

Abstract
FAK, a nonreceptor tyrosine kinase, has been recognized as a novel target class for the development of targeted anticancer agents. Overexpression of FAK is a common occurrence in several solid tumors, in which the kinase has been implicated in promoting metastases. Consequently, designing and developing potent FAK inhibitors is becoming an attractive goal, and FAK inhibitors are being recognized as a promising tool in our armamentarium for treating diverse cancers. This review comprehensively summarizes the different classes of synthetically derived compounds that have been reported as potent FAK inhibitors in the last three decades. Finally, the future of FAK-targeting smart drugs that are designed to slow down the emergence of drug resistance is discussed.
Keywords: : cancer, FAK inhibitors, focal adhesion kinase, PROTACs
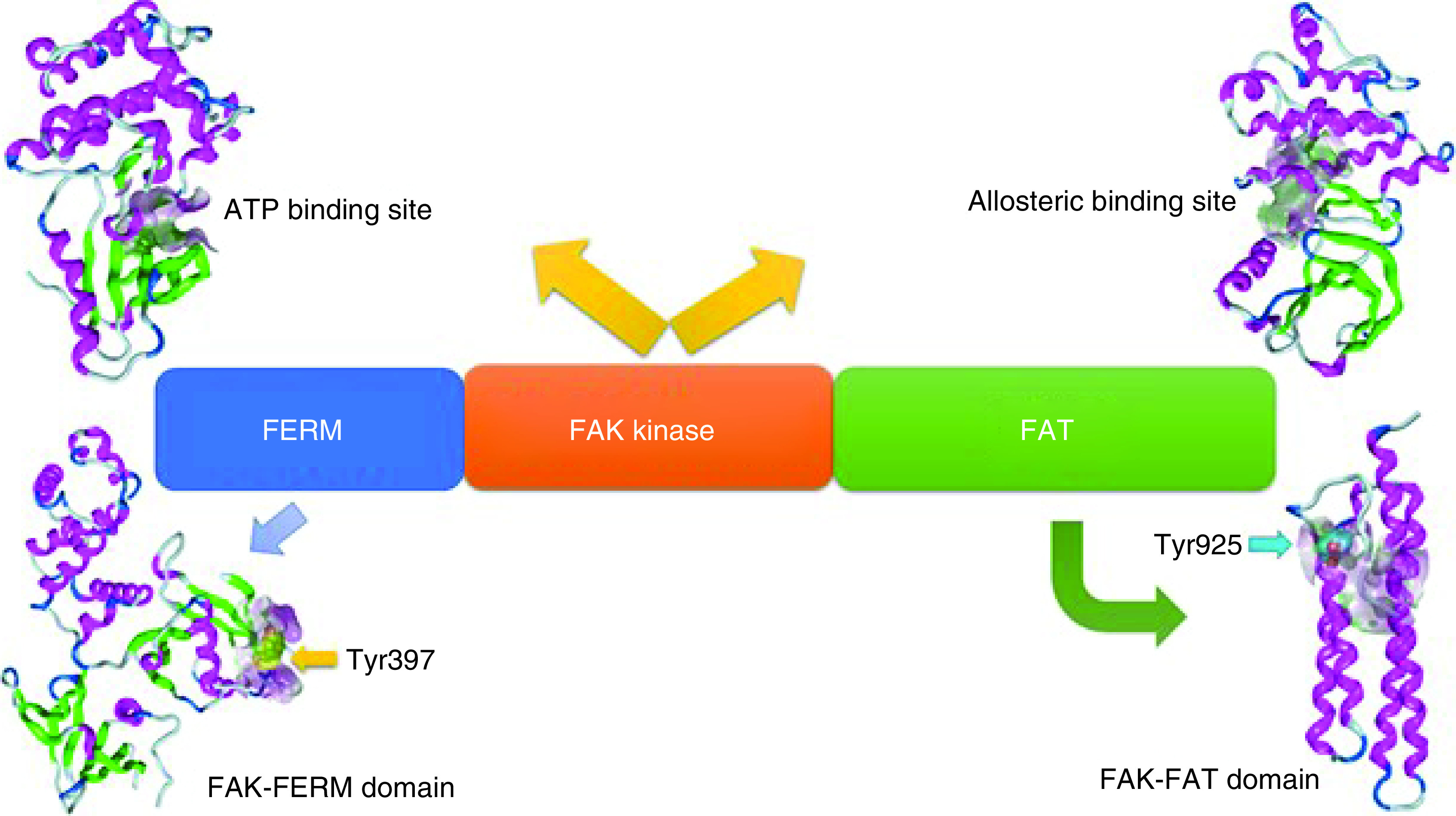
Cell migration performs significant roles not only in diverse biological processes, such as embryonic development, but also in various diseases, including fibrogenic disorders and cancer [1–3]. Cell migration involves several steps, including leading-edge protrusion, focal adhesions turnover, tractional forces generation, and tail retraction and detachment [4,5]. At each cell migration step, the integrin family, as cellular receptors for the extracellular matrix (ECM), is fundamental for cell adhesion [6]. Integrins can regulate cell migration and functions, since they can cocluster with several signaling molecules in focal adhesions. Several proteins are located in the focal adhesions, such as filamin, fibronectin, integrin, talin and FAK. FAK, also known as protein tyrosine kinase 2 (PTK2), is a nonreceptor tyrosine kinase with a weight of approximately 125 kDa. FAK is one of the most essential and the earliest identified signaling molecules. FAK was given its name in 1992 when discovered in the cell adhesion area near v-Src transformed chick embryo [3,7–10]. The interactions of FAK with growth factor receptors and integrins contribute to cell anchorage dependency, invasive growth, and motility, which are correlated with malignant overgrowth. The human FAK protein is comprised of central kinase domain, which contains the activation loop (2 tyrosine residues: Tyr576 and Tyr577) that can be phosphorylated by Src and is required for FAK kinase activity. This middle domain is flanked by an amino-terminal FERM (band 4.1-ezrin-radixin-moesin) domain that interacts with growth factor receptors and integrin (Figure 1). The third domain is a carboxy-terminal focal adhesion targeting (FAT) sequence, which obliquely connects FAK to integrins and contains Tyr925, a key phosphorylation site [11,12]. The FAT region as a tetra-helical structure provides binding of FAK to the focal adhesion proteins paxilin and talin, a necessary process for cell adhesion. FAK includes three regions rich in proline (PR1-3). Their role is to mediate binding with various molecular effectors and regulators, such as Src homology 3 (SH3)-domain-containing proteins (Figure 1) [13]. FAK is capable of enhancing cell survival in a kinase-independent manner, since FAK FERM domain forms phosphorylation-independent complexes with different proteins [14,15].
Figure 1. . The FAK structure.

PDB: Protein Data Bank.
FAK activity is described as the ability of this tyrosine kinase to transfer a phosphate group from ATP onto site-specific tyrosine residues in substrate proteins such as p130Crk-associated substrate (CAS), paxillin, cortactin, Src, VE-cadherin, α-actinin and β-catenin as a means of protein function alteration [16]. But, like other kinases, FAK's activity is spatiotemporally regulated by other cellular proteins and events. For example, the interactions of FAK with membrane receptors such as integrins are assumed to induce FAK conformational switch to expose Tyr397 phosphorylation sites [17]. This is accomplished through the binding of FERM to phosphatidylinositol-4,5-biphosphate [PI(4,5)P2], leading to transient FERM: FERM and FERM: FAT dimerization, allowing FAK autophosphorylation at Tyr397. FAK Tyr397 phosphorylation generates a binding site for the SH2 domain of Src [11,18]. FAK phosphorylation at Tyr397 can elevate the binding of FAK and the phosphorylation of acceptor proteins (Src), which play a fundamental role in cell migration. This pTyr397 affords a docking site for the acceptor protein Src, causing phosphorylation of additional sites on FAK enzyme and creating a high-affinity active site for SH2 domain of Src [19,20]. This FAK-Src complex was found to be crucial in the FAK-associated signaling process. Moreover, the binding of Src with FAK-Tyr397 residue leads to phosphorylation of two residues, Tyr576 and Tyr577, placed on FAK catalytic site and Tyr861 residue required for total catalytic activity. When FAK activates Src, various signal transduction pathways are stimulated, including PI3K-Akt, Rho/Rac/PAK, and RAF/JNK, leading to cell mobility and cell survival modulation [21,22]. FAK hyperphosphorylation and overexpression are directly associated with several kinds of solid tumors [23–25]. FAK overexpression can boost cell motility and survival as it contributes to tumorigenicity and metastasis. FAK hyperactivity improves the resistance of tumor cells to traditional anticancer therapy [26]. The N- and C-terminal sequences of FAK produce an autoinhibitory structure, and caspase cleavage of FAK results in the removal of this autoinhibition. Subsequently, FAT domain is released, leading to the dephosphorylation of FAK and induction of cell death [11,27]. Epithelial-to-mesenchymal transition (EMT) is a functional transition accompanied by multiple biochemical changes of epithelial cells during embryogenesis, organ development and repair, and various pathological processes (such as tumor invasiveness and metastasis). Overexpression of FAK was also found to be associated with tumor invasiveness and metastasis characteristics of the EMT process.
At the molecular level, the association of VE-cadherin and FAK is mediated by FERM domain binding to the VE-cadherin cytoplasmic domain. Activation of FAK at both adherens junctions (protein complexes that form epithelial and endothelial cell–cell interactions) and adhesions likely promotes crosstalk between cadherin receptors and integrin [28]. Previous studies revealed that genetic inhibition of FAK in endothelial cells lessened β-catenin and VE-cadherin phosphorylation and prevented metastasis and extravasation of tumor cells [29,30]. Additionally, many studies have shown the crucial role played by FAK enzyme in regulating adhesion dynamics. Intracellular Ca-dependent proteolysis mediated by calpain family members was proposed to regulate cell migration by modulating adhesion dynamics. Focal adhesion proteins such as FAK, talin and paxillin are targets for calpain family, and these targets' proteolysis was found to contribute to regulating both adhesion dynamics and cell migration [31]. FAK also augments the rate of proteosomal degradation of the tumor suppressor p53; this has been proposed as a key mechanism by which FAK promotes cell survival under stressful growth conditions [32–34]. FAK binds to and defends nucleostemin, a protein that binds to p53, regulates cell cycle and cell differentiation, and is a marker for cancer stem cells from stress-induced degradation [15,35]. In addition to promoting survival, FAK also suppresses apoptosis by binding to the death domain of receptor-interacting protein (RIP). Such binding leads to cell survival, enhanced cell adhesion, migration and promotion of cell cycle [36]. Tumor cells subjected to FAK attenuation experience apoptosis mediated by Fas-associated death domain protein (FADD) and caspase-8 [37,38].
Over the past three decades, evidence has solidified the role of FAK in anchorage-independent growth of tumor cells; FAK emerged as a central mediator of not just integrin signaling but also for signaling downstream for other classes of cell surface receptors [3,39]. FAK-dependent regulation of cell adhesion and migration has been established in several cell types and is widely accepted as a pathway that contributes to cancer pathogenesis and other diseases [40,41]. Consequently, FAK is regarded as a promising target for cancer treatment. Indeed, various small anti-FAK molecules have already been developed by several pharmaceutical companies for clinical trials [42–47]. Favorably, FAK inhibition affects other vital proteins, such as signal transducer and activator of transcription 3 (STAT3) and Akt. STAT3 plays a fundamental role in cell growth, and is found to be activated mostly in solid tumors (as with FAK) [48–51]. Fortunately, inhibition of FAK leads to STAT3 reduction, resulting in apoptosis induction and potent antitumor activity [52–54]. Moreover, Akt, a protein kinase, plays an important role in regulating various cellular functions such as metabolism, proliferation, survival, growth and protein synthesis [55,56]. In this respect, inhibition of Akt phosphorylation leads to significant anticancer activity. Interestingly, FAK inhibition results in Akt phosphorylation suppression [57–61]. Also, inhibition of FAK induces cell cycle arrest at G2/M phase and considerably reduces S phase. This effect is accompanied by increasing early apoptotic cells [62,63]. FAK inhibition may lead to P53 activation that subsequently leads to cell cycle arrest and apoptosis induction [64,65].
Several cancer cells express FAK protein to promote cell survival, proliferation and metastasis, which makes FAK enzyme a promising target for cancer management. These cells comprise several types of cancers such as pancreatic cancer cells [66,67], colon cells [68–70], hepatocellular carcinoma cells [62,71], lung cancer cells [72–74], ovarian cancer cells [61,75], renal cancer cells [76–78], breast cancer cells [79–82], cervical cancer [83] and prostate cancer cell lines [84]. This wide variety of cancer cells that express FAK protein shows that targeting this kinase is inevitably promising in the fight against cancer.
Binding site of FAK
The most studied and targeted site in FAK protein is the ATP binding site; this site was taken up by several research groups and was targeted using different scaffolds of FAK inhibitors. The ATP binding site of the FAK protein contains specific characteristics that allow various FAK inhibitors to bind identically. FAK inhibitory activity inside the ATP binding site depends mainly on two pivotal sites: the kinase hinge region that comprises mainly Cys502 amino acid and the helical DFG motif of the activation loop (Asp564, Phe565 and Gly566 amino acids; Figure 2). Compared with the FAK domain, the phi torsion angle rotates Asp564 of the DFG motif by 113° upon administration of different FAK inhibitors, such as TAE226 and PF562271 (Figure 3) [85,86]. Interestingly, this unusual helical DFG conformation is essential to obtain optimum anti-FAK activity. The upper part of the active site includes the gatekeeper residue Met499, while the lower part contains a solvent region. The active site contains some important amino acids that are usually involved in electrostatic interactions with different FAK inhibitors such as Arg426, Ile428, Ala452, Glu500, Leu501, Gly505, Leu553 and Leu567 (Figure 4). Among these various amino acids, Cys502 of the kinase hinge and Asp564 of the DFG motif are considered the key residues within the ATP binding site of FAK that should be targeted to obtain potent FAK inhibitory activity.
Figure 2. . The ATP binding site of FAK including the key interaction sites: hinge region and DFG motif.
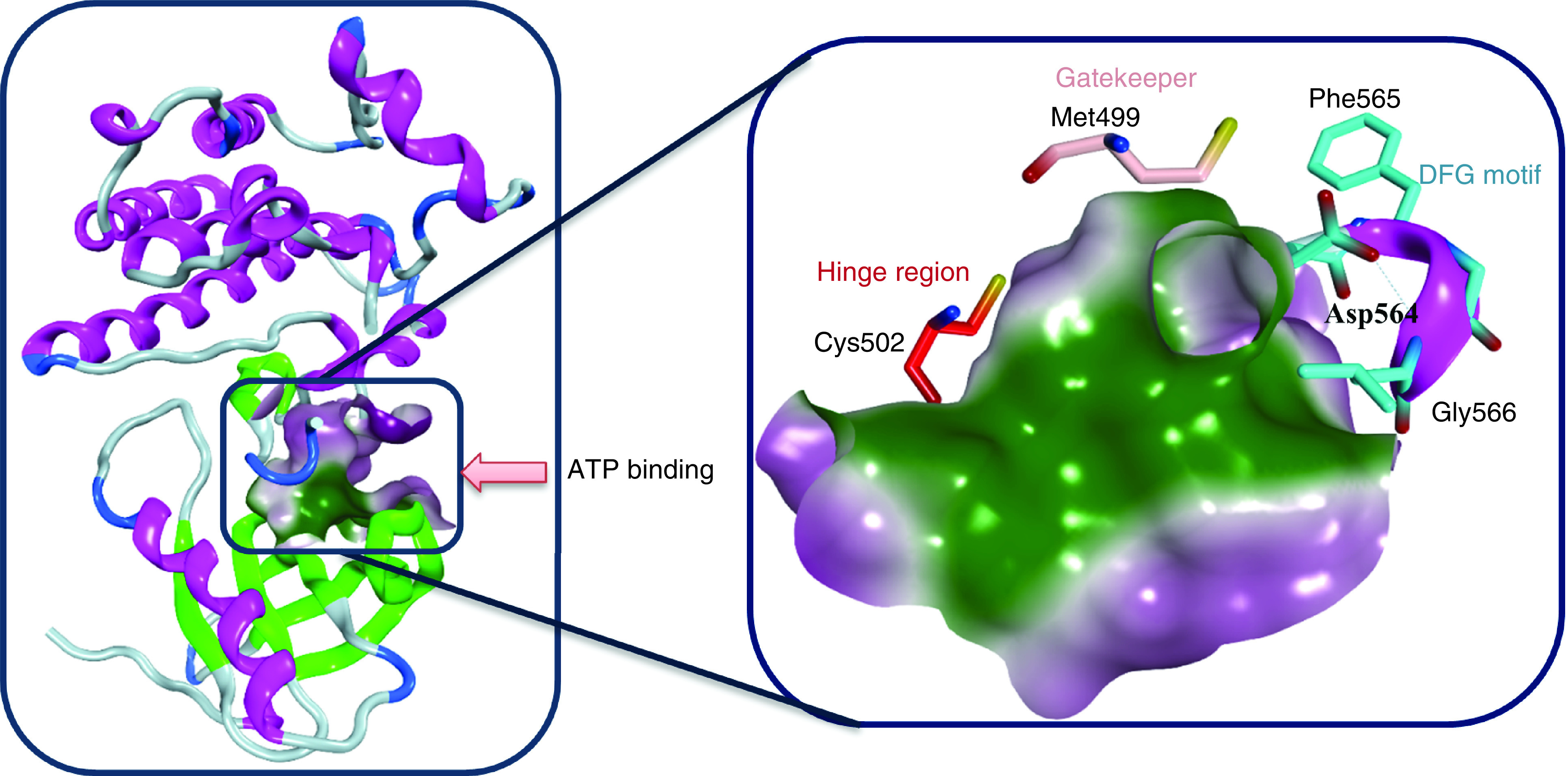
Figure 3. . Superimposition of the DFG motif illustrates the conformational change of Asp564 in FAK bound to TAE226 (cyan) compared with Asp564 in the natural FAK domain kinase (magenta).

(A) Side view and (B) front view.
Figure 4. . The ATP binding site of FAK with its key amino acids.

FAK inhibitors
The past three decades saw a significant increase in the development of new FAK inhibitors. Several research groups have worked to design appropriate scaffolds to provide the best anti-FAK activity. In the next paragraphs, the authors review the most promising findings and the status of several drugs in their clinical trial development. Various active sites in the FAK protein have been investigated in the development of anticancer agents. The majority of attempts have been directed toward targeting the ATP binding site of the kinase domain, whereas some attempts have targeted the allosteric binding site (Figure 5). Numerous computational approaches have targeted FAK-FERM and FAK-FAT domains to prevent phosphorylation at Tyr397 and Tyr925 amino acids, respectively (Figure 5). FAK inhibitors are classified as follows: six-membered heterocyclic FAK inhibitors, five-membered heterocyclic FAK inhibitors, allosteric FAK inhibitors, FAK-FAT domain inhibitors, FAK-FERM domain inhibitors, FAK degradation through proteolysis targeting chimeras (PROTACs) and miscellaneous FAK inhibitors.
Figure 5. . Methods for targeting FAK protein in the development of anticancer agents.
FAK inhibitors
Six-membered heterocyclic FAK inhibitors
One of the most well known FAK inhibitors is TAE226 (1, Table 1), which has shown significant FAK inhibitory activity with an IC50 value of 5.5 nM [86]. This inhibitory effect resulted in superior antiproliferative activity in different cancer cell lines. Compound 1 thoroughly inhibited invasion and proliferation in glioma cells in both in vitro and in vivo studies [87]. It induced apoptosis in BT474 and MCF-7 breast cancer cells, inhibited tumor growth, and reduced its weight profoundly in HeyA8 ovarian cancer cells [88,89]. Consequently, 1 decreased cell viability, induced apoptosis and arrested cell cycle at G2 phase in human neuroblastoma cells [90]. Compound 1 also had a remarkable anticancer effect on pancreatic carcinoma, squamous cell carcinoma and colorectal cancer, but it did not enter clinical trials due to severe side effects, since it affects glucose blood levels (inhibits insulin receptor) and metabolism [91–94]. Compound 1 was cocrystallized in the ATP binding pocket of the FAK enzyme (PDB codes: 4D58, 4D4S, and 2JKK) [86,95]. The cocrystallized orientation of 1 revealed the formation of two hydrogen bonds between the nitrogen of pyrimidine and o-methoxy aniline moieties and the carbonyl oxygen of Cys502 of the kinase hinge (Figure 6). Two hydrophobic contacts were observed between the carbons of the pyrimidine ring and Leu553 and Ala452 amino acids. The chloro group on C5 of the pyrimidine ring penetrated deep into the kinase binding pocket to be positioned near the gatekeeper amino acid Met499. The carbonyl group of the methyl carbamoyl moiety formed a hydrogen bond with the nitrogen of Asp564 of the DFG motif of the activation loop. The hydrophobic amino acid Leu567 that immediately follows the DFG sequence made a hydrophobic interaction with the 4-aniline ring.
Table 1. . Preclinical/clinical trial FAK inhibitors.
| No. | Compound name | Structure | FAK (IC50, nM) | Preclinical/clinical phase | Target cancer cell |
|---|---|---|---|---|---|
| 1 | TAE226 | 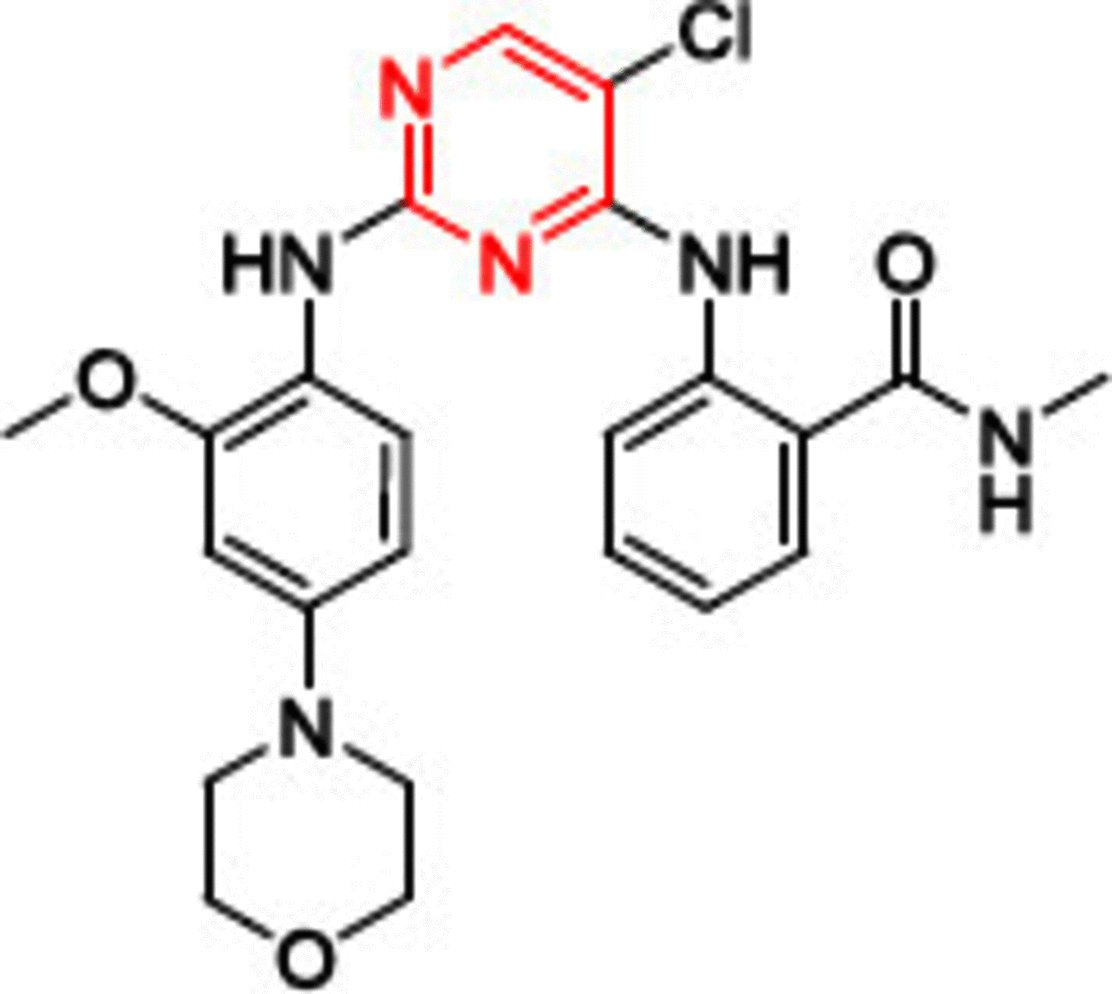 |
5.5 | Preclinical | GIST |
| 2 | PND-1186 (VS-4718) | 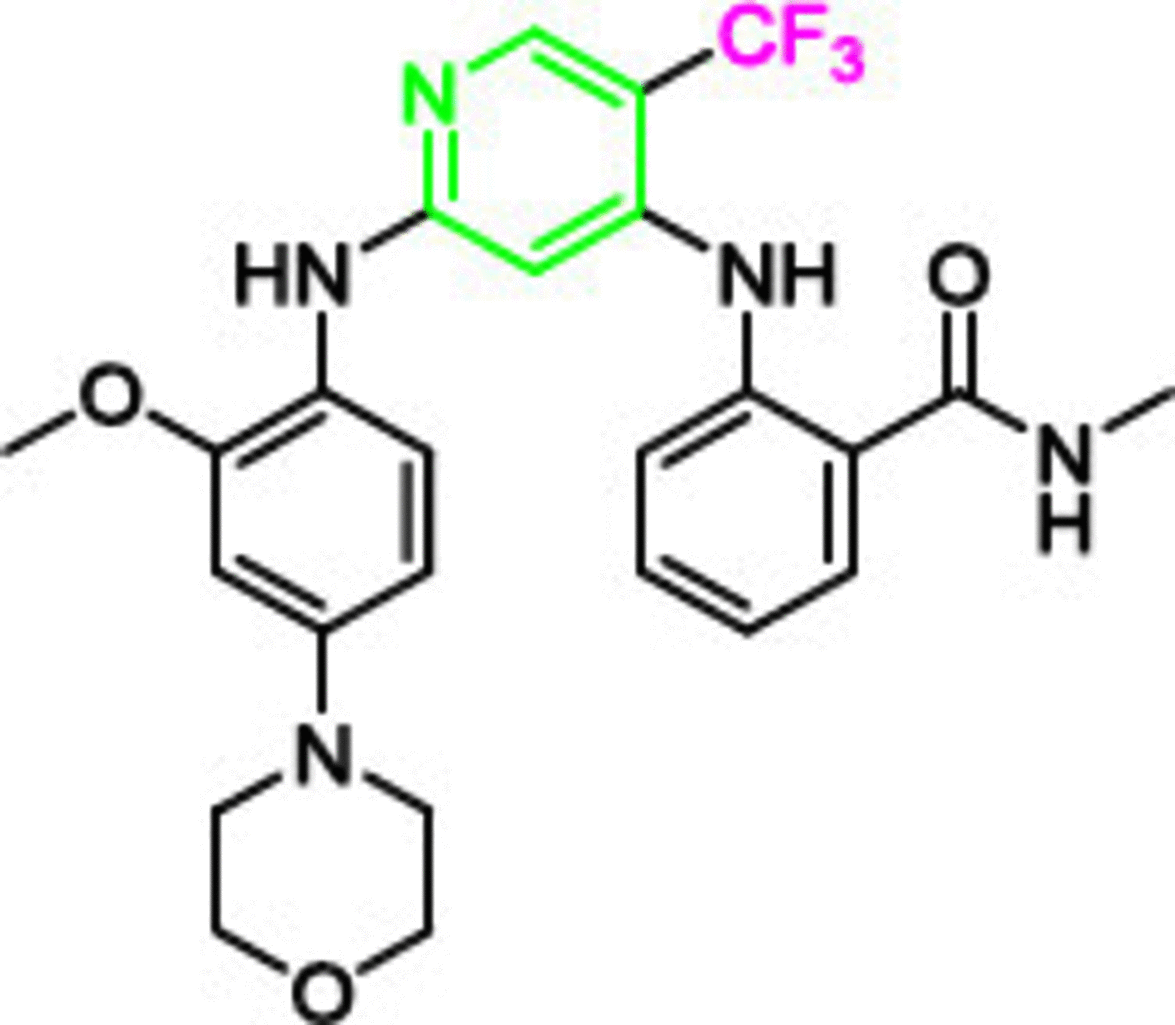 |
1.5 | Phase I | Pancreas |
| 3 | PF-573228 | 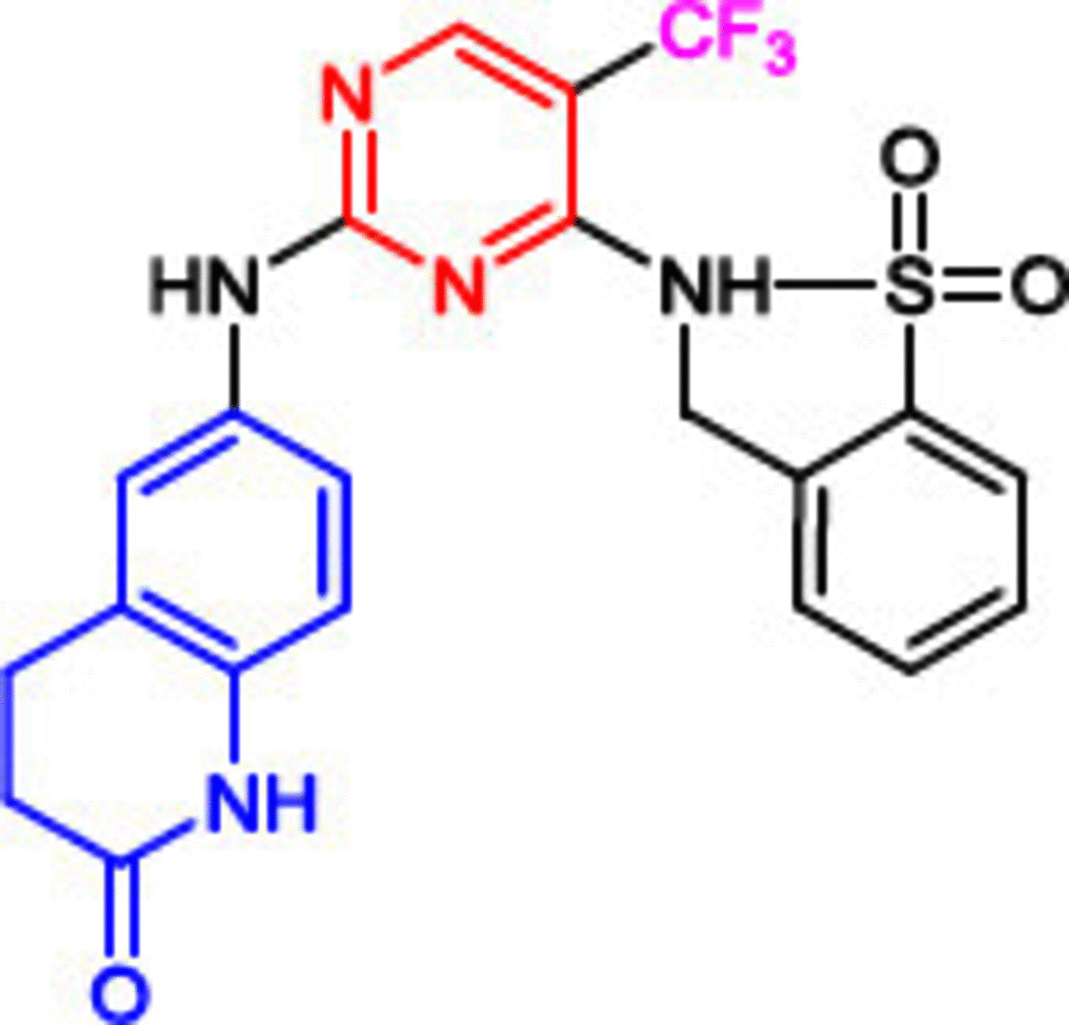 |
4 | Preclinical | Breast and lung |
| 4 | PF-562271 | 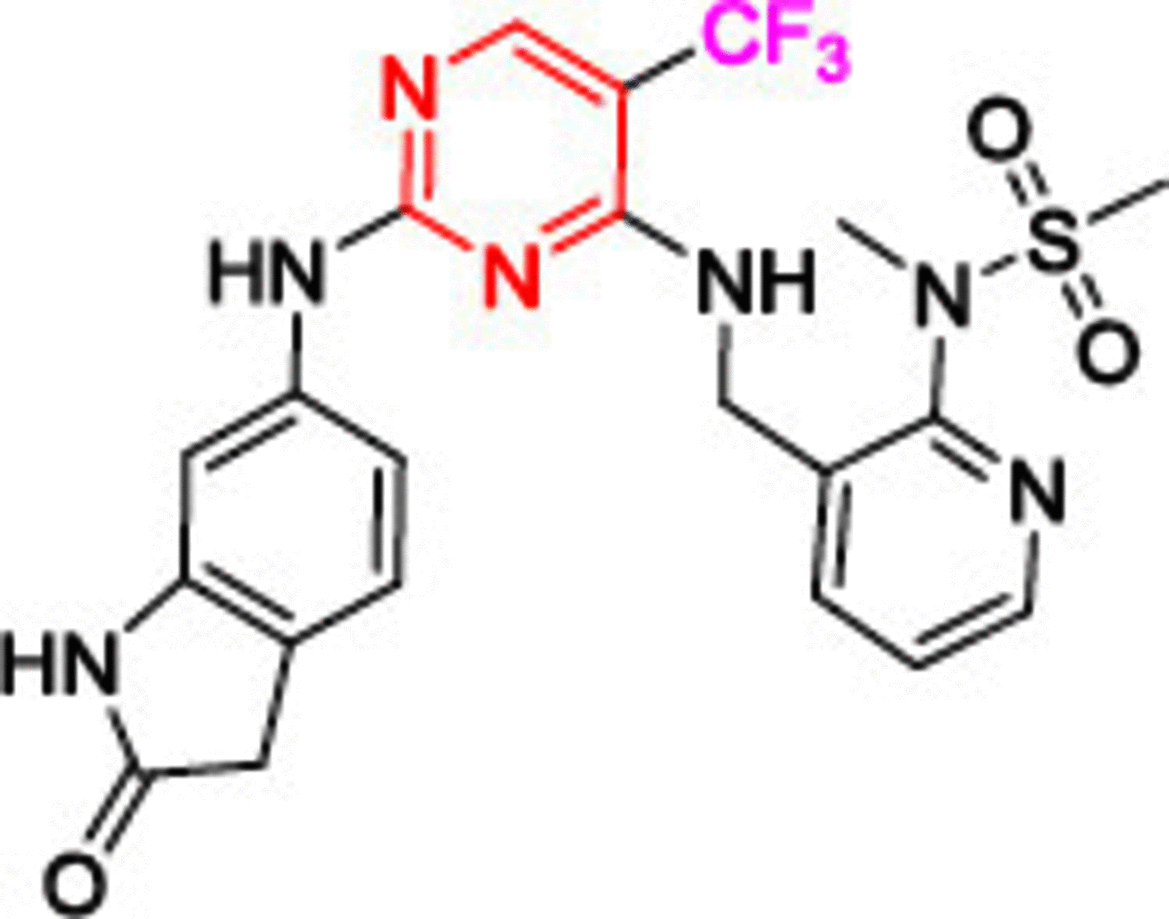 |
1.5 | Phase I | Pancreas, head, neck and prostate neoplasms |
| 5 | CEP-37440 | 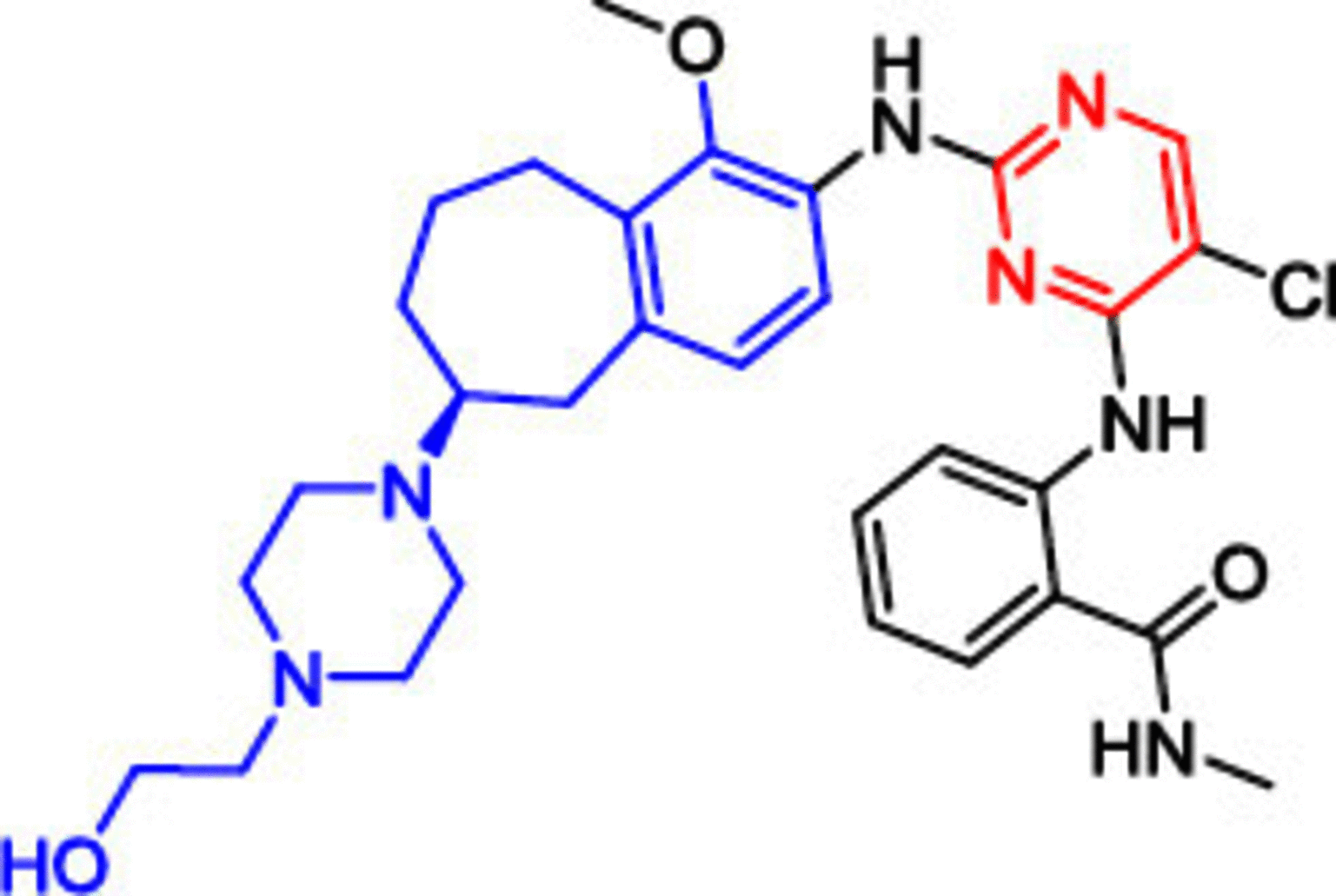 |
2 | Phase I | Solid tumors |
| 6 | Defactinib (VS-6063) | 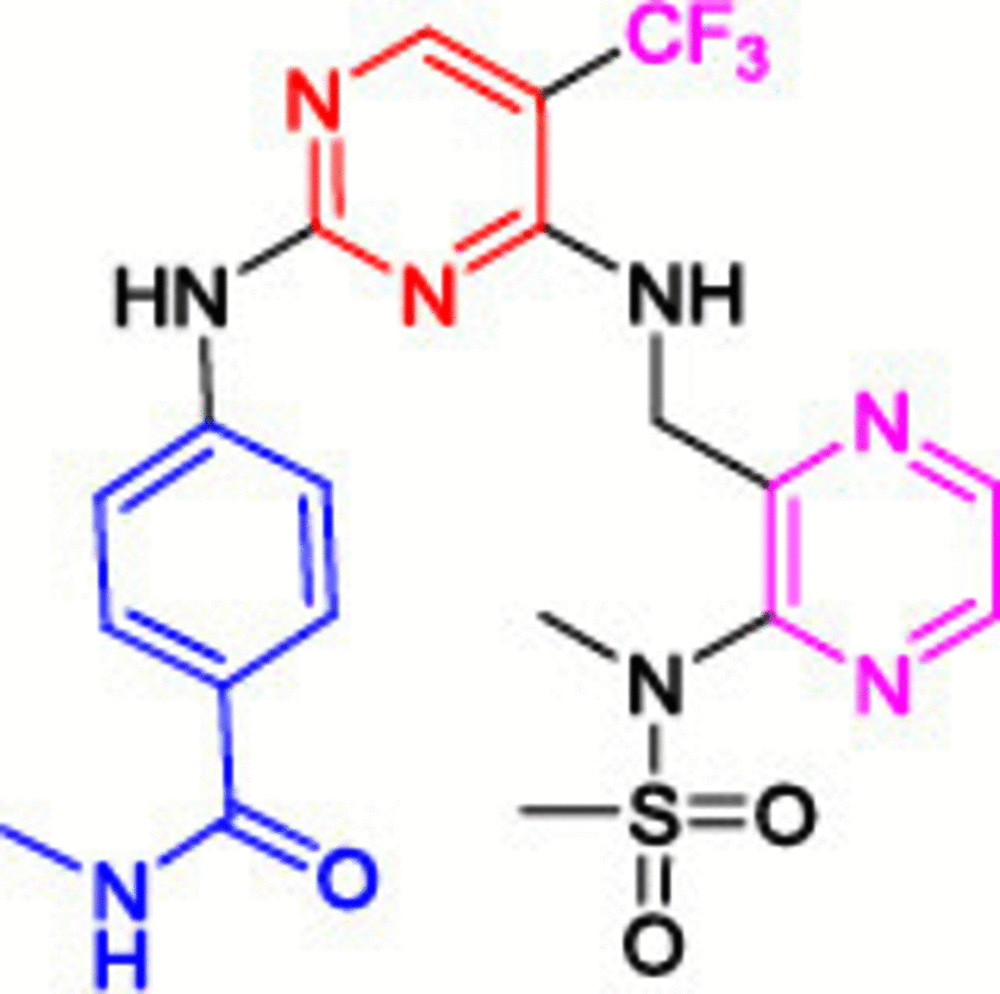 |
0.6 | Phase II | Non-small cell lung cancer |
| 7 | GSK-2256098 | 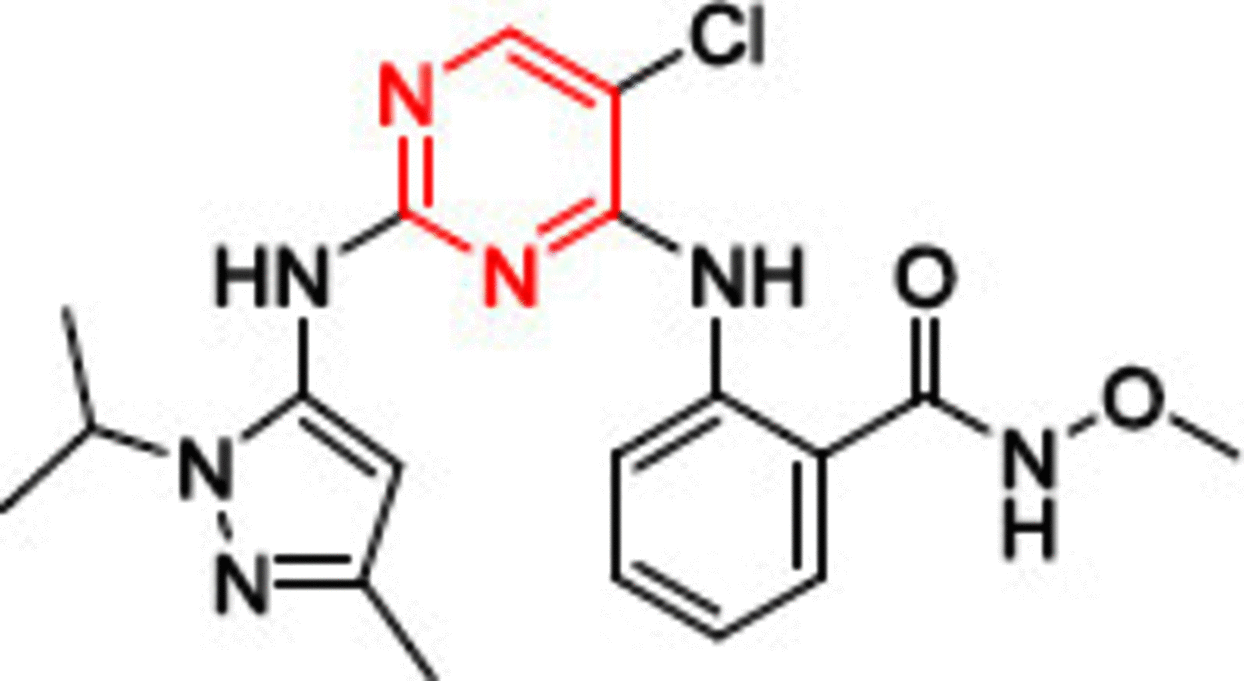 |
Ki (0.4 nM) | Phase II | All cancers, neoplasms |
| – | BI-853520 (IN-10018) | Not disclosed structure | 1 | Phase I | Neoplasm |
Figure 6. . Cocrystallized structure of TAE226 (orange) inside the ATP binding site of FAK, Protein Data Bank code: 2JKK.

PND-1186 (VS-4718, 2) was designed as a selective FAK inhibitor (IC50 = 1.5 nM) with superior phosphorylation inhibition at Tyr397 of FAK. It has the same structural features as 1; however, the pyridine moiety was introduced instead of pyrimidine, and the chlorine was replaced by trifluoromethyl as a strong electron withdrawing group. These structural modifications resulted in 3.5 times more anti-FAK activity than 1 [96]. Compound 2 revealed a strong ability to inhibit tumor growth and metastasis in 4T1 and MDA-MB-231 breast cancer cells and inhibited orthotopic ID8-IP ovarian cancer growth [75,97]. In vitro, 2 decreased the expression IL-6, which is induced by tumor necrosis factor-α, suggesting that compound 2 inhibited the progression of tumors by connecting stromal cells and tumor cells. Compared with gemcitabine, 2 profoundly extended the survival of early and late KPC mice with pancreatic ductal adenocarcinoma (PDAC) progression [98]. Compound 2 overcame PDAC fibrosis and immunosuppressive cell population; thus, immune surveillance increased significantly. Compound 2 reversed the multidrug resistance mediated by the ATP-binding cassette (ABC) transporters including ABCB1 and ABCG2 (ABC transporters decrease the intracellular concentration of different anticancer agents by pumping the anticancer compound out of the tumor cell, which is considered a drastic mechanisms in multidrug resistance) [99–103]. The combination of 2 with doxorubicin or paclitaxel considerably enhanced the IC50 values in KB-C2 and SW620/Ad300 (ABCB1-overexpressing resistant cells) in a dose-dependent manner. Despite these significant results, clinical trials of 2 were terminated and the compound was withdrawn (ClinicalTrials.gov Identifier: NCT01849744 and NCT02215629).
Another inhibitor bearing trifluoromethyl moiety (PF-373228, 3) was investigated due to its high specificity toward FAK (IC50 = 4 nM). Notably, the 4-morpholinophenyl group, found in 1 and 2, was replaced by a dihydroquinolinone moiety, and methylsulfonyl amine was introduced into the benzene ring. Despite possessing strong FAK inhibitory activity, compound 3 did not display promising apoptosis induction or proliferation inhibition [104]. PF-562271 (compound 4), the same scaffold as PF-373228 (3, Table 1), is a reversible chimeric inhibitor of proline-rich tyrosine kinase 2 (Pyk2: A cytoplasmic tyrosine kinase within the FAK subfamily contributes in multiple signaling pathways which regulate proliferation, migration and survival of cells) and FAK (IC50 = 13 and 1.5 nM, respectively) [85]. In vivo, compound 4 was able to inhibit FAK phosphorylation in a nanomolar dose-dependent manner. Moreover, the in vivo experiments revealed no morbidity, mortality or weight loss. Treating prostate cancer cells PC3M-luc-C6 in a mouse xenograft model with 25 mg/Kg of compound 4 resulted in 62% tumor growth inhibition in two weeks and showed a substantial ability to decrease metastasis [105]. Phase I clinical trials revealed that compound 4 is highly tolerable at 125 mg dose b.i.d. with food and is cleared via CYP 3A-mediated hepatic metabolism (positive metabolic response) [106]. Compound 4 was cocrystallized in the ATP binding site of FAK (PDB code: 3BZ3) and was oriented approximately the same as 1; thus, the same interactions were observed [85]. The only difference was that the oxygen atom of the amino-oxindole moiety extended out of the active site cleft to form a hydrogen bond with Arg426 amino acid.
Another highly potent derivative, CEP-37440 (5), completed phase I clinical trials with considerable anti-FAK activity (IC50 = 2 nM) [107,108]. Compound 5 showed a high selectivity on anaplastic lymphoma kinase (ALK) with an IC50 value of 3.1 nM. (ALK is an orphan receptor tyrosine kinase constitutively activated in a number of cancer types.) [109] The benzocycloheptyl ring helped to improve anti-FAK activity and maintain ALK selectivity. Moreover, 5 exhibited significant dose-dependent antitumor activity in the oral dose xenograft model. Compound 5 showed good oral bioavailability and favorable in vitro absorption, distribution, metabolism, and excretion (ADME) properties in three different species (CD-1 mouse, cynomolgus monkey and Sprague-Dawley rat). Defactinib (VS-6063, 6) is a strong FAK inhibitor, and its structural features showed a successful replacement of the phenyl ring (compounds 1, 2, 3 and 5) or the pyridine (compound 4) with a substituted pyrazine moiety. Compound 6 showed the addition of benzamide moiety instead of the morpholine (compounds 1 and 2) and δ and β lactam rings (compounds 3 and 4, respectively). Phase I clinical trials revealed that compound 6 was well tolerated when administered in combination with paclitaxel, with no dangerous side effects [110]. Combining defactinib with gemcitabine and pembrolizumab in patients with PDAC also revealed a substantial tumor regression. This combination was estimated to be well tolerated orally with few side effects [111]. Unfortunately, Phase II clinical trials showed that compound 6 alone possessed a modest activity on KRAS mutant non-small cell lung cancer [112]. These results suggest that future defactinib studies focusing on multidrug regimens are warranted.
GSK-2256098 (7), a highly selective FAK inhibitor bearing 1-isopropyl-3-methyl-1H-pyrazole, showed significant Tyr397 autophosphorylation inhibition and decreased pancreatic cancer, breast cancer, neuroblastoma, and glioblastoma tumor growth significantly [113,114]. Among 261 tested kinases, 7 showed a profound selectivity on FAK inhibition. Interestingly, 7 showed 1000-times more FAK inhibitory activity than Pyk2, the nearest FAK kinase family member [113,115]. Compound 7 showed potent FAK inhibitory activity in OVCAR8 ovarian cancer, A549 lung cancer and U87MG brain cancer cell lines at IC50 values of 8.5, 15, and 12 nM, respectively. Moreover, 7 exhibited variable FAK inhibitory activity in different (PDAC) cell lines; for example, it showed its best anti-FAK activity in L3.6P1, whereas no notable activity was observed in PANC-1 cells. Unlike PANC-1 cells, 7 induced apoptosis through caspase-9 PARP-related pathways and significantly decreased colony formation in L3.6P1 cell line. MTT assay revealed that 7 had a remarkable antiproliferative activity in L3.6P1 cell line (IC50 = 25 μM). It also inhibited ovarian cancer in 3D cultures and in vivo studies [116]. The combination of 7 with docetaxel and pazopanib resulted in a remarkable decrease in overall tumor weight [117]. Phase I clinical trial revealed that oral administration of 7 b.i.d. in a dose ranging 0.8–1.5 g resulted in 80% reduction of pFAK in patients with advanced solid tumors (melanoma and mesothelioma). Compound 7 presented an acceptable safety profile with minimal side effects, such as nausea (76%), vomiting (58%), diarrhea (65%) and decreased appetite (47%). Despite having a good safety profile, the 7/trametinib combination showed limited support for efficacy when administering 7 b.i.d. at a dose of 500 mg and trametinib at a dose of 0.375 mg q.d. [118].
BI-853520 (IN-10018, nondisclosed structure) is a highly potent FAK inhibitor (IC50 = 1 nM) that represses Tyr397 FAK phosphorylation in highly metastatic breast cancer [119]. MTS assay revealed that BI-853520 considerably reduced cell viability of PY2T, PY2T-LT and 4T1 cells grown in 2D cultures. BI-853520 inhibited growth and migration in SPC111 and SPC212 malignant pleural mesothelioma cells [120]. Western blot assay revealed a substantial ability to inhibit the FAK phosphorylation in the malignant pleural mesothelioma cells (SPC111, SPC212, M38K and P31). BI-853520 also prevented spheroid formation in P31 cells. Phase I clinical trials were performed on 21 Japanese and Taiwanese patients with different types of cancers. Unfortunately, most patients underwent disease progression (57%), while 24% showed stable disease control in various types of cancer (urachus, esophageal, meningioma, gastric cardia and intrahepatic bile duct cancer) [121]. Another Phase I clinical trial was performed on 63 patients with different types of metastatic cancers (pancreatic adenocarcinoma, ovarian cancer, esophageal cancer and soft tissue sarcoma) [122]. BI-853520 resulted in a substantial decrease in pFAK/total FAK ratio, while anticancer activity was moderate. BI-853520 showed an acceptable and manageable safety profile with favorable pharmacokinetics in both the abovementioned studies.
Recently, Ma and colleagues [44] disclosed carbonyl-substituted DPPY derivatives with potent FAK inhibitory activity. A series of nucleophilic substitution and coupling reactions were performed to obtain the target compounds. Among them, derivative 8 exhibited the highest anti-FAK activity (Figure 7). Compound 8 showed strong activity in AsPC-1, BxPC-3, PANC-1 pancreatic cancer, H1975 lung cancer and MCF-7/ADR breast cancer cell lines. The in vivo antiproliferative activity on AsPC-1 cells showed that 8 was able to inhibit tumor growth by 87% at a dose of 30 mg/kg. Furthermore, western blot analysis demonstrated that treating AsPC cells with 8 considerably repressed FAK phosphorylation at less than 50 nM concentration.
Figure 7. . Chemical structure of FAK inhibitors 8–15.
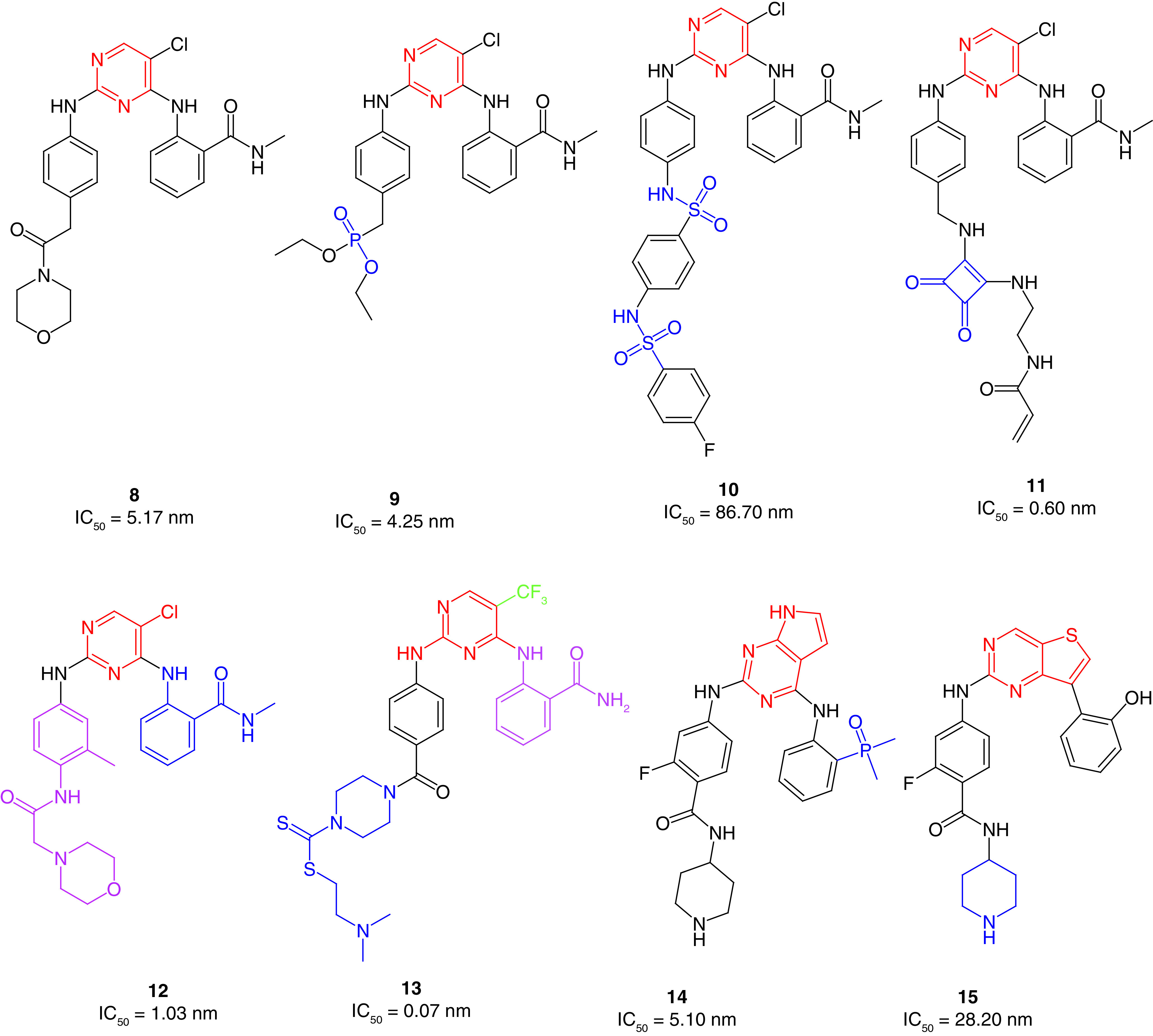
The same research group prepared phosphamide-containing diphenylpyrimidine derivatives (DPPY) as potent FAK inhibitors [123]. The ADP-Glo™ kinase assay revealed that FAK activity was substantially hindered when treated with the synthesized derivatives in nanomolar concentrations. Compound 9 exhibited better anti-FAK activity (IC50 = 4.25 nM) than the reference TAE226 (IC50 = 6.79 nM) and showed a substantial ability to inhibit the pancreatic cancer cells AsPC-1, BxPC-3 and PANC-1 (IC50 = 3.31, 0.02 and 14.76 μM, respectively) in low micromolar concentrations (Figure 7). Qu and colleagues [124] developed sulfonamide-substituted DPPY derivatives with moderate FAK inhibitory activity. Compound 10 possessed the best antiproliferative activity in cancer cells that harbor the overexpressed FAK enzyme, such as pancreatic carcinoma (AsPC-1, Panc-1, BxPC-3), leukemia (Ramos) and gefitinib-resistant lung cancer (H1975; Figure 7). The sulfonamide derivative 10 was also found to be less toxic on normal cells than TAE226. Unlike the carbonyl (8) and phosphamide (9) derivatives, using sulfonamide moiety (10) did not retain the anti-FAK activity.
Yen-Pon and colleagues[125] reported the synthesis of highly potent FAK inhibitors that were able to bind covalently to the ATP binding site of FAK. The prepared derivative 11 exhibited approximately 10-fold better anti-FAK activity than the reference TAE226 (Figure 7). Compound 11 revealed an increased FAK inhibition over 2 h as a time-dependent inhibitory approach, indicating irreversible inhibitory ability [126]. Interestingly, 11 exhibited potent multiacting inhibitory activity against eight different kinases and showed significant antiproliferative activity against squamous cell carcinoma (SCC) cells with an ED50 value of 1.73 μM (reference VS-4718, ED50 = 1.49 μM). In low concentrations, the squaramide derivative 11 inhibited FAK phosphorylation and blocked Tyr397 significantly in SCC cell line, similarly to the reversible FAK inhibitor VS-4718. Washout experiments were performed to test the covalent binding ability of 11. Unlike VS-4718, the potent inhibitor 11 retained FAK inhibitory activity after washing out.
Recently, Chen and coworkers [127] reported the synthesis of dual-acting FAK and epidermal growth factor receptor with threonine-to-methionine mutation (EGFRT790M) inhibitors containing N-alkylbenzamide-substituted diphenylpyrimidine using fragment-based drug design strategy (FBDD). Upon incorporating the FBDD approach, the researchers merged the two scaffolds of TAE226 and the previously reported EGFRT790M inhibitor Mor-DPPY in the same structure (Figure 7) [128]. MTT assay was used to assess the antiproliferative activity of the alkylbenzamide derivatives against pancreatic cancer (AsPC-1, BxPC-3 and Panc-1), breast cancer (MCF-7/adr) and lung cancer (H1975) cells. Interestingly, all the prepared derivatives possessed more potent antiproliferative activity than TAE226. Among them, 12 inhibited both FAK and EGFRT790M kinases significantly in nanomolar concentrations and was 6.5-fold more active on FAK kinase than TAE266. The promising results of 12 provoked the researchers to perform further in vivo studies on an AsPC-1 cell xenograft mouse model. The results showed 36.6% tumor growth inhibition at 60 mg/kg/q.d. for the oral dosage.
Yin and colleagues [129] synthesized novel 2,4-diarylaminopyrimidine-based FAK inhibitors bearing dithiocarbamate moiety with potent antiproliferative activity. The research team replaced the morpholine moiety of the reference TAE226 with piperazinyl dithiocarbamate. Surprisingly, 13 showed 88-fold and 11-fold more potent FAK inhibitory activity than TAE226 and defactinib, respectively (Figure 7). The dithiocarbamate derivative exhibited potent anticancer activity on HCT116 colon cancer, PC-3 prostate cancer, U87-MG glioma and MCF-7 breast cancer cells with better activity than both TAE226 and defactinib. Interestingly, 13 was 290-fold and 3000-fold more cytotoxic on HCT116 cells than TAE226 and defactinib, respectively.
Cheng and colleagues [130] designed and synthesized 7H-pyrrolo[2,3-d]pyrimidine derivatives bearing dimethylphosphine oxides as potential FAK inhibitors. Compound 14 showed the best FAK inhibitory activity (IC50 = 5.10 nM) compared with the synthesized derivatives and was better than TAE226 (IC50 = 6.20; Figure 7). It also possessed approximately the same anticancer activity on MDA-MB-231 breast cancer and A549 lung cancer cells as TAE226. Moreover, 14 expressed low cytotoxicity on the normal HK2 cells.
The same research group reported a series of 2,7-disubstituted-thieno[3,2-d]pyrimidine derivatives [131]. Compound 15 showed better IC50 values than TAE226 on U-87MG glioma, A549 lung cancer and MDA-MB-231 breast cancer cell lines (Figure 7). Derivative 15 was tenfold, fourfold and 21-fold more potent than TAE226 on U-87MG, A549 and MDA-MB-231 cells, respectively. This potent anticancer activity was accompanied by a modest ability to inhibit FAK, suggesting that 15 has an off-target mechanism. It was subsequently tested against 25 different kinase enzymes and showed significant activity on five of them (BTK, ALK, CDK2, RET and RET), indicating the multitarget kinase inhibitory activity of this scaffold. Moreover, 15 revealed considerable antimetastatic activity on MDA-MB-231 cell line in a concentration-dependent manner.
Cheng's group [132] also synthesized 7H-pyrrolo(2,3-d)pyrimidine derivatives using palladium-catalyzed Buchwald cross-coupling. Compound 16 revealed moderate anti-FAK activity (IC50 = 19.10 nM) compared with TAE226 (IC50 = 7.00 nM) and showed fourfold, sixfold, and 14-fold better antiproliferative activity than TAE226 in U-87MG, A549 and MDA-MB-231 cells, respectively (Figure 8). It also possessed significant tumor selectivity and showed considerable inhibition on 5 of 9 tested kinases (ABL1, ALK, BTK, FLT3 and KDR) at a concentration of 1 μM. Interestingly, 16 inhibited the migration of U-87MG, A549 and MDA-MB-231 cells substantially, indicating its significant ability to prevent metastasis.
Figure 8. . Chemical structure of FAK inhibitors 16–23 with their IC50 values.
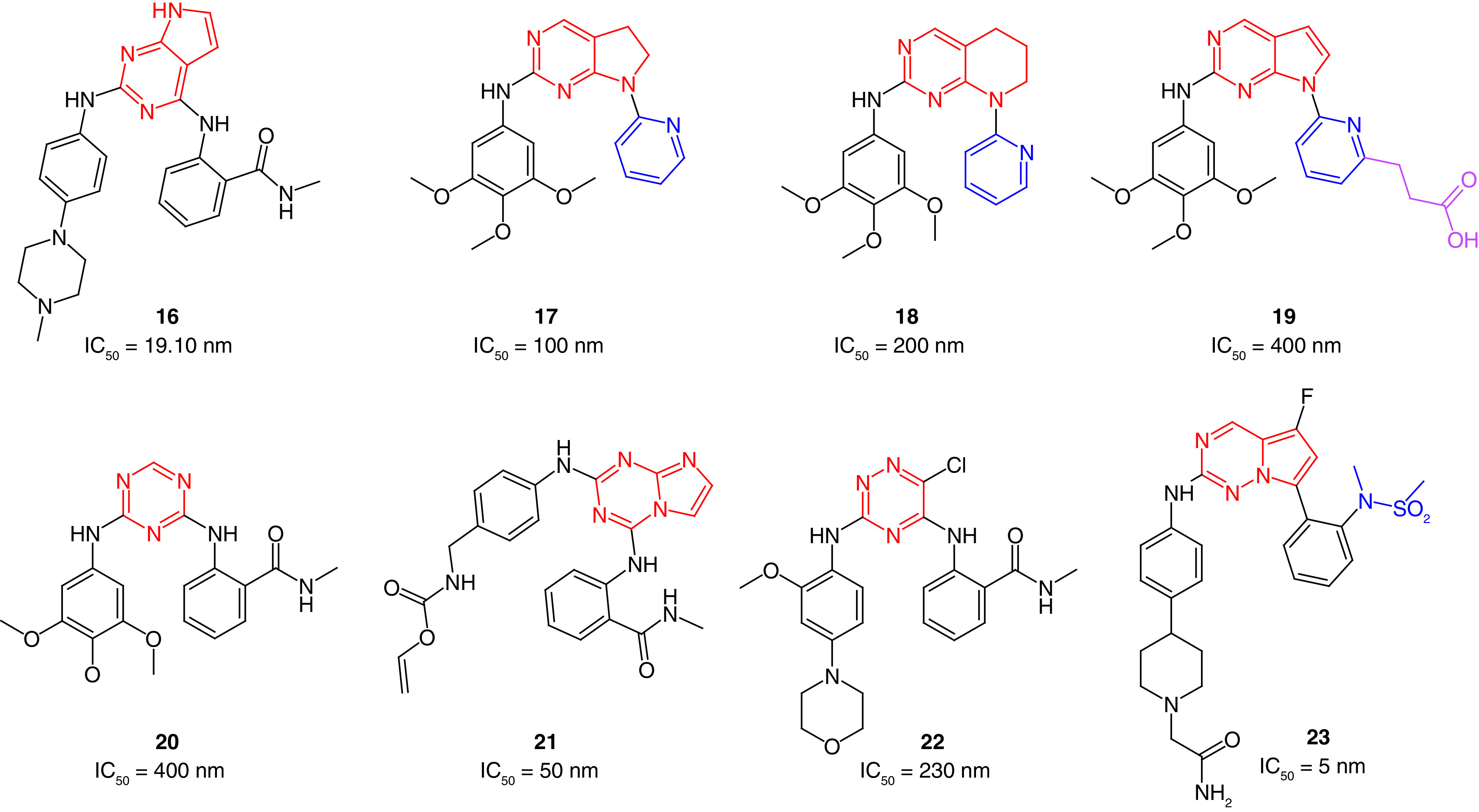
Choi and colleagues [133] designed several FAK inhibitors containing fused pyrimidines such as saturated and unsaturated scaffolds of both pyrrolopyrimidines and pyridopyrimidines. Compound 17, bearing the saturated pyrrolopyrimidine, was synthesized through Mitsunobu cyclization and showed the best anti-FAK activity among the prepared compounds (IC50 = 0.1 μM; Figure 8). The 5,6,7,8-tetrahydropyrido[2,3d]pyrimidine 18 was found to exhibit a remarkable anti-FAK activity (IC50 = 0.2 μM). Based on this study, the same research group performed further docking studies to improve the anti-FAK activity of this scaffold [134]. They suggested that adding an extension to the structure, such as aliphatic carboxylic acid moiety (19), would result in extra-interactions inside the FAK active site by forming a salt bridge with Lys454 amino acid. The substitutions in the pyrrolopyrimidine were fixed while the extension was added in the pyridine moiety. Interestingly, the newly prepared compound 19 revealed a substantial anti-FAK activity with an IC50 value of 4 nM.
In 2013, Dao and colleagues[42] applied microwave-assisted synthesis for preparing new diarylamino-1,3,5-triazines as FAK inhibitors. The prepared triazine derivatives were tested for their antiproliferative activity on HUVEC human umbilical vein endothelial cells. Compound 20 showed a comparable IC50 value to TAE226 on HUVEC cells and revealed a moderate ability to inhibit FAK (Figure 8). The blockage of endothelial-derived FAK activity was confirmed using the FACE™FAK ELISA kit. The results revealed that the autophosphorylation of FAK was inhibited, and the tyrosine 397 phosphorylation of kinase targets was significantly blocked in HUVEC cells in a dose-dependent manner. Compared with TAE226, 20 showed remarkable antiproliferative activity against U-87MG glioblastoma-astrocytoma, HCT-116 colon carcinoma, MDA-MB-231 breast cancer and PC-3 prostate cancer cells [135]. Among 10 tested kinases, 20 showed a significant selectivity on FAK and FGFR2, leading to the shutting down of their signaling pathways. In a dose-dependent manner, 20 significantly decreased cell adhesion and inhibited tube formation leading to alteration of growth, differentiation, migration and proliferation. This effect was accompanied by activation of caspase-3 and induction of apoptosis.
In 2014, Dao's research group explored fusing the imidazole ring with the 1,3,5-triazine moiety. They reported the synthesis of imidazo[1,2-a][1,3,5]triazine derivatives as potential FAK inhibitors [136]. Compound 21 exhibited more potent antiproliferative activity than TAE226 on MDA-MB-231 and PC-3 cells (Figure 8). Moreover, compound 21 substantially inhibited FAK expression and cell adhesion at a low nanomolar level. Compound 21 significantly inhibited cell migration and invasion in U87-MG cells.
In 2017, the same research group synthesized 1,2,4-triazinic FAK inhibitors with strong antiproliferative activity [137]. The researchers added a chlorine atom on position 6 of the triazine ring to simulate the structural features of TAE226 (1; Table 1), to facilitate the formation of van der Waals interactions with the gatekeeper amino acid Met499 (22; Figure 8). TR-FRET-based kinase assay revealed that 22 possessed anti-FAK activity in submicromolar concentration (IC50 = 230 nM); however, it was still less potent than TAE226 (IC50 = 7 nM). Compound 22 showed more potent antiproliferative activity than TAE226 on colon cancer HCT-116 cells, inhibited FAK autophosphorylation and blocked Tyr397 phosphorylation in U-87MG and HCT-116 cells.
Zificsak and colleagues [138] reported the synthesis of pyrrolo[2,1-f][1,2,4]triazine derivatives as potent inhibitors for FAK and Janus kinase 2 (JAK2). Compound 23 displayed FAK and JAK2 inhibitory activities at the nanomolar level (IC50 = 5 nM and 2.5 nM, respectively; Figure 8), and exhibited a remarkable ability to inhibit different kinases; 23 demonstrated more than 90% inhibition on 19 of 53 tested kinases. The phosphorylation of STAT3 decreased substantially (>75%) in a CWR22 tumor xenograft mouse model within 12 h of administration of a single oral dose (55 mg/kg) of 23. Targeting FAK and JAK2 kinases and STAT3 signaling pathways will inevitably affect tumor cells, but further studies are required to evaluate the obtained antiproliferative activity.
Heinrich and coworkers [139] used FBDD to discover a new FAK inhibitor bearing 1H-Pyrrolo[2,3-b]-Pyridine (24), which was able to induce conformational change at the helical DFG motif of the activation loop (Figure 9). N7 and 1H-NH of compound 24 formed two hydrogen bonds with the backbone N-atom and the carbonyl-O of Cys502 in the hinge region, respectively (Figure 9). The cyano group of the 7-azaindole core pointed toward the gatekeeper residue Met499 and made a hydrogen bond with the NH of Asp564 of the DFG motif. The sulfonyl group's oxygen formed a hydrogen bond with the NH of Asp564 amino acid. These two hydrogen bonds may be the reason for the induced conformational change in the DFG motif. Compound 24 exhibited potent FAK inhibitory activity with an IC50 value of 0.19 μM and showed remarkable antiproliferative activity on HT29 cells (IC50 = 1.10 μM).
Figure 9. . X-ray structure of the ATP binding site of FAK in complex with 24 (yellow), Protein Data Bank code: 4GU6.

The investigation of the structure-activity relationship (SAR) of the six-membered FAK inhibitors revealed that most compounds showed potent FAK inhibitory activity. The primary scaffold of this category comprises the presence of diphenyl pyrimidine (Figure 10). Adding various extensions to the first phenyl ring (A) such as the benzocycloheptyl ring (5), squaramide moiety (11), morpholinoacetamido phenyl (12) and dithiocarbamate (13) resulted in a dramatic increase in both the anti-FAK and antiproliferative activity. These extensions could allow the formation of extra-interactions with Glu506, Ile428, Gly429, Asn556 and Leu567 amino acids inside the ATP binding site of FAK [86]. On the other hand, ring B contained N-methylbenzamide moiety in most derivatives and did not experience modifications except for some successful trials using pyridine, pyrazine rings or adding methylsulfonyl amine substitution. Replacing the chloro with the strong electron-withdrawing trifluoromethyl group in C-5 of the pyrimidine moiety substantially boosted the activity (compounds 1, 3, 4, 6 and 13). Using fused pyrimidines, triazines or fused triazines instead of the pyrimidine ringdid not enhance the FAK inhibitory activity, whereas incorporating the pyridine moiety resulted in a remarkable increase in potency (2). Computational methods such as FBDD succeeded in retaining and enhancing anti-FAK and antiproliferative activity (compound 12). Despite promising activity, most of the prepared six-membered inhibitors were not selective on FAK; targeting covalent bond formation inside the ATP binding site of FAK, such as the squaramide derivative 11, could improve anti-FAK activity and selectivity.
Figure 10. . Common features of the six-membered FAK inhibitors.

Five-membered heterocyclic FAK inhibitors
Sun and colleagues [140] reported the synthesis of 1,3,4-thiadiazole derivatives bearing 1,4-benzodioxan as potential FAK inhibitors. The most potent derivative (25, Figure 11) showed FAK inhibitory activity with an IC50 value of 10.79 μM compared with sftaurosporine (IC50 = 11.32 μM). Moreover, it inhibited hepatocellular carcinoma HepG2 cells substantially and was more potent than 5-fluorouracil and staurosporine. Despite the enzymatic inhibitory activity, 25 did not retain the antiproliferative activity on Hela (cervical cancer), SW1116 (colorectal cancer) or BGC823 (gastric cancer) cell lines. Western blot analysis showed that 25 significantly repressed FAK phosphorylation at a concentration of 12.5 μM.
Figure 11. . Chemical structure and biological activity of FAK inhibitors 25–34.
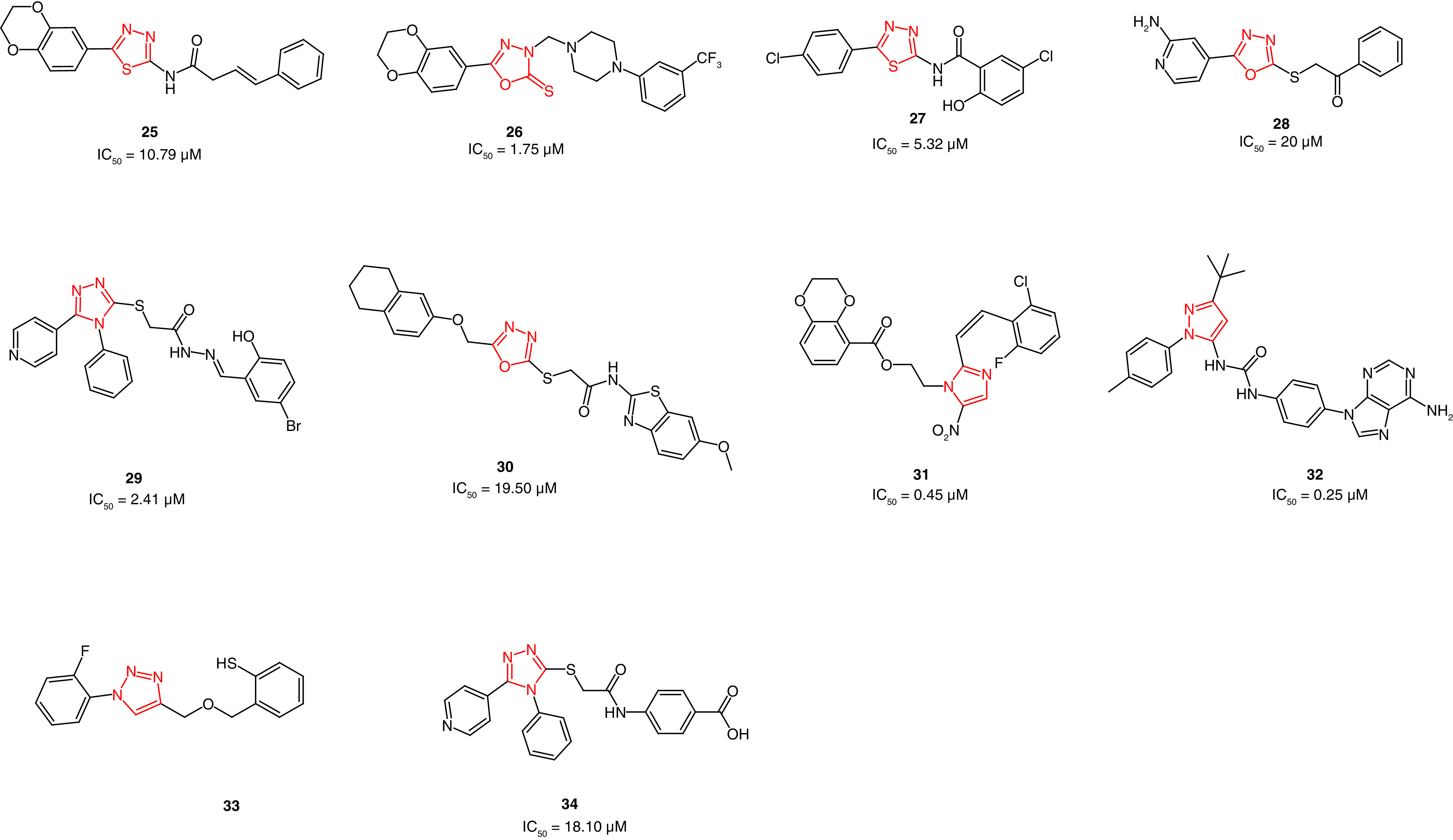
The same research group reported the synthesis of a series of 1,3,4-oxadiazole derivatives [141]. The target derivatives showed promising FAK inhibitory activity in low micromolar concentrations, in which 26 was the most active (IC50 = 0.78 μM; Figure 11). Derivative 26 retained antiproliferative activity in HepG2 cells and was twofold more active than 5-fluorouracil. However, 26 showed mild potency on Hela, SW1116 and BGC823 cell lines. Replacing the butanamide (25) with piperazine moiety, along with the presence of the trifluoromethyl group (26), enhanced the FAK inhibitory activity.
Zhu and coworkers [142] reported the synthesis of 1,3,4-thiadiazol-2-amides as potent FAK inhibitors. The obtained antiproliferative activity of the prepared derivatives revealed that compound 27 was able to inhibit the growth of MCF-7 breast cancer and B16-F10 melanoma cell lines better than staurosporine (Figure 11); 27 showed twofold better anti-FAK activity than staurosporine. Western blot analysis emphasized that 27 exerted its activity through FAK inhibition. Compound 27 is considered an excellent example of retaining both antiproliferative and anti-FAK activity.
The same research group reported the synthesis of a series 2-(1,3,4-oxadiazol-2-ylthio)-1-phenylethanones with moderate antiproliferative and anti-FAK activity[143]. The group incorporated the oxadiazole ring instead of the thiadiazole as an approach for bioisosteric replacement [142]. Unfortunately, most of the synthesized derivatives did not show potent activity, excluding 28, which showed superior activity on MCF-7 breast cancer and A431 epidermoid carcinoma cells (Figure 11). Compound 28 showed an IC50 value sevenfold better than the reference staurosporine in A431 cells. Favorably, 28 was 1.5-fold more potent than staurosporine on FAK enzyme inhibition.
Qiu and coworkers synthesized novel pyridinyl-1,2,4-triazole-based FAK inhibitors possessing acetohydrazide moiety [144]. Compound 29 showed the best potency among the prepared compounds and retained both antiproliferative and anti-FAK activities (Figure 11). The pyridinyl-1,2,4-triazole 29 possessed potent FAK inhibitory activity with an IC50 value of 2.41 μM, whereas staurosporine showed an IC50 value of 3.12 μM. Compound 29 inhibited MCF-7, HCT116 and HepG2 cells 2.3-fold, 6.6-fold and twofold better than staurosporine, respectively. The western blot analyses confirmed that the potent antiproliferative activity of this series is attributed to the significant inhibition of FAK.
Recently, Altıntop and colleagues [145] reported dual-acting Akt and FAK inhibitors bearing 1,3,4-oxadiazole moiety. Using MTT assay, compound 30 showed more potent antiproliferative activity than cisplatin and GSK690693 on A549 lung adenocarcinoma, C6 rat gliom and NIH/3T3 mouse embryonic fibroblast cells (Figure 11). Compound 30 showed potent FAK inhibitory activity with an IC50 value of 19.50 μM. and possessed better Akt inhibitory activity than cisplatin and GSK690693.
In 2014, a series of 2-styryl-5-nitroimidazole derivatives bearing 1,4-benzodioxan moiety was synthesized as potential FAK inhibitors [45]. Compound 31 showed the same ability to inhibit A549 lung cancer and hela cervical cancer cells as staurosporine (IC50 = 3.11 and 2.54 μM, respectively; Figure 11). Compared with staurosporine (IC50 = 0.50 μM), 31 exhibited potent FAK inhibitory activity (IC50 = 0.45 μM).
Grädler and colleagues[146] used fragment-based lead discovery (FBLD) to develop novel FAK inhibitors. The team screened 1920 fragments against the kinase domain of FAK using surface plasmon resonance (SPR) [147]. Protein crystallography revealed the binding mode of only 41 fragment hits in the ATP binding site of FAK. The output of this study revealed a novel pyrazole-containing compound (32) that inhibits FAK in low micromolar potency, IC50 = 0.266 μM; KD,ss = 0.111 μM (KD,ss is the steady state dissociation constant that is determined by plotting the binding levels at the end of the association phase against the concentration; Figure 11). Compound 32 inhibited FAK significant phosphorylation at Tyr397 in HT-29 human colorectal adenocarcinoma cells (IC50 = 0.75 μM). Surprisingly, 32 showed a significant multiacting kinase inhibitory activity and inhibited 14 out of 48 tested kinases.
Jia and colleagues [148] prepared thiophenol-formaldehyde-triazoles as potential antiovarian cancer derivatives with potent anti-FAK activity. After 48 h treatment, compound 33 revealed a marked lessening in FAK activation in CAOV3 ovarian cancer cells at a concentration of 0.5 μM (Figure 11). Compound 33 considerably inhibited cell viability of ES-2, CAOV3 and CAOV4 ovarian cancer cells. This compound significantly decreased CAOV3 migration in a concentration-dependent manner and decreased the infiltration of macrophages in different metastatic lesions from the intestinal, liver and pulmonary tissues.
Recently, Mustafa and colleagues [149] reported 5-pyridinyl-1,2,4-triazoles containing acetamido carboxylic acids as potent FAK inhibitors. The prepared derivative 34 showed strong antiproliferative activity in hepatocellular carcinoma HepG2 (IC50 = 3.78 μM) and Hep3B (IC50 = 4.83 μM) cells (Figure 11). Compound 34 showed better anti-FAK inhibitory activity (IC50 = 18.10 nM) than the reference GSK2256098 (IC50 = 22.14 nM). Immunoblotting analysis in HepG2 cells revealed a substantial FAK phosphorylation repression. The obtained FAK inhibition decreased the phosphorylation in PI3K, Akt, JNK and STAT3.
The five-membered nitrogen-containing FAK inhibitors are considered an important category because this scaffold is amenable to modifications that allow the molecular hybridization approach. Most prepared derivatives retained anti-FAK and antiproliferative activities, such as compounds 27, 28, 29, 32 and 34. SAR analysis revealed that the presence of the diphenyl substitution (ring A and ring B) on the five-membered heterocyclic ring is critical for activity (Figure 12). Molecular docking of most compounds revealed that the heterocyclic ring and ring A, (such asdiphenyl dioxine, substituted phenyl or pyridinyl) bind to Cys502 of the kinase hinge, while ring B attaches to the heterocyclic ring through an aliphatic or aromatic linker that allows interaction with Asp564 of the DFG motif (Figure 12). The five-membered heterocyclic ring points to the gatekeeper residue Met499. The bioisosteric replacements such as 1,3,4-oxadiazole, 1,3,4-thiadiazole, different triazoles, imidazole and pyrazole were successful in most attempts.
Figure 12. . Common features of the five-membered FAK inhibitors.

Allosteric FAK inhibitors
Another novel method of inhibiting FAK is to target the allosteric site within the kinase domain, which is distant from the ATP active site (Figure 13). Allosteric inhibition is a highly selective strategy for targeting various kinases; for example, PD184352 and Akt-I-1 are allosteric inhibitors that target mitogen-activated protein kinase (MEK) and Akt, respectively [150,151].
Figure 13. . The FAK illustrates the ATP active binding site and the allosteric site.

Tomita and colleagues [152] used high-throughput screening to report 1,5-dimethyl-1,5-dihydropyrazolo[4,3-c][2,1]benzothiazine (35) as a potent allosteric inhibitor of FAK (Figure 14). Compound 35 showed considerable selectivity on FAK enzyme upon testing eight different kinases, with an IC50 value of 0.64 μM. This effect was obtained due to the significant drop in FAK autophosphorylation level in PC-3M-luc prostate cancer cells. Compound 35 suited the allosteric FAK active site perfectly and formed several hydrogen-bonding interactions with Gly563, Asp604, Met607, Phe608, Cys611 and Arg550 (Figure 15). The methyl group on the pyrazole moiety was also subjected to the hydrophilic space, and the tertiary butyl group occupied the hydrophobic pocket. Arg550 made a cation-π interaction with the pyrazole ring.
Figure 14. . Chemical structure of the allosteric FAK inhibitors 35 and 36.

Figure 15. . Cocrystal structure of 35 within the allosteric binding site of FAK, Protein Data Bank code: 4I4F.

Miki and coworkers reported a potent inhibitor (36) with the same scaffold as 35; however, the 1,5-dihydropyrazolo[4,3-c][2,1]benzothiazine moiety was directly attached to the 4-ethylphenyl group at position 8 (Figure 14) [153]. The derivative 36 bound to the FAK allosteric site with a slow dissociation rate on unphosphorylated FAK, leading to ATP noncompetitive FAK inhibition. Compound 36 was highly selective toward FAK. However, eliminating the ethyl group from N1 of the pyrazole ring resulted in nonselective FAK inhibitory activity, giving the impression that pyrazole N substitution is necessary for achieving FAK selectivity and allosteric binding. Targeting the allosteric site of FAK is a promising approach because the obtained inhibitors revealed strong FAK selectivity. However, addditional research should be conducted to further optimize the structures and increase the anti-FAK potency.
Targeting FAT & FERM domains as potential inhibitors of FAK
Several studies reported the similarity of different kinases to the ATP binding site of FAK, which inevitably leads to off-target effects due to the lack of selectivity [154–156]. FAT domain contains Tyr925 as a key phosphorylation site that, upon its phosphorylation, metastasis and invasion of tumors,is significantly promoted [157]. Likewise, Tyr397 autophosphorylation leads to FAK activation [158,159]. Targeting these phosphorylation sites (Tyr925 and Tyr397) leads to FAK inhibition. Therefore, several trials have targeted FAT or FERM domains of FAK [160,161].
FAK-FAT domain inhibitors
Cance and coworkers [162] discovered a small molecule (chloropyramine hydrochloride, 37) as a target for FAK and VEGFR-3 binding sites with the help of the computational docking techniques (Figure 16). The histamine receptor antagonist 37 was selected out of 140,000 molecules to target the FAT domain of the FAK protein. Compound 37 induced abrogation to the phosphorylation of VEGFR-3 and FAK. This potent antikinase activity resulted in considerable antiproliferative activity on BT474 and T47D breast cancer and C8161 melanoma cells, with IC50 values lower than 5 μM. Compound 37 revealed a dramatic decrease in the FAK and VEGFR-3 colocalization in the cytoplasm of BT474 and MCF7-VEGFR3 cells. This is considered a novel method for targeting the protein–protein interactions of FAK and VEGFR-3.
Figure 16. . Chemical structure the FAK-FAT domain inhibitors 37–40.
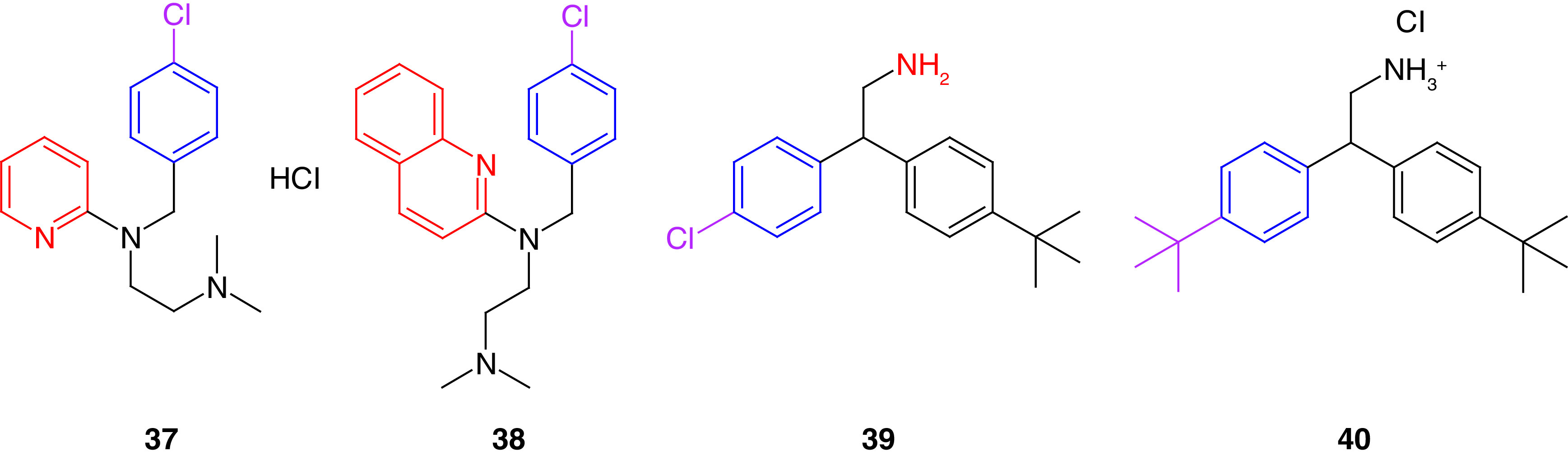
The same research group synthesized several analogs of 37 to seek better activity [163]. Their efforts revealed compound 38, which exhibited significant antiproliferative activity in MiaPaCa-2-luc and Panc-1-luc pancreatic cancer and MCF7-VEGFR-3 cell lines (Figure 16). Compound 38 considerably inhibited FAK-VEGFR-3 interaction better than 36 in MiaPaCa-2-luc cells. Compound 38 was tested against 10 different kinases; however, it did not show any significant activity, implying that 38 targets a specific FAK-VEGFR-3 interaction site. Molecular docking revealed a remarkable ability of 38 to occupy the FAK-FAT pocket with several key hydrophobic and electrostatic interactions; the quinoline ring made a cation-π interaction with Lys1032 amino acid along with a π-stacking interaction between Tyr925 and the p-chlorobenzyl group.
Recently, Kandil and colleagues[164] employed structure-based drug design and virtual screening to filter a library of 210,000 compounds against the FAK-FAT domain to identify 25 virtual hit compounds screened in the invasive MDA-MB-231 cell line. Compound 39 revealed significant antiproliferative and antimigratory properties in low micromolar concentration (Figure 16). These results exceeded those of chloropyramine, which may be attributed to the presence of the amino group that makes a salt bridge with the carboxyl group of Asp1036, and to the formation of two hydrogen bonds with the carbonyl group of Pro911 and the hydroxy group of Ser910 amino acid [162]. Compound 39 was considered a starting point that led to studying the impact of adding different substituents to the diarylamine scaffold; the prepared derivatives possessed better antiproliferative activity than chloropyramine in T47D, BT474 and MDA-MB-231 breast cancer and MIAPaca2 pancreatic cancer cell lines. The substitution variation resulted in a slight increase in the antiproliferative activity for compound 40 over compound 39 in most tested cells (Figure 16).
Interestingly, the study of the FAT-domain targeting inhibitors revealed that all the prepared compounds share the same scaffold, and only structural optimization was required. Replacing the chloro group of compounds 37–39 with trifluoromethoxy or tertiary butyl groups increased the antiproliferative activity (40). On the contrary, adding a methoxy group or aliphatic side chains in the same position led to a drastic decrease in potency. Adding quinoline moiety (38) instead of the pyridine (37), boosted the activity.
FAK-FERM domain inhibitors
Golubovskaya and colleagues [165] used a structure-based in silico molecular docking approach to target the Tyr397 phosphorylation site of FAK. A total of 140,000 small molecules were docked into the N-terminal domain of FAK kinase domain (PDB code: 2AL6) in 100 different orientations [17]. The top 35 molecules found to best fit into the Tyr397 phosphorylation site in the FERM domain with the best energy scores, were tested on six different cancer cells. Only one compound (1,2,4,5-benzenetetraamine tetrahydrochloride, 41, Y15) possessed promising antiproliferative activity in all the tested cells and was comparable to the reference TAE226 (Figure 17). Compound 41 inhibited BT474 cancer cell viability and FAK phosphorylation in a dose-dependent manner. Western blot analysis revealed that 41 specifically inhibited Tyr397 FAK phosphorylation and caused less cellular detachment than TAE226 in BT474 cells, indicating a significant ability of 41 to prevent metastasis. The in vivo studies performed in nude mice revealed that 41 blocked tumor growth and reduced tumor weight in BT474 cells at a dose of 30 mg/kg. Western blotting and immunohistochemical staining of tumors showed decreased Tyr397-FAK phosphorylation levels in 41-treated mice. The docking pose of 41 revealed the formation of four hydrogen bonds with Tyr397, Glu399, Arg57 and Thr394 amino acids, and was in higher proximity to the essential amino acid Tyr397.
Figure 17. . Chemical structure FAK-FERM domain inhibitors 41–47.
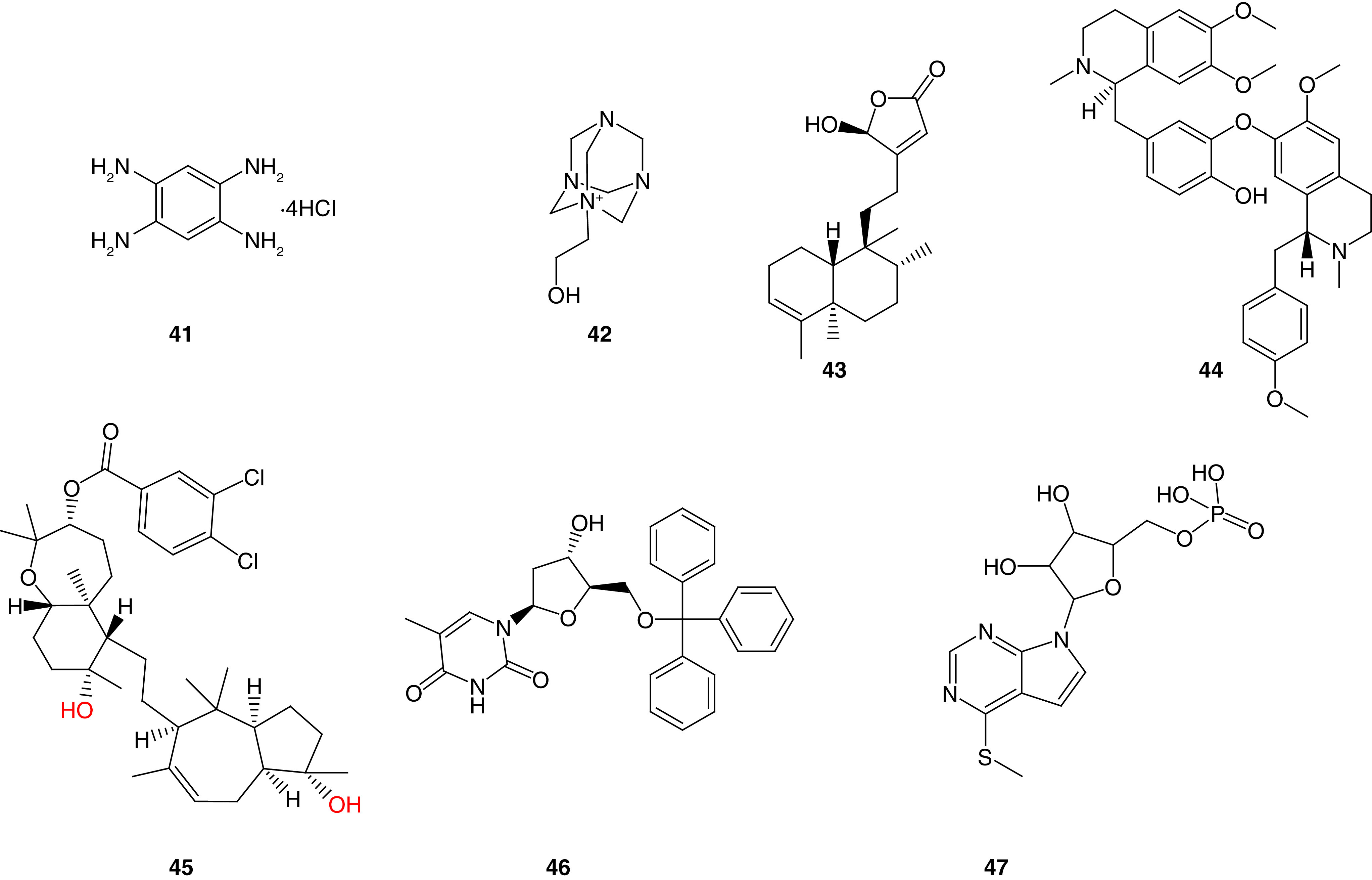
Compound 42, also known as Y11, was identified by the same research group, as a potent FAK inhibitor that targets Tyr397 site (Figure 17) [160]. The azoniatricyclodecane derivative 42 blocked the autophosphorylation of FAK effectively and significantly lessened Tyr397 phosphorylation in BT474 (breast cancer) and SW620 (colon cancer) cells. The octet binding assay confirmed the ability of 42 to bind directly to the FAK-N-terminal domain. Interestingly, 42 prevented colony formation in BT474 and SW620 cancer cells. The in vivo studies revealed that 42 (125 mg/kg) decreased tumor growth and weights in SW620 xenograft mice tumor. Additionally, it decreased Tyr397-FAK phosphorylation, and increased caspase-3 staining in tumor xenografts.
Thiyagarajan and colleagues [166] conducted docking studies to discover the natural compound 16-hydroxy-cleroda-3,13-dien-16,15-olide (HSD, 43). MTT assay revealed that 43 significantly inhibited N18 neuroblastoma and C6 glioma cells in 10 μM concentration (Figure 17). Compound 43 increased p53 expression and downregulated B-cell lymphoma 2 (Bcl-2 gene family comprises a group of related genes that regulate apoptosis) resulting in significant apoptosis induction [167]. Western blotting and immunofluorescence assays showed that 43 decreased the Tyr397-FAK expression level in C6 and N18 cells.
The same research group applied virtual screening to discover nifedipine as potent FAK and 70-kDa ribosomal S6 kinase 1 (S6K1) inhibitor [168]. Neferine (44) was able to bind perfectly to FAK-FERM domain and decreased phosphorylated FAK and S6K1 (Figure 17). Compound 44 showed better antiproliferative activity than cisplatin in C6 cells. Additional studies performed on neferine revealed that the dual FAK/S6K1 inhibitory activity of neferine results in potent anticancer activity in human neuroblastoma [169].
El Sayed and coworkers [170] studied a derivative of the natural compound sipholenol A called sipholenol A-4-O-3′,4′-dichlorobenzoate (SAP, 45) that was found to inhibit breast cancer cells with promising IC50 values, including MDA-MB-231, MCF-7, BT474 and T47D (Figure 17). Western blot analysis revealed that 45 resulted in significant FAK phosphorylation inhibition at Tyr397 site in all tested cancer cells. Moreover, 45 inhibited the phosphorylation of breast tumor kinase (a nonreceptor tyrosine kinase overexpressed in most invasive human breast tumors, with a fundamental role in promoting cancer proliferation, invasion and migration) [171,172]. In vivo experiments revealed a significant reduction in tumor volume, weight and growth (76% reduction) in a dose of 10 mg/kg. Docking 45 into the x-ray crystal structure of the FAK-FERM domain revealed the formation of two hydrogen bonds between the hydroxyl groups and Tyr394 and Glu399 amino acids, while the bicyclodecane ring formed a hydrophobic interaction with Tyr397's phenyl group.
Golubovskaya and colleagues[173] performed in silico molecular docking for more than 200,000 small molecules obtained from the NCI database to target FAK-MDM-2 interactions. The study revealed 24 optimal compounds that can target that complex. These compounds were tested for their antiproliferative activity in breast, colon, melanoma and pancreatic cancers using MTT assay. Among them, 46 had the best inhibitory activity in most tested cells (Figure 17). Compound 46 significantly inhibited the viability of BT474 breast cancer cells and increased cell detachment and apoptotic induction. Western blot analysis revealed a profound ability of 46 to decrease FAK and Tyr397 FAK, increase Mdm-2 apoptotic level, and activate P53 and caspase-8 in BT474 cells. At 100 μM, 46 decreased FAK and Mdm-2 complex formation substantially in HCT116 colon cancer and BT474 cells. In vivo experiments performed in a xenograft mouse model revealed a significant ability of 46 to decrease breast and colon tumor growths.
Deniz and colleagues[174] used INT2−31 (47) to target protein–protein interactions of FAK with cMET and IGF-1R (Figure 17). Compound 47 decreased the phosphorylation of Tyr397 in both Panc-1and Miapaca-2 pancreatic cancer cells and possessed potent anticancer activity in different types of cancer (breast, melanoma, esophageal and pancreatic cancer). This compound disrupted the binding of IGF-1R and cMET proteins and blocked the interaction of FAK with IGF-1R and cMET. In vivo studies using a xenograft mouse model revealed the substantial ability of 47 to decrease tumor growth in Miapaca2 and Panc-1 tumors. Fortunately, the combination of 47 with gemcitabine improved the activity in the pancreatic cancer xenograft.
Targeting the FAK-FERM domain revealed substantial activity. The reported compounds possessed FAK phosphorylation inhibition at the Tyr397 site and showed potent anticancer activity against various cancers. The reported compounds did not share similar structural features. Therefore, additional studies should be performed on the active site of the FAK-FERM domain to reveal the essential binding modes and interactions of the inhibitors. It might be best to cocrystallize the active inhibitors within the active site to describe these points.
FAK degradation through proteolysis targeting chimeras
PROTAC is a novel strategy that involves the chemical breakdown of the post-translational protein of interest. PROTACs consist of hetero-bifunctional molecules with two recognition moieties: one binds to a specific target protein and the other to E3 ubiquitin ligase connected by a linker (Figure 18) [175–177]. This is achieved by conjugating the E1 activating enzyme present in the ubiquitin–proteosome system (UPS) and E2 enzyme [178]. Then, E2 binds to the peptidic E3 ligase in a ternary complex that can recognize the target protein for subsequent ubiquitination and degradation by the 26S proteosome, which is a part of the UPS in eukaryotic cells [178]. Subsequently, the ligand that targets FAK protein derives the E3 ubiquitin ligase to the FAK, leading to the kinase's ubiquitination. This strategy was not developed to inhibit a specific enzyme, but to eliminate and degrade it.
Figure 18. . Targeting FAK kinase for degradation by PROTACs.
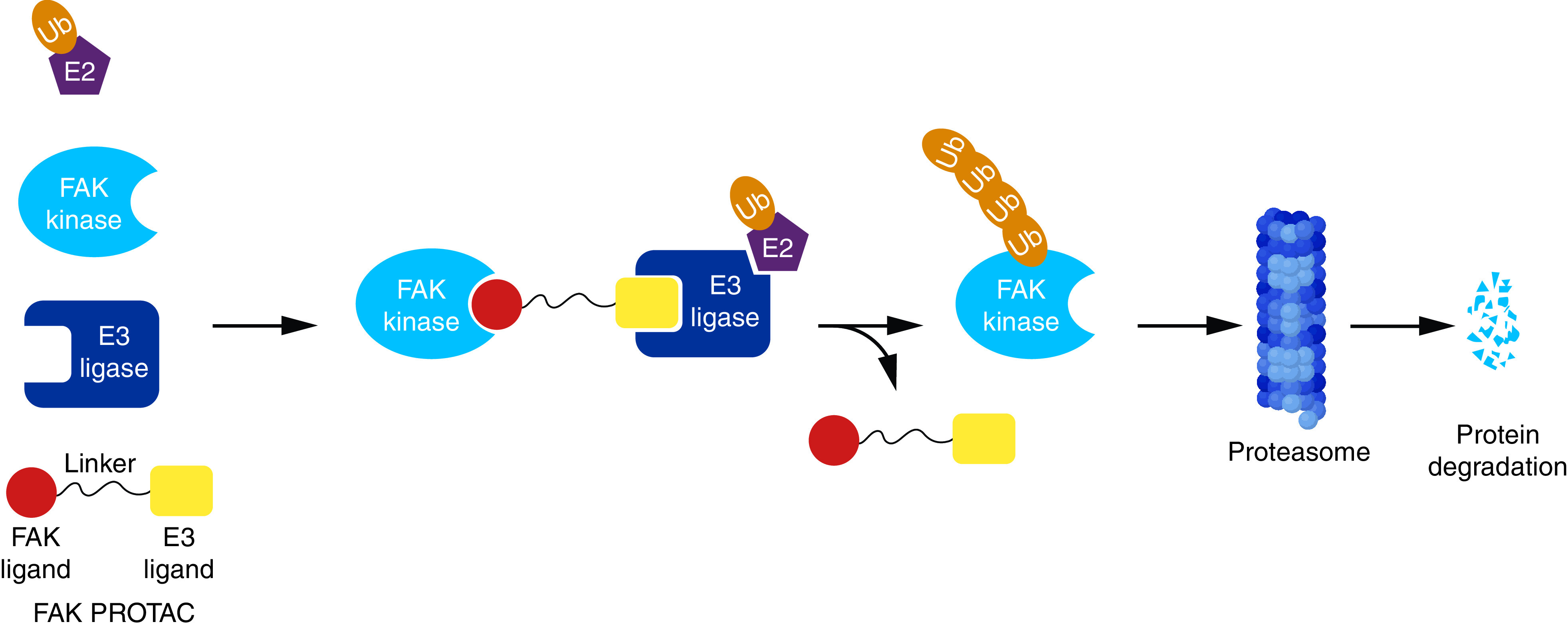
PROTAC: Proteolysis-targeting chimera.
Crews and coworkers [179] designed a potent FAK degrading PROTAC by connecting a defactinib analog and E3 recruiting element via an aliphatic ether linker. Regarding FAK inhibition, the prepared PROTAC 48 exhibited an enhanced selectivity over defactinib (Figure 19). This effect was accompanied by FAK degradation in a nanomolar concentration, which led to hampered ability of human triple-negative breast cancer MDA-MB-231 cells to migrate or invade. Hence, FAK's kinase dependent and independent signals were impaired by PROTAC.
Figure 19. . Chemical structures of FAK proteolysis targeting chimeras 48–52.
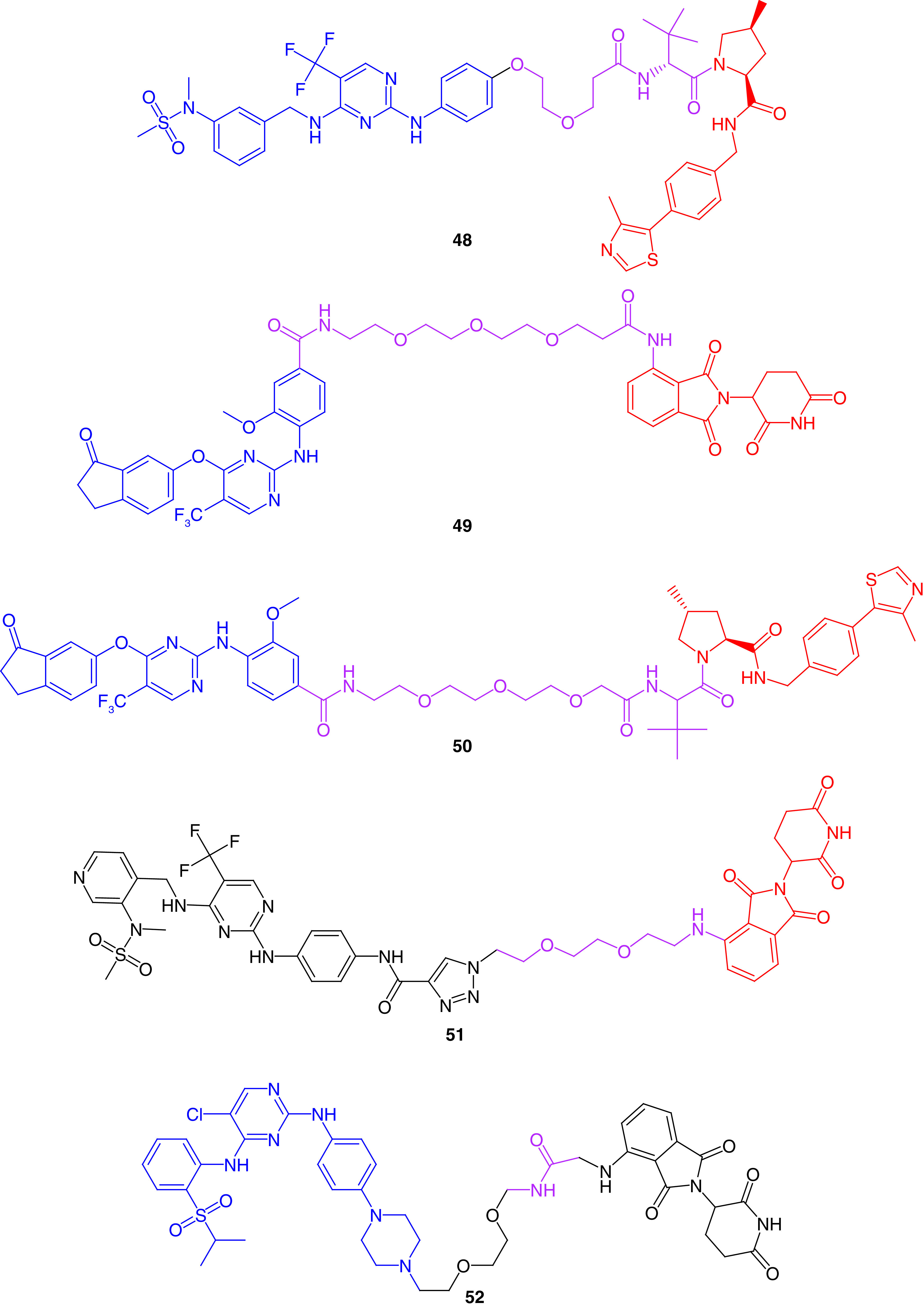
Popow and colleagues[180] used two different E3 ligases, cereblon (CRBN) and von Hippel−Lindau (VHL) complexes, and bound them to a potent FAK inhibitor via a polyethylene glycol linker (49 and 50, respectively; Figure 19). The prepared PROTACs reduced FAK levels with increased degradation activities in A549 lung cancer cells and 11 different hepatocellular carcinoma cells in nanomolar concentrations.
Rao and colleagues [181] linked an analog of the potent FAK inhibitor PF562271 to CRBN E3 ligase. The linker was flexible with two ether groups to allow swinging of the E3 ligase toward FAK. The prepared PROTAC 51 showed a substantial ability to degrade FAK in picomolar concentrations for 8 h treatment on PA1 ovarian teratocarcinoma, LNCap prostate adenocarcinoma, Ramos lymphoma and MDA-MB-436 breast cancer cells.
Multikinase degradation was accomplished by gray and his research group [182]. They prepared PROTAC 52, which was able to degrade 28 different kinases and 9 of the CDK family members (Figure 19). Compound 52 significantly degraded FAK enzyme in MOLM-14 and MOLT-4 leukemia cells.
Miscellaneous FAK inhibitors
Some FAK kinase inhibitors do not meet the typical modular structural form of the five- and six-membered FAK inhibitors. The selective FAK inhibitors in this miscellaneous category and are not easily classified in the manner outlined in this review, and are presented separately here. Ali and colleagues [183] reported the synthesis of alloxazine and 5-deazaalloxazine derivatives through nitrosative cyclization and Vilsmeier-Haack cyclization, respectively. Compounds 53, 54 and 55 revealed multiacting kinase inhibitory activity and exhibited significant potency on most tested cells, such as MCF7 breast cancer, A2780 ovarian cancer, HCT116 colon cancer and WM793B melanoma cell lines (Figure 20). Compound 53 showed approximately 13-fold and 32-fold more potent antiproliferative activity than Lapatinib on MCF7 and A2780 cells, respectively. Interestingly, 54 showed a dramatic increase in antiproliferative activity; it was 178-fold more potent than imatinib in both MCF7 and A2780 cells, while on HCT116, 54 was 393-times more active than imatinib. The synthesized compounds were tested for their ability to inhibit 20 different kinases. Compound 53 inhibited FAK (48%) and ABL1 (33%), whereas both 54 and 55 showed 34% inhibition of CDK. Compounds 54 and 55 inhibited FAK by 10% and 21%, respectively. Using a fused heterocyclic ring, such as alloxazine, may be a promising approach in inhibiting various kinases.
Figure 20. . Chemical structure and biological activity of miscellaneous FAK inhibitors 53–58.
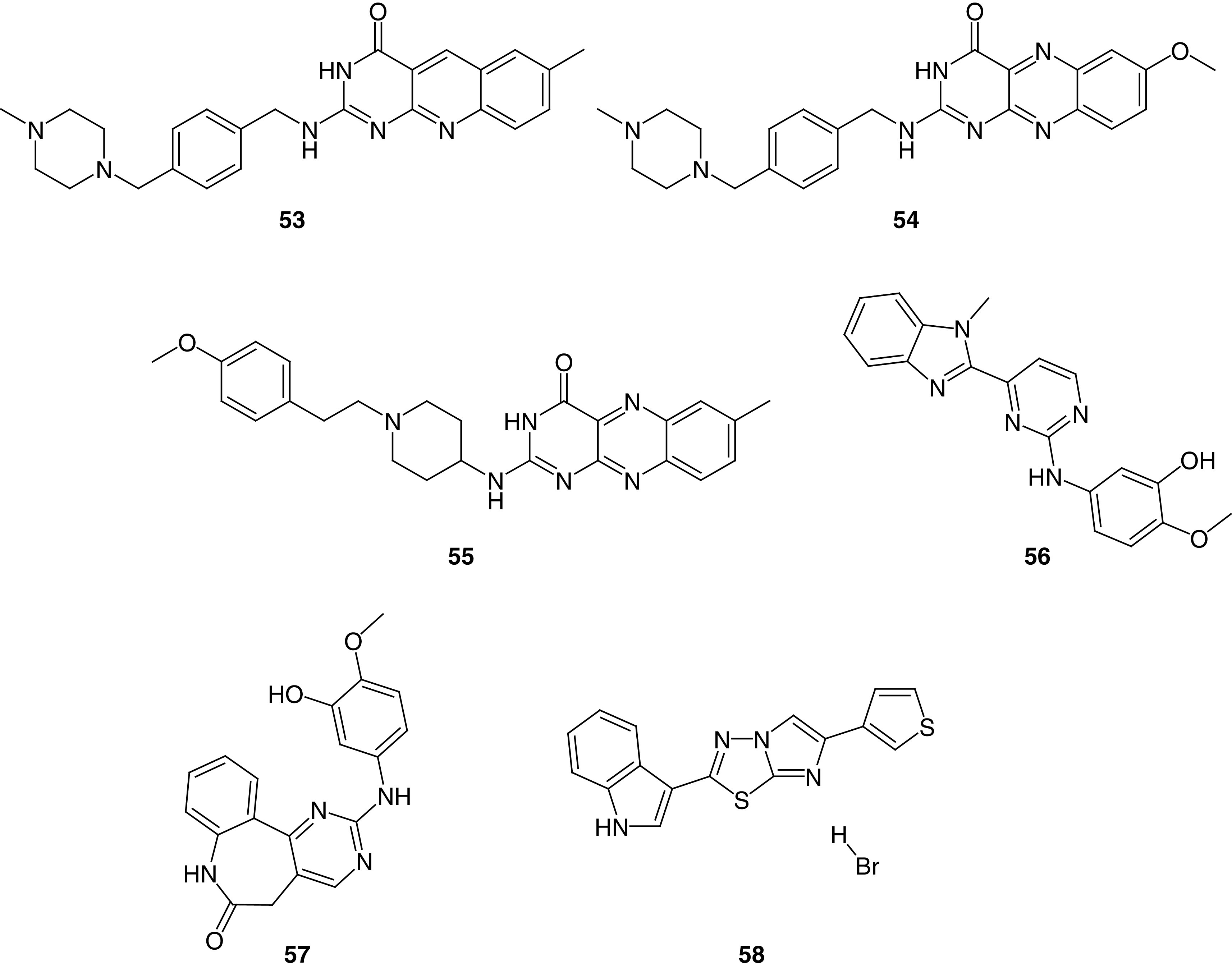
Determann and colleagues [184] prepared a series of 2-anilino-4-(benzimidazol-2-yl)-pyrimidine derivatives. Among the synthesized compounds, 56 showed considerable anti-FAK (IC50 = 3.40 μM) and antiproliferative activity (Figure 20). Using docking studies, the research team determined that their previously reported compound (57) could bind perfectly in the ATP active site in protein kinases (Figure 20) [185]. Compound 57 showed potent FAK inhibitory activity (IC50 = 0.64 μM) compared with sunitinib (IC50 = 1.60 μM). Compounds 56 and 57 inhibited three kinases efficiently (PLK1, VEGF-R2 and Aurora B). Compound 57 also showed better VEGF-R2 inhibition than sorafenib and equipotent activity to sunitinib. The antiproliferative activity of 56 and 57 was assessed on ten different cancer cells, and the best activity was obtained on MCF7, CAKI-1, MDA-MB-435, OVCAR-3 and SW620 cells.
Using Vilsmeier conditions, Diana and colleagues[186] reported a series of hybrid molecules containing imidazo[2,1-b][1,3,4]thiadiazole and indole moieties with potent FAK inhibitory activity. Compound 58 showed potent antiproliferative activity in leukemia K-562, colon cancer HT29, SW-620, and melanoma MDA-MB-435 cells (Figure 20). The sulforhodamine-B assay revealed that 58 possessed significant antiproliferative activity in PDAC cells; SUIT-2, Capan-1 and Panc-1. Compound 58 showed a significant reduction of PDAC-3 tumor sphere volume after 8 days of treatment in a dose of 8.5 μM and induced a considerable reduction in cell migration rate in SUIT-2 and Panc-1R cells. Western blot analysis revealed that 58 inhibited the phosphorylation of FAK significantly.
In 2018, Kassab and Hassan prepared hybrids of benzotriazole and N-acrylylhydrazone moieties as potent FAK inhibitors [43]. Compound 59 was approximately equipotent to the reference GSK-2256098 (IC50 = 44.60 nM) and inhibited the tyr397 phosphorylation in a nanomolar concentration (IC50 = 32.72 nM; Figure 21). Favorably, 59 revealed more than 70% growth inhibition on CCRF-CEM, HL-60 leukemia and OVCAR-3 ovarian cancer cells. Based on the obtained NCI results, OVCAR-3 and HL-60 cells were chosen for further analysis using MTT assay; 59 showed 10- and 18-times more potent antiproliferative activity on OVCAR-3 and HL-60 cells than doxorubicin, respectively.
Figure 21. . Chemical structures of miscellaneous FAK inhibitors 59–65.
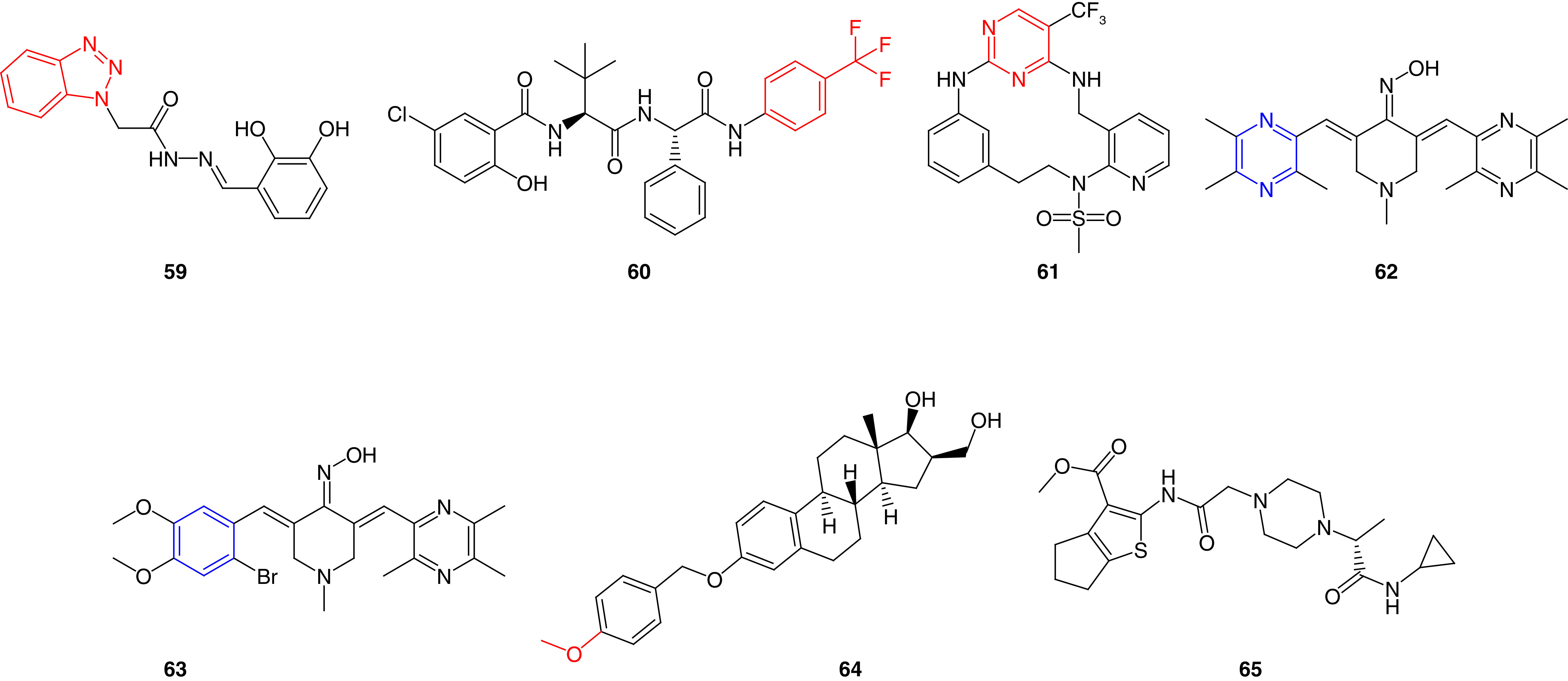
Additionally, the peptide drug discovery approach was investigated by Krystof and his research team by developing noncanonical modified leucine dipeptides [187]. At early time points, dipeptide 60 showed significant dephosphorylation of FAK, paxillin, and p130Cas at Tyr397, Tyr118 and Tyr410, respectively (Figure 21). This effect was accompanied by a gradual decrease in HCT-116 proliferating cells. This study is a excellent example of the use of dipeptides to inhibit FAK enzyme. Heck-mediated macrocyclization was implemented for preparing PF-562271 analogs by Farand and his research team [188]. The Macrocycle 60 provided enhanced selectivity on FAK and Pyk2 over the noncyclized form by 1.2-fold and 19-fold, respectively (Figure 21).
Bukhari and coworkers [189] synthesized a series of piperidin-4-one oxime derivatives bearing ligustrazine via Claisen-Schmidt condensation reaction. The ligustrazine derivatives 62 and 63 possessed the best activity among the synthesized compounds and showed more potent FAK inhibitory activity than erlotinib with IC50 values of 2.90 μM and 1.90 μM, respectively (Figure 21). Compounds 62 and 63 exhibited strong antiproliferative activity in nanomolar concentrations against A549 lung cancer, PC-3 prostate cancer, MCF7 breast cancer, PaCa-2 pancreatic cancer and HT29 colon cancer cells. The main structural difference between the two piperidin-4-one oxime derivatives is that the pyrazine moiety of 62 was replaced by a substituted phenyl ring in 63. Compounds 62 and 63 showed a wide variety of mechanistic pathways; they possessed a better ability to inhibit EGFR than erlotinib and a moderate activity on BRAFV600E kinase. Upon correlating the activity to the structural modifications, adding the oxime moiety boosted the activity dramatically. Moreover, importing pyrazine or substituted phenyl rings retained the overall activity.
Another important scaffold containing 3-benzyloxy-16-hydroxymethylene-estradiol was discovered by Zupkó and his research group [190]. Western blot assay showed that 64 significantly inhibited FAK phosphorylation in MDA-MB-231 cells in a concentration-dependent manner (Figure 21). Compound 64 possessed strong antiproliferative activity in five different breast cancer cells (MDA-MB-231, MCF7, T47D, MDA-MB-361 and MRC-5). Wound-healing assay revealed that estradiol 64 reduced the migration of 80% of MDA-MB-231 cells, indicating its substantial ability to prevent metastasis. This trial is an excellent example of incorporating the estradiol scaffold, which embodies an essential moiety used in hormonal-dependent cancers in FAK inhibition. In this respect, structural modifications could be performed for 64 to target both FAK and aromatase proteins that would fortify the anticancer activity.
Kang and coworkers [191] created two pharmacophore models to target FAK and cyclin-dependent kinase 4/6 (CDK4/6) active sites. CDKs play a crucial role in the process of mitosis and are overexpressed in various human tumors, and are considered an attractive target for the improvement of anticancer drugs [192]. Among seven million screened compounds, only six were selected from the pharmacophore model. Molecular docking and dynamics simulations performed on FAK and CDK4/6 revealed that 65 has an adequate molecular dynamics balance, stable structure and low docking energy (Figure 21). Docking of 65 within the ATP binding site of FAK revealed a hydrogen bond between the carbonyl group and Gly429, whereas the sulfur on thiazole made another hydrogen bond with Glu506. Regarding the CDK4/6 active site, the carbonyl group of 65 formed a hydrogen bond with Gly20, and the nitrogen atom on the piperazine ring made an ionic bond with Asp163 amino acid.
Conclusion
The present review article summarized many inhibitors created in the past three decades and provided an overview of the actions of FAK protein and FAK inhibitors. As an essential regulator of angiogenesis and cell migration, FAK is a proper target for cancer therapy. Numerous small-molecule FAK inhibitors were developed and assessed in preclinical trials and human clinical models. Some prepared FAK kinase inhibitors, such as 11, 13 and 34, could be ready for the preclinical trials due to their remarkable activity. Several attempts were made to enhance the anti-FAK activity, such as targeting both the ATP binding site and the allosteric binding site of FAK, FAK-FAT, and FAK-FERM active sites using different five- or six-membered nitrogen heterocyclic rings, dual and multiacting inhibitors, structure/fragment-based inhibitors, and PROTAC-mediated degradation. The strong antiproliferative nature of FAK inhibitors makes them attractive weapons against tumor cells if correctly targeted.
Future perspective: smarter FAK inhibitors
The diversity of the structural features of FAK inhibitors makes it a scalable route for the molecular hybridization approach. With the hope of slowing the emergence of drug resistance, several research groups investigated the dual or the multiacting activity of the anti-FAK derivatives. These trials targeted FAK with IGF-1R (TAE226, 1), Pyk2 (PF-373228, 3), ALK (5), EGFRT790M (12), Akt (30) and CDK4/6 (65). All these dual-acting compounds targeted FAK and another kinase. Because the structure of the ATP-binding sites of most kinases share fundamental structural similarities, these dual kinase-targeting drugs are likely to have more off-target effects and side effects. For this reason, it might be best to combine nonkinase targets with FAK (e.g., histone deacetylase, aromatase, tubulin or topoisomerase); unlike kinase-inhibitors, the compounds that inhibit these nonkinase proteins display some of the highest degrees of specificity. It is likely that in the future this approach will move in the direction of PROTAC-oriented chimeric and multiacting drugs, with anti-FAK action supported by a scaffold(s) working by a different mechanism of action. The design of the chimeric compounds should then be used in PROTAC-mediated degradation recruited to target specific proteins.
Executive summary.
FAK has been recognized as a promising target class for anticancer development.
The FAK protein is comprised of FAT, central kinase and FERM domains.
All FAK domains are targeted for FAK inhibition through binding to the ATP binding site or the phosphorylation sites.
The ATP binding site of FAK contains two key residues: Cys502 of the kinase hinge and Asp564 of the DFG motif.
The ATP binding site of FAK experiences conformational changes upon the inhibitor's binding.
Several FAK inhibitors have been developed in the last three decades.
Most of the developed inhibitors target the ATP binding site of the kinase domain (five- and six-membered inhibitors).
Several trials targeted the FAK-FERM and FAK-FAT domains and the allosteric binding site.
The developed FAK-proteolysis targeting chimeras (PROTAC) revealed promising activity and require further investigation.
Targeting FAK and other nonkinase targets in one PROTAC compound could be a successful approach to pursue in next few years.
Acknowledgments
The authors thank Ayman Mohamed, Pharmaceutical Chemistry Department, Faculty of Pharmacy, Deraya University, Minia, Egypt, for designing Figures 3 and 18.
Footnotes
Financial & competing interests disclosure
AA Abd El-Hafeez was supported by an NIH-funded Cancer Therapeutics Training Program (CT2, T32 CA121938) and P Ghosh by the NIH (CA238042, CA100768 and CA160911). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Erickson C. Cell migration in the embryo and adult organism. Curr. Opin. Cell Biol. 2(1), 67–74 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Stoker M, Gherardi E. Regulation of cell movement: the motogenic cytokines. Biochim. Biophys. Acta. Rev. Cancer 1072(1), 81–102 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Zhao X, Guan J-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Del. Rev. 63(8), 610–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell 84(3), 359–369 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Ridley AJ, Schwartz MA, Burridge Ket al. Cell migration: integrating signals from front to back. Science 302(5651), 1704–1709 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 110(6), 673–687 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl Acad. Sci. USA 89(11), 5192–5196 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sieg DJ, Hauck CR, Ilic Det al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat. Cell Biol. 2(5), 249–256 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Schaller MD, Parsons JT. Focal adhesion kinase and associated proteins. Curr. Opin. Cell Biol. 6(5), 705–710 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Hanks SK, Calalb MB, Harper MC, Patel SK. Focal adhesion protein-tyrosine kinase phosphorylated in response to cell attachment to fibronectin. Proc. Natl Acad. Sci. USA 89(18), 8487–8491 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell 129(6), 1177–1187 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadaré G, Gervasi N, Brami-Cherrier Ket al. Conformational dynamics of the focal adhesion targeting domain control specific functions of focal adhesion kinase in cells. J. Biol. Chem. 290(1), 478–491 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panera N, Crudele A, Romito I, Gnani D, Alisi A. Focal adhesion kinase: insight into molecular roles and functions in hepatocellular carcinoma. Int. J. Mol. Sci. 18(1), 99 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frame MC, Patel H, Serrels B, Lietha D, Eck MJ. The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 11(11), 802–814 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Lim S-T, Chen XL, Lim Yet al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 29(1), 9–22 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 14(9), 598–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ. Crystal structure of the FERM domain of focal adhesion kinase. J. Biol. Chem. 281(1), 252–259 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Walkiewicz KW, Girault J-A, Arold ST. How to awaken your nanomachines: site-specific activation of focal adhesion kinases through ligand interactions. Prog. Biophys. Mol. Biol. 119(1), 60–71 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18(5), 516–523 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Brunton VG, Avizienyte E, Fincham VJet al. Identification of Src-specific phosphorylation site on focal adhesion kinase: dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res. 65(4), 1335–1342 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Francois R, Iyer R, Seshadri M, Zajac-Kaye M, Hochwald SN. Current understanding of the molecular biology of pancreatic neuroendocrine tumors. J. Natl Cancer Inst. 105(14), 1005–1017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritt M, Guan JL, Sivaramakrishnan S. Visualizing and manipulating focal adhesion kinase regulation in live cells. J. Biol. Chem. 288(13), 8875–8886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sood AK, Coffin JE, Schneider GBet al. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am. J. Path. 165(4), 1087–1095 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furuyama K, Doi R, Mori Tet al. Clinical significance of focal adhesion kinase in resectable pancreatic cancer. World J. Surg. 30(2), 219–226 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Beierle EA, Massoll NA, Hartwich Jet al. Focal adhesion kinase expression in human neuroblastoma: immunohistochemical and real-time PCR analyses. Clin. Cancer Res. 14(11), 3299–3305 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Van Nimwegen MJ, Van De Water B. Focal adhesion kinase: a potential target in cancer therapy. Biochem. Pharmacol. 73(5), 597–609 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Gervais FG, Thornberry NA, Ruffolo SC, Nicholson DW, Roy S. Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J. Biol. Chem. 273(27), 17102–17108 (1998). [DOI] [PubMed] [Google Scholar]
- 28.Cavallaro U, Dejana E. Adhesion molecule signalling: not always a sticky business. Nat. Rev. Mol. Cell Biol. 12(3), 189–197 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Jean C, Chen XL, Nam J-Oet al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J. Cell Biol. 204(2), 247–263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen XL, Nam J-O, Jean Cet al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 22(1), 146–157 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan KT, Bennin DA, Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK). J. Biol. Chem. 285(15), 11418–11426 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim S-T, Miller NL, Nam J-O, Chen XL, Lim Y, Schlaepfer DD. Pyk2 inhibition of p53 as an adaptive and intrinsic mechanism facilitating cell proliferation and survival. J. Biol. Chem. 285(3), 1743–1753 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleinschmidt EG, Schlaepfer DD. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 45, 24–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tancioni I, Miller NL, Uryu Set al. FAK activity protects nucleostemin in facilitating breast cancer spheroid and tumor growth. Breast Cancer Res. 17(1), 47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hein N, Hannan KM, George AJ, Sanij E, Hannan RD. The nucleolus: an emerging target for cancer therapy. Trends Mol. Med. 19(11), 643–654 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Kurenova E, Xu L-H, Yang Xet al. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol. Cell. Biol. 24(10), 4361–4371 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu L, Owens LV, Sturge GCet al. Attenuation of the expression of the focal adhesion kinase induces apoptosis in tumor cells. Cell Growth Differ. 7(4), 413–418 (1996). [PubMed] [Google Scholar]
- 38.Xu L-H, Yang X, Bradham CAet al. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells involvement of death receptor-related signaling pathways. J. Biol. Chem. 275(39), 30597–30604 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Guan J-L, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature 358(6388), 690–692 (1992). [DOI] [PubMed] [Google Scholar]
- 40.Golubovskaya VM, Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. Int. Rev. Cytol. 263, 103–153 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Mclean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer – a new therapeutic opportunity. Nat. Rev. Cancer 5(7), 505–515 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Dao P, Jarray R, Le Coq Jet al. Synthesis of novel diarylamino-1, 3, 5-triazine derivatives as FAK inhibitors with anti-angiogenic activity. Bioorg. Med. Chem Lett. 23(16), 4552–4556 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Kassab AE, Hassan RA. Novel benzotriazole N-acylarylhydrazone hybrids: design, synthesis, anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis and FAK inhibition. Bioorg. Chem. 80, 531–544 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Ai M, Yu Jet al. Structure-based modification of carbonyl-diphenylpyrimidines (Car-DPPYs) as a novel focal adhesion kinase (FAK) inhibitor against various stubborn cancer cells. Eur. J Med. Chem. 172, 154–162 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Duan Y-T, Yao Y-F, Huang Wet al. Synthesis, biological evaluation, and molecular docking studies of novel 2-styryl-5-nitroimidazole derivatives containing 1, 4-benzodioxan moiety as FAK inhibitors with anticancer activity. Bioorg. Med. Chem. 22(11), 2947–2954 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin. Investig. Drugs 19(6), 777–788 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Wee Ma W. Development of focal adhesion kinase inhibitors in cancer therapy. Anticancer Agents Med. Chem. 11(7), 638–642 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Fagard R, Metelev V, Souissi I, Baran-Marszak F. STAT3 inhibitors for cancer therapy: have all roads been explored? Jakstat. 2(1), e22882 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frank DA. STAT signaling in the pathogenesis and treatment of cancer. Mol. Med. 5(7), 432–456 (1999). [PMC free article] [PubMed] [Google Scholar]
- 50.Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 251(2), 199–210 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Mustafa M, Abd El-Hafeez AA, Abdelhamid Det al. A first-in-class anticancer dual HDAC2/FAK inhibitors bearing hydroxamates/benzamides capped by pyridinyl-1,2,4-triazoles. Eur J. Med. Chem. 222, 113569 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Visavadiya NP, Keasey MP, Razskazovskiy Vet al. Integrin-FAK signaling rapidly and potently promotes mitochondrial function through STAT3. Cell Communication and Signaling 14(1), 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keasey MP, Kang SS, Lovins C, Hagg T. Inhibition of a novel specific neuroglial integrin signaling pathway increases STAT3-mediated CNTF expression. Cell Comm. Signal. 11(1), 35 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yue P, Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin. Investig. Drugs 18(1), 45–56 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang N, Dai Q, Su X, Fu J, Feng X, Peng J. Role of PI3K/AKT pathway in cancer: the framework of malignant behavior. Mol. Biol. Rep. 1–43 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexander W. Inhibiting the akt pathway in cancer treatment: three leading candidates. Pharm. Ther. 36(4), 225 (2011). [PMC free article] [PubMed] [Google Scholar]
- 57.Neel VA, Todorova K, Wang Jet al. Sustained Akt activity is required to maintain cell viability in seborrheic keratosis, a benign epithelial tumor. J. Invest. Dermatol. 136(3), 696–705 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Kong D, Chen F, Sima N. Inhibition of focal adhesion kinase induces apoptosis in bladder cancer cells via Src and the phosphatidylinositol 3-kinase/Akt pathway. Exp. Ther. Med. 10(5), 1725–1731 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lv J, Bai R, Wang L, Gao J, Zhang H. Artesunate may inhibit liver fibrosis via the FAK/Akt/β-catenin pathway in LX-2 cells. BMC Pharmacol. Toxicol. 19(1), 64 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Basson DM. Protein kinase B/AKT and focal adhesion kinase: two close signaling partners in cancer. Anticancer Agents Med. Chem. 11(10), 993–1002 (2011). [DOI] [PubMed] [Google Scholar]
- 61.Tancioni I, Uryu S, Sulzmaier FJet al. FAK inhibition disrupts a β5 integrin signaling axis controlling anchorage-independent ovarian carcinoma growth. Mol. Cancer Ther. 13(8), 2050–2061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gnani D, Romito I, Artuso Set al. Focal adhesion kinase depletion reduces human hepatocellular carcinoma growth by repressing enhancer of zeste homolog 2. Cell Death Differ. 24(5), 889–902 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nana FA, Lecocq M, Ladjemi MZet al. Therapeutic potential of focal adhesion kinase inhibition in small cell lung cancer. Mol. Cancer Ther. 18(1), 17–27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Golubovskaya VM, Cance WG. FAK and p53 protein interactions. Anticancer Agents Med. Chem. 11(7), 617–619 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 6(3), a026104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sawai H, Okada Y, Funahashi Het al. Activation of focal adhesion kinase enhances the adhesion and invasion of pancreatic cancer cells via extracellular signal-regulated kinase-1/2 signaling pathway activation. Mol. Cancer 4(1), 37 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huanwen W, Zhiyong L, Xiaohua S, Xinyu R, Kai W, Tonghua L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 8(1), 1–16 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buhrmann C, Shayan P, Goel A, Shakibaei M. Resveratrol regulates colorectal cancer cell invasion by modulation of focal adhesion molecules. Nutrients 9(10), 1073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Devaud C, Tilkin-Mariamé AF, Vignolle-Vidoni Aet al. FAK alternative splice mRNA variants expression pattern in colorectal cancer. Int. J. Cancer 145(2), 494–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang S-F, Chu S-C, Hsu L-S, Tu Y-C, Chen P-N, Hsieh Y-S. Antimetastatic effects of gossypol on colon cancer cells by targeting the u-PA and FAK pathways. Food Funct. 10(12), 8172–8181 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Zheng X, Liu W, Xiang Jet al. Collagen I promotes hepatocellular carcinoma cell proliferation by regulating integrin β1/FAK signaling pathway in nonalcoholic fatty liver. Oncotarget 8(56), 95586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meng X, Jin Y, Yu Yet al. Characterisation of fibronectin-mediated FAK signalling pathways in lung cancer cell migration and invasion. Br. J. Cancer 101(2), 327–334 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dragoj M, Milosevic Z, Bankovic J, Tanic N, Pesic M, Stankovic T. Targeting CXCR4 and FAK reverses doxorubicin resistance and suppresses invasion in non-small cell lung carcinoma. Cell. Oncol. 40(1), 47–62 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Shieh J-M, Cheng T-H, Shi M-Det al. α-Tomatine suppresses invasion and migration of human non-small cell lung cancer NCI-H460 cells through inactivating FAK/PI3K/Akt signaling pathway and reducing binding activity of NF-κB. Cell Biochem. Biophys. 60(3), 297–310 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Ward KK, Tancioni I, Lawson Cet al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin Exp. Metastasis 30(5), 579–594 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakamoto S, Schwarze S, Kyprianou N. Anoikis disruption of focal adhesion-Akt signaling impairs renal cell carcinoma. Eur. Urol. 59(5), 734–744 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Megison ML, Gillory LA, Stewart JE, Nabers HC, Mrozcek-Musulman E, Beierle EA. FAK inhibition abrogates the malignant phenotype in aggressive pediatric renal tumors. Mol. Cancer Res. 12(4), 514–526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Béraud C, Dormoy V, Danilin Set al. Targeting FAK scaffold functions inhibits human renal cell carcinoma growth. Int. J. Cancer 137(7), 1549–1559 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Golubovskaya VM, Zheng M, Zhang L, Li J-L, Cance WG. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC cancer 9(1), 280 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rachmi E, Purnomo BB, Endharti AT, Fitri LE. Afzelin inhibits migration of MDA-MB-231 cells by suppressing FAK expression and Rac1 activation. J. Appl. Pharm. Sci. 10(1), 077–082 (2020). [Google Scholar]
- 81.Payan I, Mcdonnell S, Torres HM, Steelant WF, Van Slambrouck S. FAK tyrosine 407 organized with integrin αVβ5 in Hs578Ts (i) 8 advanced triple-negative breast cancer cells. Int. J. Oncol. 48(5), 2043–2054 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Taliaferro-Smith L, Oberlick E, Liu Tet al. FAK activation is required for IGF1R-mediated regulation of EMT, migration, and invasion in mesenchymal triple negative breast cancer cells. Oncotarget 6(7), 4757 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Y, Hu X, Yang S. Clinical significance of focal adhesion kinase (FAK) in cervical cancer progression and metastasis. Int. J. Clin. Exp. Pathol. 13(10), 2586–2592 (2020). [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson TR, Khandrika L, Kumar Bet al. Focal adhesion kinase controls aggressive phenotype of androgen-independent prostate cancer. Mol. Cancer Res. 6(10), 1639–1648 (2008). [DOI] [PubMed] [Google Scholar]
- 85.Roberts WG, Ung E, Whalen Pet al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 68(6), 1935–1944 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Lietha D, Eck MJ. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PloS ONE 3(11), e3800 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shi Q, Hjelmeland AB, Keir STet al. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 46(6), 488–496 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Golubovskaya VM, Virnig C, Cance WG. TAE226-induced apoptosis in breast cancer cells with overexpressed Src or EGFR. Mol. Carcinog. 47(3), 222–234 (2008). [DOI] [PubMed] [Google Scholar]
- 89.Halder J, Lin YG, Merritt WMet al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 67(22), 10976–10983 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Beierle EA, Trujillo A, Nagaram A, Golubovskaya VM, Cance WG, Kurenova EV. TAE226 inhibits human neuroblastoma cell survival. Cancer Invest. 26(2), 145–151 (2008). [DOI] [PubMed] [Google Scholar]
- 91.Hatakeyama S, Tomioka D, Kawahara Eet al. Anti-cancer activity of NVP-TAE226, a potent dual FAK/IGF-IR kinase inhibitor, against pancreatic carcinoma. J. Clin. Oncol. 24(Suppl. 18), 13162–13162 (2006). [Google Scholar]
- 92.Hao H-F, Takaoka M, Bao X-Het al. Oral administration of FAK inhibitor TAE226 inhibits the progression of peritoneal dissemination of colorectal cancer. Biochem. Biophys. Res. Commun. 423(4), 744–749 (2012). [DOI] [PubMed] [Google Scholar]
- 93.Kurio N, Shimo T, Fukazawa Tet al. Anti-tumor effect of a novel FAK inhibitor TAE226 against human oral squamous cell carcinoma. Oral oncol. 48(11), 1159–1170 (2012). [DOI] [PubMed] [Google Scholar]
- 94.Liu T-J, Lafortune T, Honda Tet al. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol. Cancer Ther. 6(4), 1357–1367 (2007). [DOI] [PubMed] [Google Scholar]
- 95.Zhou J, Bronowska A, Le Coq J, Lietha D, Gräter F. Allosteric regulation of focal adhesion kinase by PIP2 and ATP. Biophys. J. 108(3), 698–705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tanjoni I, Walsh C, Uryu Set al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 9(10), 764–777 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Walsh C, Tanjoni I, Uryu Set al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. Ther. 9(10), 778–790 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jiang H, Hegde S, Knolhoff BLet al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 22(8), 851–860 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ji N, Yang Y, Cai C-Yet al. VS-4718 antagonizes multidrug resistance in ABCB1-and ABCG2-overexpressing cancer cells by inhibiting the efflux function of ABC transporters. Front. Pharmacol. 9, 1236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu Y-S, Hsu H-C, Tseng K-C, Chen H-C, Chen S-J. Lgr5 promotes cancer stemness and confers chemoresistance through ABCB1 in colorectal cancer. Biomed. Pharmacother. 67(8), 791–799 (2013). [DOI] [PubMed] [Google Scholar]
- 101.Liu F, Yang L, Zhou Xet al. Clinicopathological and genetic features of Chinese hereditary nonpolyposis colorectal cancer (HNPCC). Med. Oncol. 31(10), 223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP–dependent transporters. Nat. Rev. Cancer 2(1), 48–58 (2002). [DOI] [PubMed] [Google Scholar]
- 103.Eckford PD, Sharom FJ. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109(7), 2989–3011 (2009). [DOI] [PubMed] [Google Scholar]
- 104.Slack-Davis JK, Martin KH, Tilghman RWet al. Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 282(20), 14845–14852 (2007). [DOI] [PubMed] [Google Scholar]
- 105.Sun H, Pisle S, Gardner ER, Figg I, William D. Bioluminescent imaging study: FAK inhibitor, PF-562,271, preclinical study in PC3M-luc-C6 local implant and metastasis xenograft models. Cancer Biol. Ther. 10(1), 38–43 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Infante JR, Camidge DR, Mileshkin LRet al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J. Clin. Oncol. 30(13), 1527–1533 (2012). [DOI] [PubMed] [Google Scholar]
- 107.Ott GR, Cheng M, Learn KSet al. Discovery of clinical candidate CEP-37440, a selective inhibitor of focal adhesion kinase (FAK) and anaplastic lymphoma kinase (ALK). J. Med. Chem. 59(16), 7478–7496 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Allwein SP, Mowrey DR, Petrillo DEet al. Development of a Process Route to the FAK/ALK Dual Inhibitor TEV-37440. Org. Process Res. Dev. 21(5), 740–747 (2017). [Google Scholar]
- 109.Webb TR, Slavish J, George REet al. Anaplastic lymphoma kinase: role in cancer pathogenesis and small-molecule inhibitor development for therapy. Expert Rev. Anticancer Ther. 9(3), 331–356 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Patel MR, Infante JR, Moore KNet al. Phase 1/1b study of the FAK inhibitor defactinib (VS-6063) in combination with weekly paclitaxel for advanced ovarian cancer. (2014). [Google Scholar]
- 111.Wang-Gillam A, Lockhart AC, Tan BRet al. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer. (2018). [Google Scholar]
- 112.Gerber DE, Camidge DR, Morgensztern Det al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 139, 60–67 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang J, He D-H, Zajac-Kaye M, Hochwald SN. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell cycle 13(19), 3143–3149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mohanty A, Pharaon RR, Nam A, Salgia S, Kulkarni P, Massarelli E. FAK-targeted and combination therapies for the treatment of cancer: an overview of phase I and II clinical trials. Expert Opin. Investig. Drugs 29(4), 399–409 (2020). [DOI] [PubMed] [Google Scholar]
- 115.Auger K, Smitheman K, Korenchuk Set al. 387 the focal adhesion kinase inhibitor GSK2256098: a potent and selective inhibitor for the treatment of cancer. Eur. J. Cancer 48, 118 (2012). [Google Scholar]
- 116.Shenton DD, Mcleod K, Muir Met al. 564 Anti-tumour activity of the focal adhesion kinase inhibitor GSK2256098C in ovarian cancer. Eur. J. Cancer 48, 173 (2012). [Google Scholar]
- 117.Bottsford-Miller J, Sanguino A, Thanapprapasr Det al. Enhancing anti-angiogenic therapy by blocking focal adhesion kinase. Gynecol. Oncol. 123(2), 432 (2011). [Google Scholar]
- 118.Mak G, Soria J-C, Blagden SPet al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br. J. Cancer 120(10), 975–981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tiede S, Meyer-Schaller N, Kalathur RKRet al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis 7(9), 1–19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Laszlo V, Valko Z, Ozsvar Jet al. The FAK inhibitor BI 853520 inhibits spheroid formation and orthotopic tumor growth in malignant pleural mesothelioma. J. Mol. Med. 97(2), 231–242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Doi T, Yang JC-H, Shitara Ket al. Phase I study of the focal adhesion kinase inhibitor BI 853520 in Japanese and Taiwanese patients with advanced or metastatic solid tumors. Target Oncol. 14(1), 57–65 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.De Jonge MJ, Steeghs N, Lolkema MPet al. Phase I study of BI 853520, an inhibitor of focal adhesion kinase, in patients with advanced or metastatic nonhematologic malignancies. Target Oncol. 14(1), 43–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu H, Wu B, Ge Yet al. Phosphamide-containing diphenylpyrimidine analogues (PA-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with enhanced activity against pancreatic cancer cell lines. Bioorg. Med. Chem. 25(24), 6313–6321 (2017). [DOI] [PubMed] [Google Scholar]
- 124.Qu M, Liu Z, Zhao Det al. Design, synthesis and biological evaluation of sulfonamide-substituted diphenylpyrimidine derivatives (Sul-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with antitumor activity. Bioorg. Med. Chem. 25(15), 3989–3996 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Yen-Pon E, Li B, AcebróN-Garcia-De-Eulate Met al. Structure-based design, synthesis, and characterization of the first irreversible inhibitor of focal adhesion kinase. ACS Chem. Biol. 13(8), 2067–2073 (2018). [DOI] [PubMed] [Google Scholar]; •• The researchers formed a compound (11) that can bind covalently to the ATP binding site of FAK and showed tenfold better anti-FAK activity than the reference, TAE226. This trial was the first to target the ATP binding site through covalent bond formation.
- 126.Sanderson K. Irreversible kinase inhibitors gain traction. Nat. Rev. Drug Discov. 12, 649–651 (2013). [DOI] [PubMed] [Google Scholar]
- 127.Ai M, Wang C, Tang Zet al. Design and synthesis of diphenylpyrimidine derivatives (DPPYs) as potential dual EGFR T790M and FAK inhibitors against a diverse range of cancer cell lines. Bioorg. Chem. 94, 103408 (2020). [DOI] [PubMed] [Google Scholar]
- 128.Song Z, Huang S, Yu Het al. Synthesis and biological evaluation of morpholine-substituted diphenylpyrimidine derivatives (Mor-DPPYs) as potent EGFR T790M inhibitors with improved activity toward the gefitinib-resistant non-small cell lung cancers (NSCLC). Eur. J. Med. Chem. 133, 329–339 (2017). [DOI] [PubMed] [Google Scholar]; • The synthesis of dual-acting FAK and EGFRT790M inhibitors using fragment-based drug design strategy. The researchers merged the two scaffolds of TAE226 and Mor-DPPY (EGFRT790M inhibitor) in the same structure. Compound 12 inhibited both FAK and EGFRT790M kinases significantly in nanomolar concentrations and showed 36.6% tumor growth inhibition for 60 mg/kg/day for the oral dosage.
- 129.Su Y, Li R, Ning Xet al. Discovery of 2, 4-diarylaminopyrimidine derivatives bearing dithiocarbamate moiety as novel FAK inhibitors with antitumor and anti-angiogenesis activities. Eur. J. Med. Chem 177, 32–46 (2019). [DOI] [PubMed] [Google Scholar]; •• Researchers replaced the morpholine moiety of the reference TAE226 with piperazinyl dithiocarbamate. This substitution resulted in a significant increase in both antiproliferative and anti-FAK activity. The most potent drivative in this series (13) could be ready to enter clinical trials soon.
- 130.Wang R, Chen Y, Zhao Xet al. Design, synthesis and biological evaluation of novel 7H-pyrrolo [2, 3-d] pyrimidine derivatives as potential FAK inhibitors and anticancer agents. Eur. J. Med. Chem 183, 111716 (2019). [DOI] [PubMed] [Google Scholar]
- 131.Wang R, Yu S, Zhao Xet al. Design, synthesis, biological evaluation and molecular docking study of novel thieno [3, 2-d] pyrimidine derivatives as potent FAK inhibitors. Eur. J. Med. Chem. 188, 112024 (2020). [DOI] [PubMed] [Google Scholar]
- 132.Wang R, Zhao X, Yu Set al. Discovery of 7H-pyrrolo [2, 3-d] pyridine derivatives as potent FAK inhibitors: design, synthesis, biological evaluation and molecular docking study. Bioorganic Chemistry 102, 104092 (2020). [DOI] [PubMed] [Google Scholar]
- 133.Choi H-S, Wang Z, Richmond Wet al. Design and synthesis of 7H-pyrrolo [2, 3-d] pyrimidines as focal adhesion kinase inhibitors. Part 1. Bioorg. Med. Chem. Lett. 16(8), 2173–2176 (2006). [DOI] [PubMed] [Google Scholar]
- 134.Choi H-S, Wang Z, Richmond Wet al. Design and synthesis of 7H-pyrrolo [2, 3-d] pyrimidines as focal adhesion kinase inhibitors. Part 2. Bioorg. Med. Chem. Lett. 16(10), 2689–2692 (2006). [DOI] [PubMed] [Google Scholar]
- 135.Dao P, Jarray R, Smith Net al. Inhibition of both focal adhesion kinase and fibroblast growth factor receptor 2 pathways induces anti-tumor and anti-angiogenic activities. Cancer Lett. 348(1–2), 88–99 (2014). [DOI] [PubMed] [Google Scholar]
- 136.Dao P, Smith N, Tomkiewicz-Raulet CLet al. Design, synthesis, and evaluation of novel imidazo [1, 2-a][1, 3, 5] triazines and their derivatives as focal adhesion kinase inhibitors with antitumor activity. J. Med. Chem. 58(1), 237–251 (2015). [DOI] [PubMed] [Google Scholar]
- 137.Dao P, Lietha D, Etheve-Quelquejeu M, Garbay C, Chen H. Synthesis of novel 1, 2, 4-triazine scaffold as FAK inhibitors with antitumor activity. Bioorg. Med. Chem. Lett. 27(8), 1727–1730 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Zificsak CA, Gingrich DE, Breslin HJet al. Optimization of a novel kinase inhibitor scaffold for the dual inhibition of JAK2 and FAK kinases. Bioorg. Med. Chem. Lett. 22(1), 133–137 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Heinrich T, Seenisamy J, Emmanuvel Let al. Fragment-based discovery of new highly substituted 1 H-pyrrolo [2, 3-b]-and 3 H-imidazolo [4, 5-b]-pyridines as focal adhesion kinase inhibitors. J. Med. Chem. 56(3), 1160–1170 (2013). [DOI] [PubMed] [Google Scholar]
- 140.Sun J, Yang Y-S, Li Wet al. Synthesis, biological evaluation and molecular docking studies of 1, 3, 4-thiadiazole derivatives containing 1, 4-benzodioxan as potential antitumor agents. Bioorg. Med. Chem. Lett. 21(20), 6116–6121 (2011). [DOI] [PubMed] [Google Scholar]
- 141.Sun J, Ren S-Z, Lu X-Yet al. Discovery of a series of 1, 3, 4-oxadiazole-2 (3H)-thione derivatives containing piperazine skeleton as potential FAK inhibitors. Bioorg. Med. Chem. 25(9), 2593–2600 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Yang X-H, Xiang L, Li Xet al. Synthesis, biological evaluation, and molecular docking studies of 1, 3, 4-thiadiazol-2-amide derivatives as novel anticancer agents. Bioorg. Med. Chem. 20(9), 2789–2795 (2012). [DOI] [PubMed] [Google Scholar]
- 143.Zhang L-R, Liu Z-J, Zhang Het al. Synthesis, biological evaluation and molecular docking studies of novel 2-(1, 3, 4-oxadiazol-2-ylthio)-1-phenylethanone derivatives. Bioorg. Med. Chem. 20(11), 3615–3621 (2012). [DOI] [PubMed] [Google Scholar]
- 144.Zhang Y-B, Liu W, Yang Y-Set al. Synthesis, molecular modeling, and biological evaluation of 1, 2, 4-triazole derivatives containing pyridine as potential anti-tumor agents. Med.Chem. Res. 22(7), 3193–3203 (2013). [Google Scholar]
- 145.Altıntop MD, Sever B, Çiftçi GA, Turan-Zitouni G, Kaplancıklı ZA, Özdemir A. Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors. Eur. J. Med. Chem. 155, 905–924 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Grädler U, Bomke J, Musil Det al. Fragment-based discovery of focal adhesion kinase inhibitors. Bioorg. Med. Chem. Lett. 23(19), 5401–5409 (2013). [DOI] [PubMed] [Google Scholar]
- 147.Hennig M, Ruf A, Huber W. Combining biophysical screening and X-ray crystallography for fragment-based drug discovery. : Fragment-Based Drug Discovery and x-Ray Crystallography. Springer, 115–143 (2011). [DOI] [PubMed] [Google Scholar]
- 148.Jia Y, Si L, Lin Ret al. Thiophenol-formaldehyde triazole causes apoptosis induction in ovary cancer cells and prevents tumor growth formation in mice model. Eur. J. Med. Chem. 172, 62–70 (2019). [DOI] [PubMed] [Google Scholar]
- 149.Mustafa M, Abuo-Rahma GEA, Abd El-Hafeez AAet al. Discovery of antiproliferative and anti-FAK inhibitory activity of 1,2,4-triazole derivatives containing acetamido carboxylic acid skeleton. Bioorg Med Chem Lett 40, 127965 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Compound 34 as a strong antiproliferative and anti-FAK inhibitory derivative (IC50 = 18.10 nM). This compound revealed the best FAK-inhibitory activity of the five-membered FAK inhibitors and decreased phosphorylation in PI3K, Akt, JNK and STAT3 as a result of its strong FAK inhibition.
- 150.Ohren JF, Chen H, Pavlovsky Aet al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 11(12), 1192–1197 (2004). [DOI] [PubMed] [Google Scholar]
- 151.Lindsley CW, Zhao Z, Leister WHet al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 15(3), 761–764 (2005). [DOI] [PubMed] [Google Scholar]
- 152.Tomita N, Hayashi Y, Suzuki Set al. Structure-based discovery of cellular-active allosteric inhibitors of FAK. Bioorg. Med. Chem. Lett. 23(6), 1779–1785 (2013). [DOI] [PubMed] [Google Scholar]
- 153.Iwatani M, Iwata H, Okabe Aet al. Discovery and characterization of novel allosteric FAK inhibitors. Eur. J. Med. Chem. 61, 49–60 (2013). [DOI] [PubMed] [Google Scholar]
- 154.Chen G, Gao C, Gao Xet al. Wnt/β-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol. Cancer Ther. 17(4), 806–813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hirata E, Girotti MR, Viros Aet al. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 27(4), 574–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kang Y, Hu W, Ivan Cet al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J. Natl Cancer Inst. 105(19), 1485–1495 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ren X-D, Kiosses WB, Sieg DJ, Otey CA, Schlaepfer DD, Schwartz MA. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 113(20), 3673–3678 (2000). [DOI] [PubMed] [Google Scholar]
- 158.Hanks SK, Ryzhova L, Shin N-Y, Brábek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci. 8(1–3), d982–d996 (2003). [DOI] [PubMed] [Google Scholar]
- 159.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 14(3), 1680–1688 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Golubovskaya VM, Figel S, Ho BTet al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3. 1.1 3, 7] decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 33(5), 1004–1013 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase–targeted cancer therapeutics. Sci. Signal. 6(268), pe10 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kurenova EV, Hunt DL, He D, Magis AT, Ostrov DA, Cance WG. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J. Med. Chem. 52(15), 4716–4724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gogate PN, Ethirajan M, Kurenova EV, Magis AT, Pandey RK, Cance WG. Design, synthesis, and biological evaluation of novel FAK scaffold inhibitors targeting the FAK–vegfr3 protein–protein interaction. Eur. J. Med. Chem. 80, 154–166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kandil SB, Jones SR, Smith S, Hiscox SE, Westwell AD. Structure-based virtual screening, synthesis and biological evaluation of potential FAK-FAT domain inhibitors for treatment of metastatic cancer. Molecules 25(15), 3488 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Golubovskaya VM, Nyberg C, Zheng Met al. A small molecule inhibitor, 1, 2, 4, 5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J. Med. Chem. 51(23), 7405–7416 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thiyagarajan V, Lin S-H, Chia Y-C, Weng C-F. A novel inhibitor, 16-hydroxy-cleroda-3, 13-dien-16, 15-olide, blocks the autophosphorylation site of focal adhesion kinase (Y397) by molecular docking. Biochim. Biophys. Acta. Gen. Subj. 1830(8), 4091–4101 (2013). [DOI] [PubMed] [Google Scholar]
- 167.Boise LH, González-García M, Postema CEet al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74(4), 597–608 (1993). [DOI] [PubMed] [Google Scholar]
- 168.Thiyagarajan V, Lin SH, Chang YC, Weng CF. Identification of novel FAK and S6K1 dual inhibitors from natural compounds via ADMET screening and molecular docking. Biomed. Pharmacother. 80, 52–62 (2016). [DOI] [PubMed] [Google Scholar]
- 169.Pham DC, Chang YC, Lin SR, Fuh YM, Tsai MJ, Weng CF. FAK and S6K1inhibitor, Neferine, dually induces autophagy and apoptosis in human neuroblastoma cells. Molecules 23(12), 3110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Akl MR, Foudah AI, Ebrahim HY, Meyer SA, Sayed KaE. The marine-derived sipholenol A-4-O-3′, 4′-dichlorobenzoate inhibits breast cancer growth and motility in vitro and in vivo through the suppression of Brk and FAK signaling. Marine Drugs 12(4), 2282–2304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen H-Y, Shen C-H, Tsai Y-T, Lin F-C, Huang Y-P, Chen R-H. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol. Cell. Biol. 24(24), 10558–10572 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kamalati T, Jolin H, Fry M, Crompton M. Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signaling to PI 3-kinase and Akt, via erb B3 phosphorylation. Oncogene 19(48), 5471–5476 (2000). [DOI] [PubMed] [Google Scholar]
- 173.Golubovskaya VM, Palma NL, Zheng Met al. A small-molecule inhibitor, 5′-O-Tritylthymidine, targets FAK And Mdm-2 interaction, and blocks breast and colon tumorigenesis in vivo. Anticancer Agents Med. Chem. 13(4), 532–545 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ucar AD, Magis AT, He D-Het al. Inhibiting the interaction of cMET and IGF-1R with FAK effectively reduces growth of pancreatic cancer cells in vitro and in vivo. Anticancer Agents Med. Chem. 13(4), 595–602 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Raina K, Crews CM. Chemical inducers of targeted protein degradation. J. Biol. Chem. 285(15), 11057–11060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sun Y, Zhao X, Ding Net al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell research 28(7), 779–781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98(15), 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sun X, Gao H, Yang Yet al. PROTACs: great opportunities for academia and industry. Signal Transduct. Target Ther. 4(1), 1–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cromm PM, Samarasinghe KT, Hines J, Crews CM. Addressing kinase-independent functions of Fak via PROTAC-mediated degradation. J. Am. Chem. Soc. 140(49), 17019–17026 (2018). [DOI] [PubMed] [Google Scholar]; • A potent FAK degrader, the prepared PROTAC 48 exhibited FAK degradation in a nanomolar concentration, which led to hampered ability of human triple-negative breast cancer MDA-MB-231 cells to migrate or invade.
- 180.Popow J, Arnhof H, Bader Get al. Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions. J. Med. Chem. 62(5), 2508–2520 (2019). [DOI] [PubMed] [Google Scholar]
- 181.Gao H, Wu Y, Sun Y, Yang Y, Zhou G, Rao Y. Design, synthesis, and evaluation of highly potent FAK-targeting PROTACs. ACS Med. Chem. Lett. 11(10), 1855–1862 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Huang H-T, Dobrovolsky D, Paulk Jet al. A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell Chem. Biol. 25(1), 88–99 e86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Malki WH, Gouda AM, Ali HEet al. Structural-based design, synthesis, and antitumor activity of novel alloxazine analogues with potential selective kinase inhibition. Eur. J. Med. Chem 152, 31–52 (2018). [DOI] [PubMed] [Google Scholar]
- 184.Determann R, Dreher J, Baumann Ket al. 2-Anilino-4-(benzimidazol-2-yl) pyrimidines–a multikinase inhibitor scaffold with antiproliferative activity toward cancer cell lines. Eur. J. Med. Chem 53, 254–263 (2012). [DOI] [PubMed] [Google Scholar]
- 185.Egert-Schmidt A-M, Dreher J, Dunkel Uet al. Identification of 2-anilino-9-methoxy-5, 7-dihydro-6 H-pyrimido [5, 4-d][1] benzazepin-6-ones as dual PLK1/VEGF-R2 kinase inhibitor chemotypes by structure-based lead generation. J. Med. Chem. 53(6), 2433–2442 (2010). [DOI] [PubMed] [Google Scholar]
- 186.Cascioferro S, Petri GL, Parrino Bet al. Imidazo [2, 1-b][1, 3, 4] thiadiazoles with antiproliferative activity against primary and gemcitabine-resistant pancreatic cancer cells. Eur. J. Med. Chem 189, 112088 (2020). [DOI] [PubMed] [Google Scholar]
- 187.Jorda R, Magar P, Hendrychová Det al. Novel modified leucine and phenylalanine dipeptides modulate viability and attachment of cancer cells. Eur. J. Med. Chem 112036 (2020). [DOI] [PubMed] [Google Scholar]
- 188.Farand J, Mai N, Chandrasekhar Jet al. Selectivity switch between FAK and Pyk2: macrocyclization of FAK inhibitors improves Pyk2 potency. Bioorg. Med. Chem. Lett. 26(24), 5926–5930 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Zha G-F, Qin H-L, Youssif BGet al. Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance. Eur. J. Med. Chem. 135, 34–48 (2017). [DOI] [PubMed] [Google Scholar]
- 190.Sinka I, Kiss A, Mernyák Eet al. Antiproliferative and antimetastatic properties of 3-benzyloxy-16-hydroxymethylene-estradiol analogs against breast cancer cell lines. Eur. J. Pharm. Sci. 123, 362–370 (2018). [DOI] [PubMed] [Google Scholar]
- 191.Sun C, Feng L, Sun X, Yu R, Kang C. Design and screening of FAK, CDK 4/6 dual inhibitors by pharmacophore model, molecular docking, and molecular dynamics simulation. J. Biomol. Struct. Dyn. 1–10 (2020). [DOI] [PubMed] [Google Scholar]
- 192.Gelbert LM, Cai S, Lin Xet al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest. New Drugs 32(5), 825–837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]