including ventriculomegaly, microcephaly and developmental delays (Table 1), which occur in ~10% of maternally infected patients144. The discrepancy between fetal infection and the diverse neurological sequelae of disease has led to the separation of congenital ZIKV infection and congenital ZIKV syndrome145. Since the initial outbreak, cases of ZIKV congenital disease have waned considerably, suggesting that pre-existing immunity is important for regulating ZIKV infection of the maternal host. ZIKV infection in human placental explants and mouse models suggests clear gestational age differences in transmission efficacy and fetal sequelae, with infection in the first trimester posing the greatest risk37,53,144. Interestingly, twin discordance for placental and neonatal infections has been demonstrated with up to 50% discordance in twin pairs146, and this susceptibility can be recapitulated in vitro after delivery147, suggesting that fetally or placentally derived factors are important for limiting vertical transmission.
Like other TORCH pathogens, the mechanisms by which ZIKV crosses the placental barrier remain largely unknown (Fig. 4), despite considerable investigation. Some studies have associated placental cell tropism of ZIKV infection with receptor expression, but receptors that are important in flavivirus binding (for example, the TAM family of receptors) are not important for ZIKV infection in vivo148. In vitro mouse and ex vivo human studies have also suggested that pre-existing antibodies to the related flavivirus dengue virus (DENV), which is co-endemic with ZIKV, enhances ZIKV congenital infection149,150 and facilitates viral traversal across the placenta. However, data from human studies in endemic regions suggest that pre-existing DENV immunity is associated with reduced risk of ZIKV infection151,152, which is supported by work in non-human primate models153,154. By contrast, pre-exposure to ZIKV sensitizes to subsequent risk of severe DENV in humans155. Non-human primate models of ZIKV vertical transmission have provided important insights into the complex nature of maternal–fetal infections with this virus (reviewed in ref.156) and have also enabled studies that investigate neonatal and infant sequelae157. However, even these models have yet to elucidate the precise mechanisms by which the virus traverses the placental barrier. Both animal and in vitro models highlight the multifactorial complexity of vertical transmission of ZIKV and suggest that modelling this phenomenon may be difficult.
Fig. 4. Possible mechanisms of vertical transmission.
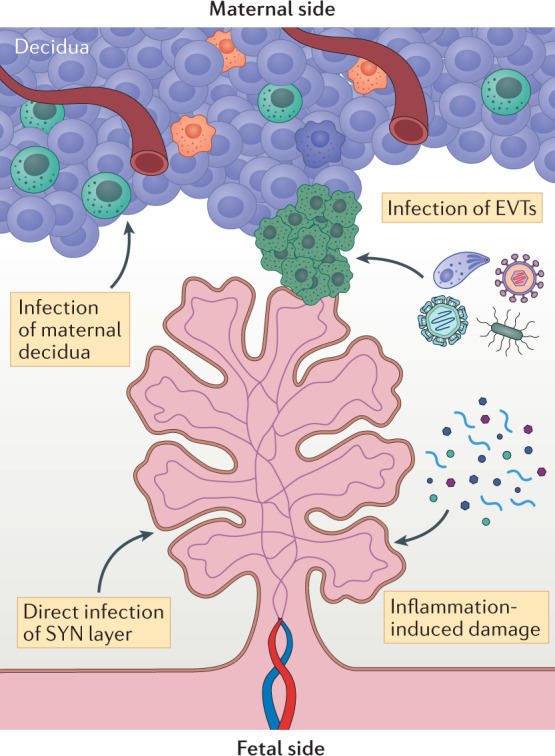
The mechanisms by which many TORCH (Toxoplasma gondii, other, rubella virus, cytomegalovirus, herpes simplex virus) pathogens access the fetus are unclear. However, studies suggest that some of these pathogens may use similar pathways to bypass the placental barrier. These mechanisms include infection of extravillous trophoblasts (EVTs; green cells) and/or infection of the maternally derived decidua, such as through direct infection of maternal immune cell populations. Other possible routes include direct infection of transmission across chorionic villi, through direct infection of the syncytium (SYN) or through inflammation-mediated damage of the syncytiotrophoblast layer that disrupts the barrier and allows transmission.
Emerging viruses
Several emerging viruses may have major impacts on the mother and fetus during pregnancy. Although pregnancy is often not well studied in the setting of emerging pandemics, there are data indicating increased severity of maternal infection with Ebola virus (EBOV) and reports of vertical transmission of Rift Valley fever virus (RVFV), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), West Nile virus (WNV) and Eastern equine encephalitis virus (EEEV).
EBOV belongs to the Filoviridae family, which are negative-strand, enveloped RNA viruses. EBOV infections are associated with maternal haemorrhage, preterm labour, miscarriage and maternal and fetal death. Maternal death occurs in 85% of cases with near 100% loss of offspring (stillbirth, miscarriage and neonatal death)158. In addition to causing severe maternal disease, there are also reports suggesting vertical transmission of EBOV. During the 2013–2016 outbreak in West Africa, pregnant women were noted to survive EBOV disease even with clear viraemia, but delivered stillborn infants with elevated viral RNA levels in placental and fetal swabs, suggesting vertical transmission of EBOV159,160. In addition, EBOV antigen has been found in the STB by immunohistochemistry, although this localization alone is insufficient to demonstrate vertical transmission and may instead suggest that the STB successfully restricted access to fetal blood159.
RVFV is an arbovirus in the Bunyaviridae family that is transmitted through an arthropod vector and is associated with severe disease in domesticated animals and livestock. In herds, RVFV outbreaks are associated with ‘abortion storms’, in which fetal loss and/or stillbirth are observed in as many as 90–100% of pregnant animals. A single case study has demonstrated vertical transmission in the third trimester in humans, and increased miscarriage and fetal demise in association with RVFV has also been described161,162. In human placental tissue ex vivo, viral replication has been demonstrated in both the STB and in CTBs163,164, but the specific route of vertical transmission in livestock and in humans, and whether these routes are similar, remains unclear.
WNV is an arthropod-borne flavivirus associated with marked neurotropic pathology. There have been several case reports of vertical transmission in humans, and a case series demonstrated an association with central nervous system abnormalities (hydrocephalus, microcephaly and lissencephaly)165, but larger case studies are needed to confirm the association, and the frequency of vertical transmission remains unknown. In mouse models, WNV induces ZIKV-like fetal death and disease, suggesting a potential to cause congenital disease in vivo166. In addition, human explant tissue including decidua, chorionic villi and fetal membrane samples are permissive to varying degrees166. In placental villi, EVTs are preferentially infected166 and the STB remains largely resistant to infection166,167, similar to what is described for ZIKV infection. Similar findings with Powassan virus (POWV) suggest that some emerging neurotropic arboviruses have the potential to affect fetal development if vertically transmitted135. By contrast, other arboviruses including chikungunya virus (CHIKV) and Mayaro virus (MAYV) did not cause fetal disease in mice and replicated inefficiently in human placental explants, suggesting differences in the teratogenic potential of emerging arboviruses135.
Most recently, the SARS-CoV-2 pandemic has highlighted the need to focus on women’s health, particularly during pregnancy. Several case reports from COVID-19-positive pregnant women describe infection of the STB by immunohistochemistry accompanied by placental pathological lesions168,169. Importantly, the vast majority of cases do not demonstrate any evidence of vertical transmission170–173, suggesting that the placenta retains its barrier function even in the setting of severe maternal infection and disease. Thus, despite the magnitude of the pandemic, the risk to the fetus as a result of vertical transmission of SARS-CoV-2 appears to be minimal and there are no data to suggest that SARS-CoV-2 is in any way teratogenic itself.
Parasites
There are several relevant parasitic infections in pregnancy that cause fetal disease globally. These parasites differ in the mode of transmission to the maternal host, but all have substantial implications for global maternal and child health.
Toxoplasma gondii
There are more than 200,000 cases of congenital toxoplasmosis globally each year174. T. gondii infects ~6–70% of fetuses, with maternal infection depending on gestational age at infection, and can be associated with devastating consequences, including ventriculomegaly, intracranial calcifications, chorioretinitis and rarely hydrocephaly174–176. Maternal symptoms occur in only ~5% of cases and clinical sequelae are thus best characterized in studies in which routine antenatal screening is performed, as maternal infection is subclinical. Fetuses infected early in pregnancy are far more likely to display clinical disease. Only 9% of women with seroconversion at the end of pregnancy deliver children with neurodevelopmental delays (in comparison with 25% of those with seroconversion in the second trimester)177.
T. gondii infection has been extensively studied at the cellular and molecular level in many non-placental cells. Infection is a multistep process that includes adhesion between T. gondii surface molecules and host cell surface proteoglycans, attachment that is driven by secretion of proteins into the host plasma membrane and invasion into the host cell that is mediated by interactions between parasite surface proteins and parasite proteins secreted onto the cell surface (reviewed in refs178,179). T. gondii also secretes protein effectors into the host cell during infection (reviewed in refs180,181). These diverse effectors alter many fundamental aspects of host cell biology, including innate immune pathways that are important for defensive signalling181. Despite characterization in non-placental cells, the mechanisms of T. gondii vertical transmission remain unclear. The STB displays distinct resistance to T. gondii infection, which occurs at the level of attachment and post-entry replication23,24,61. By contrast, CTBs and EVTs do not display the same degree of resistance23,24, suggesting cell-type specific differences in mechanisms of resistance. In addition to cell-intrinsic defences, trophoblasts also respond to T. gondii infection through the specific induction of various cytokines and chemokines, including the robust induction of the regulatory T cell chemokine CCL22 (ref.23). Remarkably, this induction is not driven by trophoblast sensing, but instead requires the delivery of the T.gondii-derived effector molecular GRA28 (refs23,182). However, it remains unclear what role placenta-derived cytokines or chemokines have in T. gondii pathogenesis and vertical transmission.
Plasmodium species
Of the four malarial species that cause disease in humans, Plasmodium falciparum causes the majority of disease in pregnancy. In areas of low transmission and low pre-existing immunity, congenital infection is symptomatic and causes cerebral malaria, respiratory distress syndrome, refractory hypoglycaemia, and miscarriage and stillbirth. In areas where the parasite is endemic and pre-existing immunity is high, malaria can cause severe maternal anaemia, preterm delivery and fetal growth restriction, and remains a major cause of neonatal mortality183–185. Congenital infection has been reported, and data suggest that perinatal acquisition through vertical transmission also occurs in endemic areas186–188.
The Plasmodium parasite has a clear predilection for the placenta. Attachment of infected erythrocytes to the placenta allows the parasite to evade the maternal immune response by changing surface antigens (described in detail below), and histopathological specimens from pathogenic malarial infection contain a substantial number of erythrocytes in the intervillous space with parasites and acute and chronic intervillositis189,190. Owing to placental malarial burden, nutrient transport across the placenta is reduced, which highlights how placental function can be compromised by infection191,192 (Fig. 5).
Fig. 5. Placental malaria in endemic areas.

Schematic of the maternal–fetal interface in an uninfected state (left panel). Schematic of the maternal–fetal interface in the setting of malaria (right panel). The parasite (yellow) accesses the placenta through the maternal circulation. The intervillous space then becomes a site of parasite replication where the parasite undergoes surface antigenic variation, ultimately leading to immune escape and further replication causing maternal anaemia. Maternal anaemia and parasitaemia substantially prevent nutrient transport across the placenta, which can lead to nutrient deprivation for the developing fetus.
The mechanisms for placenta-specific binding and sequestration of infected erythrocytes have been relatively well characterized. Plasmodium-infected erythrocytes bind to endothelial receptors for tissue sequestration in non-placental tissues through parasite-encoded variant surface antigens (VSAs) that are expressed by the infected erythrocyte (reviewed in ref.193). Expression of the VSA gene family var varies over the course of an infection. var encodes ~60 erythrocyte surface proteins annotated P. falciparum erythrocyte membrane protein 1 (PfEMP1), which are responsible for adhesion to uninfected erythrocytes and other cell types193–195. During the course of infection, antigenic variation in expression of different family members in the gene family var occurs, allowing immune evasion. During pregnancy, the parasite adapts to the presence of the placenta, allowing placental sequestration and immune evasion as well as maternal and fetal disease (Fig. 5). The Plasmodium gene encoding VSA 2CSA (var2csa) is selectively transcribed in placental malaria and allows the infected erythrocyte to adhere to the STB195–197. Outside of pregnancy, CD36 is a major receptor for infected erythrocytes, but chondroitin sulfate A (CSA) on the STB becomes the major receptor for VAR2CSA and is important for placental sequestration198–200. The upregulation of VARCS2A by the parasite in infected erythrocytes during pregnancy allows evasion of a pre-existing immune response in endemic areas and explains how primigravidity (that is, being pregnant for the first time) is a risk factor for severe disease. Accordingly, antibodies to placenta-specific VSAs including VAR2CSA are not present in men and do not develop until later in pregnancy201–203. VSAs are primary targets for maternal IgG, and elevated levels and a higher affinity for VSAs are protective from severe disease204–206. Placental sequestration of infected erythrocytes leads to an influx of phagocytes and cytokines, which is thought to damage the adjacent STB and lead to adverse outcomes in the absence of fetal parasitaemia194,207,208 (Fig. 5). Recently, placental autophagy, a mechanism to maintain normal cellular homeostasis, has been demonstrated to be dysregulated in placental malaria infection, which leads to decreased amino acid transport to the fetus and has been implicated in the development of fetal growth restriction209,210.
Trypanosoma cruzi
Chagas disease is caused by infection of T. cruzi, typically from an arthropod vector. Most women of reproductive age in endemic areas are infected and ~5% transmit the parasite vertically211. Most congenitally infected infants are asymptomatic but can develop life-threatening and disabling disease later in life. Features of symptomatic congenital Chagas disease include a low Apgar score, neonates that are small for gestational age and signs of liver failure and hydrops212,213. Different strains of T. cruzi demonstrate differential tropism for placental tissue, although the mechanistic basis for this remains unknown. High maternal parasitaemia correlates with congenital transmission214,215. Despite the fact that placental pathology demonstrates a predilection of the parasite for the decidua, ex vivo placental explants demonstrated that the parasite interacts with the STB and causes local destruction and detachment, likely through apoptosis216,217. Interestingly, T. cruzi-derived exosomes, a marker of increased parasitic burden and severe disease, cause inflammation and histopathological damage to the STB in placental explants218, suggesting that these exosomes may lead to inflammation-mediated damage and allow the parasite to cross the syncytial barrier. The parasitic mechanisms responsible for tissue damage are an area of ongoing investigation, but it is thought that after the trypomastigote gains access to the intervillous extracellular matrix, its degradation promotes parasite access to the fetal circulation216.
Conclusions
There are substantial consequences resulting from infections in pregnancy. The complexity and unique features of the maternal–fetal interface have led to the discovery of multiple pathways of host–pathogen interactions unique to this niche. Yet, the molecular mechanisms of pathogenesis remain largely uncharacterized, in part owing to the complexities of defining the interactions that occur between the pathogen and maternal and/or fetal hosts during the context of pregnancy. Moreover, modelling the unique tissue architecture and immunology of the maternal–fetal interface creates additional complexities in delineating microbial vertical transmission strategies. Although the use of mouse models has provided important insights into various aspects of pregnancy, there are substantial differences in the placental architecture between human and mouse (reviewed in refs16,219) that limit direct correlates of these findings to humans. Although the placentae of primates and guinea pigs have a more similar architecture to that of humans, these models can be difficult to establish, and their lack of genetic tractability limits some mechanistic studies. Lastly, both clinical samples and primary tissues provide human-based models to study pathogenic mechanisms or vertical transmission of TORCH pathogens; however, there can be limited access to healthy placentae to generate these models. Recently, the development of stem cell-derived organoid models of both maternal and fetal cell types220,221 at the maternal–fetal interface has opened up exciting new avenues to model this interface. However, organoid models lack immune cell components and do not recapitulate the immunological crosstalk that undoubtedly alters antimicrobial defences. The fetal and maternal immune cell phenotypes at the maternal–fetal interface are still an area of active investigation and characterization. Further understanding of the contribution of the immune cell populations enriched at the interface will be essential to further define mechanisms of pathogenesis in pregnancy.
Future studies elucidating the complexity of interactions between maternal and fetal tissues and how these interactions are modulated by pathogens are crucial to the development of targeted therapeutics. Moreover, understanding the mechanisms of microbial pathogenesis across the maternal–fetal interface has broader implications in the study of infertility, miscarriage and hypertensive disorders of pregnancy and parturition, which may share common aetiology. Although the complexity of pregnancy and the current gaps in understanding of many of the fundamental aspects of infections at the maternal–fetal interface represent a challenge, further investigation into this crucial aspect of human health will lead to strategies that could substantially improve maternal and child health.
Acknowledgements
The authors’ work on the placenta is supported by NIH AI145828 (C.B.C.) and a Magee Women’s Research Institute Clinical Trainee Research Award 4032 (C.J.M.). Additionally, C.J.M. is supported by K12 HD000849 awarded to the Reproductive Scientist Development Program. The authors apologize to any authors whose work was not included owing to space limitations.
Glossary
- Granulysins
Antimicrobial peptides that can cause pores in bacterial cell walls, leading to cell death.
- Osteochondritis
Inflammation of the cartilage.
- Hydrops
The presence of excess fluid in two or more fetal body compartments, associated with severe perinatal morbidity and death.
- Pyelonephritis
Inflammation and infection of the kidney.
- Chorioamnionitis
Inflammation of the fetal membranes, classically associated with bacterial infection.
- Intrapartum prophylaxis
The process of giving antibiotics, typically penicillins, to individuals in labour for treatment of group B Streptococcus colonization.
- Pericytes
Specialized connective cells of mesenchymal origin that classically surround endothelial cells in blood vessels.
- Aplasia cutis
A condition in which parts of the skin are missing.
- Ventriculomegaly
A condition in which the brain ventricles are abnormally large.
- Microcephaly
An abnormally small circumference.
- Chorioretinitis
Inflammation of the choroid surrounding the retina of the eye.
- Atrophy
Degenerative and small.
- Hydrocephalus
Central nervous system abnormality from pathological ventriculomegaly with excess fluid in the brain causing compression of brain parenchyma and requiring treatment.
- Lissencephaly
A brain malformation associated with severe developmental delay in which gyration of the brain is absent.
- Apgar score
A score assigned to neonates associated with neonatal health after birth. Infants are scored 0–2 points on muscle tone, skin colour, respiratory effort, pulse and reflex irritability at 1, 5 and 10 min after birth.
Author contributions
The authors contributed equally to all aspects of the article.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information
Nature Reviews Microbiology thanks S. Gaw, who co-reviewed with L. Li; N. Gomez-Lopez; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Christina J. Megli, Email: cjm232@pitt.edu
Carolyn B. Coyne, Email: carolyn.coyne@duke.edu
References
- 1.DeSilva M, et al. Congenital anomalies: case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine. 2016;34:6015–6026. doi: 10.1016/j.vaccine.2016.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyle B, et al. Estimating global burden of disease due to congenital anomaly: an analysis of European data. Arch. Dis. Child. Fetal Neonatal Ed. 2018;103:F22–F28. doi: 10.1136/archdischild-2016-311845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christianson, A., Howson, C. & Modell, B. March of Dimes. Global Report on Birth Defect. The Hidden toll of Dying and Disabled Children (March of Dimes Birth Defects Foundation, 2006).
- 4.Page JM, et al. Stillbirth associated with infection in a diverse U.S. Cohort. Obstet. Gynecol. 2019;134:1187–1196. doi: 10.1097/AOG.0000000000003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawn JE, et al. Stillbirths: rates, risk factors, and acceleration towards 2030. Lancet. 2016;387:587–603. doi: 10.1016/S0140-6736(15)00837-5. [DOI] [PubMed] [Google Scholar]
- 6.McClure EM, Goldenberg RL. Infection and stillbirth. Semin. Fetal Neonatal Med. 2009;14:182–189. doi: 10.1016/j.siny.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fouks Y, Many A, Shulman Y, Bak S, Shinar S. The contribution of an infectious workup in understanding stillbirth. Am. J. Perinatol. 2021;38:377–382. doi: 10.1055/s-0039-1697668. [DOI] [PubMed] [Google Scholar]
- 8.Fiumara NJ. A legacy of syphilis. Arch. Dermatol. 1965;92:676–678. [PubMed] [Google Scholar]
- 9.Crane J, et al. Parvovirus B19 infection in pregnancy. J. Obstet. Gynaecol. Can. 2014;36:1107–1116. doi: 10.1016/S1701-2163(15)30390-X. [DOI] [PubMed] [Google Scholar]
- 10.Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. 2014;345:760–765. doi: 10.1126/science.1251816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatesh KK, et al. Association of chorioamnionitis and its duration with neonatal morbidity and mortality. J. Perinatol. 2019;39:673–682. doi: 10.1038/s41372-019-0322-0. [DOI] [PubMed] [Google Scholar]
- 12.Stoll BJ, et al. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. N. Engl. J. Med. 2002;347:240–247. doi: 10.1056/NEJMoa012657. [DOI] [PubMed] [Google Scholar]
- 13.Oh JW, Park CW, Moon KC, Park JS, Jun JK. The relationship among the progression of inflammation in umbilical cord, fetal inflammatory response, early-onset neonatal sepsis, and chorioamnionitis. PLoS ONE. 2019;14:e0225328. doi: 10.1371/journal.pone.0225328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoll BJ, et al. Early-onset neonatal sepsis 2015 to 2017, the rise of Escherichia coli, and the need for novel prevention strategies. JAMA Pediatr. 2020;174:e200593. doi: 10.1001/jamapediatrics.2020.0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nahmias AJ, Walls KW, Stewart JA, Herrmann KL, Flynt WJ. The ToRCH complex-perinatal infections associated with toxoplasma and rubella, cytomegol- and herpes simplex viruses. Pediatr. Res. 1971;5:405–406. [Google Scholar]
- 16.Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Sci. Immunol. 2019;4:eaat6114. doi: 10.1126/sciimmunol.aat6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maltepe E, Bakardjiev AI, Fisher SJ. The placenta: transcriptional, epigenetic, and physiological integration during development. J. Clin. Invest. 2010;120:1016–1025. doi: 10.1172/JCI41211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knöfler M, et al. Human placenta and trophoblast development: key molecular mechanisms and model systems. Cell. Mol. Life Sci. 2019;76:405–406. doi: 10.1007/s00018-019-03104-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas JR, et al. Phenotypic and functional characterization of first-trimester human placental macrophages, Hofbauer cells. J. Exp. Med. 2020;218:e20200891. doi: 10.1084/jem.20200891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellery PM, Cindrova-Davies T, Jauniaux E, Ferguson-Smith AC, Burton GJ. Evidence for transcriptional activity in the syncytiotrophoblast of the human placenta. Placenta. 2009;30:329–334. doi: 10.1016/j.placenta.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guttman JA, Finlay BB. Tight junctions as targets of infectious agents. Biochim. Biophys. Acta. 2009;1788:832–841. doi: 10.1016/j.bbamem.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 22.Zeldovich VB, et al. Placental syncytium forms a biophysical barrier against pathogen invasion. PLoS Pathog. 2013;9:1–10. doi: 10.1371/journal.ppat.1003821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ander SE, et al. Human placental syncytiotrophoblasts restrict Toxoplasma gondii attachment and replication and respond to infection by producing immunomodulatory chemokines. mBio. 2018;9:e01678-17. doi: 10.1128/mBio.01678-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robbins JR, Skrzypczynska KM, Zeldovich VB, Kapidzic M, Bakardjiev AI. Placental syncytiotrophoblast constitutes a major barrier to vertical transmission of Listeria monocytogenes. PLoS Pathog. 2010;6:e1000732. doi: 10.1371/journal.ppat.1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delorme-Axford E, et al. Human placental trophoblasts confer viral resistance to recipient cells. Proc. Natl Acad. Sci. USA. 2013;110:12048–12053. doi: 10.1073/pnas.1304718110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delorme-Axford E, Sadovsky Y, Coyne CB. Lipid raft- and src family kinase-dependent entry of Coxsackievirus B into human placental trophoblasts. J. Virol. 2013;87:8569–8581. doi: 10.1128/JVI.00708-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stein KR, et al. CD46 facilitates entry and dissemination of human cytomegalovirus. Nat. Commun. 2019;10:2699. doi: 10.1038/s41467-019-10587-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemmings DG, Kilani R, Nykiforuk C, Preiksaitis J, Guilbert LJ. Permissive cytomegalovirus infection of primary villous term and first trimester trophoblasts. J. Virol. 1998;72:4970–4979. doi: 10.1128/jvi.72.6.4970-4979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vento-Tormo R, et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature. 2018;563:347–353. doi: 10.1038/s41586-018-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pique-Regi R, et al. Single cell transcriptional signatures of the human placenta in term and preterm parturition. eLife. 2019;8:e52004. doi: 10.7554/eLife.52004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinaldi SF, Makieva S, Saunders PT, Rossi AG, Norman JE. Immune cell and transcriptomic analysis of the human decidua in term and preterm parturition. Mol. Hum. Reprod. 2017;23:708–724. doi: 10.1093/molehr/gax038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamilton S, et al. Macrophages Infiltrate the human and rat decidua during term and preterm labor: evidence that decidual inflammation precedes labor1. Biol. Reprod. 2012;86:39. doi: 10.1095/biolreprod.111.095505. [DOI] [PubMed] [Google Scholar]
- 33.Kwan M, et al. Dynamic changes in maternal decidual leukocyte populations from first to second trimester gestation. Placenta. 2014;35:1027–1034. doi: 10.1016/j.placenta.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 34.Smith SD, Dunk CE, Aplin JD, Harris LK, Jones RL. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am. J. Pathol. 2009;174:1959–1971. doi: 10.2353/ajpath.2009.080995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris LK, Benagiano M, D’Elios MM, Brosens I, Benagiano G. Placental bed research: II. Functional and immunological investigations of the placental bed. Am. J. Obstet. Gynecol. 2019;221:457–469. doi: 10.1016/j.ajog.2019.07.010. [DOI] [PubMed] [Google Scholar]
- 36.Pereira L, Maidji E. Cytomegalovirus infection in the human placenta: maternal immunity and developmentally regulated receptors on trophoblasts converge. Curr. Top. Microbiol. Immunol. 2008;325:383–395. doi: 10.1007/978-3-540-77349-8_21. [DOI] [PubMed] [Google Scholar]
- 37.Weisblum Y, et al. Zika virus infects early- and midgestation human maternal decidual tissues, inducing distinct innate tissue responses in the maternal-fetal interface. J. Virol. 2017;91:e01905–e01916. doi: 10.1128/JVI.01905-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rizzuto G, Tagliani E, Manandhar P, Erlebacher A, Bakardjiev AI. Limited colonization undermined by inadequate early immune responses defines the dynamics of decidual listeriosis. Infect. Immun. 2017;85:e00153–17. doi: 10.1128/IAI.00153-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ning F, Liu H, Lash GE. The role of decidual macrophages during normal and pathological pregnancy. Am. J. Reprod. Immunol. 2016;75:298–309. doi: 10.1111/aji.12477. [DOI] [PubMed] [Google Scholar]
- 40.Jiang X, Du MR, Li M, Wang H. Three macrophage subsets are identified in the uterus during early human pregnancy. Cell. Mol. Immunol. 2018;15:1027–1037. doi: 10.1038/s41423-018-0008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crespo ÂC, et al. Decidual NK cells transfer granulysin to selectively kill bacteria in trophoblasts. Cell. 2020;182:1125–1139.e18. doi: 10.1016/j.cell.2020.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rogers LM, et al. Decidual stromal cell-derived PGE2 regulates macrophage responses to microbial threat. Am. J. Reprod. Immunol. 2018;80:e13032. doi: 10.1111/aji.13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Croxatto D, et al. Stromal cells from human decidua exert a strong inhibitory effect on NK cell function and dendritic cell differentiation. PLoS ONE. 2014;9:e89006. doi: 10.1371/journal.pone.0089006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castro-Leyva V, et al. Decidualization mediated by steroid hormones modulates the innate immunity in response to group B streptococcal infection in vitro. Gynecol. Obstet. Invest. 2017;82:592–600. doi: 10.1159/000454770. [DOI] [PubMed] [Google Scholar]
- 45.Xu X, et al. Monocyte chemoattractant protein-1 secreted by decidual stromal cells inhibits NK cells cytotoxicity by up-regulating expression of SOCS3. PLoS ONE. 2012;7:e41869. doi: 10.1371/journal.pone.0041869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guzeloglu-Kayisli O, et al. Zika virus–infected decidual cells elicit a gestational age–dependent innate immune response and exaggerate trophoblast zika permissiveness: implication for vertical transmission. J. Immunol. 2020;205:3083–3094. doi: 10.4049/jimmunol.2000713. [DOI] [PubMed] [Google Scholar]
- 47.Tabata T, Petitt M, Fang-Hoover J, Pereira L. Survey of cellular immune responses to human cytomegalovirus infection in the microenvironment of the uterine–placental interface. Med. Microbiol. Immunol. 2019;208:475–485. doi: 10.1007/s00430-019-00613-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bortolotti D, et al. Human herpes simplex 1 virus infection of endometrial decidual tissue-derived MSC alters HLA-G expression and immunosuppressive functions. Hum. Immunol. 2018;79:800–808. doi: 10.1016/j.humimm.2018.08.006. [DOI] [PubMed] [Google Scholar]
- 49.Deshmukh H, Way SS. Immunological basis for recurrent fetal loss and pregnancy complications. Annu. Rev. Pathol. Mech. Dis. 2019;14:185–210. doi: 10.1146/annurev-pathmechdis-012418-012743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reyes L, Golos TG. Hofbauer cells: their role in healthy and complicated pregnancy. Front. Immunol. 2018;9:2628. doi: 10.3389/fimmu.2018.02628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bayer A, et al. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe. 2016;19:705–712. doi: 10.1016/j.chom.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corry J, Arora N, Good CA, Sadovsky Y, Coyne CB. Organotypic models of type III interferon-mediated protection from Zika virus infections at the maternal-fetal interface. Proc. Natl Acad. Sci. USA. 2017;114:9433–9438. doi: 10.1073/pnas.1707513114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jagger BW, et al. Gestational stage and IFN-λ signaling regulate ZIKV infection in utero. Cell Host Microbe. 2017;22:366–376.e3. doi: 10.1016/j.chom.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen J, et al. Outcomes of congenital Zika disease depend on timing of infection and maternal-fetal interferon action. Cell Rep. 2017;21:1588–1599. doi: 10.1016/j.celrep.2017.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bayer A, et al. Chromosome 19 microRNAs exert antiviral activity independent from type III interferon signaling. Placenta. 2018;61:33–38. doi: 10.1016/j.placenta.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bayer A, et al. Human trophoblasts confer resistance to viruses implicated in perinatal infection. Am. J. Obstet. Gynecol. 2015;212:71.e1–71.e8. doi: 10.1016/j.ajog.2014.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dumont TMF, et al. The expression level of C19MC miRNAs in early pregnancy and in response to viral infection. Placenta. 2017;53:23–29. doi: 10.1016/j.placenta.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yockey LJ, et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018;3:eaao1680. doi: 10.1126/sciimmunol.aao1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buchrieser J, et al. IFITM proteins inhibit placental syncytiotrophoblast formation and promote fetal demise. Science. 2019;365:176–180. doi: 10.1126/science.aaw7733. [DOI] [PubMed] [Google Scholar]
- 60.Zani A, et al. Interferon-induced transmembrane proteins inhibit cell fusion mediated by trophoblast syncytins. J. Biol. Chem. 2019;294:19844–19851. doi: 10.1074/jbc.AC119.010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robbins JR, Zeldovich VB, Poukchanski A, Boothroyd JC, Bakardjiev AI. Tissue barriers of the human placenta to infection with Toxoplasma gondii. Infect. Immun. 2012;80:418–428. doi: 10.1128/IAI.05899-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Megli C, Morosky S, Rajasundaram D, Coyne CB. Inflammasome signaling in human placental trophoblasts regulates immune defense against Listeria monocytogenes infection. J. Exp. Med. 2021;218:e20200649. doi: 10.1084/jem.20200649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reis AS, et al. Inflammasome activation and IL-1 signaling during placental malaria induce poor pregnancy outcomes. Sci. Adv. 2020;6:eaax6346. doi: 10.1126/sciadv.aax6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thomas J, et al. Outbreak of listeriosis in South Africa associated with processed meat. N. Engl. J. Med. 2020;382:632–643. doi: 10.1056/NEJMoa1907462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan BT, Hohmann E, Barshak MB, Pukkila-Worley R. Treatment of listeriosis in first trimester of pregnancy. Emerg. Infect. Dis. 2013;19:839–841. doi: 10.3201/eid1905.121397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chan LM, Lin HH, Hsiao SM. Successful treatment of maternal Listeria monocytogenes bacteremia in the first trimester of pregnancy: a case report and literature review. Taiwan. J. Obstet. Gynecol. 2018;57:462–463. doi: 10.1016/j.tjog.2018.04.025. [DOI] [PubMed] [Google Scholar]
- 67.Al-Tawfiq JA. Listeria monocytogenes bacteremia in a twin pregnancy with differential outcome: fetus papyraceus and a full-term delivery. J. Microbiol. Immunol. Infect. 2008;41:433–436. [PubMed] [Google Scholar]
- 68.Mylonakis E, Paliou M, Hohmann EL, Calderwood SB, Wing EJ. Listeriosis during pregnancy: a case series and review of 222 cases. Medicine. 2002;81:260–269. doi: 10.1097/00005792-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 69.Phelps CC, et al. Relative roles of Listeriolysin O, InlA, and InlB in Listeria monocytogenes uptake by host cells. Infect. Immun. 2018;86:e00555-18. doi: 10.1128/IAI.00555-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lecuit M, et al. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: role of internalin interaction with trophoblast E-cadherin. Proc. Natl Acad. Sci. USA. 2004;101:6152–6157. doi: 10.1073/pnas.0401434101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holch A, Ingmer H, Licht TR, Gram L. Listeria monocytogenes strains encoding premature stop codons in inlA invade mice and guinea pig fetuses in orally dosed dams. J. Med. Microbiol. 2013;62:1799–1806. doi: 10.1099/jmm.0.057505-0. [DOI] [PubMed] [Google Scholar]
- 72.Disson O, et al. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature. 2008;455:1114–1118. doi: 10.1038/nature07303. [DOI] [PubMed] [Google Scholar]
- 73.Morrison HA, Lowe D, Robbins JR, Bakardjiev AI. In vivo virulence characterization of pregnancy-associated Listeria monocytogenes infections. Infect. Immun. 2018;86:e00397-18. doi: 10.1128/IAI.00397-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bakardjiev AI, Theriot JA, Portnoy DA. Listeria monocytogenes traffics from maternal organs to the placenta and back. PLoS Pathog. 2006;2:623–631. doi: 10.1371/journal.ppat.0020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Monnier A, et al. ActA is required for crossing of the fetoplacental barrier by Listeria monocytogenes. Infect. Immun. 2007;75:950–957. doi: 10.1128/IAI.01570-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Faralla C, et al. Listeria monocytogenes InlP interacts with afadin and facilitates basement membrane crossing. PLoS Pathog. 2018;14:e1007094. doi: 10.1371/journal.ppat.1007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parkash V, et al. Immunohistochemical detection of Listeria antigens in the placenta in perinatal listeriosis. Int. J. Gynecol. Pathol. 1998;17:343–350. doi: 10.1097/00004347-199810000-00008. [DOI] [PubMed] [Google Scholar]
- 78.Wolfe B, et al. Sequelae of fetal infection in a non-human primate model of listeriosis. Front. Microbiol. 2019;10:2021. doi: 10.3389/fmicb.2019.02021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Radolf JD, Kumar S. The Treponema pallidum outer membrane. Curr. Topics Microbiol. Immunol. 2018;415:1–38. doi: 10.1007/82_2017_44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gomez GB, et al. Untreated maternal syphilis and adverse outcomes of pregnancy: a systematic review and meta-analysis. Bull. World Health Organ. 2013;91:217–226. doi: 10.2471/BLT.12.107623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korenromp EL, et al. Global burden of maternal and congenital syphilis and associated adverse birth outcomes — estimates for 2016 and progress since 2012. PLoS ONE. 2019;14:e0211720. doi: 10.1371/journal.pone.0211720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Newman L, et al. Global estimates of syphilis in pregnancy and associated adverse outcomes: analysis of multinational antenatal surveillance data. PLoS Med. 2013;10:e1001396. doi: 10.1371/journal.pmed.1001396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peeling RW, Hook EW. The pathogenesis of syphilis: the Great Mimicker, revisited. J. Pathol. 2006;208:224–232. doi: 10.1002/path.1903. [DOI] [PubMed] [Google Scholar]
- 84.Wicher V, Wicher K. Pathogenesis of maternal-fetal syphilis revisited. Clin. Infect. Dis. 2001;33:354–363. doi: 10.1086/321904. [DOI] [PubMed] [Google Scholar]
- 85.Rac MWF, et al. Progression of ultrasound findings of fetal syphilis after maternal treatment. Am. J. Obstet. Gynecol. 2014;211:426.e1–426.e6. doi: 10.1016/j.ajog.2014.05.049. [DOI] [PubMed] [Google Scholar]
- 86.Kimball A, et al. Missed opportunities for prevention of congenital syphilis — United States, 2018. MMWR. 2020;69:661–665. doi: 10.15585/mmwr.mm6922a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Balaji G, Kalaivani S. Observance of Kassowitz law — late congenital syphilis: palatal perforation and saddle nose deformity as presenting features. Indian J. Sex. Transm. Dis. 2013;34:35–37. doi: 10.4103/2589-0557.112869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kassowitz, M. Die Vererbung der Syphilis (Kessinger Publishing, 1876).
- 89.Dhanaselvi H, Kalaivani S. Untreated late latent syphilis of both spouses with observation of Kassowitz law: adverse pregnancy outcomes in the postpenicillin era. Indian. J. Dermatol. 2017;62:221–222. doi: 10.4103/0019-5154.201755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wicher V, Baughn RE, Wicher K. Congenital and neonatal syphilis in guinea-pigs show a different pattern of immune response. Immunology. 1994;82:404–409. [PMC free article] [PubMed] [Google Scholar]
- 91.Wicher K, Abbruscato F, Wicher V, Baughn R, Noordhoek GT. Target organs of infection in guinea pigs with acquired or congenital syphilis. Infect. Immun. 1996;64:3174–3179. doi: 10.1128/iai.64.8.3174-3179.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sheffield JS, et al. Placental histopathology of congenital syphilis. Obstet. Gynecol. 2002;100:126–133. doi: 10.1016/s0029-7844(02)02010-0. [DOI] [PubMed] [Google Scholar]
- 93.Genest DR, et al. Diagnosis of congenital syphilis from placental examination: comparison of histopathology, steiner stain, and polymerase chain reaction for Treponema pallidum DNA. Hum. Pathol. 1996;27:366–372. doi: 10.1016/s0046-8177(96)90110-0. [DOI] [PubMed] [Google Scholar]
- 94.Myles TD, Elam G, Park-Hwang E, Nguyen T. The Jarisch–Herxheimer reaction and fetal monitoring changes in pregnant women treated for syphilis. Obstet. Gynecol. 1998;92:859–864. doi: 10.1016/s0029-7844(98)00271-3. [DOI] [PubMed] [Google Scholar]
- 95.Romero R, et al. The role of inflammation and infection in preterm birth. Semin. Reprod. Med. 2007;25:21–39. doi: 10.1055/s-2006-956773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nagy I, Pap K, Dicső F, Arany I. Chorioamnionitis is still the main cause of preterm birth. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016;206:e84. [Google Scholar]
- 97.Mendz GL, Kaakoush NO, Quinlivan JA. Bacterial aetiological agents of intra-amniotic infections and preterm birth in pregnant women. Front. Cell. Infect. Microbiol. 2013;3:58. doi: 10.3389/fcimb.2013.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Romero R, et al. Sterile and microbial-associated intra-amniotic inflammation in preterm prelabor rupture of membranes. J. Matern. Fetal. Neonatal Med. 2015;28:1394–1409. doi: 10.3109/14767058.2014.958463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schrag SJ, et al. A population-based comparison of strategies to prevent early-onset group B streptococcal disease in neonates. N. Engl. J. Med. 2002;347:233–239. doi: 10.1056/NEJMoa020205. [DOI] [PubMed] [Google Scholar]
- 100.Ayala OD, et al. Raman microspectroscopy differentiates perinatal pathogens on ex vivo infected human fetal membrane tissues. J. Biophotonics. 2019;12:e201800449. doi: 10.1002/jbio.201800449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Verani JR, McGee L, Schrag SJ, Division of Bacterial Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention (CDC). Prevention of perinatal group B streptococcal disease — revised guidelines from CDC, 2010. MMWR Recomm. Rep. 2010;59:1–36. [PubMed] [Google Scholar]
- 102.Doster RS, Sutton JA, Rogers LM, Aronoff DM, Gaddy JA. Streptococcus agalactiae induces placental macrophages to release extracellular traps loaded with tissue remodeling enzymes via an oxidative burst-dependent mechanism. mBio. 2018;9:e02084-18. doi: 10.1128/mBio.02084-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kothary V, et al. Group B Streptococcus induces neutrophil recruitment to gestational tissues and elaboration of extracellular traps and nutritional immunity. Front. Cell. Infect. Microbiol. 2017;7:19. doi: 10.3389/fcimb.2017.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Armistead B, Oler E, Adams Waldorf K, Rajagopal L. The double life of group B Streptococcus: asymptomatic colonizer and potent pathogen. J. Mol. Biol. 2019;431:2914–2931. doi: 10.1016/j.jmb.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vornhagen J, Adams Waldorf KM, Rajagopal L. Perinatal group B streptococcal infections: virulence factors, immunity, and prevention strategies. Trends Microbiol. 2017;25:919–931. doi: 10.1016/j.tim.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Galask RP, Varner MW, Rosemarie Petzold C, Wilbur SL. Bacterial attachment to the chorioamniotic membranes. Am. J. Obstet. Gynecol. 1984;148:915–928. doi: 10.1016/0002-9378(84)90534-9. [DOI] [PubMed] [Google Scholar]
- 107.Wilkie GL, et al. Microbiology and antibiotic resistance in peripartum bacteremia. Obstet. Gynecol. 2019;133:269–275. doi: 10.1097/AOG.0000000000003055. [DOI] [PubMed] [Google Scholar]
- 108.Sáez-López E, et al. Vaginal versus obstetric infection Escherichia coli isolates among pregnant women: antimicrobial resistance and genetic virulence profile. PLoS ONE. 2016;11:e0146531. doi: 10.1371/journal.pone.0146531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pathirana J, et al. Prevalence of congenital cytomegalovirus infection and associated risk of in-utero HIV acquisition in a high HIV prevalence setting, South Africa. Clin. Infect. Dis. 2019;69:1789–1796. doi: 10.1093/cid/ciz019. [DOI] [PubMed] [Google Scholar]
- 110.Fowler KB, et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N. Engl. J. Med. 1992;326:663–667. doi: 10.1056/NEJM199203053261003. [DOI] [PubMed] [Google Scholar]
- 111.Boppana SB, Rivera LB, Fowler KB, Mach M, Britt WJ. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 2001;344:1366–1371. doi: 10.1056/NEJM200105033441804. [DOI] [PubMed] [Google Scholar]
- 112.Enders G, Daiminger A, Bäder U, Exler S, Enders M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 2011;52:244–246. doi: 10.1016/j.jcv.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 113.Uenaka M, et al. Histopathological analysis of placentas with congenital cytomegalovirus infection. Placenta. 2019;75:62–67. doi: 10.1016/j.placenta.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 114.Satosar A, Ramirez NC, Bartholomew D, Davis J, Nuovo GJ. Histologic correlates of viral and bacterial infection of the placenta associated with severe morbidity and mortality in the newborn. Hum. Pathol. 2004;35:536–545. doi: 10.1016/j.humpath.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 115.Njue A, et al. The role of congenital cytomegalovirus infection in adverse birth outcomes: a review of the potential mechanisms. Viruses. 2020;13:20. doi: 10.3390/v13010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schleiss MR. Congenital cytomegalovirus infection: molecular mechanisms mediating viral pathogenesis. Infect. Disord. Drug Targets. 2011;11:449–465. doi: 10.2174/187152611797636721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Britt WJ. Human cytomegalovirus infection in women with preexisting immunity: sources of infection and mechanisms of infection in the presence of antiviral immunity. J. Infect. Dis. 2020;221:S1–S8. doi: 10.1093/infdis/jiz464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schleiss MR, McVoy MA. Guinea pig cytomegalovirus: a model for the prevention and treatment of maternal–fetal cytomegalovirus transmission. Future Virol. 2010;5:207–217. doi: 10.2217/fvl.10.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Weisblum Y, et al. Modeling of human cytomegalovirus maternal–fetal transmission in a novel decidual organ culture. J. Virol. 2011;85:13204–13213. doi: 10.1128/JVI.05749-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aronoff DM, Correa H, Rogers LM, Arav-Boger R, Alcendor DJ. Placental pericytes and cytomegalovirus infectivity: implications for HCMV placental pathology and congenital disease. Am. J. Reprod. Immunol. 2017 doi: 10.1111/aji.12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Burgos JS, et al. Hematogenous vertical transmission of herpes simplex virus type 1 in mice. J. Virol. 2006;80:2823–2831. doi: 10.1128/JVI.80.6.2823-2831.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Koi H, et al. Syncytiotrophoblast is a barrier to maternal–fetal transmission of herpes simplex virus. Biol. Reprod. 2002;67:1572–1579. doi: 10.1095/biolreprod.102.004325. [DOI] [PubMed] [Google Scholar]
- 123.Finger-Jardim F, et al. Herpes simplex virus: prevalence in placental tissue and incidence in neonatal cord blood samples. J. Med. Virol. 2014;86:519–524. doi: 10.1002/jmv.23817. [DOI] [PubMed] [Google Scholar]
- 124.Finger-Jardim F, et al. Prevalence of herpes simplex virus types 1 and 2 at maternal and fetal sides of the placenta in asymptomatic pregnant women. Am. J. Reprod. Immunol. 2017;78:e12689. doi: 10.1111/aji.12689. [DOI] [PubMed] [Google Scholar]
- 125.Gay NJ, et al. Age specific antibody prevalence to parvovirus B19: how many women are infected in pregnancy? Commun. Dis. Rep. CDR Rev. 1994;4:R104–7. [PubMed] [Google Scholar]
- 126.Li JJ, Henwood T, Van Hal S, Charlton A. Parvovirus infection: an immunohistochemical study using fetal and placental tissue. Pediatr. Dev. Pathol. 2015;18:30–39. doi: 10.2350/14-05-1495-OA.1. [DOI] [PubMed] [Google Scholar]
- 127.Jordan JA, Deloia JA. Globoside expression within the human placenta. Placenta. 1999;20:103–108. doi: 10.1053/plac.1998.0353. [DOI] [PubMed] [Google Scholar]
- 128.Wegner CC, Jordan JA. Human parvovirus B19 VP2 empty capsids bind to human villous trophoblast cells in vitro via the globoside receptor. Infect. Dis. Obstet. Gynecol. 2004;12:69–78. doi: 10.1080/10647440400009912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Moffatt S, Yaegashi N, Tada K, Tanaka N, Sugamura K. Human parvovirus B19 nonstructural (NS1) protein induces apoptosis in erythroid lineage cells. J. Virol. 1998;72:3018–3028. doi: 10.1128/jvi.72.4.3018-3028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jordan JA, Butchko AR. Apoptotic activity in villous trophoblast cells during B19 infection correlates with clinical outcome: assessment by the caspase-related M30 cytodeath antibody. Placenta. 2002;23:547–553. doi: 10.1053/plac.2002.0843. [DOI] [PubMed] [Google Scholar]
- 131.Dickinson JE, Keil AD, Charles AK. Discordant fetal infection for parvovirus B19 in a dichorionic twin pregnancy. Twin Res. Hum. Genet. 2006;9:456–459. doi: 10.1375/183242706777591353. [DOI] [PubMed] [Google Scholar]
- 132.Schiesser M, Sergi C, Enders M, Maul H, Schnitzler P. Discordant outcomes in a case of parvovirus B19 transmission into both dichorionic twins. Twin Res. Hum. Genet. 2009;12:175–179. doi: 10.1375/twin.12.2.175. [DOI] [PubMed] [Google Scholar]
- 133.Bernstein HB, Wegman AD. HIV infection: antepartum treatment and management. Clin. Obstet. Gynecol. 2018;61:122–136. doi: 10.1097/GRF.0000000000000330. [DOI] [PubMed] [Google Scholar]
- 134.Maury W, Potts BJ, Rabson AB. HIV-1 infection of first-trimester and term human placental tissue: a possible mode of maternal-fetal transmission. J. Infect. Dis. 1989;160:583–588. doi: 10.1093/infdis/160.4.583. [DOI] [PubMed] [Google Scholar]
- 135.Amirhessami-Aghili N, Spector SA. Human immunodeficiency virus type 1 infection of human placenta: potential route for fetal infection. Dis. Markers. 1991;9:348. doi: 10.1128/jvi.65.5.2231-2236.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mattern CFT, et al. Localization of human immunodeficiency virus core antigen in term human placentas. Pediatrics. 1992;89:207–209. [PubMed] [Google Scholar]
- 137.Peuchmaur M, et al. HIV proteins absent from placentas of 75 HIV-1-positive women studied by immunohistochemistry. AIDS. 1991;5:741–745. doi: 10.1097/00002030-199106000-00015. [DOI] [PubMed] [Google Scholar]
- 138.Backe E, et al. Demonstration of HIV-1 infected cells in human placenta by in situ hybridisation and immunostaining. J. Clin. Pathol. 1992;45:871–874. doi: 10.1136/jcp.45.10.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Townsend CL, et al. Low rates of mother-to-child transmission of HIV following effective pregnancy interventions in the United Kingdom and Ireland, 2000–2006. AIDS. 2008;22:973–981. doi: 10.1097/QAD.0b013e3282f9b67a. [DOI] [PubMed] [Google Scholar]
- 140.Koay WLA, et al. Prevention of perinatal HIV transmission in an area of high HIV prevalence in the United States. J. Pediatr. 2021;228:101–109. doi: 10.1016/j.jpeds.2020.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Johnson EL, Chakraborty R. HIV-1 at the placenta: immune correlates of protection and infection. Curr. Opin. Infect. Dis. 2016;29:248–255. doi: 10.1097/QCO.0000000000000267. [DOI] [PubMed] [Google Scholar]
- 142.Mlakar J, et al. Zika virus associated with microcephaly. N. Engl. J. Med. 2016;374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 143.Brady OJ, et al. The association between Zika virus infection and microcephaly in Brazil 2015–2017: an observational analysis of over 4 million births. PLoS Med. 2019;16:e1002755. doi: 10.1371/journal.pmed.1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hoen B, et al. Pregnancy outcomes after ZIKV infection in French territories in the Americas. N. Engl. J. Med. 2018;378:985–994. doi: 10.1056/NEJMoa1709481. [DOI] [PubMed] [Google Scholar]
- 145.van der Linden V, et al. Description of 13 infants born during October 2015–January 2016 with congenital Zika virus infection without microcephaly at birth — Brazil. MMWR Morb. Mortal. Wkly Rep. 2016;65:1343–1348. doi: 10.15585/mmwr.mm6547e2. [DOI] [PubMed] [Google Scholar]
- 146.Sobhani NC, et al. Discordant Zika virus findings in twin pregnancies complicated by antenatal Zika virus exposure: a prospective cohort. J. Infect. Dis. 2020;221:1838–1845. doi: 10.1093/infdis/jiz629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Amaral MS, et al. Differential gene expression elicited by ZIKV infection in trophoblasts from congenital Zika syndrome discordant twins. PLoS Negl. Trop. Dis. 2020;14:e0008424. doi: 10.1371/journal.pntd.0008424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hastings AK, et al. TAM receptors are not required for Zika virus infection in mice. Cell Rep. 2017;19:558–568. doi: 10.1016/j.celrep.2017.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Hermanns K, et al. Zika virus infection in human placental tissue explants is enhanced in the presence of dengue virus antibodies in-vitro. Emerg. Microbes Infect. 2018;7:198. doi: 10.1038/s41426-018-0199-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zimmerman MG, et al. Cross-reactive dengue virus antibodies augment Zika virus infection of human placental macrophages. Cell Host Microbe. 2018;24:731–742.e6. doi: 10.1016/j.chom.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rodriguez-Barraquer I, et al. Impact of preexisting dengue immunity on Zika virus emergence in a dengue endemic region. Science. 2019;363:607–610. doi: 10.1126/science.aav6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Pedroso C, et al. Cross-protection of dengue virus infection against congenital Zika syndrome, northeastern Brazil. Emerg. Infect. Dis. 2019;25:1485–1493. doi: 10.3201/eid2508.190113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pantoja P, et al. Zika virus pathogenesis in rhesus macaques is unaffected by pre-existing immunity to dengue virus. Nat. Commun. 2017;8:15674. doi: 10.1038/ncomms15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Breitbach ME, et al. Primary infection with dengue or Zika virus does not affect the severity of heterologous secondary infection in macaques. PLoS Pathog. 2019;15:e1007766. doi: 10.1371/journal.ppat.1007766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Katzelnick LC, et al. Zika virus infection enhances future risk of severe dengue disease. Science. 2020;369:1123–1128. doi: 10.1126/science.abb6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dudley DM, et al. Using macaques to address critical questions in Zika virus research. Annu. Rev. Virol. 2019;6:481–500. doi: 10.1146/annurev-virology-092818-015732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Koenig MR, et al. Quantitative definition of neurobehavior, vision, hearing and brain volumes in macaques congenitally exposed to Zika virus. PLoS ONE. 2020;15:e0235877. doi: 10.1371/journal.pone.0235877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bebell LM, Oduyebo T, Riley LE. Ebola virus disease and pregnancy: a review of the current knowledge of Ebola virus pathogenesis, maternal, and neonatal outcomes. Birth Defects Res. 2017;109:353–362. doi: 10.1002/bdra.23558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Muehlenbachs A, et al. Ebola virus disease in pregnancy: clinical, histopathologic and immunohistochemical findings. J. Infect. Dis. 2015;215:64–69. doi: 10.1093/infdis/jiw206. [DOI] [PubMed] [Google Scholar]
- 160.Oduyebo T, et al. A pregnant patient with ebola virus disease. Obstet. Gynecol. 2015;126:1273–1275. doi: 10.1097/AOG.0000000000001092. [DOI] [PubMed] [Google Scholar]
- 161.Baudin M, et al. Association of Rift Valley fever virus infection with miscarriage in Sudanese women: a cross-sectional study. Lancet Glob. Heal. 2016;4:e864–e871. doi: 10.1016/S2214-109X(16)30176-0. [DOI] [PubMed] [Google Scholar]
- 162.Arishi HM, Aqeel AY, Al Hazmi MM. Vertical transmission of fatal Rift Valley fever in a newborn. Ann. Trop. Paediatr. 2006;26:251–253. doi: 10.1179/146532806X120363. [DOI] [PubMed] [Google Scholar]
- 163.Oymans J, Wichgers Schreur PJ, van Keulen L, Kant J, Kortekaas J. Rift valley fever virus targets the maternal–foetal interface in ovine and human placentas. PLoS Negl. Trop. Dis. 2020;14:1–18. doi: 10.1371/journal.pntd.0007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McMillen CM, et al. Rift Valley fever virus induces fetal demise in Sprague–Dawley rats through direct placental infection. Sci. Adv. 2018;4:eaau9812. doi: 10.1126/sciadv.aau9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.O’Leary DR, et al. Birth outcomes following west nile virus infection of pregnant women in the United States: 2003–2004. Pediatrics. 2006;117:e537–e545. doi: 10.1542/peds.2005-2024. [DOI] [PubMed] [Google Scholar]
- 166.Platt DJ, et al. Zika virus-related neurotropic flaviviruses infect human placental explants and cause fetal demise in mice. Sci. Transl. Med. 2018;10:eaao7090. doi: 10.1126/scitranslmed.aao7090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Julander JG, et al. West Nile virus infection of the placenta. Virology. 2006;347:175–182. doi: 10.1016/j.virol.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 168.Chen H, et al. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: a retrospective review of medical records. Lancet. 2020;395:809–815. doi: 10.1016/S0140-6736(20)30360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pique-Regi R, et al. Does the human placenta express the canonical cell entry mediators for SARS-CoV-2? eLife. 2020;9:e58716. doi: 10.7554/eLife.58716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li Y, et al. Lack of vertical transmission of severe acute respiratory syndrome coronavirus 2, China. Emerg. Infect. Dis. 2020;26:1335–1336. doi: 10.3201/eid2606.200287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Egloff C, Vauloup-Fellous C, Picone O, Mandelbrot L, Roques P. Evidence and possible mechanisms of rare maternal-fetal transmission of SARS-CoV-2. J. Clin. Virol. 2020;128:104447. doi: 10.1016/j.jcv.2020.104447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.WAPM working group on COVID-19 . Maternal and perinatal outcomes of pregnant women with SARS-COV-2 infection. Ultrasound Obstet. Gynecol. 2021;57:232–241. doi: 10.1002/uog.23107. [DOI] [PubMed] [Google Scholar]
- 173.Flaherman VJ, et al. Infant outcomes following maternal infection with SARS-CoV-2: first report from the PRIORITY study. Clin. Infect. Dis. 2020 doi: 10.1093/cid/ciaa1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Torgerson PR, Mastroiacovo P. La charge mondiale de la toxoplasmose: une étude systématique. Bull. World Health Organ. 2013;91:501–508. doi: 10.2471/BLT.12.111732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Galanakis E, et al. Outcome of toxoplasmosis acquired during pregnancy following treatment in both pregnancy and early infancy. Fetal Diagn. Ther. 2007;22:444–448. doi: 10.1159/000106352. [DOI] [PubMed] [Google Scholar]
- 176.Berrébi A, et al. Long-term outcome of children with congenital toxoplasmosis. Am. J. Obstet. Gynecol. 2010;203:552.e1–552.e6. doi: 10.1016/j.ajog.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 177.Dunn D, et al. Mother-to-child transmission of toxoplasmosis: risk estimates for clinical counselling. Lancet. 1999;353:1829–1833. doi: 10.1016/S0140-6736(98)08220-8. [DOI] [PubMed] [Google Scholar]
- 178.Mendez OA, Koshy AA. Toxoplasma gondii: entry, association, and physiological influence on the central nervous system. PLoS Pathog. 2017;13:e1006351. doi: 10.1371/journal.ppat.1006351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bisio H, Soldati-Favre D. Signaling cascades governing entry into and exit from host cells by Toxoplasma gondii. Annu. Rev. Microbiol. 2019;73:579–599. doi: 10.1146/annurev-micro-020518-120235. [DOI] [PubMed] [Google Scholar]
- 180.English ED, Adomako-Ankomah Y, Boyle JP. Secreted effectors in Toxoplasma gondii and related species: determinants of host range and pathogenesis? Parasite Immunol. 2015;37:127–140. doi: 10.1111/pim.12166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hunter CA, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 2012;10:766–778. doi: 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rudzki EN, et al. Toxoplasma gondii GRA28 is required for specific induction of the regulatory chemokine CCL22 in human and mouse cells. bioRxiv. 2020 doi: 10.1101/2020.10.14.335802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Luxemburger C, et al. Effects of malaria during pregnancy on infant mortality in an area of low malaria transmission. Am. J. Epidemiol. 1993;154:459–465. doi: 10.1093/aje/154.5.459. [DOI] [PubMed] [Google Scholar]
- 184.Luxemburger C, et al. The epidemiology of severe malaria in an area of low transmission in Thailand. Trans. R. Soc. Trop. Med. Hyg. 1997;91:256–262. doi: 10.1016/s0035-9203(97)90066-3. [DOI] [PubMed] [Google Scholar]
- 185.Guyatt HL, Snow RW. Malaria in pregnancy as an indirect cause of infant mortality in sub-Saharan Africa. Trans. R. Soc. Trop. Med. Hyg. 2001;95:569–576. doi: 10.1016/s0035-9203(01)90082-3. [DOI] [PubMed] [Google Scholar]
- 186.Desai M, et al. Epidemiology and burden of malaria in pregnancy. Lancet Infect. Dis. 2007;7:93–104. doi: 10.1016/S1473-3099(07)70021-X. [DOI] [PubMed] [Google Scholar]
- 187.Tobian AA, et al. Frequent umbilical cord-blood and maternal-blood infections with Plasmodium falciparum, P. malariae, and P. ovale in Kenya. J. Infect. Dis. 2000;182:558–563. doi: 10.1086/315729. [DOI] [PubMed] [Google Scholar]
- 188.Ouédraogo A, et al. Transplacental transmission of plasmodium falciparum in a highly malaria endemic area of Burkina Faso. J. Trop. Med. 2012;2012:109705. doi: 10.1155/2012/109705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Beeson JG, Amin N, Kanjala M, Rogerson SJ. Selective accumulation of mature asexual stages of Plasmodium falciparum-infected erythrocytes in the placenta. Infect. Immun. 2002;70:5412–5415. doi: 10.1128/IAI.70.10.5412-5415.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Muehlenbachs A, et al. A novel histological grading scheme for placental malaria applied in areas of high and low malaria transmission. J. Infect. Dis. 2010;202:1608–1616. doi: 10.1086/656723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Okoko BJ, et al. The influence of placental malaria infection and maternal hypergammaglobulinemia on transplacental transfer of antibodies and IgG subclasses in a rural west African population. J. Infect. Dis. 2001;184:627–632. doi: 10.1086/322808. [DOI] [PubMed] [Google Scholar]
- 192.Owens S, et al. Placental malaria and immunity to infant measles. Arch. Dis. Child. 2006;91:507–508. doi: 10.1136/adc.2005.085274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Frech C, Chen N. Variant surface antigens of malaria parasites: functional and evolutionary insights from comparative gene family classification and analysis. BMC Genomics. 2013;14:427. doi: 10.1186/1471-2164-14-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Beeson JG, Duffy PE. The immunology and pathogenesis of malaria during pregnancy. Curr. Top. Microbiol. Immunol. 2005;297:187–227. doi: 10.1007/3-540-29967-x_6. [DOI] [PubMed] [Google Scholar]
- 195.Salanti A, et al. Selective upregulation of a single distinctly structured var gene in chondroitin sulphate A-adhering Plasmodium falciparum involved in pregnancy-associated malaria. Mol. Microbiol. 2003;49:179–191. doi: 10.1046/j.1365-2958.2003.03570.x. [DOI] [PubMed] [Google Scholar]
- 196.Tuikue Ndam NG, et al. High level of var2csa transcription by Plasmodium falciparum isolated from the placenta. J. Infect. Dis. 2005;192:331–335. doi: 10.1086/430933. [DOI] [PubMed] [Google Scholar]
- 197.Magistrado P, et al. VAR2CSA expression on the surface of placenta-derived Plasmodium falciparum-infected erythrocytes. J. Infect. Dis. 2008;198:1071–1074. doi: 10.1086/591502. [DOI] [PubMed] [Google Scholar]
- 198.Chotivanich K, et al. Plasmodium vivax adherence to placental glycosaminoglycans. PLoS ONE. 2012;7:e34509. doi: 10.1371/journal.pone.0034509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Fried M, Duffy PE. Adherence of Plasmodium falciparum to chondroitin sulfate A in the human placenta. Science. 1996;272:1502–1504. doi: 10.1126/science.272.5267.1502. [DOI] [PubMed] [Google Scholar]
- 200.Fried M, Domingo GJ, Gowda CD, Mutabingwa TK, Duffy PE. Plasmodium falciparum: chondroitin sulfate A is the major receptor for adhesion of parasitized erythrocytes in the placenta. Exp. Parasitol. 2006;113:36–42. doi: 10.1016/j.exppara.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 201.O’Neil-Dunne I, et al. Gravidity-dependent production of antibodies that inhibit binding of Plasmodium falciparum-infected erythrocytes to placental chondroitin sulfate proteoglycan during pregnancy. Infect. Immun. 2001;69:7487–7492. doi: 10.1128/IAI.69.12.7487-7492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ricke CH, et al. Plasma antibodies from malaria-exposed pregnant women recognize variant surface antigens on plasmodium falciparum-infected erythrocytes in a parity-dependent manner and block parasite adhesion to chondroitin sulfate A. J. Immunol. 2000;165:3309–3316. doi: 10.4049/jimmunol.165.6.3309. [DOI] [PubMed] [Google Scholar]
- 203.Oleinikov AV, et al. Effects of sex, parity, and sequence variation on seroreactivity to candidate pregnancy malaria vaccine antigens. J. Infect. Dis. 2007;196:155–164. doi: 10.1086/518513. [DOI] [PubMed] [Google Scholar]
- 204.Duffy PE, Fried M. Antibodies that inhibit plasmodium falciparum adhesion to chondroitin sulfate A are associated with increased birth weight and the gestational age of newborns. Infect. Immun. 2003;71:6620–6623. doi: 10.1128/IAI.71.11.6620-6623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Fried M, Duffy PE. Malaria during pregnancy. Cold Spring Harb. Perspect. Med. 2017;7:a025551. doi: 10.1101/cshperspect.a025551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Staalsoe T, et al. Variant surface antigen-specific IgG and protection against clinical consequences of pregnancy-associated Plasmodium falciparum malaria. Lancet. 2004;363:283–289. doi: 10.1016/S0140-6736(03)15386-X. [DOI] [PubMed] [Google Scholar]
- 207.Rogerson SJ, et al. Placental monocyte infiltrates in response to Plasmodium falciparum malaria infection and their association with adverse pregnancy outcomes. Am. J. Trop. Med. Hyg. 2003;68:115–119. [PubMed] [Google Scholar]
- 208.Moormann AM, et al. Malaria and pregnancy: placental cytokine expression and its relationship to intrauterine growth retardation. J. Infect. Dis. 1999;180:1987–1993. doi: 10.1086/315135. [DOI] [PubMed] [Google Scholar]
- 209.Lima FA, et al. Plasmodium falciparum infection dysregulates placental autophagy. PLoS ONE. 2019;14:e0226117. doi: 10.1371/journal.pone.0226117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dimasuay KG, et al. Impaired placental autophagy in placental malaria. PLoS ONE. 2017;12:e0187291. doi: 10.1371/journal.pone.0187291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Rios L, Campos EE, Menon R, Zago MP, Garg NJ. Epidemiology and pathogenesis of maternal–fetal transmission of Trypanosoma cruzi and a case for vaccine development against congenital Chagas disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165591. doi: 10.1016/j.bbadis.2019.165591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Torrico F, et al. Maternal trypanosoma cruzi infection, pregnancy outcome, morbidity, and mortality of congenitally infected and non-infected newborns in bolivia. Am. J. Trop. Med. Hyg. 2004;70:201–209. [PubMed] [Google Scholar]
- 213.Torrico F, et al. Are maternal re-infections with Trypanosoma cruzi associated with higher morbidity and mortality of congenital Chagas disease? Trop. Med. Int. Heal. 2006;11:628–635. doi: 10.1111/j.1365-3156.2006.01623.x. [DOI] [PubMed] [Google Scholar]
- 214.Siriano LDR, Luquetti AO, Avelar JB, Marra NL, De Castro AM. Chagas disease: increased parasitemia during pregnancy detected by hemoculture. Am. J. Trop. Med. Hyg. 2011;84:569–574. doi: 10.4269/ajtmh.2011.10-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Brutus L, et al. Short report: Detectable Trypanosoma cruzi parasitemia during pregnancy and delivery as a risk factor for congenital chagas disease. Am. J. Trop. Med. Hyg. 2010;83:1044–1047. doi: 10.4269/ajtmh.2010.10-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Duaso J, et al. Trypanosoma cruzi induces apoptosis in ex vivo infected human chorionic villi. Placenta. 2011;32:356–361. doi: 10.1016/j.placenta.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 217.Duaso J, et al. Trypanosoma cruzi induces tissue disorganization and destruction of chorionic villi in an ex vivo infection model of human placenta. Placenta. 2010;31:705–711. doi: 10.1016/j.placenta.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 218.Castillo C, et al. Trypanosoma cruzi exosomes increases susceptibility to parasite infection in human placental chorionic villi explants. Placenta. 2017;51:123–124. [Google Scholar]
- 219.Roberts RM, Green JA, Schulz LC. The evolution of the placenta. Reproduction. 2016;152:R179–R189. doi: 10.1530/REP-16-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Turco MY, et al. Trophoblast organoids as a model for maternal–fetal interactions during human placentation. Nature. 2018;564:263–281. doi: 10.1038/s41586-018-0753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Turco MY, et al. Long-term, hormone-responsive organoid cultures of human endometrium in a chemically defined medium. Nat. Cell Biol. 2017;19:568–577. doi: 10.1038/ncb3516. [DOI] [PMC free article] [PubMed] [Google Scholar]