

ABSTRACT
1-Deoxysphingolipids (deoxySLs) are atypical sphingolipids of clinical relevance as they are elevated in plasma of patients suffering from hereditary sensory and autonomic neuropathy (HSAN1) or type 2 diabetes. Their neurotoxicity is described best but they inflict damage to various cell types by an uncertain pathomechanism. Using mouse embryonic fibroblasts and an alkyne analog of 1-deoxysphinganine (doxSA), the metabolic precursor of all deoxySLs, we here study the impact of deoxySLs on macroautophagy/autophagy, the regulated degradation of dysfunctional or expendable cellular components. We find that deoxySLs induce autophagosome and lysosome accumulation indicative of an increase in autophagic flux. The autophagosomal machinery targets damaged mitochondria that have accumulated N-acylated doxSA metabolites, presumably deoxyceramide and deoxydihydroceramide, and show aberrant swelling and tubule formation. Autophagosomes and lysosomes also interact with cellular lipid aggregates and crystals that occur upon cellular uptake and N-acylation of monomeric doxSA. As crystals entering the lysophagosomal apparatus in phagocytes are known to trigger the NLRP3 inflammasome, we also treated macrophages with doxSA. We demonstrate the activation of the NLRP3 inflammasome by doxSLs, prompting the release of IL1B from primary macrophages. Taken together, our data establish an impact of doxSLs on autophagy and link doxSL pathophysiology to inflammation and the innate immune system.
Abbreviations: alkyne-doxSA: (2S,3R)-2-aminooctadec-17yn-3-ol; alkyne-SA: (2S,3R)-2- aminooctadec-17yn-1,3-diol; aSA: alkyne-sphinganine; ASTM-BODIPY: azido-sulfo-tetramethyl-BODIPY; CerS: ceramide synthase; CMR: clonal macrophage reporter; deoxySLs: 1-deoxysphingolipids; dox(DH)Cer: 1-deoxydihydroceramide; doxCer: 1-deoxyceramide; doxSA: 1-deoxysphinganine; FB1: fumonisin B1; HSAN1: hereditary sensory and autonomic neuropathy type 1; LC3: MAP1LC3A and MAP1LC3B; LPS: lipopolysaccharide; MEF: mouse embryonal fibroblasts; MS: mass spectrometry; N3635P: azido-STAR635P; N3Cy3: azido-cyanine 3; N3picCy3: azido-picolylcyanine 3; NLRP3: NOD-like receptor pyrin domain containing protein 3; P4HB: prolyl 4-hydroxylase subunit beta; PINK1: PTEN induced putative kinase 1; PYCARD/ASC: PYD and CARD domain containing; SPTLC1: serine palmitoyltransferase long chain base subunit 1; SQSTM1: sequestosome 1; TLC: thin layer chromatography.
KEYWORDS: Autophagy, crystal, doxSA, HSAN1, innate immunity, lipid, macrophage
Introduction
1-Deoxysphingolipids (deoxySLs) are noncanonical sphingolipids that lack the 1-hydroxy group at the lipids headgroup [1]. They derive from 1-deoxysphinganine (doxSA), a cellular product of SPTLC1 (serine palmitoyltransferase long chain base subunit 1) using L-alanine rather than L-serine in the condensation reaction with palmitoyl-CoA. Several mutants of SPTLC1 are described exhibiting an increased preference for L-alanine, leading to a buildup of deoxySLs causing health problems in the affected patients. Amongst these pathologies, slow dying of sensory and autonomic neurons is most severe and a hallmark of the rare hereditary sensory and autonomic neuropathy type 1 (HSAN1). In HSAN1 patients, plasma levels as high as 1.2 µM of deoxySLs are detected [2] compared to 0.1–0.3 µM found in healthy persons [3]. Plasma deoxySLs are also elevated in metabolic syndrome and type 2 diabetes patients [3–5].
By now, considerable knowledge on cellular deoxySL anabolism has been gathered. DoxSA is N-acylated to 1-deoxydihydroceramides (doxDHCers) by ceramide synthases [6,7] that upon further desaturation by an unknown desaturase yield 1-deoxyceramides (doxCers) [2,8–10]. Together, these members represent the most abundant fraction of the doxSL family that further includes 1-deoxysphingosine, 1-deoxymethylsphinganine, 1-deoxymethylceramides and their various hydroxylated or poly-unsaturated siblings [11]. The latter group likely results from the catabolic conversion of other doxSLs, a slow process resembling cellular detoxification procedure [1].
Given the fact that the degradation of deoxySLs takes several days, they accumulate in the interim and trigger various cellular effects. Although specific interactions of deoxySLs with proteins are still unknown, the consequences of their cellular presence have been studied. DeoxySLs are cytotoxic, and the incubation with doxSA at a low micromolar concentration causes cell death in various cell lines [1,7,12–14]. For mouse embryonic fibroblasts, MEFs, the LD50 dose of 7 μM is determined [1]. Which mechanism the cytotoxicity of doxSLs is based on is controversially discussed and may involve several cellular pathways and components [1,15]. Stimulated ceramide synthesis and PKC activation [13], triggering of atypical cell death via CASP3 and CASP12 with altered TRP53 phosphorylation [12], disrupted actin fibers [15,16] and induction of ER stress [8,17,18] and ER collapse [15,19] are proposed as the involved processes. Also, cell senescence at low doses and apoptosis and necrosis at high doses is observed [20]. Neurons are especially vulnerable to deoxySL-mediated cytotoxicity. Axon deterioration, changes to neurite morphology along with alterations in the cytoskeleton are described and extensively reviewed [1]. Apart from neurons [2,21,22] also INS (insulin)-producing cells [20] and lymphocytes [23] have been studied after their designation as physiological targets of elevated doxSL levels in HSAN1.
Owing to the lack of the 1-hydroxygroup and a double bond in the head portion of the molecule, doxySLs possess different biophysical properties than regular sphingolipids [1]. A higher hydrophobicity and reduced ability to form intra- and intermolecular H-bond networks result. In fact, doxCer shows reduced lipid miscibility in synthetic GUV bilayers, likely forming a separate gel-like phase and at elevated concentrations probably even forming aggregates [24]. In vivo such lipid aggregates are most prominently described for cholesterol that forms crystals at high concentrations. These are of physiological relevance as cholesterol crystals are an important trigger of inflammation in atherosclerotic plaques [25]. The crystals are recognized and internalized by macrophages and other phagocytes, where they induce lysosomal damage that results in the activation of the NLRP3 (nucleotide-binding domain leucine-rich repeat containing [NLR] family, pyrin domain containing 3) inflammasome [26–29]. The activated NLRP3 inflammasome, a protein complex consisting of the receptor NLRP3, the adaptor molecule PYCARD/ASC (PYD and CARD domain containing) and CASP1 cleaves the proforms of IL1B and IL18 to their mature secreted form [30,31]. The affected cells ultimately lyse, and the transmitted cytokine signals induce inflammation.
Apart from extracellular crystals and rupture of phagolysosomes the NLRP3 inflammasome can also be activated by signals derived from other damaged cell organelles such as mitochondria, Golgi apparatus and ER [32]. These signals may include ROS that is produced upon cellular stress sensing [30]. Stressed and damaged organelles routinely are subjected to macroautophagy/autophagy (hereafter called autophagy), a homeostatic mechanism for the lysosomal degradation and recycling of organelles [33]. Autophagy also directly eliminates active inflammasomes and, hence, overall limits and tempers the activation of the NLRP3 inflammasome [32,34].
Ceramides have recently been identified as an essential regulator of mitophagy, a subtype of autophagy where mitochondria are selectively degraded [35]. Mitochondrial ceramide acts as a receptor for LC3-II, the activated form of the autophagy protein LC3, and thereby recruits autophagosomes to damaged mitochondria leading to non-apoptotic lethal mitophagy [36]. Importantly, it is suggested that the mitochondrial localization of the various ceramides tested, rather than their fatty acid side chain length is important for triggering lethal mitophagy [35]. No analogous data for deoxyceramides is available.
For the analysis of the cellular fate of deoxySLs various types of labeled deoxySL probes have proven invaluable. In a previous study, we introduced alkyne-doxSA, (2S,3R)-2-aminooctadec-17-yn-3-ol, an analog of doxSA, to trace doxSL metabolism and localization [19]. Alkyne-doxSA prominently targets mitochondria, less so ER and Golgi, and neglectable signal is observed in lysosomes. Mitochondrial localization occurs within minutes after alkyne-doxSA application and like natural doxSA it impacts mitochondrial morphology. For the latter effect, micromolar concentrations of alkyne-doxSA and a ceramide synthase activity as well as extended incubation times are necessary. The induced changes in mitochondrial shape and function are accompanied by alterations in ER morphology, leading to an organelle structure resembling stressed ER, and eventually by cell collapse and death.
As stressed cell organelles are routinely subject to autophagic degradation, we now initiate dedicated experiments to study the interaction of deoxySLs and the autophagosomal apparatus. The current study aims to (i) investigate the effects of deoxySLs on the autophagosomes and lysosomes in MEF cells, (ii) provide a detailed description of the structural changes to organelles in these cells upon doxSA treatment, (iii) correlate these observations with metabolic data, and (iv) analyze effects of deoxySLs on macrophages and innate immunity.
We propose that deoxySLs, especially the deoxyceramides that build up in cells upon doxSA treatment, promote accumulation of vesicles of the autophagosomal apparatus as well as the formation of intracellular lipid aggregates and lipid crystals. In macrophages, this leads to activation of the NLRP3 inflammasome triggering the release of effector cytokines driving inflammation.
Results
DeoxySLs induce accumulation of autophagosomes and lysosomes and block cell division
To re-investigate the subcellular localization of deoxySLs, we treated MEF cells with 1 μM doxSA for 24 h. This concentration was previously found to have pronounced cellular effects [19], lies clearly below the reported LD50 concentration (7 μM) for these cells [1], matches well the reported total plasma levels of deoxySLs in HSAN1 patients [2] and by replacing 10% of it by the alkyne doxSA tracer allowed for sufficient signal intensity in all our analytics.
By microscopy, we re-confirmed a prominent localization of alkyne deoxySLs to mitochondria marked by TOMM20 immunolabelling and only minor overlap of the tracer signal with the lysosomal LAMP1 staining (Figure 1A). Lysosomes were abundant and mostly located in the perinuclear region. As before, the mitochondria generally appeared swollen and condensed, but in many cells, we also observed extended tubules interconnecting mitochondria even across the full length of the cell (Figure 1A, inset 1). These tubules were positive for alkyne doxSL and TOMM20 signals.
Figure 1.
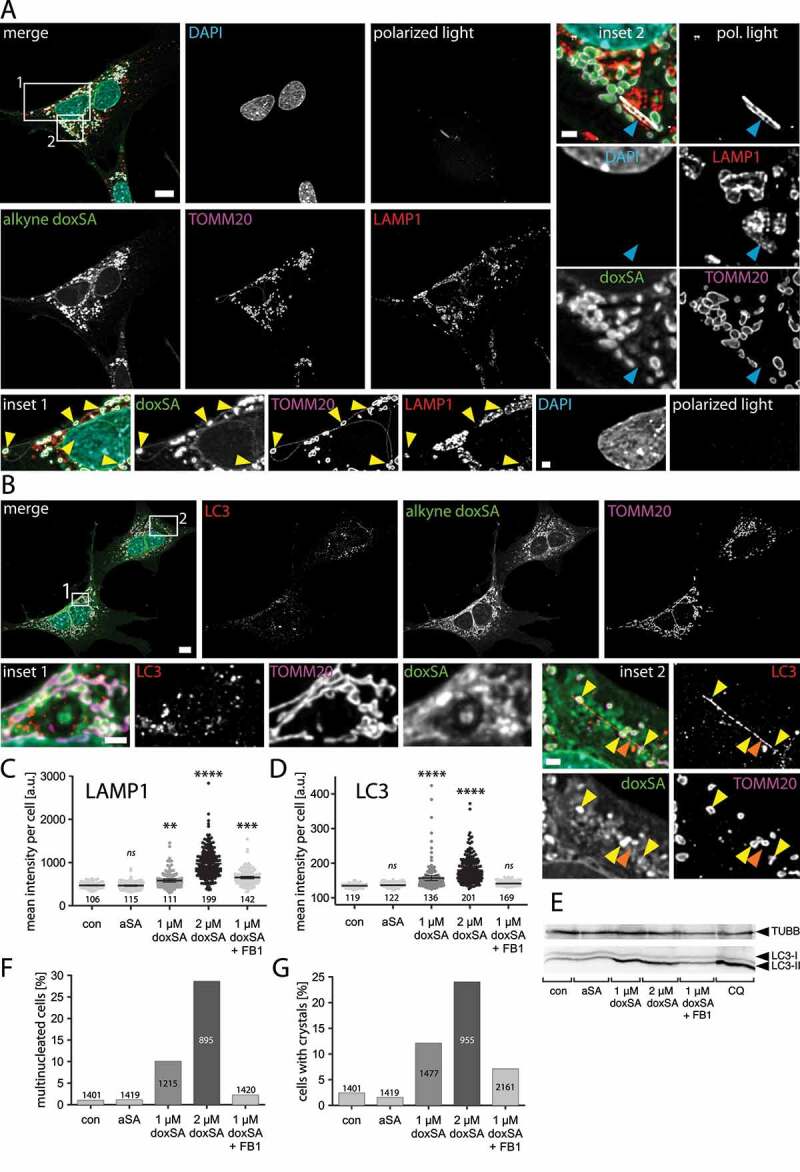
1-Deoxysphingolipids induce vesicle accumulation in the autophagosomal apparatus and block cell division. MEF cells were treated with 1 μM doxSA mix (0.9 μM doxSA + 0.1 μM alkyne doxSA tracer) for 24 h. After fixation cells were probed for various organelle markers by immunofluorescence and the alkyne moiety was reacted with N3Cy3-reporter. Micrographs were recorded using structured illumination. (A,B) Maximum image projections of z-stacks after deconvolution depict alkyne lipids (green, doxSA), nuclei (cyan, DAPI), mitochondria (magenta, TOMM20), lysosomes (red, LAMP1), autophagosomes (red, LC3) as color-merged or grayscale images. Polarized light microscopy revealing birefringent structures was also performed (A). Note the alkyne lipid-positive mitochondria interconnected by membrane tubules (A, inset 1, yellow arrowheads) and the birefringent crystal overlapping the lysosomal signal and neighboring alkyne lipid-positive mitochondria (A, inset 2, blue arrowhead). Also note the rod-like LC3-II-positive structure (B, inset 2, orange arrowhead) neighboring alkyne lipid-positive mitochondria (yellow arrowheads). Scale bars, 10 μm or 2 μm (insets). (C,D,F,G) Semi-quantitative analysis of cellular structures from micrographs exemplarily illustrated in Figures S1 and S2. MEF cells were treated with carrier (no lipid control), 1 μM alkyne sphinganine (aSA), 1 or 2 μM doxSA mix, or 1 μM doxSA mix + 25 μg/mL ceramide synthase inhibitor fumonisin B1 (1 μM doxSA + FB1) for 24 h. Mean intensity values per cell of signals for LAMP1 (C) or LC3 (D) are plotted with ±95% confidence interval. Statistical differences were calculated by ANOVA followed by Dunnett’s Multiple Comparison Test. Adjusted P values: **** p < 0.0001; *** p < 0.001; ** p < 0.0025; ns – not significant; all vs. untreated control. Percentage of multinucleated cells (F) or cells with birefringent crystals (G) are graphed. Bar number indicate the cell number considered. (E) Analysis of cellular content of LC3 and TUBB by western blotting. Cells treated with 50 μM chloroquine (CQ) served as positive control
In the vicinity of the swollen mitochondria, autophagic structures positive for LC3-II were often detectable (Figure 1B). Accumulation of mitochondria decorated by LC3-II signal could be observed (Figure 1B, inset 1). Frequently the LC3-II-positive structures displayed an elongated rod-shape and were in close approximation to mitochondria (Figure 1B). To confirm the influence on the autophagosomal apparatus as being specific for deoxySLs, we compared the labeling patterns for LAMP1 and LC3 upon incubation with either sphinganine or deoxy-sphinganine tracers (Figures S1 and S2). While cells treated with alkyne sphinganine (aSA) or untreated cells showed comparable lysosome amounts and distribution patterns, incubation with its sibling doxSA yielded a more pronounced LAMP1 signal near the nucleus, even if co-applied with fumonisin B1 (FB1), a ceramide synthase inhibitor [37] (Figure S1). Similarly, upon doxSA but not aSA treatment, the LC3-II-signal became more apparent within the cells as it clustered to numerous discrete structures (Figure S2). Co-application of doxSA and FB1 resulted in LC3 staining comparable to that of the untreated or aSA-treated controls. The doxSA-induced alterations of mitochondrial morphology were also blunted by FB1 (Figures S1 and S2).
A semi-quantitative analysis of fluorescent micrographs confirmed the visual impressions. Compared to untreated or aSA-treated controls, the incubation with doxSA increased both the mean intensity of the LAMP1 signal per cell (Figure 1C) and that of LC3 (Figure 1D). While co-application of FB1 suppressed the LC3 signal gain, it did not preclude the boost in LAMP1 fluorescence. In cells incubated with an elevated concentration of doxSA (2 μM) the observed effects were more pronounced.
To corroborate some of these findings, we also performed protein expression analyzes by western blotting (Figure 1E). In line with the microscopy data, an intensity increase of the LC3-II band, corresponding to the activated form of the LC3 protein, was observed upon doxSA treatment. Co-application of FB1 yielded LC3 levels similar to untreated or aSA-treated controls.
As the cellular accumulation of LC3-II-positive autophagosomes can result either from an increased induction or from a decreased degradation of autophagosomes, we next investigated the autophagic flux in more detail. Frequently the LC3-binding receptor SQSTM1/p62, an autophagy receptor involved in cargo transfer into the autophagosome that becomes co-degraded in the autolysosome, serves as an index of autophagic degradation [33]. Therefore, we performed a western blotting analysis and found increased SQSTM1 protein levels upon doxSA incubation, compared to untreated or aSA-treated controls (Figure S3A). Also the co-application of doxSA and FB1, or the incubation with chloroquine, an autolysosomal degradation inhibitor serving as a positive control, increased the cellular SQSTM1 content. In contrast, the amount of PINK1, a kinase involved in mitophagy, appears unchanged upon doxSA treatment.
To gain more insights into the autophagic flux under doxSA exposure, we analyzed the SQSTM1 and LC3-I/II levels in the absence and presence of a variety of flux inhibitors by western blotting (Figure S3B). Co-inhibition of lysosomal proteases by pepstatin A and E64d for 24 h did not influence the SQSTM1 levels but increased the cellular LC3 content beyond that found for doxSA treatment alone. Alike, the ratio of LC3-II over LC3-I increased further. DoxSA incubation in the presence of chloroquine, a compound that elevates the lysosomal pH, boosted the levels of SQSTM1 and LC3, as well as the ratio of LC3-II/I. However, increasing the lysosomal pH by bafilomycin A co-treatment for 2 or 24 h only minutely increased the total LC3 levels or the LC3-II/I ratio. After the SQSTM1 content first declined (2 h), it rose upon longer exposure to this inhibitor (24 h). Incubating cells for only 2 h with the combination of chloroquine and bafilomycin A as well as the protease inhibitors essentially mimicked the result of the short exposure to bafilomycin A only.
We also performed a complementary semi-quantitative analysis of the SQSTM1 levels in fluorescent micrographs (Figure S3C). Compared to untreated or aSA-treated controls, the incubation with 2 μM doxSA as well as the co-application of doxSA and FB1 or doxSA and chloroquine significantly increased the SQSTM1 mean signal. Of note, while doxSA at a concentration of 1 μM (rather than 2 μM) did not significantly increase the SQSTM1 intensity, we observed a similar change in the SQSTM1 staining patterns for both concentrations tested. Compared to controls, the SQSTM1 staining in doxSA-treated samples appeared more punctate and elongated structures became discernable. These structures, positive for SQSTM1 and LC3-II, lacked a clearly discernable doxSL labeling (Figure S3D) and were frequently seen near mitochondria (Figure S3E).
Scrutinizing the microscopy data, we also noted that upon incubation with doxSA many cells contained two or more nuclei, indicating an influence of deoxySLs on cell division specifically cytokinesis (Figures S1–S3 and 1F). Additionally, in many cells, also birefringent crystals were detectable (Figures S1, S2 and 1A, inset 2). Compared to untreated and aSA-treated control crystals were ~5-fold more frequent when cells were incubated with 1 μM doxSA, and their occurrence doubled further upon treatment with 2 μM doxSA (Figure 1G). Co-application of 1 μM doxSA and FB1 still resulted in a ~ 3-fold prevalence. These crystals occurred individually, showed no preferential localization within the cell, displayed heterogeneous length and width and were distinguishable from other birefringent structures of sub-micron diameter. The crystals often were associated to LAMP1-positive lysosomes but did not overlap with the TOMM20 signal (Figure 1A, inset 2). At their circumferential edge, the crystals showed only weak alkyne lipid signal.
DoxSA metabolism is influenced by the ceramide synthase inhibitor FB1
Upon uptake, cells metabolize the exogenously provided doxSA. Above data show that many of the observed cellular effects could be blunted by co-application of FB1. To investigate the influence FB1 exerts on doxSA metabolism, complementary analyses by TLC and quantitative MS were performed (Figure 2). As we established earlier [19], the N-acylated doxCer or dox(DH)Cer were the major cellular products (Figure 2A). Together they constituted about three-quarters of the labeled lipids after incubating the cells for 24 h (Figure 2B). The MS analysis revealed that fatty acids with 24 or 16 carbon atoms were the major N-acylation partners with a frequency of ~52% and ~32%, respectively (Figure 2C). The presence of 25 μg/mL FB1 largely prevented the cellular synthesis of doxCer and dox(DH)Cer (Figure 2A,C). Despite the block in anabolism the cellular uptake of doxSA still occurred, although total concentrations of labeled lipids dropped by a third in the inhibited cells (Figure 2B). Blocking ceramide synthases left ~98% of all labeled lipids as unprocessed doxSA in the cells. The ~2% doxCer/dox(DH)Cer still generated showed an unchanged prevalence for C24 and C16 fatty acids as N-acylation partners (Figure 2C).
Figure 2.
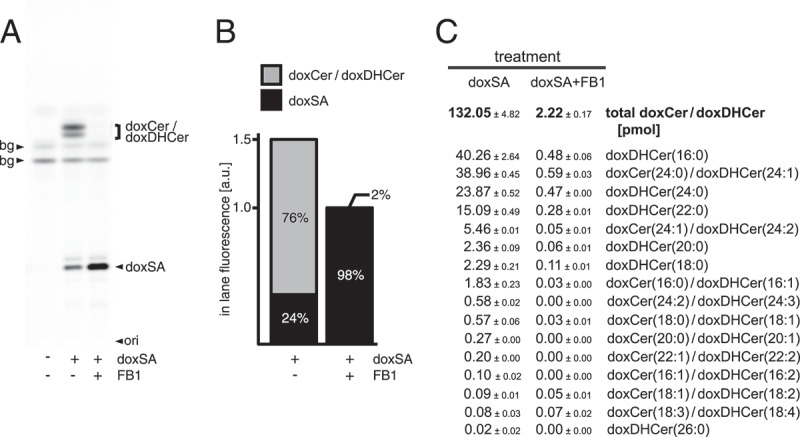
Ceramide synthase inhibition blocks 1-deoxysphingolipid anabolism. Cells were incubated with 1 μM doxSA mix in the absence or presence of the ceramide synthase inhibitor fumonisin B1 (FB1) for 24 h. Cellular lipids were extracted and extracts split. Corresponding samples were analyzed by TLC for fluorescent metabolites (A,B) or by MS for specifically fragmenting metabolites (C). Comigrating or internal standards were used for identification or quantification. Fluorescent TLC images (A) of cellular alkyne lipids were used to calculate the proportion of the alkyne deoxysphingolipids in cells (B). Note that samples lacking inhibitor are 1.5-fold brighter. Cellular alkyne metabolites were further characterized by quantitative MS analysis (C). All data are average with range, N = 2. bg, background; ori, origin
DeoxySLs induce intracellular lipid aggregates in addition to crystals
Given the fact that doxSA treatment resulted in a dose-dependent occurrence of birefringent crystals and the FB1 inhibitor reduced both N-acylation of doxSA and crystal emergence, we postulated that these crystals might be lipidic, consisting at least partially of doxSA-derived doxCer or dox(DH)Cer. As in epifluorescence micrographs, the fluorescent labeling of these crystals was rather weak (Figure 1A, inset 2), we first performed a labeling experiment in which 1 μM alkyne doxSA replaced all the non-traceable doxSA (Figure 3A). In the recorded micrographs, a stronger alkyne lipid signal resulted, that allowed for the observation of clearly resolved intracellular lipid aggregates. These lipid aggregates usually occurred as clusters of straight profiles (Figure 3A, insets 1 and 2) and looked different than the birefingent crystals (compare Figure 1A, inset 2). They were also clearly distinguishable from curved, elongated structures resembling bundled or aligned membranes (Figure 3A, inset 3).
Figure 3.
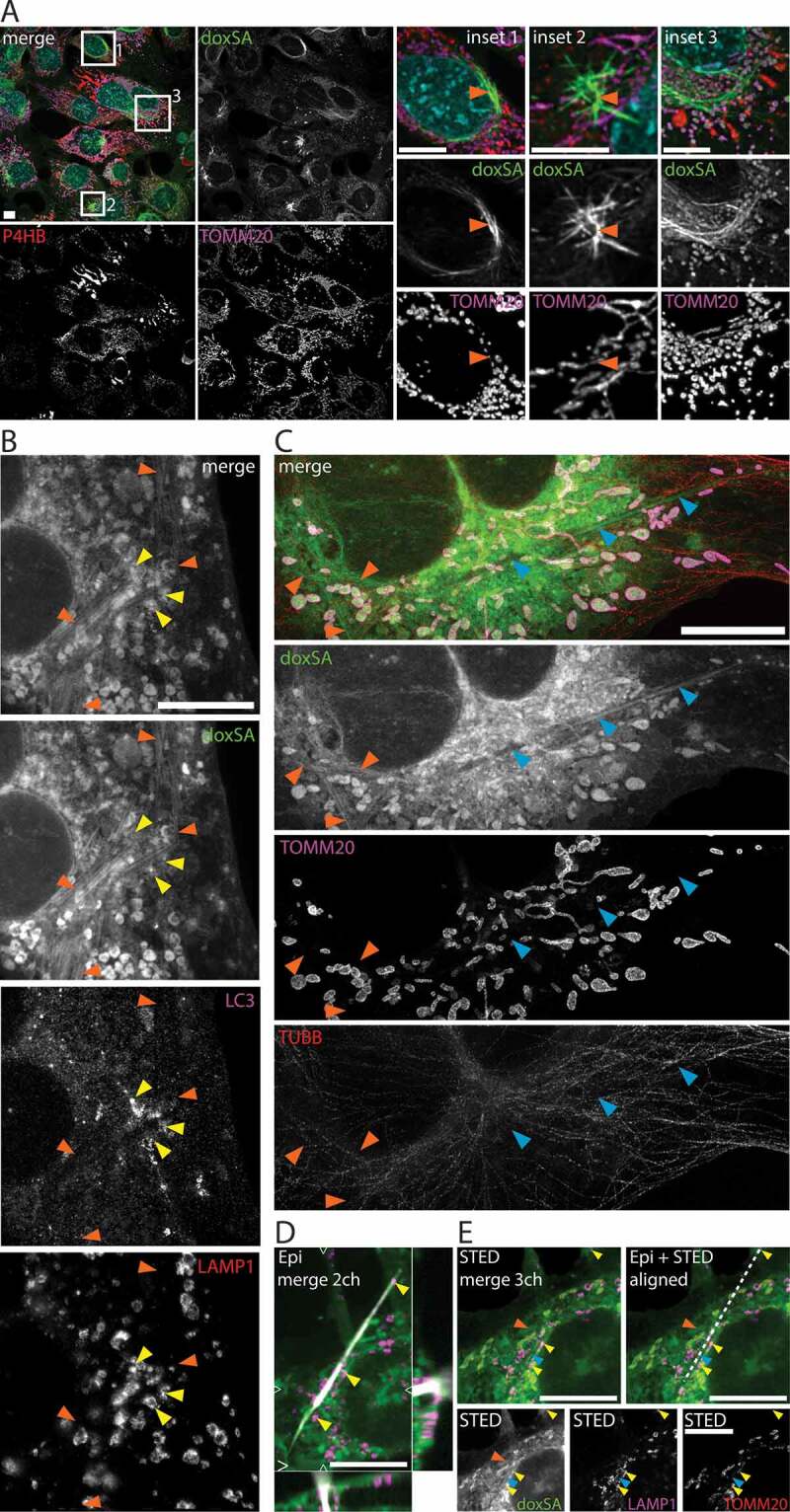
1-Deoxysphingolipids induce various cellular lipid aggregates. MEF cells were treated with (A) 1 μM alkyne doxSA or (B-E) 1 μM doxSA mix (0.9 μM doxSA + 0.1 μM alkyne doxSA tracer) for 24 h. After fixation cells were probed for various organelle markers by immunofluorescence and the alkyne moiety was reacted with ASTM-BODIPY (A) or N3635P-reporter (B–E). Micrographs were recorded using structured illumination (A,D) or STED (B,C,E) microscopy. (A) Maximum image projections of z-stacks after deconvolution depict alkyne lipids (green, doxSA), nuclei (cyan, DAPI, merge+insets), mitochondria (magenta, TOMM20), endoplasmic reticulum (red, P4HB, merge) as color-merged or grayscale images. Note the lipid aggregates (insets, orange arrowheads). (B,C) Single-layer super-resolution micrographs depict lysosomes (red, LAMP1), autophagosomes (magenta, LC3), and TUBB (red) as color-merged or grayscale images. Other channel colors are as above. Note the straight hollow profiles of lipid aggregates (orange arrowheads) distinguishable from filled crystal-like structures (blue arrowheads) both positive for alkyne lipids. Also note the autophagosomal structures positive for LC3-II and LAMP1 neighboring the lipid aggregates (yellow arrowheads). (D,E) Correlative Epifluorescence and STED microscopy images showing the same cell as (D) maximum image projection of a z-stack with side views at the indicated positions (open triangles) including the polarized light channel (gray), and (E) as a single-layer super-resolution image with superimposed birefringent crystal (dashed line), aligned using the 3 marked lysosomes (yellow arrowheads) as landmarks. Note the straight crystal-like structure (blue arrowhead), but not the lipid aggregate (orange arrowhead) exhibits birefringency. Bars, 10 μm
To study these lipid aggregates and crystals in more detail, we next visualized doxSA-treated cells by super-resolution microscopy (Figure 3B,C). Frequently the cells contained numerous straight structures of constant caliber (421 ± 67 microns, N = 15) that showed prominent lipid labeling at parallel profiles with a less stained interspace (Figure 3B,C, orange arrowheads). These lipid aggregates mostly occurred clustered. A capping structure could not be identified unequivocally. No clustered lipid aggregates were observed when FB1 was co-applied.
Occasionally also elongated, more crystal-like structures could be observed in doxSA-treated cells (Figure 3C, blue arrowheads). Straight in shape, these structures varied in caliber and often their core showed no clearly resolvable features. Performing correlative epifluorescence and STED microscopy (Figure 3D,E), we found that these crystal-like structures (blue arrowhead) exhibited birefringence, whereas no birefringence was detectable for the clustered lipid aggregates (orange arrowheads).
Lipid aggregates interact with the autophagosomal apparatus
Since deoxySLs induced vesicle accumulation in the autophagosomal apparatus and also triggered intracellular lipid aggregation, we postulated a mechanistic connection between both observations. Indeed, in super-resolution micrographs, we found vesicles stained for both LAMP1 and LC3-II in close contact with the lipid aggregates (Figures 3B and 4A). These putative autolysosomes were detected near the apparent end or in close contact to the sides of the lipid aggregates. The autolysosomes frequently showed elongated protrusions that were often straight (Figure 4A), sometimes kinked (Figure S4A,C) or curved (Figures 4B and S4B) in shape. However, they did not overlap with the lipid aggregates and were of finer caliber. Autolysosomes contacted other autolysosomes or lysosomes via such protrusion (Figures 4A,B and S4A,B) and reached sizes of several microns in diameter (Figure S4D).
Figure 4.
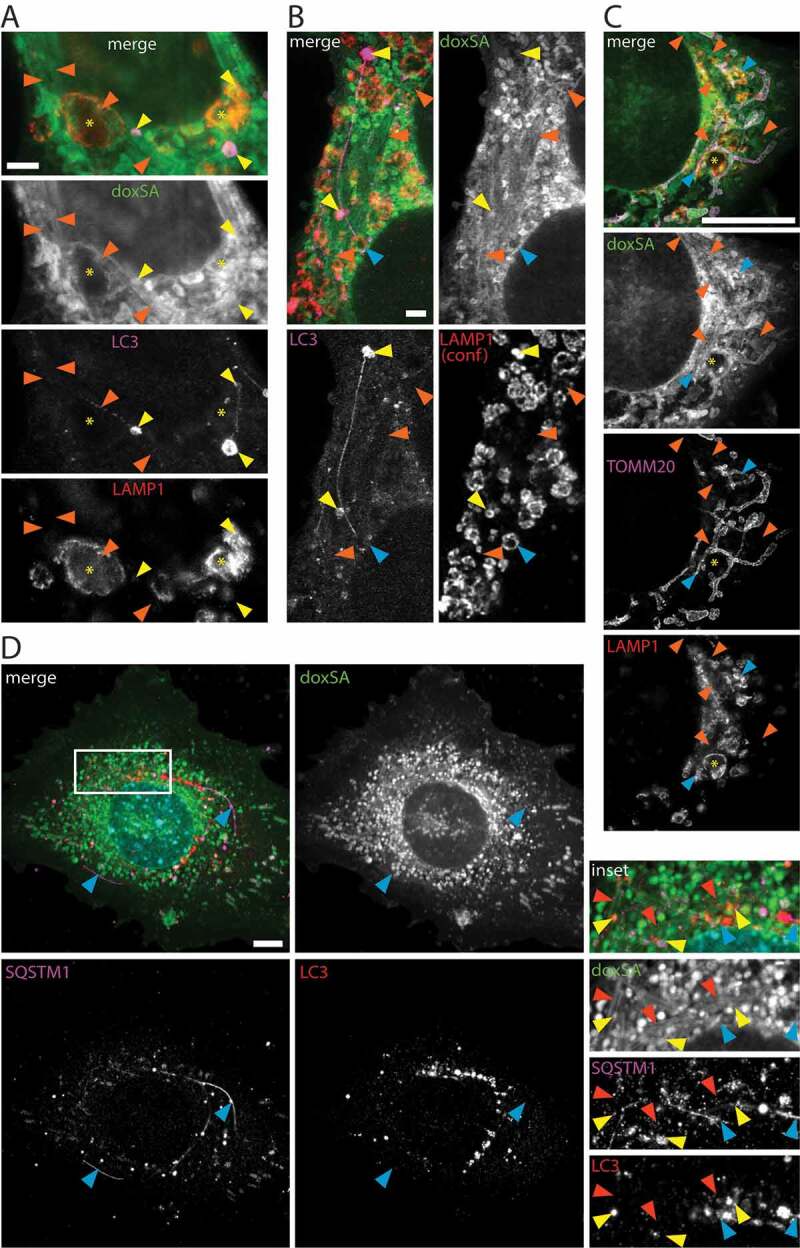
Lipid aggregates interact with the autophagosomal apparatus. MEF cells were treated with 1 μM (A-C) or 2 μM (D) doxSA mix for 24 h. After fixation cells were probed for various organelle markers by immunofluorescence and the alkyne moiety was reacted with N3635P-reporter (A–C) or N3picCy3-reporter (D). STED-micrographs (A–C) or maximum image projections of z-stacks recorded using structured illumination (D) depict alkyne lipids (green, doxSA), mitochondria (magenta, TOMM20), lysosomes (red, LAMP1), autophagosomes (magenta (A,B, LC3; D, SQSTM1) or red (D, LC3)) as color-merged or grayscale images. Note the straight lipid aggregates (orange arrowheads) neighboring large lysosomes (A,C, asterisks), autolysosomes (A,B, yellow arrowheads) and lysosomes (B, blue arrowheads) interconnected by straight and curved protrusions. Also note the close approximation of lipid aggregates and elongated SQSTM1-positive structures (D, blue arrowheads) and autophagosomes (D, yellow arrowheads). In (B) the LAMP1-signal was recorded as a confocal image. Scale bars, (A,B) 2 μm, (C,D) 10 μm
Additionally, LC3- or SQSTM1-positive structures and LAMP1-positive lysosomes, particularly large lysosomes, neighbored the lipid aggregates (Figure 4A–D) as they were found near crystals, too (Figure 1A, inset 2).
Often also mitochondria were intermingled with the lipid aggregates and LAMP1-positive vesicles of different sizes (Figure 4C). The mitochondria showed profound morphological changes, ranging from extensive tubulation (Figures 1 and S5A,B) to condensation and fragmentation (Figures S1, S2 and S5). However, while mitochondrial tubules were positive for the TOMM20 signal (Figures 1 and S5A,B) neither the lipid aggregates nor the crystals overlapped with the mitochondrial marker (Figures 4C and 1A, insert 2). Alike mitochondria putatively subject to autophagy (Figure S5C) became devoid of TOMM20 signal as degradation progressed (Figure S5D).
DeoxySLs activate the NLRP3-inflammasome in macrophages
As deoxySLs cause intracellular lipid aggregation including crystal formation and various crystals are known to trigger the NLRP3 inflammasome, we sought to investigate whether deoxySLs were capable of activating the NLPR3 inflammasome in macrophages. In general, activation of the sensor component NLRP3 leads to sequential recruitment of PYCARD and pro-CASP1, ultimately resulting in autoproteolytic cleavage of pro-CASP1. Once active CASP1 processes pro-IL1B to yield mature IL1B for subsequent release from the cell [31].
To evaluate NLRP3 inflammasome activation, we initially used an established macrophage reporter cell line, where activation leads to recruitment of fluorescent PYCARD to a single spot in the cell, called PYCARD speck, that can be visualized by microscopy [38]. We incubated these cells with different lipids and inhibitors, performed microscopy to monitor fluorescent PYCARD-speck formation (Figure 5A) and quantified micrographs by an automated procedure (Figure 5B). Upon incubation with doxSA about one-third of the cells showed aggregated PYCARD-specks, while in untreated or sphinganine-treated controls, no speck formation occurred. With co-application of doxSA and FB1 the speck formation remained at control levels, arguing for the N-acylation products doxCer or dox(DH)Cer being the causative agent for PYCARD-speck aggregation.
Figure 5.
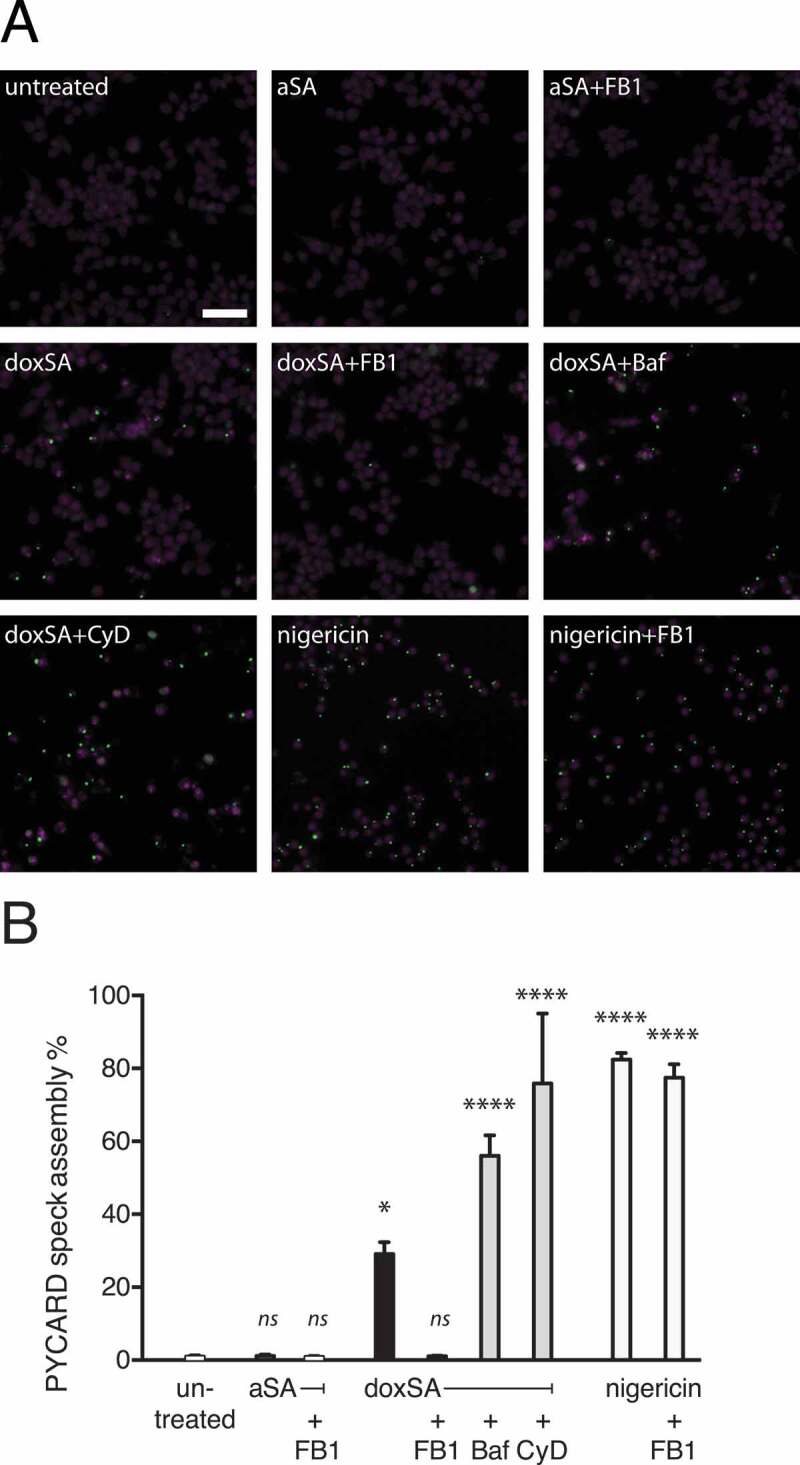
1-Deoxysphingolipids trigger PYCARD speck assembly. Clonal macrophage reporter (CMR) cells overexpressing fluorescent PYCARD were pretreated with the CASP1 inhibitor before incubation with 1 μM alkyne SA or 1 μM doxSA mix or 10 µM nigericin for 16 h or 90 min (nigericin). If applying cells were also pretreated with cytochalasin D (CyD) or bafilomycin A (Baf) or co-treated with 25 μg/mL FB1. After fixation and staining of nuclei epifluorescence microscopy was performed. (A) Micrographs depict activated PYCARD specks (green) and nuclei (magenta, DRAQ5). Note that activation usually leads to formation of one PYCARD speck per cell. Scale bar, 50 μm. (B) Semi-quantitative analysis of PYCARD speck assembly (% of all cells) upon the various treatments. Mean values ± SEM are plotted. Statistical differences were calculated by ANOVA followed by Dunnett’s multiple comparison test. Adjusted P values: **** p < 0.0001; * p = 0.0121; ns – not significant; all vs. untreated control
Inflammasome activation by crystalline agents can generally occur after phagocytic uptake of crystals from the extracellular space or as a result of intracellular precipitation [26,27]. We found that neither inhibition of lysosome acidification by vacuolar-type H(+)-ATPases with bafilomycin A, nor blocking of phagocytosis with cytochalasin D reduced PYCARD-speck formation, suggesting that inflammasome activation was secondary to intracellular deoxySL crystal formation. As expected, the PYCARD-speck frequency was highest in the positive controls treated with nigericin. Importantly, when co-applied with nigericin, the FB1 did not suppress speck formation, arguing against a general inhibitory power of FB1 for PYCARD-speck formation.
To corroborate and verify the data from the macrophage reporter cells, we next investigated IL1B secretion in response to doxSA treatment in primary murine bone marrow-derived macrophages (BMDMs). As the expression of endogenous NLRP3 and pro-IL1B in these cells is tightly regulated, the usage of primary macrophages required a priming stimulus to induce the transcription of both. Hence, LPS, a well-established priming agent, was routinely co-applied.
Starting with BMDMs from wild-type mice, we quantified the release of IL1B using established ELISA technology (Figure 6A). In line with the earlier findings, we noted that IL1B secretion from primary macrophages was substantially increased upon doxSA incubation compared to the LPS or sphinganine alone controls. Notably, this increase in IL1B was reversed when doxSA was co-applied with FB1. In the presence of FB1 the cellular formation of doxCer or dox(DH)Cer was also suppressed (Figure S6A–C), arguing that these N-acylation products are crucial for inflammasome activation. When doxSA was combined with bafilomycin or cytochalasin D, no reduction of cytokine secretion was observed (Figure 6A). Nigericin treatment served as a positive control generating a maximum output of IL1B that was not suppressed by co-application of FB1. For all conditions tested, TNF secretion remained stable, indicating that priming was unaffected (Figure 6B).
Figure 6.
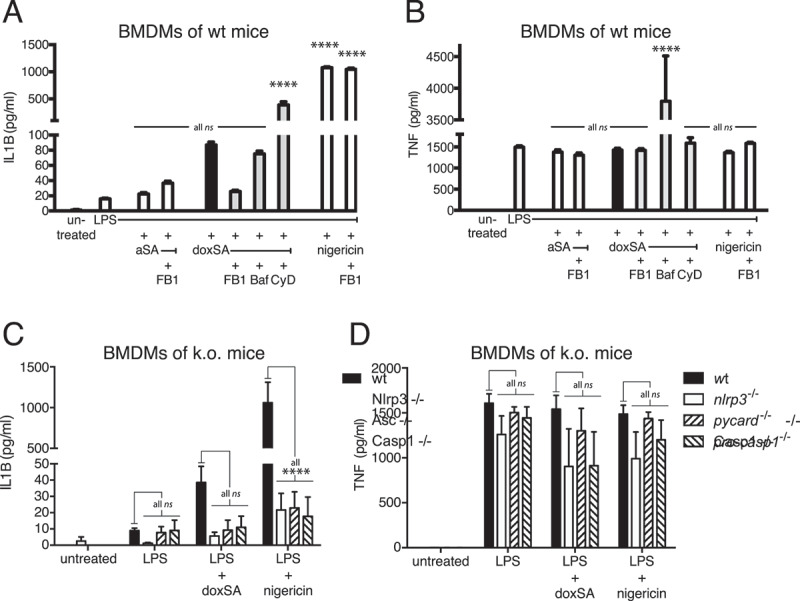
1-Deoxysphingolipids activate the NLRP3-inflammasome. Primary BMDM cells from (A,B) wild-type mice or (C,D) animals lacking expression of NLRP3 or PYCARD or pro-CASP1 were pretreated with 200 ng/mL LPS for 3 h before incubation with (A,B) 1 μM alkyne SA, (A–D) 1 μM doxSA mix or (A–D) 10 µM nigericin for 16 h or 90 min (nigericin). If applying cells were also pretreated with cytochalasin D (CyD) or bafilomycin A (Baf) or co-treated with 25 μg/mL FB1. Cytokines released to growth media were quantified by ELISA: (A,C) IL1B; (B,D) TNF. Mean values ± SEM are plotted. Statistical differences were calculated by (A,B) one-way or (C,D) two-way ANOVA followed by Tukey’s multiple comparison test. Adjusted P values: **** p < 0.0001; ns – not significant; (A,B) all vs. LPS-treated control, N = 3; (C,D) all vs. wt, N = 3
To investigate the status of autophagy in BMDMs exposed to doxSA, we performed semi-quantitative microscopy imaging of LC3. In line with the observations in MEF cells (Figure 1D), the intensity of the LC3-II signal significantly increased also in BMDM cells upon doxSA treatment (Figure S6D). Co-application of FB1 yielded LC3 levels similar to untreated controls, while incubation with chloroquine increased the cellular LC3 content. Of note, the LC3-II staining pattern in doxSA-treated BMDMs appeared more punctate than in controls and the autophagosomes often were aligned, forming elongated structures resembling those in MEF cells (Figure S6E).
To further substantiate our finding that doxSLs trigger the NLRP3-inflammasome, we repeated the analysis of IL1B secretion upon doxSA treatment now using BMDMs from genetically modified mice lacking expression of NLRP3 or its downstream effectors PYCARD or pro-CASP1 (Figure 6C). As before, the wild-type macrophages secreted substantially more IL1B upon doxSA treatment than the LPS-treated control cells. Intriguingly, BMDMs from mice lacking NLRP3 or PYCARD or pro-CASP1 secreted IL1B only at the basal level of the LPS-treated control cells. Once more, nigericin treatment served as positive control resulting in maximum cytokine release, and macrophage priming for all incubation conditions was unaffected, as illustrated by the comparable TNF release (Figure 6D).
Discussion
DoxSA is cytotoxic and the underlying mechanism is still unclear. Neurons are most vulnerable, but doxSA induces profound cellular effects in a variety of cell types. Compared to many other cell lines analyzed so far, MEF cells show the highest tolerance to doxSA, a fact that facilitates their use in studies on deoxSL action eventually leading to cell death. We use MEF cells and a doxSA concentration of 1 μM to investigate the effects that deoxySLs have on the autophagosomal apparatus. This concentration lies well below the LD50 concentration (7 μM) for MEF cells, but already exerts various cellular effects [1]. So it causes mitochondrial fragmentation and dysfunction, exerts ER stress and reduces cell viability [19]. To allow for intracellular tracing, we here replaced 10% of the doxSA by its analog alkyne doxSA.
The alkyne doxSA tracer we developed before [19] enables complementary analyses on lipids metabolism and localization benefitting from the alkyne tags versatility [39]. Previously, we already validated the use of this highly similar analog as a metabolic tracer that excellently mimics its non-traceable sibling [19]. Alkyne doxSA as the untagged doxSA becomes N-acylated to yield the expected cellular products doxCer and dox(DH)Cer. Alike, the ceramide synthase inhibitor FB1 blocks deoxyceramide synthesis from both precursors, and degradation of alkyne doxSLs and untagged doxSLs are similarly slow, leading to cellular accumulation.
A large fraction of the accumulating deoxSLs builds up in mitochondria as these organelles become prominently labeled by alkyne deoxySLs. We hypothesize that deoxyceramides and deoxydihydroceramides comprise a considerable proportion of these mitochondrial deoxySLs. This notion is based on our observation that TLC detects three-quarters of all tracer as doxCer/dox(DH)Cer. Hence, the N-acylation products probably dominate the microscopy signal over free alkyne doxSA. Additionally, it appears likely that much of the free doxSA is lost from the samples during microscopy sample preparation. Again, TLC analysis proves that cells with FB1-blocked ceramide synthase activity still readily take up doxSA, reaching two-thirds of the alkyne tracer levels seen under the uninhibited condition. Yet, the corresponding microscopy images are noticeably dimmer, indicating substantial loss of click-reacted doxSA. In essence, this would mean that the total microscopy signal and also the prominent mitochondrial signal, both result mostly from alkyne deoxyceramides and deoxydihydroceramides.
It is noteworthy, with the known CerS locating to ER [40], that it remains unknown how deoxyceramides and deoxydihydroceramides traffic to the mitochondria. An unknown mitochondrial CerS, an unacknowledged mitochondrial population of CerS2/3, or a dedicated and efficient transport mechanism shuffling deoxyceramides to mitochondria could explain our previous finding: that mitochondria are targeted by doxSL already after 5 min incubation time when hardly any alkyne doxCer/dox(DH)Cer is detectable [19]. Of note, almost all cellular effects presented here likely relate to the downstream N-acylation products doxCer/dox(DH)Cer, rather than free doxSA as they are blunted by the presence of FB1.
Based on the peculiar biophysical properties of deoxyceramides [1], their local accumulation will likely impact the mitochondrial and other membranes. In synthetic bilayers C16:0-deoxyceramides showed reduced lipid miscibility, likely formed a separate gel-like phase and at elevated concentrations probably even generated aggregates [24]. As the deoxyceramides we detected in MEF cells here and before [19] are dominated by C24 fatty acids as N-acylation partners, an even higher hydrophobicity and lower threshold concentration for aggregate formation can be expected.
With deoxyceramides and deoxydihydroceramides accumulating in the mitochondrial membranes they profoundly disturb the organelles’ morphology and physiological function [15,19,22,23]. We and others have observed swollen mitochondria with abnormal or lost cristae before [19,23] and here describe extensive mitochondrial tubule formation resulting in swollen mitochondria interconnected across large cellular distances. The aberrant mitochondria are frequently associated with SQSTM1- and LC3-II-positive autophagosomes that often extend long protrusions, or with LAMP1-positive lysosomes, all together reminiscent of mitochondria destined for autophagy. However, canonical mitophagy may not be the main autophagic process targeting doxSL-laden mitochondria, as PINK1 levels appear unchanged. Indeed, mitochondrial accumulation of various ceramides was described to facilitate recruitment of autophagosomes to damaged mitochondria leading to non-apoptotic lethal mitophagy [36]. In line with our findings is the notion that also deoxyceramide or deoxydihydroceramide may act as a receptor for LC3-II and thereby promoting lethal mitophagy. However, even our STED micrographs do not provide sufficient resolution to unequivocally identify mitophagosomes and hence our data cannot distinguish between selective mitophagy involving mitophagosomes or mitochondrial degradation during generalized autophagy.
The impact of doxSLs on the autophagosomal flux appears to be complex. The observed accumulation of autophagosomes and lysosomes upon doxSA treatment is accompanied by an increase in the cellular levels of LC3-II and SQSTM1 accumulating on autophagosomes. This may be puzzling, but there is not always a clear correlation between increases in LC3-II and decreases in SQSTM1 [33]. Blocking lysosomal degradation by protease inhibitors or neutralizing the lysosomal pH by chloroquine during doxSA exposure, resulted in LC3-II levels higher than in cells treated with doxSA only. These data are indicative of an increased autophagic flux. Noteworthy, similar to LC3-II, increased flux can be paralleled by an accumulation of SQSTM1 in the presence of flux inhibitors [41,42]. Along this, also SQSTM1 built up if lysosomal pH was manipulated by chloroquine, but SQSTM1 levels did not change when lysosomal proteases were blocked. Interfering with pH and blocking the fusion of autophagosomes with lysosomes by bafilomycin A yielded a mixed picture. Compared to doxSA-only-treated cells, this inhibitor triggered just a minor increase in LC3-II, and SQSTM1 only increased upon longer exposure to the inhibitor. Taken all together, our data demonstrate an impact of doxSLs on autophagy and support the notion that doxSLs increase the autophagic flux.
Apart from the association of autophagosomal structures and mitochondria, we also detected lipid aggregates intermingled therein. These aggregates were positive for the deoxySLs tracer but did not overlap with marker proteins for mitochondria, autophagosomes or lysosomes. They were distinguishable from birefringent lipid crystals. The notion that the lipid aggregations interact with mitochondria as well as the autophagosomal apparatus is supported by our findings. However, since no capping structure could be unequivocally identified, a final conclusion where the aggregations originate from cannot unmistakably be drawn. Likewise, no information on the precise composition of the crystals can be provided, as our attempts to purify them were not successful. They likely contain alkyne deoxySLs especially deoxyceramides/deoxydihydroceramides as they are labeled in micrographs and occur abundantly upon doxSA treatment, particularly in the absence of FB1. The fluorescent label usually is restricted to the edges, and the low signal in the crystal core likely is due to limited access to the reagents used in the click-reaction, copper catalyst and fluorescent azide probe.
Given the fact that mitochondria are the cell organelles showing the strongest labeling by deoxySLs including deoxyceramides/deoxydihydroceramides, they qualify as a source for at least some of the observed lipid aggregates. One might speculate that mitochondria progressively accumulate deoxyCer/deoxy(DH)Cer until they reach a local concentration of these poorly miscible lipids that results in the formation of lipid aggregates. With the mitochondria eventually disintegrating these aggregates become released to the cytosol. Additionally, the damaged mitochondria are subjected to autophagy, and by that, the lipid aggregates enter the autophagosomal system. Accordingly, some of the cytosolic lipid aggregates may also derive from autolysosomes upon vesicle rupture.
Rupturing phagolysosomes in the course of cholesterol crystal internalization or internal crystal formation from non-crystalline cholesterol by macrophages activates the NLRP3 inflammasome [26,27]. We find that doxSA treatment of macrophages also triggers the NLRP3 inflammasome when administered fully dissolved and not as external crystals. As the underlying mechanism, at least three possibilities must be considered. First, similar to MEF cells, the incorporated doxSA triggers the intracellular formation of deoxySL crystals that is accompanied by the rupture of autolysosomes. Second, doxSA-derived deoxySLs damage cell organelles like mitochondria, ER and Golgi apparatus leading to the generation of stress signals that are sensed by NLRP3. Third, the damaged organelles are subjected to autophagy that, in turn, acts on inflammasome activation in a complex manner. In general, autophagy was described to limit inflammasome activation by degrading damaged organelles and already assembled inflammasomes [32,34]. Yet, translocation of NLRP3 to distinct cellular sites such as mitochondria or the Golgi apparatus was shown to promote inflammasome formation [43,44]. Similarly, early autophagic vesicles that emerge upon doxSA treatment could serve as a platform to bring the inflammasome partners closer together. Indeed, induction of autophagy in the presence of inflammasome-activating stimuli, such as crystals or nigericin can lead to increased secretion of IL1B, and it was suggested that autophagosomes might act as a scaffold for inflammasome assembly [45]. If the autophagic flux is additionally affected, these effects would become even more pronounced. Indeed, we find increased LC3 levels in doxSA-treated macrophages. Based on this and our data from MEF cells, all three scenarios appear equally likely and do not mutually exclude each other. Importantly, all three pathways hold the potential to activate the NLRP3 inflammasome [32].
Increased NLRP3 expression and activation have been observed in type 2 diabetes patients [46]. The thinkable priming signals that induce NLRP3 in this context are manifold [47], yet ceramides were implicated in triggering the NLRP3 inflammasome in a mouse model of diet-induced obesity and diabetes [48]. As plasma deoxySLs are elevated in metabolic syndrome and type 2 diabetes patients [3–5], our findings point toward a potential involvement of deoxySLs in activating NLRP3 in the context of the disease. Of note, deoxyceramides seem to be more potent in triggering the NLRP3 inflammasome than ceramides or at least engage a more powerful pathway. In our hands, sphinganine administration and metabolism to ceramide does not stimulate NLRP3 activation, while doxSA does. Possible explanations for this observation could be differences in catabolism or localization between ceramides and deoxyceramides. Furthermore, deoxyceramides exhibit a higher hydrophobicity and thus might rather tend to form lipid aggregates or crystals than ceramides.
As a consequence of inflammasome activation, the macrophages secrete various cytokines that drive inflammation. While we are not aware of a study explicitly investigating macrophages or chronic inflammation in HSAN1 patients, our findings may link to related observations in affected individuals where chronic skin erosions, ulcers, or blisters in combination with impaired wound healing have been described [1]. On the basis of our data, therapeutic strategies that inhibit deoxyceramide and deoxydihydroceramide synthesis that influence autophagy or block the inflammasome pathway would be predicted to have clinical benefit. In this context, our findings also indicate novel molecular targets for the development of therapeutics to treat HSAN1.
Materials and methods
Lipid and chemical probes
Syntheses of alkyne-SA and alkyne-doxSA are described [19,49]. The alkyne-ceramide used as internal standard for MS analysis, Cer(a18:1), was synthesized by acylation of sphingosine (Cayman Chemicals, 10007907) with an NHS-ester of Octadec-9-cis-en-17-ynoic acid (a18:1, Kerafast Inc., EVU107). Unlabeled doxSA was purchased (Avanti polar lipids, 860493). The fluorescent azido-reporters N3Cy3 and N3picCy3 were obtained from Jena Bioscience (CLK-AZ119 and CLK-1178) and N3635P was from Abberior (ST635P-0004); the synthesis of ASTM-BODIPY is published [50]. The MS-reporter azides (C175-73, C175-75, C175-76, C175-77) have recently been described [51]. Fumonisin B1 (FB1) was from Cayman Chemicals (62580), nigericin (N7143), VX-765 (5313720001) and cytochalasin D (C8273) from Sigma, bafilomycin A (BVT-0252) from Adipogen and pepstatin A (ALX-260-085) as well as E64d (BML-PI107) from Enzo Life Sciences.
Animals
C57Bl/6 J wild-type mice were purchased from Charles River. Mice deficient for NLRP3, PYCARD and CASP1 were described before [52,53]. Animals were maintained in the animal facility of the University Hospital Bonn. Mice of both genders were used. All animals were held at a 12-h light/dark cycle and had ad libitum access to food and water. This study was carried out in strict accordance with the European and German guidelines for the welfare of experimental animals.
Cell lines and general cell culture procedures
Generation of wild type mouse embryonic fibroblast (MEF) cells was described previously [54]. MEF cells were cultured in DMEM medium (Gibco, 31966021) containing 10% fetal calf serum (Gibco, 11560636) and 1% penicillin/streptomycin (Gibco, 15070063).
Bone marrow-derived macrophages (BMDMs) were generated by culturing murine bone marrow in DMEM medium containing 10% fetal calf serum, 1% penicillin/streptomycin and 20% L929 cell-conditioned supernatant, prepared from cultures of L929 cell (ATCC, CCL-1TM [NCTC clone 929; L cell, L-929, derivative of Strain L]) using DMEM containing 10% fetal calf serum. On day 6 of differentiation BMDMs were collected and plated for experiments.
Immortalized NLRP3-deficient macrophages overexpressing mouse NLRP3-FLAG and human PYCARD-mCerulean termed “clonal macrophage reporter” (CMR) cells were described before [38]. CMR cells were maintained in DMEM medium containing 10% fetal calf serum and 1% penicillin/streptomycin.
Cytokine analysis
For determination of cytokine secretion, BMDM cells were primed for 3 h with 200 ng/ml LPS (Sigma, L2630) and incubated with 1 μM alkyne SA or 1 μM doxSA mix (0.9 μM doxSA + 0.1 μM alkyne doxSA tracer) or 10 µM nigericin, all ± 25 μg/mL FB1 and from ethanolic stocks, for 16 h or 90 min (nigericin). For some experiments, cells were pretreated with 10 µM cytochalasin D or 10 nM bafilomycin A. Media concentrations of TNF and IL1B were determined using commercial ELISA kits (R&D systems) according to the manufacturer’s instructions.
PYCARD speck formation analysis
CMR cells were pretreated with the CASP1 inhibitor VX-765 (10 µM) and incubated with 1 μM alkyne SA or 1 μM doxSA mix (0.9 μM doxSA + 0.1 μM alkyne doxSA tracer) or 10 µM nigericin, all ± 25 μg/mL FB1 and from ethanolic stocks, for 16 h or 90 min (nigericin). For some experiments, cells were also pretreated with 10 µM cytochalasin D or 10 nM bafilomycin A. Cells were fixed and nuclei were stained using PBS (Sigma, 806552) containing 4% paraformaldehyde and DRAQ5 (Cell Signaling Technology, 4084) before sample washing and mounting. Semi-automated epifluorescence microscopy was performed on a Zeiss observer Z1 microscope using a 20x objective and the filter sets 47 and 50 from Zeiss. Images were semi-automatically analyzed using CellProfiler 2.2.0 software [55]. For each sample nuclei and PYCARD specks were counted and the ratio of PYCARD specks to nuclei was calculated. Ratios extracted from 18 independent images (6 images per well from experimental triplicates) were averaged to calculate the final ratio PYCARD speck/nuclei for every condition.
Cell culture of MEF cells for TLC and MS analysis
The MEF culture medium was supplemented with 1 μM doxSA mix (0.9 μM doxSA + 0.1 μM alkyne doxSA tracer) ± 25 μg/mL FB1 from ethanolic stocks and cells incubated for 24 h. 400,000 MEF cells were washed with media lacking doxSA before media aspiration and addition of 600 μL methanol:chloroform 5:1 containing 72 pmol alkyne-ceramide as internal standard [51]. Culture dishes were floated on bath sonicator for 30 s before lipid collection. After centrifugation, the supernatants were retrieved and mixed 200 μL chloroform and 1.2 mL of 1% acetic acid to induce phase separation. The organic phase was collected, evaporated and redissolved in chloroform. Lipid extracts were split and stored at −20°C until analysis.
Cell culture of BMDM cells for TLC and MS analysis
The BMDM culture medium was supplemented with 1 μM doxSA mix (0.9 μM doxSA + 0.1 μM alkyne doxSA tracer) ± 25 μg/mL FB1 from ethanolic stocks and cells incubated for 16 h. 3,000,000 cells (TLC) or 1,000,000 cells (MS) were washed sequentially with PBS containing 0.5% delipidated BSA (Sigma, A8806) and with 155 mM ammonium acetate. For TLC, lipids were extracted by sequential addition of 1 mL methanol:chloroform 5:1 and 1 mL isopropanol. Upon addition of 1 mL chloroform and 1 mL 0.1% acetic acid to the extract the phases separated, and the organic phase was collected, evaporated and redissolved in chloroform. For MS, lipids were extracted by addition of 500 μL methanol:chloroform 5/1 containing 15.8 pmol alkyne-ceramide as internal standard [51]. Culture dishes were floated on bath sonicator for 30 s before lipid collection. After centrifugation the supernatants were retrieved and mixed 400 μL chloroform and 600 μL of 1% acetic acid to induce phase separation. The organic phase was collected, evaporated and redissolved in chloroform. Lipid extracts were stored at −20°C until analysis.
TLC analysis
As described before [56] dried lipids were redissolved in 7 μL chloroform and 30 μL click-reaction mix (10 μL of 2 mg/mL 3-azido-7-hydroxycoumarin (Jena Bioscience, CLK-FA047), 250 μL of 10 mM [acetonitrile]4 CuBF4 (Sigma, 677892) in acetonitrile, 850 μL ethanol) was added. The click reaction was performed at 43°C for 3 hours and then the click-reacted lipids were separated by TLC using solvent 1 (chloroform:methanol:water:acetic acid 65:25:4:1, 2/3 of the plate). TLCs were dried and then developed in solvent 2 (hexane:ethyl acetate 1:1, whole plate). Directly before fluorescence detection, TLC plates were shortly soaked in 4% N,N,-diisopropylethylamine (Sigma, 387649) in hexane (Sigma, 178918). Fluorescent images of plates were recorded using a 420 nm LED (Roithner Lasertechnik) filtered through a colored glass filter (HEBO V01, Hebo Spezialglas) for excitation and an EMCCD camera (Rolera-MGi Plus Fast 1394, Q-imaging), equipped with a 494/20 bandpass emission filter. Lipid bands were identified by their migration distances and compared to co-migrating chemically synthesized alkyne standards. Quantification was performed using ImageGauge (Fuji) software.
MS analysis
As described before [51] dried lipids were redissolved in 8 μL chloroform and 40 μL click-reaction mix (10 μl of 100 mM azido-C175-XX [51] in 50% methanol, 200 μl 5 mM [acetonitrile]4 CuBF4 in acetonitrile, 800 μl ethanol) was added. The click reaction was performed at 43°C for 16 hours. After pooling multiplex samples, 400 μl chloroform and 600 μl water were added and samples briefly shaken and centrifuged. The upper phase was removed and the organic phase dried. Spray buffer (2-propanol:methanol:water 8:5:1 + 10 mM ammonium acetate, 200–1000 μl) was added, the tubes sonicated for 5 min and the dissolved lipids analyzed. Mass spectra were recorded on a Thermo Q-Exactive Plus spectrometer equipped with a standard HESI ion source using direct injection from a Hamilton syringe driven by a syringe pump under the control of the Tune instrument control software. MS1 spectra were recorded as segmented scans with windows of m/z 150 between m/z 200–800 and windows of m/z 100 between m/z 800–1200 for 1 min total, followed by MS2 spectra by data-independent acquisition (DIA) for 10 min using the target m/z 227.7320, 228.7320, 229.7320 and an inclusion list from m/z 400.4300 to 1199.9994 at m/z 1.0006 intervals. A second scan series for double-charged species was performed in the scan range of m/z 200–700 (windows of m/z 100) with MS2 scans from m/z 224.7748–700.0648 at intervals of m/z 0.5002 and an isolation window of m/z 0.7.
MS data analysis
Raw files were converted to mzml files using MSConvert and analyzed using LipidXplorer [57]. For identifcation and quantification of labeled alkyne lipids, molecular fragment query language (mfql) files were written that identify the species by the presence of a peak corresponding to the expected masses of the labeled lipid class combined with the characteristic neutral loss. Details of peak annotation are provided in Table 1.
Table 1.
Details on peak annotation during MS data analysis
| Assigned deoxySLs | Composition (upon reaction with C175-XX) | m/z | Tolerance ± error |
|---|---|---|---|
| doxCer(16:1)/dox(DH)Cer(16:2) | H76 C42 D4 O2 N5 | 690.6556 | 7 ppm |
| doxCer(16:0)/dox(DH)Cer(16:1) | H78 C42 D4 O2 N5 | 692.6717 | 7 ppm |
| dox(DH)Cer(16:0) | H80 C42 D4 O2 N5 | 694.6866 | 7 ppm |
| doxCer(18:3)/dox(DH)Cer(18:4) | H76 C44 D4 O2 N5 | 714.6542 | 7 ppm |
| doxCer(18:1)/dox(DH)Cer(18:2) | H80 C44 D4 O2 N5 | 718.6853 | 7 ppm |
| doxCer(18:0)/dox(DH)Cer(18:1) | H82 C44 D4 O2 N5 | 720.7031 | 7 ppm |
| dox(DH)Cer(18:0) | H84 C44 D4 O2 N5 | 722.7158 | 7 ppm |
| doxCer(20:0)/dox(DH)Cer(20:1) | H86 C46 D4 O2 N5 | 748.7341 | 7 ppm |
| dox(DH)Cer(20:0) | H88 C46 D4 O2 N5 | 750.7482 | 7 ppm |
| doxCer(22:4)/dox(DH)Cer(22:5) | H82 C48 D4 O2 N5 | 768.7075 | 7 ppm |
| doxCer(22:3)/dox(DH)Cer(22:4) | H84 C48 D4 O2 N5 | 770.7141 | 7 ppm |
| doxCer(22:1)/dox(DH)Cer(22:2) | H88 C48 D4 O2 N5 | 774.7447 | 7 ppm |
| doxCer(22:0)/dox(DH)Cer(22:1) | H90 C48 D4 O2 N5 | 776.7642 | 7 ppm |
| dox(DH)Cer(22:0) | H92 C48 D4 O2 N5 | 778.7804 | 7 ppm |
| doxCer(24:2)/dox(DH)Cer(24:3) | H90 C50 D4 O2 N5 | 800.7664 | 7 ppm |
| doxCer(24:1)/dox(DH)Cer(24:2) | H92 C50 D4 O2 N5 | 802.7819 | 7 ppm |
| doxCer(24:0)/dox(DH)Cer(24:1) | H94 C50 D4 O2 N5 | 804.7975 | 7 ppm |
| dox(DH)Cer(24:0) | H96 C50 D4 O2 N5 | 806.8134 | 7 ppm |
| doxCer(26:1)/dox(DH)Cer(26:2) | H96 C52 D4 O2 N5 | 830.8122 | 7 ppm |
| doxCer(26:0)/dox(DH)Cer(26:1) | H98 C52 D4 O2 N5 | 832.8264 | 7 ppm |
| dox(DH)Cer(26:0) | H100 C52 D4 O2 N5 | 834.8369 | 9 ppm |
| Cer(a18:1) [internal standard] | H80 C44 D4 O3 N5 | 734.6804 | 7 ppm |
Peaks corresponding to the expected masses of alkyne doxCer/dox(DH)cer species after click-reaction with azido-C175-XX compounds were screened for both in MS1 and MS2 scans. A species was positively identified only, if the peak corresponding to the characteristic neutral loss was additionally detected in the MS2 scan. The intensity of the latter peak was used for all subsequent calculations. Ceramide nomenclature: dox denotes a 1-deoxy-17-alkynylsphingoid base backbone. DH indicates the dihydrogenation of the latter, followed by a description of the N-acylated fatty acid in brackets (a indicates the alkynyl moiety at the terminal end, the first digit corresponds to the number of carbon atoms and the second to the number of double bonds, separated by a colon).
Analysis of subcellular localization of alkyne lipids in MEF cells by fluorescence microscopy
MEF cells were grown on glass coverslips and culture medium was supplemented with 1 μM alkyne SA or 1 μM doxSA mix (0.9 μM doxSA + 0.1 μM alkyne doxSA tracer) ± 25 μg/mL FB1 or 2 μM doxSA mix or 1 μM alkyne doxSA tracer or carrier from ethanolic stocks and cells incubated for 24 h. Cells were fixed in 3.7% formalin for 10 min before washing sequentially with PBS, 155 mM ammonium acetate and PBS. Permeabilization and blocking were performed using 1% cold fish gelatin/0.01% saponin (Sigma, G7041, S7900)) in PBS for 1 h.
The primary antibodies and their dilutions were as follows: Anti-TOMM20 (Santa Cruz Biotechnology, sc-11415; 1:200), anti-P4HB (Stressgen, SPA891; 1:200), anti-LAMP1 (Abcam, ab25245; 1:200), anti-TUBB (tubulin, beta; Sigma, T7816; 1:500), anti-SQSTM1 (Enzo Life Science, BML-PW9860; 1:300), anti-LC3 (MBL, PM036; 1:200) or (MBL, M152-3; 1:200). Secondary antibodies were labeled with Alexa488 or 594 (for STED microscopy), or with Alexa488 or 647 (for epifluorescence microscopy), all from Invitrogen. All antibodies were diluted in 1% cold fish gelantin/0.01% saponin in PBS, except anti-P4HB that was diluted in 1% cold fish gelantin/0.1% saponin in PBS; incubation time was 4 or 16 h. After antibody staining and washing in 100 mM HEPES-KOH pH 7.4 the click reaction was performed. For that 1 mL pre-warmed 100 mM Hepes-KOH, pH 7.4, containing 10 μM fluorescent azido-reporter (N3635P for STED microscopy; N3Cy3 or N3picolylCy3 or ASTM-BODIPY for epifluorescence microscopy) were added to each well holding a coverslip. The reaction was initiated by addition of 20 μl 100 mM Cu(I)TFB in acetonitrile and the dish was incubated at 43°C for 1 h. The samples were then washed sequentially with 20 mM EDTA, 155 mM ammonium acetate, and extensively with PBS. If applying cell nuclei were stained with DAPI (Sigma, D9542). Coverslips were mounted in ProLongGold (Invitrogen, P10144) for STED microscopy or in Mowiol 4–88 (Sigma, 475904) for epifluorescence microscopy.
Epifluorescence microscopy was performed using a Zeiss Observer.Z1 microscope equipped with a Plan-Apochromat 20x (0.8 NA) and 63x (1.40 NA) and a Photometrics Coolsnap K4 camera. If applying, optical sectioning was performed using structured illumination with Zeiss ApoTome. For brightfield polarized light microscopy, two polarization filters were included in the light path, before and after the sample. The light source was a Polychrome V 150 W xenon lamp (Till Photonics). Images were processed using Fiji software [58].
STED microscopy was performed using a 4-channel easy3D super-resolution STED optics module (Abberior Instruments) coupled with an Olympus IX83 confocal microscope and equipped with an UPlanSApo 100x (1.4 NA) objective. Triple-color STED-microscopy was realized by sequential imaging of three channels, using pulsed 561 nm, 640 nm, 485 nm lasers for the excitation of Alexa 594, N3635P, Alexa 488, respectively. For depletion, pulsed 775 nm or 595 nm STED lasers were used. Signals emitted from Alexa 594, N3635P, Alexa 488 dyes were detected using 580–630 nm, 650–720 nm, or 500–550 nm filters, respectively. Pixel size was set to 25 nm.
Deconvolution of optical z-stacks was performed using Huygens software (Scientific Volume Imaging).
Localization and semi-quantification of the LC3 signal in BMDM cells by fluorescence microscopy
100,000 BMDM were grown in ibidi 8 well dishes and treated with 1 μM doxSA (0.9 μM + 0.1 μM alkyne doxSA tracer) ± 25 μg/mL FB1 for 16 h or 50 µM chloroquine for 4 h. Cells were fixed in methanol at −20°C for 15 min, washed 4 times with PBS and permeabilized using 0.1% Triton X-100 (Sigma, X100) in PBS. Blocking was performed using PBS + 10% Goat serum (Sigma, G9023), 1% FCS and 0.1% Triton X-100. Cells were probed for LC3 by incubation in primary antibody against LC3 (Cell Signaling Technology, 3868; 1:200) diluted in blocking buffer, followed by 3 PBS washes, incubation in secondary antibody (Alexa Fluor 488-F(ab’)2-Goat anti-Rabbit IgG, Invitrogen, A11070; 1:700) in blocking buffer, followed by 3 PBS washes. Nuclei were stained using Hoechst 34580 (Molecular Probes, H21486).
Epifluorescence microscopy was performed using a Zeiss Observer.Z1 microscope equipped with a Plan-Apochromat 20x (0.8 NA) and an Axiocam 506 m camera. Optical sectioning was performed using structured illumination with Zeiss ApoTome 2.0. Four images were collected per condition. Images were semi-automatically analyzed using CellProfiler 2.2.0 software [55]. Briefly, cells were identified first by nuclear stain, which was then used to identify the cells based on LC3 fluorescence. The median fluorescence intensity of each cell for the LC3 channel from each field was determined. All cells that were counted entered analysis and were blotted using GraphPad Prism version 6.0 h software.
Confocal microscopy images were collected on a Leica SP8 lightening microscope using a 63x water immersion objective (1.2 NA). The images were recorded using the lightening software wizard. Subsequent to image collection a maximal projection of the different Z planes was made and exported using the Leica LAX software.
Semi-quantification of autophagosomes, lysosomes and lipid crystals from microscopy images of MEF cells
For the semi-quantitative analysis of immunocytochemistry staining all corresponding samples were processed identically in the same dish on the same day and identical microscopy settings were applied. The Fiji software and the following procedures were used.
Nuclei were counted after tresholding the DAPI signal to minimize background. The Fiji functions “watershedding” and “analyze particles” were employed sequentially. The settings for the latter were: Size “50 μm2 to infinity”; Roundness ‘0–1ʹ. Identified nuclei directly contacting or closely neighboring each other were then manually inspected. They were classified as “multinucleated cell” if, using the full information from all other channels, no limiting cell structure between the nuclei was discernable.
Birefringent crystals were quantified using polarized light illumination. After signal thresholding, the Fiji function “analyze particles” with the following settings (Size ‘1–200 μm2ʹ; Roundness ‘0–0.6ʹ) was applied.
To quantify the mean intensity of LAMP1, LC3, or SQSTM1 signal per cell a region-of-interest (ROI) encircling the entire cell was manually drawn using the full information from all recorded channels. From the ROI the “mean signal intensity” was calculated.
Western blotting and quantification
The antibodies and their dilutions for western blotting were as follows: anti-LC3 (Novus, NB100-2220, 1:1000); anti-ACTB (Sigma, T7816, 1:2000); anti-SQSTM1 (Enzo, BML-PW9860, 1:2000); anti-PINK1 (Cell Signaling Technology, 6946; 1:1000). Secondary antibodies were labeled with HRP (Invitrogen).
Statistical analysis
Statistical differences between sample groups were calculated using GraphPad Prism version 6.0 h software. Ordinary one-way ANOVA was followed by Dunnett’s or Bonferroni’s Multiple Comparison Test with a single pooled variance by comparing the mean of each condition to that of the untreated control sample. Family-wise significance and confidence level (alpha 0.05; 95% confidence interval) settings were applied. Multiplicity adjusted P values were calculated. Ordinary two-way ANOVA was followed by Tukey’s Multiple Comparison Test by comparing the mean of each condition to that of the wild-type sample. Family-wise significance and confidence level (alpha 0.05; 95% confidence interval) settings were applied. Multiplicity adjusted P values were calculated.
Supplementary Material
Acknowledgments
We thank Gabor Horvath for support in automated image analysis and Jan-Gero Schlötel for technical assistance at the STED microscope. We would like to thank the Microscopy Core Facility of the Medical Faculty at the University of Bonn for providing their help and services. STED microscopy and MS analyses were performed at the super-resolution light microscopy and MS facilities of the LIMES Institute at the University Bonn. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - TRR83 -112927078 (to V.S., C.T., E.L. and L.K.), Germany’s Excellence Strategy - EXC2151 – 390873048, the DFG grants SFBs 645, 670, 1123, TRR 57, a grant from NIH (1R01HL112661) and by an ERC Consolidator grant (InflammAct) to E.L.
Funding Statement
This work was supported by the DFG [SFT TRR83 - 112927078]; ERC [InflammAct]; DFG [SFB 670]; DFG [SFB 1123]; DFG [SFB TRR57]; NIH [1R01HL112661]; DFG [SFB 645]; DFG [EXC2151 - 390873048].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Lone MA, Santos T, Alecu I, et al. 1-deoxy-sphingolipids. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(4):512–521. [DOI] [PubMed] [Google Scholar]
- [2].Penno A, Reilly MM, Houlden H, et al. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem. 2010;285(15):11178–11187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Othman A, Rutti MF, Ernst D, et al. Plasma deoxysphingolipids: a novel class of biomarkers for the metabolic syndrome? Diabetologia. 2012;55(2):421–431. [DOI] [PubMed] [Google Scholar]
- [4].Bertea M, Rutti MF, Othman A, et al. Deoxysphingoid bases as plasma markers in diabetes mellitus. Lipids Health Dis. 2010;9:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Othman A, Saely CH, Muendlein A, et al. Plasma 1-deoxysphingolipids are predictive biomarkers for type 2 diabetes mellitus. BMJ Open Diabetes Res Care. 2015;3(1):e000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Humpf HU, Schmelz EM, Meredith FI, et al. Acylation of naturally occurring and synthetic 1-deoxysphinganines by ceramide synthase. Formation of N-palmitoyl-aminopentol produces a toxic metabolite of hydrolyzed fumonisin, AP1, and a new category of ceramide synthase inhibitor. J Biol Chem. 1998;273(30):19060–19064. [DOI] [PubMed] [Google Scholar]
- [7].Abad JL, Nieves I, Rayo P, et al. Straightforward access to spisulosine and 4,5-dehydrospisulosine stereoisomers: probes for profiling ceramide synthase activities in intact cells. J Org Chem. 2013;78(12):5858–5866. [DOI] [PubMed] [Google Scholar]
- [8].Esaki K, Sayano T, Sonoda C, et al. L-serine deficiency elicits intracellular accumulation of cytotoxic deoxysphingolipids and lipid body formation. J Biol Chem. 2015;290(23):14595–14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gorden DL, Myers DS, Ivanova PT, et al. Biomarkers of NAFLD progression: a lipidomics approach to an epidemic. J Lipid Res. 2015;56(3):722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kramer R, Bielawski J, Kistner-Griffin E, et al. Neurotoxic 1-deoxysphingolipids and paclitaxel-induced peripheral neuropathy. Faseb J. 2015;29(11):4461–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Alecu I, Othman A, Penno A, et al. Cytotoxic 1-deoxysphingolipids are metabolized by a cytochrome P450-dependent pathway. J Lipid Res. 2017;58(1):60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Salcedo M, Cuevas C, Alonso JL, et al. The marine sphingolipid-derived compound ES 285 triggers an atypical cell death pathway. Apoptosis. 2007;12(2):395–409. [DOI] [PubMed] [Google Scholar]
- [13].Sanchez AM, Malagarie-Cazenave S, Olea N, et al. Spisulosine (ES-285) induces prostate tumor PC-3 and LNCaP cell death by de novo synthesis of ceramide and PKCzeta activation. Eur J Pharmacol. 2008;584(2–3):237–245. [DOI] [PubMed] [Google Scholar]
- [14].Zitomer NC, Mitchell T, Voss KA, et al. Ceramide synthase inhibition by fumonisin B1 causes accumulation of 1-deoxysphinganine: a novel category of bioactive 1-deoxysphingoid bases and 1-deoxydihydroceramides biosynthesized by mammalian cell lines and animals. J Biol Chem. 2009;284(8):4786–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Haribowo AG, Hannich JT, Michel AH, et al. Cytotoxicity of 1-deoxysphingolipid unraveled by genome-wide genetic screens and lipidomics in Saccharomyces cerevisiae. Mol Biol Cell. 2019;30:2814–2826. mbcE19070364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cuadros R, Montejo de Garcini E, Wandosell F, et al. The marine compound spisulosine, an inhibitor of cell proliferation, promotes the disassembly of actin stress fibers. Cancer Lett. 2000;152(1):23–29. [DOI] [PubMed] [Google Scholar]
- [17].Gable K, Gupta SD, Han G, et al. A disease-causing mutation in the active site of serine palmitoyltransferase causes catalytic promiscuity. J Biol Chem. 2010;285(30):22846–22852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Oswald MC, West RJ, Lloyd-Evans E, et al. Identification of dietary alanine toxicity and trafficking dysfunction in a Drosophila model of hereditary sensory and autonomic neuropathy type 1. Hum Mol Genet. 2015;24(24):6899–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Alecu I, Tedeschi A, Behler N, et al. Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction. J Lipid Res. 2017;58(1):42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zuellig RA, Hornemann T, Othman A, et al. Deoxysphingolipids, novel biomarkers for type 2 diabetes, are cytotoxic for insulin-producing cells. Diabetes. 2014;63(4):1326–1339. [DOI] [PubMed] [Google Scholar]
- [21].Guntert T, Hanggi P, Othman A, et al. 1-deoxysphingolipid-induced neurotoxicity involves N-methyl-d-aspartate receptor signaling. Neuropharmacology. 2016;110(Pt A):211–222. [DOI] [PubMed] [Google Scholar]
- [22].Wilson ER, Kugathasan U, Abramov AY, et al. Hereditary sensory neuropathy type 1-associated deoxysphingolipids cause neurotoxicity, acute calcium handling abnormalities and mitochondrial dysfunction in vitro. Neurobiol Dis. 2018;117:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Myers SJ, Malladi CS, Hyland RA, et al. Mutations in the SPTLC1 protein cause mitochondrial structural abnormalities and endoplasmic reticulum stress in lymphoblasts. DNA Cell Biol. 2014;33(7):399–407. [DOI] [PubMed] [Google Scholar]
- [24].Jimenez-Rojo N, Sot J, Busto JV, et al. Biophysical properties of novel 1-deoxy-(dihydro)ceramides occurring in mammalian cells. Biophys J. 2014;107(12):2850–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abela GS.Cholesterol crystals piercing the arterial plaque and intima trigger local and systemic inflammation. J Clin Lipidol. 2010;4(3):156–164. [DOI] [PubMed] [Google Scholar]
- [26].Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rajamaki K, Lappalainen J, Oorni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5(7):e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Samstad EO, Niyonzima N, Nymo S, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. 2014;192(6):2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nakayama M. Macrophage recognition of crystals and nanoparticles. Front Immunol. 2018;9:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. [DOI] [PubMed] [Google Scholar]
- [31].Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev. 2018;281(1):62–73. [DOI] [PubMed] [Google Scholar]
- [33].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dany M, Ogretmen B. Ceramide induced mitophagy and tumor suppression. Biochim Biophys Acta. 2015;1853(10 Pt B):2834–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8(10):831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Riley RT, Merrill AHJ. Ceramide synthase inhibition by fumonisins: a perfect storm of perturbed sphingolipid metabolism, signaling, and disease. J Lipid Res. 2019;60(7):1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stutz A, Horvath GL, Monks BG, et al. ASC speck formation as a readout for inflammasome activation. Methods Mol Biol. 2013;1040:91–101. [DOI] [PubMed] [Google Scholar]
- [39].Kuerschner L, Thiele C. Multiple bonds for the lipid interest. Biochim Biophys Acta. 2014;1841(8):1031–1037. [DOI] [PubMed] [Google Scholar]
- [40].Levy M, Futerman AH. Mammalian ceramide synthases. IUBMB Life. 2010;62(5):347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Navarro-Yepes J, Anandhan A, Bradley E, et al. Inhibition of protein ubiquitination by paraquat and 1-methyl-4-phenylpyridinium impairs ubiquitin-dependent protein degradation pathways. Mol Neurobiol. 2016;53(8):5229–5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhan L, Chen S, Li K, et al. Autophagosome maturation mediated by Rab7 contributes to neuroprotection of hypoxic preconditioning against global cerebral ischemia in rats. Cell Death Dis. 2017;8(7):e2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Subramanian N, Natarajan K, Clatworthy MR, et al. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564(7734):71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J. 2011;30(23):4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee HM, Kim JJ, Kim HJ, et al. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lauterbach MA, Wunderlich FT. Macrophage function in obesity-induced inflammation and insulin resistance. Pflugers Arch. 2017;469(3–4):385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gaebler A, Milan R, Straub L, et al. Alkyne lipids as substrates for click chemistry-based in vitro enzymatic assays. J Lipid Res. 2013;54(8):2282–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hofmann K, Thiele C, Schott HF, et al. A novel alkyne cholesterol to trace cellular cholesterol metabolism and localization. J Lipid Res. 2014;55(3):583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Thiele C, Wunderling K, Leyendecker P. Multiplexed and single cell tracing of lipid metabolism. Nat Methods. 2019;16(11):1123–1130. [DOI] [PubMed] [Google Scholar]
- [52].Kanneganti TD, Ozoren N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–236. [DOI] [PubMed] [Google Scholar]
- [53].Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. [DOI] [PubMed] [Google Scholar]
- [54].Claas RF, Ter Braak M, Hegen B, et al. Enhanced Ca2+ storage in sphingosine-1-phosphate lyase-deficient fibroblasts. Cell Signal. 2010;22(3):476–483. [DOI] [PubMed] [Google Scholar]
- [55].Kamentsky L, Jones TR, Fraser A, et al. Improved structure, function and compatibility for cell profiler: modular high-throughput image analysis software. Bioinformatics. 2011;27(8):1179–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Thiele C, Papan C, Hoelper D, et al. Tracing fatty acid metabolism by click chemistry. ACS Chem Biol. 2012;7(12):2004–2011. [DOI] [PubMed] [Google Scholar]
- [57].Herzog R, Schwudke D, Schuhmann K, et al. A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 2011;12(1):R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.