

ABSTRACT
Biogenesis of autophagosomes is the early step of macroautophagy/autophagy and requires membrane acquisition mainly from ER-Golgi-sourced precursor vesicles. Matured autophagosomes fuse with lysosomes for final degradation. However, how this selective fusion is determined remains elusive. Here, we identified Sac1 by a high throughput screen in Saccharomyces cerevisiae to show it was critical for autophagosome-lysosome fusion through its PtdIns4P phosphatase activity. Sac1 deficiency caused a dramatic increase of PtdIns4P at early Golgi apparatus and abnormal incorporation of PtdIns4P into Atg9 vesicles and autophagosomes, which caused failure to recruit SNARE proteins for autophagosome fusion with vacuoles. Sac1 function in autophagy was highly conserved from yeast to mammalian cells. Our work thus suggested that correct upstream lipid incorporation was important for downstream fusion step of autophagy and that Sac1 played a critical and ancient role in this surveillance of lipid integration.
Abbreviations: Ape1: aminopeptidase Ι; ATG: autophagy related; EBSS: Earle’s balanced salt solution; ER: endoplasmic reticulum; ERGIC: Golgi apparatus and ER-Golgi intermediate compartment; HOPS: homotypic fusion and protein sorting complex; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; PtdIns3P: phosphatidylinositol-3-phosphate; PtdIns4K: phosphoinositide-4-kinase; PtdIns4P: phosphatidylinositol-4-phosphate; SD-N: nitrogen starvation medium; SNARE: soluble N-ethylamide-sensitive factor attachment protein receptor
KEYWORDS: autophagosome-lysosome fusion, ER, Golgi, PtdIns4P, Sac1
Introduction
The macroautophagy/autophagy process is highly conserved among eukaryotes from yeast to humans [1] and plays fundamental roles in a variety of both physiological and pathological conditions [2]. Autophagy is initiated at a special region associated with the endoplasmic reticulum (ER), Golgi apparatus and ER-Golgi intermediate compartment (ERGIC) [3]. The autophagosomes fuse with the lysosomes (in mammalian cells) or the vacuoles (in yeast and plants) for final degradation [4,5].
Autophagosomes are special as they are generated de novo. This unique formation indicates the importance of controlling lipids, mainly phosphatidylinositol, for use in autophagosome formation and final fusion by specific membranes. The concerted function of various phosphatidylinositol kinases and phosphatases generates the unique PIP signature of distinct membrane compartments [6]. In addition to PtdIns3P, PtdIns4P is another abundant PI phosphate isoform in cells and is generated by PtdIns4K. A steep PtdIns4P gradient from the Golgi (high) to the ER (low) promotes the PtdIns4P-dependent post-Golgi secretory trafficking pathway [7].
Here, we embarked on a high throughput screen for new autophagy genes and identified Sac1, a PtdIns4P hydrolase, that exerted a critical and ancient role in the autophagy pathway. Our findings clearly demonstrated that controlling lipid incorporation into autophagosomes at the biogenesis step was crucial for their downstream fusion with lysosomes.
Results
Screening for novel, essential components of autophagy
Approximately 40 ATG genes have been found in S. cerevisiae [1,8]. Using cell viability assay, we classified the ATG genes into two categories: essential factors and nonessential ones (Figure 1A). The mitochondria regulator Vms1 was used as control, showing its effect on cell viability but not in autophagy [9–11]. In wild-type (WT) and mutant cells lacking nonessential ATG genes, autophagic degradation of cytoplastic GFP-fusion proteins such as GFP-Atg8 proceeded normally based on GFP cleavage assays [12]; but in mutants lacking essential factors Atg1 or Atg7 (atg1∆, atg7∆), the autophagic degradation was completely blocked (Figure 1B). We performed a genome-wide screen with the yeast knockout collection that covers 4857 genes (Figure 1C and S1A). Using this screen, we identified Sac1, a lipid phosphatase that dephosphorylates phosphatidylinositol-4-phosphate (PtdIns4P), as a novel candidate for autophagic essential factor.
Figure 1.
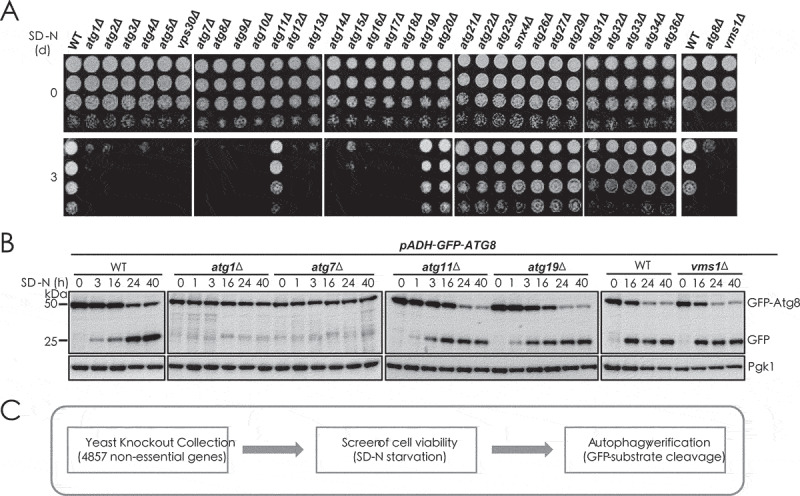
Screening for novel essential factors of autophagy process. (A) WT and indicated ATG gene and VMS1 gene-deleted yeast cells were starved for 3 d in SD-N medium and then detected for cell viability by serial dilution spotting and growth on YPD plates. (B) GFP cleavage assays were performed to determine the vacuole degradation of cytosolic GFP-Atg8 by autophagy in WT and indicated yeast cells. Immunoblotting was done with anti-GFP and anti-Pgk1 antibodies. The positions of full-length GFP-Atg8 and cleaved free GFP are indicated. The Pgk1 protein was used as loading control. The cleaved GFP moiety indicates autophagic degradation. (C) Flow chart of the genome-wide screening for novel essential factors of autophagy in yeast cells
Identification of Sac1 as a novel, essential component for autophagy
Yeast Sac1 has been first identified as a type II transmembrane protein in the ER (Figure 2A). The N-terminal catalytic domain (termed the SAC domain) has PtdIns4P phosphatase activity and faces to the cytosol [13–15]. Deficiency of Sac1 caused a total blockage of autophagy similar to that observed with the loss of known essential factors Pep4, Atg1, Atg9 and Ypt7 (Figure 2B–E, S2A and S2B). Yeast cells lacking Sac1 showed an obvious accumulation of dot signal of GFP-Atg8 (autophagosome marker) that was abolished by the additional loss of Atg1, but not Atg15 (Figure 2F). Protease degradation protection assays suggested that the accumulated autophagosomes in SAC1-deleted cells were closed (Figure S2C). This phenomenon prompted us to determine whether Sac1 functioned as part of SNARE or HOPS complex to mediate the fusion of autophagosomes with vacuoles [16,17]. Mostly, SNARE and HOPS complex components also function in the fusion of endocytic vesicles with vacuole. We found that Sac1 was not involved in the transfer of endocytic substrate GFP-Sna3 into vacuole for degradation, showing that Sac1 was different from Ypt7 or SNARE/HOPS complex components (Figure S2D–F). Interestingly, there was no dramatic blockage of autophagy in mutants lacking one, or even two, of the other types of phosphatidylinositol-phosphate hydrolases (Figure 2G and S2G).
Figure 2.
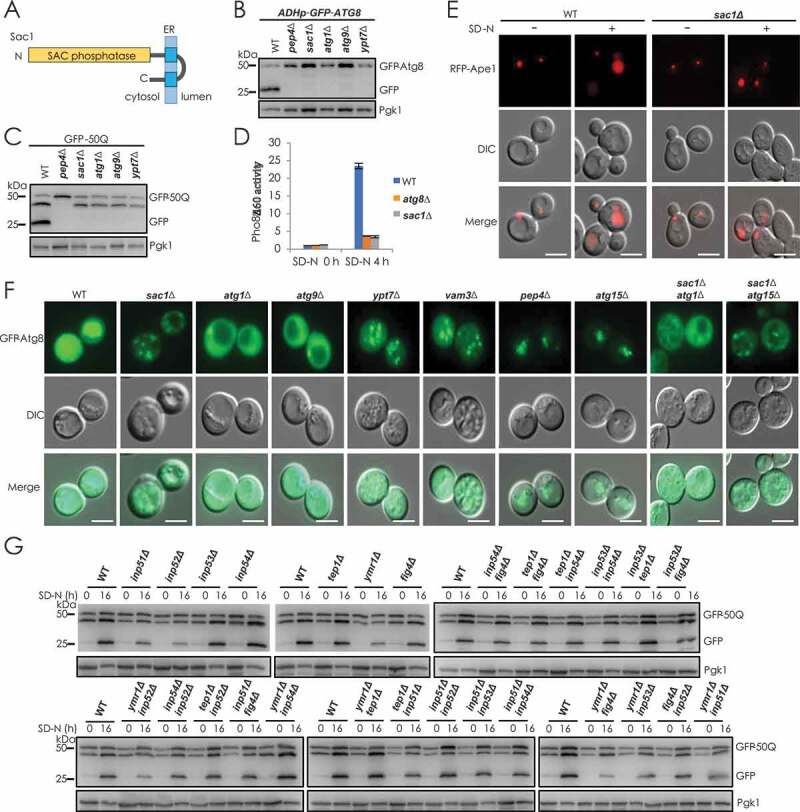
Identification of yeast Sac1 being a novel and essential factor for autophagy. (A) Domain organization of yeast Sac1. (B) N-terminally GFP-tagged Atg8 under control of ADH promoter was checked for its autophagic degradation in indicated yeast cells by GFP-cleavage assays after 16 h starvation. Yeast cells with PEP4, ATG1, ATG9 or YPT7 gene deleted were used as positive controls. (C) Validation of Sac1-dependent degradation of autophagic substrate GFP-50Q (50 poly-glutamine) as in B. (D) Detection of Sac1-dependent transport of autophagic substrate Pho8Δ60 by ALP assays. (E) Autophagic transport of substrate Ape1 was blocked in Sac1-deficient cells. Autophagy substrate Ape1 was labeled with RFP and expressed in WT and sac1Δcells. Its translocation to vacuole by the autophagy process leading to the detection of RFP signal in vacuoles and blocked autophagy in sac1∆ cells caused cytosolic retention of Ape1 aggregates. Experiments were conducted for at least three times and representative images were shown. Scale bars: 5 μm. (F) Deficiency of Sac1 causes accumulation of autophagosomes in yeast cells. Autophagosome marker GFP-Atg8 expressed in indicated cells was observed by fluorescence examination after 16 h starvation. In WT cells, GFP-Atg8 was transferred to vacuole by autophagy leading to the detection of GFP signal in vacuoles. The GFP-Atg8 dots in ypt7∆, vam3∆ or in sac1∆ cells showed blockage of autophagosome fusion and diffused GFP-Atg8 in atg1∆ or atg9∆ cells showed blockage of autophagosome formation. The accumulated autophagosomes in sac1∆ cells were abolished by further loss of Atg1. Scale bars: 5 μm. (G) WT and single or double deletion of indicated PI phosphatase yeast cells were determined for autophagic degradation of GFP-50Q
From all these lines of evidence, we concluded that Sac1, indeed, exerted a pivotal role in autophagy process, and its deficiency caused total blockage of autophagy and dramatic accumulation of autophagosomes in cells.
Golgi location and PtdIns4P phosphatase activity of Sac1 was indispensable for its function in autophagy
To clarify the role of Sac1 in autophagy, we tested different variants of Sac1 either with catalytic domain deleted or with transmembrane domain and Golgi-targeting sequence deleted (Figure 3A). The full-length of Sac1 restored autophagic degradation and starvation resistance in sac1∆ cells (Figure S3A–D). However, Sac1 with the catalytic SAC domain truncated or lacking either the TMs or the Golgi-targeting sequence could not recover autophagy in sac1 null cells (Figure 3A–C, S3E and S3F). Similarly, the catalytic point mutant of Sac1 could not restore autophagic degradation or starvation resistance in yeast cells lacking Sac1 (Figure 3D,E and S3G). PtdIns4P-specific probe GFP-PHPLEKHA3 showed obvious punctate localization, and a higher number and intensity of punctate was observed in cells lacking Sac1 (Figure 3F).
Figure 3.
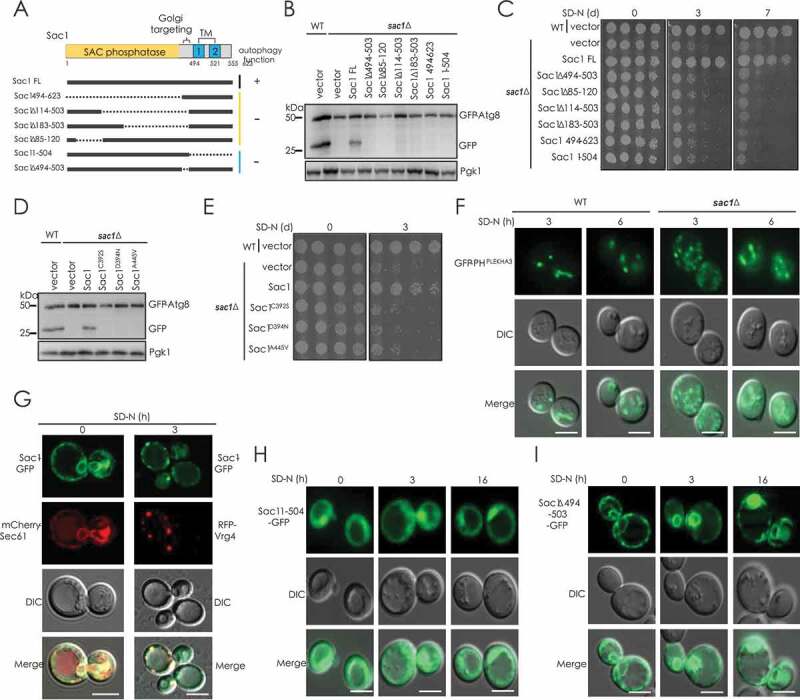
Both Golgi localization and PtdIns4P phosphatase activity of Sac1 were key for its function in autophagy. (A) Schematic representation of yeast Sac1 and indicated truncates checked for function in autophagy. (B and C) Sac1 mutants with catalytic domain truncated or with localization domain truncated could not restore autophagy and cell resistance to nitrogen starvation in sac1∆ cells. (D and E) Sac1 mutants with a point mutation in catalytic domain lost the ability to restore autophagy and cell resistance to nitrogen starvation in sac1∆ cells. (F) PtdIns4P level is increased in sac1∆ cells detected by PtdIns4P-specific probe GFP-PHPLEKHA3. Scale bars: 5 μm. (G) Sac1 was tagged with GFP and its co-localization with ER marker Sec61 and Golgi marker Vrg4 was detected before and after starvation. (H and I) Localization of differently truncated Sac1 was observed before and after nitrogen starvation. Scale bars: 5 μm
Sac1 is previously shown to be an ER-localized enzyme that could transfer to Golgi in certain situations, such as in cells arriving late log phase [18]. Indeed, we observed that nitrogen starvation induced the absence of Sac1 from ER and greatly promoted its early Golgi translocation (Figure 3G). Two Sac1 mutants, showing either diffuse cytosolic distribution (TM truncated, 1–504) or ER retention (Golgi-targeting motif truncated, ∆494-503), could not restore autophagy in SAC1-deleted cells (Figure 3A,H,J). When searching for Sac1-interacting proteins at starvation condition, Sec31, a component of ER-Golgi trafficking COP-II complex, was found to bind Sac1 strongly (Figure S3H-J). This was in line with previous high throughput studies that also identify Sac1-Sec31 interaction [19,20]. Knockdown of Sec31 caused loss of Golgi-localization of Sac1 and blockage of autophagy (Figure S3K–M).
Together, these data thus suggested that Sac1 function in autophagy depends on its PtdIns4P phosphatase activity and specific early Golgi localization. This is interesting as Sac1 deficiency blocked the fusion of autophagosomes with vacuoles, a late step in autophagy process, while this PtdIns4P hydrolase exerted its role at Golgi apparatus that generally provides membrane precursor for autophagosome formation at the early step of autophagy process.
SAC1 deletion caused abnormal integration of PtdIns4P into autophagosomes and blockage of SNARE recruitment
We then analyzed how Sac1 deficiency blocked the fusion of autophagosomes with vacuoles. PtdIns4P has been found to be located at late Golgi to facilitate secretion [21,22], and SAC1 deletion had little or no effect on that (Figure S4A and S4B). However, it showed accumulation of higher levels of PtdIns4P at early Golgi apparatus in SAC1-deleted yeast cells compared to WT cells (Figure 4A and S4C). Since Golgi is considered as membrane sources for autophagosomes through trafficking of Atg9 vesicles that provide membrane resources for autophagosomes [23,24], we analyzed whether Sac1 deficiency caused PtdIns4P could integrate into Atg9 vesicles that transfer lipid for autophagosomes. Dramatically, more PtdIns4P was found with Atg9 vesicles in sac1∆ cells (Figure 4B and S4D). The co-localization of PtdIns4P with Atg9 vesicles in sac1∆ cells was suggested not to be caused by retention of Atg9 at Golgi, as no co-localization of Atg9 with Golgi markers was found (Figure S4E and S4F). The integration of PtdIns4P into Atg9 vesicles was in line with the observation that more PtdIns4P was found with autophagosomes in sac1∆ cells compared to ypt7∆ cells (Figure 4C and S4G). These results indicated that Sac1 deficiency caused abnormal incorporation of PtdIns4P into Atg9 vesicles and autophagosomes. As Sac1 deficiency caused blockage of autophagosome fusion with vacuoles, we detected the autophagosome recruitment of Vam7 and Ykt6, two components of SNARE complex mediating the fusion of autophagosomes with vacuoles [25–28]. The Vam7 and Ykt6 were observed in co-localization with autophagosomes (Atg8 dots) in ypt7∆ cells but not in ypt7Δsac1∆ cells (Figure 4D and S4H-J). Unlike the deletion of SNARE PEP12, GTPase YPT7 and HOPS components (PEP3, PEP5, VPS16 and VPS33) that caused blockage of both autophagy and endocytosis pathways, SAC1 deletion had no effect on endocytosis, which suggested that these proteins functioned normally in sac1∆ cells (Figure S4K). This was further strengthened by the observation that these components distributed similarly in WT and sac1∆ cells (Figure S4L-O).
Figure 4.
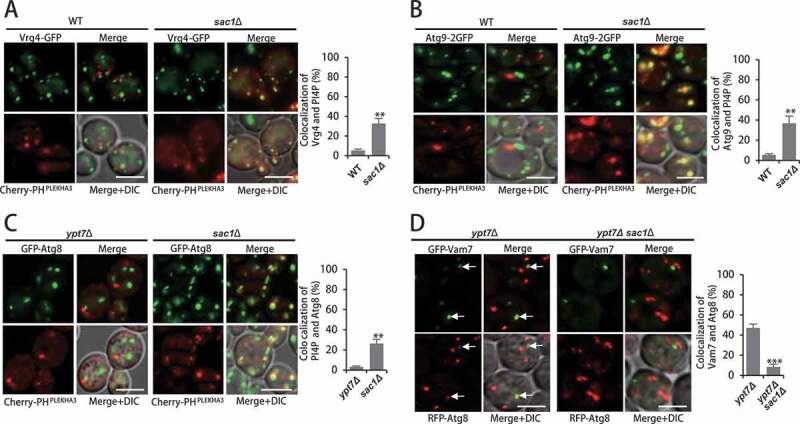
SAC1 deletion caused abnormal integration of PtdIns4P into autophagosomes and blockage of SNARE recruitment. (A) Sac1 deficiency caused accumulation of PtdIns4P at early Golgi apparatus. PtdIns4P probe Cherry-PHPLEKHA3 and GFP-tagged early Golgi marker Vrg4 were observed in WT and sac1∆ cells after 2 h starvation. Experiments were conducted for at least three times and representative images were shown. Quantification of co-localization was shown on the right. Scale bars: 5 μm. **P < 0.01. (B) Sac1 deficiency caused incorporation of PtdIns4P into Atg9 vesicles. PtdIns4P probe Cherry-PHPLEKHA3 and C-terminal chromosomally 2GFP-tagged Atg9 were observed in WT and sac1∆ cells after 2 h starvation. Quantification of co-localization was shown on the right. Scale bars: 5 μm. **P < 0.01. (C) Sac1 deficiency caused abnormal integration of PtdIns4P into autophagosomes. PtdIns4P probe Cherry-PHPLEKHA3 and GFP-tagged Atg8 were observed in ypt7∆ and sac1∆ cells after 2 h starvation. Representative images were shown. Quantification of co-localization was shown on the right. Scale bars: 5 μm. **P < 0.01. (D) Sac1 deficiency blocked recruitment of SNARE component Vam7. GFP-tagged Vam7 and RFP-tagged Atg8 were observed for co-localization in ypt7∆ and ypt7Δsac1∆ cells after 2 h starvation. Representative images were shown. Quantification of co-localization was shown on the right. Scale bars: 5 μm. ***P < 0.001
Combining the above results with the fact that both Golgi location and PtdIns4P phosphatase activity of Sac1 were needed for its function in autophagy, we speculated Sac1 hydrolyzed PtdIns4P at early Golgi to make sure the membrane sources for autophagosome formation with low levels of PtdIns4P. The autophagosome with correct lipid ingredients (no or less PtdIns4P) then recruited SNARE for its fusion with vacuoles.
Sac1 function in autophagy was highly conserved across eukaryotes
We next searched for Sac1 orthologs in other higher eukaryotes through bioinformatics analyses by sequence homology algorithm (protein-protein BLAST, BlastP) [29,30]. The search revealed highly conserved orthologs of Sac1 in diverse eukaryotic species (Figure S5A). The widespread existence of such putative orthologs suggested the evolutionary conservation of Sac1 sequence in eukaryotic cells (Figure S5B). All Sac1 orthologs presented the same architecture, with a SAC phosphatase catalytic domain at the N-terminus and two TM domains at the C-terminus (Figure 5A). A simple phylogenetic tree was constructed, showing the evolutionary conservation of Sac1 proteins in different organisms (Figure 5B). Strikingly, when expressed in yeast, all of the Sac1 orthologs not only restored the autophagic degradation but also suppressed the hypersensitivity of Sac1-deficient yeast cells subjected to starvation (Figure 5C,D). The function of human SACM1L in autophagy in yeast was shown to be dependent on its hydrolytic activity (Figure 5E,). The constantly Golgi-localized SACM1LK2A mutant was as potent as WT (Figure 5E,F). Knockdown of SAC-1 in Caenorhabditis elegans also caused blockage of autophagic degradation (Figure 5G).
Figure 5.
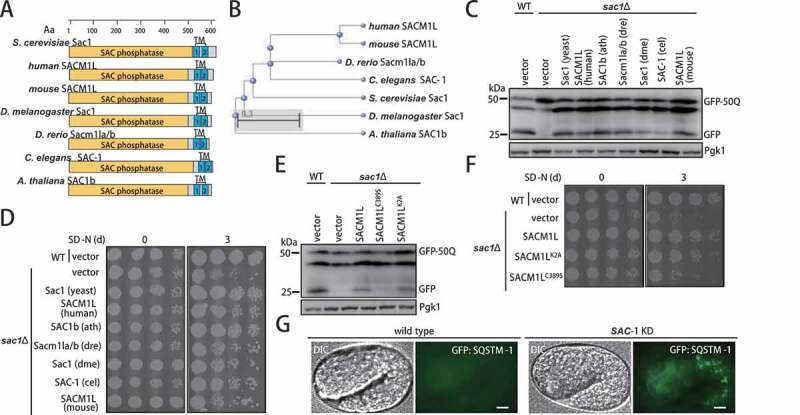
Sac1 was highly conserved across eukaryotes to function in autophagy. (A) Schematic map of protein domains of Sac1 proteins in yeast and higher eukaryotic organisms. S. cerevisiae: Saccharomyces cerevisiae, human: Homo sapiens, mouse: Mus musculus, D. melanogaster: Drosophila melanogaster, D. rerio: Danio rerio, C. elegans: Caenorhabditis elegans, A. thaliana: Arabidopsis thaliana. (B) Phylogenetic tree of Sac1 orthologs in different species. (C) Sac1 orthologs from complex eukaryotic species could restore autophagy in yeast sac1∆ cells. yeast: Saccharomyces cerevisiae, human: Homo sapiens, ath: Arabidopsis thaliana, dre: Danio rerio, dme: Drosophila melanogaster, cel: Caenorhabditis elegans, mouse: Mus musculus. (D) Expression of Sac1 orthologs rescued yeast cell from vulnerability to nitrogen starvation caused by lacking Sac1. (E) Human SACM1L depended on its phosphatase enzyme activity and Golgi-localization for restore of autophagy. GFP cleavage assays were performed using catalytic mutant (C389S) or Golgi-retention mutant (K2A) of human SACM1L. (F) Human SACM1L restored yeast cell resistance to starvation depending on its phosphatase enzyme activity and Golgi localization. (G) Autophagic substrate GFP::SQSTM1-labeled aggregates were ectopically accumulated in C. elegans embryos with SAC-1 knockdown. Representative images were shown. Scale bars: 5 μm
Thus, SAC1 proteins from more complex eukaryotes fulfilled both bioinformatics and functional criteria for being true orthologs of S. cerevisiae Sac1.
Human SACM1L regulates autophagosome fusion with lysosomes
Expression of SACM1L was knocked down in human HeLa cells by siRNA (Figure S6A and S6B). We observed significant accumulation of SQSTM1/p62 (a substrate receptor degraded by autophagy) and LC3-II (autophagosome membrane marker protein) caused by SACM1L knockdown in both resting cells and autophagy-stimulated cells with EBSS starvation treatment (Figure 6A,B and S6C). Higher levels of PtdIns4P was detected in HeLa cells transfected with SACM1L-targeting siRNA compared to control cells (Figure 6C). An abundance of LC3-positive dots indicated accumulated autophagosomes in SACM1L knockdown cells (Figure 6D), which was confirmed by EM observation of autophagosomes (Figure 6E). Furthermore, autophagy flux assays were conducted by measuring the localization of fusion protein GFP-RFP-LC3. Increased autophagy induced by EBSS starvation caused high intensity of RFP signal and low GFP signal (Figure 6F). These results suggested that the fusion step was blocked by SACM1L deficiency, which was probably caused by abnormal integration of PtdIns4P into autophagosomes shown by dramatic co-localization signal of PtdIns4P and LC3 dots (Figure S6D).
Figure 6.
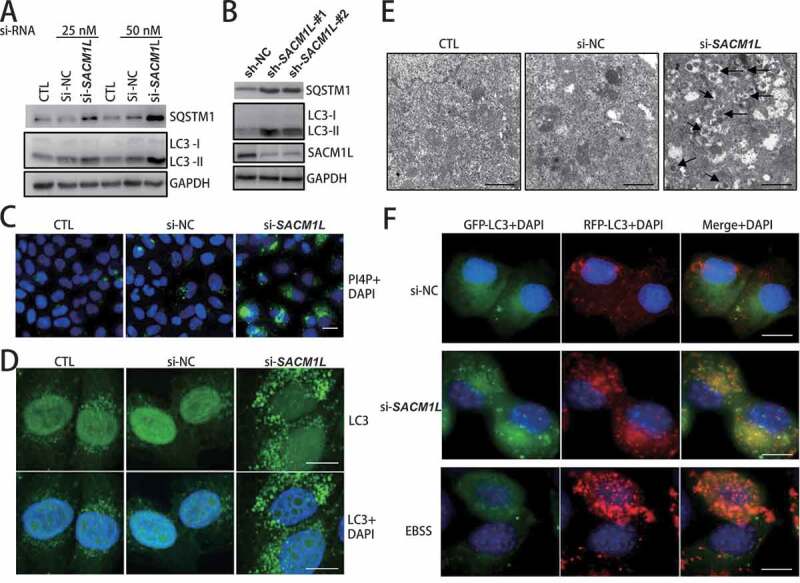
Human SACM1L functioned in autophagy through regulating autophagosome fusion with lysosomes. (A and B) Knockdown of human SACM1L in HeLa cells resulted in accumulation of SQSTM1 and LC3. siRNA- or shRNA-targeting human SACM1L were transfected into HeLa cells for 48 h and protein levels of autophagic substrate SQSTM1 and autophagosome marker LC3 (human homolog of yeast Atg8) were detected with specific antibodies. siRNA or shRNA with scrambled sequence transfected cells (NC) or no transfection cells (CTL) were used as negative controls. (C) SACM1L deficiency caused a dramatic increase of PtdIns4P levels in HeLa cells. HeLa cells were transfected with siRNAs indicated for 48 h. PtdIns4P-specific antibodies were used for immuno-fluorescence observation. Experiments were conducted for at least three times and representative images were shown. Scale bars: 10 μm. (D) Knockdown of human SACM1L caused accumulated autophagosomes in HeLa cells. HeLa cells were transfected with siRNAs indicated for 48 h and LC3 dots (representing autophagosomes) were observed through immunofluorescence observation. Representative images were shown. Scale bars: 10 μm. (E) Knockdown of human SACM1L caused accumulated autophagosomes in HeLa cells. Control cells and SACM1L knockdown cells were analyzed by transmission electron microscopy. Arrows point to the accumulated autophagosomes in cells. Representative images from three replicated experiments were shown. Scale bars: 0.5 μm. (F) Knockdown of human SACM1L blocked autophagosome fusion with lysosomes in HeLa cells. GFP-RFP-LC3-expressing HeLa cells were transfected with indicated siRNAs or subject to starvation with EBSS media. Autophagosome marker LC3 was monitored by fluorescence microscope. Higher level of RFP-LC3 signal and lower level of GFP-LC3 signal indicated the stimulated autophagy flux while the similar signal levels of RFP-LC3 and GFP-L3 (showing yellow in merge) indicated the blocked fusion step of autophagy flux. Representative images were shown. Scale bars: 10 μm
From these lines of evidence, we thus concluded that SACM1L indeed played an essential role in mammalian autophagy pathway and that its deficiency blocked fusion of autophagosomes with lysosomes resulting from abnormal incorporation of PtdIns4P into autophagosomes.
Discussion
Given that autophagy is highly conserved, we were motivated to screen for novel factors of autophagy core machinery in yeast cells. The motivation was also coupled with the hope that the potentially identified core factors in yeast might have hitherto overlooked mammalian homologs with pivotal roles in autophagy. We were particularly interested in Sac1 based on its PtdIns4P phosphatase activity and unique intracellular localization and found both were prerequisites for Sac1 function in autophagy. Upon autophagy activation, yeast Sac1 and human SACM1L traffics to Golgi and ERGIC [31], where membrane lipid resources are generated for the formation of autophagosomes. Interestingly, deficiency of yeast Sac1 or its vertebrate orthologs evidently showed the blockage of autophagosome fusion with vacuole/lysosome, which is a late step in the autophagy process. This finding was explained by the observation that loss of Sac1 caused accumulated higher levels of PtdIns4P at early Golgi/ERGIC and its abnormal integration into autophagosomes, which resulted in defects in the recruitment of SNARE components for fusion.
Another potential reason for restricting PtdIns4P from autophagosomes may involve avoidance of changing lipid homeostasis of lysosome membrane after fusion, as increased PtdIns4P on lysosome membrane disrupts its morphology and function [32–34]. Interestingly, it has been reported that PtdIns4Ks catalyzing PtdIns4P generation are important for the autophagy pathway [35,36]. The dynamic localization of PtdIns4P at endoplasmic reticulum exit site (ERES) promotes the production and trafficking of COPII vesicles to ERGIC [37], where they serve as membrane precursors for autophagosome formation. We thus proposed an “ER in Golgi out” working model of PtdIns4P in autophagy. Dynamic PtdIns4P at ER integrated into and facilitated the budding of COPII vesicles and its subsequent movement to ERGIC, where Sac1 then dephosphorylated PtdIns4P into PI to ensure the absence of PtdIns4P in precursor vesicles thereby promoting the formation of autophagosomes that could finally fuse with lysosomes. This special “ER in Golgi out” regulation of PtdIns4P thereby opened new avenues for a clear understanding of autophagy initiation. Sac1 was perhaps tailored for this dynamic and spatial regulation of PtdIns4P since it was the only PI phosphatase embodying the critical TM domains [14].
Interestingly, it has been shown recently that in mammalian cells, the PtdIns4-kinase PI4KB/PI4KIIIβ could interact with ATG9A vesicles and promotes its delivery of PtdIns4P to the autophagosome initiation sites [38]. Human SACM1L is also found to bind ATG9A vesicles in this study. We speculated that the PtdIns4-kinases and hydrolase (Sac1) might be coordinately involved in the autophagy process in a temporal and/or spatial manner through regulating PtdIns4P integration into Atg9 vesicles. At the Golgi apparatus, where both PtdIns4-kinases and Sac1 localized, PtdIns4P was generated by PtdIns4-kinases and integrated into Atg9 vesicle to promote its exit from and trafficking through Golgi. However, before the Atg9 vesicles leaving Golgi, Sac1 functioned to lower down PtdIns4P levels, so that Atg9 vesicles carried appropriate lipid to sites of autophagosome formation. This surveillance of PtdIns4P by Sac1 possibly happened at early Golgi since Sac1 located thereafter autophagy activation, and specifically, PtdIns4P in this region was accumulated by SAC1 deletion. The long flexible linker between the catalytic domain and the membrane anchors of Sac1 allows it to hydrolyze PtdIns4P in nearby membranes [15], possibly Atg9 vesicles.
Abnormal PtdIns4P homeostasis is linked with neurodegeneration diseases, pathogen invasions and cancers [39,40]. Sac1/SACM1L is the only hydrolase catalyzing dephosphorylation of PtdIns4P [14]. Our studies here provided much-needed insight into the pivotal roles of SAC1 in neurodegeneration progression through function in autophagy. The expression of the human SACM1L gene is coordinated with the aging of neural systems [41]. In a genome-wide association study (GWAS) used to search for genetic variations affecting the susceptibility to side effects of antidepressant therapy, the top 10 SNPs (p = 4.98r10x7, q = 0.023) are found to be located in the SACM1L gene [42]. These data emphasized the potential neuro-disease-related role of SACM1L.
Taken together, our study provided a detailed molecular mechanism and strong genetic evidence revealing the indispensable and conserved role of Sac1/SACM1L in the autophagy pathway. We proposed the role of Sac1 in autophagy as a dynamic and potent “PtdIns4P guardian” functioning in autophagy (Figure 7A). When autophagy was induced by starvation in wild type cells, Sac1 trafficked to early Golgi mediated by COP-II vesicles to hydrolyze PtdIns4P there. Then the Golgi-sourced Atg9 vesicles with low levels of PtdIns4P were incorporated into autophagosomes. SNARE complex was next recruited to autophagosomes for their fusion with lysosomes. However, when Sac1 was depleted, the higher level of PtdIns4P in the early Golgi led to abnormal integration of PtdIns4P-containing Atg9 vesicles into autophagosomes. The PtdIns4P-enriched autophagosomes failed to recruit SNARE complex and thus could not fuse with lysosomes, which eventually led to blockage of autophagy process.
Figure 7.
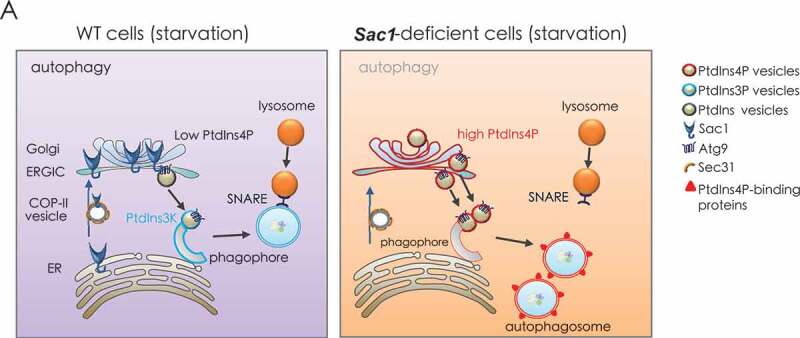
A proposed model for Sac1 function in autophagy through restraining PtdIns4P from incorporation into autophagosomes. Left, when wild type cells were under starvation conditions, Sac1 trafficked to Golgi by COP-II vesicles to hydrolyze PtdIns4P there. Then the Golgi sourced lipid vesicles with low levels of PtdIns4P were incorporated into autophagosomes. SNARE complex was next recruited to autophagosomes for their fusion with lysosomes. Right, when Sac1 was depleted, the higher level of PtdIns4P in the Golgi led to abnormal integration of PtdIns4P-containing vesicles into autophagosomes. The PtdIns4P enriched autophagosomes s failed to recruit SNARE complex and thus could not fuse with lysosomes
Materials and methods
Yeast strains and manipulations and constructs
Yeast strains and constructs used in this study are listed in Table S1 and S2. Standard protocols were used for yeast manipulations. Yeast cultures were inoculated from overnight cultures, grown using standard growth conditions and media. If not indicated otherwise, cells were cultured at 30°C in YP (Sangon Biotech, A610961 for yeast extract and A505247 for peptone) D-media containing glucose (Sangon Biotech, A600219) (2%) as carbon source. For autophagy induction by starvation, cells were cultured to exponential growth phase in YPD-media and then switched into SD-N medium (synthetic minimal medium lacking nitrogen; 0.17% yeast nitrogen base, without amino acids and ammonium sulfate (Sangon Biotech, A600505), supplemented with 2% glucose) for indicated times (hours, h). Chromosomally knockout strains were constructed by a PCR-based strategy. Standard cloning and site-directed mutagenesis techniques were used. For observation of fluorescent tag-labeled proteins, yeast cells were grown in synthetic complete (SC) medium (0.17%YNB, 2% glucose, and 0.2% amino acids) and observed by fluorescence microscopy before and after SD-N starvation. Images were taken by a fully automated Zeiss inverted microscope (Observer 7 or LSM880). Yeast cells were first cultured in YPD-media to the log phase and then switched to synthetic minimal medium lacking nitrogen (SD-N) for indicated times. For GFP-cleavage assays, autophagy substrates are GFP-tagged (e.g. GFP-Atg8 or GFP-50Q) and their vacuolar degradation upon starvation is monitored by the accumulation of the released GFP moiety, which is highly stable and escapes autophagic degradation. For cell viability determination assays, yeast cells were subject to nitrogen starvation (SD-N) for indicated days before spotted to YPD-plates for re-growth.
Cell culture
Standard cell culture techniques were used. HeLa (ATCC, CCL-2) cells were grown in DMEM (Invitrogen, 10569044) supplemented with 10% FBS (Gibco, 26140–079), 2 mM L-glutamine (Gibco, 25030081), and 100 U/ml penicillin-streptomycin (Gibco, 15070063) in a humidified incubator at 37°C with 5% CO2. Plasmid and siRNA transfections were carried out using Lipofectamine 2000 (Invitrogen, 11668027) according to the manufacturer’s protocol.
Immunoblotting techniques
Total cell protein extracts were prepared by TCA precipitation, and immunoprecipitated samples were prepared in HU lysis buffer (4M urea (Sangon Biotech, A600148), 2.5% SDS (Sangon Biotech, A600485), 100 mM Tris-pH 6.8, 0.05M DTT (Sangon Biotech, A620058)). Proteins were resolved on SDS-PAGE gels and analyzed by standard immunoblotting techniques using indicated specific antibodies and HRP-conjugated secondary antibodies (Bio-Rad,1706515 and 1706516). Protein bands were detected within the linear range based on loading samples with appropriate protein concentrations (with total amount less than 10 μg per lane in 15-well/1-mm gels), application of antibodies according to manufacturer’s documents, and serial time exposure with signal saturation avoidance (saturated signal will be labeled with red color in the ChemiDoc MP Imaging System; Bio-Rad). Monoclonal antibodies against HA-epitope (F-7) and GFP (B-2) were purchased from Santa Cruz Biotechnology (sc-7392 and sc-9996). Monoclonal Pgk1 antibody (22C5; ab113687) was from Abcam. Monoclonal LC3 antibody (4108) was from Cell Signaling Technology. Monoclonal anti-SQSTM1/p62 (109102) was from Abcam. Rabbit polyclonal anti-SACM1L (13033) and rabbit polyclonal anti-GAPDH (60004) were from Proteintech. Mouse monoclonal anti-PtdIns4P (P004) was from Echelon.
Co-immunoprecipitation
Native yeast extracts were prepared by cell disruption on a multitube bead-beater (SCIENTZ) in lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 10% glycerol (Sangon Biotech, A600232), protease inhibitors [Roche, 5892970001]) with zirconia/silica beads (BioSpec, 11079105z). To avoid protein degradation and loss of PTMs, buffers were supplemented with protease inhibitors: 20 mM NEM and EDTA-free complete cocktail (Roche, 5892970001). GFP-Trap agarose beads (Chromotek, gta-20) were used. IPs were performed for 2 h with head-over-tail rotation at 4°C and were followed by stringent washing steps to remove nonspecific background binding to the beads.
Transmission electron microscopy
HeLa cells or tissues from mouse models were fixed in 2.5% glutaraldehyde in PBS (Sangon Biotech, E607008) overnight at 4°C and washed three times for 15 min with 0.1M phosphate buffer, pH 8.0 and then fixed in 2% aqueous osmium tetraoxide for 1 h followed by washing three times each for 15 min with deionized water. Samples were then dyed with 2% uranyl acetate for 30 min, and dehydrated through graded alcohols (50%–100%) and 100% acetone each for 15 min. After that, Samples were embedded in EPON 812 resin (Merck, 45345) and cured for 24 h at 37°C, 45°C, 60°C, respectively. Ultrathin (70 nm) sections were obtained by ultra-thin slicer machine and stained with 2% uranyl acetate and 0.3% lead citrate. Electron microscopy images of the samples were taken using Tecnai G2 Spirit transmission electron microscope (FEI Company). Representative images of at least three independent replicates are shown.
Fluorescence and immunofluorescence assays
Yeast cells were cultured in SC medium (0.17%YNB, 2% glucose, and 0.2% amino acids) at 30°C. Colonies were first inoculated into SC medium and cultured overnight. On the second day, optical density at 600 nm (OD600) of the culture was adjusted to approximately 0.1 and cultured to an OD600 of approximately 0.8 to 1.0. Yeast cells were washed twice in stilled water and then cultured in SD-N media for indicated times. Aliquots (200 μl) of liquid culture were collected and allowed to precipitate on concanavalin A (Solarbio, C8110)-coated cover glass for five min. Images were collected on a Zeiss Observer 7 inverted fluorescence microscope equipped with Apotome. The intensity of the excitation was set to 100%, and the exposure time was 30 ms (for GFP, except 10 ms for capturing Atg9-2GFP) or 50 ms (for red fluorescent protein RFP and Cherry). For immunofluorescence microscopy, cells were seeded onto precision cover glass. Cells were then fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 (Sangon Biotech, A110694). Following block with 1% BSA (Solarbio, A8010), cells were incubated with primary antibodies followed by incubation with Alexa Fluor-conjugated secondary antibodies (Abcam, ab150077, ab150113 and ab150080). Images were acquired on a Zeiss LSM 880 microscopes. Representative images of at least three independent replicates are shown.
Protease K protection assays
Conventional protease K (PK) protection assays were performed. Briefly, yeast cells expressing GFP-Atg8 were grown to mid-log phase in YPD medium and shifted to SD-N for 2 h. The yeast cells were then spheroplasted and lysed. Cell lysates were centrifuged at 5,000 × g for 5 min, and then membrane components were collected by another centrifugation at 10,000 × g for 10 min. The pellet fraction was resuspended and treated with PK (40 μg/ml; Sangon Biotech, A510453) with or without 0.4% Triton X-100 (Sangon Biotech, A110694). After the PK treatment, proteins were precipitated with TCA (Sangon Biotech, A600968) before immunoblot analyses.
Statistical analysis
All experiments were independently repeated at least three times with consistent conclusions and representative results were shown. Statistical analyzes were performed using the Student’s t test in Prism software. Values are expressed as mean ± SD of at least three independent experiments unless otherwise noted. Images were quantified with ImageJ (NIH, 64-bit Java 1.8.0_112). Each time, a region of approximately 700 by 700 pixels was chosen, which contained around 30 cells. The total intensity in each color channel was counted as the denominators before analyzed for quantification of co-localization ratios. This counting was repeated three times with samples from replicated experiments. Standard deviations between these counting were calculated.
Supplementary Material
Acknowledgments
We thank Ivan Psakhye for early discussions, Jochen Rech and Alexander Strasser for technical assistance on yeast screen, Zhiping Xie and Cong Yi for materials and technical assistance. We thank the Imaging Center of Sichuan University School of Medicine for assistance with confocal microscopy.
Funding Statement
This study was supported by the National Key R&D Program of China under grant [2017YFA0506300] (to K.L.) and the National Natural Science Foundation under grants [31770820] (to K.L.).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10(7):458–467. [DOI] [PubMed] [Google Scholar]
- [2].Levine B, Kroemer G.. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ge L, Melville D, Zhang M, et al. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife. 2013;2:e00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wen X, Klionsky DJ. An overview of macroautophagy in yeast. J Mol Biol. 2016;428(9 Pt A):1681–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. [DOI] [PubMed] [Google Scholar]
- [6].Burke JE. Structural basis for regulation of phosphoinositide kinases and their involvement in human disease. Mol Cell. 2018;71(5):653–673. [DOI] [PubMed] [Google Scholar]
- [7].Mesmin B, Antonny B. The counterflow transport of sterols and PI4P. Biochim Biophys Acta. 2016;1861(8):940–951. [DOI] [PubMed] [Google Scholar]
- [8].Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Heo J-M, Livnat-Levanon N, Taylor EB, et al. A stress-responsive system for mitochondrial protein degradation. Mol Cell. 2010;40(3):465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baek GH, Cheng H, Kim I, et al. The Cdc48 protein and its cofactor Vms1 are involved in Cdc13 protein degradation. J Biol Chem. 2012;287(32):26788–26795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Heo J-M, Nielson JR, Dephoure N, et al. Intramolecular interactions control Vms1 translocation to damaged mitochondria. Mol Biol Cell. 2013;24(9):1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Whitters EA, Cleves AE, McGee TP, et al. SAC1p is an integral membrane protein that influences the cellular requirement for phospholipid transfer protein function and inositol in yeast. J Cell Biol. 1993;122(1):79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Del BL, Brill JA. Sac1, a lipid phosphatase at the interface of vesicular and nonvesicular transport. Traffic. 2018;19(5):301–318. [DOI] [PubMed] [Google Scholar]
- [15].Manford A, Xia T, Saxena AK, et al. Crystal structure of the yeast Sac1: implications for its phosphoinositide phosphatase function. Embo J. 2010;29(9):1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moreau K, Renna M, Rubinsztein DC. Connections between SNAREs and autophagy. Trends Biochem Sci. 2013;38(2):57–63. [DOI] [PubMed] [Google Scholar]
- [17].van der Beek J, Jonker C, van der Welle R, et al. CORVET, CHEVI and HOPS-multisubunit tethers of the endo-lysosomal system in health and disease. J Cell Sci. 2019;132(10):jcs189134. [DOI] [PubMed] [Google Scholar]
- [18].Faulhammer F, Konrad G, Brankatschk B, et al. Cell growth-dependent coordination of lipid signaling and glycosylation is mediated by interactions between Sac1p and Dpm1p. J Cell Biol. 2005;168(2):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kuzmin E, VanderSluis B, Wang W, et al. Systematic analysis of complex genetic interactions. Science. 2018;360(6386):6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hoppins S, Collins SR, Cassidy-Stone A, et al. A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol. 2011;195(2):323–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shin J, Liu P, Chan LJ, et al. pH biosensing by PtdIns4P regulates cargo sorting at the TGN. Dev Cell. 2020;52(4):461–476.e4. [DOI] [PubMed] [Google Scholar]
- [22].Waugh MG. The great escape: how phosphatidylinositol 4-kinases and PtdIns4P promote vesicle exit from the Golgi (and drive cancer). Biochem J. 2019;476(16):2321–2346. [DOI] [PubMed] [Google Scholar]
- [23].Mari M, Griffith J, Rieter E, et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190(6):1005–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yamamoto H, Kakuta S, Watanabe TM, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198(2):219–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu X, Mao K, Yu AH, et al. The Atg17-Atg31-Atg29 complex coordinates with Atg11 to recruit the Vam7 SNARE and mediate autophagosome-vacuole fusion. Curr Biol. 2016;26(2):150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bas L, Papinski D, Licheva M, et al. Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome-vacuole fusion. J Cell Biol. 2018;217(10):3656–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Matsui T, Jiang P, Nakano S, et al. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol. 2018;217(8):2633–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gao J, Reggiori F, Ungermann C. A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J Cell Biol. 2018;217(10):3670–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Boratyn GM, Schäffer AA, Agarwala R, et al. Domain enhanced lookup time accelerated BLAST. Biol Direct. 2012;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yu H, Luscombe NM, Lu HX, et al. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14(6):1107–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bajaj PK, Wang J, Blagoveshchenskaya A, et al. Phosphoregulatory protein 14-3-3 facilitates SAC1 transport from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2015;112(25):E3199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Foti M, Audhya A, Emr SD. Sac1 lipid phosphatase and Stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol Biol Cell. 2001;12(8):2396–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moorhead AM, Jung J-Y, Smirnov A, et al. Multiple host proteins that function in phosphatidylinositol-4-phosphate metabolism are recruited to the chlamydial inclusion. Infect Immun. 2010;78(5):1990–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Venditti R, Masone MC, Wilson C, et al. PI(4)P homeostasis: who controls the controllers? Adv Biol Regul. 2016;60:105–114. [DOI] [PubMed] [Google Scholar]
- [35].Yamashita S, Oku M, Wasada Y, et al. PtdIns4P-signaling pathway for the synthesis of a nascent membrane structure in selective autophagy. J Cell Biol. 2006;173(5):709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang K, Yang Z, Liu X, et al. Phosphatidylinositol 4-kinases are required for autophagic membrane trafficking. J Biol Chem. 2012;287(45):37964–37972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Blumental-Perry A, Haney CJ, Weixel K, et al. Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev Cell. 2006;11(5):671–682. [DOI] [PubMed] [Google Scholar]
- [38].Judith D, Jefferies HBJ, Boeing S, et al. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIbeta. J Cell Biol. 2019;218(5):1634–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zubenko GS, Stiffler JS, Hughes HB, et al. Reductions in brain phosphatidylinositol kinase activities in Alzheimer’s disease. Biol Psychiatry. 1999;45(6):731–736. [DOI] [PubMed] [Google Scholar]
- [40].Wu B, Kitagawa K, Zhang N-Y, et al. Pathophysiological concentrations of amyloid beta proteins directly inhibit rat brain and recombinant human type II phosphatidylinositol 4-kinase activity. J Neurochem. 2004;91(5):1164–1170. [DOI] [PubMed] [Google Scholar]
- [41].Rapoport SI, Primiani CT, Chen CT, et al. Coordinated expression of phosphoinositide metabolic genes during development and aging of human dorsolateral prefrontal cortex. PLoS One. 2015;10(7):e0132675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Clark SL, Adkins DE, Aberg K, et al. Pharmacogenomic study of side-effects for antidepressant treatment options in STAR*D. Psychol Med. 2012;42(6):1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.