

ABSTRACT
Macroautophagy/autophagy is a host natural defense response. Viruses have developed various strategies to subvert autophagy during their life cycle. Recently, we revealed that autophagy was activated by binding of Avibirnavirus to cells. In the present study, we report the inhibition of autophagy initiated by PIK3C3/VPS34 via the PDPK1-dependent AKT-MTOR pathway. Autophagy detection revealed that viral protein VP3 triggered inhibition of autophagy at the early stage of Avibirnavirus replication. Subsequent interaction analysis showed that the CC1 domain of VP3 disassociated PIK3C3-BECN1 complex by direct interaction with BECN1 and blocked autophagosome formation, while the CC3 domain of VP3 disrupted PIK3C3-PDPK1 complex via directly binding to PIK3C3 and inhibited both formation and maturation of autophagosome. Furthermore, we found that PDPK1 activated AKT-MTOR pathway for suppressing autophagy via binding to AKT. Finally, we proved that CC3 domain was critical for role of VP3 in regulating replication of Avibirnavirus through autophagy. Taken together, our study identified that Avibirnavirus VP3 links PIK3C3-PDPK1 complex to AKT-MTOR pathway and inhibits autophagy, a critical step for controlling virus replication.
Abbreviations
ATG14/Barkor: autophagy related 14; BECN1: beclin 1; CC: coiled-coil; ER: endoplasmic reticulum; hpi: hours post-infection; IBDV: infectious bursal disease virus; IP: co-immunoprecipitation; mAb: monoclonal antibody; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MOI: multiplicity of infection; MTOR: mechanistic target of rapamycin kinase; PDPK1: 3-phosphoinositid-dependent protein kinase-1; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PtdIns3K: phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol-3-phosphate; SQSTM1: sequestosome 1; vBCL2: viral BCL2 apoptosis regulator.
KEYWORDS: AKT-MTOR, autophagy, Avibirnavirus VP3, CC domain, PIK3C3-BECN1
Introduction
Autophagy plays a crucial role in maintaining cell homeostasis by recycling damaged proteins or organelles, and even clearing intracellular pathogens [1]. This process involves the engulfment of intracellular cargo in autophagosomes, which then fuse with lysosomes for final cargo degradation. Many autophagy-related proteins acting in this process have been identified. One protein necessary for autophagy is MAP1LC3/LC3 (microtubule associated protein 1 light chain 3). The lipidated form of LC3 (LC3-II) localizes to the membrane of growing phagophores and closed autophagosomes. LC3 has been widely used as a marker to track the formation of autophagosomes and to monitor autophagy. In addition, the polyubiquitin binding protein SQSTM1 (sequestosome 1) is specifically digested through the autophagy-lysosome process. Therefore, SQSTM1 could be used as a marker to assess autophagic flux [2].
BECN1 (beclin 1), a mammalian homolog of yeast Vps30/Atg6, participates in the formation of the PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3) complex and recruits additional proteins, such as ATG14/Barkor (autophagy related 14) and UVRAG (UV radiation resistance associated) [3,4]. BECN1 is a coiled-coil (CC) protein that plays a central role in autophagosome formation and autophagosome-lysosome fusion [5]. Several viruses inhibit autophagy by the binding of viral proteins to BECN1. Herpes simplex virus-1 encoded ICP34.5 (infected cell protein 34.5) [6], γ-herpesviruses encoded viral protein, Kaposi sarcoma-associated herpesvirus encoded orf16, and murine γ-herpesvirus 68 encoded M11 inhibit autophagy by blocking BECN1 incorporation into the class III PtdIns3K (phosphatidylinositol 3-kinase) complex with PIK3C3 and ATG14 [7–9].
PIK3C3, belonging to the PtdIns3K family, forms several complexes with different proteins and is involved in a variety of cellular functions, such as the multivesicular body pathway, retrograde trafficking from endosomes to the Golgi, phagosome maturation, and autophagy [8]. In mammals, the core PIK3C3 complex comprises PIK3C3, BECN1, PIK3R4/VPS15 (phosphoinositide-3-kinase regulatory subunit 4), and ATG14 [10]. This is directed to the autophagosome formation nucleation site along the endoplasmic reticulum (ER) membrane by ATG14 to promote local synthesis of PtdIns3P (phosphatidylinositol-3-phosphate) and initiate autophagosome biogenesis, probably at sites of contact between the ER and mitochondria [11]. Although PIK3C3 plays important roles in autophagy and other critical cell functions, knowledge about its role during virus infection is still limited.
PDPK1 (3-phosphoinositide dependent protein kinase 1) is a master kinase, which is crucial for the activation of AKT/PKB (AKT serine/threonine kinase) and many other AGC (group A, G, and C) kinases including PRKC (protein kinase C), RPS6KB1 (ribosomal protein S6 kinase B1) and SGK (serum/glucocorticoid regulated kinase) [12]. An important role for PDPK1 is in the class I phosphoinositide 3-kinases (PtdIns3Ks)-PDPK1-AKT signaling pathway that is activated by growth factors, hormones, or pathogen infection [13,14]. The pathway regulates various cellular processes, including cell growth, survival, autophagy, apoptosis, and proliferation. Accordingly, the dysregulation of this pathway has been implicated in several human cancers and immunological diseases, and the components of this pathway are attractive targets of current therapeutic strategies [14]. Phosphorylation of AKT by PDPK1 depends on PtdIns(3,4,5)P3, which is generated by class I PtdIns3K. However, PDPK1 itself is not activated by class I PtdIns3Ks [15]. The mechanism that controls the activity of PDPK1 is still unknown.
Avibirnavirus, infectious bursal disease virus (IBDV), a member of Birnaviridae family, contains a bi-segmented dsRNA genome within a non-enveloped icosahedral capsid [16], and encodes an RNA-dependent RNA polymerase VP1, two structural proteins VP2 and VP3, a viral protease VP4, and a nonstructural protein, VP5 [17–22]. VP2 interacts with VP3 during particle morphogenesis [21]. An early report describes the link between autophagy and Avibirnavirus in autophagic vesicles using electron microscopy [23]. Recent research shows that autophagy is induced by Avibirnavirus protein VP2 at the immediately early stage upon infection, and is then inhibited at early stage after infection [24]. However, the molecular mechanism by which Avibirnavirus infection inhibits autophagy is unknown.
In the present study, we showed that Avibirnavirus inhibits autophagy through viral protein VP3. VP3 inhibited autophagosome formation by interrupting both the PIK3C3-BECN1 complex and the PIK3C3-PDPK1 complex through its CC1 and CC3 domains. However, VP3 blocked autophagosome maturation by destroying the PIK3C3-PDPK1 complex through its CC3 domain only. These findings identified a novel complex mediated by virus to control the formation and maturation of autophagy.
Results
Cellular autophagy was inhibited by viral protein VP3 during Avibirnavirus infection
A previous study suggests that autophagy is inhibited at the early stage of IBDV infection [24]; therefore, we further explored which viral protein was responsible for blocking autophagy. 293T or DF-1 cell lines with stable expression of GFP-LC3 were transfected with vectors expressing VP1, VP2, VP3, VP4 or VP5, respectively, 293T cells with VP3 expression showed fewer GFP puncta than did the control cells, while VP1, VP2, and VP5 expressing 293T cells revealed more GFP puncta (Figure 1A). Consistently, DF-1 cells with VP3 expression but not VP1, VP2, VP4 and VP5 expressions, also showed fewer GFP puncta than did the control cells (Figure 1B). To further investigate the effect of VP3 on autophagy, we examined the conversion of endogenous LC3-I to LC3-II. As shown in Figure 1C, VP3 expression inhibited the formation of LC3-II significantly and increased level of SQSTM1 compared with those cells transfected with the empty vector or expressing other viral proteins. In addition, we also tested if VP3 inhibited the formation of PtdIns3P using 2× FYVE as the marker to track PtdIns3P. As shown in Figure S1A, the formation of GFP-puncta was decreased in IBDV-infected cells at 6 h post-infection (hpi) or in VP3-transfected cells. Co-localization analysis showed that VP3 expression led to the dissociation of BECN1 from the ER (Figure S1B). Taken together, these results suggest that the Avibirnavirus VP3 protein could inhibit the formation of autophagosomes.
Figure 1.
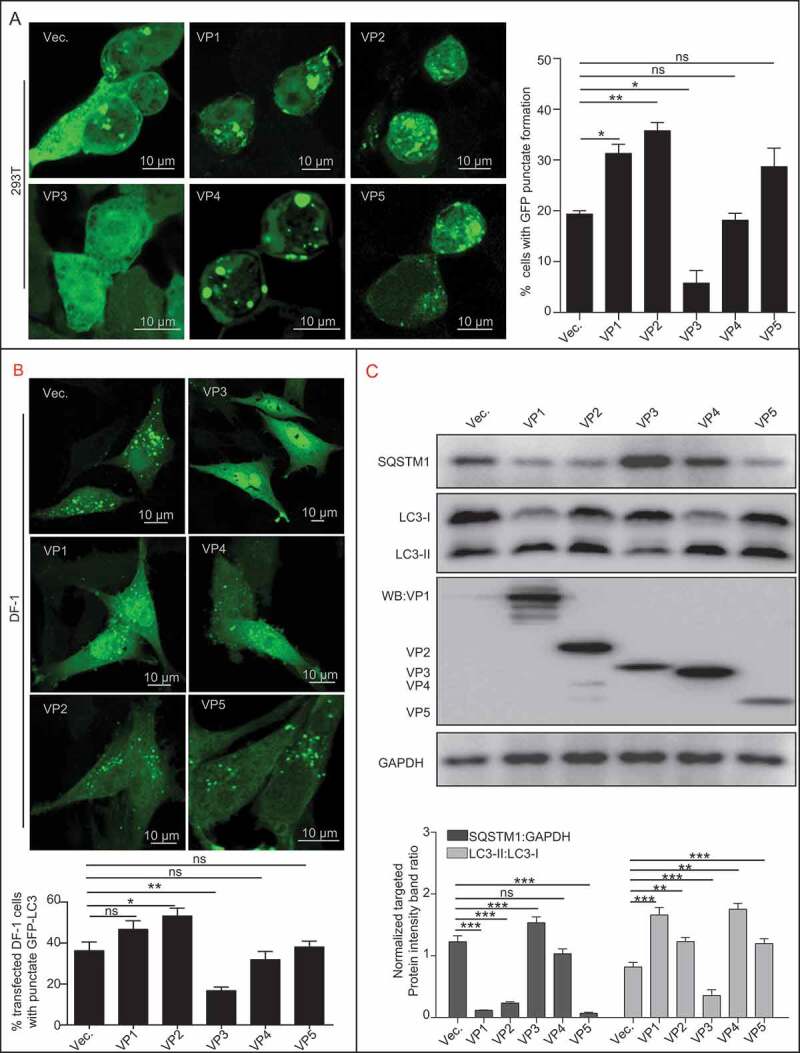
Avibirnavirus VP3-dependent autophagy inhibition. (A) The effect of Avibirnavirus proteins VP1, VP2, VP3, VP4, or VP5 on the basal autophagic vacuole formation in HEK293T cells. HEK293T cells stably expressing GFP-LC3 were transfected with empty vector pCI-neo, or vectors respectively expressing VP1, VP2, VP3, VP4, and VP5. Cells were treated with starvation for 4 h before collection. The cells were fixed and observed under confocal microscopy. The numbers of puncta were counted. Scale bar: 10 μm. (B) DF-1 cells stably expressing GFP-LC3 were transiently transfected with VP3 or empty vector pCI-neo. At 20 h post-transfection, DF-1 cells were starved for 4 h. The cells were then fixed and observed using confocal microscopy. Scale bar: 10 μm. (C) Cell samples in (A) were lysed and subjected to immunoblotting using indicated antibody. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001 compared to control.
CC1 or CC3 domain of VP3 was required for its interaction with BECN1 or PIK3C3/VPS34 respectively
Since VP3 was able to inhibit autophagy, we investigated whether PIK3C3-BECN1, a complex required for autophagy process, is a target of VP3. Anti-VP3 co-immunoprecipitation (IP) and immunoblotting with anti-PIK3C3 and anti-BECN1 antibody were performed. The results indicated that VP3 was able to interact with endogenous PIK3C3 and BECN1 (Figure 2A). Consistently, FLAG-tagged BECN1 or PIK3C3 could also interact with MYC-tagged VP3 (Figure S2). Furthermore, the GST affinity isolation revealed a direct interaction of VP3 with BECN1 or PIK3C3 (Figure 2B,C). Considering that PIK3R4 is a major component of BECN1-PIK3C3 complex, we test whether PIK3R4 mediates the interaction between VP3 and BECN1 or PIK3C3/VPS34. As shown in Figure S3, either overexpression or knockdown of PIK3R4 had no effect on interaction between VP3 and BECN1 or PIK3C3, indicating that VP3 interaction with BECN1 or PIK3C3 was irrelevant to PIK3R4. Next, we predicted the potential coiled-coil (CC) domains of VP3 by using the software (http://embnet.vital-it.ch/software/COILS_form.html). The prediction results showed that VP3 contained three CC domains, CC1, CC2, and CC3 (Figure 2D). Subsequently, the vectors expressing CC domain-deleted VP3 mutants, ∆CC1, ∆CC2, and ∆CC3 were constructed and each VP3 mutant was co-transfected into 293T cells together with FLAG-BECN1 or FLAG-PIK3C3. As shown in Figure 2E,F, the deletion of CC1 domain, but not CC2 or CC3, abrogated the VP3 interaction with BECN1 in IP, while a lack of the CC3 domain, but not CC1 or CC2, destroyed VP3 binding to PIK3C3, indicating that the CC1 of VP3 is the binding domain for BECN1 and CC3 of VP3 is the binding domain for PIK3C3.
Figure 2.
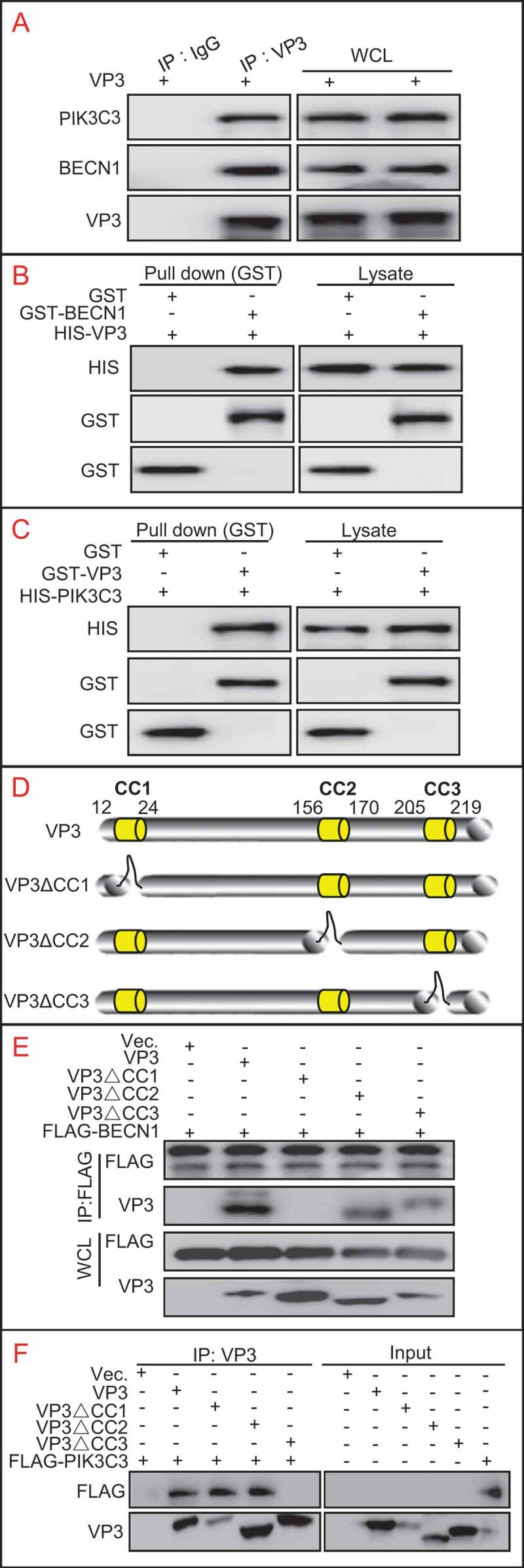
VP3 interacts with BECN1 and PIK3C3. (A)VP3 interacts with endogenous PIK3C3 or BECN1. HEK293T cells was transfected with pCI-neo vector expressing VP3 for 48 h. Anti VP3-precipitation and immunoblotting analyses using anti-PIK3C3 or anti-BECN1 antibodies were performed. (B)Verification of direct interaction on VP3 and BECN1 by GST affinity-isolation assays. GST, GST-BECN1 and HIS-VP3 were expressed in E. coli. GST affinity isolation and immunoblotting assays were then performed using anti-GST or anti-HIS antibodies. (C) Verification of direct interaction on VP3 and PIK3C3 by GST affinity-isolation assays. GST, GST-VP3 and HIS-PIK3C3 were expressed in E. coli. GST affinity isolation and immunoblotting assays were then performed using anti-GST or anti-HIS antibodies. (D) Schematic representation of CC domains of VP3 and its deletion mutants. (E) The CC1 domain of VP3 is necessary for interacting with BECN1. HEK293T cells were transiently co-transfected with VP3, VP3∆CC1 (∆CC1), VP3∆CC2 (∆CC1), VP3∆CC3 (∆CC1), and FLAG-BECN1. Cellular lysates were subjected to FLAG-precipitation and immunoblotting analysis of VP3, ∆CC1, ∆CC2, ∆CC3 and BECN1. * indicated heavy chain of antibody. (F) The CC3 domain of VP3 is necessary for interacting with PIK3C3. HEK293T cells were transfected with VP3, ∆CC1, ∆CC2, ∆CC3, and FLAG-PIK3C3 separately. Anti-VP3 IP and immunoblotting analysis using indicated antibodies were performed by the mixture of two indicated cell lysates.
PIK3C3 binding to BECN1 was disrupted by the CC1 domain of VP3
In addition, the IP assay showed that IBDV infection reduced the PIK3C3 interaction with BECN1 in Avibirnavirus-infected cells (Figure 3A). Further detection showed that only deletion of the CC1 domain of VP3 rescued the interaction between BECN1 and PIK3C3 (Figure 3B), indicating that the CC1 domain of VP3 disrupted PIK3C3’s interaction with BECN1 by binding competitively to BECN1. Furthermore, confocal microscope analysis of colocalization between PIK3C3 and BECN1 was performed upon expression of VP3 or its CC domain-deletion mutants (Figure 3C). Quantitative analysis of colocalization coefficient between VP3 or its mutants, PIK3C3 and BECN1 showed that VP3 or VP3∆CC2 separated BECN1 from PIK3C3 and colocalized with them respectively. And the co-localization coefficient of VP3∆CC1 and BECN1, but not and PIK3C3, decreased greatly compared with VP3 and BECN1, and VP3∆CC1 could not disassociate BECN1 from PIK3C3. By contrast, the colocalization coefficient of VP3∆CC3 and PIK3C3, but not and BECN1, reduced significantly. Interestingly, VP3∆CC3 was still capable of detaching BECN1 from PIK3C3. The above results suggest that CC1 domain of VP3 is necessary for interfering binding of PIK3C3 to BECN1.
Figure 3.
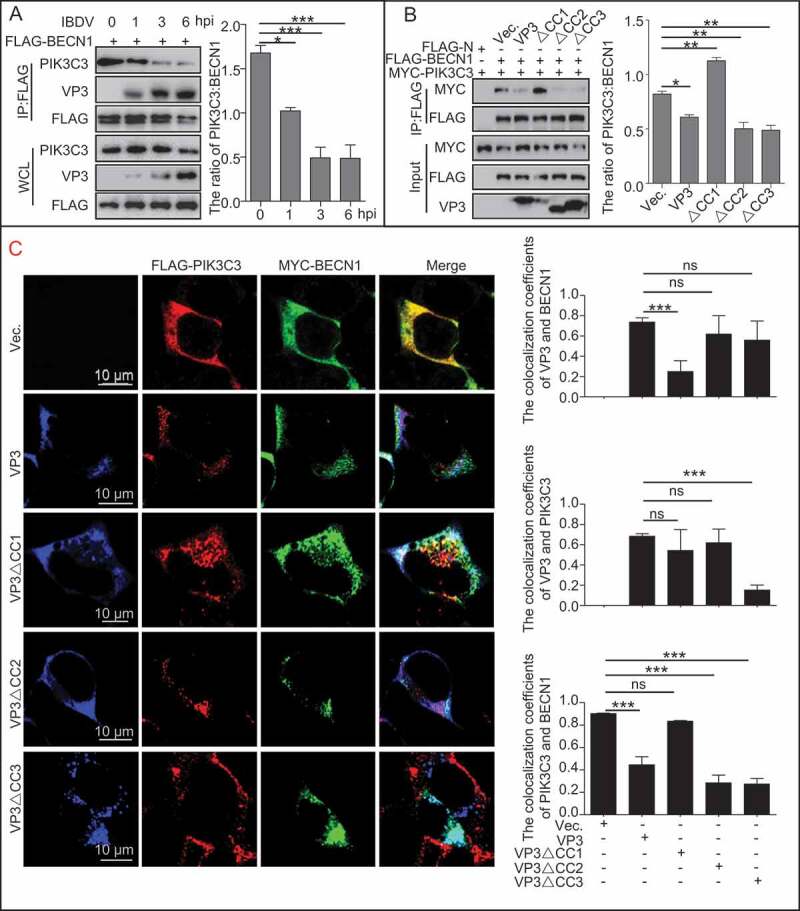
The effect of VP3 coiled-coil domains on interaction between PIK3C3 and BECN1. (A) PIK3C3’s interaction with BECN1 was gradually inhibited during Avibirnavirus infection. HEK293T cells were transfected with FLAG-BECN1 for 24 h, and then infected with Avibirnavirus at MOI = 10 for 0, 1, 3, and 6 h. The resultant cell lysates were subjected to FLAG-precipitation and immunoblotting analysis. (B and C) CC1 domain is necessary for disrupting the interaction between PIK3C3 and BECN1. HEK293T cells were transiently co-transfected with empty N-terminal FLAG-tagged vector (FLAG-N), FLAG-BECN1, MYC-PIK3C3 and VP3, ∆CC1, ∆CC2 or ∆CC3 for 24 h in 293T cells. On the one hand, cell lysates were subjected to analysis of FLAG-precipitation and immunoblotting analysis (B). On the other hand, cells were fixed and performed confocal analysis. 20 cells for each colocalization were counted. Quantitative analysis of colocalization coefficient was performed (C). Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001 compared to control.
The CC3 domain of VP3 is necessary for inhibition of complete autophagy
To detect the role of other CC domains of VP3 in autophagic inhibition, the vectors expressing various VP3 CC domain-deleted mutants were separately transfected into 293T cell lines with stable expression of GFP-LC3. As shown in Figure 4A,B, in comparison with wild-type VP3 and VP3∆CC2, VP3∆CC1 and VP3∆CC3 restored the formation of GFP-LC3 puncta by confocal analysis. Consistently, expression of wild-type VP3 and VP3∆CC2, but not VP3∆CC1 and VP3∆CC3, decreased conversion of LC3-I to LC3-II significantly in 293T cells either in absence or presence of chloroquine (CQ) by western blotting (WB) analysis (Figure 4C), suggesting that VP3 inhibited LC3-II formation, rather than promoting excessive degradation of LC3-II, through either CC1 or CC3 domain. However, only VP3∆CC3, but not VP3∆CC2 and VP3∆CC1, lost the ability to increase the level of SQSTM1 (Figure 4C). Moreover, transcriptional levels of LC3 or SQSTM1 had no significant differences between cell lines transfected with empty vector and vector expressing VP3 or its mutants by qPCR (Figure 4D). In order to verify the effects of VP3 CC domain-deleted mutants on autophagy, IBDV with VP3 CC domain-deleted mutants were constructed. Regrettably, these virus mutants were not able to be rescued. Alternatively, we generated various recombinant adenovirus (rAdV) containing Avibirnavirus VP3 and its CC deleted mutants to test the in vivo effects of VP3 CC domain on autophagy. As shown in Figure S4, in comparison to infection of 293T cells with control rAdV, infection of cells with rAdV containing VP3 or VP3∆CC2, but not VP3∆CC1 or VP3∆CC3, decreased conversion of LC3-I to LC3-II significantly. Similarly, only infection of 293T cells with virus containing VP3∆CC3, but not VP3∆CC2 and VP3∆CC1, failed to increase the level of SQSTM1. The above results suggest that the CC3 domain was required for VP3-mediated inhibition of complete autophagy. Subsequently, we analyzed whether the CC domain affected the fusion between autophagosomes and lysosomes. As shown in Figure 4E, in comparison to empty vector transfection, expression of VP3∆CC3, but not VP3∆CC1, increased fusion between autophagosomes and lysosomes. Consistently, expression of VP3, VP3∆CC2 and VP3∆CC1, but not VP3∆CC3, decreased the ratio of red dots to orange dots (Figure S5A and B). Moreover, expressions of VP3, VP3∆CC2 and VP3∆CC1, but not VP3∆CC3, decreased level of free GFP from GFP-LC3 (Figure S5C), suggesting that the CC3 domain of VP3 was critical for inhibition of autophagosome maturation. Collectively, the CC1 domain contributes to inhibition of autophagosomes formation, while CC3 domain contributes to inhibition of both autophagosome formation and maturation.
Figure 4.
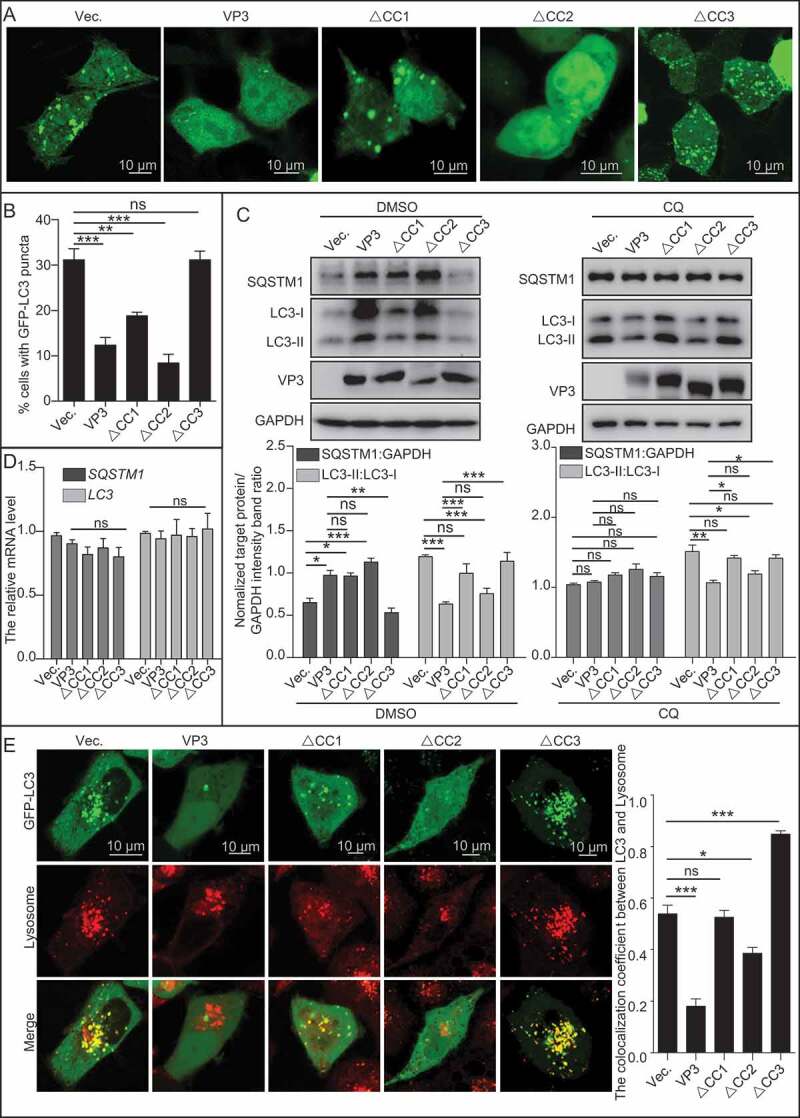
VP3 inhibits complete autophagy through CC3 domain. (A) HEK293T cells were transiently transfected with pCI-neo empty vector or vectors expressing VP3, ∆CC1, ∆CC2, and ∆CC3. At 20 h post-transfection, the cells were starved for 4 h and fixed for observation using confocal microscopy. Scale bar: 10 μm. (B) The quantitative analysis of the numbers of puncta in (A). (C) HEK293T cells were transiently transfected with vector, VP3, ∆CC1, ∆CC2 and ∆CC3 in presence of DMSO or CQ (50 μM). The cell samples were subjected to immunoblotting analysis with the indicated antibody. (D) Total RNA extracted from cell samples from (C) were subjected to qPCR analysis. (E) HEK293T cells were transiently transfected with empty vector (Vec.), VP3, ∆CC1, ∆CC2 and ∆CC3 for 24 h. HEK293T cells were then treated with LysoTracker for 30 min. The cells were observed under a confocal microscope. Scale bar: 10 μm. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001 compared to control.
VP3 disrupts the PIK3C3 interaction with PDPK1 via its CC3 domain binding to PIK3C3
Since PIK3C3-BECN1 complex was not involved in VP3-mediated inhibition of autophagy, we considered if other components interacting with PIK3C3 were involved in this process. PIK3C3 is implicated in controlling PDPK1-AKT-MTOR signaling pathway [8]. We then performed anti-PIK3C3 IP and the immunoblotting analysis. The results showed that endogenous PDPK1 could be detected (Figure 5A), suggesting that PIK3C3 was able to interact with PDPK1. Furthermore, affinity-isolation assay confirmed the direct interaction between PIK3C3 and PDPK1 (Figure 5B). Anti-FLAG IP showed that VP3 expression decreased the level of PIK3C3 precipitated by PDPK1, suggesting VP3 impaired the affinity of PIK3C3 to PDPK1 (Figure 5C). As shown in Figure 5D, as expected, confocal microscope analysis showed that PDPK1 colocalized together with PIK3C3 and the ER in cells co-transfected with PDPK1 and PIK3C3; however, the expression of VP3 resulted in disassociation of PDPK1 from PIK3C3 and the ER, and translocation of PDPK1 to the membrane (Figure 5D). Considering that VP3 interacts with PIK3C3 via its CC3 domain, we further established whether the CC3 domain affected the affinity of the PIK3C3 to PDPK1. Indeed, PDPK1’s interaction with PIK3C3 was attenuated in cells expressing wild-type VP3, mutant lacking CC1 or CC2, rather than CC3 domain, suggesting that the CC3 domain was critical for VP3-mediated abrogation of PIK3C3’s interaction with PDPK1 (Figure 5E). Collectively, VP3 blocks PIK3C3 interaction with PDPK1 via its CC3 domain binding competitively to PIK3C3.
Figure 5.
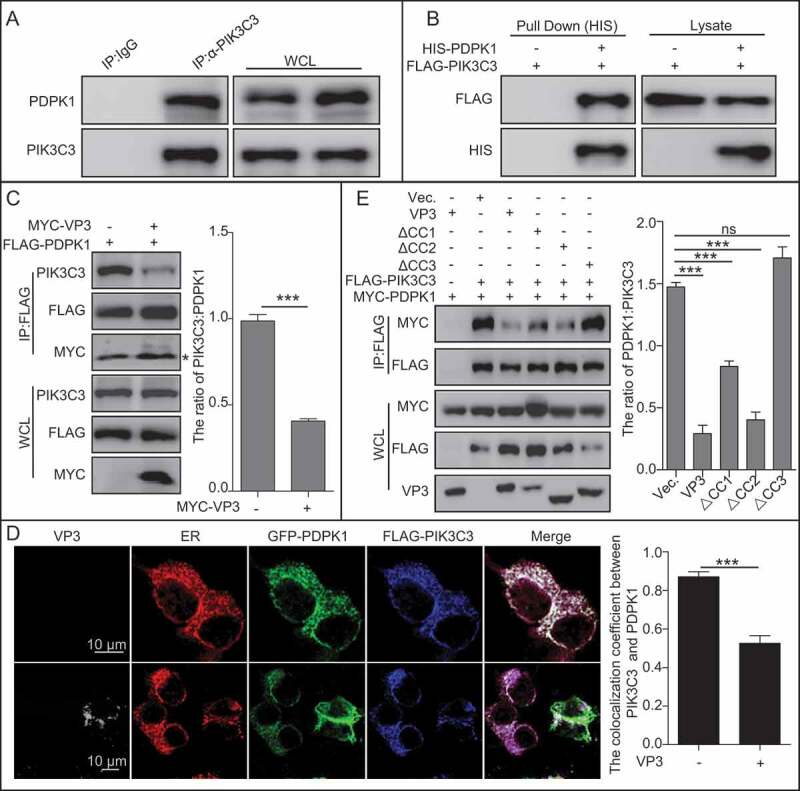
VP3 inhibits interaction between PIK3C3 and PDPK1 through CC3 domain. (A) Endogenous PIK3C3 interacts with endogenous PDPK1. Anti-PIK3C3 IP and immunoblotting analyses using anti-PIK3C3 or anti-PDPK1 antibodies were performed with whole 293T cell lysate. (B) Affinity-isolation analysis of interaction between PIK3C3 and PDPK1. HIS-PDPK1 expressed in prokaryotic cells was purified and then incubated with FLAG-PIK3C3 extracted from its expression 293T cells. In vitro affinity isolation and immunoblotting analyses using anti-FLAG or anti-HIS antibodies were performed. (C) The interaction of PIK3C3 with PDPK1 was inhibited by VP3. HEK293T cells were transiently co-transfected with FLAG-PDPK1 and MYC-VP3 for 48 h. Cellular lysates was subjected to FLAG-precipitation and immunoblotting analysis of PIK3C3, PDPK1, and VP3. * indicated light chain of antibody. (D) Colocalization analysis of VP3 with PDPK1, and PIK3C3 and the ER, in HEK293T cells. HEK293T cells were transiently co-transfected with four vectors DsRed-ER, GFP-PDPK1, FLAG-PIK3C3, and pCI-neo-VP3 (VP3) for 24 h. Cells were immunostained with anti-FLAG and anti-VP3 monoclonal antibodies and were observed using confocal microscopy. Scale bar: 10 μm. (B) Quantitative analysis of the numbers of puncta. (E) The CC3 domain of VP3 is critical for blocking PIK3C3 interaction with PDPK1. HEK293T cells were transiently co-transfected with FLAG-PIK3C3, MYC-PDPK1, pVP3, pVP3△CC1, pVP3△CC2 and pVP3△CC3 for 48 h. Cellular lysates were subjected to FLAG-precipitation and immunoblotting analysis of PIK3C3, PDPK1, VP3, VP3△CC1, VP3△CC2, and VP3△CC3. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001 compared to control.
PDPK1 contributed to VP3-induced autophagic inhibition by activating the AKT-MTOR pathway
To investigate the influence of PDPK1 on the MTOR (mammalian target or rapamycin) pathway under the condition of VP3 expression, we next checked the activity of the MTOR pathway. Super-resolution imaging assay showed that cytoplasmic PDPK1 colocalized with the ER but not with AKT close to the cell membrane in mock-transfected cells. By contrast, VP3 expression resulted in translocation of PDPK1 from the ER to the membrane, the attachment of PDPK1 to AKT close to the cell membrane, and the significant downregulation of the colocalization between PDPK1 and the ER in the cytoplasm, as assessed using Pearson’s correlation coefficient analysis (Figure 6A and S6, p < 0.001). This indicated that VP3 expression promoted translocation of PDPK1 to the cell membrane. IP assays further indicated an increased interaction between AKT and PDPK1 in VP3-expressing cells (Figure 6B). Similarly, at 3 h after IBDV infection, PDPK1 showed an attenuated interaction with PIK3C3 and an enhanced interaction with AKT (Figure 6C,D), indicating that VP3 promoted PDPK1 binding to AKT.
Figure 6.
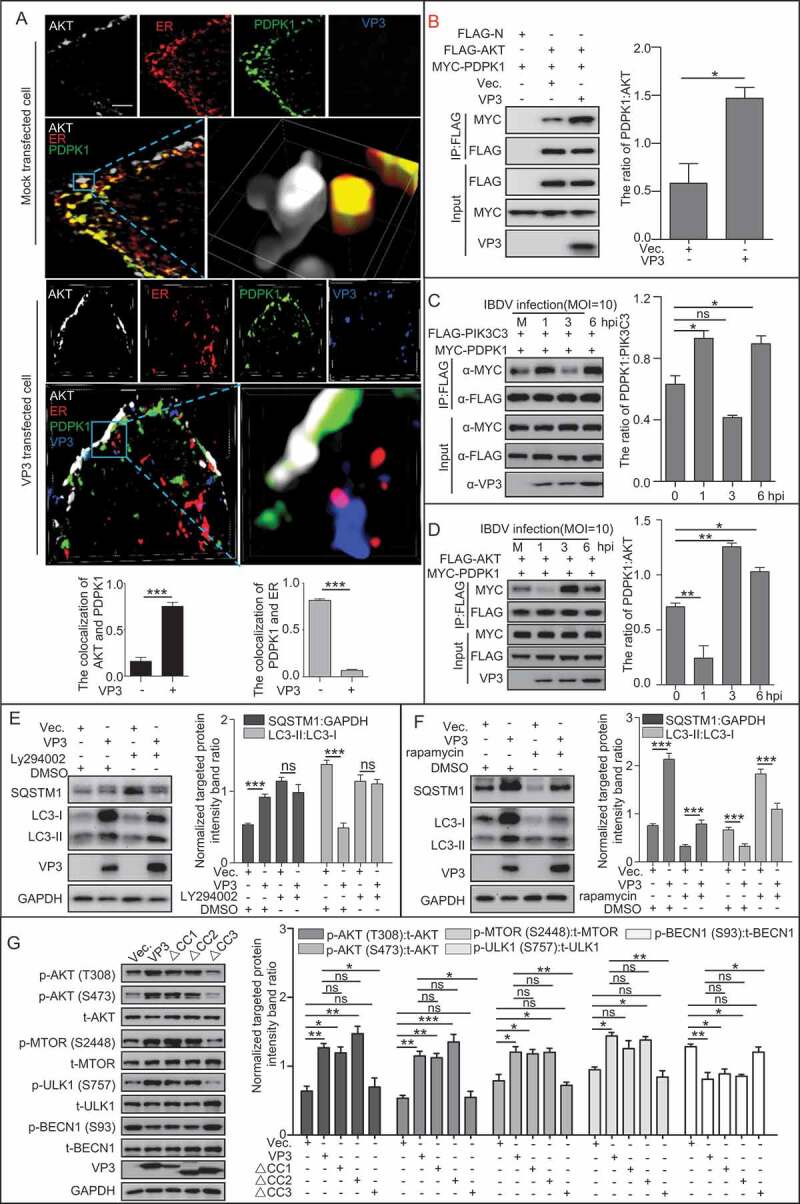
VP3 enhances the interaction between AKT and PDPK1 and the phosphorylation of AKT and MTOR. (A) Three-dimensional image analysis of the colocalization between PDPK1, AKT, the ER, and VP3 using a super resolution microscope. HEK293T cells were transiently co-transfected with four vectors DsRed-ER, GFP-PDPK1, FLAG-AKT, and pCI-neo-VP3 or empty vector pCI-neo for 24 h. The cells were then immunostained with anti-FLAG and anti-VP3 mAbs and observed under a structured illumination microscope. Scale bar: 1 μm. (B) VP3 expression promoted interaction between PDPK1 and AKT. HEK293T cells were transiently co-transfected with vectors expressing FLAG-AKT, MYC-PDPK1, and pCI-neo-VP3 or empty vector pCI-neo for 48 h. The cell lysates were subjected to anti-FLAG IP and immunoblotting analysis of AKT, PDPK1, and VP3 using the indicated antibodies. (C) PIK3C3 binding to PDPK1 was decreased at 3 h post-infection. HEK293T cells were co-transfected with FLAG-PIK3C3 and MYC-PDPK1 for 24 h. The resultant cells were then infected with Avibirnavirus at MOI = 10 for 0, 1, 3, and 6 h and subjected to IP and immunoblotting assays with the indicated antibodies. (D) PDPK1 binding to AKT was enhanced at 3 h post-infection. The HEK293T cells were co-transfected with FLAG-AKT and MYC-PDPK1 for 24 h. The resultant cells were then infected with Avibirnavirus at MOI = 10 for 0, 1, 3, and 6 h and subjected to IP and immunoblotting assays using the indicated antibodies. (E and F) HEK293T cells were transiently transfected with VP3 and vector. After 20 h, the cells were separately treated with DMSO, LY294002 (10 μg/ml), or rapamycin (5 μM) for 4 h. Immunoassays of extracts of HEK293T cells were then performed using the indicated antibodies. (G) CC3 domain is required for VP3 mediating activation of AKT-MTOR pathway. HEK293T cells were transfected with VP3, VP3∆CC1, VP3∆CC2, VP3∆CC3, and vector, respectively. At 20 h post-transfection, HEK293T cells were starved for 4 h. The cellular extracts were then subjected to immunoblotting assay using the indicated antibodies. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.001 compared to control.
Considering that MTOR regulates autophagy negatively, we investigated the contribution of VP3 to the MTOR pathway by testing the SQSTM1 level and the conversion of LC3-I to LC3-II in VP3 transfected cells in the absence or presence of a PtdIns3K inhibitor (Ly294002) or MTOR inhibitor (rapamycin). As shown in Figure 6E, Ly294002 treatment recovered the conversion of LC3-I to LC3-II and the level of SQSTM1 remarkably in VP3-transfected cells (p < 0.001), indicating that VP3 inhibited autophagy through the class III PtdIns3K-MTOR pathway. As shown in Figure 6F, in comparison to DMSO-treated and empty vector-transfected cells, rapamycin treatment increased the conversion of LC3-I to LC3-II in empty vector- or VP3-transfected cells (p < 0.001), but VP3 expression indeed blunted the conversion of LC3-I to LC3-II induced by rapamycin treatment, confirming that VP3 inhibited autophagy through MTOR. Subsequently, we further transfected 293T cells with vectors expressing VP3 or its various ∆CC mutants, separately. Phosphorylation detection showed the significantly increase of phosphorylated AKT at T308 and S473 sites, MTOR at S2448 site, ULK1 at S757 site, and remarkably decrease of phosphorylated BECN1 at S93 site in 293T cells expressing wild-type VP3 or its mutant lacking CC1 or CC2, but not CC3 domain, suggesting that the CC3 domain of VP3 plays a critical role in activating AKT-MTOR pathway (Figure 6G). Collectively, these data demonstrate that PDPK1 binding to AKT contribute to activation of the AKT-MTOR pathway in VP3-induced autophagic inhibition and that the CC3 domain of VP3 is the target to induce autophagy inhibition.
CC3 domain is critical for VP3 mediating virus replication through autophagy
Since CC3 domain is required for VP3-mediated inhibition of autophagy, and autophagy plays critical role in regulating virus replication, we next tested whether CC3 domain of VP3 impacts on replication of virus through autophagy. Firstly, we tested the role of autophagy on replication of Avibirnavirus. As shown in Figure 7A, titers of Avibirnavirus increased greatly in PIK3C3 KO DF-1 cell lines and decreased significantly upon starvation treatment in WT DF-1 cell lines, suggesting that autophagy inhibits replication of Avibirnavirus. Next, we examined the role of VP3 or its mutants on replication of Avibirnavirus. Because mutated Avibirnavirus with expression of VP3 lacking CC1, CC2 or CC3 was unobtainable, detection of mutated virus titers is consequently unavailable. We alternatively calculated titers of Avibirnavirus collected from wild-type (WT) or PIK3C3 KO DF-1 cell lines stably expressing VP3, VP3∆CC1, VP3∆CC2 or VP3∆CC3 respectively. As shown in Figure 7A, titers of Avibirnavirus increased greatly in VP3, VP3∆CC1 and VP3∆CC2, but not in VP3∆CC3 expressing WT DF-1 cell lines compared with that in empty vector-transfected DF-1 cell lines. By contrast, titers of Avibirnavirus collected in VP3 or its mutants expressing PIK3C3 KO DF-1 cell lines had no differences from that in empty vector-transfected PIK3C3 KO cell lines, indicating that PIK3C3 KO counteracted the effects of VP3 and its mutants on replication of Avibirnavirus. The above results suggest that CC3 domain contributes to effect of VP3 on regulating replication of Avibirnavirus through autophagy.
Figure 7.
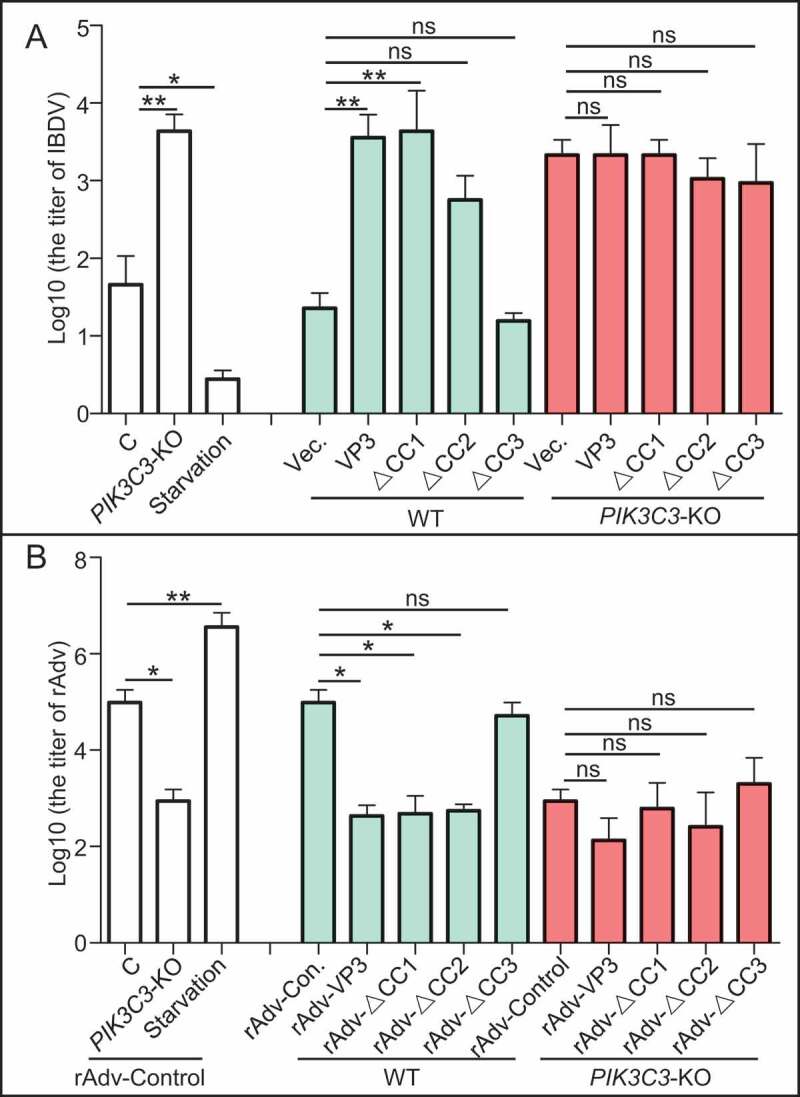
Effects of different coiled-coil domains of VP3 on replication of virus. (A) CC3 domain is critical for VP3 promoting replication of Avibirnavirus. WT or PIK3C3 KO DF-1 cell lines stably expressing VP3, VP3∆CC1, VP3∆CC2, VP3∆CC3 or not were infected Avibirnavirus at MOI = 0.01 for 48 h. The virus was collected for titer detection. (B) CC3 domain is critical for VP3 inhibiting replication of rAdV. The rAdV expressing VP3, VP3∆CC1, VP3∆CC2, VP3∆CC3 or not were rescued from 293T cells. WT or PIK3C3 KO 293T cell lines were infected with these rescued rAdVs. Titers of progeny virus were then tested in 293T cells. Error bars: Mean ± SD of 3 independent tests. One-way ANOVA; *p < 0.05; ** p < 0.01; ***p < 0.001 compared to control.
Similarly, considering CC3 domain is necessary for inhibition of autophagy by infection of rAdV containing VP3, we want to examine whether CC3 domain affected role of VP3 produced by virus during infection on replication of rAdV through autophagy. To begin with, we tested the role of autophagy on replication of control rAdV (without expression of any Avibirnavirus protein). As shown in Figure 7B, titer of control rAdV decreased greatly in PIK3C3 KO 293T cell lines and increased significantly upon starvation treatment in WT 293T cell lines, suggesting that autophagy promoted replication of control rAdV. Next, we examined titers of rAdVs containing VP3 or various VP3 mutants and respectively collected from WT or PIK3C3 KO 293T cell lines. In comparison to the titer of control rAdV, titers of rAdvs containing VP3 or its mutants lacking CC1 or CC2, but not lacking CC3, decreased greatly (Figure 7B). However, in PIK3C3 KO 293T cell lines, there was no significant difference between titer of control rAdV and rAdV containing VP3 or any VP3 mutants, suggesting that PIK3C3 KO counteracted the effects of VP3 or its mutants on replication of rAdV (Figure 7B). The above results confirm that CC3 domain is critical for effect of virus producing VP3 on regulating replication of virus through autophagy.
Discussion
Different pathogens inhibit autophagy through different strategies. In our study, we showed that Avibirnavirus inhibited autophagy by its protein VP3 targeting the PIK3C3-PDPK1-MTOR pathway (Figure 8). Briefly, the CC1 domain of VP3 induced dissociation of BECN1-PIK3C3 complex by binding competitively to BECN1 which resulted in decreased formation of PtdIns3P and autophagosomes. By contrast, the CC3 domain of VP3 promoted the release of PDPK1 from the PDPK1-PIK3C3 complex by competitively attaching to PIK3C3. The released PDPK1 then bind to AKT and activated AKT-MTOR pathway for subsequently inhibiting both autophagosome formation and maturation.
Figure 8.
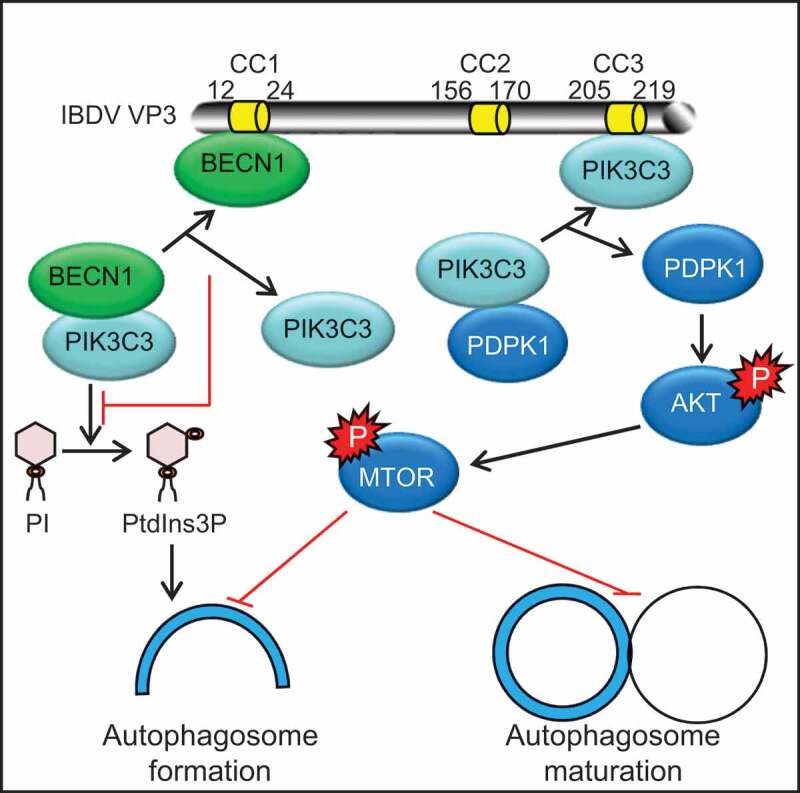
Effects of different coiled-coil domains of VP3 on autophagosome formation and maturation. The CC1 domain of VP3 interacts competitively with BECN1 and destroys BECN1 interaction with PIK3C3. Dissociation of BECN1-PIK3C3 complex in turn inhibits the formation of PI3P and autophagosomes. Meanwhile, the CC3 domain of VP3 interacts competitively with PIK3C3. Released PDPK1 from the PDPK1-PIK3C3 complex then binds to AKT and phosphorylates AKT and MTOR. Activation of the AKT-MTOR pathway subsequently inhibits both autophagosome formation and maturation.
BECN1 plays a critical role in regulating autophagy and is emerging as a limiting host factor for some viruses. However, Chu et al. showed that knockdown of BECN1 has no effect on the parkinsonian neurotoxin MPP+(1-methyl-4-phenylpyridinium)-induced autophagy, but could be blocked by knockdown of Atg5, Atg7, and Atg8. Moreover, gossypol induces BECN1-independent autophagy through ATG5 . In our study, although the CC1 domain was required for VP3 inhibiting the interaction between PIK3C3 and BECN1 (Figure 2B,C), as well as the production of PtdIns3P (Figure S1A), the deletion of CC1 domain still restored the ability of VP3 to inhibit autophagy (Figure 3C), suggesting that the BECN1 pathway was not only the target of Avibirnavirus to regulate autophagy.
After colonizing cells, pathogens need to evade intracellular host defense responses, such as autophagy, an effective way to remove pathogens from cells. Meanwhile, pathogens have evolved clever ways to escape from this defense pathway. PIK3C3-BECN1 and MTOR are two classical pathways to initiate autophagy. Especially, as a common virus target, BECN1-dependent autophagy is often inhibited by viruses, i.e. herpes simplex virus-1 encoded ICP34.5, HCMV (human cytomegalovirus) encoded TRS1 and IRS1, HIV-1 (human immunodeficiency virus)-encoded NEF, murine γ-herpes virus 68 encoded vBCL2 and Kaposi sarcoma-associated herpesvirus inhibit autophagy by binding to BECN1 [30,31]. However, BECN1-independent autophagy would be activated by inhibiting MTOR signaling and result in cell death in response to influenza A virus H5N1 infection [32]. Therefore, host cells may initiate autophagy through an alternative signal pathway. Although VP3 impaired autophagosome formation through inhibiting PIK3C3-BECN1 complex, it inhibited both formation and maturation of autophagosome through disruption of PIK3C3-PDPK1, which regulated autophagy via MTOR pathway, suggesting that MTOR might be a more effective way in controlling autophagy in response to virus infection. Moreover, considering the role of MTOR in inhibiting cell apoptosis, we inferred that MTOR pathway might be an alternative pathway utilized by VP3 to inhibit apoptosis, although Avibirnavirus VP3 was already reported to inhibit cell apoptosis through the protein kinase R pathway [33]. Therefore, we concluded that autophagy signaling initiating from PIK3C3-PDPK1-MTOR may play an important role in innate immunity and that PIK3C3 was a limiting host factor for Avibirnavirus replication.
Many CC domains act as molecular spacers in the scaffolding of large macromolecular complexes. Omp-α is an outer membrane protein from Thermotoga maritime that is predicted to contain a CC domain and observed to span the periplasmic space . Spc110 joins the inner and central plaques of the spindle pole body through its CC domain . Deletion of the CC domain in Spc110 leads to changes to the spindle pole body structure. Viral protein VP3 localizes at the inner surface of the Avibirnavirus particle and links the viral genome and the VP1 protein within the virus particle. VP3 also provides a scaffold for Avibirnavirus capsid assembly. In the present study, three CC domains of VP3 were identified. Therefore, we hypothesized that the CC domains within VP3 might play a vital role in virus particle assembly, except when it interacts with BECN1 and PIK3C3 during virus replication to inhibit the formation and maturation of autophagosomes.
Rodriguez-Rocha et.al suggested that autophagy promotes adenoviruses replication. Consistently, according to our data, autophagy promoted replication of rAdV. Interestingly, recombinant rAdV expressing VP3 or its mutants lacking CC1 or CC2, but not CC3, inhibited replication of virus itself, confirming that the role of CC domain on autophagy indeed affected the replication of virus and rAdV should be a very powerful tool that could be used to assess the role of certain exogenous protein on replication of virus.
Collectively, we inferred a model of PDPK1-mediated autophagy during virus infection. That is, under physiological conditions, PDPK1 stayed in the ER pool by interacting with PIK3C3, and PDPK1-mediated AKT-MTOR pathway activation did not occur. When cells were stimulated by the virus, PDPK1 was released from the ER and translocated to the cell membrane, leading to AKT phosphorylation and activation of the AKT-MTOR pathway, ultimately inhibiting autophagy.
Materials and methods
Cells, antibodies, reagents, and virus
HEK293T (CRL-11268) and DF-1 (CRL-12203) cells from the ATCC (American Type Culture Collection) were cultured in Dulbecco’s modified Eagle medium (Life Technologies, 12100–038) supplemented with 10% fetal bovine serum (Sigma Life Technologies, F2442). Earle’s Balanced Salt Solution (14155063) was purchased from Thermo Fisher. Anti-PDPK1 (3062), anti-phospho-PDPK1 (Ser241; 3061), anti-LC3B (2775), anti-phospho-AKT (Ser473; 4060), anti-phospho-AKT (Thr308; 4056), anti-phospho-BECN1 (Ser93; 14717), anti-phospho-ULK1 (Ser757; 14202), anti-ULK1 (8054), anti-PtdIns3K/PI3 kinase class III (3811) for WB and anti-PtdIns3K/PI3 kinase class III (4263) for IP mAbs (monoclonal antibodies) were purchased from Cell Signaling Technology. Anti-AKT1 (ab32505), anti-phospho-MTOR (Ser2448; ab109268), anti-MTOR (ab83495), anti-SQSTM1 (ab109012), anti-BECN1 (ab62557) mAbs were purchased from Abcam. Anti-FLAG (F1804, F7425) and anti-MYC (M5546) mAbs, and rapamycin (R8781) and dimethyl sulfoxide (DMSO, 472301) were purchased from Sigma-Aldrich. Anti-MYC antibody (R1208-1) for western blotting was purchased from Hangzhou HuaAn Biotechnology. Anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (AB-P-R001) was purchased from GoodHere Technology. Ni-NTA agarose (30210) was purchased from QIAGEN. Chicken antiserum against IBDV viral protein VP3 (for IFA) and murine mAbs against IBDV viral proteins VP1 (1D4 for WB), VP2 (4C12 for WB), VP3 (2D6 for WB and IFA), VP4 (6H8 for WB), and VP5 (5C1 for WB) were produced by our lab. DAPI (2-[4-amidinophenyl]-1H-indole-6-carboxamidine; 10236276001) was purchased from Roche Life Science. Fluorescein isothiocyanate-labeled goat anti-chicken IgG antibodies (02-24-06), peroxidase-labeled goat anti-mouse IgG antibodies (074–1806), and peroxidase-labeled goat anti-rabbit IgG antibodies (074–1506) were purchased from Kirkegaard & Perry Laboratories Inc. Dylight 405 goat anti-mouse IgG antibodies (A23110) were obtained from Abbkine Scintific Co., Ltd. Alexa Fluor® 488 donkey anti-rabbit IgG antibodies (A21206), Alexa Fluor 546 donkey anti-rabbit IgG antibodies (A21085), Alexa Fluor® A647 goat anti-mouse IgG antibody (A21236), and LysoTracker Red DND-99 (L7528) were purchased from Invitrogen. Ly294002 (S1737) and NP-40 Lysis Buffer (P0013F) were purchased from Beyotime Biotechnology. Protein A/G PLUS Agarose (sc-2003) and anti-GFP mAb (sc9996) for WB were purchased from Santa Cruz Biotechnology. Human PIK3R4 SiRNA (SR309370) was purchased from ORIGENE. ExFect transfection Reagent (T101-01/02), TRIzol reagent (R401-01), reverse transcription kit (R211-01) and AceQ qPCR SYBR Green Master Mix (Q111-02) were purchased from Vazyme Biotech Co., Ltd. Phenylmethylsulfonyl fluoride (P8340) was purchased from Solarbio® Life Sciences. Puromycin (ant-pr) was purchased from Invivogen. IBDV strain NB was generated in DF-1 cell lines.
Plasmid construction
LC3, cloned from the DF-1 cells genome (NM_001031461), was ligated into pEGFP-C3 (Clontech, 6082–1) and named eGFP-LC3. The eGFP-LC3 gene was then ligated into pCI-neo (Promega, E1841) and named PCI-eGFP-LC3. Subsequently, VP1, VP2, VP3, VP4, and VP5, cloned from IBDV strain NB genome, were constructed into PCI-eGFP-LC3. The resultant plasmids were VP1, VP2, VP3, VP4, and VP5. VP3 truncated variants were amplified from PCI-eGFP-LC3-VP3 (pVP3) using specific primers (VP3COIL 1S: GAGACCCCCGAACTCGAGGACCCACTATTCCAATC; VP3COIL 1R: GATTGGAATAGTGGGTCC TCGAGTTCGGGGGTCTC; VP3COIL 2S: CTATCTAGACTACGTGCATGCAAAGGGCAGCTACGTC GATC; VP3COIL 2R: GATCGACGTAGCTGCCCTTTGCATGCACGTAGTCTAGATAG; VP3COIL 3S: CATGGACGTGGCCCAAACCAAATGAAGCATCGCAATC; VP3COIL 3R: GATTGCGATGCTTCATT TGGTTTGGGCCACGTCCATG) and then cloned into vector PCI-eGFP-LC3. The resultant plasmids were VP3 ∆CC1, VP3 ∆CC2, and VP3 ∆CC3.
Cell transfection and treatment
For cell transfection, 293T or DF-1 cells were seeded on the designated plates or glass coverslips at a suitable density according to the experimental scheme. When cells were 70–80% confluent, they were transfected with ExFect™ Transfection Reagent and finally immunoblotting was used to detect the protein levels.
For the treatment experiment, after transfection with pVP3 and vector for 24 h, the cells were treated with DMSO, Ly294002 (10 μg/ml), or rapamycin (5 μM) for 4 h post-transfection. The cells were then collected for subsequent detection.
PIK3C3 cell line construction
Generation of PIK3C3-knockout 293T and DF-1 cell lines. The human PIK3C3 gene target sequence 5ʹ CTACATCTATAGTTGTGACC 3ʹ and the chicken PIK3C3 gene target sequence 5ʹ TTATGTGTATAGCT GCGACC 3ʹ were separately inserted into the guide RNA expression plasmid PX 459 (Addgene, 62988; deposited by Feng Zhang lab, Broad Institute of Massachusetts Institute of Technology), a vector or also expressing Cas9 enzyme. The recombinant vector constructs were separately transfected into HEK293T cells and DF-1 cells for 48 h and then selected under puromycin (4 μg/ml for HEK293T cells and 2 μg/ml for DF-1 cells) for another 72 h. The cells were then diluted to 50 cells/100 ml and inoculated into 96-well plates for colony formation. Each colony was separately transferred into 24-well plates. Knockout of PIK3C3 was confirmed by western blot.
Construction of 293T or DF-1 cell lines with stable expression of VP3, VP3∆CC1, VP3∆CC2 and VP3∆CC3
The VP3 and its truncated fragment VP3∆CC1, VP3∆CC2, VP3∆CC3 were cloned into overexpression lentivirus vector PCDH-CMV-MCS-EF1 (System Biosciences, CD510B-1). The plasmids were transiently transfected into DF-1 cells to make sure that the expressions were successful by western blot. Then the plasmids were separately co-transfected with the ViraPower™ Lentiviral Packaging Mix (Invitrogen, K497500) respectively into 293T cells to produce a lentiviral stock according to the manufacturer’s guide, empty vector as control. After 72 h transfection, viral particles were harvested from the medium by ultracentrifugation. After lentiviral preparation, WT or PIK3C3 KO cells were seeded into the 6-wells plate and grown overnight to 80% confluence. On the second day, WT or PIK3C3 KO cells were separately infected with 2 ml PCDH-CMV-VP3-EF1, PCDH-CMV- VP3∆CC1-EF1, PCDH-CMV- VP3∆CC2-EF1, PCDH-CMV-VP3∆CC3-EF1 or PCDH-CMV-MCS-EF1 virus for 6 h. After transduction, cells were cultured for another 24 h with complete medium, then screened positive cells by 2 μg/ml puromycin. Expression of VP3 and its mutated proteins were detected in these cell lines were detected by WB.
Recombinant adenovirus production
The recombinant adenovirus was generated following the protocol (Adenovirus User Manual-Genemedi). Briefly, VP3, VP3∆CC1, VP3∆CC2, and VP3∆CC3 were respectively cloned into pDC316- mCMV- EGFP (YRgene, VXY0585). The constructs were seperately co-transfected into 293 cell lines with pBHGlox (delta) E1, 3Cre (Biovector NTCC, Inc, BioVector 912752). Virus was collected by freeze-thaw cycle for 3 times and then infected fresh 293 cells. After 5 rounds expansion in fresh 293 cells, titration of purified virus was analyzed by MOI (moltiplicity of infection) test.
Western blotting
Cells were harvested and lysed immediately in lysis buffer (2% sodium dodecyl sulfate [SDS], 1% Triton X-100 [Sigma-Aldrich, T9284], 50 mM Tris-HCl, 150 mM NaCl, pH 7.5). The lysate protein concentration was measured using a bicinchoninic acid (BCA) assay (Pierce, 23227). Equal amounts of total protein from different samples were separated on SDS-polyacrylamide gel electrophoresis and the protein bands were transferred onto nitrocellulose blotting membrane (GE Healthcare Life Science, 10600001). After blocking with 5% nonfat dry milk containing 0.1% Tween 20 (Amresco, 077) for 30 min at 37°C, the membranes were incubated with primary antibodies for 2 h at 37°C, followed horseradish peroxidase-conjugated anti-mouse/rabbit IgG (Kirkegaard & Perry Laboratories, Inc., 074–1506), and visualized using a SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, 34094).
Immunofluorescence staining
The indicated 293T cells were plated on 35 mm glass bottom cell culture dishes at a density of 6 × 104 cells/ml. Within 24 h, the cells were transfected with the indicated plasmids, treated with the indicated drugs, or cultured in starvation medium. After 24 h, the cells were fixed with 4% paraformaldehyde in PBS buffer (Sangon Biotech, E607008) for 10 min at room temperature, and then blocked in 5% nonfat milk for 1 h. After washing three times with PBST, the cells were incubated with the indicated antibodies. The cells were washed with PBS and then incubated with FITC, Dylight 405, Alexa Fluor 488, Alexa Fluor 546, or Alexa Fluor 647- labeled IgG antibodies for 1 h. After washing three times with PBST, the cells were observed under a Nikon A1R/A1 laser scanning confocal microscope (Nikon, Tokyo, JPN) or under an N-structured illumination microscopy Super-Resolution microscope (Nikon, Tokyo, JPN). Only cells with at least five GFP dots or ring like structures were scored as positive.
Co-immunoprecipitation assay
Cell lines were separately co-transfected with various vectors. Cells were lysed using NP40 lysis buffer with phenylmethylsulfonyl fluoride (1 mM). Lysates from cells were incubated with anti-FLAG antibody and Protein A/G beads for 4 h at 4°C. After centrifugation, the supernatant was removed, and the pellets was suspended in washing buffer. Final pellets were lysed in lysis buffer for WB analysis.
Affinity-isolation assay
Various proteins were expressed separately in 293T cells. Lysates from cell lines transfected with empty vector or vector expressing the indicated genes were mixed separately with each other overnight at 4°C. The mixtures were incubated with the indicated antibodies and Protein A/G beads for 4 h at 4°C. After centrifugation, the supernatant was removed, and the pellets were suspended in washing buffer. Final pellets were lysed in lysis buffer for WB analysis.
RNA extraction and quantitative real-time PCR
Total RNAs were isolated following the instructions of the TRIzol reagent and the RNAs were reverse transcribed into cDNA using the reverse transcription kit. Quantitative real-time PCR was performed on a Roche LC96 real-time PCR system using AceQ qPCR SYBR Green Master Mix with specific primers. The primers were LC3 S: 5ʹ CTTCGCCGACCGCTGTAA 3ʹ, LC3 R: 5ʹ AAGCCGTCCTCGTCTTTCT 3ʹ, SQSTM1 S: 5ʹ GCAATGGGCCTGTGGTAG 3ʹ, SQSTM1 R: 5ʹ CCCGAAGTGTCCGTGTTT 3ʹ, GAPDH S: 5ʹ AACGGATTTGGTCGTATTG 3ʹ and GAPDH R: 5ʹ GGAAGATGGTGATGGGATT 3ʹ. GAPDH was used as an internal control for normalization. The results were calculated using the 2−∆∆Ct method.
Statistical analysis
For quantitative analysis of GFP-LC3 puncta formation, only cells with at least 5 GFP dots or ring like structures were scored as positive. Red-positive ring-like structures were calculated from 20 cells. The statistical significance of differences between groups was determined using the Student t-test. p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We gratefully acknowledge Ms. Yunqin Li for technical assistance on laser confocal microscopy.
Funding Statement
This study is supported by grants from National Science Fundation of China [Grant No.31630077], National Key technology R & D Program of China [Grant No.2015BAD12B01] and China Agriculture Research System [Grant No. CARS-40-K13].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary data
Supplemental data for this article can be accessed here.
References
- [1].Mehrpour M, Esclatine A, Beau I, et al. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748. [DOI] [PubMed] [Google Scholar]
- [2].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].McKnight NC, Zhenyu Y.. Beclin 1, an essential component and master regulator of PI3K-III in health and disease. Curr Pathobiol Rep. 2013;1:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Decuypere JP, Parys JB, Bultynck G.. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells. 2012;1:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sun T, Li X, Zhang P, et al. Acetylation of Beclin 1 inhibits autophagosome maturation and promotes tumour growth. Nat Commun. 2015;6:7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23. [DOI] [PubMed] [Google Scholar]
- [7].Sinha S, Colbert CL, Becker N, et al. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4:989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1. [DOI] [PubMed] [Google Scholar]
- [9].Cavignac Y, Esclatine A. Herpesviruses and autophagy: catch me if you can! Viruses. 2010;2:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Obara K, Ohsumi Y. Atg14: a key player in orchestrating autophagy. Int J Cell Biol. 2011;2011:713435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang L, Wang H, Liu L, et al. The role of Insulin/IGF-1/PI3K/Akt/GSK3beta signaling in Parkinson’s disease dementia. Front Neurosci. 2018;12:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fruman DA, Chiu H, Hopkins BD, et al. The PI3K pathway in human disease. Cell. 2017;170:605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Alessi DR, Deak M, Casamayor A, et al. 3-phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol. 1997;7:776. [DOI] [PubMed] [Google Scholar]
- [16].Luque D, Saugar I, Rodriguez JF, et al. Infectious bursal disease virus capsid assembly and maturation by structural rearrangements of a transient molecular switch. J Virol. 2007;81:6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zheng XJ, Hong LL, Shi LX, et al. Proteomics analysis of host cells infected with infectious bursal disease virus. Mol Cell Proteomics. 2008;7:612. [DOI] [PubMed] [Google Scholar]
- [18].Wu YP, Peng CE, Xu L, et al. Proteome dynamics in primary target organ of infectious bursal disease virus. Proteomics. 2012;12:1844. [DOI] [PubMed] [Google Scholar]
- [19].Wu YP, Hong LL, Ye JX, et al. The VP5 protein of infectious bursal disease virus promotes virion release from infected cells and is not involved in cell death. Arch Virol. 2009;154:1873. [DOI] [PubMed] [Google Scholar]
- [20].Wang YZ, Wu XP, Li HB, et al. Antibody to VP4 protein is an indicator discriminating pathogenic and nonpathogenic IBDV infection. Mol Immunol. 2009;46:1964. [DOI] [PubMed] [Google Scholar]
- [21].Wang SY, Hu BL, Si WY, et al. Avibirnavirus VP4 protein is a phosphoprotein and partially contributes to the cleavage of intermediate precursor VP4-VP3 polyprotein. PloS One. 2015;10:e0128828. doi: 10.1371/journal.pone.0128828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ye C, Jia L, Sun Y, et al. Inhibition of antiviral innate immunity by birnavirus VP3 protein via blockage of viral double-stranded RNA binding to the host cytoplasmic RNA detector MDA5. J Virol. 2014;88:11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kaufer I, Weiss E. Electron-microscope studies on the pathogenesis of infectious bursal disease after intrabursal application of the causal virus. Avian Dis. 1976;20:483. [PubMed] [Google Scholar]
- [24].Hu B, Zhang Y, Jia L, et al. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy. 2015;11:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liang XH, Kleeman LK, Jiang HH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li F, Zhang C, Li Y, et al. Beclin1 restricts RNA virus infection in plants through suppression and degradation of the viral polymerase. Nat Commun. 2018;9:1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chu CT, Zhu JH, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress. Autophagy. 2007;3:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gao P, Bauvy C, Souquere S, et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J Biol Chem. 2010;285:25570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lian J, Karnak D, Xu L. The Bcl-2-Beclin 1 interaction in (-)-gossypol-induced autophagy versus apoptosis in prostate cancer cells. Autophagy. 2010;6:1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chaumorcel M, Lussignol M, Mouna L, et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J Virol. 2012;86:2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mouna L, Hernandez E, Bonte D, et al. Analysis of the role of autophagy inhibition by two complementary human cytomegalovirus BECN1/Beclin 1-binding proteins. Autophagy. 2016;12:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ma JH, Sun Q, Mi RF, et al. Avian influenza A virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J Genet Genome. 2011;38:533. [DOI] [PubMed] [Google Scholar]
- [33].Busnadiego I, Maestre AM, Rodriguez D, et al. The infectious bursal disease virus RNA-binding VP3 polypeptide inhibits PKR-mediated apoptosis. PloS One. 2012;7:e46768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Truebestein L, Leonard TA. Coiled-coils: the long and short of it. BioEssays. 2016;38:903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Engel AM, Cejka Z, Lupas A, et al. Isolation and cloning of Omp alpha, a coiled-coil protein spanning the periplasmic space of the ancestral eubacterium Thermotoga maritima. Embo J. 1992;11:4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lupas A, Muller S, Goldie K, et al. Model structure of the Omp alpha rod, a parallel four-stranded coiled coil from the hyperthermophilic eubacterium Thermotoga maritima. J Mol Biol. 1995;248:180. [DOI] [PubMed] [Google Scholar]
- [37].Kilmartin JV, Dyos SL, Kershaw D, et al. A spacer protein in the Saccharomyces cerevisiae spindle poly body whose transcript is cell cycle-regulated. J Cell Biol. 1993;123:1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bottcher B, Kiselev NA, Stel’Mashchuk VY, et al. Three-dimensional structure of infectious bursal disease virus determined by electron cryomicroscopy. J Virol. 1997;71:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maraver A, Ona A, Abaitua F, et al. The oligomerization domain of VP3, the scaffolding protein of infectious bursal disease virus, plays a critical role in capsid assembly. J Virol. 2003;77:6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rodriguez-Rocha H, Gomez-Gutierrez JG, Garcia-Garcia A, et al. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology. 2011;416:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zheng X, Hong L, Li Y, et al. In vitro expression and monoclonal antibody of RNA-dependent RNA polymerase for infectious bursal disease virus. DNA Cell Biol. 2006;25:646. [DOI] [PubMed] [Google Scholar]
- [42].Wang XN, Zhang GP, Zhou JY, et al. Identification of neutralizing epitopes on the VP2 protein of infectious bursal disease virus by phage-displayed heptapeptide library screening and synthetic peptide mapping. Viral Immunol. 2005;18:549. [DOI] [PubMed] [Google Scholar]
- [43].Wang YZ, Wu XP, Li HB, et al. Antibody to VP4 protein is an indicator discriminating pathogenic and nonpathogenic IBDV infection. Mol Immunol. 2009;46:1964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.