

ABSTRACT
Macroautophagy/autophagy plays a pivotal role in cytoplasmic material recycling and metabolic turnover, in which ATG4B functions as a “scissor” for processing pro-LC3 and lipidated LC3 to drive the autophagy progress. Mounting evidence has demonstrated the tight connection between ROS and autophagy during various pathological situations. Coincidentally, several studies have shown that ATG4B is potentially regulated by redox modification, but the underlying molecular mechanism and its relationship with autophagy is ambiguous. In this study, we verified that ATG4B activity was definitely regulated in a reversible redox manner. We also determined that Cys292 and Cys361 are essential sites of ATG4B to form reversible intramolecular disulfide bonds that respond to oxidative stress. Interestingly, we unraveled a new phenomenon that ATG4B concurrently formed disulfide-linked oligomers at Cys292 and Cys361, and that both sites underwent redox modifications thereby modulating ATG4B activity. Finally, increased autophagic flux and decreased oxidation sensitivity were observed in Cys292 and Cys361 double site-mutated cells under normal growth conditions. In conclusion, our research reveals a novel molecular mechanism that oxidative modification at Cys292 and Cys361 sites regulates ATG4B function, which modulates autophagy.Abbreviations: Air-ox: air-oxidation; ATG4B: autophagy related 4B cysteine peptidase; BCNU: 1,3-bis(2-chloroethyl)-1-nitrosourea; CBB: Coomassie Brilliant Blue; CM: complete medium; CQ: chloroquine; DTT: dithiothreitol; GSH: reduced glutathione; GSNO: S-nitrosoglutathione; GSSG: oxidized glutathione; HMW: high molecular weight; H2O2: hydrogen peroxide; NAC: N-acetyl-L-cysteine; NEM: N-ethylmaleimide; PE: phosphatidylethanolamine; PTM: post-translational modification; ROS, reactive oxygen species; WT: wild type
KEYWORDS: ATG4B, autophagy, disulfide bond, oligomer, oxidative modification, redox
Introduction
Macroautophagy, usually termed as “autophagy”, is a catalytic process where aggregated macromolecules and dysfunctional organelles are degraded and recycled through autolysosomes in response to specific stress stimuli to maintain cellular homeostasis [1,2]. Several reports have stated that autophagy is involved in diverse pathological diseases, such as neurodegeneration, cancer, cardiovascular diseases, and is proposed to be a therapeutic target [3 4 5 7–8].
Initiation and development of autophagy is modulated by several autophagy-related (ATG) genes, which have been identified in yeast and human [9,10]. Autophagy begins with the formation and maturation of the double-membraned autophagosomes which engulf and deliver cargoes to lysosomes for degradation. The assembling process of autophagosomes is mediated by a family of core ATG proteins, including two ubiquitin-like systems [11]. First, inactive precursor MAP1LC3/LC3 is cleaved by protease ATG4B at the conserved C-terminal glycine residue. After a set of transfer and activation by ATG7, ATG3 and ATG12–ATG5-ATG16L1, the processed LC3 (LC3-I) finally conjugates with phosphatidylethanolamine (PE) at the exposed glycine site via a covalent bond [12,13]. So far, lipidated LC3, called LC3-II, has become a biomarker of autophagy because of its characteristic of anchoring into the autophagosome membrane [14]. Upon fusion with lysosomes, ATG4B can, in turn, deconjugate LC3-PE to release LC3-I to the cytoplasm for reuse [15]. Hence, ATG4B serves as a priming and delipidation enzyme whose fine regulation is essential for autophagy process.
Studies have revealed that atg4b knockout represses basal autophagy due to impaired LC3 substrate processing [16,17]. Besides, defects in ATG4B function are also associated with specific pathologies and diseases. Marino G et al. observe impaired autophagy and disequilibrium in atg4b−/- mice [16]. Another study discovers that knockdown of ATG4B arrests the cell cycle and causes abnormal cell proliferation [18]. Recently, studies have highlighted the mechanisms of post-translational modifications (PTMs) of ATG4B and revealed several modification mechanisms. For instance, Jo et al. reported that O-GlcNAcylation of ATG4B could positively regulate autophagy [19]. In addition, phosphorylation of ATG4B plays an important role in modulating its hydrolase activity [20 21–22]. A recent study reveals a novel mechanism that S-nitrosation of ATG4B impairs autophagy, which leads to neurotoxicity under high glucose levels [23]. In 2007, Scherz-Shouval et al. show that reactive oxygen species (ROS) are necessary for autophagy induction and ATG4A activity is regulated by ROS at the site of Cys81 near the catalytic site Cys77 [24]. In Saccharomyces cerevisiae, Chlamydomonas reinhardtii, and Arabidopsis, Atg4 are similarly redox-regulated. Yeast Atg4 and green alga Atg4 have been found to form intramolecular disulfide bond upon oxidation [25–27]. Meanwhile, alga Atg4 can simultaneously aggregate and form oligomers which regulate autophagy [27]. Although Scherz-Shouval et al. have also identified site Cys78, which is conserved with Cys81 in ATG4A, as the oxidation-sensitive cysteine of human ATG4B [24], more in-depth studies are still required to determine how human ATG4B is regulated by oxidative stress.
Accumulating evidence has demonstrated the association of autophagy with the generation of ROS. Moreover, perturbations in cellular redox have been implicated in aging and many diseases [28 29 30 31 32 33–35]. Yet, the precise molecular mechanisms of these processes are largely unknown. Here, we explored the relationship between redox processes and ATG4B, and revealed a novel molecular mechanism that Cys292 and Cys361 are essential cysteines for the formation of intra- and intermolecular disulfide bonds which are regulated by redox signals to influence ATG4B activity and autophagy.
Results
ATG4B activity is redox-regulated in vitro
To establish the in vitro ATG4B proteolytic assay, we constructed prokaryotic expressed plasmids and purified the recombinant ATG4B with a His-tag at the N terminus. In addition, we employed a recombinant LC3-GST to imitate pro-LC3. Results showed that ATG4B cleaved LC3-GST in a dose-dependent and time-dependent manner (Figure 1A,B). To determine how the human ATG4B was regulated under redox conditions, we treated ATG4B with dithiothreitol (DTT) and oxidant hydrogen peroxide (H2O2) at given concentrations. As predicted, ATG4B activity was increased in the presence of increasing DTT and inhibited by H2O2 (Figure 1C,D). We also performed air oxidation assay to induce a mild oxidation [36,37]. Consequently, ATG4B lost most of its activity when exposed to air for up to 3 h, however, the activity of ATG4B was partly restored with an addition of DTT (Figure 1E). The increase in ATG4B activity caused by preincubation with a low concentration of DTT (0.2 mM) was suppressed by H2O2, but a higher concentration of DTT (2 mM) efficiently recovered ATG4B activity (Figure 1F), further proving the reversible modification of ATG4B. Additional tests were carried out for other oxidants, such as oxidized glutathione (GSSG), and GSNO, an endogenous S-nitrosothiol. Results showed that the inhibitory effects of the two oxidants on ATG4B were reversed upon reduction (Figure S1A,B). Taken together, these results support the idea that there exists a redox post-translational modification mechanism that controls ATG4B activity.
Figure 1.
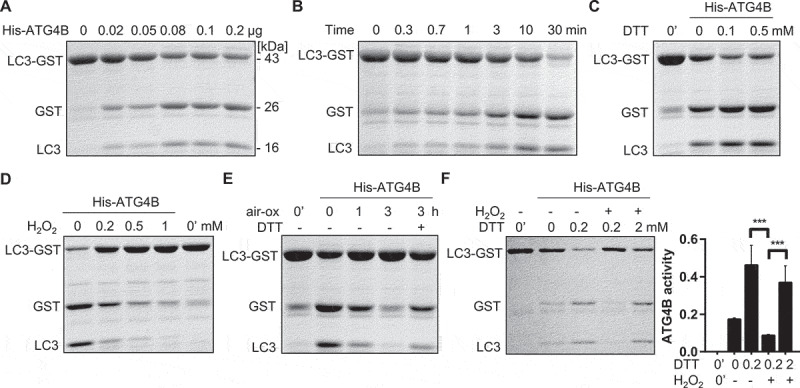
ATG4B activity is promoted by reductant and reversibly inhibited by oxidant. (A-B) Activity of recombinant His-ATG4B was monitored by quantitative cleaved GST product in a dose-dependent (A) and time-dependent manner (B). (C) His-ATG4B (0.015 µg) was pre-treated with different concentration of DTT for 10 min, after which ATG4B was incubated with LC3-GST (5 µg) in reaction buffer at 37℃ for 30 min. 0ʹ represents LC3-GST only without ATG4B or any other treatments. (D) His-ATG4B (0.025 µg) was incubated in the presence of indicated H2O2 for 30 min, after which LC3-GST (5 µg) was added and reaction proceeded at 37℃ for 30 min. (E) His-ATG4B (0.015 µg) was laid and exposed to air on ice for up to 3 h, then ATG4B cleaved LC3-GST (5 µg) in the absence or presence of DTT (1 mM). (F) His-ATG4B (0.015 µg) was pretreated with 0.2 mM of DTT for 10 min (lane 3); then, H2O2 (1 mM) was added for 30 min (lane 4); finally, oxidized ATG4B was treated with 2 mM of DTT for another 30 min (lane 5). After each specific procedure except blank control (lane 1), ATG4B was incubated with LC3-GST to assess activity. All the reaction mixtures were subject to SDS-PAGE, followed by Coomassie Brilliant Blue (CBB) staining. Data are presented as mean ± SEM from 4 independent assays. *** P < 0.001.
ATG4B activity is redox-regulated in cells
It has been proven that ATG4A activity is strongly inhibited by either starvation or H2O2 treatment in CHO cells [24]. Similarly, we found that ATG4B was strongly inhibited in processing LC3-GST substrate in a lysate of HEK293 cells treated with H2O2 (Figure 2A). Under the same condition, the level of lipidated LC3 was increased in HEK293 cells and HeLa cells (Figure 2B,C). Hence, we inferred that the strong oxidant might block the delipidation activity of endogenous ATG4B, subsequently leading to the loss of cleavage of LC3-GST substrate in vitro and the accumulation of LC3-II in vivo.
Figure 2.
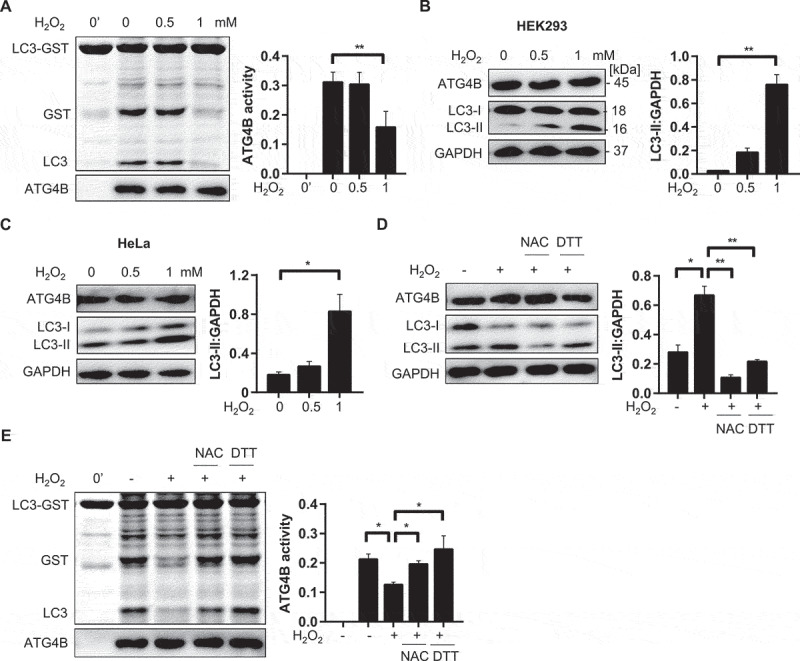
Endogenous ATG4B activity is inhibited by H2O2. (A) HEK293 cells were treated with designated concentration of H2O2 for 1 h, then cells were harvested in non-denatured buffer. Cell lysate (8 µg) was incubated with substrate LC3-GST (3 µg) at 37℃ for 30 min. The cleavage of LC3-GST by ATG4B was presented by CBB staining and relevant expression level of ATG4B was analyzed by immunoblot with anti-ATG4B antibody. (B-C) Deconjugation activity of endogenous ATG4B in vivo. HEK293 (B) and HeLa (C) cells were exposed to indicate H2O2 for 1 h, respectively. Relevant LC3-II level was detected by immunoblot analysis with anti-LC3B antibody. (D) HeLa cells pretreated with NAC (10 mM) for 15 min were incubated with H2O2 (1 mM) for 1 h, or DTT (1 mM) was added for 15 min after cells were exposed to H2O2 for 1 h. LC3-II level was determined as described in (B-C). (E) Cell lysates (8 µg) from cells processed in (D) were utilized to test processing activity toward LC3-GST (3 µg), followed by CBB staining. Graphical data are presented as mean ± SEM from 3 individual experiments. * P < 0. 05; ** P < 0.01.
To examine whether the attenuation of ATG4B could be recovered, we tested the effect of N-acetyl-L-cysteine (NAC), a well-known antioxidant that decomposes H2O2, and DTT on ATG4B oxidative inhibition. HeLa cells were pre-treated with NAC (10 mM) followed by H2O2. In another group, cells were treated with H2O2 after which DTT was added. As expected, antioxidant reagent NAC promoted ATG4B activity, enhancing more delipidation of LC3-II compared to H2O2 group. Similarly, after treatment with H2O2, addition of DTT abolished all the H2O2-induced LC3-II bands and restored the original state of the cells probably by relieving ATG4B inhibition (Figure 2D). In agreement with results mentioned above, the activity of endogenous ATG4B to process LC3-GST in vitro was restored by NAC and DTT (Figure 2E). A similar cleavage pattern of LC3-GST was observed as well by immunoblot analysis (Figure S2A).
HeLa cells stably expressing GFP-LC3 were treated with 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU), an inhibitor of cellular glutathione reductase that prevents the transition of GSSG to reduced glutathione (GSH) in order to alter the intracellular redox potential levels. Fluorescence analysis displayed increased GFP puncta in BCNU-treated group, indicating the accumulation of GFP-LC3 labeled autophagic vacuoles. On the contrary, the addition of DTT diminished the accumulation of LC3 puncta (Figure S2B). Since oxidation can affect ATG3 and ATG7 and thus lipidation of LC3 as well [34], we further compared HeLa or HEK293 with ATG4B KO HeLa cells expressing GFP-LC3[G120] which mimic the I form of LC3 (LC3-I) which can be conjugated to PE independent of ATG4B. As shown in Figure S2C, GFP-LC3 puncta only accumulated in ATG4B KO HeLa cells due to the impaired delipidation function. After BCNU or H2O2 treatment, GFP-LC3 puncta significantly increased in HeLa and HEK293 cells, while no such changes were observed in cells lacking ATG4B. These results suggested that the decreased ATG4B delipidation activity might be responsible for the accumulation of GFP-LC3 puncta instead of ATG3 or ATG7 inhibition. These data indicate that ATG4B undergoes reversible modification following oxidative stress, which acts as an “activity switch” to control the dual activities of ATG4B.
Formation of intramolecular disulfide bonds of ATG4B requires Cys292 and Cys361 upon oxidation
To explore the changes in ATG4B when exposed to oxidants and to determine the mechanism for the loss of ATG4B activity, we next investigated the state of ATG4B molecule. There are four mammalian homologs in ATG4 cysteine protease family. Among them, only ATG4A and ATG4B have been proposed to be reversibly regulated by oxidation in terms of their proteolytic activity [24]. ATG4 is rich in cysteine residues, which are vulnerable to oxidation attack. Oxidative PTM of cysteine has recently become a common mechanism in which reactive cysteine residues function as redox-switches to sense the changes in redox-environment [38 39–40]. Normally, reactive sulfhydryl is alternatively oxidized to sulfenic acid (Cys-SOH), a labile intermedium which forms intramolecular disulfide bonds with proximal thiol or could be S-glutathionylated. In some cases, it can be oxidized to irreversible sulfinic acid (Cys-SO2H) or sulfonic acid (Cys-SO3H) (Figure 3A) [41]. To examine whether S-glutathionylation modification occurred, HEK293 cells were treated with H2O2 or not, after which immunoprecipitation with anti-GSH was performed to pull down S-glutathionylated ATG4B. Unfortunately, unlike the glutathionylated protein nucleoplasmic translocation of the nucleolar protein NPM1 (nucleophosmin 1), no signal of glutathionylation was observed for endogenous or overexpressed ATG4B (Figure S3A) [42]. This result excluded the possibility of glutathionylation modification of ATG4B in the present assay. Thus, we paid attention to the formation of disulfide bonds. β-mercaptoethanol or DTT are general reducing agents widely used in SDS-PAGE to reduce protein disulfide bonds or other disulfides in case non-linear proteins mobilize independently of molecular weight of the monomer. To trace the original redox state of ATG4B, we prepared reductant-free loading buffer to perform non-reduced SDS-PAGE and used 8% polyacrylamide gel to seek out subtle difference. After 24 h of transfection with Flag-tagged ATG4B plasmid into HEK293 cells, interestingly, results showed two bands in the absence of DTT compared to the reduced group (Figure 3B).
Figure 3.

ATG4B forms intramolecular disulfide bonds upon oxidation. (A) Reversible and irreversible cysteine modifications. (B) HEK293 cells transiently transfected with Flag-ATG4B or not were resolved in reduced (containing DTT) or non-reduced (DTT free) loading buffer and analyzed with anti-FLAG antibody. The relative level of Flag-ATG4B expression compared to endogenous control was determined with anti-ATG4B antibody. (C) Cell lysates (25 µg) from (B) were incubated with DTT ranging from 0.1 mM to 5 mM at 4℃ for 15 min, after which non-reduced SDS-PAGE was performed with anti-FLAG antibody. (D-E) Cell lysates (25 µg) from HEK293 cells transiently transfected with Flag-ATG4B (D) and HEK293 cells (E) were subject to non-reduced immunoblot assay after these procedures: treatment with 0.2 mM of DTT at 4℃ for 15 min (lane 1 in D, lane 2 in E); pretreatment with 0.2 mM of DTT followed by 1 mM of H2O2 for 30 min (lane 2 in D, lane 3 in E); 2 mM of DTT was added to mixture as detailed in lane 2 (lane 3 in D, lane 4 in E); no treatment (lane 4 in D, lane 1 in E). Reduced form of ATG4B (R) and oxidized form of ATG4B (O) were labeled. (F) HEK293 cells transiently transfected with plasmids of Flag-ATG4B or 13 single-site mutants (cysteine to serine) of ATG4B were lysed and subject to non-reduced immunoblot with anti-FLAG antibody after 24 h adherent growth. (G) Lysates from HEK293 cells overexpressing Flag-ATG4B or single-site mutant Flag-ATG4BC292S, Flag-ATG4BC361S or double-site mutant Flag-ATG4BC292,361S (Flag-ATG4B2CS) were incubated with H2O2 (1 mM) for 30 min. (H) ATG4B KO HeLa cells stably transfected with Flag-ATG4B or Flag-ATG4BC292,361S were treated with H2O2 (1 mM) at 37℃ for 1 h, or cell lysates extracted from ATG4B KO HeLa cells stably transfected with Flag-ATG4B or Flag-ATG4BC292,2361S were exposed to H2O2 (1 mM) for 30 min. Samples were subject to non-reduced SDS-PAGE and analyzed with anti-FLAG antibody.
To rule out the presence of undesired nonspecific bands, we tested a series of concentration gradients of DTT and found that both bands gradually converted into a single band that migrated upward as the concentration of DTT increased (Figure 3C). For further confirmation, an exchange of incubation between DTT and H2O2 was constituted to test the reversible transformation of both bands under oxidation and reduction. In this assay, preincubation with low concentration of DTT (0.2 mM) exacerbated the upper band (lane 1), whereas, H2O2 treatment increased the lower band (lane 2), but this effect was reversed by high concentration of DTT (2 mM, lane 3) (Figure 3D). Similarly, endogenous ATG4B showed the same conversion pattern in HEK293 cells (Figure 3E). These results suggest that under non-reduced condition, ATG4B exists in both reduced and oxidized forms in vivo, in which the reduced ATG4B containing free reactive thiols tends to form a reversible disulfide linkage with other -SH groups which is referred to as the oxidized form.
There are 13 cysteine residues within ATG4B sequence (Figure S3B), among which, Cys74 has been identified to be one of the key catalytic sites of ATG4B. Mutation of Cys74 causes complete loss of ATG4B activity [43,44]. To explore the sites involved in the redox modification of ATG4B, we performed single site-directed mutagenesis on the 13 cysteines with a Flag tag fused in C-terminal. In vitro cleavage assays proved that mutation of any of these cysteine residues hardly affected the ATG4B activity except catalytic Cys74 (Figure S3C). Cells transfected with these mutant plasmids were harvested and then subjected to non-reduced SDS-PAGE. As shown in Figure 3F, most of the mutants exhibited both reduced and oxidized bands as wild type (WT) ATG4B did, including ATG4BC74S. However, ATG4BC292S and ATG4BC361S showed a single reduced band. H2O2 was then added to test the response of both mutants to oxidant stimulation. Interestingly, ATG4BC292S or ATG4BC361S was oxidizable although the appearance of oxidized bands was relatively weaker than that of other ATG4B mutants (Figure 3G). However, double-site mutant ATG4BC292,361S (depicted as 2CS), maintained as a completely reduced form despite H2O2 treatment (Figure 3G). To determine the interference of endogenous ATG4B, plasmid encoding ATG4B or ATG4BC292,361S was transfected to ATG4B KO HeLa cells respectively. In agreement with previous results, a remarkable exchange between reduced and oxidized forms was observed in ATG4B strains while ATG4BC292,361S maintained as a highly reduced form after H2O2 treatment (Figure 3H). Meanwhile, a randomly employed mutant ATG4BC246S was oxidized similar to ATG4BC246,292S (Figure S3D). These results suggest that Cys292 and Cys361 are essential cysteines involved in the formation of the oxidized form of ATG4B with intramolecular disulfide bonds.
ATG4B forms oligomers linked by intermolecular disulfide bonds in response to oxidation
In the above studies, we have demonstrated that ATG4B is redox regulated by forming intra-disulfide bonds. However, it was noted that in the presence of an oxidant, only a fraction of the reduced form was oxidized. In theory, however, the oxidized form and reduced form should be conserved, except when there are other modifications that are not detected. To test this possibility, the intact PVDF membrane was reanalyzed. Indeed, after modification and enrichment with N-ethylmaleimide (NEM) which blocked the free cysteine residues and prevented further reductive or oxidative modifications during sample preparation, immunoblot analysis revealed that ATG4B formed high molecular weight (HMW) oligomers (of approximately 250 kDa, indicated as oATG4B) which difficultly entered into separated gel, whereas the 45-kDa species (indicated as mATG4B) were located on the position where the monomer ATG4B was expected to be. Moreover, the oligomers gradually enlarged and accumulated as the concentration of H2O2 increased (Figure 4A). Furthermore, we confirmed that these oligomers formed in a redox-dependent manner since either the addition of DTT to the oxidized lysates or pre-incubation with antioxidant NAC, remarkably attenuated the HMW oligomers when analyzed with anti-FLAG antibody (Figure 4B) or anti-ATG4B antibody (Figure S4A). To examine whether ATG4B oligomers can be reduced by the cellular antioxidant system, we applied auranofin, an inhibitor of thioredoxin reductase, and found that treatment of auranofin sustained ATG4B oligomers (Figure S4B), suggesting that thioredoxin system is required to protect ATG4B from forming considerate poor oligomers.
Figure 4.

ATG4B forms disulfide-linked intermolecular oligomers in response to oxidation. (A) HEK293 cells stably expressing Flag-ATG4B were incubated with increasing concentration of H2O2 for 1 h, then were lysed and modified with 10 mM of NEM to avoid undesirable modifications on other free sulfydryls during sample preparation, after which non-reduced immunoblot was performed. oATG4B, oligomeric ATG4B. (B) HEK293 cells stably transfected with Flag-ATG4B were incubated with H2O2 (10 mM) for 1 h, after which the cell lysates were treated with DTT (3 mM) for another 30 min or HEK293 cells stably transfected with Flag-ATG4B were pretreated with 10 mM of NAC followed by H2O2 (10 mM) incubation for 1 h, reaction mixture were resolved for non-reduced immunoblot analysis using anti-FLAG antibody. The oligomeric ATG4B (oATG4B) and monomeric ATG4B (mATG4B) were marked on the left. (C-E) Reaction mixture containing His-ATG4B (4 µg) were terminated with 10 mM of NEM and analyzed by CBB after these procedures respectively: treatment with given concentration of H2O2 at 4℃ for 30 min (C); pretreatment with 5 mM of NEM for 10 min to block all cysteine residues, after which H2O2 (10 mM) was added (D); incubation with 20 mM of DTT for 20 min after H2O2 oxidation (E). (F) Recombinant His-ATG4B or His-ATG4BC292S, His-ATG4BC361S and His-ATG4BC292,361S were treated with incremental H2O2 in the presence or absence of DTT (20 mM), then reaction was stopped by addition of 10 mM of NEM. (G) Contrast between His-ATG4B and His-ATG4B2CS with or without NEM alkylation. (H) His-ATG4B and His-ATG4BC292,361S treated with H2O2 (10 mM) with or without DTT (20 mM) incubation were separated in 2D-diagonal SDS-PAGE. BSA was represented as positive control. (I) untreated His-ATG4B; (ii) oxidized His-ATG4B; (iii) oxidized His-ATG4B followed by reduction; (iv) untreated His-ATG4B2CS; (V) oxidized His-ATG4B2CS; (vi) oxidized His-ATG4BC292,361S followed by reduction. The horizontal direction is performed by non-reduced PAGE, with the vertical direction reduced.
To monitor the formation of ATG4B oligomers in vitro, we treated the recombinant ATG4B protein with H2O2. Similarly, we observed a high number of mixed HMW species as the concentration of H2O2 increased (Figure 4C). To examine whether the HMW complexes formed at cysteine residues, we pre-treated ATG4B with NEM to block all the 13 cysteines. The results showed that HMW complexes disappeared immediately even after the treatment of H2O2 (Figure 4D), indicating that cysteine residues were responsible for the formation of these oligomers. As expected, the addition of DTT abolished the oxidation of proteins to monomers (Figure 4E), suggesting the reversibility of these oxidized forms and the presence of mixed disulfides between HMW complexes. Next, we postulated that Cys292 and Cys361 were likely to participate in the formation of HMW oligomers. To validate this hypothesis, we compared the recombinant ATG4BC292S and ATG4BC361S, using ATG4BC246S as a negative control. SDS-PAGE analysis showed that ATG4BC246S group displayed a similar pattern as WT ATG4B (Figure S4C). On the other hand, most of the HMW complexes disappeared in His-ATG4BC292,361S even after a high concentration of oxidant (Figure 4F,G). In contrast, ATG4BC292S or ATG4BC361S merely showed decreased HMW species (Figure 4F). These results were further confirmed by immunoblot analysis (Figure S4D). It was also found that H2O2 treatment did not alter the intrinsic fluorescence intensity but causing a slight blue shift in the maximal emission wavelength of ATG4B from 332 nm to 334 nm, while there was no change in ATG4BC29,361S protein (Figure S4E), indicating that the mutations of Cys292 and Cys361 may strengthen the conformational stability of ATG4B.
For further validation, we introduced a general approach named “2D-diagonal” SDS-PAGE, which was applied to identify intra- and inter-molecular disulfide bonds in the proteins [45,46]. Basal state of recombinant ATG4B protein shifted to the diagonal plane of the gel (Figure 4Hi). After separation in the first phase of non-reduced electrophoresis, an oxidized fraction of ATG4B was reduced and shifted at a similar speed beneath the diagonal in the second completely reduced dimension electrophoresis (Figure 4Hii), indicating that these HMW complexes were linked by intermolecular disulfide bonds. Finally, after the addition of DTT, the oxidized ATG4B settled on the diagonal (Figure 4Hiii). Next, we investigated the characteristics of ATG4BC292,361S mutant on the diagonal gel after oxidation. We found that ATG4BC292,361S protein was consistently anchored to the diagonal after either oxidation or reduction (Figure 4Hiv-vi), suggesting that the fully reduced state of ATG4BC292,361S resisted the oxidative modification. Taken together, this study shows that in addition to forming intramolecular disulfide bonds, ATG4B may reversibly aggregate and form high molecular weight oligomers possibly via the intermolecular disulfide linkage at Cys292 and Cys361 residues. These two sites may co-regulate the ATG4B protease activity in response to oxidative attack.
Mutation of Cys292 and Cys361 reduces the redox sensitivity of ATG4B and promotes autophagic flux
These results confirmed that Cys292 and Cys361 are the key cysteines targeted for oxidation modification of ATG4B under oxidative stress. Thus, we took a deep insight into the impact of the candidate mutants on ATG4B function. As previously stated, H2O2 treatment resulted in a significant accumulation of lipidated LC3 and impaired the cleaving activity toward LC3-GST. Here, we explored the reaction of ATG4B strains and ATG4BC292,361S strains to H2O2. We observed that ATG4BC292,361S mutant decreased about 80% of lipidated LC3 levels following H2O2 stimulation (Figure 5A). The external processing activity was subsequently tested. Lysates from both cell strains treated with H2O2 were collected and incubated with LC3-GST substrate. Results showed that ATG4BC292,361S had an inferior sensitivity to oxidant and eliminated the inhibitory effect of H2O2 on ATG4B activity (Figure 5B). However, single-site mutant ATG4BC292S or ATG4BC361S failed to abolish the H2O2-induced inhibition (Figure S5A). At the same time, the recombinant ATG4BC292,361S exposed to air for 2 h similarly exhibited strong resistance to air oxidation, WT ATG4B lost almost half of its activity on the contrary (Figure 5C). To rule out the effects of endogenous ATG4B, we compared the redox sensitivity of ATG4B KO HeLa transfected with different mutants by detecting the cleavage efficiency and the delipidation function of ATG4B. As shown in Figure S5B, the cleavage activity of most mutants were suppressed by H2O2 except ATG4BC74S. Importantly, ATG4BC292,361S displayed a strong resistance to oxidation to the extent that there was no inhibition of ATG4B activity. Since pro-LC3 in ATG4B KO HeLa cells could be easily primed to LC3-I and then LC3-II when basal ATG4B activity was rescued, the reduction in ATG4B activity caused by H2O2 could be measured by blocking the delipidation process. As shown in Figure S5C, in consistent with cells expressing WT ATG4B, the delipidation function of most mutants was strongly suppressed once H2O2 was added. In contrast, only pro-LC3 was left in ATG4BC74S cells due to the loss of enzymatic activity of ATG4B. Importantly, the cleavage activity of ATG4BC292,361S was hardly suppressed by H2O2, suggesting that the double mutant of Cys292 and Cys361 is unique with inferior oxidant sensitivity. At the same time, all ATG4B mutants was transfected into HEK293 cells individually to perform the non-reduced SDS-PAGE after H2O2 treatment. Results showed that most of the mutants were oxidized except for the double mutant ATG4BC292,361S (Figure S5D), indicating that the formation of intramolecular disulfide bonds may have effect on ATG4B activity. In addition, we also tested Cys78, formerly suspected to be a redox-regulated site of ATG4B. Surprisingly, ATG4BC78S was unable to rescue ATG4B impairment following oxidation as measured by LC3-GST cleavage in our model (Figure S5E). The starvation experiments reported previously were repeated in this study with similar treatments [24]. However, no dramatic decrease of LC3 puncta in ATG4BC78S mutant was found compared to WT ATG4B (Figure S5F). Based on these data, it can be inferred that Cys292 and Cys361 are responsible for the oxidative modification of ATG4B, which mainly accounts for the impairment of ATG4B function under oxidative stress in vitro and in vivo.Finally, the roles of Cys292 and Cys361 in the modulation of autophagy process were studied. To determine whether ATG4BC292,361S played a positive role in autophagy, we performed autophagy flux experiments with a universal autophagy inhibitor, CQ, a lysosomotropic agent which impacts the late stage of autophagy by averting the fusion of autophagosomes with lysosomes. Results showed that ATG4BC292,361S strains increased the level of LC3-II after CQ treatment in contrast with cells transfected with ATG4B only (Figure 5D). Immunofluorescence analysis showed that the amount of LC3-II puncta located on autophagosome membrane was increased after treatment of CQ in ATG4BC292,361S strains, but no puncta was observed in ATG4B KO cells with or without CQ treatment (Figure 5E). HEK293 cells stably transfected with tandem-LC3 were used to test the lysosome delivery function. As shown in Figure S6, the number of autophagosomes (red only dots) of both ATG4B constructs decreased after CQ treatment. In contrast, more autolysosomes (yellow dots) were formed in ATG4BC292,361S cells than in cells overexpressing ATG4B. Based on these results, we conclude that the ATG4BC292,361S strains growing exponentially in medium display stronger autophagy activity. In brief, mutation of both Cys292 and Cys361 promotes autophagic flux.
Figure 5.
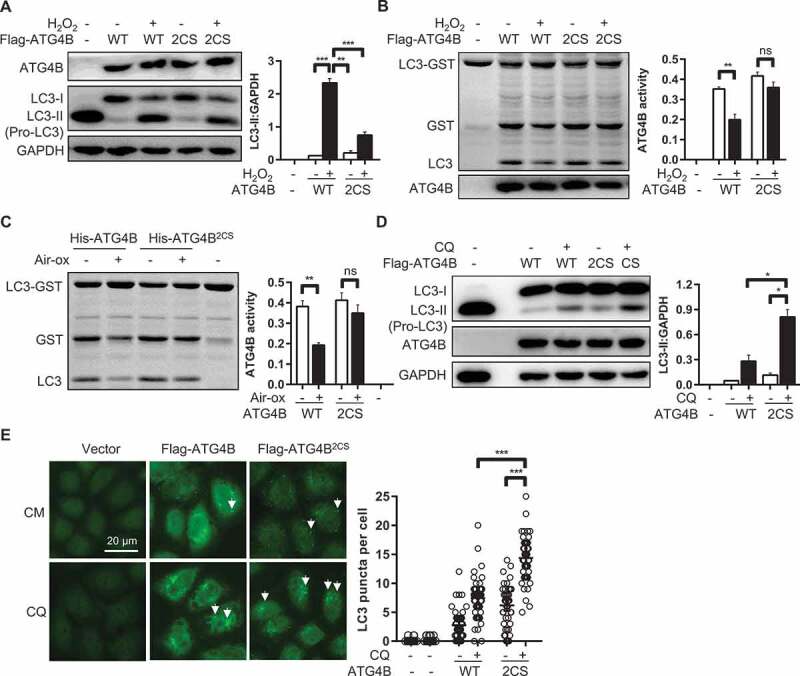
Double-site mutation of Cys292 and Cys361 compensates for ATG4B impairment upon oxidation and leads to increasing autophagic flux. (A) ATG4B KO HeLa cells stably expressing Flag-ATG4B or Flag-ATG4BC292,361S were stimulated by H2O2 (1 mM) for 1 h, ATG4B KO HeLa cells expressed with empty vector was used as control, corresponding LC3-II level were monitored. (B) Cell lysates (5 µg) from cells in (A) were employed to test ATG4B activity by cleaving LC3-GST substrate, results were represented by CBB analysis. (C) Recombinant His-ATG4B and His-ATG4BC292,361S (0.015 µg) were respectively exposed to air on ice for 2 h, after which enzyme assay was performed. (D-E) ATG4B KO HeLa cells stably expressing Flag-ATG4B or Flag-ATG4BC292,361S growing exponentially in medium were treated with or without CQ (40 µM) for 2 h, followed by immunoblot analysis with anti-LC3B antibody (D), and immunofluorescence using the same antibody (E). N = 44, 49, 54, 47, 51, 55, the number of LC3 puncta were quantified and at least 3 independent experiments were performed. Data are presented as mean ± SEM. * P < 0.05; ** P < 0.01; *** P < 0.001; ns, not significant.
Discussion
Accumulating evidence indicates that there is a strong link between ROS and autophagy. Indeed, a series of molecular mechanisms have been proposed to show how ATG proteins modulate autophagy process under intracellular redox conditions. ATG4B, a mammalian core autophagy gene, is reported to be the most efficient proteolytic enzyme in cleaving precursor LC3 and lipidated LC3 substrate [47]. Although many studies have reported that ATG4B protease is redox-regulated, more details into its molecular machinery are required. Our study proposed a novel oxidative post-translational modification in which ATG4B formed intramolecular disulfide bonds and oligomers in response to oxidation, thereby controlling its activity. Furthermore, we identified Cys292 and Cys361 as the main sites targeted for oxidative modifications. These two sites were involved in the regulation of ATG4B redox state and function. Importantly, we proved that the expression of oxidation-insensitive ATG4B mutant continuously activated autophagy flux, indicating that ATG4B may be a promising target in the mechanism of ROS regulating autophagy.
These results also showed that ATG4B activity was redox-regulated as the treatment of different oxidants and reducing reagents altered its activity in vitro and in vivo (Figures 1 and 2). An important finding of this study is that the oxidized form of ATG4B was characterized by formation of intramolecular disulfide bonds and the reduced form harbored free thiols under non-reducing condition (Figure 3B). Due to the reduced environment in cytoplasm, we inferred that most of ATG4B are in a reduced state in vivo. Once extracted from cultured cells or exposed to oxidants, the dehydrogenation of reactive -SH groups easily occurs which then form a reversible disulfide linkage with other -SH groups, thereby forming the oxidized band. Notably, disulfide bonds can reduce the hydrodynamic radius of polypeptides, thereby making the structure more compact and increasing the migration rate of protein in the gel [48,49]. Our study utilized a general method of non-reduced SDS-PAGE to explore ATG4B negative redox state. In this experiment, given the subtle difference in molecular weight of any oxidative modification between the modified protein and the primary form, we used 8% polyacrylamide gel to magnify the variation. Our analysis revealed that the oxidized form containing intramolecular disulfide bonds exchanged with the reduced form in a redox-dependent manner (Figure 3D,E). In addition, our study showed that the increase in the oxidized form due to oxidation was accompanied by formation of mixed oligomers. The formation of intramolecular disulfide bonds and oligomers were mostly in charge of ATG4B function impairments under oxidative stress. Indeed, human ATG4B homologs, Atg4 in Saccharomyces cerevisiae, Chlamydomonas reinhardtii, and Arabidopsis, have been reported to be redox-regulated, and their regulatory mechanisms have been described [25–27]. For instance, previous biochemical studies demonstrate that yeast Atg4 activity is redox controlled by forming a disulfide bond between Cys338 and Cys394 through thiol/disulfide exchange mediated by 2 electrons transfer [26]. In another case, alga Atg4 is regulated by a single disulfide bond and forms oligomers by aggregating in vitro and in vivo [27]. Although multiple sequence alignments have shown that Cys338 and Cys394 of yeast Atg4 are conserved with Cys400 and Cys473 in alga Atg4, Cys400 is identified as the key site participating in Atg4 redox regulation, whereas Cys473 is not involved [27]. Our findings are in agreement with these results, demonstrating the redox-regulated ATG4 is widely conserved among eukaryotes.Site-directed mutagenesis revealed that Cys292 and Cys361, which are not conserved with any of these sites identified as redox-regulated in other species, were key cysteine residues responsible for the formation of intramolecular disulfide bonds. Our results showed that simultaneous mutation of Cys292 and Cys361 to serine preserved the reduced form of ATG4B inert toward oxidation (Figure 3G,H). Based on the spatial structure of ATG4B, it is noted that Cys292 and Cys361 are located on a flexible and exposed region, with minor space resistance (Figure 6A). Therefore, both cysteines are susceptible to oxidation attack. Although the distance between Cys292 and Cys361 is too far to form disulfide linkage, it is possible that folded proteins can adapt themselves to the liquid phase of cytoplasm due to their flexibility and instability. Therefore, in response to a stimuli, ATG4B is likely to change its conformation, which may bring active residues adjacent to each other thereby forming disulfide bonds. Indeed, we have observed the shift of maximal emission wavelength of ATG4B after H2O2 treatment, suggesting the possible alteration of ATG4B conformation (Figure S4E). Notably, the N-terminal regulatory loop in which the catalytic triad (Cys74, Asp278, and His280) is located, lies between Cys292 and Cys361. Hence, we deduced that the compulsive formation of Cys292-Cys361 disulfide bond readily warps the regulatory loop and eventually shields the catalytic sites, which partially explains why oxidation decreased ATG4B activity. Since single-site mutant Cys292 or Cys361 failed to thoroughly disrupt the putative intramolecular disulfide bonds (Figure 3G), it is reasonable to conclude that besides Cys292-Cys361, there may be other forms of intramolecular disulfide bonds between Cys292 or Cys361 and any other cysteine residues (marked as X) (Figure 6B). However, regardless of which form of disulfide bond ATG4B forms, Cys292 or Cys361 is always required, suggesting that Cys292 and Cys361 are essential cysteines for the formation of intramolecular disulfide bonds. Meanwhile, we found that the recombinant double-mutant ATG4BC292,361S significantly abolished ATG4B aggregation and oligomeric complexes caused by treatment with hydrogen peroxide, indicating that Cys292 and Cys361 acted as an inter-disulfide bond linking moieties of ATG4B monomers. However, unlike typical simple oligomers, ATG4B tended to form oligomers with complex and interlaced structures, probably owing to the naked and pure system environment in which the purified proteins were proximal to each other. Here, we propose a model in which ATG4B monomers are randomly paired between Cys292 and Cys361 to form disulfide-linked oligomers. In addition, a small proportion of Cys292 or Cys361 are likely to link with another cysteine residue (marked as X) since there were still few oligomers observed when only Cys292 or Cys361 was mutated, indicating the high flexibility and reactivity of these two oxidation-sensitive sites (Figure 6B). Interestingly, Cys292 has been recently identified as an S-nitrosylation site engaged in autophagy impairment-related neurotoxicity under hyperglycemia, showing the high reactivity and particularity of Cys292 [23].
Figure 6.
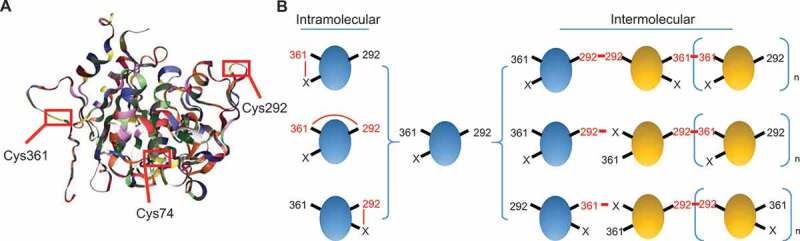
Proposed model of ATG4B oxidation. (A) 3D structure of human ATG4B. (B) A proposed model for the intramolecular disulfide bonds (left) and oligomeric modification of ATG4B (right) in vivo.
ATG4B is an indispensable enzyme which has a proteolytic activity for LC3 precursor and a deconjugation activity for lipidated LC3. These two processes are pivotal for the initiation and maturation of autophagosomes, a complex and fundamental step of autophagy process [14]. Thus, the regulation of ATG4B is tightly connected with the control of autophagy. In Chlamydomonas, norflurazon promotes Atg4 oxidation and oligomerization by generating intracellular ROS level, which in turn triggers autophagy because the inhibition of Atg4 causes the lipidation of Atg8 for autophagosome biosynthesis [27]. However, in yeast, mutation of Atg4 redox-sensitive sites increases Atg4 activity, thereby accelerating the formation of phagophore assembly site [26]. These conclusions reveal the diverse functions of ATG4 as a regulatory machinery of autophagy under oxidative stress, which may be due to the species differences. ATG4A, an orthologue of ATG4B, having high sequence similarity with ATG4B, was reported to be redox-regulated at the catalytic cysteine Cys77 and Cys81. It has been shown that mutation of Cys81 promotes ATG4A deconjugation activity, leading to decreased GFP-GATE-16 labeled autophagosomes under nutrient starvation [24]. In the same survey, mutation of Cys78 in human ATG4B, which is conserved with Cys81 in ATG4A, exhibits a similar autophagic dysfunction. However, in our study, an oxidized form was detected in ATG4BC78S mutant (Figure 3F). Moreover, mutation of Cys78 was unable to compromise the inhibitory effect of hydrogen peroxide (Figure S5B-E), indicating that, in our model, Cys78 was not involved in the oxidative inhibition. Moreover, we also repeated the same starvation experiment to study the effect of C78S mutant on the formation of LC3 puncta. Unfortunately, we haven’t observed the significant decrease in LC3 puncta under starvation, which may be attributed to the different expression level of ATG4B or other unknown factors (Figure S5F). We have previously identified Cys292 and Cys361 as the two oxidation-prone sites through a series of biochemical analysis. We compared the function of WT ATG4B and oxidation-inactive mutant in ATG4B KO HeLa cells. Results showed that in basal conditions, ATG4BC292,361S mutant displayed some activity, which was further increased under oxidative inhibition, suggesting that ATG4B activity was slightly attenuated even though exposed to negligible air (Figure 5B,C). Interestingly, the level of LC3-II in ATG4BC292,361S mutant was increased under normal growth, which was correlated with continuously-active autophagic flux evidenced by the assay of lysosome inhibitor treatment (Figures 5 and S6). Although ROS has been reported to be involved in aging-related central nervous system diseases, such as neurodegenerative disorder, there are no effective approaches for blocking the deleterious effects of ROS [6]. According to our findings, we propose that blocking ROS-targeted sites on ATG4B protease can modulate its activity and hence provide a promising therapeutic strategy to prevent ROS toxicity by initiating autophagy-protective mechanism.
In this study, we explored the specific molecular mechanism of ATG4B oxidative modification. We unraveled that intramolecular disulfide bond and intermolecular oligomerization are novel oxidative PTM to control ATG4B activity. Besides, we also identified Cys292 and Cys361 as key oxidation targets synergistically mediating ATG4B function and subsequent autophagy progress. However, oxidative stress is a complex pathological condition, involving multiple factors that coordinate cellular signals. In this study, although Cys292 and Cys361 are primarily responsible for ATG4B oxidative inhibition, it is likely that other unidentified inhibitory factors driven by oxidation pressure may be involved, since ablation of these two sites was not sufficient to completely rescue the ATG4B activity. In particular, Cys74 may make difference to some extent in the oxidation of ATG4B due to its highly catalytic and active characteristic.
Materials and methods
Reagents
Hydrogen peroxide (H2O2; D0136874) was purchased from Calbiochem; dithiothreitol (DTT; A100281), oxidized glutathione (GSSG; A100524), N-acetylcysteine (NAC; A1127), N-ethylmaleimide (NEM; EB0450) and chloroquine (CQ; A506569) were all purchased from Sangon Biotech; 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU; MB1303) was from Meilun Biotech; S-nitrosoglutathione (GSNO; 82240) was purchased from Cayman; auranofin (T1303) was obtained from TargetMol.
Antibodies
Antibodies in this study used in western blot and immunofluorescence analysis were as follow: anti-ATG4B (Cell Signaling Technology, 5299S), anti-FLAG (Medical & Biological Laboratories, M185-32), anti-LC3B for western blot (Sigma, L7543), anti-LC3B for immunofluorescence (Medical & Biological Laboratories, PM036), anti-GST (GenScript, A00097), anti-GAPDH (Santa Cruz Biotechnology, sc-365062), anti-His (Thermo Fisher Scientific, MA1-135), anti-GSH (Santa Cruz Biotechnology, sc-52399), anti-NPM1 (Proteintech, 60096–1). Secondary antibodies conjugated with Alex Fluor 488, and Alexa Fluor 594 were purchased from Life Technologies (35503, 35511, 35553, and 35561).
Plasmid construction and site mutation
The E. coli strain BL21 (DE3) (TransGen Biotech, CD601-01) were used as the host for prokaryotic expression and DH5ɑ cells (TransGen Biotech, CD201-01) for cloning. ATG4B was amplified by high-fidelity PCR and subcloned between XhoI and BamHI sites of modified pLVX vector (Clontech, 632154) for mammalian overexpression. For prokaryotic recombinant protein expression, His-tagged human ATG4B and GST-tagged human LC3B were cloned between EcoRI and XhoI sites of pET-28a (+) vector (Novagen, 69864). All the ATG4B cysteine mutants including double mutant ATG4BC292,361S were created by following the instruction of overlap Fast Mutagenesis kit (TransGen Biotech, FM111-01) and confirmed by gene sequencing. Construction of these mutants were performed according to the same methods stated previously. For lentivirus-based overexpression, PsPAX2 (Addgene, 12260) and PMD2.G (Addgene, 12259) were cloned and extracted from DH5ɑ following TIANprep Mini Plasmid Kit protocol (Tiangen Biotech, DP118-02).
Protein expression and purification
Recombinant plasmids encoding His-tagged WT and all cysteine-mutated ATG4B, and LC3-GST were transformed into E. coli BL21 (DE3) after induction of isopropyl β-D-1-thiogalactopyranoside (IPTG [Sangon Biotech, A600168], 0.5 mM, 16°C, 20 h). Then proteins were purified with Ni2+-NTA resin affinity chromatography (Weishibohui, CS-A01b-01). All preparations were desalted by PD-10 desalting columns (GE Healthcare, 52-1308-00). Proteins eluted with 200 mM of imidazole (Sangon Biotech, A600277) were preserved in desalting buffer (20 mM Na3PO4, 100 mM NaCl, pH 7.4) containing 20% glycerol at −80°C.
Cell culture and plasmid transfection
HEK293 (ATCC, CRL-1573) and HeLa cells (ATCC, CCL-2) were grown in Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific, 11965092) supplemented with 10% fetal bovine serum (Gibco, 10270) and 1% penicillin-streptomycin (Gibco, 15140–122) in 37℃ thermostatic incubator containing 5% CO2. For protein expression, plasmids were transfected to cells with indicated Lipo2000 (Life Technologies, 11668–019) or lentivirus according to manufacturer’s instructions. To construct stably transfected cell lines, after 24 h viral infection and expression, a pool of cells was cultured in medium containing 5 µg/ml (ATG4B KO HeLa cells) or 2 µg/ml (HEK293 cells) puromycin (Alomone Labs, P638) for 3 d, then half of the concentration of puromycin was continuously kept for long-term cell culture. The ATG4B KO HeLa cell was generated by CRISPR/Cas9 methods before and stored in our lab [17].
Immunoblotting assay
Cells were harvested and lysed in RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100 [Sangon Biotech, A110694], 0.1% SDS, 5% glycerol, 1 mM EDTA [Sigma, E6758]) containing protease inhibitor (Thermo Fisher Scientific, A32959). For reduced SDS-PAGE, samples were resolved in reduced loading buffer (25 mM Tris-HCl, pH 6.8, 50% [v:v] glycerol, 5% bromophenol blue, 5 mM DTT [Sangon Biotech, A100281]). Appropriate amount of lysates was loaded on and separated by 10% SDS-PAGE, then transferred to PVDF membranes (Millipore, ISEQ00010). After incubation with primary antibodies for at least 12 h at 4℃ and following HRP-conjugated secondary antibodies at room temperature, signal was detected with ImageQuat LAS 4000 mini (GE Healthcare, Uppsala, Sweden). For non-reduced SDS-PAGE, samples were prepared in the same loading buffer without DTT and separated in 8% gel to study ATG4B oxidation, subsequent procedures were according to the same protocols described above.
Immunoprecipitation
Cells grown on 60-mm Petri dish were washed with cold PBS and collected in 300 μl lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA) containing protease inhibitor and 10 mM NEM to avoid unnecessary modifications. After centrifugation, 600 μg of the cell lysate was mixed and incubated with anti-GSH antibody (1:100) in gyratory shaker overnight at 4℃. Next, 1 mg of Dynabeads Protein G (Invitrogen, 10004D) were added for another 3 h at 4℃. Then, the mixture was put on a magnetic rack, after discarding the supernatant, the beads were washed with wash buffer (0.01 M phosphate-buffered saline [Thermo Fisher Scientific, 10010023], pH 7.4, 0.02% Tween 20 [Scientific Chemical, VA29901]) 3 times. Finally, 20 μl elution buffer (50 mM glycine, pH 7.4) and loading buffer without DTT was added. Samples were subject to western blot and analyzed with anti-ATG4B antibody to detect glutathionylation modification.
Immunofluorescence assay
Cells cultured on a Petri dish were fixed with 4% paraformaldehyde for 30 min, followed by permeabilization (0.1% Triton X-100) and then blocked in 5% goat serum (Boster Biological Technology, AR0009) for 1 h at room temperature. Cells were incubated with primary antibodies (1:150) at 4℃ overnight. After washing with PBS 3 times, Alexa Fluor 488- or 594-conjugated secondary antibodies (1:500) were applied subsequently. Fluorescence were examined using EVOS FL Auto (Life Technologies). At least 40 cells in each experiment were analyzed for quantification.
Enzymatic assay
Cells were collected and homogenized in non-denaturing lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA). Purified ATG4B or cell lysates were incubated with LC3-GST substrate in reaction buffer (25 mM Tris, pH 7.4, 50 mM KCl) for 30 min at 37℃. Then the reaction was stopped by addition of 5X reduced loading buffer and separated by SDS-PAGE. Results were analyzed by CBB staining. The remained amounts of LC3-GST and the cleaved GST and LC3 products were quantified by gel densitometry to determine the ATG4B activity, which is equal to ODLC3-GST(cleaved)/(ODLC3-GST(remained) + ODLC3-GST(cleaved)) x 100%. In this equation, ODLC3-GST(cleaved) = ODGST + ODLC3.
Sequential 2D-diagonal SDS-PAGE
For carrying out 2D-diagonal SDS-PAGE, prepare two 10% SDS-PAGE polyacrylamide gels, one of which was prepared according to standard protocols, the other was only equipped with separation gel without stacking gel. In first dimension electrophoresis, samples were resolved in non-reduced loading buffer and separated in the regular 10% SDS-polyacrylamide gel. Then, the entire lane containing separated proteins was excised and incubated with 10 mM of DTT in gyratory shaker for 1 h at room temperature. Then the gel slice was rotated 90° and laid on top of another particular 10% SDS-polyacrylamide gel for the second electrophoresis. Gels were analyzed by staining with Coomassie Brilliant Blue (0.25% w:v CBB R-250 [Sangon Biotech, A610037], 45% methanol, 45% deionized water, 10% hydrochloric acid).
Statistics
All results were expressed as the mean ± SEM of at least 3 independent experiments. Statistic difference between 2 groups were determined by the Student t test. Statistical analysis was performed using Graphpad prism 8.0. In these analysis, P < 0.05 was considered significant, *** as P < 0.001, ** as P < 0.01, * as P < 0.05, and ns as not significant.
Supplementary Material
Funding Statement
This work was supported in part by the National Natural Science Foundation of China [31970699, 31671437], the Natural Science Foundation of Guangdong Province, China [2019A1515011030], and the Guangdong Provincial Key Laboratory of Construction Foundation [2017B030314030].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
The Supplemental data for this article can be accessed here.
References
- [1].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. PMID: 18305538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].He C, Klionsky DJ.. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. PMID: 19653858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Galluzzi L, Bravo-San Pedro JM, Blomgren K, et al. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467–484. PMID: 27256553. [DOI] [PubMed] [Google Scholar]
- [4].Michaud M, Martins I, Sukkurwala AQ, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. PMID: 22174255. [DOI] [PubMed] [Google Scholar]
- [5].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. PMID: 16625205. [DOI] [PubMed] [Google Scholar]
- [6].Galluzzi L, Bravo-San Pedro JM, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511. PMID: 28529316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. PMID: 16625204. [DOI] [PubMed] [Google Scholar]
- [8].Yoshii SR, Kuma A, Akashi T, et al. Systemic analysis of Atg5-null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev Cell. 2016;39:116–130. PMID: 27693508. [DOI] [PubMed] [Google Scholar]
- [9].Tsukada M, Ohsumi Y.. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174. PMID: 8224160. [DOI] [PubMed] [Google Scholar]
- [10].Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. PMID: 20811353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. PMID: 11100732. [DOI] [PubMed] [Google Scholar]
- [12].Tanida I, Sou YS, Ezaki J, et al. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem. 2004;279:36268–36276. PMID: 15187094. [DOI] [PubMed] [Google Scholar]
- [13].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. PMID: 21801009. [DOI] [PubMed] [Google Scholar]
- [14].Kirisako T, Ichimura Y, Okada H, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–276. PMID: 11038174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nakatogawa H, Ishii J, Asai E, et al. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy. 2012;8:177–186. PMID: 22240591. [DOI] [PubMed] [Google Scholar]
- [16].Marino G, Fernandez AF, Cabrera S, et al. Autophagy is essential for mouse sense of balance. J Clin Invest. 2010;120:2331–2344. PMID: 20577052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fu Y, Hong L, Xu J, et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy. 2019;15:295–311. PMID: 30176161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu PF, Hsu CJ, Tsai WL, et al. Ablation of ATG4B suppressed autophagy and activated AMPK for cell cycle arrest in cancer cells. Cell Physiol Biochem. 2017;44:728–740. PMID: 29169176. [DOI] [PubMed] [Google Scholar]
- [19].Jo YK, Park NY, Park SJ, et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget. 2016;7:57186–57196. PMID: 27527864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yang Z, Wilkie-Grantham RP, Yanagi T, et al. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J Biol Chem. 2015;290:26549–26561. PMID: 26378241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ni Z, He J, Wu Y, et al. AKT-mediated phosphorylation of ATG4B impairs mitochondrial activity and enhances the Warburg effect in hepatocellular carcinoma cells. Autophagy. 2018;14:685–701. PMID: 29165041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pengo N, Agrotis A, Prak K, et al. A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat Commun. 2017;8:294. PMID: 28821708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Li Y, Zhang Y, Wang L, et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy. 2017;13:1145–1160. PMID: 28633005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26:1749–1760. PMID: 17347651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Woo J, Park E, Dinesh-Kumar SP. Differential processing of Arabidopsis ubiquitin-like Atg8 autophagy proteins by Atg4 cysteine proteases. Proc Natl Acad Sci U S A. 2014;111:863–868. PMID: 24379391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Perez-Perez ME, Zaffagnini M, Marchand CH, et al. The yeast autophagy protease Atg4 is regulated by thioredoxin. Autophagy. 2014;10:1953–1964. PMID: 25483965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perez-Perez ME, Lemaire SD, Crespo JL. Control of autophagy in chlamydomonas is mediated through redox-dependent inactivation of the ATG4 protease. Plant Physiol. 2016;172:2219–2234. PMID: 27756818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Navarro-Yepes J, Burns M, Anandhan A, et al. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal. 2014;21:66–85. PMID: 24483238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lavandero S, Chiong M, Rothermel BA, et al. Autophagy in cardiovascular biology. J Clin Invest. 2015;125:55–64. PMID: 25654551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. PMID: 21248839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. PMID: 18978800. [DOI] [PubMed] [Google Scholar]
- [32].Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. PMID: 11089981. [DOI] [PubMed] [Google Scholar]
- [33].Burgoyne JR, Mongue-Din H, Eaton P, et al. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–1106. PMID: 23023511. [DOI] [PubMed] [Google Scholar]
- [34].Frudd K, Burgoyne T, Burgoyne JR. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun. 2018;9:95. PMID: 29311554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Carroll B, Otten EG, Manni D, et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat Commun. 2018;9:256. PMID: 29343728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mukai K, Ouchi A, Nagaoka S, et al. Pyrroloquinoline quinone (PQQ) is reduced to pyrroloquinoline quinol (PQQH2) by vitamin C, and PQQH2 produced is recycled to PQQ by air oxidation in buffer solution at pH 7.4. Biosci Biotechnol Biochem. 2016;80:178–187. PMID: 26264520. [DOI] [PubMed] [Google Scholar]
- [37].Silveira-Dorta G, Monzon DM, Crisostomo FP, et al. Oxidation with air by ascorbate-driven quinone redox cycling. Chem Commun (Camb). 2015;51:7027–7030. PMID: 25805569. [DOI] [PubMed] [Google Scholar]
- [38].Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med. 2015;80:148–157. PMID: 25433365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev. 2013;113:4633–4679. PMID: 23514336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhong X, He T, Prashad AS, et al. Mechanistic understanding of the cysteine capping modifications of antibodies enables selective chemical engineering in live mammalian cells. J Biotechnol. 2017;248:48–58. PMID: 28300660. [DOI] [PubMed] [Google Scholar]
- [41].Cremers CM, Jakob U. Oxidant sensing by reversible disulfide bond formation. J Biol Chem. 2013;288:26489–26496. PMID: 23861395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang K, Wang M, Zhao Y, et al. A redox mechanism underlying nucleolar stress sensing by nucleophosmin. Nat Commun. 2016;7:13599. PMID: 27886181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kumanomidou T, Mizushima T, Komatsu M, et al. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J Mol Biol. 2006;355:612–618. PMID: 16325851. [DOI] [PubMed] [Google Scholar]
- [44].Sugawara K, Suzuki NN, Fujioka Y, et al. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J Biol Chem. 2005;280:40058–40065. PMID: 16183633. [DOI] [PubMed] [Google Scholar]
- [45].Brennan JP, Wait R, Begum S, et al. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J Biol Chem. 2004;279:41352–41360. PMID: 15292244. [DOI] [PubMed] [Google Scholar]
- [46].Winger AM, Taylor NL, Heazlewood JL, et al. Identification of intra- and intermolecular disulphide bonding in the plant mitochondrial proteome by diagonal gel electrophoresis. Proteomics. 2007;7:4158–4170. PMID: 17994621. [DOI] [PubMed] [Google Scholar]
- [47].Li M, Hou Y, Wang J, et al. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286:7327–7338. PMID: 21177865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Braakman I, Hoover-Litty H, Wagner KR, et al. Folding of influenza hemagglutinin in the endoplasmic reticulum. J Cell Biol. 1991;114:401–411. PMID: 1650370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Heldman N, Vonshak O, Sevier CS, et al. Steps in reductive activation of the disulfide-generating enzyme Ero1p. Protein Sci. 2010;19:1863–1876. PMID: 20669236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.