

ABSTRACT
Age-related impairment of macroautophagy/autophagy and loss of cardiac tissue homeostasis contribute significantly to cardiovascular diseases later in life. MTOR (mechanistic target of rapamycin kinase) signaling is the most well-known regulator of autophagy, cellular homeostasis, and longevity. The MTOR signaling consists of two structurally and functionally distinct multiprotein complexes, MTORC1 and MTORC2. While MTORC1 is well characterized but the role of MTORC2 in aging and autophagy remains poorly understood. Here we identified TGFB-INHB/activin signaling as a novel upstream regulator of MTORC2 to control autophagy and cardiac health during aging. Using Drosophila heart as a model system, we show that cardiac-specific knockdown of TGFB-INHB/activin-like protein daw induces autophagy and alleviates age-related heart dysfunction, including cardiac arrhythmias and bradycardia. Interestingly, the downregulation of daw activates TORC2 signaling to regulate cardiac autophagy. Activation of TORC2 alone through overexpressing its subunit protein rictor promotes autophagic flux and preserves cardiac function with aging. In contrast, activation of TORC1 does not block autophagy induction in daw knockdown flies. Lastly, either daw knockdown or rictor overexpression in fly hearts prolongs lifespan, suggesting that manipulation of these pathways in the heart has systemic effects on longevity control. Thus, our studies discover the TGFB-INHB/activin-mediated inhibition of TORC2 as a novel mechanism for age-dependent decreases in autophagic activity and cardiac health.
Abbreviations: AI: arrhythmia index; BafA1: bafilomycin A1; BMP: bone morphogenetic protein; CQ: chloroquine; CVD: cardiovascular diseases; DI: diastolic interval; ER: endoplasmic reticulum; HP: heart period; HR: heart rate; MTOR: mechanistic target of rapamycin kinase; NGS: normal goat serum; PBST: PBS with 0.1% Triton X-100; PDPK1: 3-phosphoinositide dependent protein kinase 1; RICTOR: RPTOR independent companion of MTOR complex 2; ROI: region of interest; ROUT: robust regression and outlier removal; ROS: reactive oxygen species; R-SMAD: receptor-activated SMAD; SI: systolic interval; SOHA: semi-automatic optical heartbeat analysis; TGFB: transformation growth factor beta; TSC1: TSC complex subunit 1
KEYWORDS: Atg8a, autophagic flux, dawdle, INHB/activin ligand, semi-automatic optical heartbeat analysis (SOHA), TOR complex 2
Introduction
Aging is associated with an exponential increase in the incidence of cardiovascular diseases (CVD) [1,2]. Age-related changes in cardiovascular structure and output have been linked to increased risk of coronary heart disease, sudden cardiac death and stroke in the elderly population [3]. During normal aging, the left ventricular wall of human hearts becomes thickened and the diastolic filling rate of left ventricle gradually decreases, while the left ventricular systolic performance at rest remains no change with age [3]. Several mechanisms underlying these age-associated changes in cardiovascular structure and function have been proposed, for example, changes in growth factor signaling, decreased cellular quality control, altered calcium handling, elevated extracellular matrix deposition or fibrosis, increased mitochondria damage, and the production of reactive oxygen species (ROS) [2]. Resolving the contributing mechanisms underlying the age-dependent decline of cardiovascular function is critical for the development of therapeutic interventions for the treatment of cardiovascular diseases.
Cellular quality control systems, such as macroautophagy (hereinafter referred to as autophagy), are essential to the maintenance of tissue homeostasis during aging [4]. Autophagy is a highly conserved process that is responsible for the degradation and recycling of damaged organelles, protein aggregates, and other cytoplasmic substances [5]. It is generally accepted that autophagy activity declines with age [6]. However, how autophagic activity is altered in aging hearts remains unclear. Disruption of autophagy pathways often leads to loss of tissue homeostasis and tissue dysfunction. For example, knocking out autophagy gene Atg5 in the mouse heart accelerates cardiac aging, including an increase in left ventricular hypertrophy and decrease in fractional shortening [7]. As a key autophagy regulator, MTOR (mechanistic target of rapamycin kinase) plays an important role in the regulation of cardiac tissue homeostasis [2]. The MTOR signaling consists of two structurally and functionally distinct protein complexes, MTORC1 and MTORC2 [8]. Rapamycin-mediated suppression of MTORC1 induces autophagy and protects mouse cardiomyocytes from oxidative stress [9]. Drosophila genome contains evolutionarily conserved Tor kinase and several major TOR complex subunits (e.g., raptor and Lst8 for TORC1, rictor, Sin1 and Lst8 for TORC2) [10]. Drosophila MTORC1 promotes electrical pacing-induced heart failure [11], and high-fat-diet-induced cardiomyopathy [12]. Unlike MTORC1, the role of MTORC2 in autophagy, tissue homeostasis, and cardiac aging remains poorly understood. Two recent studies show that disruption of MTORC2 subunit RICTOR (RPTOR independent companion of MTOR complex 2) induces cardiac dysfunction in mice [13,14]. Interestingly, 2 MTOR complexes exhibit distinct regulation on lifespan. Inhibition of MTORC1 is known to prolong lifespan [15], while deletion of RICTOR, the key component of MTORC2, shortens lifespan of male mice [16]. However, the molecular basis for MTORC2-regulated aging and tissue homeostasis remains to be further examined.
TGFB (transforming growth factor beta) is an evolutionarily conserved signaling pathway that regulates a wide variety of cellular processes, such as proliferation, differentiation, apoptosis, inflammation, and fibrosis [17]. Recent studies demonstrate that TGFB factors promote cardiac fibrosis during aging [18], while reduction of TGFB signaling improves cardiac health in aged mice through the regulation of microRNA MIR29 [19]. As in mammals, TGFB family in Drosophila has 2 major branches, BMP (bone morphogenetic protein) and TGFB-INHB/activin signaling pathways [17]. In both TGFB pathways, signaling starts with ligand binding to a receptor complex composed of type I and type II receptor kinases, followed by phosphorylation of receptor-activated SMAD (R-SMAD). Recently, we and other groups show that TGFB-INHB/activin signaling regulates muscle proteostasis and longevity in Drosophila [20,21]. However, it remains to be determined whether TGFB family proteins, in particular, INHB/activin, regulate cardiac homeostasis and function.
While investigating the role of Drosophila TGFB-INHB/activin signaling in the regulation of autophagy and age-related cardiomyopathy, we find that TGFB-INHB/activin signaling genetically interacts with TORC2, but not TORC1 to control age-dependent decreases in autophagy and cardiac health. We further show that the activation of TORC2 alone can induce autophagy and preserve cardiac function during aging. Thus, our studies uncover a novel crosstalk between TGFB-INHB/activin signaling and TORC2 that is essential for the maintenance of autophagic activity, tissue homeostasis, and cardiac health with aging.
Results
Heart-specific knockdown of TGFB-INHB/activin-like ligand daw slows cardiac aging
Our previous study demonstrated that the TGFB-INHB/activin-like protein daw regulates tissue homeostasis (especially in indirect flight muscle) and longevity in Drosophila [20]. Interestingly, we found that the mRNA expression of daw was significantly upregulated in aging fly hearts (29.7-fold) (Figure 1A), suggesting that TGFB-INHB/activin signaling may play a role in regulating cardiac aging. To investigate this possibility, we knocked down the expression of daw specifically in the heart with a binary GAL4/UAS system where we crossed UAS-daw RNAi lines into a cardiac-specific tissue driver (Hand-gal4 or Hand4.2-gal4) (Figure S1A,B). We analyzed cardiac performance in young and old flies using a high-speed video imaging system (semi-automatic optical heartbeat analysis, SOHA) [22]. M-Mode traces from the SOHA analysis indicated that knockdown of daw preserved cardiac contractility at advanced ages (Figure 1B). In wild-type flies, aged hearts showed increased arrhythmia (Figures 1C, S1I and S1J, data from three wild-type genotypes), lengthened diastolic intervals and heart period (bradycardia, slow heart rate) (Figure 1D,E), and decreased cardiac output (Figure 1F). Systolic intervals normally remain unchanged (Figure S1E). There is a small age-related reduction in fractional shortening (Figure S1F), but the diastolic and systolic diameters were not significantly altered (Figure S1G,H). Interestingly, cardiac-specific knockdown of daw attenuated the age-dependent increase in arrhythmia, diastolic intervals, and heart period (Figure 1C–E). Knockdown of daw also maintained relatively normal cardiac output at advanced ages (Figure 1F). Similar results were observed from three independent daw RNAi lines (the knockdown efficiency of daw RNAi was verified by qRT-PCR previously [20], as well as reconfirmed in the present study) (Figure S1C,D). However, cardiac-specific overexpression of daw did not lead to premature cardiac aging phenotypes (Figure S1K), which may be because the ectopic expression of daw in the heart did not generate active daw proteins.
Figure 1.
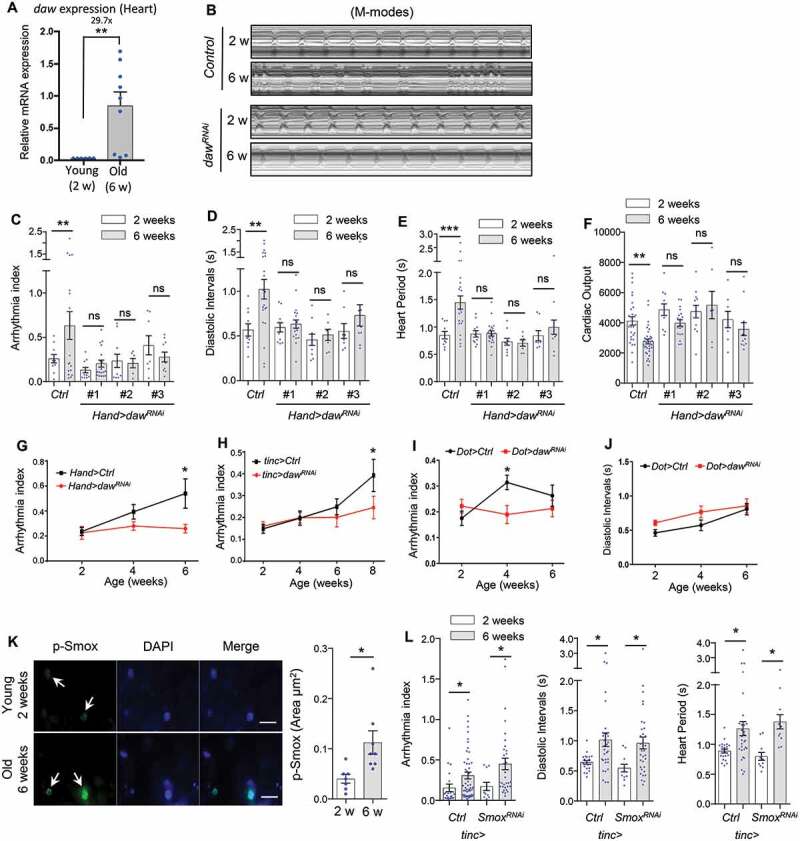
Heart-specific knockdown of daw slows cardiac aging. (A) QRT-PCR analysis of daw expression in young and old fly hearts. N = 3. Student t-test (** p < 0.01). (B) Representative M-mode traces (8 s) showing age-dependent movement of heart wall in control and heart-specific daw knockdown flies (Hand-gal4> UAS-dawRNAi). (C-F) Age-related changes in arrhythmia index, diastolic intervals, heart period, and cardiac output in control (Ctrl) and cardiac-specific daw knockdown flies (dawRNAi). Flies were cultured at 40% relative humidity. Hand-gal4 driver was used to knockdown gene expression specifically in cardiac tissues (cardiomyocytes and pericardial cells). Results from three independent UAS-dawRNAi lines are shown (RNAi #1: BDSC, 34974, RNAi #2: Vienna Drosophila Resource Center, 105309, RNAi #3: BDSC, 50911). N = 7 ~ 20. Two-way ANOVA followed by Tukey multiple comparisons test (* p < 0.05, ** p < 0.01, *** p < 0.001, ns = not significant). The interaction between genotype and age is statistically significant for heart period (p = 0.0041) and diastolic interval (p = 0.0243). (G-I) Age-dependent changes in cardiac arrhythmia between control and daw knockdown using various tissue drivers, Hand-gal4 (cardiomyocytes and pericardial cells), tinc-gal4 (cardiomyocytes), and Dot-gal4 (pericardial cells). N = 25 ~ 31. Student t-test (* p < 0.05, ** p < 0.01, *** p < 0.001). (J) Age-dependent changes in diastolic intervals between control and daw knockdown in pericardial cells (Dot-gal4). N = 13 ~ 26. Student t-test (* p < 0.05, ** p < 0.01, *** p < 0.001). (K) Immunostaining of p-Smox in fly hearts at young (2 weeks) and old ages (6 weeks). Arrows indicate cardiomyocyte nuclei and positive p-Smox staining. Scale bar: 20 μm. Quantification shown on the right. N = 8. Student t-test (* p < 0.05). Data are represented as mean ± SEM in all figures. (L) Age-dependent changes in arrhythmia index, diastolic intervals, heart period between control and Smox knockdown (tinc-gal4). N = 11 ~ 31. Student t-test (* p < 0.05).
The Hand-gal4 line drives gene expression in both cardiomyocytes and pericardial cells (Figure S1A). To test the cardiomyocyte-specific role of daw in cardiac aging, we then crossed daw RNAi into a cardiomyocyte-specific driver tinc-gal4 (or tincΔ4-gal4) (Figure S1A). Similar to the results from Hand-gal4 (Figure 1C,G), cardiomyocyte-specific knockdown of daw prevented the age-related increase in cardiac arrhythmia (Figure 1H). We also tested whether daw expression in pericardial cells can regulate cardiac aging using a pericardial cell-specific driver Ugt36A1/Dot-gal4. Interestingly, knockdown of daw in pericardial cells also alleviated age-induced arrhythmia (Figure 1I), but not diastolic interval (Figure 1J). Thus, reduction of daw signaling can alleviate age-related cardiac dysfunction, particularly the age-induced arrhythmia, through both cardiomyocyte-specific regulation and non-autonomous signaling from pericardial cells.
It is known that TGFB-INHB/activin signaling typically functions through its downstream transcription factor SMAD2 (or Smox in Drosophila). We noticed that during aging the phosphorylation of Smox/SMAD2 was elevated in the fly heart nuclei (Figure 1K). To test the role of Smox in cardiac aging, we performed SOHA analysis on flies with cardiac-specific Smox knockdown. Surprisingly, knockdown of Smox in the heart did not attenuate the age-dependent increase in arrhythmia, diastolic intervals, and heart period (Figure 1L). These findings suggest that daw may regulate cardiac aging through a Smox-independent pathway (e.g., MAPK/p38 signaling [23]).
Cardiomyocyte-specific knockdown of INHB/activin receptor babo delays cardiac aging
In Drosophila, INHB/activin-like ligand daw signals through the type I receptor babo, the fly homolog of mammalian ACVR1B/ALK4 (activin A receptor, type 1B) [17]. To investigate whether daw acts through its receptor babo to modulate cardiac aging, we first tested if babo plays a similar role in preventing the age-dependent increase in cardiomyopathy. Consistent with the results from daw knockdown, we found that cardiomyocyte-specific knockdown of babo blocked the age-related increase in cardiac arrhythmia, diastolic intervals, and heart period (Figure 2A–C). Two independent babo RNAi lines were used and both gave similar results (the knockdown efficiency of babo RNAi was verified by qRT-PCR in our previous study [20]. Knockdown of babo did not affect age-dependent changes in cardiac output (Figure S1L), which may be attributed to the similar diastolic diameter and systolic diameter between wild-type and babo knockdown flies (Figure S1M,N). These results suggest that babo and daw regulate both common and distinct aspects of cardiac function.
Figure 2.
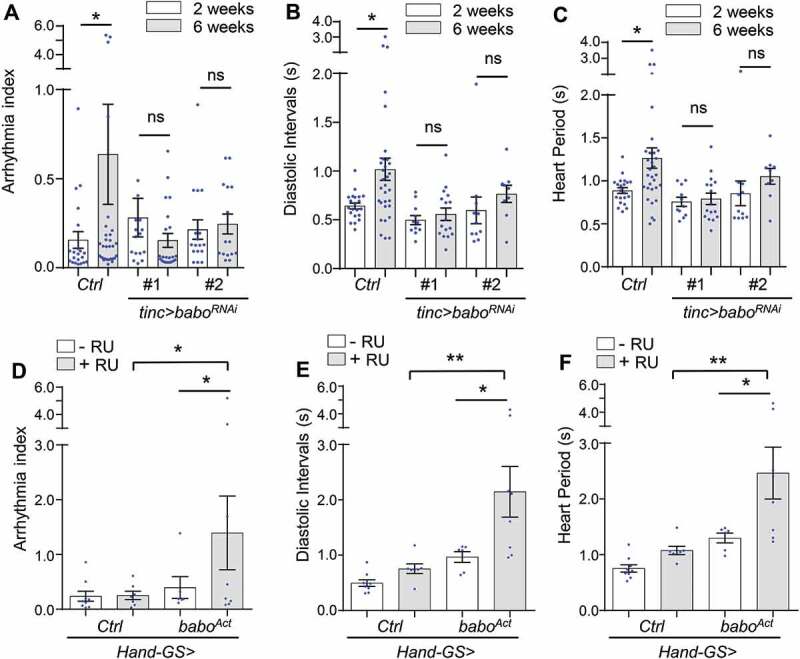
Cardiomyocyte-specific knockdown of INHB/activin receptor babo delays cardiac aging. (A-C) Age-dependent changes in cardiac arrhythmia, diastolic intervals, and heart period in control (Ctrl) and cardiomyocyte-specific babo knockdown flies (babo RNAi). Flies were cultured at 40% relative humidity. tinc-gal4 driver was used. Results from two independent UAS-baboRNAi lines are shown (RNAi #1: BDSC, 25933, RNAi #2: BDSC, 40866). N = 15 ~ 30. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (D-F) Changes in cardiac arrhythmia, diastolic intervals, and heart period in fly hearts expressing constitutively activated babo (baboAct). GeneSwitch heart driver Hand-GS-gal4 was used to induce adult-onset babo activation. RU: RU486 (Mifepristone). Flies were cultured at 40% relative humidity. N = 6 ~ 9. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant).
In a reciprocal experiment, we used a GeneSwitch heart driver (Hand-GS-gal4) to express a constitutively active form of babo (baboAct, or baboQ302D) specifically in adult hearts. GeneSwitch heart driver was used because constitutively expressing active babo in the heart (Hand>baboAct) led to severe developmental arrest at pupal stages (data not shown). We found that cardiac expression of activated babo significantly increased cardiac arrhythmia, diastolic intervals, and heart period already at young ages (Figure 2D–F). Taken together, these results suggest that INHB/activin type I receptor babo plays an important role in age-related cardiomyopathy, especially cardiac arrhythmia, diastolic function, and heart rate.
daw negatively regulates autophagy in fly hearts
Autophagy is one of the key mechanisms in maintaining tissue hemostasis, and its activity normally declines with age [4,6,7]. Our previous studies showed that TGFB-INHB/activin signaling regulates autophagosome formation and Atg8a transcription in Drosophila flight muscle [20]. It is likely that daw regulates cardiac aging by modulating autophagic activity. To first verify the role of daw in autophagy-lysosome activity, we used LysoTracker staining, a well-established autophagy marker in flies [24], to monitor the lysosomal acidification in adult fat body. Consistent with our previous finding, heterozygous daw[11] loss-of-function mutants showed increased LysoTracker staining, suggesting an elevated basal autophagy/lysosome activity (Figure 3A). We furthermore showed that mutation of daw increased autophagic flux, indicated by the induction of lipidated Atg8a (Atg8a-II) upon the treatment of lysosome inhibitor bafilomycin A1 (BafA1) (Figure 3B). Atg8a is the fly homolog of mammalian LC3-GABARAP family proteins. A rabbit monoclonal antibody for human GABARAP (E1J4E) [25 26–27] was used in the lipidation assay. The specificity of the antibody has been previously verified [28] and further confirmed in the present study (Figure S2, also see Methods). Finally, the mosaic analysis revealed that somatic cell clones in larval fat body expressing RNAi against either daw or babo (marked by GFP) showed increased numbers of autophagosome, indicated by mCherry-Atg8a-positive puncta (Figure 3C). These data suggest that TGFB-INHB/activin signaling negatively regulates autophagy and lysosome activities.
Figure 3.
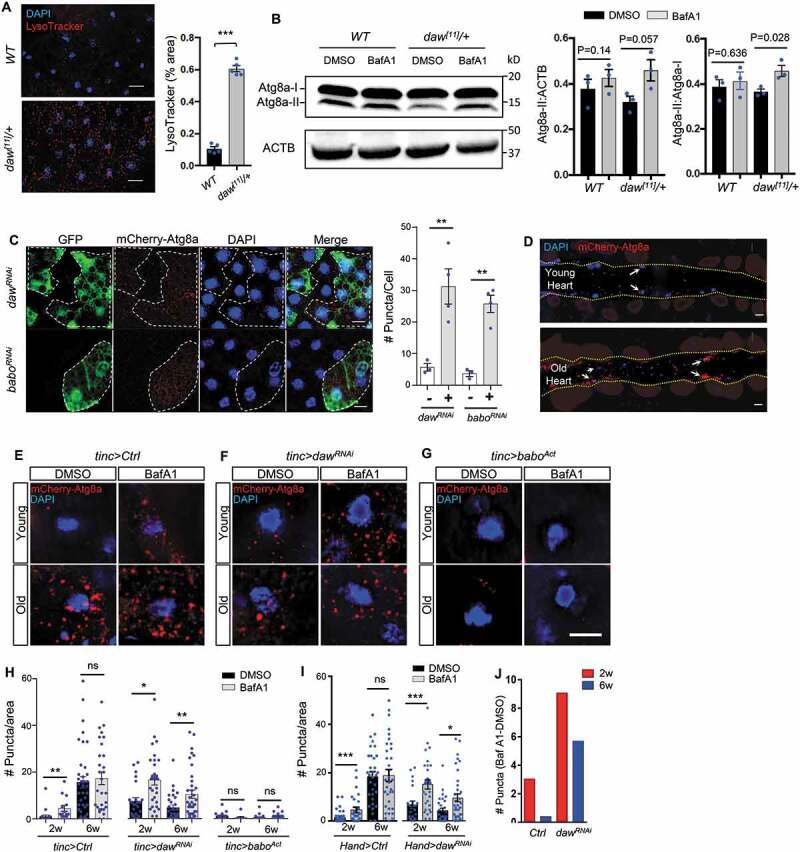
Daw negatively regulates autophagy in fly hearts. (A) Representative images of LysoTracker staining of young adult fat body between wild-type (WT) and heterozygous daw[11]/+ mutants. Scale bar: 20 μm. Quantification shown on the right. N = 5. Student t-test (*** p < 0.001). (B) Western blot analysis on Atg8a lipidation between WT and daw[11]/+ mutants. GABARAP antibodies were used to detect Atg8a. The lower band represents lipidated Atg8a (Atg8a-II), and the upper band represents non-lipidated Atg8a (Atg8a-I). Abdominal fly carcass is incubated with 5 μM of BafA1 prior to western blots. ACTB (actin beta) is used as the loading control. Quantification of band intensity shown on the right. N = 3. Student t-test (*** p < 0.001). (C) Representative images of mosaic analysis on autophagosome staining in larval fat body. Fat body clones were generated by crossing dawRNAi and baboRNAi lines into a FLPout line carrying mCherry-Atg8a reporter (hs-flp; endogenous P-3xmCherry-Atg8a, UAS-GFP/Cyo; Act>CD2> Gal4,UAS-Dcr-2). RNAi Clones are GFP-positive cells (dashed lines). Scale bar: 20 μm. Quantification of autophagosome puncta shown on the right. N = 5. Student t-test (** p < 0.01). (D) Representative images of fly hearts expressing autophagosome reporter mCherry-Atg8a at both young and old ages. Arrows indicate cardiomyocyte nuclei and surrounding autophagosomes. Abdominal segments A2-A3 are shown. Heart tube is located between two yellow dashed lines. (E-G) Representative images of mCherry-Atg8a reporter surrounding cardiomyocyte nuclei in control, dawRNAi, and baboAct flies with or without BafA1 treatment. Semi-intact hearts were incubated with 100 nM of bafilomycin A1 (BafA1) for 2 h prior to immunostaining. Scale bar: 10 μm. (H) Quantification of age-dependent changes in autophagic flux in control, dawRNAi, and baboAct fly hearts. tinc-gal4 was used to drive the expression of mCherry-Atg8a reporter and gene knockdown. N = 5 ~ 7. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (I) Quantification of age-dependent changes in autophagic flux in control and dawRNAi fly hearts. Hand -gal4 was used to drive the expression of mCherry-Atg8a reporter and daw knockdown. N = 5 ~ 7. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (J) Quantification of BafA1-induced mCherry-Atg8a-positive puncta between control and dawRNAi. The data represent the differences in the number of puncta between BafA1 and DMSO treatments.
Next, we examined whether daw plays a similar role in modulating autophagy in fly hearts. Immunostaining with GABARAP (E1J4E) antibodies showed that the number of autophagosome in the heart was higher in daw[11] mutants compared to wild-type (Figure S3A,B). Because autophagy is a dynamic process, the increased number of autophagosome could result from elevated autophagy activities, or blockage of lysosomal degradation [29]. To test these possibilities, we next examined the role of daw in the regulation of autophagic flux (the turnover of autophagosome). We exposed semi-intact fly hearts to BafA1, the V-ATPase inhibitor that blocks both lysosomal acidification and autophagosome-lysosome fusion [30]. We found that BafA1 treatment resulted in a higher induction in the number of Atg8a-positive autophagosome in daw[11] hearts compared to wild-type hearts (Figure S3A–C). Thus, the results suggest that daw negatively regulates autophagic flux in fly hearts.
Knockdown of daw preserves autophagic activity during cardiac aging
Aging tissues show reduced autophagy and accumulated cellular damages. We next examined whether inhibition of INHB/activin-daw signaling in fly hearts can alleviate age-dependent deregulation of autophagy. Using a mCherry-Atg8a reporter line, we observed a large accumulation of Atg8a-positive autophagosome in aged hearts of wild-type flies (Figure 3D,E,H,I). The age-related accumulation of autophagosome was not due to increased expression of the cardiac driver. Because both Hand-gal4 and tinc-gal4 drivers showed either reduced or no change of expression during aging (Figure S1A,B). In fact, the accumulation of autophagosome is likely due to the blockage of autophagosome turnover (autophagic flux), because BafA1 treatment induced Atg8a-positive puncta in young hearts, but not in the old hearts (Figure 3E,H,I,J). To confirm this finding, we monitored autophagosome using an alternative method, the immunostaining with GABARAP antibody (Figure S4A,B). We observed a similar accumulation of autophagosome in aged hearts, and the autophagic flux was also reduced during aging (Figure S4A,B). Furthermore, we found that the receptor protein ref(2)P/SQSTM1/p62, a marker for defective aggrephagy [31], was accumulated in aging hearts and Atg8a mutants (Figure S5), suggesting an age-related impairment in autophagy/lysosome degradation system. We also tested autophagic flux using mCherry-GFP-Atg8a tandem reporter. However, we hardly observed any GFP signals in fly hearts, even after BafA1 treatment (Figure S4C). This is quite different from fat body tissue, where BafA1 treatment significantly induced GFP signals when the tandem reporter was expressed using r4-gal4 (Figure S4D).
Intriguingly, flies with heart-specific daw knockdown maintained high levels of cardiac autophagosome turnover (or active autophagic flux) at both young and old ages, as indicated by the induction of autophagosome number upon BafA1 treatment (Figure 3F,H,I,J). Notably, constitutively activated babo suppressed the autophagic flux in both young and old hearts (Figure 3G,H). Activation of babo also blocked the age-dependent accumulation of autophagosome in old fly hearts (Figure 3G,H), which suggests that besides the regulation of autophagosome turnover (degradation), TGFB-INHB/activin signaling also plays an important role in autophagosome initiation and formation. Taken together, our findings suggest that reduction of INHB/activin-daw signaling promotes autophagosome turnover and preserves autophagic activity during cardiac aging.
Inhibition of autophagy, but not activation of TORC1, blocks daw knockdown-mediated cardioprotection
Since we observed an induction of autophagy in daw knockdown flies, we next asked whether autophagy activity is required for daw-regulated cardiac aging. First, we fed control and daw knockdown flies with a lysosomal inhibitor chloroquine (CQ). As expected, CQ treatment increased cardiac arrhythmia in daw knockdown flies back to the similar levels seen in old wild-type flies (Figure 4A), while CQ treatment is nontoxic and did not lead to any mortality within two weeks of feeding (Figure S5D). To directly test whether autophagy plays any role in daw-regulated cardiac aging, we generated double knockdown flies by combining UAS-daw RNAi and UAS-Atg1 RNAi fly lines. Atg1 is a serine/threonine-protein kinase that is essential for autophagy initiation, and has been previously shown to play a role in regulating cardiac function [32]. Cardiac-specific knockdown of Atg1 increased both cardiac arrhythmia and diastolic intervals at young and old ages (Figure 4B), similar to the fly heart with activated babo (Figure 2D,E). Consistent with CQ treatment, cardiac-specific knockdown of Atg1 attenuated cardioprotective effects of daw RNAi. Simultaneously knockdown of Atg1 and daw in the heart led to an age-dependent increase in arrhythmia similar to control flies (Figure 4C). Atg1 knockdown in daw RNAi background also slightly increased arrhythmia at young ages (Figure 4C). The knockdown efficiency of daw was not affected by combining daw RNAi and Atg1 RNAi lines (Figure S5E). Thus, these results suggest that autophagic activity and Atg1 are required for daw-regulated cardiac aging.
Figure 4.
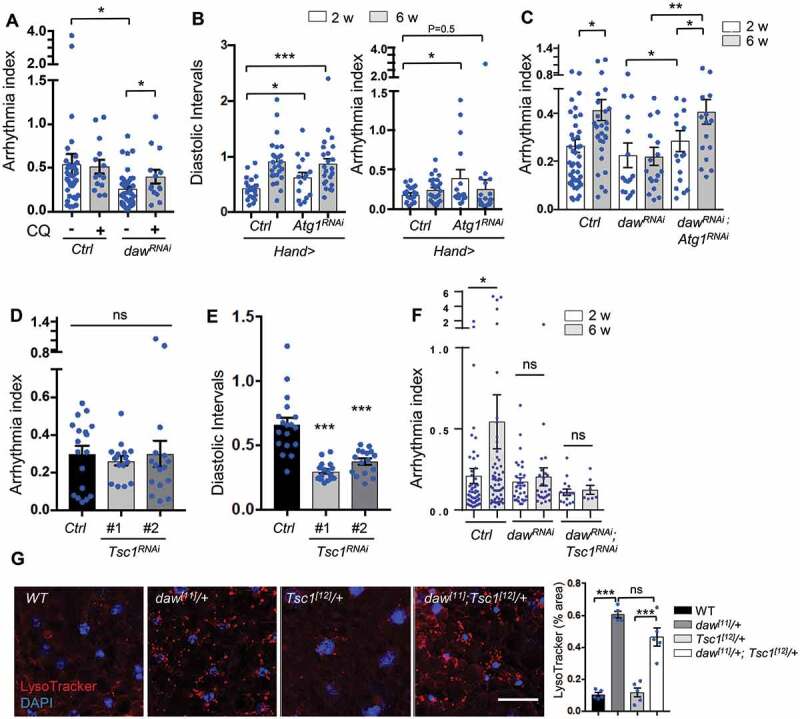
Inhibition of autophagy, but not activation of TORC1, blocks daw knockdown-mediated cardioprotection. (A) Cardiac arrhythmia of chloroquine-treated (20 mM, 24 h) 6-week-old control and daw knockdown flies (Hand-gal4 used). N = 14 ~ 38. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (B) Cardiac diastolic intervals and arrhythmia in heart-specific knockdown of Atg1 at young and old ages (Hand-gal4 used). N = 16 ~ 19. One-way ANOVA (*** p < 0.001, * p < 0.05). (C) Age-dependent changes in cardiac arrhythmia in control, dawRNAi, and dawRNAi; Atg1RNAi flies (tinc-gal4 used). N = 16 ~ 35. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (D-E) Cardiac arrhythmia and diastolic intervals of Tsc1 knockdown flies (tinc-gal4 used). Two independent Tsc1 RNAi lines were used (RNAi-1: BDSC, 52931, RNAi-2: BDSC, 54034). N = 16 ~ 18. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (F) Age-dependent changes in cardiac arrhythmia in control, dawRNAi, and dawRNAi; Tsc1RNAi flies (tinc-gal4 used). N = 7 ~ 30. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (G) Representative images of LysoTracker staining in adult fat body of WT, daw[11]/+, Tsc1[12]/+ and double mutants daw[11]/+; Tsc1[12]/+. Scale bar: 10 μm. Quantification shown on the right. N = 5. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant).
TORC1 is a key negative regulator of autophagy (Figure S6A) [24]. We found that TORC1 activity, indicated by phosphorylation of Thor/EIF4EBP1, was reduced in daw knockdown flies (Figure S6B). We then tested whether daw could regulate autophagy and cardiac aging through TORC1 signaling. If TORC1 acts as a downstream effector of daw to regulate autophagy and cardiac aging, one would expect that activation of TORC1 can attenuate daw knockdown-mediated cardiac protection and autophagy induction. To test this possibility, we first generated double knockdown flies by combining UAS-daw RNAi and UAS-Tsc1 RNAi. Drosophila Tsc1 (TSC complex subunit 1) is a negative regulator of TORC1, knockdown of Tsc1 increased the phosphorylation of Thor/EIF4EBP1 in fly hearts (Figure S6B). The knockdown efficiency of Tsc1 is shown in Figure S6C. Surprisingly, activation of TORC1 via Tsc1 knockdown had no effects on cardiac arrhythmia (Figure 4D), but reduced diastolic intervals at young age (Figure 4E), which is very different from the cardiomyopathy resulted from cardiac-specific activation of INHB/activin receptor babo (Figure 2D,E). Furthermore, knockdown of Tsc1 did not affect the cardioprotective role of daw silencing (Figure 4F). Simultaneously knockdown of Tsc1 and daw showed similar arrhythmia index as daw RNAi alone. In addition, loss-of-function mutant Tsc1[12] did not rescue the elevated LysoTracker staining in daw[11] mutants (Figure 4G). These results suggest that activation of TORC1 is not required for daw-regulated autophagy and cardiac aging.
It is known that inhibition of TORC1 prolongs lifespan [15,33] and attenuates stress-induced cardiomyopathies [11,34 35–36]. However, the specific role of TORC1 in cardiac aging is not well understood. Here, we observed unique alternations of cardiac function in response to activation of TORC1 (via Tsc1 knockdown), including decreased heart period (Tachycardia, fast heart rate), and reduced diastolic intervals (Figures 4E, S6D,E) These cardiac functional changes are not the same as age-induced cardiomyopathies, rather they resemble high-fat-diet-induced cardiac complications where TORC1 is known to play an important role [12]. Intriguingly, flies with cardiac-specific knockdown of Tsc1 showed no age-dependent increases in heart period, diastolic intervals, and arrhythmia (Figure S6D–F). Similar cardiomyopathy phenotypes were also observed in other TORC1 activation mutants, such as 1) knockdown of REPTOR (Repressed by TOR), a recently identified bZIP transcription factor that is negatively regulated by TORC1 [37] (Figure S6G–I), and 2) overexpression of Rheb (Ras Homolog Enriched in Brain), a positive regulator of TORC1 (Figure S6J–L). Thus, it seems that INHB/activin-daw signaling and TORC1 exhibit different effects on cardiac function.
daw genetically interacts with TORC2 (rictor) to control autophagy and cardiac aging
Besides the regulation of p-Thor (Figure S6B), cardiac-specific knockdown of daw increased the phosphorylation of Akt1 (at Ser505) in the heart, the upstream regulator of TORC1 (Figure 5A). On the other hand, activated receptor babo decreased cardiac Akt1 phosphorylation (Figure 5B). It is interesting that the p-Akt1 appeared as fluorescence puncta in fly hearts. The p-Akt1 puncta might be associated with mitochondria, since it has been shown that MTORC2 and Akt1 are localized on mitochondria-associated endoplasmic reticulum (ER) membrane [38]. We further confirmed the regulation of Akt1 phosphorylation by TGFB-INHB/activin signaling using immunoblotting (Figure 5C). Although the activation of babo did not alter Akt1 phosphorylation in western blotting analysis, we think that the lack of changes in Akt1 phosphorylation might be due to the contamination of adjacent tissue (e.g., fat body). It is known that Akt1 can be activated and phosphorylated by two kinases at distinct positions, PDPK1/Pdk1 (3-phosphoinositide dependent protein kinase 1) at Thr308 and MTORC2 at Ser473 (Ser505 in flies) [39]. Cardiac-specific overexpressing rictor can also induced Akt1 phosphorylation (Figure 5D). Interestingly, knockdown of daw and babo upregulated the mRNA expression of rictor in fly hearts (Figure 5E). These results suggest that INHB/activin-daw is a negative regulator of MTORC2 (rictor).
Figure 5.
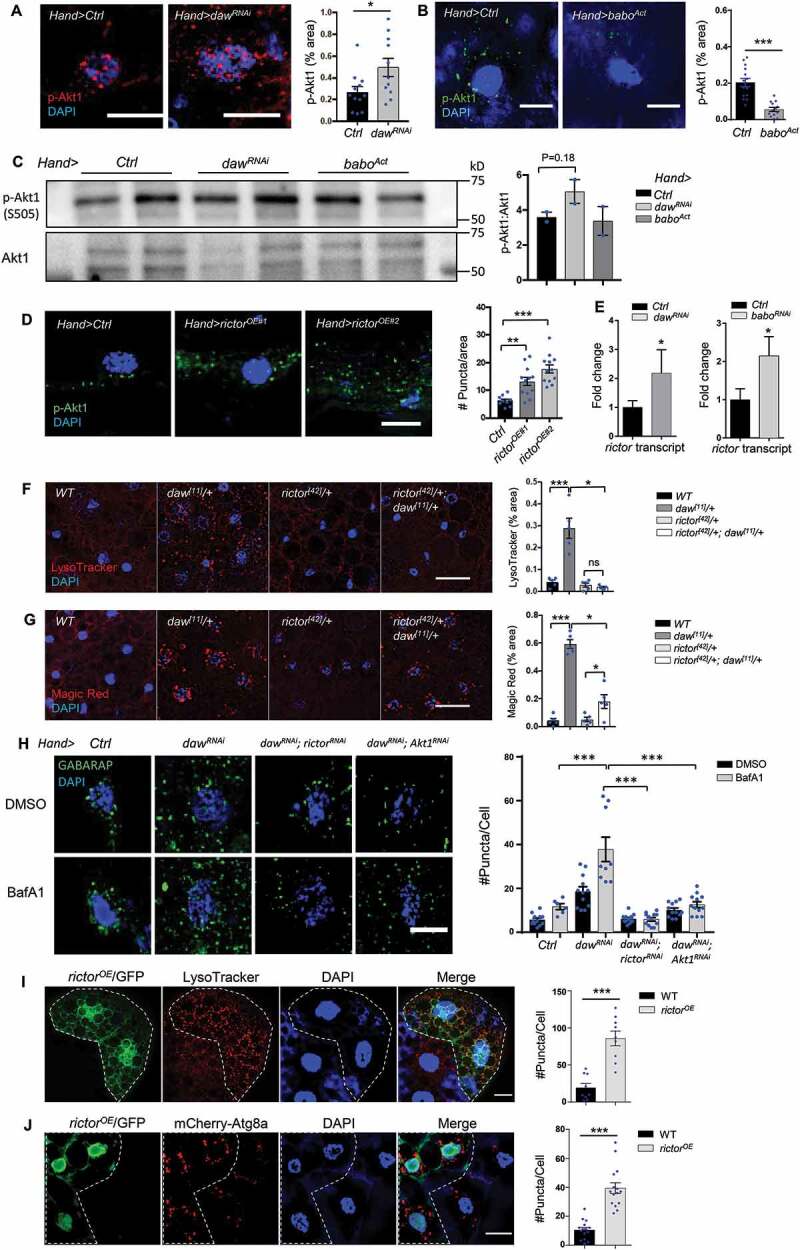
Daw genetically interacts with TORC2 (rictor) to control autophagy. (A) Representative images of p-Akt1 staining in cardiomyocytes of control and daw knockdown flies (Hand-gal4). Scale bar: 10 μm. Quantification shown on the right. N = 5. Student t-test (* p < 0.05). (B) Representative images of p-Akt1 staining in cardiomyocytes of control and babo-activated flies (Hand-gal4). Scale bar: 10 μm. Quantification shown on the right. N = 5. Student t-test (*** p < 0.001). (C) Western blot analysis on Akt1 phosphorylation of the hearts dissected from control, daw knockdown and babo-activated flies (Hand-gal4). Quantification of band intensity shown on the right. N = 3. Student t-test. (D) Representative images of p-Akt1 staining in cardiomyocytes of control and rictor overexpression (Hand-gal4). Two independent rictor overexpression used (#1: UAS-rictor, #2: UAS-HA-rictor [72]). Scale bar: 10 μm. Quantification shown on the right. N = 5. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (E) QRT-PCR analysis of rictor expression in fly hearts with daw and babo knockdown (Hand-gal4). N = 3. Student t-test (* p < 0.05). (F) Representative images of LysoTracker staining in adult fat body of WT, daw[11]/+, rictor[42]/+ and double mutants rictor[42]/+; daw[11]/+. Scale bar: 20 μm. Quantification shown on the right. N = 5. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (G) Representative images of Magic Red staining in adult fat body of WT, daw[11]/+, rictor[42]/+ and double mutants rictor[42]/+; daw[11]/+. Scale bar: 20 μm. Quantification shown on the right. N = 5. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (H) Representative images of GABARAP immunostaining in cardiomyocytes of BafA1-treated control, dawRNAi alone, dawRNAi; rictorRNAi, and dawRNAi; Akt1RNAi flies. Scale bar: 20 μm. Quantification shown on the right. N = 15. Student t-test (*** p < 0.001). (I) Representative images of mosaic analysis of LysoTracker staining in larval fat body. Fat body clones were generated by crossing rictor overexpression lines into a FLPout line (yw, hs-flp, UAS-CD8::GFP; Act>y+>Gal4,UAS-GFP.nls;UAS-Dcr-2). Clones with rictor overexpression are GFP-positive cells (dashed lines). Scale bar: 20 μm. (J) Representative images of mosaic analysis of autophagosome staining in larval fat body. Fat body clones were generated by crossing rictor overexpression lines into a FLPout line carrying mCherry-Atg8a reporter (hs-flp; endogenous P-3x mCherry-Atg8a, UAS-GFP/Cyo; Act>CD2> Gal4,UAS-Dcr-2). Clones with rictor overexpression are GFP-positive cells (dashed lines). Scale bar: 20 μm.
Unlike TORC1, the role of TORC2 in the regulation of autophagy is unclear. Intriguingly, we found that loss-of-function mutants rictor[42] suppressed the high lysosomal activities in daw[11] mutants (Figure 5F,G). Similarly, mutations in Sin1, another core component of TORC2, blocked the elevated LysoTracker staining in daw mutants (Figure S7A,B). Cardiac-specific knockdown of either rictor or Akt1 attenuated the induction of autophagosome formation by daw RNAi in fly hearts (Figure 5H). Furthermore, the mosaic analysis revealed that overexpression of rictor induced lysosome activity (Figure 5I) and increased the number of Atg8a-positive autophagosome in fat body (Figure 5J). Interestingly, unlike Tsc1 mutants, rictor[42] did not suppress starvation-induced LysoTracker staining (Figure S7C). Similarly, activated TGFB/activin signaling (baboAct) did not block starvation-induced lysosome activity (Figure S7D). Taken together, these data suggest that TGFB-INHB/activin signaling and TORC2 (rictor) regulate autophagy differently from TORC1, and rictor acts downstream of TGFB-INHB/activin signaling in the regulation of autophagy.
Lastly, we noticed that the levels of p-Akt1 decreased in aging hearts (Figure 6A), while overexpression of rictor induced autophagic flux in old fly hearts (Figure 6B). Given that rictor positively regulates autophagy, we predict that rictor is a cardiac protective factor. Indeed, cardiac-specific overexpression of rictor prevented age-related increases in diastolic intervals and arrhythmia (Figure 6C,D), while knockdown of rictor induced arrhythmia, especially at old ages (Figure 6E). Knockdown of rictor also attenuated the cardioprotection by daw RNAi (Figure 6F). Furthermore, overexpression of rictor attenuated the elevated diastolic intervals and arrhythmia in fly hearts with constitutively activated babo (Figure 6G–I), suggesting that fly TGFB/activin signaling regulates cardiac aging through TORC2 (rictor) (Figure 6J).
Figure 6.
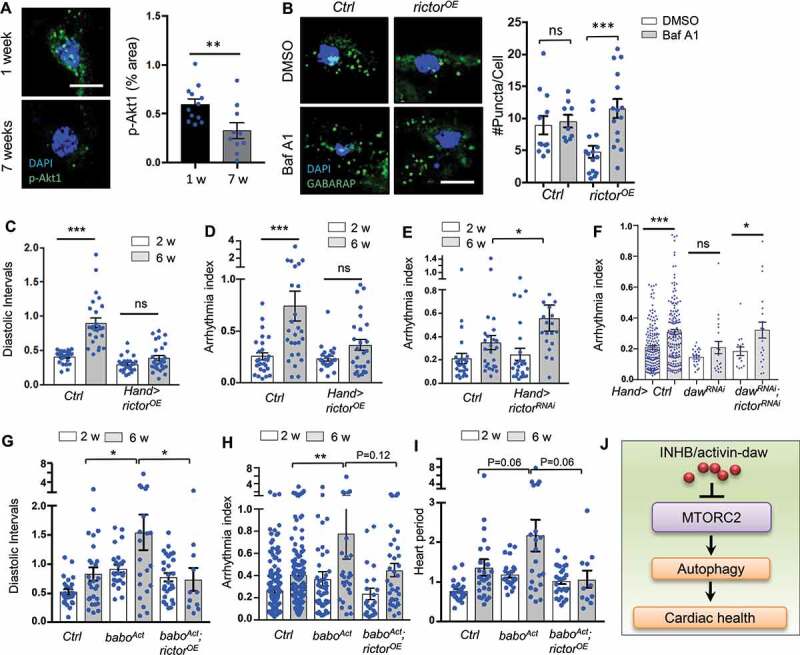
Heart-specific overexpression of rictor preserved cardiac function during aging. (A) Representative images of p-Akt1 staining in young and old cardiomyocytes of wild-type flies. Scale bar: 10 μm. Quantification shown on the right. N = 12. Student t-test (** p < 0.01). (B) Representative images of autophagic flux in control and rictor overexpressing cardiomyocytes at old ages (Hand-gal4). Scale bar: 10 μm. Quantification shown on the right. N = 15. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (C-D) Diastolic intervals and arrhythmia index in flies with heart-specific overexpression of rictor (Hand-gal4). N = 19 ~ 26. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (E) Cardiac arrhythmia index in flies with heart-specific knockdown of rictor. N = 24. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (F) Cardiac arrhythmia index in flies with heart-specific knockdown of daw alone or daw; rictor double knockdown. N = 15 ~ 30. Student t-test (*** p < 0.001, * p < 0.05, ns = not significant). (G-I) Diastolic intervals, arrhythmia index, and heart period in flies with heart-specific expression of baboAct, or baboAct; rictorOE (Hand-gal4). N = 11 ~ 27. One-way ANOVA (*** p < 0.001, ** p < 0.01, * p < 0.05, ns = not significant). (J) Proposed model: INHB/activin-mediated inhibition of MTORC2 as a novel mechanism for age-dependent regulation of autophagy and cardiac health.
Cardiac-specific reduction of INHB/activin-daw and overexpression of rictor prolong lifespan
Tissue-specific hormonal signaling in systemic aging control has been previously reported [40 41–42]. However, whether prolonged organismal lifespan can be achieved by maintaining healthy hearts remains less clear. Here we tested the effects of cardiac-specific knockdown of INHB/activin signaling in longevity control. Knockdown of daw in the heart using two cardiac tissue drivers (tinc-gal4 and Hand-gal4) significantly prolongs lifespan (36.2% and 23.2% extension of mean lifespan compared to control lines) (Figure 7A,B,D). Because daw is a hormonal factor, it remains to be determined whether daw regulates lifespan through cell-autonomous or non-autonomous mechanisms. Interestingly, cardiac-specific overexpression of rictor also extended lifespan (12% extension of mean lifespan) (Figure 7C,D). Together, our findings suggest that cardiac-specific INHB/activin-daw and TORC2 (rictor) signaling pathways play important roles in regulating autophagy, cardiac health, as well as longevity.
Figure 7.
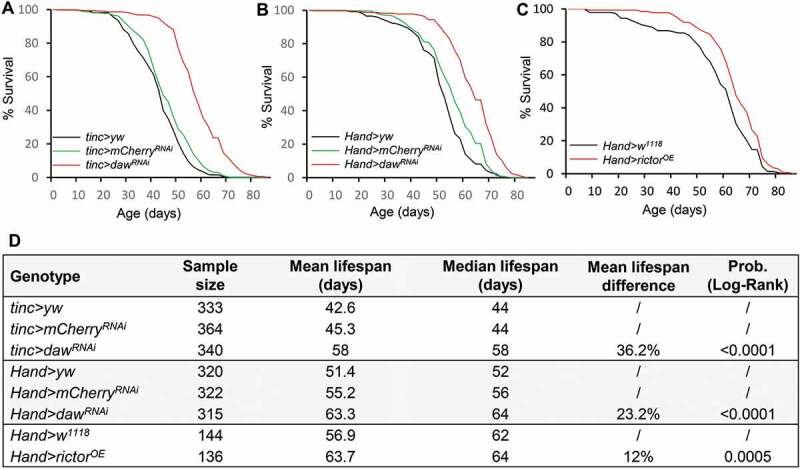
Cardiac-specific knockdown of daw and overexpression of rictor prolong lifespan. (A-B) Survival analysis of cardiac-specific (tinc-gal4 and Hand-gal4) knockdown of daw. Two control lines used (yw and mCherry RNAi) were performed. Log-Rank test, p < 0.0001. (C) Survival analysis of heart-specific (Hand-gal4) overexpression of rictor. Log-Rank test, p = 0.0005. (D) Lifespan table to show sample size, mean and median lifespan of the survival analysis.
Discussion
TGFB signaling plays vital roles in a wide range of human diseases, including cancer and cardiovascular diseases [43]. Originally identified as a reproductive hormone, INHB/activin, a TGFB subfamily member, has become an emerging target for the treatment of many human diseases [44]. In the present study, we discover a novel crosstalk between TGFB-INHB/activin and TORC2 in the regulation of autophagy and cardiac aging in Drosophila. Similar to our previous study in Drosophila flight muscle [20], reduction of TGFB-INHB/activin signaling alleviates age-related cardiac dysfunction, mainly through the regulation of cardiac autophagic activity. Mutant daw flies exhibit high autophagic flux and elevated lysosome activities. Importantly, we show that activation of TORC1 does not block daw knockdown-mediated cardioprotective effects. Instead, INHB/activin-daw signaling interacts with TORC2 subunit rictor to regulate autophagy and cardiac function. Activation of TORC2 alone can promote cardiac autophagy and maintain cardiac function at old ages. Taken together, our studies identify TGFB-INHB/activin signaling as a novel upstream regulator of TORC2, and INHB/activin-mediate inhibition of TORC2 might be a novel mechanism for age-related autophagy impairment and cardiomyopathy.
Although previous studies have linked TGFB family proteins to longevity regulation [20,21,45], the role of TGFB-INHB/activin signaling in cardiac aging is still not well understood. It has been shown that INHBA/ACTIVIN A serum levels significantly increase with age [46]. The mRNA expression of INHBA/ACTIVIN A, as well as serum INHBA/ACTIVIN A levels are positively correlated with heart failure [47,48], and hypertension in the elderly [49]. These findings suggest that activation of TGFB-INHB/activin signaling at old ages might be detrimental to the heart. It is known that INHBA/ACTIVIN A promotes cardiac fibrosis and myocardial damage after ischemia reperfusion [50]. Consistently, mice with heterozygous mutations of ACVR1B/ALK4 are protected from pressure overload-induced fibrosis and dysfunction [51,52]. Inhibition of TGFBR1/ALK5 also reduces myocardial infarction-induced systolic dysfunction and left ventricular remodeling in rats [53]. However, other studies suggest that INHBA/ACTIVIN A protects hearts from hypoxia/reoxygenation- and ischemia/reperfusion-induced cell death [54], even though it can promote cardiac apoptosis at higher concentrations [55]. The findings from our study support the detrimental effects of TGFB-INHB/activin signaling in cardiac function at old ages. This is consistent with the pro-aging action of TGFB and INHB/activin [20,45]. In addition, our results also suggest that daw-regulated cardiac aging through a Smox/SMAD2-independent pathway (Figure 1).
TGFB-INHB/activin signaling has been implicated in the regulation of a wide range of cellular processes, including cell death and proliferation, inflammation, fibrosis, and metabolic homeostasis [44,56 57–58]. The role of TGFB-INHB/activin signaling in the control of autophagy, a key tissue maintenance process, has only been carefully characterized recently [20,59,60]. In Caenorhabditis elegans, the reduction of INHB/activin-like protein DAF-7 suppresses beta-amyloid peptide-induced autophagosome accumulation [59]. Another recent study found that INHBA/ACTIVIN A blocked oxygen-glucose deprivation-induced autophagy through the inhibition of MAPK8/JNK and MAPK14/p38 MAPK pathways in neuronal PC12 cells [60]. Our previous studies show that knockdown of Drosophila INHB/activin-like protein daw in flight muscle induces autophagosome formation and autophagy gene expression [20]. However, how TGFB-INHB/activin signaling regulates autophagy and how this regulation contributes to tissue homeostasis during aging remain largely unknown. Our findings suggest that fly INHB/activin-like protein daw promotes cardiac aging through inhibition of autophagy and TORC2 signaling. Importantly, fly hearts with reduced daw expression maintain high levels of autophagosome turnover (or autophagic flux) throughout the life, whereas the autophagosome turnover is largely blocked in aged hearts of wild-type flies. The active autophagosome turnover at old ages is likely responsible for the healthy hearts seen in daw knockdown flies. Intriguingly, TORC2 but not TORC1 is required for daw-regulated cardiac autophagy. These findings reveal a novel mechanism for age-dependent autophagy regulation that is mediated through the interplay between INHB/activin and TORC2. On the other hand, our results further suggest that TGFB-INHB/activin acts as a key upstream regulator of TORC2 signaling.
The MTOR signaling, in particular, MTORC1, is a well-known autophagy regulator and it inhibits autophagosome formation by increasing the phosphorylation of the serine/threonine protein kinase Atg1 (ULK1/ULK2 in mammals), the initiator of the autophagy machinery [5]. Intriguingly, activation of TORC1 does not block the induction of autophagosome formation in daw knockdown flies, suggesting that there might be unknown TORC1-independent mechanisms involved in daw-regulated autophagy and cardiac aging. TORC1-independent autophagy regulation has been observed in several recent studies [61 62–63]. A genome-wide screen study identifies an important role of class III phosphatidylinositol 3-kinase in autophagy control under normal nutritional conditions [61]. The screen also revealed that growth factor signaling pathways, such as MAPK/ERK and AKT-FOXO3, negatively regulate autophagy by inhibiting the class III phosphatidylinositol 3-kinase cascade. Interestingly, we show that knockdown of daw leads to elevated Akt1 phosphorylation and increased expression of TORC2 subunit rictor, suggesting TORC2-AKT signaling may be involved in daw-regulated autophagy and cardiac aging. Indeed, mutation of rictor blocks the activation of autophagy in daw knockdown flies. MTORC2 is one of the two MTOR complexes that is insensitive to short-term rapamycin treatment, and is involved in glucose homeostasis and actin cytoskeleton reorganization [64,65]. The direct regulatory role of MTORC2 (RICTOR) in autophagy has not been previously established, although several studies suggest that mammalian RICTOR may be involved in autophagy activation under certain circumstances. For example, RICTOR is required for resveratrol-induced autophagy in rat myocardium and H9c2 cardiac myoblast cells [66]. In rat kidney NRK-52E cells, silencing Rictor blocks cisplatin-induced autophagy [67]. Our findings align with these studies suggesting that Drosophila rictor is a positive regulator of autophagy. However, conflicting findings are reported in other studies showing RICTOR negatively regulates autophagosome formation in mouse skeletal muscle [68], and LC3-II levels in human senescent endothelial cells [69]. Together, our results suggest that Drosophila TORC2 (rictor) is a positive regulator of autophagy, which is opposite to what TORC1 does. The distinct regulation of autophagy by 2 MTOR complexes may explain why MTORC1 and MTORC2 differentially control lifespan.
The positive regulation of autophagy by rictor identified in the present study suggests that activation of TORC2 may be cardioprotective. Indeed, we show that cardiac-specific overexpression of rictor slows cardiac aging, similar to daw knockdown. These findings are consistent with two recent studies showing that disruption of mammalian RICTOR induces cardiac dysfunction [13,14]. Cardiac-specific deletion of Rictor in mice induces dilation and decreases fractional shortening by activating MST1 kinase and hippo pathway [14]. Deletion of Rictor decreases lifespan in male mice [16], although conflicting results are reported in C. elegans [70,71]. Interestingly, we show that cardiac-specific overexpression of rictor prolongs Drosophila lifespan, which is consistent with shortened lifespan seen in rictor deletion mice [16]. Besides longevity regulation, it has been shown that Drosophila mutants of rictor and Sin1 decrease the tolerance to heat stress [72] and overexpression of rictor rescues Pink1 knockdown-induced mitochondrial aggregation in Drosophila indirect flight muscle [73]. Conversely, overexpression of PINK1 increases AKT phosphorylation in human SH-SY5Y neuroblastoma cells [74]. The rictor-Pink1 interaction suggests that rictor could be a potential positive regulator for mitophagy, a key process for the mitochondria quality control during aging. Interestingly, we found that p-Akt1 exhibits a puncta pattern in fly hearts. We speculate that the p-Akt1 puncta may be associated with mitochondria and ER subcompartment, since it has been shown that MTORC2 (RICTOR) is localized at the mitochondria-associated ER membrane [38]. Future work is needed to examine whether and how TORC2 (rictor) regulates mitophagy activity to maintain mitochondrial quality in the heart.
Different from TORC2, TORC1 is known to promote age-dependent increases in cardiac dysfunction in Drosophila [11,36]. Cardiac-specific overexpression of TORC1 increases stress-induced heart failure, whereas overexpression of Tsc1 and Tsc2 prevents the age-related increase of stress-induced heart failure [11]. Interestingly, we show that activation of TORC1 produced unique cardiac changes that are different from aging fly hearts. Cardiac-specific activation of TORC1 via knockdown of Tsc1 induced heart rate and decreased heart period, which is similar to cardiac dysfunction caused by high-fat-diet feeding [12]. A recent study found that MTORC1 activity does not increase with age in most tissues of wild-type mice, arguing that the assumption about increased MTOR signaling driving age-related pathology may be overly simplified [75]. Intriguingly, cardioprotective effects of MTOR were previously observed in a mouse model with cardiac-specific overexpression of MTOR (MTOR-Tg) [76]. The MTOR-Tg mice show attenuated inflammatory response and cardiomyopathy upon left ventricular pressure overload induced by transverse aortic constriction. It is unclear whether MTOR-Tg affects only MTORC1 or both MTOR complexes. Thus, the distinct role of MTORC1 and MTORC2 in stress- and aging-induced cardiac dysfunction remains to be further examined.
During normal aging, the heart undergoes complex phenotypic changes such as progressive myocardial remodeling, reduced myocardial contractile capacity, increased left ventricular wall thickness and chamber size, prolonged diastole as well as increased arrhythmia [3]. All of these biological changes can gradually alter cardiac functions and confer vulnerability of the heart to various cardiovascular stresses, thus increasing the chance of developing cardiovascular disease dramatically. The present study reveals an important role of TGFB-INHB/activin signaling pathway in the regulation of autophagy and cardiac aging in Drosophila. We also find an intriguing interaction between INHB/activin and TORC2 in the regulation of cardiac autophagy, as well as a new role of rictor in cardiac aging and longevity control in Drosophila. It remains to be determined how INHB/activin regulates TORC2 (rictor) in fly hearts. It is possible that TGFB-INHB/activin signaling regulates rictor through both transcriptional activation (e.g., INS-FOXO signaling [77]) and post-translational modification. Additionally, cardiac-specific daw knockdown and rictor overexpression extend fly lifespan, suggesting that longevity can be achieved by maintaining a healthy heart. Our findings also suggest that daw itself or other unidentified hormonal factors secreted from cardiomyocytes may play an important role in systemic aging control. Hence, it remains to be determined the mechanistic basis for INHB/activin-TORC2-mediated control of cardiac aging and longevity, which will eventually help the development of therapeutic interventions targeting INHB/activin and TORC2 for the treatment of age-related cardiovascular diseases.
Materials and methods
Fly husbandry and stocks
Flies were maintained at 25°C, 60% relative humidity and 12 h light/dark (40% relative humidity was used in the experiments from Figures 1 and 2, which were performed in Bodmer laboratory). Adults were reared on agar-based diet with 0.8% cornmeal, 10% sugar, and 2.5% yeast (unless otherwise noted). Fly stocks used in the present study are: UAS-daw RNAi (Bloomington Drosophila Stock Center [BDSC], 34974, BDSC, 50911, and Vienna Drosophila Resource Center [105309]), UAS-daw (dawOE, Gift from Michael O’Connor, University of Minnesota Twin Cities), UAS-Smox RNAi (BDSC, 26756), UAS-babo RNAi (BDSC, 25933, 40866), UAS-baboAct/baboQ302D [78], UAS-Tsc1 RNAi (BDSC, 52931, 54034), UAS-REPTOR RNAi (BDSC, 25983), UAS-Rheb (BDSC, 9688), UAS-Atg1 RNAi (BDSC, 26731), UAS-rictor RNAi (BDSC, 36699), daw[11] [79], Tsc1[12] [80], rictor[42] [81], Sin1[e03756] (BDSC, 18188), UAS-rictor [72], UAS-HA-rictor [72], UAS-Akt1 RNAi (BDSC, 31701), Hand/Hand4.2-gal4 [82], and tinc/tinc∆4-gal4 [83], Ugt36A1/Dot-gal4 [84], Hand-GS-gal4 [85], UAS-mCherry-Atg8a (BDSC, 37750), Atg8a∆4 [86], UAS-GFP.nls (BDSC, 4775), fln/IFM-gal4, da-GS-gal4 [87], UAS-mCherry-GFP-Atg8a (Gift from Gábor Juhász, Eotvos Lorand University, r4-gal4 (Gift from Marc Tatar, Brown University).
UAS-daw RNAi (BDSC, 34974) and UAS-babo RNAi (BDSC, 25933) were backcrossed into ywR background for 5 ~ 7 generations, and ywR flies were used as control or wild-type (WT) flies in most of the experiments. For other UAS-RNAi lines that were not backcrossed to ywR, following genotypes were used as control: y1 sc* v1; P[VALIUM20-mCherry]attP2 (BDSC, 35785), y1 v1; P[CaryP]attP2 (BDSC, 36303), or y1 v1; P[CaryP]attP40 (BDSC, 36304). Female flies were used in all experiments. RU486 (Mifepristone; Cayman Chemical, 84371-65-3) were used to activate Hand-GS-Gal4 at a final concentration of 200 µM mixed in food.
Mosaic analysis
Two FLPout lines were used: 1). hs-flp; endogenous P-3x mCherry-Atg8a, UAS-GFP/Cyo; Act>CD2> Gal4,UAS-Dcr-2/TM6) (kindly provided by Gábor Juhász, Eotvos Lorand University). 2). yw, hs-flp, UAS-CD8::GFP; Act>y+>Gal4,UAS-GFP.nls;UAS-Dcr-2/SM6::TM6B (kindly provided by Jun-yuan Ji, Texas A&M University, originally generated by Bruce Edgar, University of Utah). To generate clones, freshly laid eggs (within 4–6 h) were heated in a 37°C water bath for 45 min. Early L3 larvae (84 h after egg laying) were used in the experiments.
Fly heartbeat analysis
To measure cardiac function parameters, semi-intact Drosophila adult fly hearts were prepared according to previously described protocols [22]. High-speed 3000 frames movies were taken at a rate of 100 frames per second using a Hamamatsu ORCA-Flash4.0 digital CMOS camera (Hamamatsu Photonics) on an Olympus BX51WI microscope with a 10X water immersion lens. Hamamatsu EM-CCD 9300 camera was used in the experiments from Figures 1 and 2, which were performed in Bodmer laboratory. The live images that contain heart tub within abdominal A3 segment were processed using HCI imaging software (Hamamatsu Photonics). M-modes and cardiac parameters were generated using SOHA, a MATLAB-based image application as described previously [22]. The M-mode provides a snapshot of the movement of heart wall over time. Cardiac parameters were calculated as below: Diastolic interval (DI) is the duration for each heart relaxation phase (diastole). Heart period (HP) is the pause time between the two consecutive diastoles (HP is the reciprocal of heart rate, HR). Systolic interval (SI) was calculated as the HP minus the DI. Arrhythmia index (AI) is the standard deviation of all HP in each fly normalized to the median HP. Cardiac output was calculated using following equation: (π r[d]2 – π r[s]2) x HR. r(d) is the radius of the heart tube at diastolic phase, while r(s) is the radius of the heart tube at systolic phase. Fractional shortening was calculated as (diastolic diameters – systolic diameters)/diastolic diameters.
Quantitative RT-PCR
RNA extraction and cDNA synthesis were performed using a Cells-to-CT kit (Thermo Fisher Scientific, 44-029-54) from ~15 dissected adult hearts. QRT-PCR was performed with a Quantstudio 3 Real-Time PCR System (Thermo Fisher Scientific). mRNA abundance of each gene was normalized to the expression of RpL32 (Ribosomal protein L32) by the method of comparative CT. Primer sequences are listed in Table S1.
Antibodies and immunostaining
Since commercial antibodies for Drosophila Atg8a show nonspecific staining in immunostaining of fly tissues (data not shown), we used a human GABARAP (E1J4E) antibody (1:300) (Cell Signaling Technology [CST], 13733) to stain and detect endogenous Drosophila Atg8a. The epitope used to generate GABARAP antibody has identical amino acid sequences to Drosophila Atg8a protein (personal communication with Cell Signaling Technology and see sequence alignment in Figure S2A). This antibody has been previously used in several Drosophila autophagy studies [25–27], and verified by Kim et al. [28]. We further verified the specificity of the GABARAP antibody using an Atg8a loss-of-function mutant Atg8a∆4 (Figure S2B–D). Other antibodies used for immunostaining are list below: ref(2)P (1:1000) (previously generated by the Nezis laboratory) [31], p-Smox/SMAD2 antibody (1:500; CST, 3108), p-EIF4EBP1 (1:300; CST, 2855), p-AKT1 (Ser473, 1:300; CST, 4060), Alexa Fluor 488-conjugated phalloidin (or Alexa Fluor 594) for F-actin staining (Thermo Fisher Scientific, A12379, A12381). All fluorescence-conjugated secondary antibodies were from Jackson ImmunoResearch (Alexa Fluor 488, 711-545-152; Alexa Fluor 594, 711-585-152).
For immunostaining, adult female flies were collected and dissected in PBS (Fisher Scientific, AAJ61196AP). Hearts were fixed in 4% paraformaldehyde for 15 min at room temperature (RT). After washing in PBS with 0.1% Triton X-100 (Fisher Scientific, BP151-100) (PBST), the fixed hearted were blocked in 5% normal goat serum (NGS; Jackson ImmunoResearch, 005-000-121) diluted in PBST for 1 h at RT. Hearts were then washed with PBST and incubated overnight at 4°C with primary antibodies diluted in 5% NGS. After washing with PBST, the samples were incubated for 2 h at RT with appropriate fluorescence-conjugated secondary antibodies. Hearts were mounted in ProLong Gold anti-fade reagent (Thermo Fisher Scientific, P36930) before being imaged using an epifluorescence-equipped Olympus BX51WI microscope.
Imaging analysis and quantification
For imaging analysis and quantification, fluorescence images were first processed using the deconvolution module in Olympus cellSens Dimensions software, and then the number of puncta or fluorescent area/intensity in a selected region of interest (ROI, ~707 µm2) surrounding cardiomyocyte nuclei were measured with the “Measure and Count” module in Olympus cellSens software. To quantify autophagosome in fat body, the number of puncta per cell was measured. The imaging quantifications were done single- or double-blind.
LysoTracker and Magic Red staining
Acidic organelles (including autolysosome) were monitored by staining tissue with 100 nM LysoTracker Red DND-99 (Thermo Fisher Scientific, L7528) for 5 min at room temperature. Lysosomal CtsB1 (cathepsin B1) activities were monitored using Magic Red Cathepsin-B Assay kit (ImmunoChemistry Technologies, 938) following the manufacturer’s manual. Nuclei were stained with either DAPI or Hoechst 33342 (1 µg/ml) (ImmunoChemistry Technologies, 938).
Western blotting for Atg8a lipidation and Akt1 phosphorylation
Antibodies for western blot included: ACTB (actin beta) antibody (1:2000) (CST, 4967), GABARAP (E1J4E) antibody (1:2000) (CST, 13733), Drosophila p-Akt1 (Ser505) (1:1000) (CST, 4054), AKT1 (Pan) (1:2000) (CST, 4691), and HRP-conjugated secondary antibodies (Jackson ImmunoResearch, 711-035-152). For Atg8a lipidation, flies were first transferred to centrifuge tubes containing 1x laemmli buffer (Bio-Rad Laboratories, 1610737) (10 µl buffer for each milligram of fly), and heated at 100°C for 3 min. The samples were then homogenized and heated again at 100°C for 3 min. After the centrifuge at 14000 x g for 5 min, supernatants were collected and loaded onto Mini-PROTEAN precast gels (Bio-Rad Laboratories, 456–1095). Following incubation with primary and secondary antibodies, the blots were visualized with Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, PI34577). For Akt1 phosphorylation, RIPA lysis buffer (Thermo Fisher Scientific, PI36978) was used to extract protein samples.
Bafilomycin A1 and chloroquine treatment
For bafilomycin A1 treatment, semi-intact hearts were incubated with 100 nM of bafilomycin A1 (Fisher Scientific, AAJ61835MCR) in artificial hemolymph (buffer receipt in [22]) for 2 h at room temperature prior to the appropriate immunostaining. DMSO was used as a control.
For chloroquine treatments, 100 µl of 20 mM chloroquine diphosphate salt, CQ (Fisher Scientific, ICN19391925) was added onto the fly food. Flies were fed with chloroquine for at least 24 h prior to the cardiac analysis or the western blots. To test the toxicity of CQ treatment, we performed a survival analysis by feeding ywR flies with 20 mM CQ and observed the mortality within a period of within two weeks (about 100 flies per treatment). Flies were transferred to fresh CQ food every day and fly mortality was recorded daily.
Demography and survival analysis
Newly enclosed female flies were allowed to mate for two days, then separated from males and assigned to replicate one-liter demography cages at a density of 100–125 flies per cage. Three independent cages were set-up per genotype. Food was changed every 2 d, at which time dead flies were removed from the cage and counted. Survival analysis was conducted with JMP statistical software (SAS Institute), and data from replicate cages were combined. Mortality distributions were compared by Log-rank test.
Statistical analysis
GraphPad Prism (GraphPad Software) was used for statistical analysis. To compare the mean value of treatment groups versus that of control, either student t-test or one-way ANOVA was performed using Tukey multiple comparison. The effects of mutants during aging were analyzed by two-way ANOVA, including Tukey multiple comparisons test. In SOHA analysis, the outliers were identified using Robust regression and Outlier removal (ROUT) method (Q = 1%) prior to the data analysis.
Supplementary Material
Acknowledgments
We thank Bloomington Drosophila Stock Center, Harvard Medical School, and Drosophila Genomics Resource Center for fly stocks and cDNA clones. We thank Michael O’Connor for INHB/activin reagents and fly stocks, Duojia Pan, Stocker Hugo, Jongkyeong Chung, Gábor Juhász, Jun-yuan Ji, Bruce Edgar, Marc Tatar for fly stocks. Marc Tatar for help with Demography and survival analysis.
Funding Statement
This work was supported by NIH/NIA R00 AG048016, R01 AG058741 and AFAR Research Grants for Junior Faculty to HB, NIH/NHLBI R01 HL132241 to KO, NIH/NHLBI R01 HL054732 to RB, BBSRC grants BB/L006324/1 and BB/P007856/1 to IPN.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Dai DF, Chen T, Johnson SC, et al. Cardiac aging: from molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2012;16:1492–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].North BJ, Sinclair DA.. The intersection between aging and cardiovascular disease. Circ Res. 2012;110:1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lakatta EG, Levy D.. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. [DOI] [PubMed] [Google Scholar]
- [4].Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. [DOI] [PubMed] [Google Scholar]
- [5].Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res. 2014;24:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chang JT, Kumsta C, Hellman AB, et al. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. Elife. 2017;6:e18459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Taneike M, Yamaguchi O, Nakai A, et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy. 2010;6:600–606. [DOI] [PubMed] [Google Scholar]
- [8].Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dutta D, Xu J, Kim JS, et al. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity. Autophagy. 2013;9:328–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Takahara T, Maeda T. Evolutionarily conserved regulation of TOR signalling. J Biochem. 2013;154:1–10. [DOI] [PubMed] [Google Scholar]
- [11].Wessells R, Fitzgerald E, Piazza N, et al. d4eBP acts downstream of both dTOR and dFoxo to modulate cardiac functional aging in Drosophila. Aging Cell. 2009;8:542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Birse RT, Choi J, Reardon K, et al. High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metab. 2010;12:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shende P, Xu L, Morandi C, et al. Cardiac mTOR complex 2 preserves ventricular function in pressure-overload hypertrophy. Cardiovasc Res. 2016;109:103–114. [DOI] [PubMed] [Google Scholar]
- [14].Sciarretta S, Zhai P, Maejima Y, et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 2015;11:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kaeberlein M, Powers RW 3rd, Steffen KK, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. [DOI] [PubMed] [Google Scholar]
- [16].Lamming DW, Mihaylova MM, Katajisto P, et al. Depletion of rictor, an essential protein component of mTORC2, decreases male lifespan. Aging Cell. 2014;13:911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Upadhyay A, Moss-Taylor L, Kim MJ, et al. TGF-β family signaling in drosophila. Cold Spring Harb Perspect Biol. 2017;9(9). pii: a022152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis. 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- [19].Lyu G, Guan Y, Zhang C, et al. TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat Commun. 2018;9:2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bai H, Kang P, Hernandez AM, et al. Activin signaling targeted by insulin/dFOXO regulates aging and muscle proteostasis in Drosophila. PLoS Genet. 2013;9:e1003941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Demontis F, Patel VK, Swindell WR, et al. Intertissue control of the nucleolus via a myokine-dependent longevity pathway. Cell Rep. 2014;7:1481–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fink M, Callol-Massot C, Chu A, et al. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques. 2009;46:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hu J, Wang X, Wei SM, et al. Activin A stimulates the proliferation and differentiation of cardiac fibroblasts via the ERK1/2 and p38-MAPK pathways. Eur J Pharmacol. 2016;789:319–327. [DOI] [PubMed] [Google Scholar]
- [24].Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. [DOI] [PubMed] [Google Scholar]
- [25].Ratliff EP, Barekat A, Lipinski MM, et al. Brain trauma and autophagy: what flies and mice can teach us about conserved responses. Autophagy. 2016;12:2256–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ratliff EP, Kotzebue RW, Molina B, et al. Assessing basal and acute autophagic responses in the adult drosophila nervous system: the impact of gender, genetics and diet on endogenous pathway profiles. PLoS One. 2016;11:e0164239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Barekat A, Gonzalez A, Mauntz RE, et al. Using Drosophila as an integrated model to study mild repetitive traumatic brain injury. Sci Rep. 2016;6:25252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kim M, Semple I, Kim B, et al. Drosophila Gyf/GRB10 interacting GYF protein is an autophagy regulator that controls neuron and muscle homeostasis. Autophagy. 2015;11:1358–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mauvezin C, Nagy P, Juhasz G, et al. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nezis IP, Simonsen A, Sagona AP, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee JH, Budanov AV, Park EJ, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shioi T, McMullen JR, Tarnavski O, et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–1670. [DOI] [PubMed] [Google Scholar]
- [35].Ding Y, Sun X, Huang W, et al. Haploinsufficiency of target of rapamycin attenuates cardiomyopathies in adult zebrafish. Circ Res. 2011;109:658–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Luong N, Davies CR, Wessells RJ, et al. Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metab. 2006;4:133–142. [DOI] [PubMed] [Google Scholar]
- [37].Tiebe M, Lutz M, De La Garza A, et al. REPTOR and REPTOR-BP regulate organismal metabolism and transcription downstream of TORC1. Dev Cell. 2015;33:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Betz C, Stracka D, Prescianotto-Baschong C, et al. Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A. 2013;110:12526–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. [DOI] [PubMed] [Google Scholar]
- [40].Hwangbo DS, Gershman B, Tu MP, et al. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–566. [DOI] [PubMed] [Google Scholar]
- [41].Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang G, Li J, Purkayastha S, et al. Hypothalamic programming of systemic ageing involving IKK-β, NF-kappaB and GnRH. Nature. 2013;497:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Tsuchida K, Nakatani M, Hitachi K, et al. Activin signaling as an emerging target for therapeutic interventions. Cell Commun Signal. 2009;7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shaw WM, Luo S, Landis J, et al. The C. elegans TGF-β dauer pathway regulates longevity via insulin signaling. Curr Biol. 2007;17:1635–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Baccarelli A, Morpurgo PS, Corsi A, et al. Activin A serum levels and aging of the pituitary-gonadal axis: a cross-sectional study in middle-aged and elderly healthy subjects. Exp Gerontol. 2001;36:1403–1412. [DOI] [PubMed] [Google Scholar]
- [47].Yndestad A, Ueland T, Oie E, et al. Elevated levels of activin A in heart failure: potential role in myocardial remodeling. Circulation. 2004;109:1379–1385. [DOI] [PubMed] [Google Scholar]
- [48].Wei Q, Wang YN, Liu HY, et al. The expression and role of activin A and follistatin in heart failure rats after myocardial infarction. Int J Cardiol. 2013;168:2994–2997. [DOI] [PubMed] [Google Scholar]
- [49].Tsai YL, Chang CC, Liu LK, et al. The association between serum activin A levels and hypertension in the elderly: a cross-sectional analysis from I-Lan longitudinal aging study. Am J Hypertens. 2018;31(3):369–374. [DOI] [PubMed] [Google Scholar]
- [50].Chen Y, Rothnie C, Spring D, et al. Regulation and actions of activin A and follistatin in myocardial ischaemia-reperfusion injury. Cytokine. 2014;69:255–262. [DOI] [PubMed] [Google Scholar]
- [51].Li CY, Chen YH, Wang Q, et al. Partial inhibition of activin receptor-like kinase 4 attenuates pressure overload-induced cardiac fibrosis and improves cardiac function. J Hypertens. 2016;34:1766–1777. [DOI] [PubMed] [Google Scholar]
- [52].Chen YH, Wang Q, Li CY, et al. Haplodeficiency of activin receptor-like kinase 4 alleviates myocardial infarction-induced cardiac fibrosis and preserves cardiac function. J Mol Cell Cardiol. 2017;105:1–11. [DOI] [PubMed] [Google Scholar]
- [53].Tan SM, Zhang Y, Connelly KA, et al. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;298:H1415–1425. [DOI] [PubMed] [Google Scholar]
- [54].Oshima Y, Ouchi N, Shimano M, et al. Activin A and follistatin-like 3 determine the susceptibility of heart to ischemic injury. Circulation. 2009;120:1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu M, Mao C, Li J, et al. Effects of the activin A-follistatin system on myocardial cell apoptosis through the endoplasmic reticulum stress pathway in heart failure. Int J Mol Sci. 2017;18(2). pii: E374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ghosh AC, O’Connor MB. Systemic activin signaling independently regulates sugar homeostasis, cellular metabolism, and pH balance in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2014;111:5729–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chng WB, Bou Sleiman MS, Schupfer F, et al. Transforming growth factor β/activin signaling functions as a sugar-sensing feedback loop to regulate digestive enzyme expression. Cell Rep. 2014;9:336–348. [DOI] [PubMed] [Google Scholar]
- [58].Werner S, Alzheimer C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006;17:157–171. [DOI] [PubMed] [Google Scholar]
- [59].Florez-McClure ML, Link CD. Erratum to: florez-McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD. Decreased insulin-receptor signaling promotes the autophagic degradation of β-amyloid peptide in C. elegans. Autophagy 2007; 3:569-80. Autophagy. 2009;5:132–133. [DOI] [PubMed] [Google Scholar]
- [60].Xue LX, Xu ZH, Wang JQ, et al. Activin A/Smads signaling pathway negatively regulates Oxygen Glucose Deprivation-induced autophagy via suppression of JNK and p38 MAPK pathways in neuronal PC12 cells. Biochem Biophys Res Commun. 2016;480:355–361. [DOI] [PubMed] [Google Scholar]
- [61].Lipinski MM, Hoffman G, Ng A, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010;18:1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Li Y, Xu M, Ding X, et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat Cell Biol. 2016;18:1065–1077. [DOI] [PubMed] [Google Scholar]
- [63].Henson SM, Lanna A, Riddell NE, et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8(+) T cells. J Clin Invest. 2014;124:4004–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gaubitz C, Prouteau M, Kusmider B, et al. TORC2 structure and function. Trends Biochem Sci. 2016;41:532–545. [DOI] [PubMed] [Google Scholar]
- [66].Gurusamy N, Lekli I, Mukherjee S, et al. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Li J, Xu Z, Jiang L, et al. Rictor/mTORC2 protects against cisplatin-induced tubular cell death and acute kidney injury. Kidney Int. 2014;86:86–102. [DOI] [PubMed] [Google Scholar]
- [68].Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. [DOI] [PubMed] [Google Scholar]
- [69].Xiong Y, Yepuri G, Forbiteh M, et al. ARG2 impairs endothelial autophagy through regulation of MTOR and PRKAA/AMPK signaling in advanced atherosclerosis. Autophagy. 2014;10:2223–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Soukas AA, Kane EA, Carr CE, et al. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 2009;23:496–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Robida-Stubbs S, Glover-Cutter K, Lamming DW, et al. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Jevtov I, Zacharogianni M, van Oorschot MM, et al. TORC2 mediates the heat stress response in Drosophila by promoting the formation of stress granules. J Cell Sci. 2015;128:2497–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wu Z, Sawada T, Shiba K, et al. Tricornered/NDR kinase signaling mediates PINK1-directed mitochondrial quality control and tissue maintenance. Genes Dev. 2013;27:157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Murata H, Sakaguchi M, Jin Y, et al. A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via mTORC2. J Biol Chem. 2011;286:7182–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Baar EL, Carbajal KA, Ong IM, et al. Sex- and tissue-specific changes in mTOR signaling with age in C57BL/6J mice. Aging Cell. 2016;15:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Song X, Kusakari Y, Xiao CY, et al. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299:C1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chen CC, Jeon SM, Bhaskar PT, et al. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell. 2010;18:592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Brummel T, Abdollah S, Haerry TE, et al. The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev. 1999;13:98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Serpe M, O’Connor MB. The metalloprotease tolloid-related and its TGF-β-like substrate Dawdle regulate Drosophila motoneuron axon guidance. Development. 2006;133:4969–4979. [DOI] [PubMed] [Google Scholar]
- [80].Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001;15:1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lee G, Chung J. Discrete functions of rictor and raptor in cell growth regulation in Drosophila. Biochem Biophys Res Commun. 2007;357:1154–1159. [DOI] [PubMed] [Google Scholar]
- [82].Han Z, Olson EN. Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Development. 2005;132:3525–3536. [DOI] [PubMed] [Google Scholar]
- [83].Lo PC, Frasch M. A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identities in the dorsal vessel of Drosophila. Mech Dev. 2001;104:49–60. [DOI] [PubMed] [Google Scholar]
- [84].Zhang F, Zhao Y, Han Z. An in vivo functional analysis system for renal gene discovery in Drosophila pericardial nephrocytes. J Am Soc Nephrol. 2013;24:191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Monnier V, Iche-Torres M, Rera M, et al. dJun and Vri/dNFIL3 are major regulators of cardiac aging in Drosophila. PLoS Genet. 2012;8:e1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pircs K, Nagy P, Varga A, et al. Advantages and limitations of different p62-based assays for estimating autophagic activity in Drosophila. PLoS One. 2012;7:e44214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Tricoire H, Battisti V, Trannoy S, et al. The steroid hormone receptor EcR finely modulates Drosophila lifespan during adulthood in a sex-specific manner. Mech Ageing Dev. 2009;130:547–552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.