

ABSTRACT
Macroautophagy/autophagy plays key roles in development, oncogenesis, and cardiovascular and metabolic diseases. Autophagy-specific class III phosphatidylinositol 3-kinase complex I (PtdIns3K-C1) is essential for autophagosome formation. However, the regulation of this complex formation requires further investigation. Here, we discovered that STYK1 (serine/threonine/tyrosine kinase 1), a member of the receptor tyrosine kinases (RTKs) family, is a new upstream regulator of autophagy. We discovered that STYK1 facilitated autophagosome formation in human cells and zebrafish, which was characterized by elevated LC3-II and lowered SQSTM1/p62 levels and increased puncta formation by several marker proteins, such as ATG14, WIPI1, and ZFYVE1. Moreover, we observed that STYK1 directly binds to the PtdIns3K-C1 complex as a homodimer. The binding with this complex was promoted by Tyr191 phosphorylation, by means of which the kinase activity of STYK1 was elevated. We also demonstrated that STYK1 elevated the serine phosphorylation of BECN1, thereby decreasing the interaction between BECN1 and BCL2. Furthermore, we found that STYK1 preferentially facilitated the assembly of the PtdIns3K-C1 complex and was required for PtdIns3K-C1 complex kinase activity. Taken together, our findings provide new insights into autophagy induction and reveal evidence of novel crosstalk between the components of RTK signaling and autophagy.
Abbreviations: AICAR: 5-aminoimidazole-4-carboxamide ribonucleotide; AMPK: adenosine 5‘-monophosphate (AMP)-activated protein kinase; ATG: autophagy related; ATP: adenosine triphosphate; BCL2: BCL2 apoptosis regulator; BECN1: beclin 1; Bre A: brefeldin A; Co-IP: co-immunoprecipitation; CRISPR: clustered regularly interspaced short palindromic repeats; DAPI: 4′,6-diamidino-2-phenylindole; EBSS: Earle’s balanced salt solution; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescent protein; GSEA: gene set enrichment analysis; MAP1LC3/LC3, microtubule associated protein 1 light chain 3; MAPK8/JNK1: mitogen-activated protein kinase 8; mRFP: monomeric red fluorescent protein; MTOR: mechanistic target of rapamycin kinase; MTT: 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4: phosphoinositide-3-kinase regulatory subunit 4; qRT-PCR: quantitative reverse transcription PCR; RACK1: receptor for activated C kinase 1; RUBCN: rubicon autophagy regulator; siRNA: small interfering RNA; SQSTM1: sequestosome 1; STYK1/NOK: serine/threonine/tyrosine kinase 1; TCGA: The Cancer Genome Atlas; Ub: ubiquitin; ULK1: unc-51 like autophagy activating kinase 1; UVRAG: UV radiation resistance associated; WIPI1: WD repeat domain, phosphoinositide interacting 1; ZFYVE1: zinc finger FYVE-type containing 1
KEYWORDS: ATG14-BECN1-PIK3C3 complex, autophagy, BCL2, dimerization, STYK1, Tyr191
Introduction
Cellular homeostasis is important for preventing cellular injuries that could lead to impaired cellular function and malignant transformation. Macroautophagy/autophagy, a crucial mechanism for cellular homeostasis, is an evolutionarily conserved intracellular degradation process from yeast to mammals. Cytosolic proteins and organelles are engulfed into autophagosomes and digested in lysosomes by acidic hydrolases [1]. The large capacity of the autophagosomes enables the removal of damaged organelles and protein aggregates, thus maintaining organelle function and protein quality [2]. To date, more than 41 yeast-specific, autophagy-related (ATG) genes have been identified [3]. Their roles in autophagy, especially in the early stages of autophagosome formation, which is considered the most complex stage, have been reported [4]. In particular, autophagy is activated during metabolic stress and is important for the survival of eukaryotic cells under various pathological conditions, such as tumorigenesis and infection [5]. Therefore, therapeutic approaches that target and intervene in autophagy are considered promising for treating pathological cellular functions, such as oncogenesis and cancer therapy resistance, as well as cardiovascular, metabolic, and neurodegenerative disorders [6,7]. Indeed, a January 2019 search of the ClinicalTrials.gov website using the search term “Autophagy” returned hits on 52 studies focused on intervening in autophagy to improve patient outcomes. The key to improving the management of these pathophysiological disorders is the identification and characterization of the potential drug targets among the regulators of autophagy.
Class III phosphatidylinositol 3-kinase (PtdIns3K) is important in both the initiation of autophagosomal particles and their eventual fusion with lysosomes [1]. PtdIns3K forms at least 2 distinct complexes, known as autophagy-specific complex I (PtdIns3K-C1) and II (PtdIns3K-C2). Both complexes contain the catalytic subunit PIK3C3/VPS34, the putative protein kinase PIK3R4/VPS15 and BECN1/Beclin 1. The PtdIns3K-C1 complex contains ATG14, whereas PtdIns3K-C2 contains UVRAG (UV radiation resistance associated) [8]. The PtdIns3K-C1 complex plays a central role in the initiation stage of autophagy, which is regulated by AMPK activation or MTOR inhibition in response to nutrient depletion [9–11]. Briefly, upon binding with BECN1 and ATG14, PIK3C3 generates a phagophore-specific pool of phosphatidylinositol-3-phosphate (PtdIns3P), leading to the nucleation of the phagophore. The PtdIns3K-C1 complex then recruits other autophagy-related proteins and autophagy-specific PtdIns3P effectors, such as the ZFYVE1 (zinc finger FYVE-type containing 1) and WIPI1 (WD repeat domain, phosphoinositide interacting 1) proteins [12] for autophagy initiation. Many other BECN1-PIK3C3 interaction partners and their regulatory functions in autophagy have been characterized in recent years [13,14], and they represent an increasing number of potentially druggable targets. For example, through binding to the BH3 domain of BECN1, BCL2 facilitates BECN1 release from the PtdIns3K-C1 complex and, in turn, inhibits the activity of autophagy [13]. This process is inhibited after the phosphorylation of BCL2 at Thr69 and Ser70/87 by MAPK8/JNK1 (mitogen-activated protein kinase 8) in response to starvation [15]. RUBCN contributes to the efficient inhibition of the lipid kinase activity of the PtdIns3K-C2 complex through direct interaction with PIK3C3 via its RUN domain, which is post-transcriptionally upregulated by supplementation with the saturated fatty acid palmitate [14].
Moreover, DACT1/Dapper1 plays a critical role in PtdIns3K-C1 complex promoted autophagy in the central nervous system [16]. In addition, based on the current understanding of the PtdIns3K-C1 complex structure in yeast [17], more subunits of the complex have been identified, such as NRBF2, which mediates the PtdIns3K-C1 complex dimerization through its central coiled-coil domain, and enhances the lipid kinase activity of PIK3C3 by approximately 10-fold [18,19]. Additionally, PAQR3 (progestin and adipoQ receptor family member 3) was recently identified as an receptor of the PtdIns3K-C1 complex and a promoter of autophagosome formation, which is activated through phosphorylation at Thr32 by AMPK in response to nutrient starvation [20].
STYK1 (serine/threonine/tyrosine kinase 1), also called NOK, is a newly identified oncogenic protein that belongs to the receptor tyrosine kinase (RTK) family. Similar to other RTK members, STYK1 contains a single transmembrane domain and an intracellular tyrosine kinase domain, but an almost completely truncated extracellular N-terminal domain [21]. STYK1 has been reported to promote cervical cancer and the development and metastasis of hepatocellular carcinoma [21,22] and facilitate the genesis and remodeling of blood and lymphatic vessels during tumor progression [23]. A mutation in the Tyr327 or Tyr356 residue of the STYK1 kinase domain to phenylalanine (STYK1Y327F,Y356F) reduces tyrosine phosphorylation levels and dramatically suppresses tumor progression [24]. However, the mechanisms by which STYK1 regulates tumor progression and other biological processes remain largely unclear.
In the present study, by investigating several autophagy markers and events in several tumor cell lines and zebrafish, we identified STYK1 as a novel positive regulator of autophagy. Moreover, we revealed the existence of an interaction between STYK1 and PtdIns3K-C1 complex via confocal microscopy and GST-affinity-isolation assays. Furthermore, our study revealed that the tyrosine kinase activity of STYK1 plays an essential role in autophagy induction. Finally, we identified that Tyr191 is important for STYK1 dimerization and the assembly of the PtdIns3K-C1 complex. Collectively, these results provide new insights into and evidence for the STYK1 as a regulator of autophagy.
Results
STYK1 depletion impairs autophagy flux
The molecular mechanism of STYK1 that promotes cancer oncogenesis and metastasis remain elusive. To better understand the role of STYK1 in cancer development, gene set enrichment analysis (GSEA) was used with The Cancer Genome Atlas (TCGA) database of liver cancer to analyze differences in the genes that are enriched in patients with high and low levels of STYK1. The results indicate that many genes that correlated with metabolism were enriched in the group that expressing high levels of STYK1 (Fig. S1A-E), suggesting a metabolic function of STYK1 in tumor development. Moreover, we noticed that the genes involved in autophagy, from which diverse metabolic fuel sources can be produced [25], were also co-expressed with STYK1 (Figure 1A). Next, we set out to explore the role of STYK1 in autophagy. Thus, we first examined the effect of STYK1 depletion with siRNA knockdown in HepG2 and HeLa tumor cells, which endogenously express high levels of STYK1 (Fig. S2A) on the level of MAP1LC3/LC3 and SQSTM1/p62, the most commonly used autophagic flux markers for autophagosome formation [26]. We found that LC3-II levels were significantly reduced and SQSTM1 levels were increased in STYK1-depleted HepG2 cells (Fig. S2B). We also found that STYK1 knockdown suppressed LC3-II levels in the presence or absence of the autophagy inducer rapamycin and with or without the lysosomal inhibitor chloroquine (CQ), whereas it had the opposite effect on SQSTM1 levels (Figure 1B, S2C-E). Similar results were obtained under both normal and serum starvation conditions, the latter of which is known to induce autophagy [27] (Figure 1C).
Figure 1.
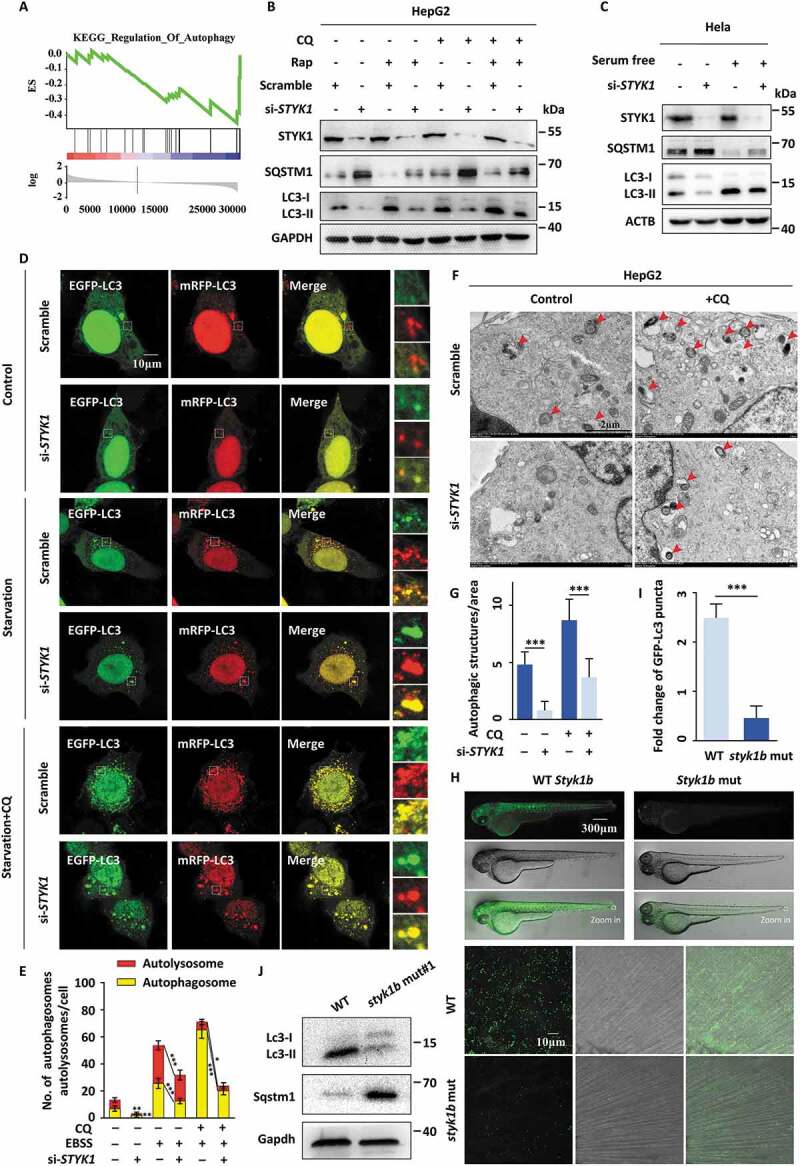
STYK1 depletion impairs autophagy flux. (A) Many genes correlated with autophagy are enriched in liver cancer patients who express high levels of STYK1 expression, as determined by gene set enrichment analysis (GSEA) using the TCGA database. (B) HepG2 cells were transfected with STYK1-specific siRNA for 36 h; the cells were then treated with rapamycin (10 nM) with or without CQ (10 μM) treatment for another 12 h. Then, the cell lysates were sent to western blotting using indicated antibodies. (C) HeLa cells were transfected with STYK1 siRNA (hereafter referred to as siRNA#2) for 36 h. Then, the complete cell culture medium was replaced with serum-free low glucose (1000 mg/L) medium. After another 12 h, the cells were lysed and sent to western blotting using indicated antibodies. (D-E) Autophagic flux was detected with the mRFP-GFP-LC3 reporters. STYK1 siRNA and scramble RNA were separately co-transfected with mRFP-GFP-LC3 under both normal and serum starvation conditions along with or without CQ (10 μM) treatment. Representative confocal images are shown, and the number of cells showing an accumulation of yellow or red puncta was quantified (n = 10). Scale bars: 10 µm. (F-G) Representative electronic micrographs of the autophagosomes or the autolysosomes of the HepG2 cells transfected with STYK1 siRNA with or without CQ (10 μM) treatment. Red arrows indicate autophagic structures. The number of autophagic structures per area was quantified (n = 10). Scale bars: 2 µm. (H-I) styk1b-mutated GFP-Lc3 transgenic zebrafish were generated using CRISPR-Cas9 technology, and representative confocal images of the GFP-Lc3 puncta in the defined region of the tail fin of the zebrafish embryos are shown. The white square indicates the defined region. The fold change of the number of GFP-Lc3 puncta was quantified (n = 10). (J) Western blotting analysis of the whole lysates of normal and styk1b mutant zebrafish embryos with the indicated antibodies. Four dpf of embryos were isolated for protein preparation, (10 > embryos/samples). *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± SD.
Furthermore, the levels of ubiquitinated protein were increased after STYK1 knockdown (Fig. S2F). As GFP-LC3 puncta were also used to assess autophagosome formation, we carried out immunofluorescence experiments in HeLa cells and found that serum starvation or treatment with CQ substantially increased GFP-LC3 puncta formation, while the number of GFP-LC3 puncta was significantly reduced after STYK1 knockdown (Fig. S2G). We further detected the effect of STYK1 knockdown on the autophagic flux with the mRFP-GFP-LC3 tandem reporter, which enables observations of the differences between autophagosomes (GFP-positive/RFP-positive, yellow puncta) and autolysosomes (GFP-negative/RFP-positive, red puncta) [28]. We found that STYK1 depletion inhibited the generation of autophagosomes and autolysosomes under both normal and serum starvation conditions either with or without CQ treatment (Figure 1D,E). By utilizing transmission electron microscopy, we observed a significant decrease in autophagic vacuoles in the STYK1-depleted HepG2 cells (Figure 1F,G), confirming that STYK1 mediates the formation of autophagic vacuoles. These findings show that STYK1 is a positive regulator of autophagy and is required for autophagosome formation.
Next, we investigated whether STYK1 functions in the regulation of autophagy in vivo. We utilized the zebrafish Tg (CMV: GFP-Lc3) autophagy reporter line to demonstrate the role of Styk1 in autophagy [29]. We used CRISPR/Cas9 genome editing technology to generate styk1b mutant zebrafish embryos or larvae. We designed short guide RNAs for targeting sites at the beginning of the coding exons 2 and 3 of the styk1b gene (ENSDART00000139664.1 styk1-001), which are upstream of the exons that encode the functional transmembrane domain regions. The predicted effect of the CRISPR mutation indicated a complete loss of Styk1b protein function or depletion (Fig. S2H). A mixture of sgRNA and Cas9 protein was injected into zebrafish embryos at the one-cell stage, and the effect of the CRISPR/Cas9 was verified by Sanger sequencing (Fig. S2I). The mRNA levels of styk1b were significantly decreased (Fig. S2J). We imaged the GFP-Lc3 puncta signals in vivo and performed western blotting detection on whole embryo extracts to detect Lc3 and Sqstm1. The results indicated that the styk1b mutation significantly decreased the GFP-LC3 puncta in zebrafish embryos at 4 d post-fertilization (dpf) (Figure 1H,I). We further found that the Lc3 levels decreased and Sqstm1 levels increased in the styk1b mutant zebrafish, which is consistent with the GFP-Lc3 puncta results (Figure 1J and S1K).
We next examined whether STYK1 regulates the initial steps of vesicle nucleation during which PtdIns3P is generated by the activated PtdIns3K-C1 complex. For this, we quantified the number of autophagic structures as indicated by the levels of ATG14 and ULK1, which are recruited to the phagophore assembly site and contribute to early stage autophagosome formation. The number of ATG14 and ULK1 puncta dramatically decreased with or without serum starvation in STYK1 knockdown HeLa cells (Figure 2A–D). Collectively, these results indicate that STYK1 depletion impairs autophagy initiation both in vivo and in vitro.
Figure 2.
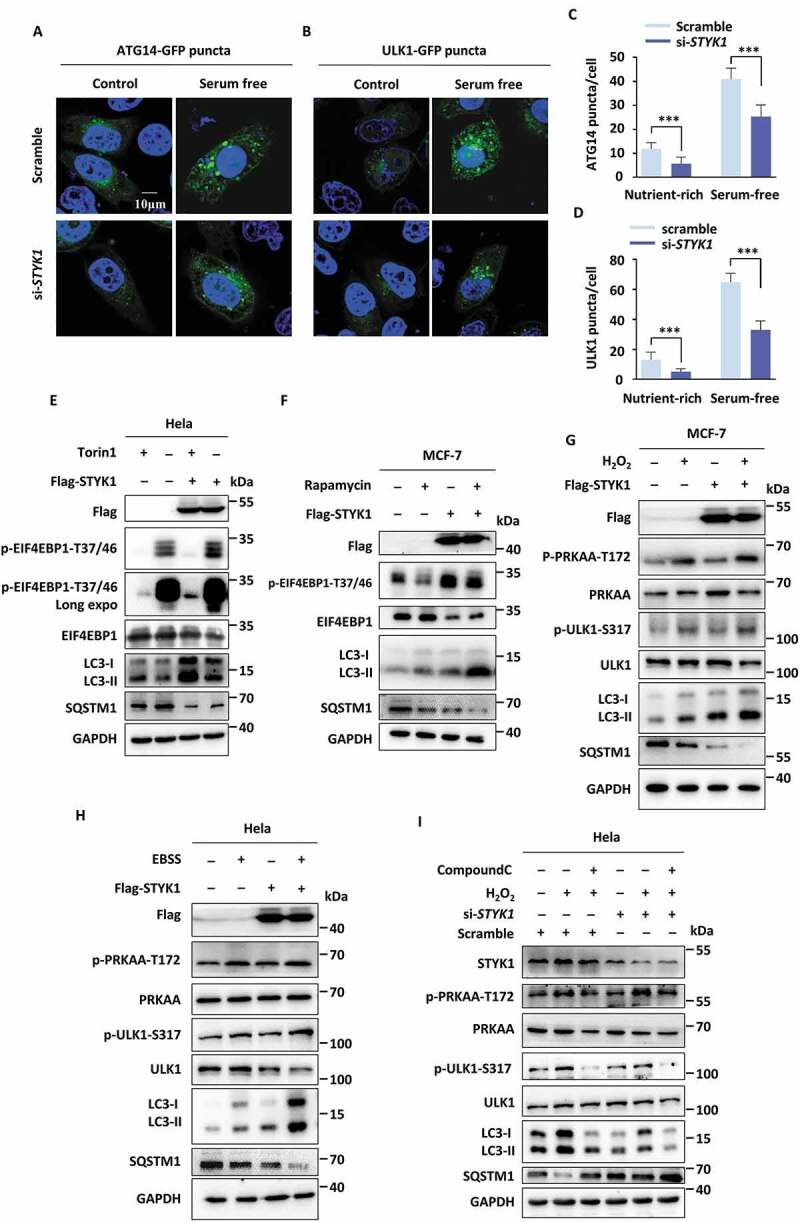
STYK1 promotes autophagy without altering MTOR or AMPK signaling. (A-D) Representative confocal images of ATG14-GFP and ULK1-GFP puncta in the HeLa cells transfected with STYK1 siRNA under nutrient-rich and serum-free starvation conditions. The number of ATG14-GFP and ULK1-GFP puncta was quantified using ImageJ software. (n = 10). Scale bars: 10 µm. (E-H) HeLa or MCF-7 cells were transfected with empty or Flag-STYK1 expression plasmids for 48 h and treated with or without torin1 (10 nM) for the later 6 h (E), rapamycin (10 nM) for the later 12 h (F), H2O2 (0.75 mM) for the later 12 h (G), and EBSS starvation for the later 2 h (H). The cells were harvested and the cell lysates were subjected to western blotting assay using the antibodies indicated. (I) HeLa cells were transfected with scramble RNA and siSTYK1 for 48 h and treated with or without H2O2 simultaneously with or without compound C (10 μM). The cells were harvested and the cell lysates were subjected to western blotting assay using the indicated antibodies. *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± SD.
STYK1 promotes autophagy without altering MTOR or AMPK signaling
Next, we utilized STYK1 overexpression to further verify the effects of STYK1 on autophagy. For this, we transfected HeLa cells with Flag-STYK1 and monitored GFP-LC3 puncta. We found that STYK1 overexpression significantly increased the number of GFP-LC3 puncta and western blotting revealed that LC3-II levels significantly increased in the background of STYK1 overexpression in both HeLa and MCF-7 cells, whereas the SQSTM1 levels were significantly decreased (Fig. S3A and B). MTOR activity is known to inhibit autophagy through the phosphorylation of ULK1 and ATG13 [9,10], while AMPK stimulates autophagy by directly phosphorylating ULK1 and BECN1 [10,30,31]. We investigated whether STYK1 promotes autophagy through the regulation of MTOR or AMPK, or through a pathway that is independent of any of the two pathways. The results indicated that STYK1 overexpression in both HeLa and MCF-7 cells led to a slight increase in the phosphorylation of EIF4EBP1, which indicating increased activity of MTORC1, however, with consistently elevated LC3-II levels in either the presence or absence of torin1 and/or rapamycin, two structurally distinct compounds that inhibit MTOR activity (Figure 2E,F). These results indicated that another pathway can reverse the inhibitory effects of MTOR on autophagy. Next, we assessed whether STYK1 can regulate AMPK activity by assessing the level of PRKAA/AMPKα phosphorylation at Thr172 [32] and by measuring the phosphorylation levels of the AMPK substrate ULK1 (phosphorylated at Ser317 [33]). We found that STYK1 overexpression in HeLa and MCF-7 cells did not lead to altered PRKAA and ULK1 phosphorylation levels in the presence of the AMPK activator, H2O2 or Earle’s balanced salt solution (EBSS) or under normal or serum-free starvation conditions. However, the effects of STYK1 and AMPK activation on LC3-II and SQSTM1 were both as expected (Figure 2G, H and S3C). It was also shown that knockdown of STYK1 inhibited H2O2-induced LC3-II accumulation, even in the presence of compound C, an inhibitor of AMPK (Figure 2I). Therefore, our data support the hypothesis that STYK1 regulates autophagy without altering AMPK or MTOR activity.
STYK1 colocalizes to autophagosomes and stimulates their formation
The data presented above suggest that STYK1 may upregulate genes that function in the autophagosome formation steps or localize to the autophagosomes and further enhance their formation. To investigate these possibilities, we assessed the expression levels of the PtdIns3K-C1 complex components after STYK1 overexpression in HeLa and knockdown in HepG2 cells, respectively. We found that the expression levels of ATG14, BECN1, PIK3C3 and ULK1 were not altered after STYK1 overexpression or knockdown (Fig. S4A). Next, the physical proximity of STYK1 to the autophagosome was investigated. To this end, the subcellular localization of STYK1 with some components of the PtdIns3K-C1 complex was examined. As shown in Figure 3A, ATG14-GFP was mostly distributed in the cytoplasm of HeLa cells under nutrient-rich conditions in the control cells. However, after STYK1 transfection, a strong colocalization of ATG14-GFP and STYK1-mCherry in the perinuclear structures was noticed (Figure 3A). Furthermore, in the cells co-transfected with ATG14-GFP and STYK1-mCherry, the number of ATG14-GFP puncta dramatically increased (Figure 3B). The colocalization between STYK1-mCherry and ULK1-GFP was further confirmed. As expected, the results indicated that many ULK1-GFP puncta were STYK1-positive. Furthermore, overexpression of STYK1 increased the number of ULK1 puncta (Fig. S4B and C).
Figure 3.
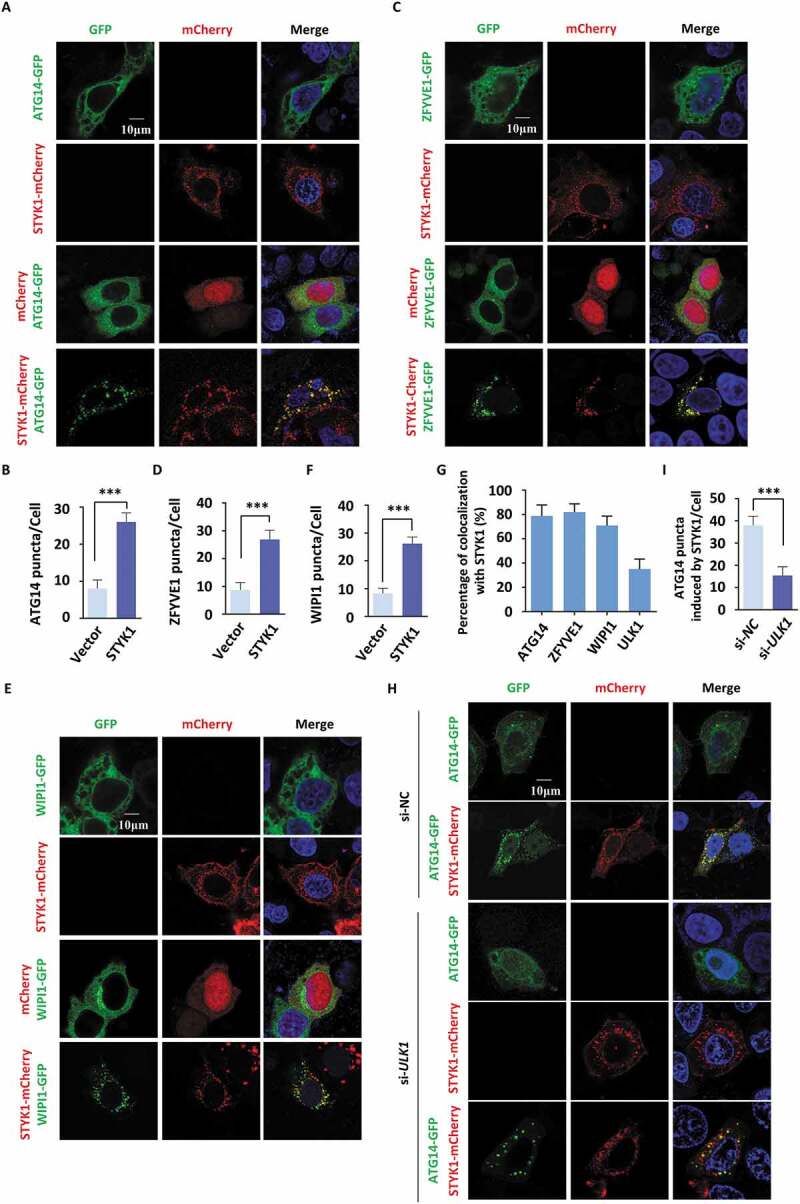
STYK1 colocalizes to autophagosomes and stimulates their formation. (A-F) Colocalization of STYK1-mCherry and ATG14-GFP (A), ZFYVE1-GFP (C), and WIPI1-GFP (E) in HeLa cells. The number of ATG14-GFP (B), ZFYVE1-GFP (D), and WIPI1-GFP (F) puncta before or after STYK1 overexpression was quantified using ImageJ software (n = 10, respectively). Scale bars: 10 µm. (G) The colocalization of ATG14-GFP, ZFYVE1-GFP, WIPI1-GFP and STYK1-mCherry was quantified. Scale bars: 10 µm. (H) Immunofluorescence analysis of ATG14-GFP puncta after co-transfection with ATG14-GFP, STYK1-mCherry and ULK1 siRNA into the HeLa cells for 48 h. Scramble siRNA was used as a negative control. The number of ATG14 puncta was quantified (n = 10). Scale bars: 10 µm. *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± SD.
ZFYVE1 is widely used as a marker of the omegasomes formed by phagophore-associated PtdIns3P-enriched endoplasmic reticulum (ER) membranes [34,35]. WIPI1 is a PtdIns3P-binding protein that is recruited to omegasomes during the initiation of autophagosome formation [36]. Next, the possible colocalization of ZFYVE1 and WIPI1 with STYK1 was investigated. We found that ZFYVE1-GFP and WIPI1-GFP formed puncta upon STYK1-mCherry expression and colocalized with STYK1-mCherry in the HeLa cells (Figure 3C–G). As ULK1 acts as an upstream effector of the PtdIns3K-C1 complex with respect to the autophagy activation, the role of ULK1 in STYK1-induced ATG14 puncta formation was investigated. We found that ULK1 knockdown suppressed the induction of ATG14 puncta by STYK1 (Figure 3H,I), indicating that ULK1 is critical for mediating the effects of STYK1 on ATG14 puncta formation. Collectively, these data suggest that STYK1 colocalizes with autophagosomes and stimulates their formation.
STYK1 interacts with the PtdIns3K-C1 complex
To investigate whether STYK1 interacts with the PtdIns3K-C1 complex, HeLa cells were simultaneously transfected with four plasmids harboring Flag-STYK1, MYC-ATG14, HA-BECN1 and HA-PIK3C3. The lysates were pulled down using the Flag antibody, and the precipitates were subjected to an SDS-PAGE and Coomassie brilliant blue staining. The results revealed three bands that corresponded to the molecular weights of HA-PIK3C3, MYC-ATG14 and HA-BECN1, which were equimolar to the three subunits in the Flag-STYK1 coprecipitate (Figure 4A). Further co-immunoprecipitation (co-IP) assays showed that Flag-STYK1 interacts with endogenous PIK3C3, BECN1, ATG14 and PIK3R4 but not with RUBCN. We also identified that Flag-STYK1 interacts with RACK1 (receptor for activated C kinase 1), which was recently reported to be functionally important in the assembly of the PtdIns3K-C1 complex [37], and NRBF2, which has been identified as the fifth component of the PtdIns3K-C1 complex [38] (Figure 4B). Additionally, a co-IP assay using BECN1 antibody was performed after the transfection of Flag-STYK1 expression plasmid into HeLa cells. The results showed that endogenous BECN1 was bound to Flag-STYK1 (Figure 4C). We also found that endogenous STYK1 co-precipitated with endogenous BECN1, PIK3C3, PIK3R4 and ATG14. Further, endogenous BECN1 and ATG14 co-precipitated with endogenous STYK1 (Figure 4D,E). The interactions between STYK1 and the PtdIns3K-C1 complex were further confirmed by co-IP assays performed with HEK293T cells (Figure 4F, Fig. S5A and B). Immunofluorescence using confocal microscopy showed that BECN1, PIK3C3 and PIK3R4 colocalized with STYK1 in HeLa cells under nutrient-rich conditions (Figure 4G). To further examine the simultaneous presence of BECN1, ATG14 and STYK1 within the same complex, 2-step co-IP assays were performed using the HeLa cell lysates harboring four plasmids encoding HA-BECN1, Flag-BECN1, MYC-ATG14 and non-tagged STYK1. The first immunoprecipitation of the lysate with the Flag antibody, followed by a second immunoprecipitation with a MYC antibody revealed the presence of STYK1 in the immunoprecipitate (Figure 4H). These observations supported the hypothesis that STYK1 physically interacts with the PtdIns3K-C1 complex.
Figure 4.
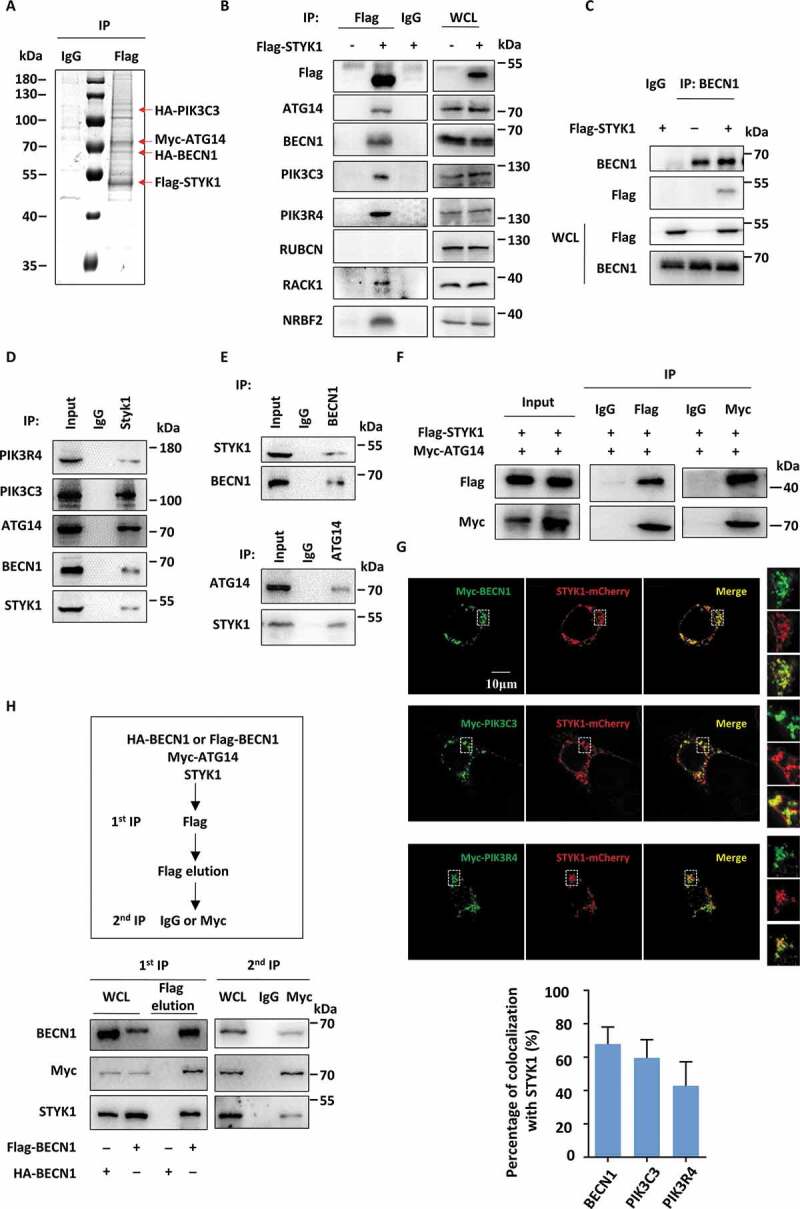
STYK1 interacts with the PtdIns3K-C1 complex. (A) HeLa cells were simultaneously transfected with four plasmids containing Flag-STYK1, HA-PIK3C3, HA-BECN1 and MYC-ATG14 for 48 h, then immunoprecipitated with an anti-Flag antibody and subjected to SDS-PAGE, followed by Coomassie blue staining. (B) The interaction between Flag-STYK1 and the components of the PtdIns3K-C1 complex in HeLa cells. The cell lysates were used for IP and western blotting with the indicated antibodies. (C) The interaction between endogenous BECN1 and Flag-STYK1. (D and E) The interactions between endogenous BECN1, ATG14 and endogenous STYK1. (F) The interaction between Flag-STYK1 and MYC-ATG14 in HEK293T cells. (G) Colocalization between STYK1-mCherry and BECN1, PIK3C3, PIK3R4. Scale bars: 10 µm. (H) Two-step co-IP assay in HEK293T cells to determine the complex containing STYK1, BECN1, and ATG14. The procedures of the two-step co-IP are outlined at the top. The samples were subjected to western blotting with the indicated antibodies.
STYK1 promotes the assembly of the PtdIns3K-C1 complex
Based on the interaction of STYK1 with the PtdIns3K-C1 complex and its stimulatory effect on early autophagic processes, we examined whether STYK1 affected the assembly of the PtdIns3K-C1 complex. The results indicated that STYK1 overexpression enhanced the interaction of endogenous PIK3C3 with BECN1 and ATG14, as well as with the exogenous expressed Flag-BECN1 in HeLa cells (Figure 5A, B and S6A). STYK1 knockdown decreased the interaction of endogenous PIK3C3 with BECN1, ATG14 and the interaction of exogenous expressed Flag- ATG14 with HA-PIK3C3 (Figure 5C, S6B and C). It has been reported that the kinase activity of the PtdIns3K-C1 complex is negatively regulated by the BCL2 family proteins that bind to the BH3 domain of BECN1, and by RUBCN that binds to PIK3C3 via its RUN domain [2,13,39]. Therefore, the effects of STYK1 on the interactions among BECN1, BCL2 and RUBCN were further investigated. The results revealed that the BECN1-BCL2 interaction was decreased and less RUBCN associated with the complex during STYK1 overexpression (Figure 5D, E and S6D).
Figure 5.
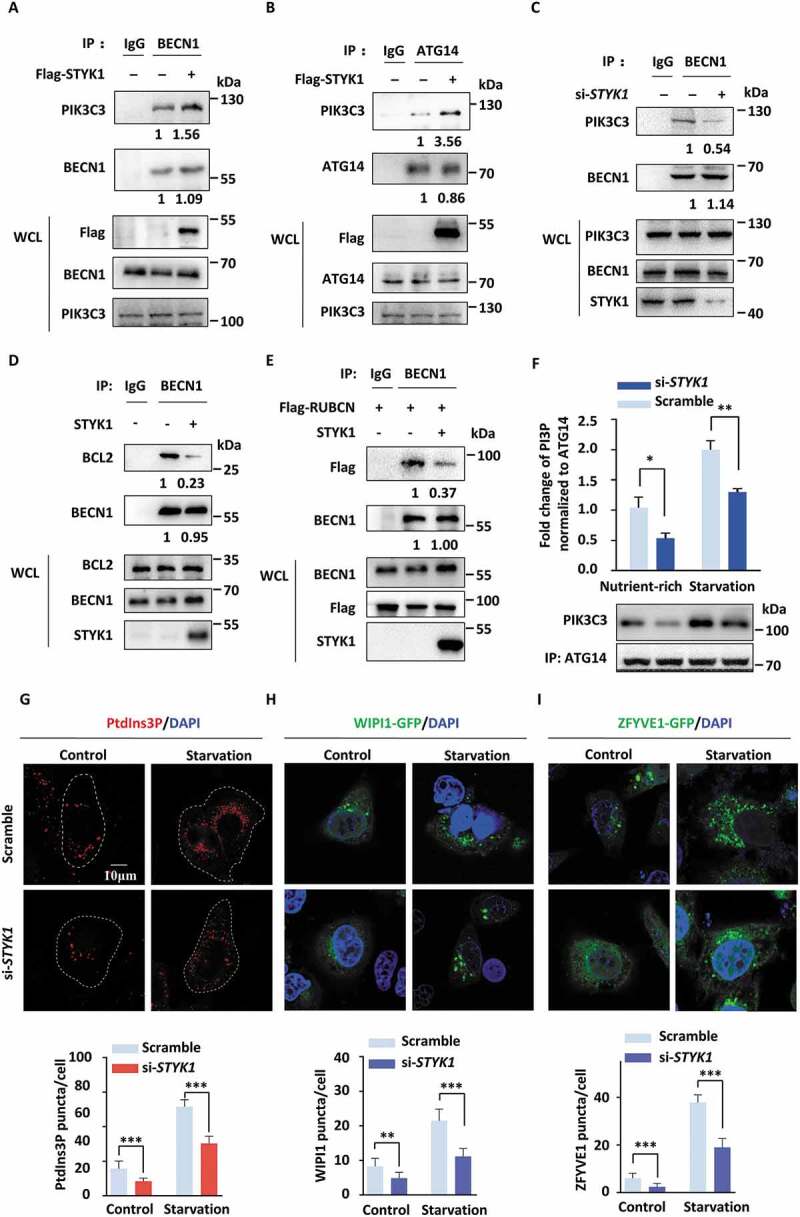
STYK1 promotes the assembly of the PtdIns3K-C1 complex. (A-B) The exogenously expressed Flag-STYK1 enhanced BECN1-PIK3C3 and ATG14-PIK3C3 interactions in HeLa cells. The immunoprecipitated endogenous PIK3C3 was quantified using Image lab software and normalized against the amount of PIK3C3 in the whole-cell lysates (WCL). (C) STYK1 knockdown inhibits the interaction between endogenous BECN1 and HA-PIK3C3. The immunoprecipitated PIK3C3 was quantified and normalized against the amount of PIK3C3 in WCL. (D) The exogenously expressed nontagged STYK1 inhibits the interaction between BECN1 and BCL2. The immunoprecipitated BCL2 was quantified and normalized against the amount of BCL2 in WCL. (E) The exogenously expressed nontagged STYK1 inhibits the interaction between BECN1 and Flag-RUBCN. The immunoprecipitated Flag-RUBCN was quantified and normalized against the amount of Flag-RUBCN in WCL. (F) Different PtdIns3K-C1 complex components from the control or STYK1 knockdown HeLa cells were immunoprecipitated by the ATG14 antibody under either nutrient-rich or starvation conditions. PIK3C3 activity was measured by analyzing PtdIns3P production using the ELISA assay described in the Materials and Methods. The PtdIns3P fold change was calculated based on the concentration of PtdIns3P and was normalized to the amount of ATG14 used in the assay. (G-I) Confocal microscopy of either the nutrient-rich or the starvation-induced (EBSS 2 h) PtdIns3P puncta (G), WIPI1-GFP puncta (H), and ZFYVE1-GFP puncta (I) in the control and STYK1 knockdown HeLa cells. The numbers of PtdIns3P, ZFYVE1-GFP and WIPI1-GFP puncta was quantified (n = 10). Scale bars: 10 µm. *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± SD.
PtdIns3P in omegasomes produced by class III PtdIns3K are crucial for autophagy initiation [40]. To determine whether STYK1 directly modulates the activity of PtdIns3K-C1 complex, the effects of STYK1 on three key downstream effectors: PtdIns3P, ZFYVE1 and WIPI1 were analyzed. After transfecting Flag-ATG14 in HeLa cells for 48 h with or without co-transfection with STYK1 interfering RNA in nutrient-rich or EBSS starvation conditions and followed by an immunoprecipitation (IP) assay with anti-Flag antibody, we incubated the anti-ATG14 immunoprecipitate with phosphatidylinositol as the substrate through a quantitative ELISA assay. We found that ATG14-linked PtdIns3K kinase activity was dramatically reduced after STYK1 depletion in both the nutrient-rich and EBSS starvation conditions (Figure 5F). Moreover, the immunofluorescence analysis indicated that knockdown of STYK1 in HeLa cells led to a reduced number of PtdIns3P, GFP-WIPI1 and GFP-ZFYVE1 puncta in nutrient-rich or EBSS starvation conditions (Figure 5G–I). These data indicate that STYK1 is essential for the activation of the ATG14-dependent class III PtdIns3K activity and the assembly of the PtdIns3K-C1 complex during the initiation phase of autophagy.
STYK1 dimerization involves the PtdIns3K-C1 complex
The ligand-dependent or ligand-independent oligomerization of RTKs is a key step in receptor activation and intracellular signaling [41]. As we noticed that Flag-STYK1 precipitated HA-STYK1 (Fig. S7A), indicating the presence of STYK1 dimers, which is in agreement with a previous report [42]. Therefore, we investigated whether STYK1 dimerization is involved in the PtdIns3K-C1 complex and plays a role in autophagy induction. To this end, a 2-step co-IP was performed by using HEK293T cell lysates harboring three plasmids containing HA-STYK1, MYC-STYK1 and Flag-BECN1. HA-STYK1 was detected in the immunoprecipitate after two rounds of co-IP using the Flag and MYC antibodies (Figure 6A). A similar result was observed in another 2-step co-IP, in which the cells were co-transfected with HA-STYK1, MYC-STYK1 and Flag-ATG14 (Fig. S7B), indicating that HA-STYK1 and MYC-STYK1 occupy the PtdIns3K-C1 complex. These data suggest that STYK1 interacts with the PtdIns3K-C1 complex in the form of STYK1 dimers.
Figure 6.
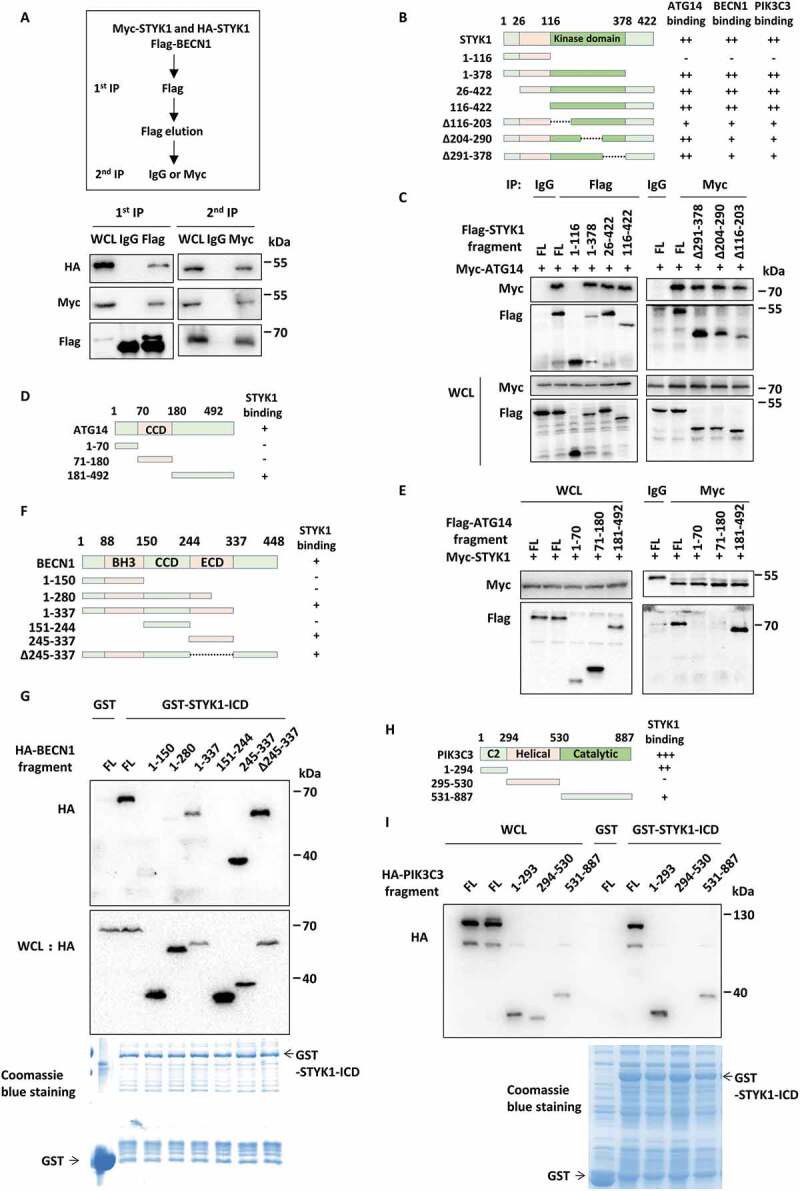
STYK1 dimerization involves the PtdIns3K-C1 complex. (A) A two-step co-IP assay using the HEK293T cells was performed to determine the complex containing MYC-STYK1, HA-STYK1, and Flag-BECN1. The procedures of the two-step co-IP are outlined at the top. The samples were subjected to IP and western blotting with the indicated antibodies. (B) The schematic diagram of STYK1 truncation mutations generation and the domains mediated the STYK1-ATG14, STYK1-BECN1, and STYK1-PIK3C3 interactions. (C) HEK293T cells were transfected with the STYK1 truncations indicated for 48 h and then harvested for IP and western blotting analysis. (D) The schematic diagram of ATG14 truncation mutations generation and ATG14-STYK1 interacting domain. (E) HEK293T cells were transfected with the ATG14 truncation mutations indicated for 48 h and then harvested for IP and western blotting analysis. (F) The schematic diagram of BECN1 truncation mutations generation and the BECN1-STYK1 interacting domain. (G) HEK293T cells were transfected with the BECN1 truncation mutations indicated for 48 h and then harvested for in vitro GST-affinity-isolation assay. (H) The schematic diagram of PIK3C3 truncation mutations generation and the PIK3C3-STYK1 interacting domain. (I) HEK293T cells were transfected with the PIK3C3 truncation mutations indicated for 48 h and then harvested for in vitro GST-affinity-isolation assay.
The domains of STYK1 involved in interactions with ATG14, BECN1 and PIK3C3 were then examined. Four STYK1 truncation constructs were constructed for the co-IP assays in HEK293T cells. The results showed that the STYK1 kinase domain (aa 116–378) mediated interactions with ATG14, BECN1 and PIK3C3 (Figure 6B,C, S7C and D). Using further deletion mutations within the kinase domain for similar experiments, we found that the interaction between ATG14 and STYK1 decreased upon the deletion of aa 116–203 compared with the other two truncations, whereas interactions between BECN1, PIK3C3 and STYK1 were impaired upon each deletion (Figure 6C, S7C and D). It is suggested that the region aa 116–203 within the kinase domain of STYK1 is important for STYK1 binding to the PtdIns3K-C1 complex. The domains of ATG14, BECN1 and PIK3C3 involved in the interactions with STYK1 were then investigated. As shown in Figure 6D,E, STYK1 interacted with full-length ATG14 and the region containing aa 181–492 in the co-IP assay. By using purified GST-tagged N-terminal ectodomain and transmembrane domain-truncated STYK1, which we called STYK1-ICD, we found that STYK1 interacts with BECN1 through the aa 281–448 (Figure 6F,G). Similarly, STYK1 interacts with PIK3C3 through both the C2 and the catalytic domain (Figure 6H,I). Furthermore, we also determined that STYK1 formed dimers through the kinase domain (aa 116–378) (Fig. S7E). An in vitro affinity-isolation assay was performed using externally purified proteins to investigate the direct binding between STYK1 and the PtdIns3K-C1 complex. However, we could hardly purify full-length ATG14, BECN1 and PIK3C3. Alternatively, we successfully purified GST-tagged ATG14 aa 181–492, BECN1 aa 151–450, PIK3C3 aa 1–293 and PIK3C3 aa 531–887. After incubation of the aforementioned GST tagged proteins with externally purified His-STYK1, we found that His-STYK1 was detectable in all the precipitates of the GST-tagged proteins (Fig. S7F). These results indicate that STYK1 directly binds to the PtdIns3K-C1 complex.
The phosphorylation of STYK1 Tyr191 contributes to its dimerization and BECN1 phosphorylation
Next, the role of STYK1 kinase activity in autophagy induction was investigated. This was achieved by transfecting HepG2 and HeLa cells with the STYK1K147R (kinase-dead mutation), which has impaired ATP binding ability. The results showed that the levels of LC3-II were decreased, while the levels of SQSTM1 were increased compared to those in cells that were transfected with WT STYK1 in the presence or absence of CQ (Figure 7A and S8A). Moreover, we also noticed that the loss of the transmembrane domain led to similar effects to autophagy compared with K147R mutation (Figure 7A and S8A). Whether any tyrosine residues in STYK1 are involved in the phosphorylation or dimerization and subsequent kinase activity was investigated as STYK1 is known to be phosphorylated in the cell cytoplasm [24] and tyrosine phosphorylation is known to initiate the dimerization of many proteins [43,44]. Toward this end, the possible phosphorylated tyrosine sites were predicted using NetPhos 3.1 (http://www.cbs.dtu.dk/services/NetPhos/). Four candidate sites were identified: Y24, Y229, Y327 and Y404 (Fig. S8B). ClustalW alignment analysis revealed that the STYK1 tyrosine residue Y191 is conserved among the members of the RTK subfamilies (Fig. S8B). Therefore, the effects of these STYK1 tyrosine residues and Y356, which has been reported to be involved in tumorigenesis [24], on the autophagic processes of HeLa cells were investigated. We found that mutations Y24F or Y191F led to significantly decreased LC3-II levels and increased SQSTM1 levels, which indicated impaired autophagy in HeLa cells, whereas mutations Y229, Y327, Y356 and Y404 showed no significant effects (Figure 7B,C). The effects of Y24F and Y191F on autophagy were further confirmed in MCF-7 cells in the presence or absence of CQ (Figure 7D).
Figure 7.
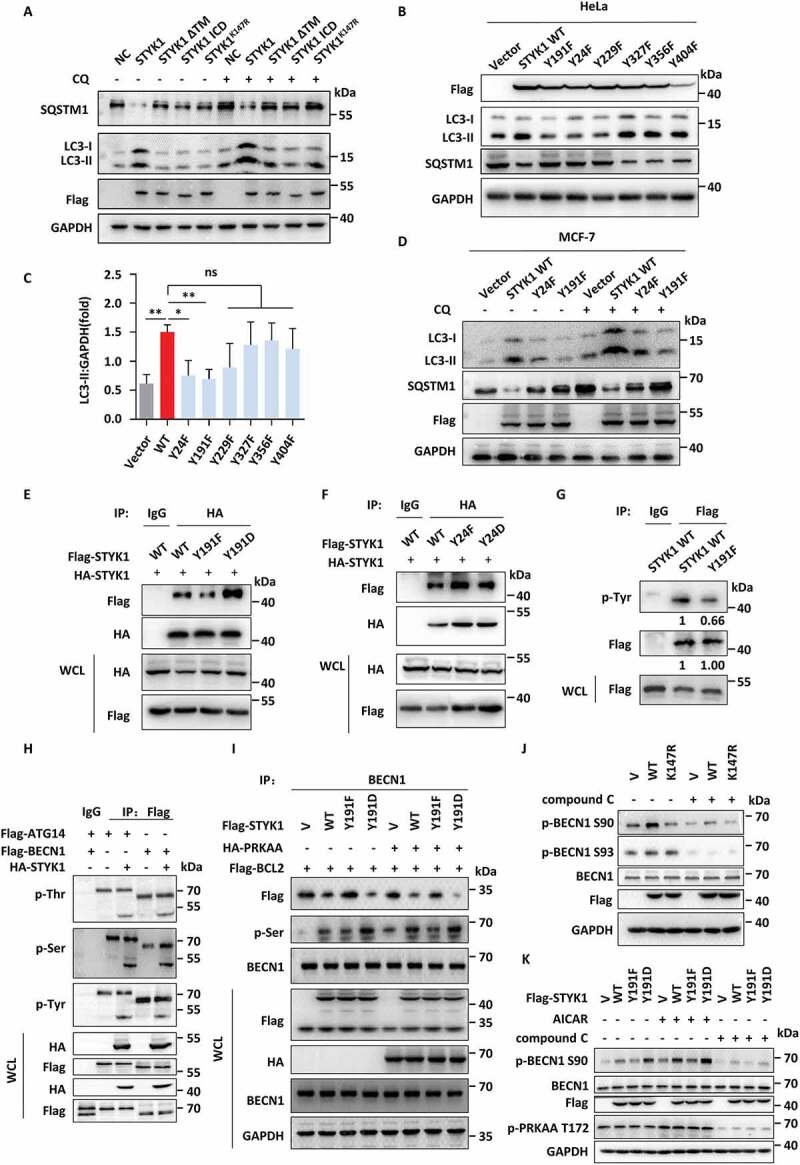
The phosphorylation of STYK1 Tyr191 contributes to its dimerization and BECN1 phosphorylation. (A) Western blotting analysis of LC3-II and SQSTM1 in HepG2 cells transfected with WT STYK1, transmembrane domain deletion mutation (ΔTM), N-terminal 1–26 aa deletion mutation (ICD) and STYK1K147R mutation treated with or without CQ (10 μM). (B and C) Western blotting analysis of LC3-II and SQSTM1 in HeLa cells transfected with WT STYK1 and its mutations as indicated. The amount of LC3-II was quantified using Image Lab (Bio-Rad) and normalized to GAPDH. (D) Western blotting analysis of LC3-II and SQSTM1 in MCF-7 cells transfected with WT STYK1, Y24F and Y191F mutations in the treatment with or without CQ (10 μM). (E and F) HA-STYK1 was co-transfected with Flag-STYK1Y191F, Y191D, Y24F and Y24D mutations into HeLa cells, and after 48 h cells were harvested for the STYK1 dimerization assay. (G) HeLa cells were transfected with either WT STYK1 or STYK1Y191F mutation for 48 h and harvested for the co-IP assay using Flag antibody, then analyzed by western blotting assay using the antibody indicated. (H) 293T cells were co-transfected with either Flag-ATG14 or Flag-BECN1 with HA-STYK1 for 48 h were harvested for the co-IP assay using Flag antibody, then analyzed by western blotting assay using the indicated antibodies. (I) 293T cells were transfected with either empty vector (V), WT STYK1, STYK1Y191F or STYK1Y191D mutations simultaneously transfected with or without HA-AMPK for 48 h. The cells were then harvested for the co-IP assay using the BECN1 antibody, then analyzed by western blotting assay using the indicated antibodies. (J) 293T cells were transfected with either empty vector (V), WT STYK1, kinase-dead STYK1K147R mutant for 48 h with or without compound C treatment. The cells were then harvested and then analyzed by western blotting assay using the indicated antibodies. (K) 293T cells were transfected with either empty vector (V), WT STYK1, STYK1Y191F or STYK1Y191D mutations for 48 h with or without AICAR/compound C treatment. The cells were then harvested and then analyzed by western blotting assay using the indicated antibodies. *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± SD.
In addition, we found that the Y191F mutation, but not Y24F mutation significantly decreased STYK1 dimerization and that the Y191D mutation enhanced dimerization (Figure 7E,F). These results are consistent with the results of the GST-affinity-isolation assays showing that the aa 116–378 fragment is required for STYK1 dimerization. Homology modeling analysis of STYK1 dimerization also suggested that STYK1 Y191 is close to the STYK1 dimerization site (Fig. S8C). Additionally, we found that the Y191F mutant reduces the phosphorylation of STYK1, indicating that the Y191 residue is a phosphorylation site (Figure 7G).
Whether STYK1 can phosphorylate one or more components of the PtdIns3K-C1 complex was examined as STYK1 binds directly to the complex. This mechanism was investigated by co-transfecting 293T cells with ATG14, BECN1, PIK3C3 and STYK1 respectively. We found that STYK1 increased the total serine phosphorylation level of BECN1 (Figure 7H). We also found that the total serine phosphorylation level of BECN1 was lower in the STYK1Y191F and higher in STYK1Y191D-transfected 293T cells compared with the levels in the WT STYK1 transfected cells. Moreover, the serine phosphorylation level of BECN1 was much higher after AMPK co-transfection (Figure 7I), suggesting that AMPK is an upstream kinase for STYK1. BECN1 has been reported as being phosphorylated at S93 and S96 by AMPK [31] and at S90 by several kinases [45]. We then examined the effects of STYK1 to BECN1 S90 and S93 using commercial specific antibodies. The results indicated that the level of BECN1 S90 phosphorylation significantly increased after STYK1 overexpression, but not STYK1K147R mutant. And treatment with compound C inhibited the level of STYK1-induced BECN1 S90 phosphorylation (Figure 7J). However, the results showed there were little effects of STYK1 on BECN1 S93 phosphorylation (Figure 7J). Furthermore, we found that the level of BECN1 S90 phosphorylation was lower in STYK1Y191F and higher in STYK1Y191D-transfected 293T cells compared with WT STYK1 transfected cells and the effects were elevated or depressed after AICAR treatment or compound C treatment (Figure 7K). Moreover, STYK1 depletion had a limited effect on the total phosphorylation of BECN1 after both Ser90 and 93 were mutated to alanine (Fig. S8D).
As we showed that STYK1 overexpression significantly decreased the interactions between BECN1 and BCL2 (Figure 5D), and the BECN1S90A/S93A/T388A mutant was more strongly bound to BCL2 compared with the binding of the wild-type control [46]. We also demonstrated that the interaction between BECN1 and BCL2 increased upon STYK1 depletion with a concurrent reduction in BECN1 serine phosphorylation, and decreased upon STYK1 overexpression concurrent with elevated BECN1 serine phosphorylation with or without AICAR and/or compound C treatment (Figure 7I, S8D and E). Additionally, using an in vitro tyrosine kinase activity assay, we discovered that both the Y191F and Y24F mutations inhibited STYK1 kinase activity. In contrast, the Y191D mutation promoted STYK1 kinase activity (Fig. S8F). We also found that STYK1 overexpression accelerated the total tyrosine phosphorylation of PIK3C3 (Fig. S8G). Taken together, these data indicate that the phosphorylation of STYK1 Tyr191 contributes to its dimerization and BECN1 phosphorylation.
STYK1 Tyr191 phosphorylation contributes to the assembly of the PtdIns3K-C1 complex
As the above results show that STYK1 Tyr191 affects the dimerization of STYK1, which is involved in the PtdIns3K-C1 complex, and Tyr191 located in aa 116–203, deletion of which decreased the interactions between STYK1 and ATG14, BECN1 and PIK3C3, we next assessed whether STYK1 Tyr191 phosphorylation affects the assembly of the PtdIns3K-C1 complex. We found that an increased amount of endogenous ATG14, BECN1, PIK3C3 and PIK3R4 was co-precipitated along with the STYK1Y191D mutant, whereas a significant reduction in the amount of these coprecipitated endogenous proteins was obtained with the STYK1Y191F mutant compared with the results obtained with the WT STYK1 (Figure 8A). Moreover, the STYK1Y191F mutation significantly suppressed the interaction of BECN1 with endogenous ATG14, PIK3C3 and PIK3R4, and it also reduced the interaction of ATG14 with BECN1, PIK3C3, and PIK3R4 (Figure 8B,C). Consistently, the Y191F and Y191D mutations decreased and increased the formation of WIPI1 and ZFYVE1 puncta, respectively (Figure 8D, E and S9A). An in vitro PtdIns3P generation ELISA assay was performed, and we found that ATG14-linked PtdIns3K kinase activity dramatically increased after STYK1 overexpression under nutrient-rich or starvation conditions or in the background of AICAR treatment (Figure 8F). Furthermore, cells transfected with the STYK1Y191D mutant showed an increased amount of PtdIns3K kinase activity compared with the level shown in cells transfected with WT STYK1, whereas cells with the Y191F mutant showed fewer effects (Figure 8F). These findings indicate that the phosphorylation of STYK1 Y191 is essential for the PtdIns3K-C1 complex assembly.
Figure 8.
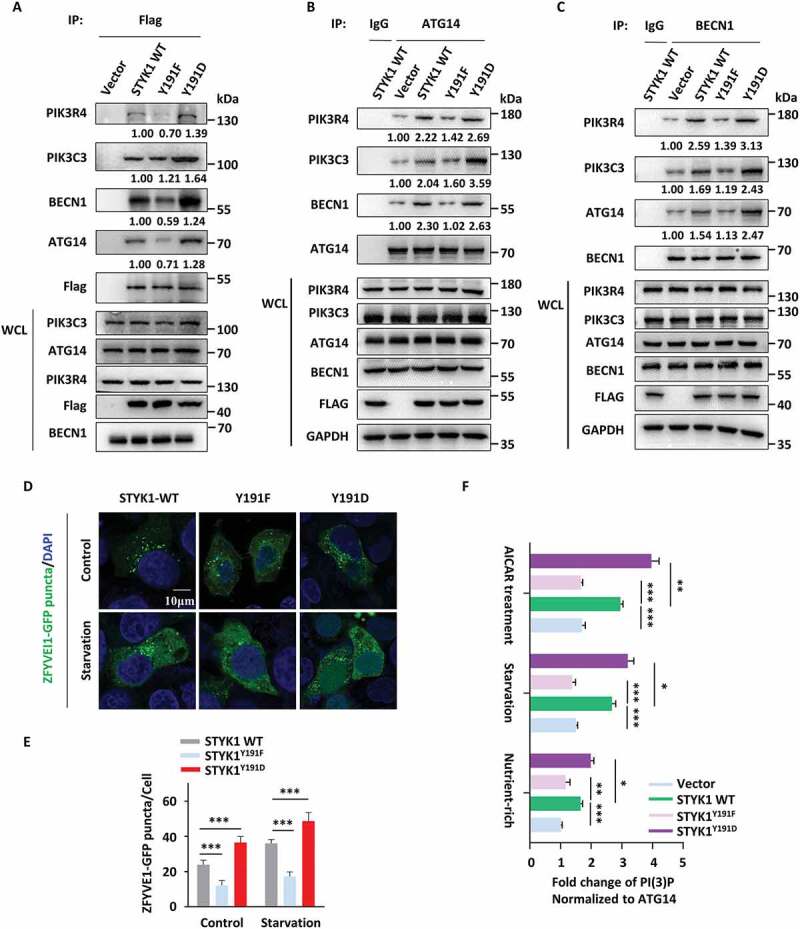
STYK1 Tyr191 phosphorylation contributes to the assembly of the PtdIns3K-C1 complex. (A-C) HeLa cells were transfected with WT STYK1 and its mutations for 48 h, then cell lysates were subjected to IP with Flag- (A), BECN1- (B) and ATG14- (C) antibodies. Precipitates were then subjected to western blotting analysis. Empty vector or rabbit IgG was used as the control. (D and E) Confocal microscopy of either nutrient-rich or starvation-induced (EBSS 2 h) ZFYVE1-GFP puncta in WT STYK1 and its mutant transfected HeLa cells. The number of GFP-ZFYVE1 puncta was quantified (n = 10). Scale bars: 10 µm. (F) Different PtdIns3K-C1 complex components from the empty vector, WT STYK1 or its mutant transfected HeLa cells were immunoprecipitated using the ATG14 antibody, either in nutrient-rich, starvation or AICAR treatment conditions. PIK3C3 activity was measured by analyzing PtdIns3P production in the ELISA assay described in the Materials and Methods section. The PtdIns3P fold change was calculated based on the concentration of PtdIns3P and was normalized to the amount of ATG14 used in the assay. *P < 0.05; **P < 0.01; ***P < 0.001. Data are presented as mean ± SD.
STYK1 affects autophagy-mediated starvation resistance, suppression of lipid degradation and protein aggregate clearance
It has been recognized that autophagic responses constitute a means to cope with intracellular and environmental stress, thus favoring tumor progression in some cases [47]. Therefore, whether STYK1 deficiency influences cell viability upon nutrient starvation stress was investigated. We found that STYK1-deficient HepG2 cells cultured in EBSS, a growth factor-free medium, exhibited lower cell viability in a time-dependent manner compared with normal cells (Figure 9A). Additionally, the cells treated with activators of autophagy, rapamycin and brefeldin A (simulation of endoplasmic reticulum stress) [48] dramatically recovered the cell viability (Figure 9A,B). In contrast, treatment with the autophagy inhibitor CQ produced an opposite effect (Figure 9C), indicating that STYK1 influences cell viability during nutrient starvation partly through autophagy. In addition, the function of STYK1 in hepatic lipid accumulation and protein clearance was investigated. The results showed that STYK1 depletion led to significant lipid accumulation in hepatocytes (Figure 9D,E), a finding that is similar to those in which the phenotypes were attributed to ATG5 or ATG7 deletion in hepatocytes [49,50]. We also found that the STYK1Y191F and STYK1K147R mutations were associated with increased lipid accumulation in hepatocytes compared with levels observed in the WT STYK1 and the STYK1Y191D mutant (Figure 9D,E), thereby presenting an essential role for STYK1 in maintaining lipid homeostasis as an autophagy regulator. Puromycin-induced protein aggregates and SQSTM1-positive aggresome-like induced structures (ALISs) are known to be selectively recognized by ubiquitin-binding autophagy receptors and subsequently removed via autophagy [51,52]. Therefore, the dynamics of puromycin-induced Ub (ubiquitin) and SQSTM1-positive ALIS clearance upon STYK1 depletion in HepG2 cells were assessed. We found that STYK1 depletion caused the appearance of Ub-positive (Ub+) puncta in untreated cells that were rarely present in control cells (Figure 9E, NT). Moreover, the puromycin-induced SQSTM1+/Ub+ protein aggregates were efficiently cleared in control cells, while the recovery of the increased protein aggregates was not apparent even after 6 h in STYK1-depleted cells (Figure 9F,G). In addition, STYK1 overexpression, but not STYK1Y191F mutation, decreased the protein aggregates induced by puromycin compared with those produced in control cells (Figure 9F,G), indicating that STYK1 might be involved in clearing protein aggregates.
Figure 9.
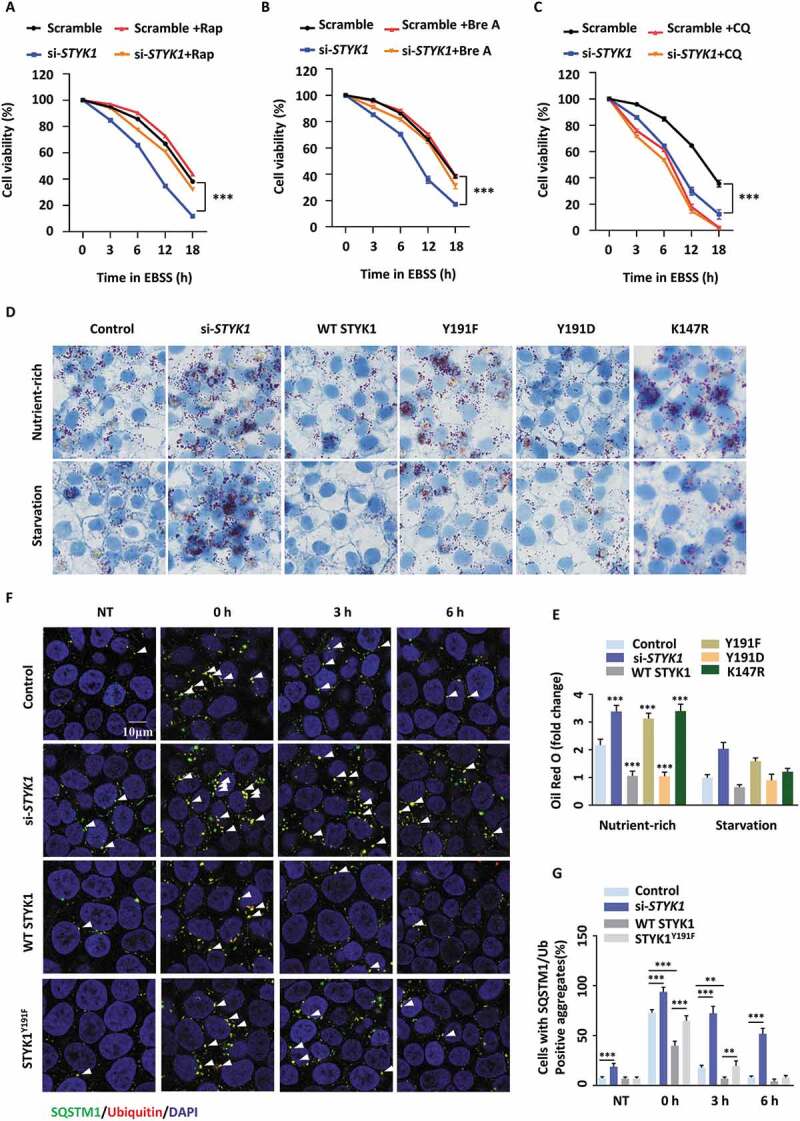
STYK1 affects the autophagy-mediated starvation resistant, suppression of lipid degradation and protein aggregate clearance. (A-C) HepG2 cells were transfected with scramble or STYK1 siRNA cultured in EBSS in the presence of rapamycin (Rap, 10 nM) (A), brefeldin A (Bre A, 5 mM) (B), CQ (10 μM) (C) or the control solution for 0, 3, 6, 12 or 18 h, respectively. Cell viability was assessed via MTT assay. (D and E) HepG2 cells were transfected with scramble RNA, STYK1 siRNA, and STYK1 overexpression plasmid and its mutations for 48 h were subjected to starvation and then fixed and analyzed by oil red O staining. The fold change of intracellular oil red O content was quantified. (F and G) HepG2 cells transfected with STYK1 siRNA, STYK1 overexpression plasmid and the STYK1Y191F mutation for 48 h were treated with 5 mg/mL puromycin for 2 h. Cells were either fixed in 4% paraformaldehyde (PFA) (0 h) or washed three times in DMEM (10% fetal bovine serum) without puromycin, then incubated for a further 3 h or 6 h to fixation and immunofluorescence analysis. NT represents HepG2 cells without puromycin treatment. The number of SQSTM1 and Ub double-positive puncta was quantified using ImageJ software (n = 10). Scale bars: 10 µm. **P < 0.01; ***P < 0.001. Data are presented as mean ± SD.
Discussion
In this study, we discovered that STYK1 is a novel positive regulator of autophagy. Specifically, STYK1 depletion impaired autophagosome formation both in vivo and in vitro. Furthermore, autophagy is significantly inhibited after Styk1b depletion in GFP-Lc3 transgenic zebrafish. Our study also revealed that STYK1, as a unique member of the RTK family with the ability to promote cancer development, but with such negligible extracellular domain, physically and functionally interacts with the PtdIns3K-C1 complex, providing a novel example of a crosstalk between RTK signaling and autophagy.
Cumulatively, our data indicate that STYK1 is required for the early steps of autophagosome formation. The amount of autophagosome biogenesis indicated by ATG14, ZFYVE1 and WIPI1 was reduced in STYK1-depleted HeLa cells. Moreover, LC3-II levels clearly showed that the activity of autophagosome biogenesis was decreased after STYK1 knockdown. We also revealed that ATG14-linked PIK3C3 activity was significantly reduced in STYK1 knockdown cells, which is consistent with the notion that PtdIns3P is generated by PIK3C3 upon binding with ATG14 in the phagophore nucleation phase. Furthermore, our data reveal that STYK1 promotes the assembly of the PtdIns3K-C1 complex, which is important for initiating autophagosome formation under both nutrient starvation and basal conditions. Collectively, our data suggest a model in which STYK1 is required in the early steps between autophagosome formation.
It has been previously reported that the insulin receptor TKD, an RTK member, undergoes RTK auto-inhibition by its kinase domain in a cis-auto-inhibitory interaction manner. Phosphorylation of the key tyrosine Y1162, along with phosphorylation of two other tyrosine sites, in the activation loop of the TKD kinase domain could release the activation loop from the active site, allowing it to be accessed by both ATP and protein substrates, leading to kinase activation [53,54]. A conceptually similar mechanism was also found in FGFR1 [55], which shares 30% sequence similarity with STYK1 in the intracellular domain [56], suggesting that STYK1 may also share this method of kinase activation. Indeed, our findings of the importance of STYK1 Tyr191 for promoting its dimerization and the assembly of the PtdIns3K-C1 complex, together with previously reported phosphorylation sites at Tyr327 and 356 [24], suggests that the three tyrosine sites may play important roles in STYK1 kinase activation. Our data also revealed that autophagy is impaired when we transfected HeLa and HepG2 cells with the STYK1K147R kinase-dead mutation with impaired ATP binding ability, which shows the same effect as STYK1Y191F mutation transfection compared with WT STYK1, presenting the enormous possibility that the substrate of STYK1 also functions in STYK1 induced autophagy activity.
AMPK and MTOR are the two central modulators of autophagy regulation [33]. However, our data showed that STYK1 promotes autophagy initiation without altering the activity of AMPK and MTOR, indicating that neither AMPK nor MTOR is a downstream effector of STYK1. As the phosphorylation of BECN1, ATG14 and PIK3C3 have been reported to function in autophagy induction [57] and STYK1 directly binds to ATG14, BECN1 and PIK3C3, our findings revealed that STYK1 can increase the Ser90 phosphorylation of BECN1. Moreover, our data indicate that the effects of STYK1 to the level of BECN1 Ser90 phosphorylation could be enhanced and reduced upon the treatment of AICAR and compound C, suggesting that AMPK is an upstream kinase for STYK1. BECN1 has been reported as being phosphorylated at Ser90 by MAPK1/3 [58], and STYK1 had a positive role in the activation of the RAS/MAPK signaling pathway [42], suggesting that STYK1 elevated the phosphorylation of BECN1 Ser90 through MAPK pathway. However, it is also worth investigating whether STYK1 can phosphorylate either ATG14, BECN1 or PIK3C3 directly in future studies.
The transmembrane domain of STYK1 has been reported as critical for STYK1 oligomerization and subsequent kinase activation autoinhibition [58]. However, our results showed that loss of the STYK1 transmembrane domain significantly inhibits the induction of autophagy, suggesting that the localization of STYK1 may play an essential role in regulating autophagy initiation. Indeed, the loss of the STYK1 transmembrane domain caused the distribution of the STYK1 protein in the nucleus [59]. Moreover, STYK1 has been reported to distribute in a dot and aggregation pattern (DP and AP, respectively), our investigation into the cellular colocalization of the autophagosome were mainly with the aggregation pattern of STYK1, which was also identified the most in cancer cells [59].
Basal autophagy plays an essential role in the maintenance of tissue homeostases, such as hepatocyte lipid accumulation and protein quality control [14]. For example, the receptor for RACK1 (encoded by gene GNB2/1), which is indispensable for the formation of the PtdIns3K-C1 complex in nutrient starvation conditions and has an effect on basal autophagy activity, prevents increased lipid storage in hepatocytes by promoting autophagy [37]. DACT1/DAPPER1, interacts and functions in the PtdIns3K-C1 complex, impairs basal autophagy and the accumulation of ubiquitinated aggregates in the central nervous system and liver tissues [16]. Our data also identify STYK1 depletion results in impaired basal autophagy activity. Furthermore, STYK1 depletion increased hepatocyte lipid accumulation and protein aggregates. As expected, in nutrient-rich conditions, RACK1 was precipitated by STYK1 (Figure 4). Those regulators or scaffold proteins of the PtdIns3K-C1 complex that promote basal autophagy activity are implicated in several autophagy-related pathological disorders. Indeed, STYK1 plays important roles in promoting the development of different types of cancer cells, but the underlying regulatory mechanisms are not well understood.
In some cases, there is a controversy between autophagy and cell proliferation, as well as tumor development, such as pancreatic cancer with activating KRAS mutations and high basal autophagy, autophagy plays an essential role in tumor progression [60]. Consistently, through gene set enrichment analysis (GSEA) using the cancer genome atlas (TCGA) database of pancreatic cancer, we found that STYK1 levels were correlated with the metabolism of several amino acids including glycine, serine, threonine, cysteine and methionine. STYK1 levels were also correlated with the increased metabolism of pyruvate and enrichment of genes essential for the citrate cycle (Fig. S1).
In summary, our study reveals a novel role of STYK1 in autophagy and identifies STYK1 modulating PtdIns3K-III activity by stabilizing BECN1, ATG14, PIK3C3 and PIK3R4 proteins in the assembly of the PtdIns3K-C1 complex for autophagosome formation. Future functional and structural studies would allow for an increased understanding of the cross-talk between STYK1 and autophagy initiation.
Materials and methods
Plasmids construction
DNA fragments encoding STYK1, BECN1, ATG14, PIK3C3, ZFYVE1, WIPI1, BCL2, RUBCN, PRKAA1 and PRKAA2 amplified by PCR were gifts from Prof. Jiahuai Han (Xiamen University, China). The PCR products of STYK1 and BECN1 were both cloned into pCMV-3 × Flag (Sigma, E4401), pCMV-Myc (Clontech, 631,604), and pcDNA3.0-3 × HA (Miaolingbio, P9394). The PCR product of ATG14 was cloned into pCMV-3 × Flag and pCMV-Myc. The PCR products of BCL2 and RUBCN were cloned into pCMV-3 × Flag. The PCR products of ZFYVE1, WIPI1, and STYK1 were cloned into pCMV-N1-EGFP (Clontech, 6085–1). pCMV-N1-ATG14 and pCMV-Myc-UVRAG were gifts from Prof. Yan Chen (Shanghai Institutes for Biological Sciences, China). GFP-LC3 was a gift from Prof. QingKenneth Wang (Huazhong University of Science and Technology, China). ptfLC3 (mammalian expression of rat LC3 fused to mRFP and GFP) was a gift from Prof. Tamotsu Yoshimori (Addgene, 21074). To make STYK1 mutants, a series of forwarding primers harboring the desired mutations were used in PCRs to generate the STYK1 K147R, Y24F, Y24D, Y191F, 191D, Y24F/Y191F, Y24D/Y191D, Y229F, Y327F and Y356F mutations. To make deletion mutations, overlap extension PCR was used to generate DNA sequences encoding STYK1 1–116, STYK1 1–378, STYK1 26–422, STYK1 116–378, STYK1 ΔTM, STYK1-ICD, STYK1 Δ116-203, STYK1 Δ204-290, STYK1 Δ291-378, ATG14 1–70, ATG14 71–180, ATG14 181–492, PIK3C3 1–293, PIK3C3 294–530, PIK3C3 531–887, BECN1 1–150, BECN1 1–280, BECN1 1–337, BECN1 151–244, BECN1 245–337, and BECN1 Δ245-337. To generate pCDNA3.0-STYK1-mCherry, two DNA fragments that encode STYK1 and mCherry were fused by PCR, mCherry was cloned from 7TGC and 7TGC was a gift from RoelNusse (Addgene, 24304). Lentiviral construct containing Flag-tagged human coding sequence of STYK1 was cloned into pCDH-CMV-MCS-EF1-Puro system, which was a gift from Dr. Xiaorong Zhang (Institute of Biophysics, Chinese Academy of Sciences, China). psPAX2 and pMD2.G were also gifts from Dr. Xiaorong Zhang. The plasmids were purified using the EndoFree Mini Plasmid Kit II (TIANGEN, DP118). All complete plasmids were verified by sequencing.
Antibodies and reagents
The following commercially available antibodies and the corresponding dilutions were used: anti-STYK1 (Abcam, ab97451; 1:1000 dilution), anti-ACTB/β-actin (Proteintech, 60008-1-Ig; 1:1000 dilution), anti-GAPDH (Proteintech, 60004-1-Ig; 1:1000 dilution), anti-LC3B (Proteintech, 18725-1-AP; 1:1000 dilution), anti-SQSTM1/p62 (Boster, BM4385; 1:1000 dilution), anti-Ubiquitin (Enzo, BML-PW8810; 1:1000 dilution), anti-mouse Flag (EMD Millipore, M1805-3L; 1:1000 dilution), anti-rabbit Flag (EMD Millipore, PM020A; 1:1000 dilution), anti-mouse HA (EMD Millipore, M180-3; 1:1000 dilution), anti-rabbit HA (Proteintech, 51064-2-AP; 1:1000 dilution), anti-mouse MYC (EMD Millipore, M047-3; 1:1000 dilution), anti-rabbit MYC (Sigma, SAB2702192; 1:1000 dilution), anti-p-PRKAA1/2-T172 (Cell Signaling Technology, 2535; 1:1000 dilution), anti-PRKAA2 (Boster, A30453; 1:500 dilution), anti-pEIF4EBP1-T37/46 (Cell Signaling Technology, 2855; 1:1000 dilution), anti-EIF4EBP1 (Proteintech, 60246-1-Ig; 1:1000 dilution), anti-p-ULK-S317 (Cell Signaling Technology, 12,753; 1:1000 dilution), anti-ULK1 (Boster, BM5170; 1:500 dilution), anti- ATG14/Barkor (Proteintech, 19,491-1-AP; 1:1000 dilution), anti-BECN1 (Proteintech, 11,306-1-AP; 1:1000 dilution), anti- PIK3C3/VPS34 (Proteintech, 10,828-1-AP; 1:1000 dilution), anti-RACK1 (Santa Cruz Biotechnology, sc-17,754; 1:500 dilution), anti-UVRAG (Proteintech, 19,571-1-AP; 1:1000 dilution), anti- PIK3R4/VPS15 (Santa Cruz Biotechnology, sc-100,798; 1:500 dilution), anti-RUBCN (Proteintech, 21,444-1-AP; 1:1000 dilution), anti-GFP (EMD Millipore, 598; 1:1000 dilution), anti-PtdIns3P (Echelon, Z-P003; 1:200 dilution), anti-BCL2 (Proteintech, 12,789-1-AP; 1:1000 dilution), anti-phosphotyrosine (Sigma, P4110; 1:1000 dilution), anti-p-BECN1-S90 (Cell Signaling Technology, 86,455; 1:1000 dilution), anti-p-BECN1-S93 (Cell Signaling Technology, 14,717; 1:1000 dilution). Commercially reagents used are as follows: chloroquine diphosphate salt (CQ; 10 μM; Sangon Biotech, A506569), rapamycin (10 nM; Selleck, S1039), torin1 (10 nM; MedChemExpress, HY-13,003), AMPK inhibitor compound C (10 μM; Selleck, S7306). Absolute dimethyl sulfoxide (DMSO; Sangon Biotech, A100231) was used as a solvent to make stock solutions, which were typically diluted at 1:1000 to obtain final solutions. Identical concentrations of DMSO were used as vehicle controls. Pharmacological agents were typically replenished every 3 days.
Cell culture, siRNAs, and transfection
LO2, HeLa, HEK293T, HepG2 and HPD E6-C7 cell lines were gifts from Dr. HaiMing Hu (Wuhan Institute of Virology, Chinese Academy of Sciences, China). The MCF-7 cell line was a gift from Prof. Xuan Cao (Tongji Medical College, Huazhong University of Science and Technology, China). SiHa and PANC-1 cells were obtained from the Cell Culture Center (Chinese Academy of Medical Science, TCHu113 and TCHu 98). Cells were cultured in high glucose (4500 mg/L) Dulbecco’s modified Eagle’s medium (DMEM; Hyclone, SH30022.01) containing 10% heat-inactivated fetal bovine serum (Biological Industries, 04-001-1ACS), 100 units/ml penicillin, 100 μg/ml streptomycin (Biosharp, BL505A) and 4 mM L-glutamine at 37°C with 5% CO2 in a humidified atmosphere. The medium was replaced every 2–3 d and the cell was subcultured and used for an experiment at 80–90% confluence. DNA transfection with GenJetVer.II (SignaGen, SL100489) and interference RNA with Lipofectamine 2000 (Invitrogen, 11,668,019) according to the manufacturer’s instructions. siRNA for STYK1 #1 was 5ʹ- CCAUCUUUCGAGCCAAUAUTT −3ʹ, #2 was 5ʹ- GGAUGGUCUUCUCUAUGAUTT −3ʹ, #3 was 5ʹ- GGUGGUACCUGAACUGUAUTT −3ʹ, siRNA for BECN1 was 5ʹ-CCGUGGAAUGGAAUGAGAUUAAUTT-3ʹ, siRNA for ULK1 was: 5ʹ-CGCGGUACCUCCAGAGCAATT-3ʹ. Starvation in this study was carried out to switch the culture medium from the complete medium to low glucose (1000 mg/L) DMEM medium without fetal bovine serum or EBSS (Hyclone, SH30024.01) medium.
Crispr/Cas9 mediated mutagenesis of zebrafish styk1b
Single guide RNAs (sgRNAs) targeting the second and third coding exon of zebrafish styk1b (ENSDART00000139664.1) were designed using the chop-chop website [61]. To generate the sgRNAs, the single-strand DNA (ssDNA) (122 bases) template was obtained by PCR complementation and amplification of full-length ssDNA oligonucleotides. Oligonucleotides up to 81 nucleotides were purchased from Sangon Biotech (Shanghai) using standard synthesis procedures (10 nmol concentration, purification with page method). The final PCR products were purified with a DNA clean up kit (CWBIO, CW2301M). The purified PCR products were confirmed by gel electrophoresis and Sanger sequencing by Sangon Biotech (Shanghai). For in vitro transcription of sgRNAs, 0.5 µg template DNA was used to generate sgRNAs using the MEGA short script ®T7 kit (ThermoFisher, AM1354) and purified by RNA Clean up kit (TIANGEN, DP412). The Cas9 Nuclease was purchased from New England (NEB, M0646). A mixture of sgRNA and Cas9 mRNA was injected into one-cell stage WT (AB line) embryos (sgRNA 150 pg/embryo and Cas9 mRNA 300 pg/embryo). The effect of CRISPR injection was confirmed by PCR and Sanger sequencing.
Qrt-PCR assay
Total RNA was extracted using Trizol reagent (ThermoFisher, 15,596,026) according to the manufacturer’s instructions and purified with RNeasy Min Elute Clean up kit (TIANGEN, DP412). RNAs were quantified using a NanoDrop 2000c instrument (ThermoFisher). cDNA was reverse transcribed using a HiScript cDNA synthesis kit (Vazyme, R323-01). Quantitative PCR was performed using the Universal SYBR qPCR Master Mix (Vazyme, Q511-02). The primers for styk1b were: Forward: 5ʹ- CATAGCACTGGACACAACCACT −3ʹ, Reverse: 5ʹ-TCCGTTAAAGTTCTCCGGCAGT-3ʹ. The primers for Gapdh were: Forward: 5ʹ- TCGGTCGCATTGGC −3ʹ, Reverse: 5ʹ-CCGCCTTCTGCCTTA-3ʹ.
Immunoprecipitation and western blotting
Cells or zebrafish larvae (10> larva/group) were washed twice with phosphate-buffered saline (PBS; Servicebio, WGSH30256-01) and lysed with RIPA lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100 [Sangon Biotech, 9002-93-1], 10 mM NaF, 1 mM EDTA and proteinase inhibitor cocktail [Biomake, B14001] and Halt phosphatase inhibitor cocktail [Thermofisher Scientific, 78,420]) for 30 min. The lysate was centrifuged at 12,000 × g for 30 min at 4°C, and the supernatant was incubated overnight with primary antibodies according to each individual experiment, and then with protein A/G agarose beads (Santa Cruz Biotechnology) for 2 h at 4°C. The beads were washed twice with PBS buffer, twice with RIPA lysis buffer and twice with TBST buffer (Servicebio, G0004). Protein concentration was measured using the Protein Assay kit (BioRad, 5,000,006), then the immunoprecipitate was eluted from the beads by adding and boiling in 2 × SDS loading buffer (100 mM Tris-HCl, pH 6.8, 4% [wt:vol] SDS, 200 mM dithiothreitol, 0.2% [wt:vol] bromophenol blue, and 20% [vol:vol] glycerol) for 8 min at 98°C and was then subjected to SDS-PAGE and western blotting. Western blotting was performed as described [62]. Briefly, cells were lysed in RIPA lysis for 30 min on ice and the lysate was centrifugated at 4°C. The supernatant was then removed to new 1.5 ml microcentrifuge tubes, boiled for 8 min at 98°C after adding 2 × SDS loading buffer and loaded on SDS-PAGE gels. For all western blotting, gels were blotted on 0.45-µm polyvinylidene fluoride membrane (PVDF; Millipore, IPFL85R). Transferred membranes were blocked in TBST 5% skimmed milk (Coolaber, CN7861) TBST buffer or TBS-T 3% BSA (Sangon Biotech, A600903-0005) for phospho-specific antibodies for 2 h. Primary antibodies were diluted in TBST buffer and incubated for either 1 h at room temperature or 4°C overnight. Secondary antibodies were also diluted in TBST buffer. Mild stripping buffer (2 M glycine [ThermoFisher, A37730IP] pH 2.2, 1% SDS, 10% Tween20 [Sangon Biotech, A100777]) was used for stripping antibodies from membranes. Detection was performed using supersignal west pico plus (Invitrogen, 34,580) according to the manufacturer’s instructions.
PIK3C3/VPS34 kinase assay
PIK3C3 activity was measured with quantitative and competitive ELISA assays according to the manufacturer’s manual (Echelon, K-3000). Briefly, PIK3C3 was immunoprecipitated using anti-ATG14 or anti-UVRAG antibody with or without STYK1 interfering RNA under nutrient-rich and starvation conditions. PIK3C3 was also obtained using ATG14 antibody after WT STYK1, STYK1Y191F or STYK1Y191D transfection respectively in normal, starvation and AICAR treatment conditions. Twenty-one µl kinase reaction buffer (100 mM HEPES [ThermoFisher, 15,630,080] pH 7.5, 300 mM NaCl, 2 mM CHAPS [ThermoFisher, 28,300], 10 mM MnCl2, 2 mM DTT, 100 µM ATP [Sangon Biotech, A600020]) and 4 µl of 500 µM phosphatidylinositol substrate (Echelon, K-3000) were added to the beads immunoprecipitated with PIK3C3 and incubated at room temperature for 2 h. The reaction mixture was quenched with 5 µl of 100 mM EDTA diluted with 90 µl H2O, and then added 80 µl PtdIns3P detection buffer (provided by the Kit, K-3004). After the PtdIns3K reactions were complete and quenched, reaction products and PtdIns3P detector protein (provided by the Kit) were then added to the PtdIns3P-coated microplate, for competitive binding to the PtdIns3P detector protein. The amount of PtdIns3P detector protein bound to the plate was determined through colorimetric detection of absorbance at 450 nm. The concentration of PtdIns3P in the reaction mixture was calculated as reversed to the amount of PtdIns3P detector protein bound to the plate.
Immunostaining and confocal microscopy
HeLa cells transfected with the appropriate plasmids were grown on glass chambers at 60% density and incubated in normal medium, serum-free medium for 12 h or EBSS medium at the indicated time, either untreated or treated with 10 μM CQ for 2 h. Cells were then fixed with 4% paraformaldehyde in PBS for 10 min. After cells were permeabilized with 0.5% Triton X-100 in PBS for 15 min, samples were washed three times with PBS and blocked with 10% goat serum (Sangon Biotech, E510009), 0.1% Triton X-100 in PBS for 2 h at room temperature. Cells were then incubated with 1:100 or 1:200 dilution of corresponding primary antibodies overnight at 4°C and 1:100 dilution of fluorescence-labeled secondary antibodies diluted in immunostaining buffer (2% FBS plus1% BSA in PBS) for 2 h. DAPI (Solarbio, C0065) was used for nuclei staining. Finally, cells were washed again with PBS and mounted on glass slides and examined with a confocal laser-scanning microscope (Leica SP8, Wetzlar, Germany) using a 63× oil immersion objective. Data analysis was performed using the Leica LAS AF Lite software. The numbers of GFP-LC3, ULK1, ATG14-GFP, ZFYVE1-GFP and WIPI1-GFP puncta per cell were counted in 10 non-overlapping fields.
GST affinity-isolation assay
The cDNA encoding STYK1 ICD (aa 26–422) was cloned into pGEX4T-1 (GE, 27-4580-01) and pET-28a (EMD Biosciences, 69,864–3). The cDNAs encoding ATG14 (aa 181–492), BECN1 (aa 151–450) and PIK3C3 (aa 1–293, 531–887) were cloned into pGEX4T-1. GST, GST-STYK1-ICD, His-STYK1-ICD, truncated ATG14 (aa 181–492), BECN1 (aa 151–450) and PIK3C3 (aa 1–293, 531–887) proteins were then purified from the bacteria cells (BL21 strain; TaKaRa, 9126) that were transformed with pGEX4T-1 and respective pGEX4T1 expression vector, using standard glutathione-agarose beads according to the manufacturer’s instruction (Thermofisher, 16,100). For the direct interaction assay, we use externally expressed truncated GST- ATG14 181–492, GST-BECN1 151–450, GST-PIK3C3 1–293 and 531–887 to incubate with externally expressed His-STYK1, and then detect the STYK1 signal by western blotting assay.
GSEA analysis
Global mRNA expression profiles of human liver cancer tissue obtained from The Cancer Genome Atlas (TCGA) database were subject to Gene Set Enrichment Analysis (GSEA) using the GSEA v2.2.4 software (http://www.broadinstitute.org/gsea/) to identify the association of STYK1 expression with cell metabolism signature.
In vitro tyrosine kinase activity assay
The HEK293T cells cultured in 10 cm dishes were transiently transfected with STYK1 and STYK1 mutants for 48 h. The cells were collected and washed with cold PBS and lysed in lysis buffer supplemented with protease and phosphatase inhibitors. The cell lysates were subjected to IP assay using the Flag antibody, and the immunoprecipitate was used for determining the tyrosine kinase activity using the universal tyrosine kinase assay kit (TaKaRa, MK410). Briefly, 40 μl of the diluted immunoprecipitated sample was incubated for 30 min at 37°C with substrate peptides that were bound to streptavidin present a microtiter plate containing 10 μl of kinase reaction buffer with 1 mM ATP in each well. Then, 50 μl of anti-phosphotyrosine (PY20)-HRP solution was added into each well and incubated for 30 min at 37°C. After washing the sample four times, 100 μl of TMB substrates was also added, and the samples were incubated for up to 15 minutes. The reaction was stopped by adding 2 M sulfuric acid (TaKaRa, MK410). The absorbance in each well was measured using a spectrophotometric plate reader (BioTek, ELx800) at a wavelength of 450 nm.
Cell viability detection
Cell viability was determined by MTT assay. Briefly, HepG2 cells (1 × 105 cells/well) were seeded into 12 – well plates for 12 h to adhere. After the replacement of the complete culture medium to EBSS with or without rapamycin (Rap, 10 nM), brefeldin A (Bre A, 5 mM), CQ (10 μM) or the control solution, cells were stained at the indicated time points with 100 μl sterile MTT dye (0.5 mg/ml; Sigma, M2128) for 4 h at 37°C, followed by removal of the culture medium and the addition of 150 μl DMSO (Sigma, W387520). The number of viable cells was assessed by measurement of the absorbance at 490 nm by a microplate reader. All experiments were performed in triplicate.
Statistical analysis
All experiments were independently performed at least three times. All statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad, La Jolla, CA, USA). All data are presented as mean ± SD (standard deviation) from triplicates. P values <0.05 were statistically significant. Statistical analysis was done using paired Student’s t-test; ***, ** and * indicate P < 0.001, P < 0.01 and P < 0.05, respectively.
Supplementary Material
Acknowledgments
We are grateful to Prof. Daniel Klionsky for sharing the GFP-Lc3 transgenic zebrafish line. We also thank Hubei University of Technology and Wuhan University for financial and equipment support to this research.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was supported by the National Natural Science Foundation of China (31871420 and 81602448 to J.F.T., 31701228 to C.F.Z., 31871176 and 81570648 to X.Z.C.), Wuhan Science and Technology Project (2019020701011475 to J.F.T).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Galluzzi L, Pietrocola F, Levine B, et al. Metabolic control of autophagy. Cell. 2014;159(6):1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Maejima Y, Kyoi S, Zhai P, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19(11):1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ohsumi Y.Historical landmarks of autophagy research. Cell Res. 2014;24(1):9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: a troika governing cancer and its treatment. Cell. 2016;166(2):288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shchors K, Massaras A, Hanahan D. Dual targeting of the autophagic regulatory circuitry in gliomas with repurposed drugs elicits cell-lethal autophagy and therapeutic benefit. Cancer Cell. 2015;28(4):456–471. [DOI] [PubMed] [Google Scholar]
- [8].Kihara A, Noda T, Ishihara N, et al. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152(3):519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Egan D, Kim J, Shaw RJ, et al. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7(6):643–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhou C, Wang M, Yang J, et al. Integral membrane protein 2A inhibits cell growth in human breast cancer via enhancing autophagy induction. Cell Commun Signal. 2019;17(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–774. [DOI] [PubMed] [Google Scholar]
- [13].Sun Q, Zhang J, Fan W, et al. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J Biol Chem. 2011;286(1):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64(6):1994–2014. [DOI] [PubMed] [Google Scholar]
- [15].Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ma B, Cao W, Li W, et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014;24(8):912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rostislavleva K, Soler N, Ohashi Y, et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science. 2015;350(6257):aac7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Young LN, Cho K, Lawrence R, et al. Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy. Proc Natl Acad Sci U S A. 2016;113(29):8224–8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu J, He L, Behrends C, et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun. 2014;5:3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xu DQ, Wang Z, Wang CY, et al. PAQR3 controls autophagy by integrating AMPK signaling to enhance ATG14L-associated PI3K activity. Embo J. 2016;35(5):496–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu L, Yu XZ, Li TS, et al. A novel protein tyrosine kinase NOK that shares homology with platelet- derived growth factor/fibroblast growth factor receptors induces tumorigenesis and metastasis in nude mice. Cancer Res. 2004;64(10):3491–3499. [DOI] [PubMed] [Google Scholar]
- [22].Wang Z, Qu L, Deng B, et al. STYK1 promotes epithelial-mesenchymal transition and tumor metastasis in human hepatocellular carcinoma through MEK/ERK and PI3K/AKT signaling. Sci Rep. 2016;6:33205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu Y, Li T, Hu D, et al. NOK/STYK1 promotes the genesis and remodeling of blood and lymphatic vessels during tumor progression. Biochem Biophys Res Commun. 2016;478(1):254–259. [DOI] [PubMed] [Google Scholar]
- [24].Chen Y, Li YH, Chen XP, et al. Point mutation at single tyrosine residue of novel oncogene NOK abrogates tumorigenesis in nude mice. Cancer Res. 2005;65(23):10838–10846. [DOI] [PubMed] [Google Scholar]
- [25].Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25(5):1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wei Y, Zou Z, Becker N, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1269–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. [DOI] [PubMed] [Google Scholar]
- [29].He C, Bartholomew CR, Zhou W, et al. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy. 2009;5(4):520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hawley SA, Davison M, Woods A, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271(44):27879–27887. [DOI] [PubMed] [Google Scholar]
- [33].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Aguilera MO, Beron W, Colombo MI. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy. 2012;8(11):1590–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zientara-Rytter K, Subramani S. Role of actin in shaping autophagosomes. Autophagy. 2016;12(12):2512–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhao Y, Wang Q, Qiu G, et al. RACK1 promotes autophagy by enhancing the Atg14L-Beclin 1-Vps34-Vps15 complex formation upon phosphorylation by AMPK. Cell Rep. 2015;13(7):1407–1417. [DOI] [PubMed] [Google Scholar]
- [38].Ohashi Y, Soler N, Garcia Ortegon M, et al. Characterization of Atg38 and NRBF2, a fifth subunit of the autophagic Vps34/PIK3C3 complex. Autophagy. 2016;12(11):2129–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. [DOI] [PubMed] [Google Scholar]
- [40].Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186(6):773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li YH, Wang YY, Zhong S, et al. Transmembrane helix of novel oncogene with kinase-domain (NOK) influences its oligomerization and limits the activation of RAS/MAPK signaling. Mol Cells. 2009;27(1):39–45. [DOI] [PubMed] [Google Scholar]
- [43].Hu J, Liu J, Ghirlando R, et al. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol Cell. 2003;12(6):1379–1389. [DOI] [PubMed] [Google Scholar]
- [44].Chen X, Vinkemeier U, Zhao Y, et al. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93(5):827–839. [DOI] [PubMed] [Google Scholar]
- [45].Menon MB, Dhamija S. Beclin 1 phosphorylation - at the center of autophagy regulation. Front Cell Dev Biol. 2018;6:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang D, Wang W, Sun X, et al. AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy. 2016;12(9):1447–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. Embo J. 2015;34(7):856–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Carpio MA, Michaud M, Zhou W, et al. BCL-2 family member BOK promotes apoptosis in response to endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2015;112(23):7201–7206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Karsli-Uzunbas G, Guo JY, Price S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4(8):914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Martinez-Lopez N, Garcia-Macia M, Sahu S, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23(1):113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Szeto J, Kaniuk NA, Canadien V, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2(3):189–199. [DOI] [PubMed] [Google Scholar]
- [52].McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57(1):39–54. [DOI] [PubMed] [Google Scholar]
- [53].Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5(6):464–471. [DOI] [PubMed] [Google Scholar]
- [54].Cao X, Kaneko T, Li JS, et al. A phosphorylation switch controls the spatiotemporal activation of Rho GTPases in directional cell migration. Nat Commun. 2015;6:7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mohammadi M, Schlessinger J, Hubbard SR. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell. 1996;86(4):577–587. [DOI] [PubMed] [Google Scholar]
- [56].Pitulescu ME, Adams RH. Regulation of signaling interactions and receptor endocytosis in growing blood vessels. Cell Adh Migr. 2014;8(4):366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ohashi Y, Tremel S, Williams RL. VPS34 complexes from a structural perspective. J Lipid Res. 2019; J Lipid Res. 60(2):229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wei Y, An Z, Zou Z, et al. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ding X, Jiang QB, Li R, et al. NOK/STYK1 has a strong tendency towards forming aggregates and colocalises with epidermal growth factor receptor in endosomes. Biochem Biophys Res Commun. 2012;421(3):468–473. [DOI] [PubMed] [Google Scholar]
- [60].Rosenfeldt MT, O’Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. [DOI] [PubMed] [Google Scholar]
- [61].Labun K, Montague TG, Gagnon JA, et al. CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016;44(W1):W272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhou C, Yu J, Wang M, et al. Identification of glycerol-3-phosphate dehydrogenase 1 as a tumour suppressor in human breast cancer. Oncotarget. 2017;8(60):101309-101324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.