

ABSTRACT
Interferon-induced BST2 (bone marrow stromal cell antigen 2) inhibits viral replication by tethering enveloped virions to the cell surface to restrict viral release and by inducing the NFKB-dependent antiviral immune response. However, the mechanism by which BST2 uses the selective autophagy pathway to inhibit viral replication is poorly understood. In this study, we showed that BST2 expression was significantly increased during porcine epidemic diarrhea virus (PEDV) infection of Vero cells by IRF1 targeting its promoter. We also showed that BST2 suppressed PEDV replication by binding and degrading the PEDV-encoded nucleocapsid (N) protein. The downregulation of N protein was blocked by macroautophagy/autophagy inhibitors but not a proteasome inhibitor, implying that the N protein was degraded via the selective autophagy pathway. Both the BST2 and N protein interacted with the E3 ubiquitin ligase MARCHF8/MARCH8 and the cargo receptor CALCOCO2/NDP52, and the ubiquitination of N protein was necessary for the degradation of N mediated by the BST2-MARCHF8 axis. The knockdown of MARCHF8 or ATG5 with small interfering RNAs blocked the selective autophagy pathway, rescued the protein abundance of PEDV N in 293T cells, and prevented the inhibition of PEDV replication by BST2 in Vero cells. Together, our data demonstrate the novel mechanism of BST2-mediated virus restriction, in which BST2 recruits MARCHF8 to catalyze the ubiquitination of the PEDV N protein. The ubiquitinated N protein is then recognized by CALCOCO2/NDP52, which delivers it to autolysosome for degradation through the selective autophagy pathway.
Abbreviations: 3MA: 3-methyladenine; ATG: autophagy-related; Baf A1: bafilomycin A1; BST2: bone marrow stromal cell antigen 2; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CC: coiled-coil; ChIP: chromatin immunoprecipitation; Co-IP: co-immunoprecipitation; CQ: chloroquine; CT: cytoplasmic tail; DAPI: 4′,6-diamidino-2-phenylindole; GPI: glycosyl-phosphatidylinositol; hpi: hours post infection; IRF1: interferon regulatory factor 1; ISG: IFN-stimulated gene; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MARCHF8/MARCH8: membrane-associated ring-CH-type finger 8; MOI: multiplicity of infection; N protein: nucleocapsid protein; PED: porcine epidemic diarrhea; PEDV: porcine epidemic diarrhea virus; RT: room temperature; siRNA: small interfering RNA; STAT: signal transducer and activator of transcription; TCID50: 50% tissue culture infectious doses; TM: transmembrane.
KEYWORDS: BST2, CALCOCO2/NDP52, IRF1, MARCHF8/MARCH8, nucleocapsid protein, PEDV, replication, selective autophagy
Introduction
Porcine epidemic diarrhea virus (PEDV), a single-stranded positive-sense RNA virus, belongs to the genus Alphacoronavirus in the family Coronaviridae [1]. Porcine epidemic diarrhea (PED) was first recognized in England in 1971 [2], and a highly virulent PEDV emerged in China in 2010 [3], causing acute diarrhea, vomiting, and high mortality rates in neonatal piglets. This highly virulent PEDV was detected and spread rapidly in the USA in May 2013, causing enormous economic losses to the swine industry worldwide [4 5–6]. The viral genome is approximately 28 kb long and encodes 2 polyproteins (pp1a and pp1ab), an accessory protein (open reading frame 3, ORF3), and 4 structural proteins (spike, S; envelope, E; membrane, M; and nucleocapsid, N) [7,8]. The innate immune response is the first line of defense against viral invasion. Consequently, many viruses, such as PEDV, have evolved complex evasion strategies to modulate the host innate immune response during infection. It has been reported that about 10 PEDV-encoded proteins suppress the IFN (interferon) signaling pathway, including both nonstructural proteins and structural proteins [9 10 12–13]. Although many studies have investigated the pathogenesis and immune evasion strategies of PEDV, the underlying mechanisms of PEDV replication, and the innate immune response to it are still unclear.
In eukaryotic cells, macroautophagy/autophagy is a major degradative process that maintains cellular homeostasis through the degradation and recycling of damaged organelles and misfolded or long-lived cytoplasmic proteins and is mediated by a unique double-membrane autophagosome [14–16]. It is activated by many intracellular and extracellular stresses, including damaged organelles, cellular starvation, endoplasmic reticulum (ER) stress, and viral infection [17 18–19]. During selective autophagy, damaged organelles or proteins are modified with ubiquitin and then recognized by cargo-specific autophagy receptors, such as SQSTM1/p62, OPTN (optineurin), NBR1 (NBR1, autophagy cargo receptor) and CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2). The complexes of cargo receptors and specific substrates interact directly with the Atg8-family proteins and are sequestered within double-membrane vesicles called “autophagosomes”, which fuse with lysosomes to degrade their engulfed contents [20 21–23]. As well as the physiological functions of autophagy, it plays an important role in viral replication [24,25]. For example, influenza A virus and porcine reproductive and respiratory syndrome virus infections can trigger autophagosome formation, but prevent the fusion of lysosomes and autophagosomes, benefitting viral replication through the accumulation of viral RNA and proteins [26,27]. However, herpes simplex virus replication is inhibited by autophagy through EIF2AK2/protein kinase R-dependent autophagic degradation [28], and the replication of zika virus is restricted to mature neurons by autophagy, through its degradation by RELA/NFKB-dependent STING autophagy [29]. Autophagy during the viral life cycle increases not only viral replication, but also inhibits viral proliferation at different growth phases. For instance, influenza virus replication is significantly enhanced in the early phase of infection and is inhibited in the late phase of infection by autophagy [30]. It has been demonstrated that PEDV utilizes autophagy to facilitate its replication in Vero cells [31]. However, another study suggested that rapamycin-induced autophagy restricted PEDV infectivity in porcine intestinal epithelial cells (IECs) [32]. Therefore, the effects of autophagy on PEDV replication may differ in different cells.
BST2/tetherin/CD317/HM1.24 (bone marrow stromal cell antigen 2) is an IFN-induced type II transmembrane protein consisting of an N-terminal cytoplasmic tail (CT) domain, a transmembrane (TM) domain, a coiled-coil (CC) ectodomain, and a C-terminal glycosyl-phosphatidylinositol (GPI) anchor [33,34]. Studies have shown that BST2 inhibits the release of a large number of enveloped viruses, including retroviruses [35,36], rhabdoviruses [37], and herpesviruses [38,39]. BST2 is also reported to inhibit viral reproduction by activating the proinflammatory transcription factor NFKB through the recruitment of TRAF2 and/or TRAF6, TAB and the mitogen-activated kinase MAP3K7/TAK1 [40,41]. BST2-deficient mice are more susceptible to viral infection than normal mice, indicating that BST2 plays a direct antiviral role in vivo [42]. Evidence has emerged that BST2 recruits the E3 ubiquitin ligase MARCHF8 (membrane-associated ring-CH-type finger 8) to catalyze the ubiquitin chains to MAVS, which is then recognized by the cargo receptor CALCOCO2, which delivers MAVS to autolysosomes for degradation, thereby inhibiting type I IFN signaling in a negative feedback loop [20]. In this study, we showed that BST2, an intrinsic antiviral protein, inhibited PEDV replication by targeting and degrading the PEDV N protein through selective autophagy. Upon PEDV infection, BST2 was upregulated by IRF1 (interferon regulatory factor 1) and recruited the E3 ubiquitin ligase MARCHF8 to catalyze the ubiquitination of the PEDV N protein, thereby causing the cargo receptor CALCOCO2 to deliver the N protein to autophagosomes for selective degradation. Our findings demonstrate a new antiviral mechanism of BST2, by which it suppresses PEDV replication through selective autophagy.
Results
PEDV infection upregulates the expression of BST2 in vero and LLC-PK1 cells
We screened potential proteins for their ability to inhibit PEDV replication using the proteomics approach of isobaric tagging for relative and absolute quantitation (iTRAQ). Vero cells were collected and assayed after infection with PEDV (strain JS-2013) at an MOI (multiplicity of infection) of 1, as previously described [43]. We found that an IFN-inducible restriction factor, BST2, was upregulated in Vero cells during PEDV infection. To confirm the expression of BST2, Vero cells (Figure 1A and B) and porcine kidney cells (LLC-PK1) (Figure 1C and D) were infected with PEDV at an MOI of 1 and harvested at the indicated times. The cell lysates were analyzed with western blotting and quantitative real-time PCR (qRT-PCR). The protein and mRNA levels of BST2 were both upregulated in the Vero cells and LLC-PK1 cells compared with their levels in uninfected cells. These results indicate that BST2 expression is upregulated by PEDV infection in Vero cells and LLC-PK1 cells.
Figure 1.
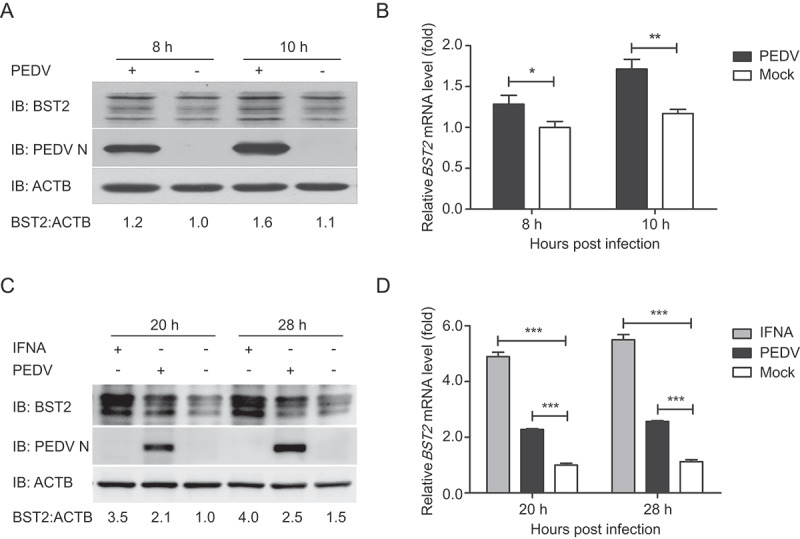
PEDV infection upregulated BST2 expression in Vero cells and LLC-PK1 cells. (A) Vero cells were infected or mock-infected with PEDV at an MOI of 1 and harvested at the indicated times. The expression of BST2 and PEDV N proteins were analyzed by western blotting. ACTB/β-Actin was used as the sample loading control. (B) BST2 mRNA levels in the same samples (A) were analyzed by real-time PCR. (C) LLC-PK1 cells were treated with universal type I interferon (1,000 U/mL) or infected with PEDV at an MOI of 1 and harvested at the indicated times. The lysates were analyzed by western blotting. (D) BST2 mRNA levels in the same samples (C) were analyzed by real-time PCR. Data are means ± SD of triplicate samples. *p < 0.05, **p < 0.01, ***p < 0.001 (two-tailed Student’s t test).
IRF1 plays a direct role in the transcriptional regulation of BST2
The expression of BST2, an IFN-inducible gene, can be induced by multiple stimuli. The promoter of the human BST2 gene contains binding sites for the transcription factors IRF9/ISGF3G (interferon regulatory factor 9), IRF1, and STAT3 (signal transducer and activator of transcription 3) [34]. Some IFN-independent stimulants of BST2 expression have also been reported, such as virus-activated IRF7, which induces the expression of BST2 independently of IFN signaling [44]. To investigate the boundaries of the minimal porcine BST2 promoter, we amplified 1906 base pairs (bp) of the BST2 promoter sequence (Table S2) and cloned it into a luciferase vector (pGL3-Basic) (Fig. S1A). A series of truncated promoters (designated M1-M9) were cloned into the luciferase vector, and their ability to direct luciferase expression in 293T cells was tested. The promoter deletion mutants containing nucleotides from −243 to −1 (M4, M7, and M9) induced the same luciferase expression as the full-length promoter (Fig. S1A). In contrast, the constructs that lacked the sequence from −243 to −1 of the BST2 promoter induced very low or undetectable basal luciferase activity. These results suggest that the BST2 core promoter region lies at positions −243 to −1. To confirm the boundaries of the minimal BST2 promoter, a series of truncated promoters (designated D1-D8), based on the sequence of BST2 promoter nucleotides −243 to −1, were cloned into the luciferase vector and the luciferase activity induced was measured. The constructs containing nucleotides from −182 to −138 (D1, D2, D5, D6, and D7) showed increased luciferase expression (Figure 2A). Together, these results indicate that the boundaries of the minimal BST2 core promoter are at positions −182 to −138.
Figure 2.
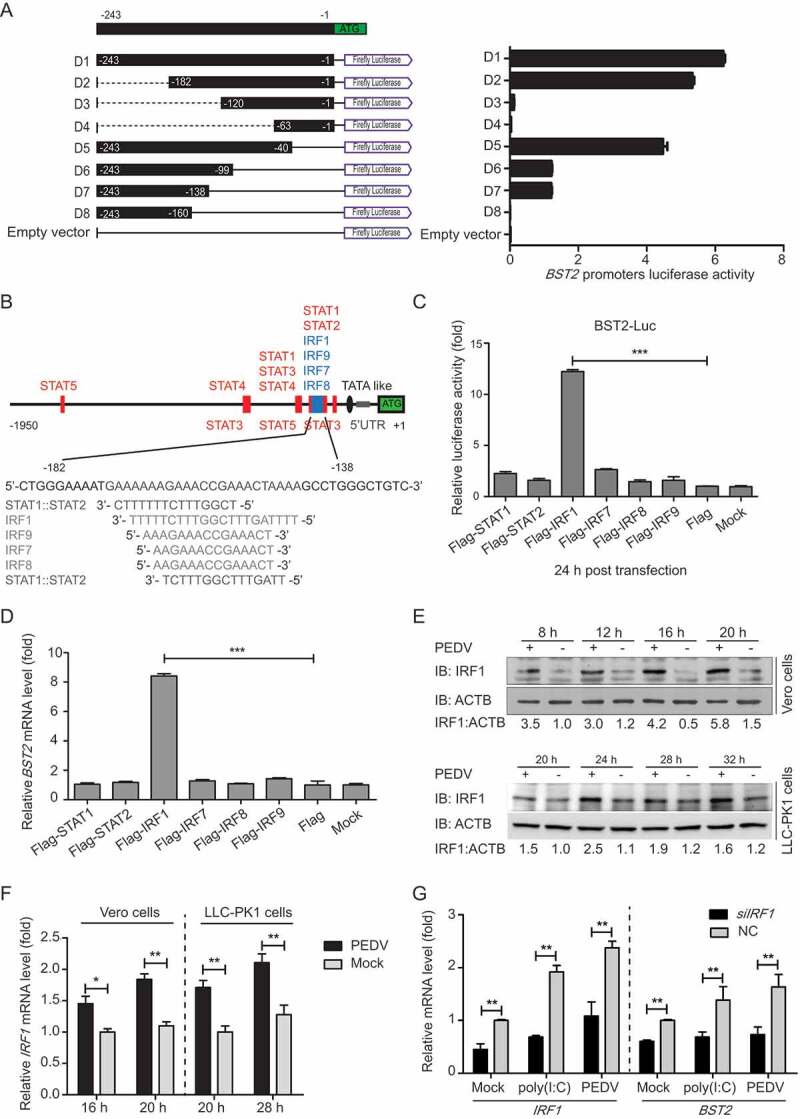
IRF1 directly controls the transcription of BST2. (A) 293T cells were co-transfected with a series of truncated BST2 promoter constructs (−243 to −1, D1 to D8), together with Renilla luciferase reporter vector (pRL-TK-luc), and were analyzed for dual luciferase activity. (B) Regulatory elements in the BST2 promoter region were predicted with the JASPAR vertebrate database (http://jaspar.genereg.net/). Red indicates putative regulatory elements for STAT (signal transducer and activator of transcription), and blue indicates those for IRF (IFN regulatory factor). (C) 293T cells were transfected with BST2 promoter-driven luciferase vector, pRL-TK-luc vector, and plasmids encoding Flag-tagged putative transcription factors (Flag-STAT1, Flag-STAT2, Flag-IRF1, Flag-IRF7, Flag-IRF8, or Flag-IRF9). Samples were collected at 24 h post-transfection and analyzed for dual luciferase activity. (D) 293T cells were transfected with plasmid encoding Flag-STAT1, Flag-STAT2, Flag-IRF1, Flag-IRF7, Flag-IRF8, or Flag-IRF9, followed by real-time PCR to measure the levels of BST2 transcription. (E and F) Vero cells and LLC-PK1 cells were infected or mock-infected with PEDV at an MOI of 0.01, and harvested at the indicated times. The expression of IRF1 was analyzed with western blotting and real-time PCR. ACTB was used as the sample loading control. (G) Vero cells were transfected with IRF1 siRNA or negative control siRNA. The cells were then treated with poly (I:C) (low molecular weight) (10 mg/mL) or PEDV (MOI = 0.01) and the transcription levels of IRF1 and BST2 were analyzed with real-time PCR. Data are means ± SD of triplicate samples. *p < 0.05, **p < 0.01, ***p < 0.001 (two-tailed Student’s t test).
Studies have demonstrated that the human BST2 promoter region contains multiple cis-acting regulatory elements for transcription factors STATs, IRFs, NFKB, and NFAT [38]. To analyze the transcriptional regulation of the porcine BST2 gene, we examined the promoter for potential transcription factor binding sites (TFBS) using the JASPAR vertebrate database (http://jaspar.genereg.net/) [45]. We found that the minimal BST2 core promoter region, spanning nucleotide positions −182 to −138, contains several TFBS, including STAT1-, STAT2-, IRF1-, IRF7-, IRF8-, and IRF9-binding sites (Figure 2B). To assess the effects of different transcription factors on the BST2 promoter in directing the expression of the gene, sequences encoding all the putative transcription factors were cloned into a mammalian expression vector and transfected with the BST2 promoter-driven luciferase vector to test their ability to direct luciferase expression in 293T cells. Cells overexpressing STAT1, STAT2, IRF7, IRF8, or IRF9 protein showed very low luciferase activity. In contrast, cells overexpressing IRF1 showed significantly increased luciferase expression from the BST2 promoter (Figure 2C). When we overexpressed the putative transcription factors to investigate the inducible expression of endogenous BST2, we found that only constructs encoding IRF1 activated the transcription of the BST2 gene more than 8-fold compared with its transcription in 293T cells transfected with the empty vector (Figure 2D). To confirm, a chromatin immunoprecipitation (ChIP) assay was performed with Flag-IRF1 to immunoprecipitate the BST2 core promoter region. The results showed that IRF1 directly binds to the BST2 core promoter region (Fig. S1B). We also observed that the abundances of IRF1 protein and mRNA were upregulated during PEDV infection (Figure 2E and F). To confirm that IRF1 is involved in the regulation of BST2 expression, we selected small interfering RNAs (siRNAs) that targeted the IRF1 mRNA sequence. A qRT-PCR analysis revealed that the IRF1 mRNA level was significantly reduced in the Vero cells transfected with IRF1 siRNA, with a concomitant reduction in BST2 expression (Figure 2G). Consistent with this, treatment with intracellular (IC) poly(I:C) (low molecular weight) or PEDV induced endogenous IRF1 and BST2 expression in the cells, and the induced endogenous gene expression was also reduced by IRF1 siRNA (Figure 2G). Overall, these results indicate that IRF1 plays a direct role in inducing BST2 expression by binding to the BST2 promoter.
BST2 inhibits PEDV replication in vero and LLC-PK1 cells
To investigate whether BST2 inhibits PEDV replication, the porcine BST2 (pBST2) and simian BST2 (mBST2) proteins were overexpressed in Vero cells for 24 h, and the cells were then infected with PEDV at an MOI of 0.01. The cells and supernatants of the infected cell cultures were collected at various time points and assayed for the PEDV viral loads. The PEDV N protein levels were significantly lower in the Vero cells previously transfected with Flag-pBST2 or Flag-mBST2 than in the cells previously transfected with the control vector and transfection reagent (Figure 3A). As expected, the viral titers in the supernatants of the Vero cells overexpressing pBST2 or mBST2 were significantly lower than those in the cells transfected with the empty vector (Figure 3B). To confirm these results, we synthesized and selected siRNAs targeting mBST2 (Fig. S2A). Vero cells were transfected with mBST2 siRNA and infected with PEDV at an MOI of 0.01. The viral yield was measured at 16 hpi with western blotting (Figure 3C), qPCR (Figure 3D), and the median tissue culture infective dose TCID50 (Fig. S2B). As expected, the viral titers were significantly higher in the Vero cells transfected with the mBST2 siRNA. We also observed that pBST2 overexpression inhibited PEDV replication while increased by BST2 silencing in LLC-PK1 cells (Figure 3E and F). Together, these results indicate that BST2 plays a role in the inhibition of PEDV replication.
Figure 3.
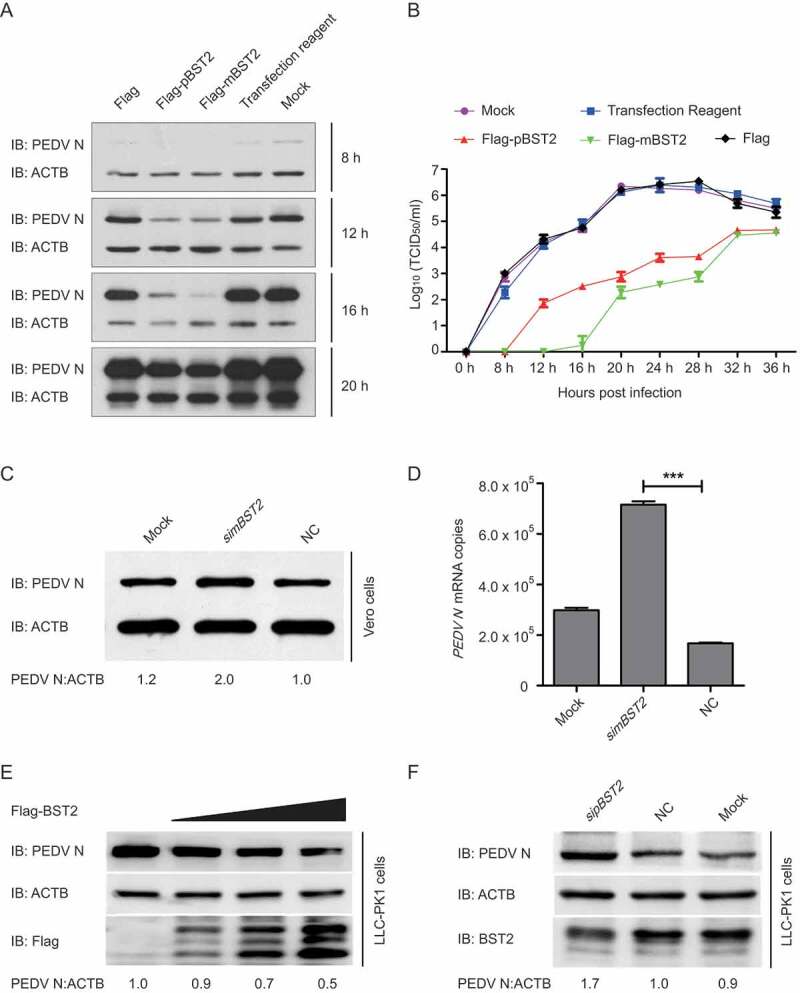
BST2 inhibits PEDV replication in Vero and LLC-PK1 cells. (A) Vero cells were transfected with plasmid encoding Flag-pBST2, Flag-mBST2, or the Flag tag. At 24 h post-transfection, the cells were infected with PEDV at an MOI of 0.01 and harvested at the indicated times. PEDV N protein expression was analyzed with western blotting. ACTB was used as the sample loading control. (B) PEDV titers in the culture supernatants of the Vero cells treated described in (A) were measured as TCID50. (C and D) Vero cells were transfected with mBST2 siRNA or negative control siRNA. At 24 h post-transfection, the cells were infected with PEDV at an MOI of 0.01, and the cell lysates and culture supernatants were then collected for analysis of PEDV N protein expression and viral RNA levels with western blotting and real-time PCR, respectively. (E and F) LLC-PK1 cells were transfected with increasing concentrations of a vector expressing Flag-BST2 (wedge) or pBST2 siRNA. At 24 h post-transfection, the cells were infected with PEDV at an MOI of 1, and the cell lysates were collected for analysis of PEDV N protein expression with western blotting. ACTB was used as the sample loading control. Data are means ± SD of triplicate samples. *p < 0.05, **p < 0.01, ***p < 0.001 (two-tailed Student’s t test).
BST2 interacts with PEDV n protein
It has been reported that BST2 physically tethers enveloped viruses to inhibit the release of budding virions from infected cells [35]. To determine the molecular mechanisms by which BST2 suppresses PEDV replication, we performed a coimmunoprecipitation (co-IP) analysis. We found that BST2 protein efficiently coimmunoprecipitated with PEDV, which was detected with a monoclonal antibody (mAb) directed against the PEDV N protein (Fig. S3). We then cloned the genes for 5 PEDV structural proteins (S1, S2, E, M, and N) into the mammalian expression vector pCAGGS and co-transfected cells with the individual plasmids together with a plasmid encoding Flag-BST2 to identify the PEDV structural protein that interacts directly with BST2. The co-IP analysis showed that only the hemagglutinin (HA)-tagged N protein interacted efficiently with Flag-BST2 (Figure 4A). GST pull-down assay also verified the binding between PEDV N and BST2. GST-fused to the PEDV N (GST-N) bound to the BST2, but GST protein did not (Figure 4B), indicating that PEDV N protein directly binds to BST2. We next investigated the colocalization of BST2 and the PEDV N protein with confocal microscopy. 293T cells were co-transfected with plasmids encoding enhanced green fluorescent protein (EGFP)-BST2 (green fluorescence) and mCherry-N (red fluorescence), and protein localization were examined after 24 h. A confocal immunofluorescence assay showed that the BST2 and N proteins colocalized in the cytoplasm (Figure 4C). In summary, these data demonstrate that BST2 coimmunoprecipitates with the PEDV N protein.
Figure 4.
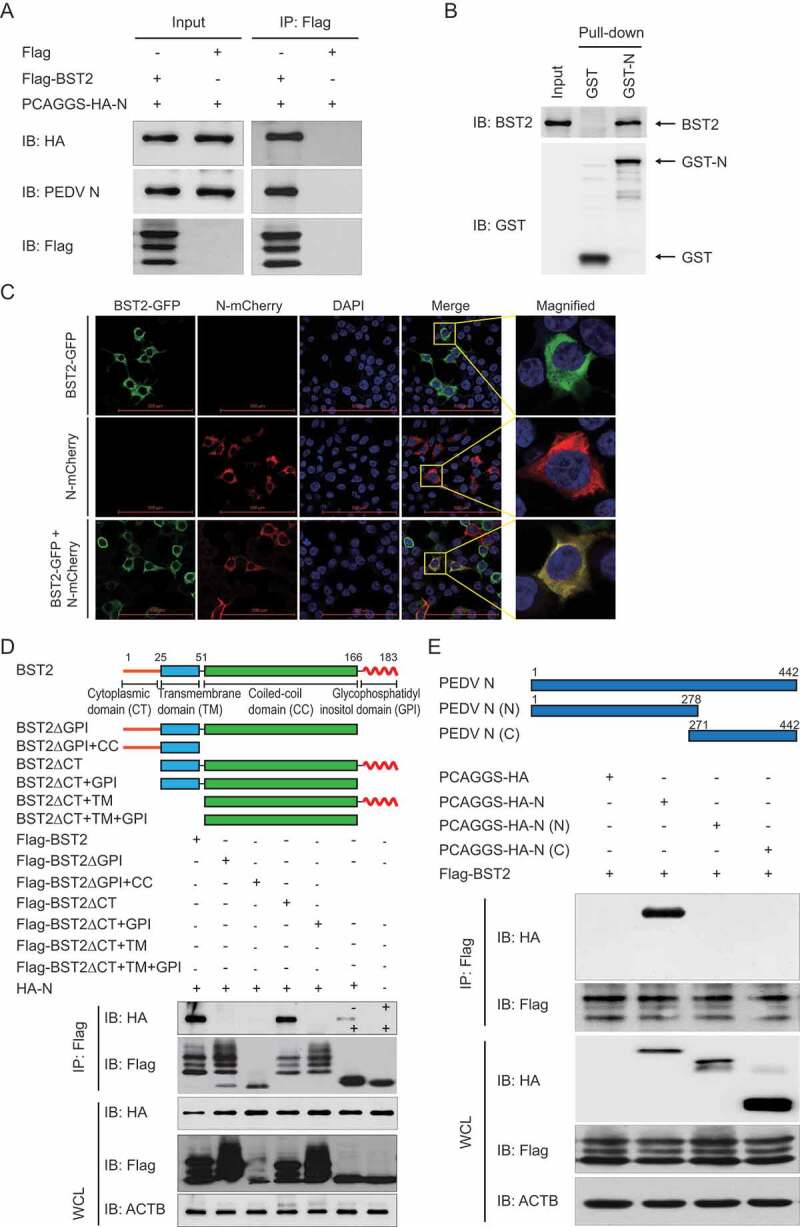
BST2 interacts with PEDV N protein. (A) Vero cells were transfected with plasmids encoding HA-N and Flag-BST2 or empty vectors for 24 h, followed by Co-IP with anti-Flag binding beads and a western blotting analysis with anti-HA, anti-PEDV N, and anti-Flag antibodies. (B) The full length of the BST2 gene and PEDV N gene were cloned into pCold TF plasmid and pCold GST plasmid, respectively. Recombinant proteins were expressed in bacterial strain BL21 (DE3) and purified for the GST pull-down analysis. After adequate washing, proteins eluted from beads were analyzed by western blotting. Input, BST2. (C) 293T cells were transfected with plasmids encoding BST2-GFP and N-mCherry for 24 h. Cell nuclei were labeled with DAPI, and the fluorescent signals were observed with confocal immunofluorescence microscopy. 293T cells were transfected with plasmids encoding BST2-GFP or N-mCherry as control. Scale bars: 100 μm. (D) 293T cells were transfected with plasmids encoding HA-N and Flag-BST2 or the indicated BST2 mutants. They were then analyzed with Co-IP with anti-Flag binding beads and western blotting with anti-HA and anti-Flag antibodies. Throughout was the western blot analysis of whole-cell lysates (WCLs) without immunoprecipitation. ACTB was used as the sample loading control. (E) Co-IP and western blotting analyses of 293T cells transfected with PEDV N or the indicated N mutants, together with a vector encoding Flag-BST2.
Structurally, BST2 consists of a short N-terminal cytoplasmic tail (CT) domain, an alpha-helical transmembrane (TM) domain, a labile coiled-coil (CC) ectodomain, and a C-terminal GPI anchor [34]. The unusual topology of BST2, with 2 membrane anchors, is necessary to suppress viral replication by directly tethering the viral membrane to the plasma membrane of the host cell [35]. To determine the domains of BST2 involved in its interaction with the PEDV N protein, we constructed several deletion mutants of BST2 and co-transfected 293T cells with the plasmids and a plasmid encoding the HA-N protein. A co-IP analysis showed that full-length BST2 (amino acids 1–183), BST2ΔCT (amino acids 26–183), and BST2ΔCT+TM (amino acids 52–183) interacted with the full-length N protein, whereas BST2ΔGPI (amino acids 1–166), BST2ΔGPI+CC (amino acids 1–51), BST2ΔCT+GPI (amino acids 26–166), and BST2ΔCT+TM+GPI (amino acids 52–166) did not (Figure 4D). These results indicate that the GPI domain of BST2 is important for its interaction with the PEDV N protein. We also generated several truncated mutants of the N protein and performed a co-IP experiment to identify the domain of the N protein required for its interaction with BST2. BST2 only interacted with the full-length N protein (amino acids 1–442), but not with N (N) (amino acids 1–278) or N (C) (amino acids 271–442) (Figure 4E). These results indicate that the integrity of the N protein is essential for its ability to bind BST2.
BST2 promotes PEDV n protein degradation by autophagy
The PEDV N protein is abundantly produced in infected cells and plays an important role in enhancing the efficiency of viral replication. For instance, the PEDV N protein inhibits IFNB production and ISG (IFN-stimulated gene) expression [11] and upregulates CXCL8/IL8 (C-X-C motif chemokine ligand 8) expression [46]. Because BST2 is one of the key players in the innate immune response against a variety of enveloped viruses, we speculated that BST2 inhibits PEDV replication by changing the abundance of N protein via the BST2-N interaction. We co-transfected 293T cells with plasmids encoding Flag-BST2 and HA-N and examined the protein abundances with a western blotting analysis. The expression of BST2 strongly reduced the production of PEDV N protein, and higher levels of BST2 had a stronger inhibitory effect (Figure 5A). Conversely, an analysis of the lysates of cells co-transfected with BST2-directed siRNA and a plasmid encoding HA-N showed that the knockdown of BST2 expression in Vero cells upregulated N protein production (Figure 5B).
Figure 5.
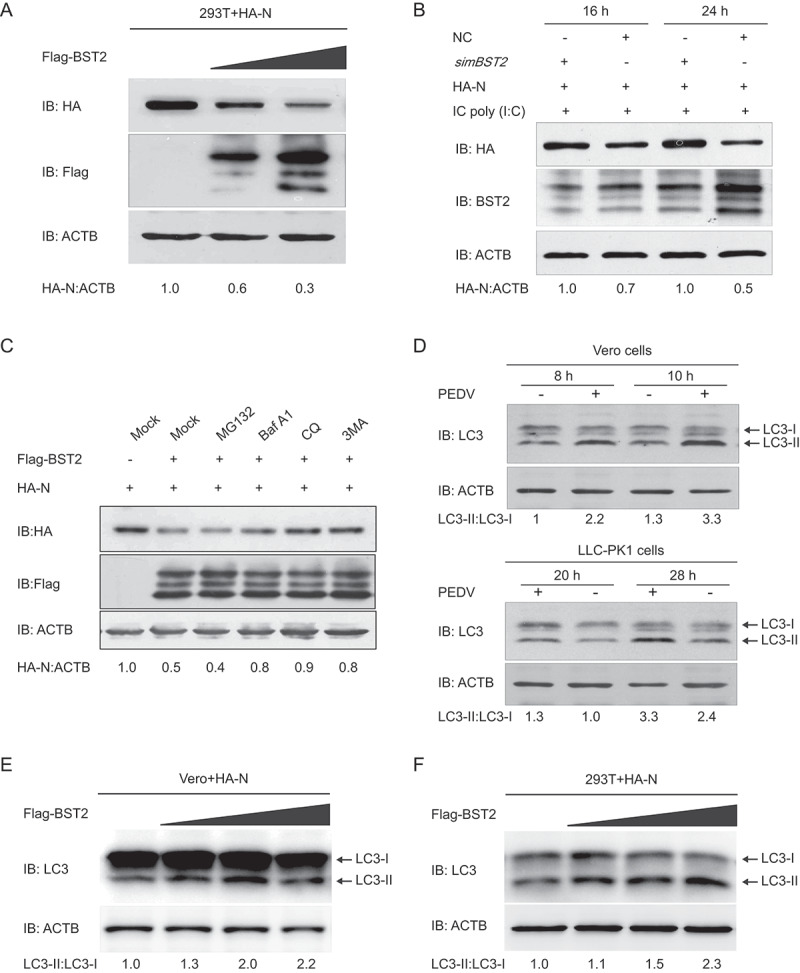
BST2 promotes the autophagic degradation of PEDV N protein. (A) 293T cells were transfected with the vector expressing HA-N and increasing concentrations of a vector expressing Flag-BST2 (wedge) for 24 h. The cell lysates were analyzed with western blotting. ACTB was used as the sample loading control. (B) Vero cells were transfected with HA-N-expressing vector and mBST2 siRNA or negative control siRNA. The cells were then treated with poly(I:C) (low molecular weight) (10 mg/mL) for the indicated times. The cell lysates were analyzed with western blotting. (C) 293T cells were transfected with plasmids encoding Flag-BST2 and HA-N, and the cells were then treated with MG132 (5 mM), bafilomycin A1 (Baf A1; 0.1 mM), chloroquine (CQ; 10 mM), or 3-methyladenine (3MA; 0.5 mM) for 8 h. The cell lysates were then analyzed with western blotting. (D) Vero cells and LLC-PK1 cells were infected or mock-infected with PEDV at an MOI of 1 and harvested at the indicated times. The expression of LC3 was analyzed by western blotting. Vero cells (E) and 293T cells (F) were transfected with the vector expressing HA-N and increasing concentrations of a vector expressing Flag-BST2 (wedge) for 24 h. The cell lysates were analyzed with western blotting.
The ubiquitin-proteasome system and autolysosome pathway are 2 major intracellular protein degradation pathways in eukaryotic cells [47]. Therefore, to identify the system that predominantly mediates the degradation of PEDV N protein by BST2, the amount of N protein in 293T cells co-transfected with plasmids encoding Flag-BST2 and HA-N was examined, and the cells were then treated with the protease inhibitor MG132 or the autophagy inhibitor 3-methyladenine (3MA), chloroquine (CQ), or bafilomycin A1 (Baf A1). As shown in Figure 5C, the degradation of N protein mediated by BST2 was blocked by the autophagy inhibitors 3MA, CQ, and Baf A1, but not by the proteasome inhibitor MG132 (Figure 5C). To explore whether autophagy was induced by PEDV infection, the levels of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), a hallmark of autophagy [48], was examined in PEDV-infected Vero cells or LLC-PK1 cells. We found that LC3-II protein was significantly increased at different times post-PEDV infection, and the ratio of LC3-II to LC3-I was much higher in PEDV-infected cells when compared to the control (Figure 5D), which is consistent as previously described [31]. Meanwhile, we co-transfected plasmids encoding Flag-BST2 and HA-N in Vero or 293T cells and found that the BST2 and N protein co-expressed in Vero or 293T cells increased the conversion of LC3-I to LC3-II in a dose-dependent manner (Figure 5E and F), which suggested that BST2 could increase the levels of autophagy in cells expressing N protein. Together, these results indicated that BST2 degrades the PEDV N protein via autophagy.
PEDV n protein degraded by autophagy through the bst2-marchf8-calcoco2-autophagosome pathway
During selective autophagy, the substrate proteins are modified with ubiquitin by an E3 ubiquitin ligase and recognized by cargo receptors. These deliver the substrates to and interact with the ATG8 family proteins to form the autophagosome that degrades the substrates [23]. Recent studies have shown that BST2 recruits the E3 ubiquitin ligase MARCHF8 to attach K27-linked ubiquitin chains onto MAVS to cause its CALCOCO2-dependent autophagic degradation during RNA viral infections [20]. In this study, we found that BST2 promotes PEDV N protein degradation via autophagy. Co-IP and immunofluorescence analyses showed that BST2 efficiently coimmunoprecipitates and colocalizes with both MARCHF8 and CALCOCO2 (Fig. S4A and B), which is consistent with a previous report [20]. To confirm that the PEDV N protein interacts with MARCHF8 and CALCOCO2, co-IP assays were performed with an anti-HA mAb in 293T cells co-transfected with plasmids encoding HA-N and MYC-MARCHF8 or MYC-CALCOCO2. The results showed that N protein interacts with MARCHF8 and CALCOCO2 (Figure 6A). GST pull-down assay indicating that PEDV N protein directly binds to MARCHF8 (Figure 6B) and CALCOCO2 (Figure 6C). We also investigated the colocalization of N-MARCHF8 and N-CALCOCO2 and found that PEDV N protein interacts with MARCHF8 and CALCOCO2 (Figure 6D). In mammalian cells, ubiquitin chains attached to substrate proteins act as a major signal recognized by cargo receptors [49]. Upregulating or downregulating MARCHF8 showed that the overexpression of MARCHF8 significantly increased the polyubiquitination of N protein. While interrupting the expression of MARCHF8 reduced the polyubiquitination of N protein (Figure 6E). These data suggested that BST2 links the PEDV N protein and MARCHF8 to the cargo receptor CALCOCO2 for autophagic degradation.
Figure 6.
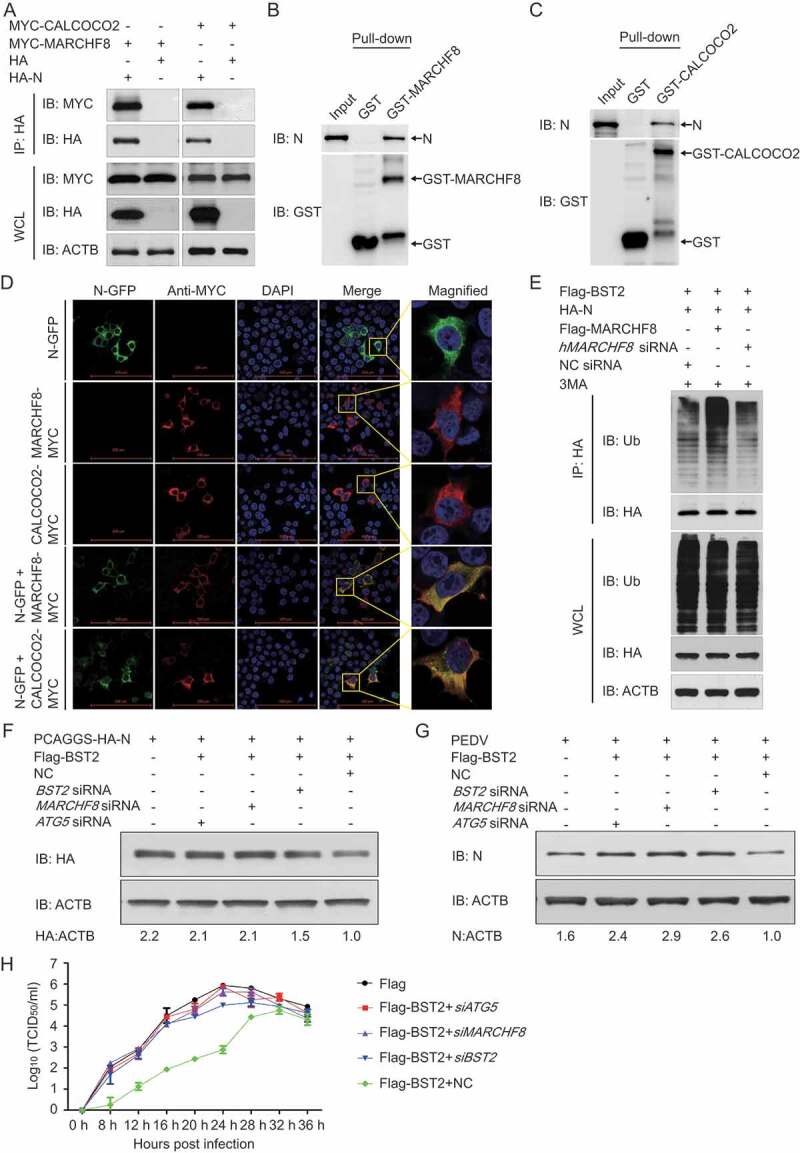
PEDV N protein is degraded by autophagy through the BST2-MARCHF8-CALCOCO2-autophagosome pathway. (A) 293T cells were transfected with plasmids encoding HA-N and MYC-MARCHF8 or MYC-CALCOCO2 for 24 h, and then analyzed with Co-IP with anti-HA binding beads and western blotting with anti-MYC and anti-HA antibodies. Throughout was the western blot analysis of whole-cell lysates (WCLs) without immunoprecipitation. ACTB was used as the sample loading control. (B) The full length of PEDV N gene and MARCHF8 gene were cloned into pCold TF plasmid and pCold GST plasmid, respectively. Recombinant proteins were expressed in bacterial strain BL21 (DE3) and purified for the GST pull-down analysis. After adequate washing, proteins eluted from beads were analyzed by western blotting. Input, PEDV N. (C) The full length of PEDV N gene and CALCOCO2 gene were cloned into pCold TF plasmid and pCold GST plasmid, respectively. Recombinant proteins were expressed in bacterial strain BL21 (DE3) and purified for the GST pull-down analysis. After adequate washing, proteins eluted from beads were analyzed by western blotting. Input, PEDV N. (D) 293T cells were transfected with plasmids encoding N-GFP and MYC-MARCHF8 or MYC-CALCOCO2 for 24 h, and then MYC-MARCHF8 and MYC-CALCOCO2 were labeled with specific primary antibodies and secondary antibodies (red). Cell nuclei were stained with DAPI (blue). The fluorescent signals were observed with confocal immunofluorescence microscopy. 293T cells were transfected with plasmids encoding N-GFP, MARCHF8-MYC, or CALCOCO2-MYC as control. Scale bars: 100 μm. (E) 293T cells were co-transfected with HA-N plasmid, Flag-BST2 plasmid, and Flag-MARCHF8 plasmid or siRNA (hMARCHF8 siRNA or negative control siRNA). Cell lysates were harvested after 3MA (0.5 mM) treatment for 8 h. The proteins were immunoprecipitated with anti-HA antibody, and analyzed with western blotting with anti-ubiquitin (Ub) and anti-HA antibodies. (F) 293T cells were co-transfected with plasmids encoding Flag-BST2 and HA-N and small interfering RNA (BST2 siRNA, MARCHF8 siRNA, ATG5 siRNA, or negative control siRNA), and then analyzed with western blotting with an anti-HA antibody. (G) Vero cells were co-transfected with plasmids encoding Flag-BST2 and small interfering RNA (BST2 siRNA, MARCHF8 siRNA, ATG5 siRNA, or negative control siRNA), and then infected with PEDV (MOI = 0.01) and harvested at the indicated times. Western blotting analysis was performed with a monoclonal antibody against PEDV N protein. (H) PEDV titers in the culture supernatants of the Vero cells treated described in (G) were measured as TCID50.
To determine whether the MARCHF8-CALCOCO2-triggered autophagy pathway is necessary for the BST2-induced degradation of N protein, we transfected 293T cells with plasmids encoding HA-N and Flag-BST2 together with small interfering RNAs (BST2 siRNA, MARCHF8 siRNA, ATG5 siRNA, or negative control siRNA). After 24 h, we used western blotting to analyze the cell lysates and found that interrupting BST2, MARCHF8, or ATG5 expression effectively prevented the degradation of N protein induced by BST2 (Figure 6F). We also showed that the inhibition of BST2, MARCHF8, or ATG5 protein expression by their respective siRNAs effectively prevented the inhibition of PEDV replication by BST2 in Vero cells (Figure 6G). Consistently, the viral titers were significantly increased when the autophagy pathway was interrupted by MARCHF8-directed siRNA or ATG5-directed siRNA (Figure 6H), indicating that the ability of BST2 to inhibit PEDV proliferation significantly decreased when interrupting the autophagy pathway. These results suggested that the PEDV N protein is degraded by autophagy through the BST2-MARCHF8-CALCOCO2-autophagosome pathway.
Discussion
The IFN-inducible restriction factor BST2 has emerged as an important component of the antiviral immune response. BST2 was initially shown to inhibit human immunodeficiency virus replication by inhibiting the release of budding virions from infected cells [35,36]. Recently, BST2 has been shown to be a signaling molecule that activates the NFKB signaling pathway and, thus, the innate immune responses to infection [22,41]. Increasing evidence show that BST2 inhibits the replication of a large number of enveloped viruses, including retroviruses [50], herpesviruses [39], filoviruses [11], and rhabdoviruses [51]. In this study, we have extended the repertoire of BST2-restricted viruses to include PEDV, an enveloped single-stranded positive-sense RNA virus belonging to the family Coronaviridae. We have also demonstrated a novel function of the BST2-mediated virus-restriction mechanism, in which BST2 suppresses PEDV replication by degrading the viral nucleocapsid protein through the BST2-MARCHF8-CALCOCO2-autophagosome pathway.
During viral infection, pathogen-associated molecular patterns (PAMPs) activate the innate immune system to produce IFN and establish an antiviral state [52]. To establish a productive infection, many viruses have developed several strategies to counteract IFN signaling and the host’s innate immune response. Previous studies have shown that PEDV infection suppresses type I IFN production and the expression of IFN-inducible genes (ISGs) in MARC-145 cells and porcine small intestine epithelial cells [13,53]. The PEDV nsp1 protein promotes the degradation of the host CREB-binding protein via the proteasome-dependent pathway to suppress type I IFN production [13]. The PEDV N protein suppresses IRF3 activation and downregulates the production of type I IFN by sequestering the interaction between IRF3 and TBK1 [22]. The IFN response is also antagonized by PLP2 (papain-like proteinase 2) of PEDV, by deubiquitinating DDX58/RIG-I and STING [54]. Our results indicate that BST2 is significantly upregulated by PEDV infection in Vero cells. Therefore, other molecular mechanisms must control BST2 transcription in the absence of functional IFN signaling. Type I and II IFN induce BTS2 expression in different mouse and human cell types [55,56]. Consistent with these findings, an analysis of the mouse and human BST2 promoter region found several potential TFBS that were related to the IFN signaling pathway [34,44]. In this study, we amplified the porcine BST2 promoter sequence and found that the boundaries of the minimal BST2 core promoter region occur at positions −182 to −138. We then analyzed the porcine BST2 core promoter region and observed multiple cis-acting regulatory elements for transcription factors such as STATs and IRFs, which is consistent with previous reports [38]. The induction of BST2 expression by IFN is dependent on STAT1 phosphorylation, and cells lacking STAT1 or unable to phosphorylate STAT1 cannot upregulate BST2 expression in the presence of IFN [44].
Furthermore, an analysis of the human BST2 promoter, which identified the regulatory elements, indicated that virus-activated IRF7 directly stimulates the BST2 promoter and upregulates BST2 protein expression in the absence of IFN signaling and STAT1 in HEK293T cells [44]. By analyzing the TFBS in the porcine BST2 core promoter, we showed that the expression of IRF1 is sufficient to upregulate BST2 expression. Our data show that the expression of BST2 in PEDV-infected cells in response to virus-induced IRF1 ensures that the cells actively maintain an abundance of BST2 to suppress viral replication as it is required, even though the initial IFN response is interrupted by PEDV.
The unique topology of BST2, which contains 2 membrane anchors, allows it to tether enveloped viruses to the surfaces of infected cells to restrict the release of budding virions. During a viral release, one of the BST2 membrane anchors is attached to the plasma membrane of the infected cell, and the other membrane anchor is inserted into the viral membrane of the budding enveloped virus to tether it to the surface of the infected cell [15,35,57]. The overall configuration of BST2, with a flexible ectodomain and 2 membrane anchors, is sufficient to inhibit virion release. When an artificial protein was expressed that lacked any sequence homology to BST2 but had a topology that included 2 membrane anchors and a flexible ectodomain, it was also able to inhibit virion release [17]. The activation of NFKB is another antiviral function of BST2. The cytoplasmic tail is the BST2 domain essential for activation of NFKB by the recruitment of TRAF2, TRAF6, and MAP3K7 [40,41]. Our results indicate that the GPI domain of BST2 is the domain essential for its interaction with the PEDV N protein and that BST2 promotes PEDV N protein degradation through autophagy. A recent study has shown that BST2 recruits the E3 ubiquitin ligase MARCHF8 to catalyze the ubiquitination of MAVS, which induces its selective autophagic degradation [20]. Our results demonstrate that BST2 recruits MARCHF8 to catalyze the ubiquitination of the PEDV N protein for its selective autophagic degradation. However, no interaction was observed between BST2 and the N proteins of other coronaviruses, such as Transmissible gastroenteritis virus, middle east respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV) (data not shown).
Autophagy is a cellular catabolic process that is important in maintaining intracellular homeostasis through the degradation of damaged organelles and misfolded or long-lived proteins. Recent evidence indicates that autophagy plays a dual role in the antiviral innate immune responses [27,58]. Viral components can be directly recognized and subjected to autophagic degradation. The spread of the Sindbis virus is limited by selective autophagy through the recruitment of the E3 ligase SMURF1 and the ubiquitination of the viral capsid, allowing its recognition by SQSTM1, because SQSTM1 recruits capsids proteins to autophagosomes [59,60]. Conversely, autophagy also plays a positive role in viral replication.
On the one hand, autophagy degrades components of the innate immune systems or mitochondria to reduce the release of pro-inflammatory signals [61]. On the other hand, autophagy reportedly provides a membrane platform for viral replication or to mediate virion assembly and release [62]. It has been reported that autophagy was triggered by the effective replication of PEDV and benefited PEDV replication by using autophagy regulators and RNA interference in Vero cells [31,63]. Still, rapamycin-induced autophagy reduces PEDV infection in porcine IECs [32]. Here, we demonstrated that BST2 and PEDV could induce autophagy in Vero or LLC-PK1 cells, and BST2 could increase the levels of autophagy in cells expressing N protein. Furthermore, BST2 was able to suppress PEDV replication by directly targeting PEDV N protein for degradation in Vero and LLC-PK1 cells, and the ability of BST2 to inhibit PEDV proliferation was significantly decreased when interrupting the autophagy pathway. We speculate that the new function of the BST2-mediated viral restriction mechanisms might be dependent on selective autophagy to degrade viral nucleocapsid protein.
Selective autophagy is mediated by cargo receptors, which link substrate proteins to LC3 on the autophagosome, where they are packaged into the autophagosome and degraded by lysosome [20]. Protein modification by ubiquitination plays a critical role in recognition of substrate proteins by cargo receptors during selective autophagy. Here, we have shown that the E3 ubiquitin ligase MARCHF8 catalyzes the polyubiquitination of the PEDV N protein to allow its recognition by CALCOCO2. Previous research showed that the cargo receptor CALCOCO2 directly targets MAVS, which is involved in type I IFN signaling, for degradation by autophagy to negatively regulates type I IFN signaling [20]. In this study, we have demonstrated that the PEDV N protein is ubiquitinated by the E3 ubiquitin ligase MARCHF8 and interacts with the cargo receptor CALCOCO2 for degradation. The PEDV N protein is a highly conserved RNA-binding protein with important roles in viral replication and cellular infection. The PEDV N protein suppresses IRF3-mediated IFN production by interacting with TBK1 to disrupt the interaction between IRF3 and TBK1 [11]. Another study showed that the PEDV N protein activates the NFKB signaling pathway during the viral infection of intestinal epithelial cells [64]. Our results indicate that BST2 promotes PEDV N protein degradation via autophagy and that the abundance of the PEDV N protein and PEDV replication increase when the selective autophagy pathway is disrupted.
In summary, we have shown that BST2 binds and degrades the PEDV N protein by selective autophagy to suppress PEDV replication and that PEDV infection upregulated IRF1 expression. IRF1 directly binds the BST2 promoter to upregulate the abundance of BST2 protein. BST2 recruits the E3 ubiquitin ligase MARCHF8 to catalyze the ubiquitination of the PEDV N protein. Then the cargo receptor CALCOCO2 recognizes the ubiquitin chains on the N protein and delivers N to autolysosomes for degradation. Thus, BST2 downregulates the protein abundance of the PEDV N protein by selective autophagy to suppress viral replication in cells (Figure 7). Our findings provide evidence that IRF1 directly binds the BST2 promoter to regulate BST2 expression, independently of IFN signaling. We also provide evidence to support a new function of the BST2-mediated viral restriction mechanisms in which BST2 suppresses PEDV replication by targeting and degrading the viral nucleocapsid protein by selective autophagy.
Figure 7.
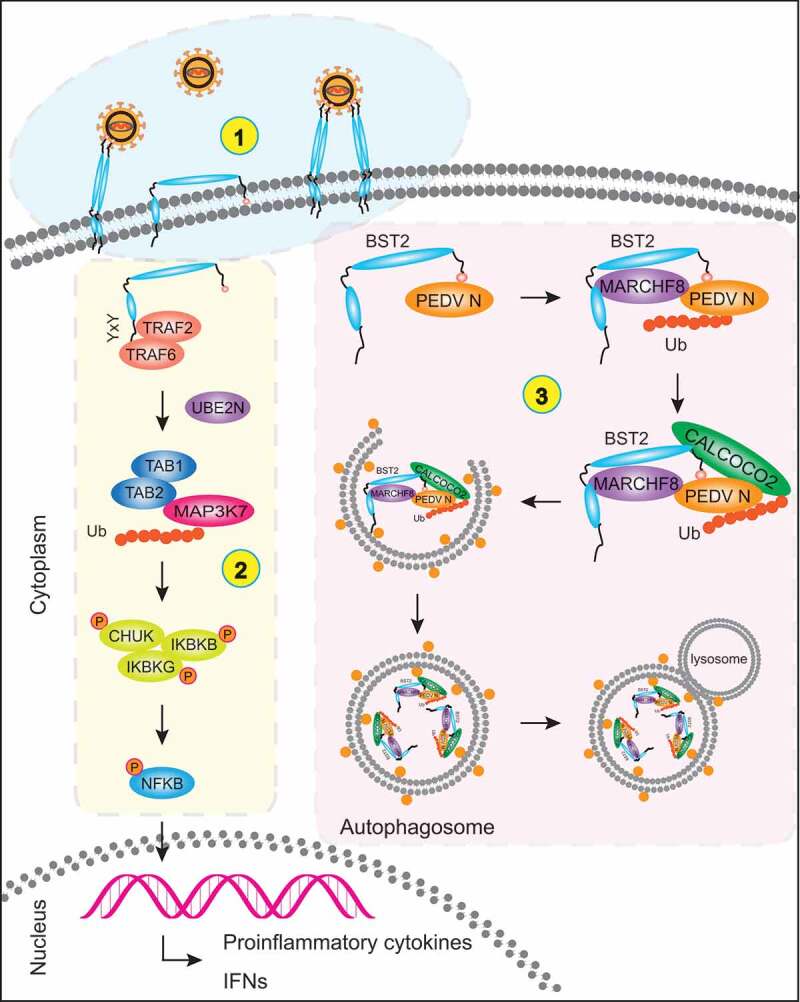
Antiviral mechanism of BST2. (1) BST2 homodimers restrict the release of mature virions from infected cells by positioning one transmembrane domain in the cellular membrane and the other in the virion membrane. (2) YXY motif in the cytoplasmic tail of BST2 interacts with TRAF2/TRAF6. Together, they recruit TAB1, TAB2, and the activated MAP3K7 kinase complex to induce the phosphorylation of CHUK, IKBKB, and IKBKG, thereby activating the canonical NFKB pathway. (3) BST2 recruits the E3 ubiquitin ligase MARCHF8 to catalyze the ubiquitination of PEDV N protein. The cargo receptor CALCOCO2 then recognizes the ubiquitin chains on N protein and delivers the N protein to autolysosome for degradation.
Materials and methods
Antibodies, reagents, and plasmids
Anti-IRF1 antibody (8478), anti-LC3A/B antibody (12,741), anti-ATG5 antibody (12,994) and anti-HA-tag antibody (3724) were purchased from Cell Signaling Technology. Anti-BST2 antibody (ab88523) was purchased from Abcam. Anti-Flag M2 antibody produced in mouse (F1804) and anti-MYC antibody (M4439) were obtained from Sigma-Aldrich. Anti-ubiquitin antibody (SC-8017) was purchased from Santa Cruz Biotechnology. Anti-ACTB/β-actin antibody (60,008–1), anti-GST-tag antibody (10,000-0-AP), anti-MARCHF8 antibody (14,119-1-AP), horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody (SA00001-1), and horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody (SA00001-2) were purchased from the Proteintech Group. The monoclonal antibody directed against the PEDV (PEDV JS-2013) N protein and against porcine BST2 protein were prepared in our laboratory [65,66]. Poly(I:C) LMW (low molecular weight) was purchased from InvivoGen. Universal type I interferon (112,001) was purchased from PBL InterferonSource. 3-Methyladenine (3-MA, M9281), chloroquine phosphate (CQ, PHR1258), and MG132 (M7449) were purchased from Sigma-Aldrich. Bafilomycin A1 (Baf A1, 54,645) was purchased from Cell Signaling Technology.
Simian BST2 siRNA, IRF1 siRNA, ATG5 siRNA, and MARCHF8 siRNA were designed and purchased from GenePharma (Shanghai, China), together with the control siRNA (Table S1). Human BST2 siRNA was purchased from GenePharma, according to a previous report [20]. Human ATG5 siRNA (SC-41,445) and human MARCHF8 siRNA (SC-90,432) were purchased from Santa Cruz Biotechnology. The mammalian expression vector p3× FLAG-CMV-7.1 (P1118) was purchased from Sigma-Aldrich. The pGL3-Basic vector (E1751), pRL-TK luciferase reporter plasmid (E2241), and Dual-Glo Luciferase Assay System (E1910) were purchased from Promega. The pEGFP-N1 plasmid, pCold TF plasmid (3365) and pCold GST plasmid (3372) were purchased from Clontech Laboratories, Inc. The pCMV-N-mCherry plasmid (D2711) was purchased from Beyotime Biotechnology. All recombinant plasmids were constructed by homologous recombination with the ClonExpress II One Step Cloning Kit (Vazyme Biotech, C112-02), according to the manufacturer’s instructions.
Cell cultures, transfection, and viral infection
African green monkey kidney cells (Vero cells; ATCC, CCL-81) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, 12,430,054) supplemented with 10% fetal bovine serum (FBS; Gibco, 10,099,141). Porcine kidney cells (LLC-PK1 cells) were obtained from Dr. Rui Luo (Huazhong Agricultural University, Wuhan, China) and maintained in modified Eagle’s medium (MEM; Invitrogen, 11,095,080). Human embryonic kidney cells (293T cells; ATCC, CRL-11,268) and porcine kidney cells (PK-15 cells; ATCC, CCL-33) were cultured in DMEM (Sigma-Aldrich, D6429) supplemented with 10% FBS. All cells were incubated at 37°C with 5% CO2. Cells grown to approximately 80%-90% confluence in 6-well cell culture plates were transfected with plasmids using Lipofectamine 3000 Reagent (Invitrogen, L3000015). Cells grown to 50%-60% confluence were transfected with siRNA using Lipofectamine RNAiMAX (Invitrogen, 13,778,150), according to the manufacturer’s instructions.
The PEDV variant strain JS-2013 was isolated and stored in our laboratory [43]. According to the requirements of different experiments, Vero cells were infected with PEDV at an MOI of 1 or 0.01, or were mock infected with DMEM. Vero cells were grown to approximately 80%-90% confluence in 6-well culture plates and infected with PEDV at varying MOIs, together with 5 μg/mL trypsin (Invitrogen, 15,050,065), and then washed 3 times with PBS (Gibco, C20012500BT). After incubation for 1 h, the unbound virus was removed by washing the cells 3 times with PBS. They were then cultured in serum-free DMEM with 5 μg/mL trypsin at 37°C for different times before harvesting. The viral titers were calculated with Kaerber’s method and expressed as 50% tissue culture infectious doses (TCID50) per milliliter.
Quantitative real-time PCR (qPCR)
Total RNA was extracted from cells with the QIAamp Viral RNA Mini Kit (Qiagen, 52,906) or RNeasy Mini Kit (Qiagen, 74,104). For the reverse transcription (RT)-qPCR analysis, cDNA was generated with the PrimeScript™ RT reagent Kit (Takara, RRO47A) and analyzed by qPCR using SYBR Premix Ex Taq™ (Takara, RR420A), according to the manufacturer’s protocol. The human BST2 and GAPDH primer sequences were designed according to a previous report [20]. Other primer sequences used for the qPCR analysis are listed in Table S1. ACTB or GAPDH were used as the reference genes for normalization.
Western blotting analysis
Cells were washed twice with cold PBS and incubated on ice with RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, 89,901) containing Phosphatase Inhibitor Cocktail (Bimake, B15001) and Protease Inhibitor Cocktail (Bimake, B14001). The lysates were denatured for 5 min in 5 × SDS-PAGE loading buffer and separated with SDS-PAGE, and the proteins were then transferred to nitrocellulose western blotting membranes (GE Healthcare, 10,600,001). The membranes were blocked with PBS containing 5% nonfat dry milk (BD, 232,100) and 0.2% Tween 20 (Sigma-Aldrich, P1379) for 1 h at room temperature and then incubated with the primary antibody at room temperature for 1 h. After the membranes were washed with PBS containing 0.1% Tween 20 (0.1% PBST), they were incubated with the corresponding secondary antibody (conjugated with HRP) at room temperature for 1 h and detected with enhanced chemiluminescence (ECL) (Thermo Fisher Scientific, 34,580). The protein bands were quantified with the ImageJ software (National Institutes of Health).
Co-ip assay
Cells were transfected with the indicated plasmids for 24 h and lysed with ice-cold NP40 Cell Lysis Buffer (Life Technologies, FNN0021) containing Phosphatase Inhibitor Cocktail and Protease Inhibitor Cocktail. The cell lysates were centrifuged and incubated with an anti-HA- or anti-Flag-antibody-bound Dynabeads Protein G (Life Technologies, 10004D) for 30 min at room temperature. The Dynabeads were washed 4 times with 0.02% PBST and resuspended in elution buffer (50 mM glycine, pH 2.8). The proteins were analyzed with immunoblotting using the indicated antibodies.
Luciferase reporter assay
293T cells were plated in 12-well culture plates and transfected with plasmids using Lipofectamine 3000, according to the manufacturer’s protocol. The plasmids transfected differed in each experiment. For the promoter experiments (Figure 2A), cells were co-transfected with a luciferase reporter plasmids (encoding firefly luciferase) containing the full BST2 promoter or a truncated promoter, together with the pRL-TK plasmid (encoding Renilla luciferase, as the internal reference control). To identify the BST2 activators (Figure 2C and D), cells were co-transfected with plasmid encoding IRF1, IRF7, IRF8, IRF9, STAT1, or STAT2, the luciferase reporter plasmid (containing the BST2 promoter), and the pRL-TK plasmid. The cells were collected in 24 h after transfection and their fluorescence was measured with the Dual-Luciferase Reporter Assay System (Promega, E1910), according to the manufacturer’s protocol.
GST affinity-isolation assays
The full-length BST2 gene, PEDV N gene, MARCHF8 gene and ATG5 gene were cloned into pCold GST plasmid (Clontech Laboratories, Inc, 3372) or pCold TF plasmid (Clontech Laboratories, Inc, 3365), respectively. Recombinant proteins were expressed in BL21 (DE3) competent cells (Vazyme Biotech, C504-03) and purified for the GST affinity-isolation analysis with the GST Protein Interaction Pull-Down Kit (Thermo, 21,516), according to the manufacturer’s instructions. The bait proteins and prey proteins were incubated together with GST glutathione agarose resin. The bound proteins were eluted by glutathione and analyzed with immunoblotting using the indicated antibodies.
Chip assay
PK-15 cells were plated in 12-well culture plates and transfected with plasmids encoding Flag or Flag-IRF1. After 24 h, the cells were harvested and processed for the ChIP analysis with the SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling Technology, 9003), according to the manufacturer’s instructions. Normal rabbit IgG (Cell Signaling Technology, 2729) or anti-Flag antibody (Sigma-Aldrich, F1804) was coupled with protein G magnetic beads (Cell Signaling Technology, 9006) to immunoprecipitate the chromatin fragments. The chromatin fragments were quantified with qPCR. The sequences of the primers used for the qPCR analyses are listed in Table S1.
Confocal immunofluorescence assay
Cells were seeded on coverslips in 6-well plates and transfected with specific expression plasmids. After 24 h, the cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, P6148) in PBS for 15 min, and then washed 3 times with PBS and permeabilized with 0.1% Triton™ X-100 (Sigma-Aldrich, T9284) for 10 min at room temperature. After the cells were washed 3 times with PBS, they were blocked with 5% bovine serum albumin (BSA; Cell Signaling Technology, 9998) for 1 h at 37°C, and then incubated with primary antibody (anti-MYC) diluted in PBS containing 5% BSA for 1 h at 37°C. The cells were washed 3 times, and incubated with a fluorescently labeled secondary antibody (Alexa-Fluor-568-conjugated donkey anti-mouse IgG antibody; Thermo Fisher Scientific, A10037) for 1 h at 37°C in the dark. After the cells were washed 3 times with PBS, they were stained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime Biotechnology, C1002) at room temperature for 5 min. The cells were observed under a confocal immunofluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Statistical analysis
All results are representative of 3 independent experiments. Data are presented as means ± standard deviations (SD) and were analyzed with the two-tailed Student’s t test using the GraphPad Prism 5 software (GraphPad Software, USA). P values of < 0.05 were considered statistically significant.
Supplementary Material
Funding Statement
This study was supported by the National Key Research and Development Programs of China (no. 2016YFD0500103), The Natural Science Foundation of Shanghai (no. 19ZR1469100), The National Natural Science Foundation of China (no. 31872478) and The China Postdoctoral Science Foundation (no. 2017M611074)
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Chen J, Wang C, Shi H, et al. Molecular epidemiology of porcine epidemic diarrhea virus in China. Arch Virol. 2010;155:1471–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wood EN.An apparently new syndrome of porcine epidemic diarrhoea. Vet Rec. 1977;100:243–244. [DOI] [PubMed] [Google Scholar]
- [3].Sun RQ, Cai RJ, Chen YQ, et al. Outbreak of porcine epidemic diarrhea in suckling piglets, China. Emerg Infect Dis. 2012;18:161–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li W, Li H, Liu Y, et al. New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis. 2012;18:1350–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Debouck P.Pensaert M Experimental infection of pigs with a new porcine enteric coronavirus, CV 777. Am J Vet Res. 1980;41:219–223. [PubMed] [Google Scholar]
- [6].Stevenson GW, Hoang H, Schwartz KJ, et al. Emergence of Porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Invest. 2013;25:649–654. [DOI] [PubMed] [Google Scholar]
- [7].Kocherhans R, Bridgen A, Ackermann M, et al. Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes. 2001;23:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Duarte M, Gelfi J, Lambert P, et al. Genome organization of porcine epidemic diarrhoea virus. Adv Exp Med Biol. 1993;342:55–60. [DOI] [PubMed] [Google Scholar]
- [9].Zhang Q. Yoo D Immune evasion of porcine enteric coronaviruses and viral modulation of antiviral innate signaling. Virus Res. 2016;226:128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guo L, Luo X, Li R, et al. Porcine Epidemic Diarrhea Virus Infection Inhibits Interferon Signaling by Targeted Degradation of STAT1. J Virol. 2016;90:8281–8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ding Z, Fang L, Jing H, et al. Porcine epidemic diarrhea virus nucleocapsid protein antagonizes beta interferon production by sequestering the interaction between IRF3 and TBK1. J Virol. 2014;88:8936–8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang Q, Ke H, Blikslager A, et al. Type III interferon restriction by porcine epidemic diarrhea virus and the role of viral protein nsp1 in IRF1 signaling. J Virol. 2018;92:e01677–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang Q, Shi K, Yoo D. Suppression of type I interferon production by porcine epidemic diarrhea virus and degradation of CREB-binding protein by nsp1. Virology. 2016;489:252–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Arias JF, Evans DT. Tethering viral restriction to signal transduction. Cell Host Microbe. 2014;16:267–269. [DOI] [PubMed] [Google Scholar]
- [16].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].JS K, DM V, Insall RH. The induction of autophagy by mechanical stress. Autophagy. 2011;7:1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sun P, Zhang S, Qin X, et al. Foot-and-mouth disease virus capsid protein VP2 activates the cellular EIF2S1-ATF4 pathway and induces autophagy via HSPB1. Autophagy. 2018;14:336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jin S, Tian S, Luo M, et al. Tetherin suppresses type i interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol Cell. 2017;68:308–322 e304. [DOI] [PubMed] [Google Scholar]
- [21].Deng Z, Purtell K, Lachance V, et al. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 2017;27:491–504. [DOI] [PubMed] [Google Scholar]
- [22].Katsuragi Y, Ichimura Y. Komatsu M p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. Febs J. 2015;282:4672–4678. [DOI] [PubMed] [Google Scholar]
- [23].Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. [DOI] [PubMed] [Google Scholar]
- [24].Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Deretic V. Autophagy in infection. Curr Opin Cell Biol. 2010;22:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu G, Zhong M, Guo C, et al. Autophagy is involved in regulating influenza A virus RNA and protein synthesis associated with both modulation of Hsp90 induction and mTOR/p70S6K signaling pathway. Int J Biochem Cell Biol. 2016;72:100–108. [DOI] [PubMed] [Google Scholar]
- [27].Sun MX, Huang L, Wang R, et al. Porcine reproductive and respiratory syndrome virus induces autophagy to promote virus replication. Autophagy. 2012;8:1434–1447. [DOI] [PubMed] [Google Scholar]
- [28].Talloczy Z, Virgin H, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–29. [DOI] [PubMed] [Google Scholar]
- [29].Liu Y, Cherry S. Zika virus infection activates sting-dependent antiviral autophagy in the Drosophila brain. Autophagy. 2019;15:174–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Feizi N, Mehrbod P, Romani B, et al. Autophagy induction regulates influenza virus replication in a time-dependent manner. J Med Microbiol. 2017;66:536–541. [DOI] [PubMed] [Google Scholar]
- [31].Guo X, Zhang M, Zhang X, et al. Porcine epidemic diarrhea virus induces autophagy to benefit its replication. Viruses. 2017;9:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ko S, Gu MJ, Kim CG, et al. Rapamycin-induced autophagy restricts porcine epidemic diarrhea virus infectivity in porcine intestinal epithelial cells. Antiviral Res. 2017;146:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kupzig S, Korolchuk V, Rollason R, et al. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic. 2003;4:694–709. [DOI] [PubMed] [Google Scholar]
- [34].Sauter D. Counteraction of the multifunctional restriction factor tetherin. Front Microbiol. 2014;5:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Neil SJ, Zang T, Bieniasz PD. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature. 2008;451:425–430. [DOI] [PubMed] [Google Scholar]
- [36].Van Damme N, Goff D, Katsura C, et al. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe. 2008;3:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Weidner JM, Jiang D, Pan XB, et al. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J Virol. 2010;84:12646–12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Blondeau C, Pelchen-Matthews A, Mlcochova P, et al. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J Virol. 2013;87:13124–13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liu Y, Luo S, He S, et al. Tetherin restricts HSV-2 release and is counteracted by multiple viral glycoproteins. Virology. 2015;475:96–109. [DOI] [PubMed] [Google Scholar]
- [40].Galao RP, Le Tortorec A, Pickering S, et al. Innate sensing of HIV-1 assembly by Tetherin induces NFkappaB-dependent proinflammatory responses. Cell Host Microbe. 2012;12:633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tokarev A, Suarez M, Kwan W, et al. Stimulation of NF-kappaB activity by the HIV restriction factor BST2. J Virol. 2013;87:2046–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Liberatore RA, Bieniasz PD. Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108:18097–18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kong N, Wu Y, Meng Q, et al. Suppression of virulent porcine epidemic diarrhea virus proliferation by the PI3K/Akt/GSK-3alpha/beta Pathway. PLoS One. 2016;11:e0161508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bego MG, Mercier J, Cohen EA. Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J Virol. 2012;86:3513–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Antell GC, Dampier W, Aiamkitsumrit B, et al. Utilization of HIV-1 envelope V3 to identify X4- and R5-specific Tat and LTR sequence signatures. Retrovirology. 2016;13:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xu X, Zhang H, Zhang Q, et al. Porcine epidemic diarrhea virus N protein prolongs S-phase cell cycle, induces endoplasmic reticulum stress, and up-regulates interleukin-8 expression. Vet Microbiol. 2013;164:212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [48].Chen Q, Fang L, Wang D, et al. Induction of autophagy enhances porcine reproductive and respiratory syndrome virus replication. Virus Res. 2012;163:650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jouvenet N, Neil SJ, Zhadina M, et al. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J Virol. 2009;83:1837–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sarojini S, Theofanis T. Reiss CS Interferon-induced tetherin restricts vesicular stomatitis virus release in neurons. DNA Cell Biol. 2011;30:965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. [DOI] [PubMed] [Google Scholar]
- [53].Cao L, Ge X, Gao Y, et al. Porcine epidemic diarrhea virus inhibits dsRNA-induced interferon-beta production in porcine intestinal epithelial cells by blockade of the RIG-I-mediated pathway. Virol J. 2015;12:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xing Y, Chen J, Tu J, et al. The papain-like protease of porcine epidemic diarrhea virus negatively regulates type I interferon pathway by acting as a viral deubiquitinase. J Gen Virol. 2013;94:1554–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Blasius AL, Giurisato E, Cella M, et al. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. 2006;177:3260–3265. [DOI] [PubMed] [Google Scholar]
- [56].Yoo H, Park SH, Ye SK, et al. IFN-gamma-induced BST2 mediates monocyte adhesion to human endothelial cells. Cell Immunol. 2011;267:23–29. [DOI] [PubMed] [Google Scholar]
- [57].Perez-Caballero D, Zang T, Ebrahimi A, et al. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell. 2009;139:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sharma M, Bhattacharyya S, Nain M, et al. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy. 2014;10:1637–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Orvedahl A, MacPherson S, Sumpter R Jr., et al. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Orvedahl A, Sumpter R Jr., Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015;210:973–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chiramel AI, Brady NR, Bartenschlager R. Divergent roles of autophagy in virus infection. Cells. 2013;2:83–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zou D, Xu J, Duan X, et al. Porcine epidemic diarrhea virus ORF3 protein causes endoplasmic reticulum stress to facilitate autophagy. Vet Microbiol. 2019;235:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cao L, Ge X, Gao Y, et al. Porcine epidemic diarrhea virus infection induces NF-kappaB activation through the TLR2, TLR3 and TLR9 pathways in porcine intestinal epithelial cells. J Gen Virol. 2015;96:1757–1767. [DOI] [PubMed] [Google Scholar]
- [65].Pan X, Kong N, Shan T, et al. Monoclonal antibody to N protein of porcine epidemic diarrhea virus. Monoclon Antib Immunodiagn Immunother. 2015;34:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kong N, Meng Q, Wu Y, et al. Monoclonal antibody to bone marrow stromal cell antigen 2 protein of swine. Monoclon Antib Immunodiagn Immunother. 2016;35:172–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.