

ABSTRACT
Macroautophagy/autophagy is an intracellular degradation process that delivers cytosolic materials and/or damaged organelles to lysosomes. De novo synthesis of the autophagosome membrane occurs within a phosphatidylinositol-3-phosphate-rich region of the endoplasmic reticulum, and subsequent expansion is critical for cargo encapsulation. This process is complex, especially in mammals, with many regulatory factors. In this study, by utilizing PRKN (parkin RBR E3 ubiquitin protein ligase)-mediated mitochondria autophagy (mitophagy)-inducing conditions in conjunction with chemical crosslinking and mass spectrometry, we identified human BCAS3 (BCAS3 microtubule associated cell migration factor) and C16orf70 (chromosome 16 open reading frame 70) as novel proteins that associate with the autophagosome formation site during both non-selective and selective autophagy. We demonstrate that BCAS3 and C16orf70 form a complex and that their association with the phagophore assembly site requires both proteins. In silico structural modeling, mutational analyses in cells and in vitro phosphoinositide-binding assays indicate that the WD40 repeat domain in human BCAS3 directly binds phosphatidylinositol-3-phosphate. Furthermore, overexpression of the BCAS3-C16orf70 complex affects the recruitment of several core autophagy proteins to the phagophore assembly site. This study demonstrates regulatory roles for human BCAS3 and C16orf70 in autophagic activity.
Abbreviations: AO: antimycin A and oligomycin; Ash: assembly helper; ATG: autophagy-related; BCAS3: BCAS3 microtubule associated cell migration factor; C16orf70: chromosome 16 open reading frame 70; DAPI: 4‘,6-diamidino-2-phenylindole; DKO: double knockout; DMSO: dimethyl sulfoxide; ER: endoplasmic reticulum; fluoppi: fluorescent-based technology detecting protein-protein interactions; FIS1: fission, mitochondrial 1; FKBP: FKBP prolyl isomerase family member 1C; FRB: FKBP-rapamycin binding; hAG: humanized azami-green; IP: immunoprecipitation; IRES: internal ribosome entry site; KO: knockout; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MFN2: mitofusin 2; MS: mass spectrometry; MT-CO2: mitochondrially encoded cytochrome c oxidase II; mtDNA: mitochondrial DNA; OPTN: optineurin; PFA: paraformaldehyde; PE: phosphatidylethanolamine; PtdIns3K: phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol-3-phosphate; PtdIns(3,5)P2: phosphatidylinositol-3,5-bisphosphate; PINK1: PTEN induced kinase 1; PRKN/Parkin: parkin RBR E3 ubiquitin protein ligase; PROPPIN: β-propellers that bind polyphosphoinositides; RB1CC1/FIP200: RB1 inducible coiled-coil 1; TOMM20: translocase of outer mitochondrial membrane 20; ULK1: unc-51 like autophagy activating kinase 1; WDR45B/WIPI3: WD repeat domain 45B; WDR45/WIPI4: WD repeat domain 45; WIPI: WD repeat domain, phosphoinositide interacting; WT: wild type; ZFYVE1/DFCP1: zinc finger FYVE-type containing 1
KEYWORDS: Mitophagy, phagophore, parkin, pink1, starvation, wd40
Introduction
Autophagy is an intracellular degradation pathway critical for cell homeostasis that delivers cytosolic materials and/or damaged organelles to lysosomes in response to various stressors and signals [1,2]. Three types of autophagy have been identified: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy (hereafter referred to as autophagy) is distinguished by de novo membrane synthesis. In mammals, autophagy activation triggers synthesis of an phagophore, within a particular region of the endoplasmic reticulum (ER). The phagophore then elongates and encapsulates cellular components targeted for degradation by forming an enclosed double-membrane structure called the autophagosome. Fusion of the autophagosome with lysosomes results in hydrolytic degradation of the autophagic cargo and subsequent recycling.
More than 35 proteins are essential for autophagy [3]. The earliest steps in the autophagy process require the ULK (unc-51-like autophagy activating kinase) complex, which is composed of ULK1/2, RB1CC1 (RB1 inducible coiled-coil 1), ATG13 (autophagy related 13) and ATG101 [4,5]. Under nutrient-rich conditions, this complex is maintained in an inactivated state by MTOR (mechanistic target of rapamycin kinase) complex 1. Under starvation conditions, the inhibitory block is removed and the activated ULK complex promotes the recruitment of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, which consists of PIK3C3/VPS34, BECN1 (beclin 1), ATG14, NRBF2 and PIK3R4/VPS15, to the ER [6]. The phosphatidylinositol-3-phosphate (PtdIns3P) generated by the PtdIns3K complex forms a PtdIns3P-enriched ER subdomain (i.e. the omegasome) that functions as a platform for nucleation and elongation of the phagophore as well as recruitment of a number of PtdIns3P-binding proteins. Although ZFYVE1 (zinc finger FYVE-type containing 1), which binds PtdIns3P through double FYVE domains, is used as a marker for the omegasome, its contribution to autophagy activity (at least in starvation-induced autophagy) remains unknown [7]. In contrast, the PROPPIN (β-propellers that bind polyphosphoinositides) family proteins, WIPI1 (WD repeat domain, phosphoinositide interacting 1) and WIPI2 (yeast Atg18 homologs), and WDR45B (WD repeat domain 45B) and WDR45 (yeast Hsv2 homologs) [8] coordinately function in expansion and maturation of the phagophore [9]. Initial formation of the autophagosome likewise involves ATG9A vesicles. Atg8-family proteins, which associate with the phagophore membrane following lipidation via an the ATG12–ATG5-ATG16L1 complex, are critical for closure of the phagophore [10] as well as lysosomal degradation of the inner membrane [11].
Autophagy proteins are evolutionarily conserved from yeast to humans. They were initially identified by genetic screens in the budding yeast Saccharomyces cerevisiae [12–14], and Caenorhabditis elegans [15]. More recently, siRNA-mediated gene silencing [16] and knockout (KO)-based genome-wide screens [17] have been carried out in mammalian cells. However, accessory components, whose single gene deletions only manifest as mild defects in autophagy activity, might be missed by these types of genetic screens.
Our understanding of both starvation-induced autophagy (representative of non-selective autophagy) and ubiquitin-dependent selective autophagy has been greatly expanded over the past decade. Furthermore, mitochondrial autophagy (mitophagy) mediated by PRKN (parkin RBR E3 ubiquitin protein ligase) and PINK1 (PTEN induced kinase 1) is one of the most extensively characterized selective autophagy pathways [18–21]. The E3 ligase PRKN and the mitochondrial kinase PINK1, which are causal for familial Parkinson disease, coordinately function to generate poly-ubiquitin chains on damaged mitochondria through ubiquitin phosphorylation. In response to a loss in the mitochondrial membrane potential, PINK1 accumulates on the outer mitochondrial membrane (OMM) [22,23] and phosphorylates ubiquitin [24–27]. Cytosolic PRKN is then recruited to the damaged mitochondria through direct interactions with phosphorylated ubiquitin. PINK1 further phosphorylates the ubiquitin-like domain in PRKN to activate its E3 ligase activity. Through the action of a positive feedback amplification loop, a number of OMM proteins are subsequently ubiquitinated [28] and recognized by autophagy receptors. Two such autophagy receptors, CALCOCO2 (calcium binding and coiled-coil domain 2) and OPTN (optineurin), were recently shown to recruit the ULK complex and the ATG9A vesicles, respectively, as well as the ATG8 family proteins via their LC3-interacting region motifs [29–32].
Although the requirement of autophagy receptors is unique to ubiquitin-dependent autophagy, the molecular mechanisms of the later step (i.e. autophagosome formation) are thought to be common to both selective and non-selective autophagy. PRKN-PINK1 mitophagy is thus an ideal system for elucidating biogenesis of the phagophore because mitophagy can be spatiotemporally regulated, at least in the context of cell biology. For example, nucleation of the phagophore can be induced by addition of an uncoupler that dissipates mitochondrial membrane potential such that elongation of the phagophore is limited to regions near damaged mitochondria.
In this study, we took advantage of this experimental system in conjunction with mass spectrometry analysis to identify human BCAS3 (BCAS3 microtubule associated cell migration factor) and C16orf70 (chromosome 16 open reading frame 70) as novel proteins that associate with the phagophore assembly site in both non-selective and selective autophagy. We demonstrate that BCAS3 and C16orf70 form a complex, and that this interaction is mutually essential for association with the phagophore assembly site. Based on structural modeling, mutational analyses and lipid binding assays, we show that critical PtdIns3P-binding pockets reside in the WD40 repeat domain of human BCAS3. Furthermore, we report that overexpression of the BCAS3-C16orf70 complex impedes recruitment of ATG13 and ATG16L1 to the phagophore assembly site. These results suggest human BCAS3 and C16orf70 regulate autophagy activity.
Results
Mass spectrometric identification of BCAS3 and C16orf70 as WIPI1 co-immunoprecipitating proteins
Both ZFYVE1 and WIPI1 localize to the phagophore assembly site in response to autophagy induction. We and others previously showed that ZFYVE1 and WIPI1 translocate from the cytosol to the phagophore assembly site in response to PRKN-mediated mitophagy [33,34]. Therefore, we sought to identify novel mammalian proteins that associate with damaged mitochondria and/or the phagophore assembly site during PRKN-mediated mitophagy by immunoprecipitating ZFYVE1 or WIPI1. For this purpose, single clones of HeLa cells stably expressing YFP-PRKN and 3FLAG-tagged ZFYVE1 (3FLAG-ZFYVE1) or WIPI1 (3FLAG-WIPI1) were isolated and mitophagy-dependent recruitment of ZFYVE1 and WIPI1 to damaged mitochondria was confirmed (Figure 1A). Cells treated with or without valinomycin were then crosslinked with 0.1% paraformaldehyde (PFA), co-immunoprecipitated with an anti-FLAG antibody, and the immunoprecipitated proteins were analyzed by mass spectrometry (MS) and label-free quantification. PFA-mediated crosslinking facilitated the capture of proteins that directly interact with ZFYVE1 and WIPI1 as well as proteins localized adjacent to damaged mitochondria and/or the phagophore assembly site. Significant enrichment was observed in a number of proteins (i.e. those with high abundance ratios) in the valinomycin-treated ZFYVE1-IP and WIPI1-IP samples relative to control (untreated) samples (Figure 1B). The top 35 most abundant proteins in the valinomycin-treated samples included core autophagy proteins (ATG2B, WDR45, ATG14, ATG101, ATG16L1 and WIPI2), autophagy-related proteins (WHAMM [WASP homolog associated with actin, golgi membranes and microtubules] and JMY [junction mediating and regulatory protein, p53 cofactor]) [35,36], autophagy receptors (OPTN) [37,38], and the ubiquitin-related proteins (UBB [ubiquitin B] and USP4 [ubiquitin specific peptidase 4]). The presence of these proteins in our screen confirmed the utility of our approach for efficiently capturing proteins in close proximity to the mitophagy-specific phagophore assembly site (Figure 1C). We also identified a number of mitochondrial proteins (Figure 1C) that likely correspond to precursor proteins that had accumulated on the outer mitochondrial surface rather than the matrix as valinomycin-induced loss in membrane potential blocks protein translocation into the matrix. To validate the MS-based results and identify novel autophagy-related proteins, non-mitochondrial candidate proteins were individually tested for mitochondrial recruitment during PRKN-mediated mitophagy (Fig. S1). Although the subcellular localization of most of the proteins tested did not change (Fig. S1), signals for several small GTPases, including RAB1A (RAB1A, member RAS oncogene family), RAB1B, RAB24, and SAR1B (secretion associated Ras related GTPase 1B), were detected in close proximity to the damaged mitochondria as reported previously [39–41]. Signals for 3FLAG-tagged BCAS3/Rudhira and C16orf70 dispersed throughout the cytosol under normal conditions, but accumulated around damaged mitochondria in response to valinomycin treatment (Figure 2A and Fig. S2A). Mammalian BCAS3 is a cytoskeletal protein essential for development of angiogenesis [42–44] and C16orf70 gene has been reported to undergo hypermethylation in male schizophrenia patients [45]. The molecular function of these proteins in autophagy, however, is unknown.
Figure 1.
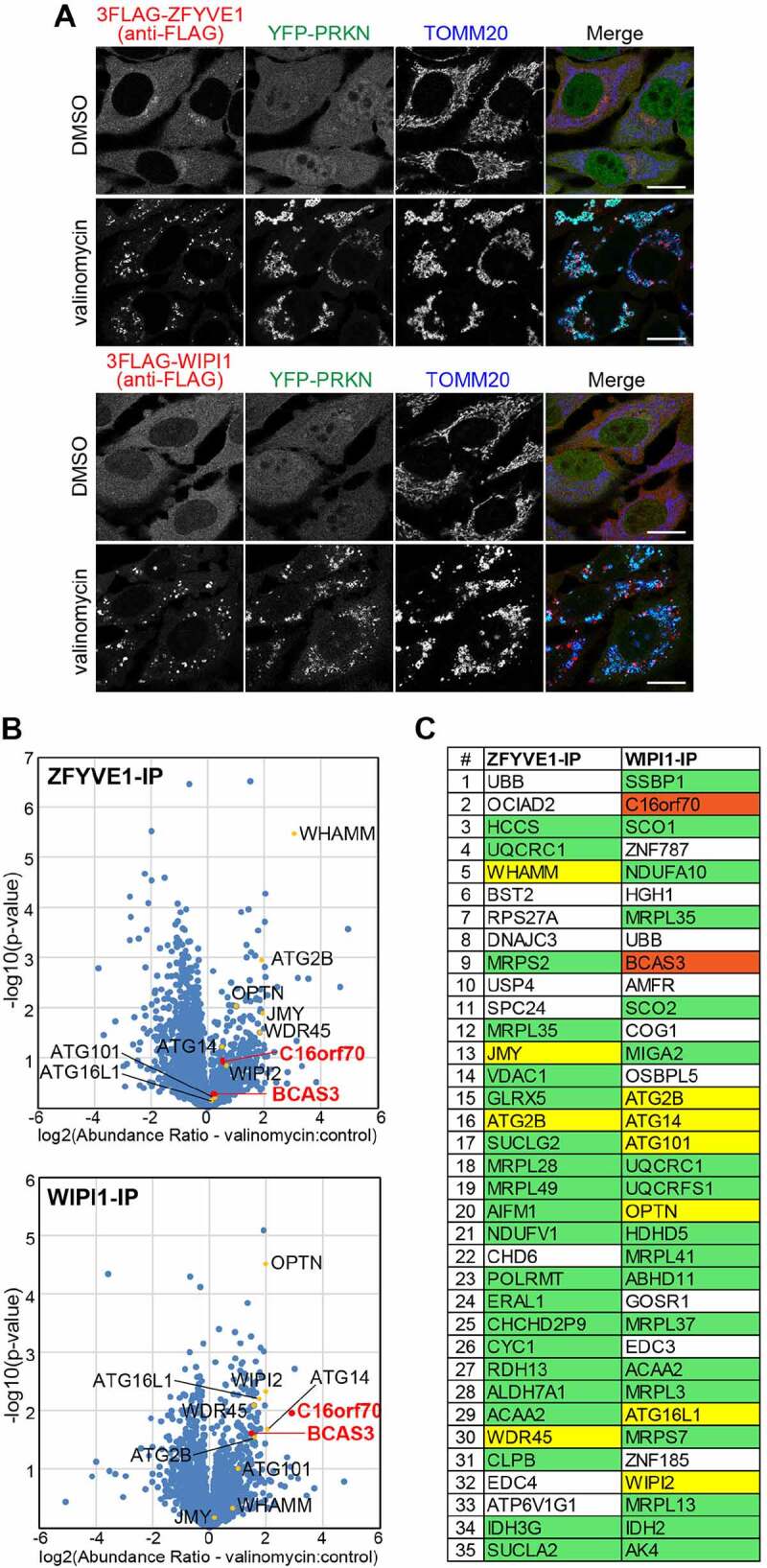
Co-IP followed by MS analysis of WIPI1-associated proteins during mitophagy identified human BCAS3 and C16orf70. (A) HeLa cells stably expressing YFP-PRKN and 3FLAG-ZFYVE1 or 3FLAG-WIPI1 were treated with dimethyl sulfoxide (DMSO; control) or valinomycin for 3 h and then immunostained. Bars: 20 µm. (B) After treatment with DMSO or valinomycin, the cells in (A) were treated with 0.1% PFA, solubilized, and co-immunoprecipitated with an anti-FLAG/DDDDK antibody and then analyzed by MS. The volcano plots (ZFYVE1-IP – upper panel; WIPI1-IP – lower panel) show the abundance ratios (valinomycin versus control) plotted against the p-value in a – log10 scale for three independent experiments. Yellow dots indicate autophagy-related proteins listed in (C) and red dots indicate BCAS3 and C16orf70. (C) Co-IP proteins in (B) were ranked according to the Proteome Discoverer software-based quantification abundance ratio (valinomycin versus control). Protein designations are based on the UniProt database. Mitochondrial and autophagy-related proteins are highlighted in green and yellow, respectively
Figure 2.
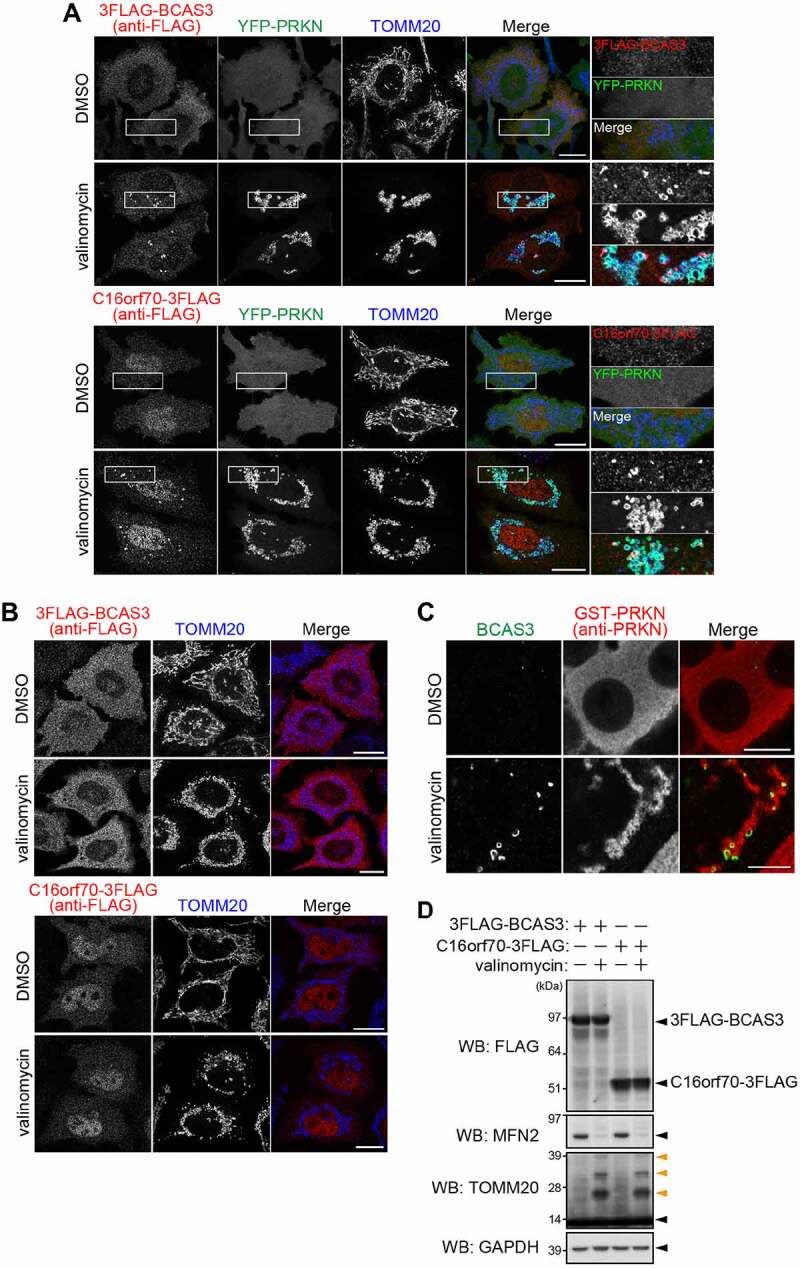
BCAS3 and C16orf70 are recruited to the phagophore assembly site upon mitophagy in a PRKN-PINK1 dependent manner. (A and B) HeLa cells stably expressing 3FLAG-BCAS3 or C16orf70-3FLAG with (A) or without YFP-PRKN (B) were treated with DMSO or valinomycin for 3 h and then immunostained. Bars: 20 µm. (A) Magnified images are shown in the rightmost panels. (C) HCT116 cells stably expressing GST-PRKN were treated with DMSO or valinomycin for 3 h and then immunostained. Endogenous BCAS3 was detected with an anti-BCAS3 antibody. Bars: 10 µm. (D) Total cell lysates in (A) were analyzed by western blotting. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Orange arrowheads denote ubiquitinated TOMM20
Although both BCAS3 and C16orf70 formed punctate structures adjacent to the damaged mitochondria in response to mitophagy stimulation, these structures did not appear in HeLa cells in the absence of PRKN overexpression or in siRNA PINK1-suppressed cells even after 3 h of valinomycin treatment (Figure 2B; Fig. S2B and S2C). This suggests that the mitochondrial recruitment of BCAS3 and C16orf70 depends on PRKN and PINK1. To gain insights into the mitochondrial recruitment of endogenous BCAS3, we produced an anti-BCAS3 antibody. Immunocytochemistry with this antibody showed that both exogenous and endogenous BCAS3 concentrated near damaged mitochondria in a PRKN-mediated mitophagy-dependent manner (Figure 2C; Fig. S2D). Furthermore, unlike the mitochondrial proteins; TOMM20 (translocase of outer mitochondrial membrane 20) and MFN2 (mitofusin 2), BCAS3 and C16orf70 are not ubiquitinated or degraded by mitophagy induction (Figure 2D).
BCAS3 and C16orf70 form a heterodimer
While determining the subcellular localization of BCAS3 and C16orf70, we noticed that under basal conditions the BCAS3 and C16orf70 signals resolved as weak dot-like structures that were almost completely merged (Figure 3A), suggesting that they form a complex. To test this possibility, we performed a fluoppi (fluorescent-based technology detecting protein-protein interactions) assay [25,46,47]. When a protein fused with a homo-tetrameric humanized azami green (hAG) interacts with another protein that is fused with a homo-oligomeric assembly helper (Ash), they form large fluorescent foci in cells through multivalent interactions. hAG-tagged BCAS3 (hAG-BCAS3) generates a diffuse cytosolic signal when expressed with the HA-Ash vector (Figure 3B). In sharp contrast, when hAG-BCAS3 was expressed with HA-Ash-tagged C16orf70 (HA-Ash-C16orf70), foci were formed (Figure 3B). To further examine this interaction, we also used the FRB (FKBP-rapamycin binding)-FKBP (FKBP prolyl isomerase family member 1C) system in which the two proteins dimerize in the presence of rapamycin [48]. We anchored the FRB domain to the mitochondria by fusing it with the transmembrane segment of FIS1 (fission, mitochondrial 1) and forced heterotropic mitochondrial localization of FKBP-tagged BCAS3 (BCAS3-2FKBP) by addition of rapamycin-analog (rapalog) (Figure 3C). In the absence of BCAS3-2FKBP, C16orf70-3FLAG localizes in the cytosol irrespective of rapalog treatment (Figure 3C). However, when cytosolic BCAS3-2FKBP was forced to associate on mitochondria after 3 h of rapalog treatment, C16orf70-3FLAG also moved to the mitochondria (Figure 3C), indicating that BCAS3 and C16orf70 interact with each other even under basal conditions. We also performed co-immunoprecipitation assays to confirm the BCAS3-C16orf70 interaction occurs regardless of mitophagy induction (Figure 3D and 3E). It should be noted that although ZFYVE1 and WIPI1 were used as baits in the initial MS-IP analysis, neither was pulled down with FLAG-tagged BCAS3 or C16orf70 without a chemical crosslinker (Figure 3D and 3E). Based on these assays, we conclude that BCAS3 and C16orf70 form a heterodimer under both normal growing and mitophagy-inducing conditions.
Figure 3.
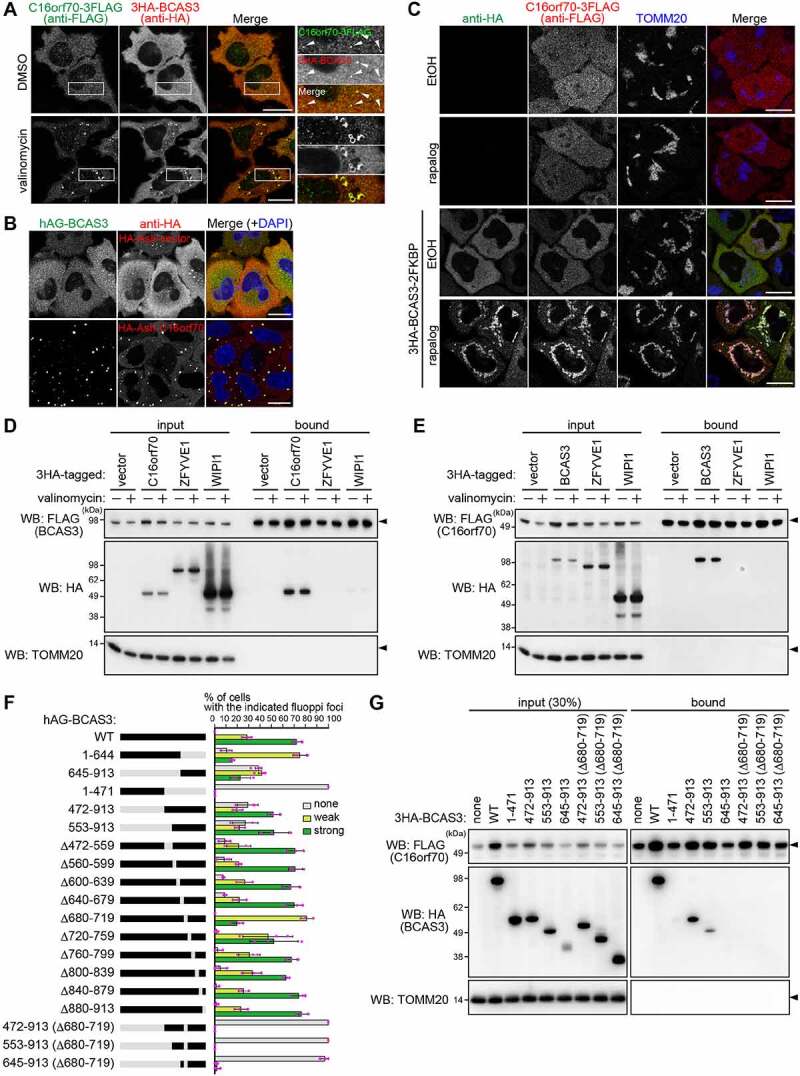
BCAS3 and C16orf70 interact with each other. (A) 3HA-BCAS3, C16orf70-3FLAG and GST-PRKN were stably expressed in HeLa cells. Cells were treated with DMSO or valinomycin for 3 h and then immunostained. Magnified images are shown in the rightmost panels. Bars: 20 µm. (B) hAG-BCAS3 was expressed with either an HA-Ash vector or HA-Ash-C16orf70 in HeLa cells. Cells were immunostained 24 h post-transfection. Nuclei were stained with DAPI. Bars: 20 µm. (C) The indicated proteins were co-expressed with FRB-FIS1TM in HeLa cells. Cells were treated with ethanol (EtOH, control) or rapalog for 3 h and then immunostained. Bars: 20 µm. (D and E) 3HA-tagged C16orf70, BCAS3, ZFYVE1, or WIPI1 were transiently expressed in HeLa cells stably expressing 3FLAG-BCAS3 or C16orf70-3FLAG. After 24 h of transfection, cells were immunoprecipitated with an anti-FLAG antibody. The input (30% against the bound fraction) and the bound fractions were analyzed by western blotting. TOMM20 was used as a negative control. (F) Summary of results from fluoppi assays using the BCAS3 constructs. Truncated or deleted regions in BCAS3 are shown in gray. hAG-tagged WT BCAS3 and associated mutants were transiently expressed with HA-Ash-C16orf70 in HeLa cells and immunostained (Fig. S4B). Graphs show the percentage (%) of cells with the indicated fluoppi foci (none, weak, or strong). Error bars represent mean ± s.d. with > 100 cells counted in each of three independent experiments. (G) HEK293 cells expressing 3FLAG-C16orf70 and the indicated 3HA-BCAS3 truncated mutants were immunoprecipitated using an anti-FLAG antibody. The input (30% against the bound fraction) and the bound fractions were analyzed by western blotting. TOMM20 was used as a negative control
Taking further advantage of the fluoppi assay, we next used BCAS3 truncation mutants to determine the region of BCAS3 responsible for C16orf70 interactions. In general, the fluorescence intensity of the fluoppi foci correlates with the strength of the protein-protein interaction. Thus, we classified BCAS3-C16orf70 interactions based on the size of the resulting foci as “none” for no foci, “weak” for small foci, and “strong” for large foci. The structure prediction algorithm HHpred suggested residues 58–644 of BCAS3 correspond to a WD40 repeat domain. To assess the potential functional role of this region, we generated two BCAS3 mutants (residues 1–471 and 1–644) that lose the C-terminal parts but retain the predicted WD40 repeat domain, as well as three BCAS3 mutants (residues 472–913, 553–913, and 645–913) that lose the N-terminal parts. hAG fusion with full-length wild type (WT) BCAS3 efficiently formed fluoppi foci with HA-Ash-C16orf70. Similarly, all of the N-terminal truncations (i.e. residues 472–913, 553–913, and 645–913) formed foci albeit less efficiently than WT (Figure 3F; Fig. S3). In contrast, no foci were formed with the 1–471 mutant, and the 1–644 mutant only formed small foci, suggesting that the C-terminal region of BCAS3 is important for C16orf70 interactions (Figure 3F; Fig. S3). To further narrow down the interaction region, we generated a series of 40 aa deletions spanning residues 472–913 of full-length BCAS3. With the exception of Δ680–719, all of the constructed deletion mutants (Δ472–559, Δ560–599, Δ600–639, Δ640–679, Δ680–719, Δ720–759, Δ760–799, Δ800–839, Δ840–879, and Δ880–913) formed foci similar to WT. Although the Δ680–719 deletion did not abolish foci formation, the foci size was clearly reduced. Furthermore, inserting the Δ680–719 deletion into the N-terminal truncation mutants completely inhibited foci formation (Figure 3F; Fig. S3). These results suggest that residues 680–719 of BCAS3 are important for C16orf70 interactions. We further analyzed the BCAS3-C16orf70 interaction by co-IP. HA-tagged WT BCAS3 and the mutants shown in Figure 3F were co-expressed with FLAG-tagged C16orf70 in HeLa cells and cell extracts were immunoprecipitated with an anti-FLAG antibody. We first confirmed that full-length WT BCAS3 and the 472–913, but not the 1–471, were efficiently pulled down with FLAG-C16orf70 (Figure 3G). Furthermore, consistent with the fluoppi results, interactions of the BCAS3 472–913 and 553–913 mutants with C16orf70 were completely lost when the Δ680–719 deletion was introduced. We also noticed that the protein level of exogenous C16orf70 was stabilized by interactions between BCAS3 and C16orf70 (Figure 3G input of FLAG-C16orf70). From these two independent assays (i.e. the fluoppi assay and co-IP), we concluded that residues 680–719 of BCAS3 are crucial for C16orf70 interactions and that other regions in the BCAS protein likely stabilize those interactions.
Interactions between BCAS3 and C16orf70 are essential for accumulating around damaged mitochondria
Since BCAS3 and C16orf70 form a complex, we next tested whether the interaction is required for their recruitment around mitochondria in response to PRKN-mediated mitophagy. For this purpose, we established BCAS3-/- and C16orf70-/- HCT116 cell lines using CRISPR-Cas9 gene editing methods (see details in Materials and Methods), and then stably expressed C16orf70-3FLAG and 3HA-BCAS3 in the cell lines along with GST-PRKN. In WT HCT116 cells, BCAS3 and C16orf70 were observed as punctate structures around mitochondria under mitophagy-inducing conditions (Figure 4A and 4B). In C16orf70-/- cells, however, 3HA-BCAS3 did not accumulate around the damaged mitochondria (Figure 4A and 4C). Similar defects in the mitochondrial recruitment of C16orf70 were observed in BCAS3-/- cells (Figure 4B and 4C). These defective phenotypes were observed in all of the BCAS3-/- and C16orf70-/- clones generated (Fig. S4A and S4B). Importantly, exogenous BCAS3 expression in BCAS3-/- cells and exogenous C16orf70 expression in C16orf70-/- cells recovered the accumulation of C16orf70 and BCAS3, respectively, around the damaged mitochondria (Figure 4A – 4C). These results indicate that BCAS3 and C16orf70 are mutually dependent for their accumulation around damaged mitochondria.
Figure 4.
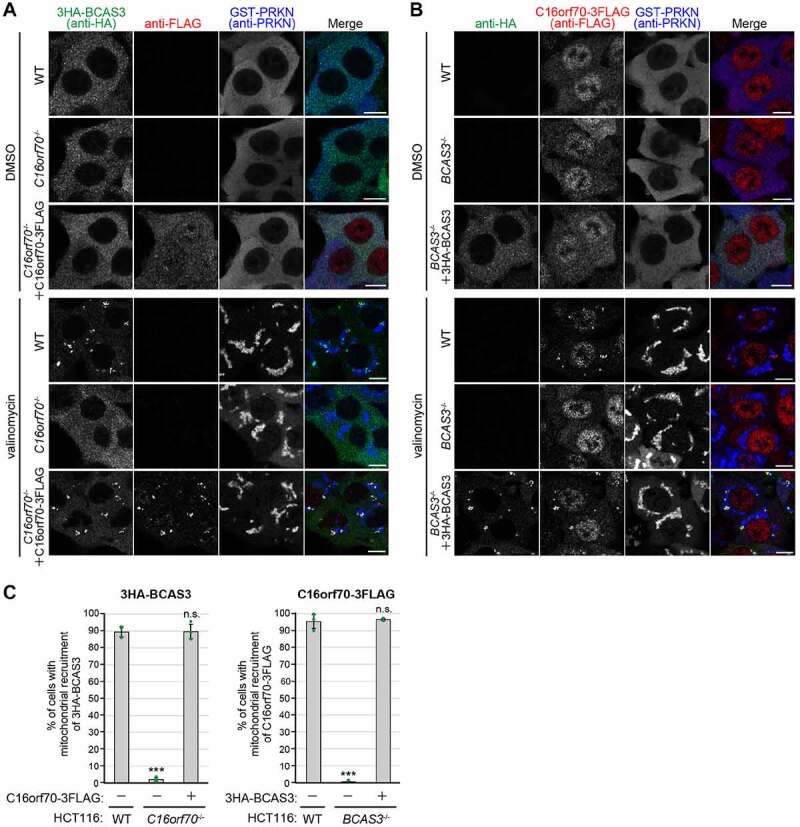
BCAS3 and C16orf70 are mutually dependent for accumulating around damaged mitochondria. (A and B) GST-PRKN and the indicated proteins were stably expressed in WT, C16orf70-/-, and BCAS3-/- HCT116 cells. Cells were treated with or without valinomycin for 3 h and then immunostained. 3HA-BCAS3, C16orf70-3FLAG and GST-PRKN were detected with anti-HA, anti-FLAG/DDDDK, and anti-PRKN antibodies, respectively. Bars: 10 µm. (C) The degree of mitophagy-dependent mitochondrial recruitment of 3HA-BCAS3 in (A) and C16orf70-3FLAG in (B) was quantified. Each dot represents the mean value determined from >100 cells and error bars represent mean ± s.d. in three independent experiments. Statistical differences were determined by student’s t-test (n.s.: not significant; ***P < 0.001)
BCAS3-C16orf70 complex is associated with the phagophore assembly site during PRKN-mediated mitophagy
Following a loss in membrane potential, PRKN molecules associated with damaged mitochondria colocalize with TOMM20 [49]. In contrast, signals for BCAS3 near damaged mitochondria appeared as dot-like and/or cup-shaped structures (Figure 5A), raising the possibility of direct association with phagophores or the phagophore assembly site rather than mitochondria. To address this possibility, we next used fluorescent microscopy methods to examine colocalization of 3 HA-BCAS3 with autophagy core proteins. The autophagy-initiation complex, which consists of ULK1, RB1CC1, ATG13 and ATG101, is essential for autophagy activation [33,50,51]. GFP-ULK1 formed mitophagy-dependent foci that clearly merged with some 3HA-BCAS3 puncta (Figure 5B). We next examined colocalization between BCAS3 and autophagy proteins containing PtdIns3P-binding domains. Following mitophagy stimulation, overexpressed GFP-ZFYVE1 and GFP-WIPI1 as well as endogenous WIPI2 were often found in cup-shaped structures that colocalized with the 3HA-BCAS3 signals (Figure 5C – 5E). Since PtdIns3P is generated within a particular region of the ER membrane, the cup-shaped structures are thought to comprise the phagophore assembly site [7]. ATG2A is known to act as a bridge between the ER and the phagophore via direct interactions with WIPIs [52,53]. Colocalization of BCAS3 and ATG2AΔC, a C-terminal deletion that enables selective association with the phagophore by inhibiting localization on lipid droplets [54], also suggested that BCAS3 is recruited to the phagophore assembly site (Figure 5F). A similar colocalization pattern was observed with BCAS3 and ATG16L1; however, ATG16L1 signals were more frequently observed with BCAS3 as an enclosed cup-shape (Figure 5G). MAP1LC3B is known to associate with elongating phagophores as well as enclosed autophagosomal membranes. Although a portion of the MAP1LC3B signal colocalized with BCAS3, not all MAP1LC3B signals aligned with BCAS3 (Figure 5H). To further examine colocalization of the BCAS3-C16orf70 complex with endogenous WIPI2 or MAP1LC3B, we constructed a HeLa cell line stably expressing C16orf70-3HA and untagged BCAS3 using the IRES (internal ribosome entry site) system and used a super-resolution confocal microscope to image the immunostained cells. As shown in Figure 5I and 5J, the BCAS3-C16orf70 complex completely merged with WIPI2, but not MAP1LC3B, suggesting preferential localization of the BCAS3-C16orf70 complex to the phagophore assembly site rather than enclosed autophagosomal membranes.
Figure 5.
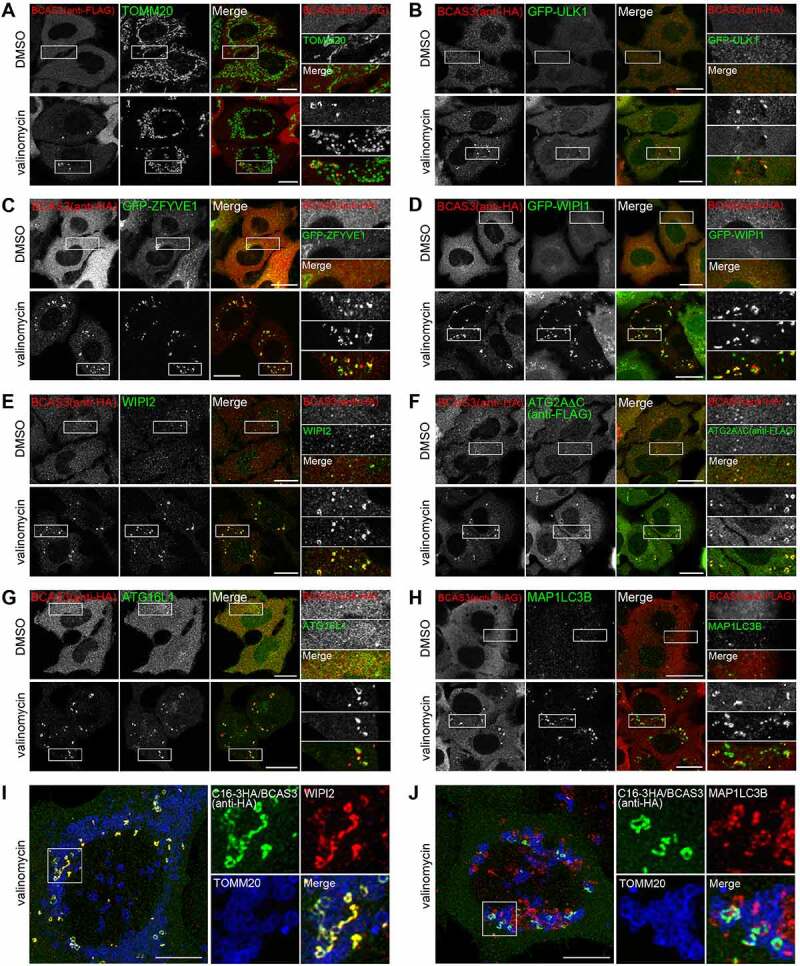
BCAS3 colocalizes with autophagy proteins in response to mitophagy. (A-H) HeLa cells stably expressing 3FLAG-BCAS3 and HA-PRKN (A), 3HA-BCAS3, GFP-ULK1 and GST-PRKN (B), 3HA-BCAS3, GFP-ZFYVE1 and GST-PRKN (C), 3HA-BCAS3, GFP-WIPI1 and GST-PRKN (D), 3HA-BCAS3 and GST-PRKN (E and G), 3HA-BCAS3, 3FLAG-ATG2AC and GST-PRKN (F), and HCT116 cells stably expressing 3FLAG-BCAS3 and HA-PRKN (H) were treated with DMSO or valinomycin for 3 h and then immunostained. Magnified images are shown in the rightmost panels. Bars: 20 µm. (I and J) HeLa cells stably expressing untagged PRKN and the C16orf70-3HA-BCAS3 complex (C16-3HA/BCAS3) using an IRES system were treated with valinomycin for 3 h and then immunostained. The z-stack images were taken with an SP8 confocal microscope and processed for deconvolution and maximum projection. Magnified images are shown to the right. Bars: 10 µm
BCAS3-C16orf70 complex is recruited to the phagophore assembly site in response to starvation
We next sought to determine if BCAS3 and C16orf70 recruitment to the phagophore assembly site is mitophagy specific or if the two proteins can also be recruited in response to non-selective autophagy conditions. To address this question, we examined the localization of BCAS3 and C16orf70 under amino-acid starvation condition. When the cells were incubated in media without amino acids for 1 h, a number of BCAS3 and C16orf70 puncta appeared (Figure 6A). In addition, MAP1LC3B was consistently observed adjacent to the BCAS3-C16orf70 puncta (Figure 6B). Furthermore, endogenous BCAS3 clearly colocalized with GFP-ULK1 in response to starvation (Figure 6C). These results indicate that the BCAS3-C16orf70 complex is recruited to the phagophore assembly site irrespective of cargo during both non-selective and selective autophagy.
Figure 6.
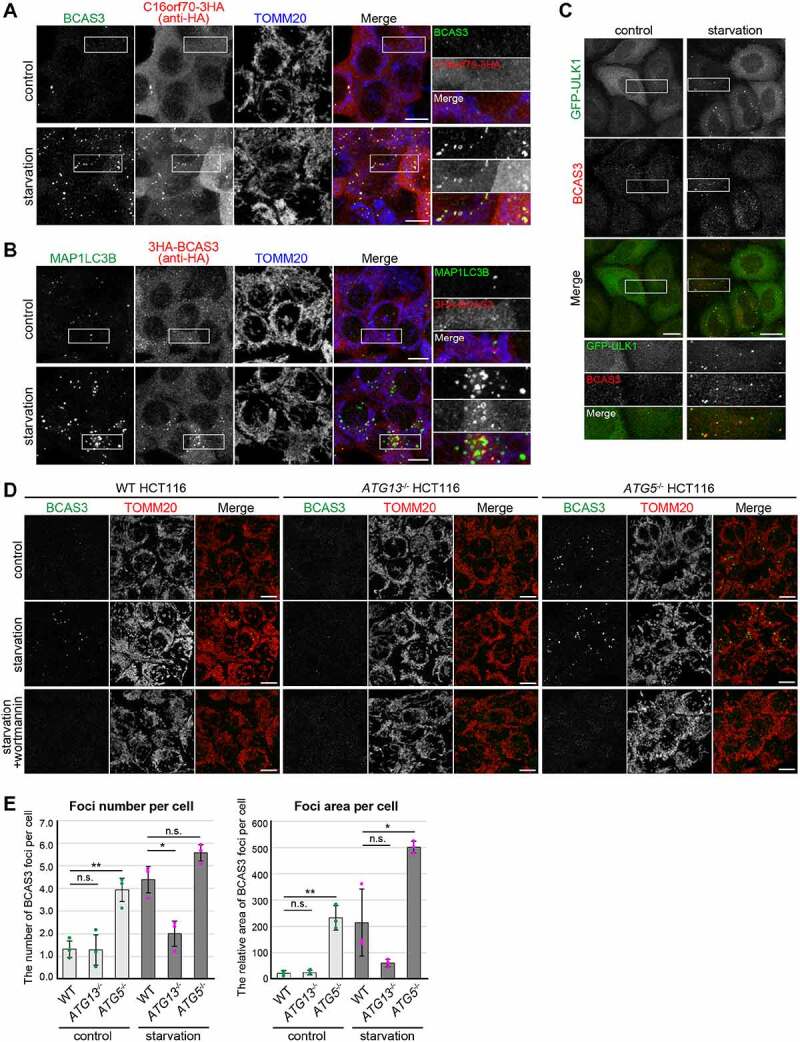
BCAS3 and C16orf70 colocalize with MAP1LC3 during starvation-induced autophagy. (A and B) HCT116 cells stably expressing BCAS3 and C16orf70-3HA from an IRES system (A) or 3HA-BCAS3 (B) were incubated in normal media (control) or amino-acid free media (starvation) for 1 h before immunostaining. Images are displayed as z-stacked confocal slices. Magnified images are shown in the rightmost panels. Bars: 10 µm. (C) HeLa cells stably expressing GFP-ULK1 were incubated as in (A). Bars: 20 µm. (D) WT, ATG13-/- and ATG5-/- HCT116 cells were incubated in normal media (control), amino-acid free media (starvation), or with wortmannin for 1 h prior to immunostaining. Endogenous BCAS3 was detected using an anti-BCAS3 antibody. Images are displayed as z-stacked confocal slices. Bars: 10 µm. (E) The number (left panel) and relative area (right panel) of BCAS3 foci in (D) were quantified. Each dot represents the mean value determined from >120 cells and error bars represent mean ± s.d. in three independent experiments. Statistical differences were determined by student’s t-test (n.s.: not significant; *P < 0.05; **P < 0.01)
When autophagy is induced, autophagy core proteins are assembled at the phagophore assembly site in a hierarchical manner. Previous studies showed that deletion of the most upstream unit ULK complex impairs the assembly of the downstream factors such as WIPIs, whereas genetic depletion of ATG5, which is involved in the later steps of autophagosome formation, results in an accumulation of WIPIs as enlarged foci at the phagophore assembly site [33]. Furthermore, STX17 (syntaxin 17), a SNARE (soluble N-ethylmaleimide sensitive fusion protein attachment protein receptor) for fusion of the autophagosome with the lysosome, is recruited to the autophagosome only after the autophagosomal membrane has enclosed [55]. Additionally, in ubiquitin-dependent selective autophagy, the autophagy receptors, CALCOCO2 (calcium binding and coiled-coil domain 2) and OPTN, hierarchically precede the ULK complex [31]. We thus assessed the pathway effects of BCAS3 under starvation conditions using autophagy deficient ATG KO HCT116 cell lines and a PtdIns3K inhibitor. ATG13, the most upstream core ATG protein, is a critical subunit in the ULK complex. In ATG13-/- HCT116 cells, starvation-induced BCAS3 puncta were reduced compared to WT HCT116 cells (Figure 6D and 6E). Previous reports showed that deletion of the ATG5 gene abrogates MAP1LC3 lipidation [56] and that several ATG proteins accumulate in ATG5 KO cells even under basal conditions [57]. Similar to WIPI1 [57], BCAS3 puncta were observed in ATG5-/- HCT116 cells and their quantity increased after 1 h of starvation (Figure 6D and 6E). In sharp contrast, no BCAS3 puncta were observed when cells were treated with the PtdIns3K inhibitor wortmannin prior to starvation (Figure 6D). These analyses strongly suggest that BCAS3 is positionally situated between ATG13 and ATG5 and is recruited to either the phagophore assembly site or the elongating phagophore in a PtdIns3P-dependent manner similar to WIPI family proteins.
Structural modeling predicts that the WD40 repeat domain in BCAS3 contains phosphoinositide-binding pockets
As indicated above, human BCAS3 has a predicted N-terminal WD40 repeat domain. WD40 repeat domains are characterized by tandem repeats of 40–60 amino acids terminating in a Trp-Asp (WD) dipeptide that form an encircled β-propeller structure and which act as scaffolds for protein-protein interactions as well as protein-DNA and protein-lipid interactions [58]. The WD40 repeat domain is one of the most abundant domains in eukaryotes and is conserved across diverse proteins including signaling factors, transcription factors and autophagy proteins [59]. HHpred [60] predicts structural similarities between BCAS3 and Pichia angusta Atg18 (homolog of human WIPI1 and WIPI2) and Kluyveromyces lactis Hsv2 (homolog of human WDR45B and WDR45) with high probability (e-value: 1.2e-18 and 2.4e-18, respectively) (Figure 7A). Atg18 and Hsv2 function in autophagy and interact with PtdIns3P and/or PtdIns(3,5)P2 through their WD40 repeat domains [61,62]. Furthermore, the crystal structure of the Hsv2 WD40 repeat domain (PDB: 4EXV) complexed with sulfate ions has been determined [63,64]. We thus modeled the human BCAS3 WD40 repeat domain on the Hsv2 structure (Figure 7A and 7B). The predicted structure consists of a 7-bladed β-propeller with two inserts in blades 4 and 6 (Figure 7A and 7B), the latter of which includes predicted disordered regions. The insertion pattern is similar to that of yeast Atg18. Furthermore, the structural model suggests that the BCAS3 WD40 repeat domain binds two sulfate ions via interactions in sites 1 and 2 (Figure 7B – 7D). Both sites correspond to a cluster of polar and charged residues: site 1 consists of H350, D370, L372, H377, and H400 in blade 5, whereas site 2 is formed by R401 of blade 5 and R426, T428 and H430 of blade 6 (Figure 7C and 7D). These results and chemical similarities between the sulfate and phosphate ions imply that the two sites comprise the phosphoinositide-binding pockets (Figure 7D). When compared with K. lactis Hsv2 sites 1 and 2 [63], the critical phospholipid binding residues in Hsv2 are more conserved in site 2 than site 1 (Figure 7D).
Figure 7.
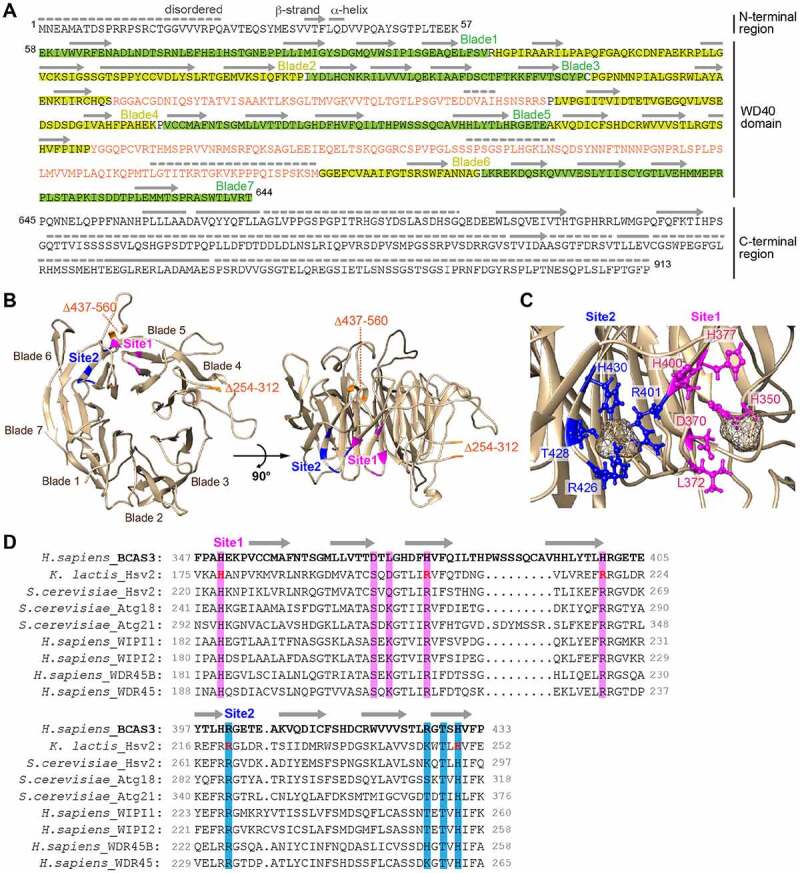
Two phospholipid-binding sites are predicted in the BCAS3 WD40 repeat domain. (A) Predicted secondary structure for human BCAS3. The secondary structure in the WD40 repeat domain was assigned based on the model shown in (B) and secondary structure features in the N- and C-terminal regions were determined using PSIPRED [76]. Disordered regions were predicted using DISOPRED3 [77]. Lines, arrows, and dotted lines indicate helix, strand and disordered region, respectively. The two inserts in blades 4 and 6 are colored orange. (B) Predicted 3D structure of the human BCAS3 WD40 repeat domain. The two inserts in WD40 (i.e. residues 254–312 and 437–560) are deleted. Sites 1 and 2 (the predicted phospholipid-binding pockets) are colored magenta and blue, respectively. (C) Detailed sulfate binding depicted for sites 1 and 2. Side chains of the indicated residues in site 1 (magenta) and site 2 (blue) are depicted in ball-and-stick mode, whereas the sulfate ions are depicted as van der Waals spheres. (D) Alignment of the amino acid sequence around sites 1 and 2 of human BCAS3 and various Atg18 family proteins. Residues critical for phospholipid binding in K. lactis Hsv2 [63] are colored in red. Amino acid residues in sites 1 and 2 that were functionally examined in Figure 8 are highlighted in magenta and blue, respectively
Phosphoinositide binding is important for BCAS3 association with the phagophore assembly site
Based on the structural prediction, we hypothesized that phosphoinositide phosphates are recognized by sites 1 and/or 2. To determine if the predicted phosphoinositide-binding pockets are important for BCAS3 association with the phagophore assembly site, we replaced amino acid residues in the two sites with alanine. The resultant 3HA-tagged BCAS3 mutants were expressed in BCAS3-/- cells and recruitment to the phagophore assembly site assessed in response to mitophagy. Under normal growing conditions, all of the mutants localized to the cytosol (Fig. S5A). Following mitophagy induction, two of the site 1 mutants, D370A and H400A, exhibited decreased efficiency of recruitment, whereas recruitment of the other site 1 mutants (H350A, L372A, and H377A) was comparable to WT (Figure 8A and 8B). In contrast, site 2 mutants were largely defective in mitophagy-dependent accumulation of BCAS3 on the phagophore assembly site; minimal inhibition was seen with the R426A mutant and near complete inhibition was seen with the R401A, T428A, and H430A mutants (Figure 8A and 8B). The alanine substitutions, however, may have adversely impacted C16orf70 interactions, which are essential for BCAS3 association with the phagophore assembly site (Figure 4). Therefore, to rule out this possibility, fluoppi assays were performed with the BCAS3 alanine mutants. As shown in Fig. S5B, all hAG-tagged BCAS3 mutants formed foci with HA-Ash-C16orf70. Consequently, dissociation of the BCAS3 mutants from the phagophore assembly site does not result from defective C16orf70 interactions. We also tested whether the insertions in the WD40 repeat domain are critical for association with the phagophore assembly site. The WD40 repeat domain was predicted to contain two insertions encompassing residues 254–312 in blade 4 and residues 437–560 in blade 6. Deletion of the blade 4 insertion (Δ254–312) or the blade 6 insertion (Δ437–560) completely abolished BCAS3 association with the phagophore assembly site during mitophagy (Figure 8A and 8B) without affecting C16orf70 interactions (Fig. S5B). Computational modeling and targeted mutation of predicted sites demonstrated that BCAS3 association with the phagophore assembly site depends on phosphoinositide-binding pockets (mainly site 2) and the two insertions in the WD40 repeat domain.
Figure 8.
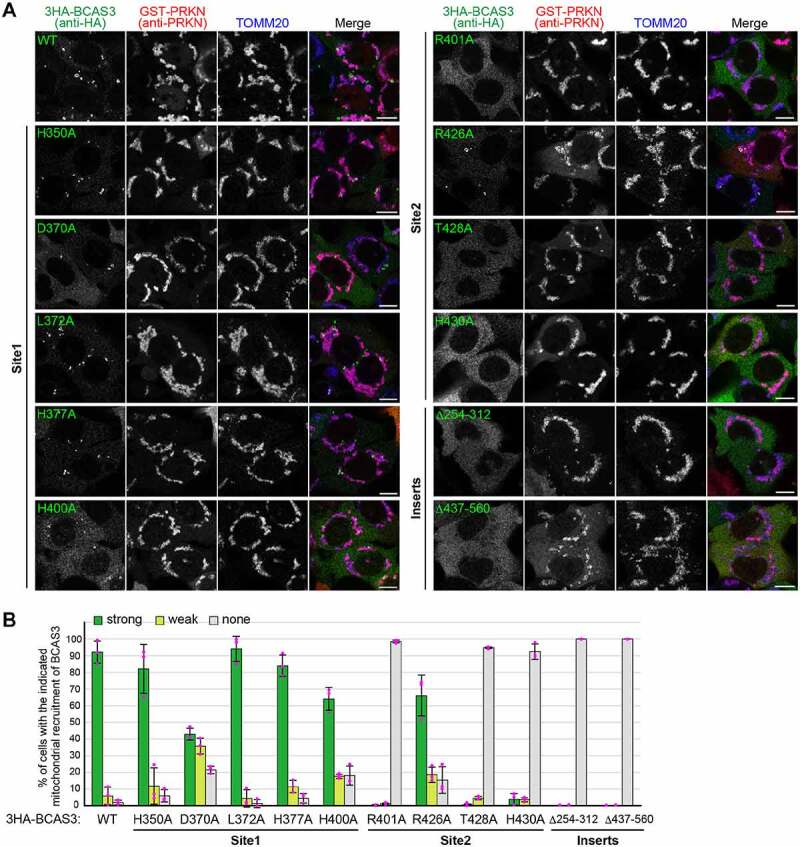
Predicted BCAS3 phospholipid-binding sites and the two insert regions are essential for associating with the phagophore assembly site. (A) BCAS3-/- HCT116 cells transiently expressing the indicated 3HA-BCAS3 construct with GST-PRKN were treated with valinomycin for 3 h and then immunostained. Bars: 10 µm. (B) The degree of BCAS3 mitochondrial recruitment in (A) was quantified. Each dot represents the mean value determined from >100 cells and error bars represent mean ± s.d. in three independent experiments
Overexpression of the BCAS3-C16orf70 complex retards recruitment of ATG13 and ATG16L1 to the phagophore assembly site
Are BCAS3 and C16orf70 required for autophagy? To address this question, we monitored mitophagy using a FACS (fluorescence-activated cell sorting) assay and DKO (double knockout) cell lines. For this purpose, YFP-PRKN and mitochondria-targeted Keima (mt-Keima) were stably expressed in WT and BCAS3-/-C16orf70-/- DKO HCT116 cells. Mitophagy-dependent delivery of mt-Keima to lysosomes can be monitored by FACS based on the pH-sensitive spectral shift of Keima [31,65]. Under basal growing conditions, all cell lines exhibited less than 2% mitophagy (Figure 9A and 9B). In contrast, when treated with antimycin A and oligomycin (AO) for 9 h, 83% of WT cells were mitophagy positive, and DKO clones #1-1-7 and #1-1-17 exhibited 90–92% mitophagy (Figure 9A and 9B). In this assay, AO rather than valinomycin, was used to minimize the inhibition of lysosomal acidification. Since the slight enhanced mitophagy efficiencies in BCAS3-/- C16orf70-/- DKO cells did not change following the introduction of exogenous BCAS3-C16orf70 complex (3HA-BCAS3 and C16orf70-3FLAG), the differences are likely attributable to clonal differences (Figure 9A and 9B). Mitophagy in the DKO cells was also detected by degradation of mitochondrial matrix proteins and mitochondrial DNA (mtDNA). AO treatment for 24 h decreased the OMM protein TOMM20 as well as the matrix proteins MT-CO2 (mitochondrially encoded cytochrome c oxidase II) and PDHA1 (pyruvate dehydrogenase E1 subunit alpha 1) in a PRKN-dependent manner (Fig. S6A). The reduced degradation of MT-CO2 and PDHA1 in ATG5-/- cells suggests that the process is autophagy dependent (Fig. S6A). Under these conditions, the mitochondrial degradation observed in two different BCAS3-/-C16orf70-/- DKO cell lines was comparable to the WT HCT116 cell line (Fig. S6A). Furthermore, genetic deletion of the BCAS3-C16orf70 complex did not affect mtDNA degradation in response to PRKN-mediated mitophagy (Fig. S6B). We thus concluded that mitophagy is not affected by BCAS3 and/or C16orf70 gene deletions at least in cultured cells. We next tested if autophagy was impacted by overexpression of the BCAS3-C16orf70 complex. Although both western blotting (Fig. S6C) and immunocytochemistry (Fig. S6D) indicated that overexpression of the BCAS3-C16orf70 complex had little to no effect on mitochondrial degradation over 24 h, we found the subcellular distributions of some core autophagy proteins were altered. Endogenous ATG13 and ATG16L1 formed foci near damaged mitochondria following mitophagy stimulation in WT HeLa cells. However, under the same conditions, IRES-based overexpression of the BCAS3-C16orf70 complex resulted in severely decreased foci formation of both proteins (Figure 10A and 10B). This inhibitory effect is dependent on BCAS3 phosphoinositide binding since overexpression of the BCAS3R401A mutant with C16orf70 did not affect the recruitment of either ATG13 or ATG16L1 to the phagophore assembly site (Figure 10A and 10B). In contrast, overexpression of the BCAS3-C16orf70 complex had no effect on the recruitment of the phosphoinositide-binding protein WIPI2 or MAP1LC3B-labeled autophagosome formation (Figure 10C – 10E). Of note, the composition of the ULK complex was not altered by overexpression of the BCAS3-C16orf70 complex under basal or mitophagy-inducing conditions (Figure 10F). A number of studies have reported that ZFYVE1 overexpression impedes phosphoinositide-binding protein recruitment by concealing phosphoinositides on the phagophore assembly site [7,66]. Therefore we compared the degree of endogenous ATG13, ATG16L1, WIPI2 and MAP1LC3B recruitment in cells in which the BCAS3-C16orf70 complex was overexpressed with cells in which ZFYVE1 and WIPI1 were overexpressed. The recruitment of ATG13 and ATG16L1 was reduced following ZFYVE1 overexpression as was the association of WIPI2 with the phagophore assembly site (Fig. S7A – S7C), whereas MAP1LC3B-labeled autophagosomes accumulated (Fig. S7D). WIPI1 overexpression, on the other hand, inhibited ATG13 foci formation, but had no effect on the recruitment of ATG16L1, WIPI2 or MAP1LC3B during mitophagy (Fig. S7A – S7E). WIPI2 recruitment was only inhibited when ZFYVE1 was overexpressed, suggesting that the BCAS3-C16orf70 complex does not simply compete with WIPI proteins for PtdIns3P on the phagophore assembly site.
Figure 9.
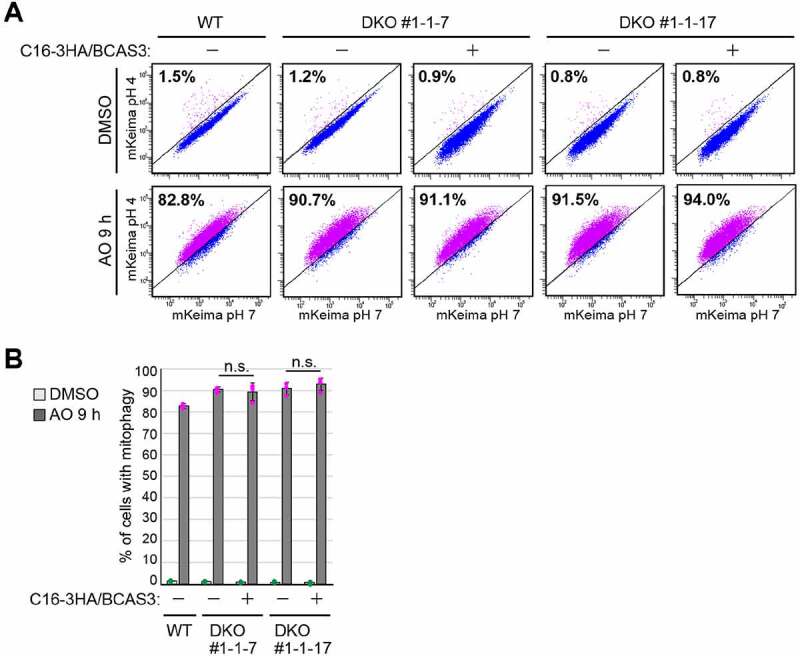
Loss of the BCAS3-C16orf70 complex does not affect overall mitochondrial degradation in response to mitophagy. (A) WT and BCAS3-/-C16orf70-/- DKO clones #1-1-7 and #1-1-17 HCT116 cells stably expressing YFP-PRKN and mt-Keima were treated with DMSO or antimycin A and oligomycin (AO) for 9 h and then analyzed by FACS. Representative FACS data with the percentage of cells exhibiting lysosomal mt-Keima signals are shown. (B) Quantification of the FACS-based mitophagy in (A). Each dot represents the mean value and error bars represent mean ± s.d. in three independent experiments. Statistical differences were determined by student’s t-test (n.s.: not significant)
Figure 10.
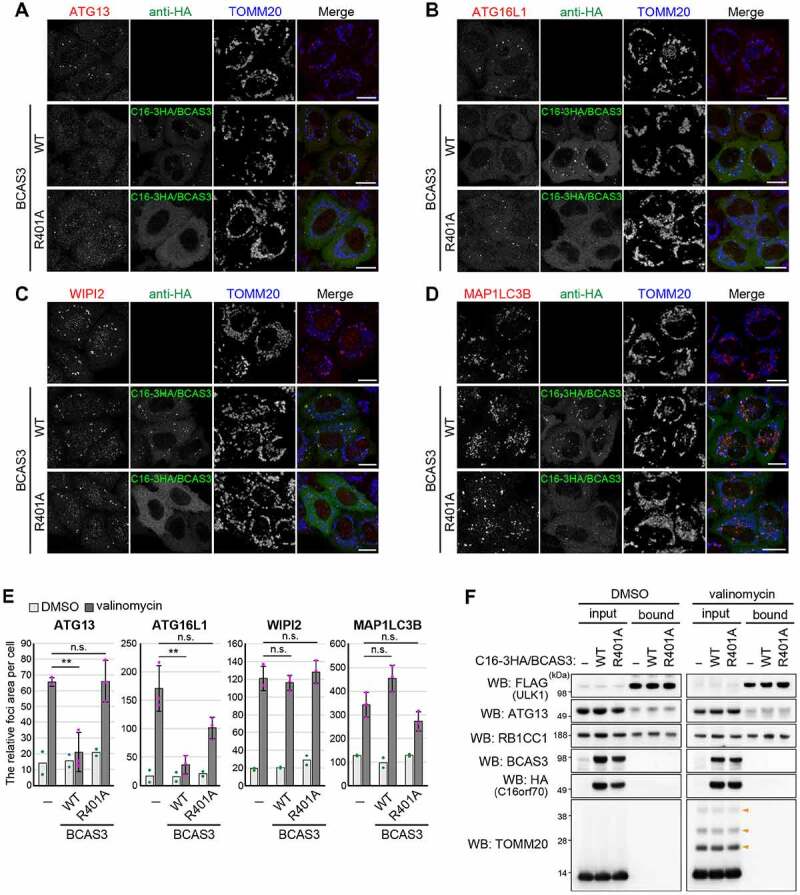
Overexpression of the BCAS3-C16orf70 complex affects recruitment of core autophagy proteins to the phagophore assembly site. (A-D) HeLa cells stably expressing PRKN alone (upper panel) or with IRES-based overexpression of C16orf70-3HA/BCAS3 (C16-3HA/BCAS3), WT (middle panel), or R401A (lower panel) were treated with valinomycin for 3 h and then immunostained with the indicated antibodies. Images are displayed as z-stacked confocal slices. Bars: 20 µm. (E) The relative foci areas for endogenous ATG13, WIPI2, ATG16L1 and MAP1LC3B in cells shown in (A-D) were quantified. Each dot represents the mean value determined from >100 cells and error bars represent mean ± s.d. in two (for DMSO) and three (for valinomycin) independent experiments. Statistical differences were determined by student’s t-test (n.s.: not significant; **P < 0.01). (F) HeLa cells stably expressing PRKN and 3FLAG-ULK1 with or without IRES-based overexpression of C16orf70-3HA/BCAS3 (C16-3HA/BCAS3), WT, or R401A were treated with DMSO or valinomycin for 3 h. The cell lysates were co-immunoprecipitated with an anti-FLAG antibody and 30% of the input and bound fractions were analyzed by western blotting. Orange arrowheads denote ubiquitinated TOMM20
The BCAS3-C16orf70 complex binds PtdIns3P in vitro
Structural modeling (Figure 7) and cellular analyses (Figure 8) strongly suggest that the BCAS3-C16orf70 complex is recruited to phagophores via BCAS3 phosphoinositide-binding. To test this, we performed an in vitro membrane flotation assay using PtdIns3P-containing liposomes. GFP-BCAS3-His and GFP-C16orf70-His were purified from HEK293T cells along with the control proteins, GFP-His and GFP-WIPI1-His (Figure 11A). Liposomes composed of phosphatidylcholine (PC) and phosphatidylethanolamine (PE) with or without PtdIns3P or PtdIns(3,5)P2 were incubated with the purified proteins, and then centrifuged using an OptiPrep step gradient (Figure 11B). Consistent with a previous report [63], WIPI1 strongly bound the PtdIns3P- and PtdIns(3,5)P2-containing liposomes, whereas GFP alone did not (Figure 11C). Furthermore, WT BCAS3 was found in the liposome-containing fraction (T, top fraction) when it contained PtdIns3P or PtdIns(3,5)P2 (Figure 11C). BCAS3R401, which is not recruited to the phagophore assembly site in response to PRKN-mediated mitophagy (Figure 8B), interacted with the high percentage (15%) PtdIns3P liposomes similar to that of WT BCAS3. However, in the presence of reduced (5%) PtdIns3P, BCAS3R401 more readily dissociated from the liposome than WT BCAS3 (Figure 11D). These results indicate that BCAS3-C16orf70 complex interactions with PtdIns3P-labeled membranes are partially dependent on the phosphoinositide binding pocket in the BCAS3 WD40 repeat domain.
Figure 11.
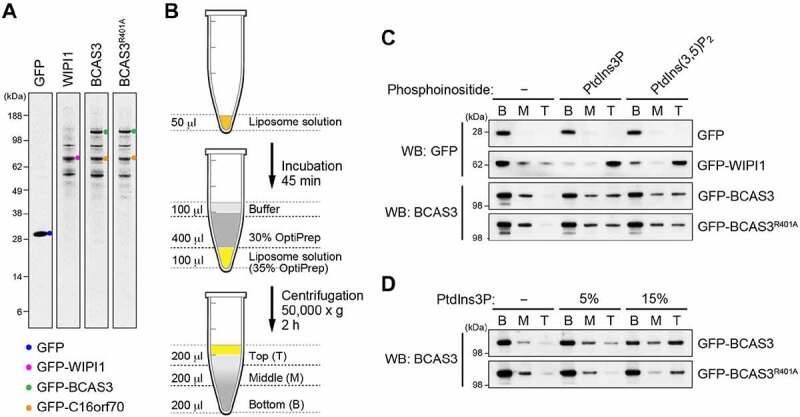
The BCAS3-C16orf70 complex binds to PtdIns3P in vitro. (A) CBB staining of purified GFP-His, GFP-WIPI1-His, GFP-BCAS3-His/GFP-C16orf70-His and GFP-BCAS3R401A-His/GFP-C16orf70-His. (B) Schematic diagram of the liposome flotation assay. After incubating the liposome solution with purified proteins, OptiPrep was added to a final concentration of 35% and then 30% OptiPrep and buffer were sequentially layered on top. After centrifugation, the Bottom (B), Middle (M) and Top (T) fractions are collected and analyzed by western blotting. (C) Flotation assays using the purified proteins and liposomes containing the indicated phosphoinositides were performed. GFP-His and GFP-WIPI1-His were detected with an anti-GFP antibody, and GFP-BCAS3-His was detected with an anti-BCAS3 antibody. (D) Flotation assays using liposomes with various concentrations of PtdIns3P were performed. GFP-BCAS3-His was detected with an anti-BCAS3 antibody
Discussion
The transfer of intracellular materials to lysosomes requires de novo autophagic membrane synthesis. In mammals, the biogenesis of autophagosome formation is complicated, requiring a number of accessory and/or regulatory components. In this study, using PRKN-mediated mitophagy as an autophagy-inducing model and MS-based analyses, we identified human BCAS3 and C16orf70 as novel proteins that associate with the phagophore assembly site.
BCAS3 and C16orf70 have no known counterparts in yeast (e.g. Saccharomyces cerevisiae). Our homology predictions, which considered secondary structure and the presence of disordered regions in the WD40 repeat domain, however, suggested structural similarities between human BCAS3 and yeast PROPPIN family proteins. Subsequent structural modeling revealed the presence of two phosphoinositide-binding pockets in the human BCAS3 WD40 repeat domain. Although the FRRG motif, which is diagnostic of WD40 repeat domains in PROPPINs, is not conserved in BCAS3, structural analyses have shown that the second and third arginine in the PROPPIN FRRG motif correspond to two different phosphoinositide-binding pockets [63,64]. Our mutational analysis showed that the site 2 pocket, a part of which includes the arginine in the LHRG motif (i.e. R401 in human BCAS3), is critical for association with phagophores. We also showed that BCAS3 recruitment to the phagophore assembly site is dependent on the insertions in two of the propeller blades structures, a finding that is consistent with previous results showing that insertions in the WD40 repeat domain support membrane associations [63,64]. In addition, although BCAS3 localizes to the cytosol under basal growing conditions, no BCAS3 associations with endosomes or Golgi membranes, which are typically rich in phosphoinositides, were observed. Therefore, taken together, the evidence strongly suggests that BCAS3 is a PROPPIN subtype specific for the autophagic membrane.
To clarify the function of BCAS3 and C16orf70 in autophagy, we knocked out each individually and in tandem. The absence of mitophagy defects in BCAS3-/-C16orf70-/- cells was not unexpected as all known autophagy-specific phosphoinositide-binding proteins are thought to more or less function cooperatively in elongation of the phagophore. siRNA-mediated knockdown of ZFYVE1 likewise generated no obvious autophagy defects [7]. Alfy, a 400 kDa protein with both WD40 and FYVE domains that directly interacts with ATG5, is also not required for starvation-induced autophagy [67]. Furthermore, autophagy defects in single wdr45 KO mice neurons [68] are much milder than those in either atg5 [69] or atg7 KOs [70]. On the other hand, we found that overexpressing an active BCAS3-C16orf70 complex inhibited the recruitment of ATG13 and ATG16L1 without affecting the association of WIPI2 with the phagophore assembly site. Therefore, the inhibitory effect generated by BCAS3-C16orf70 overexpression likely does not arise from simple competition with endogenous WIPI proteins, which differs from that of ZFYVE1 overexpression (Figure 10E and Fig. S7E). The BCAS3-C16orf70 complex may thus fine-tune autophagy activity by either regulating the association of other core proteins with the phagophore assembly site or functioning cooperatively with the core proteins.
During the preparation of this manuscript, Yamada et al. identified KinkyA and DDB_G0272949, homologs of mammalian C16orf70 and BCAS3, respectively, as genes involved in spore formation in Dictyostelium, a species of soil-dwelling amoeba [71]. Although the phenotypes are much weaker than those observed following gene deletion of core autophagy proteins such as atg5 or atg7, the study showed that autophagic activity (monitored by the efficiency of Atg8 delivery to lysosomes) is impaired following deletion of either KinkyA or DDB_G0272949 in Dictyostelium.
Although the molecular functions of the mammalian BCAS3-C16orf70 complex remain enigmatic, mouse BCAS3 has been proposed to function in angiogenesis [42,72]. While we did not observe the BCAS3-microtubule associations suggested by the previous report, it will be interesting in the future to determine if any autophagic defects are observed in animals following depletion of BCAS3 (or C16orf70). Similarly, it would be worthwhile to investigate any potential relationships between the BCAS3-C16orf70 complex and autophagic phenotypes in various animal models including angiogenesis.
Materials and methods
Cell culture
HeLa [32], HEK293 [32], and HEK293T (ATCC, CRL-3216) cell lines were cultured in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, D5796; DMEM) supplemented with 10% (v:v) fetal bovine serum (Biowest, S1820; FBS), 1× sodium pyruvate (Gibco, 11360–070), 1× nonessential amino acids (Gibco, 11140–050; NEAA), and 1× Penicillin-Streptomycin-Glutamine (Gibco, 10378–016; PSG) or 1× GlutaMax (Gibco, 35050–061). HCT116 cell lines were cultured in McCoy’s 5A medium (Gibco, 16600108) supplemented with 10% (v:v) FBS, NEAA, and PSG. All cells were incubated at 37°C in a 5% CO2 incubator.
To induce mitophagy, the mitochondrial membrane potential was dissipated with 10 µM valinomycin (Sigma-Aldrich, V0627). To induce mitophagy without lysosomal neutralization, cells were treated with a mixture of 10 µM oligomycin (Calbiochem, 495455) and 4 µM antimycin A (Sigma-Aldrich, A8674). To induce amino-acid starvation, cells were washed with phosphate-buffered saline (PBS; 10 mM phosphate, pH 7.4, 137 mM NaCl, 2.7 mM KCl) and incubated with DMEM without amino acids (Wako, 048–33575) for 1 h. Wortmannin (Sigma-Aldrich, W1628) was used at a final concentration of 100 nM to inhibit PtdIns3P synthesis. A/C Heterodimerizer (Clonetech, AP21967) was used at a final concentration of 500 nM. Q-VD-OPh (AdipoGen Life Sciences, AG-CP3-0006-M005) was used at a final concentration of 10 µM to prevent apoptosis.
Plasmid construction and mutagenesis
For gene cloning, genes of interest were amplified from HeLa and HCT116 cell cDNA libraries and inserted into pTOPO-vector using a TOPO TA cloning kit (Thermo Fisher Scientific, 45–0030).
For 3FLAG and 3HA tags, the genes were amplified from pTOPO-plasmids or plasmids purchased from Addgene (36456 and 24302; deposited by Noboru Mizushima, 19922; deposited by Yue Xiong, 11185; deposited by William Sellers, 70446; deposited by Dominic Esposito, 19723; deposited by Channing Der, 38318; deposited by Quan Lu, 11771; deposited by Sankar Ghosh, 20648; deposited by William Hahn and Jean Zhao) or RIKEN BioResource Research Center (IRAL043P19, IRAL002B07, IRAL012J06, IRAK026K16, ARiS122O22) using a Phusion High-Fidelity PCR system (Thermo Fisher Scientific, F-530L). Products were inserted using the Gibson Assembly system (New England Biolabs, E2611S) or SLiCE [73] into the BamHI and EcoRI sites of vectors containing 3FLAG-TEV (TEV protease cleavage site: ENLYFQS), 3HA-TEV, TEV-3FLAG, or TEV-3HA coding sequences [32]. To express hAG or Ash fusion proteins for the fluoppi assay, genes encoding BCAS3 and C16orf70 were inserted into the BamHI and NotI sites of phAG-MCL (Medical & Biological Laboratories, AM-8011M) and pHA-Ash-MCL [32]. To construct BCAS3-2FKBP-HA for the FRB-FKBP dimerization assay, the BCAS3 coding sequence was inserted into the EcoRI and XbaI sites of the PINK1Δ110-YFP-FKBP plasmid [48], which has two tandem FKBP followed by a single HA-tag; insertion of the BCAS3 coding sequence between the EcoRI and XbaI sites replaced the PINK1Δ110-YFP sequence. The plasmid expressing FRB-FIS1 (i.e. the mitochondrial transmembrane domain of FIS1 fused to FRB) was a kind gift from Dr. Richard J. Youle. Point mutations or partial deletions were introduced either by primer-based direct PCR mutagenesis or by 2-step PCR followed by ligation using a DNA Ligation Kit (TaKaRa Bio, 6023). All the plasmids used in this study are listed in Table 1.
Table 1.
Plasmid DNAs used in this study
| # | Plasmid DNA | SOURCE |
|---|---|---|
| 1 | pMXs-puro retroviral vector | Cell Biolabs, RTV-012 |
| 2 | pBABE-puro retroviral vector | Addgene, 1764 (Depositing Labs: Hartmut Land, Jay Morgenstern, Bob Weinberg) |
| 3 | pcDNA3.1(+) | Invitrogen, V79020 |
| 4 | pMXs-puro_YFP | Yamano et al. 2020 |
| 5 | pMXs-puro_3FLAG-TEV | Yamano et al. 2020 |
| 6 | pMXs-puro_3HA-TEV | Yamano et al. 2020 |
| 7 | pMXs-puro_TEV-3FLAG | Yamano et al. 2020 |
| 8 | pMXs-puro_TEV-3HA | Yamano et al. 2020 |
| 9 | pBABE-puro_3FLAG-TEV | Yamano et al. 2020 |
| 10 | pBABE-puro_3HA-TEV | Yamano et al. 2020 |
| 11 | pBABE-puro_TEV-3FLAG | Yamano et al. 2020 |
| 12 | pBABE-puro_TEV-3HA | Yamano et al. 2020 |
| 13 | pcDNA3.1(+)_3FLAG-TEV | Yamano et al. 2020 |
| 14 | pcDNA3.1(+)_3HA-TEV | Yamano et al. 2020 |
| 15 | pcDNA3.1(+)_TEV-3FLAG | Yamano et al. 2020 |
| 16 | pcDNA3.1(+)_TEV-3HA | Yamano et al. 2020 |
| 17 | pAsh-MCL | Medical & Biological Laboratories, AM-8011M |
| 18 | pHA-Ash-MCL | Yamano et al. 2020 |
| 19 | phAG-MCL | Medical & Biological Laboratories, AM-8011M |
| 20 | pUMVC (Gag-Pol) | Kindly gifted from Dr. R.Youle Lab. |
| 21 | pCMV-VSV-G (VSV-G) | Kindly gifted from Dr. R.Youle Lab. |
| 22 | pCHAC/EYFP-MAP1LC3B-IRES-MCS2 | Yamano et al. 2014 |
| 23 | FRB-FIS1 | Lazarou et al. 2012 |
| 24 | pBMNz-YFP-PRKN | Lazarou et al. 2015 |
| 25 | pMXs-puro_3FLAG-ZFYVE1 | This study |
| 26 | pMXs-puro_3FLAG-WIPI1 | This study |
| 27 | pMXs-puro_3HA-ZFYVE1 | This study |
| 28 | pMXs-puro_3HA-WIPI1 | This study |
| 29 | pcDNA3.1(+)_3HA-ZFYVE1 | This study |
| 30 | pcDNA3.1(+)_3HA-WIPI1 | This study |
| 31 | pMXs-puro_GST-PRKN | This study |
| 32 | pMXs-puro_HA-PRKN | Matsuda et al. 2010 |
| 33 | pMXs-puro_3FLAG-BCAS3 | This study; gene cloned from cDNA library |
| 34 | pMXs-puro_BCAS3-3FLAG | This study |
| 35 | pMXs-puro_3FLAG-C16orf70 | This study; gene cloned from cDNA library |
| 36 | pMXs-puro_C16orf70-3FLAG | This study |
| 37 | pMXs-puro_3HA-BCAS3 | This study |
| 38 | pMXs-puro_C16orf70-3FLAG | This study |
| 39 | pMXs-puro_3FLAG-ATG2A | This study; gene cloned from pEGFP-C1-hAtg2A (Addgene, 36456; Depositing Lab: Noboru Mizushima) |
| 40 | pMXs-puro_3FLAG-ATG2AΔC | This study |
| 41 | pMXs-puro_3FLAG-CFAP298 | This study; gene cloned from cDNA library |
| 42 | pMXs-puro_3FLAG-CUL4B | This study; gene cloned from pcDNA3-myc3-CUL4B (Addgene, 19922; Depositing Lab: Yue Xiong) |
| 43 | pMXs-puro_3FLAG-DYNLL1 | This study; gene cloned from cDNA library |
| 44 | pMXs-puro_3FLAG-GGA3 | This study; gene cloned from 1574 pSG5L Flag HA GGA3 (Addgene, 11185; Depositing Lab: William Sellers) |
| 45 | pMXs-puro_3FLAG-PEBP1 | This study; gene cloned from R777-E162 Hs.PEBP1-nostop (Addgene, 70446; Depositing Lab: Dominic Esposito) |
| 46 | pMXs-puro_3FLAG-RALA | This study; gene cloned from pBabe puro RalA (short) wt (Addgene, 19723; Depositing Lab: Channing Der) |
| 47 | pMXs-puro_3FLAG-TRAPPC2 | This study; gene cloned from cDNA library |
| 48 | pMXs-puro_3FLAG-TSG101 | This study; gene cloned from pEF6.mCherry-TSG101 (Addgene, 38318; Depositing Lab: Quan Lu) |
| 49 | pMXs-puro_3FLAG-SORBS3 | This study; gene cloned from cDNA library |
| 50 | pMXs-puro_3FLAG-EHMT2 | This study; gene cloned from cDNA library |
| 51 | pMXs-puro_3FLAG-OCIAD2 | This study; gene cloned from cDNA library |
| 52 | pMXs-puro_3FLAG-PDCD10 | This study; gene cloned from cDNA library |
| 53 | pMXs-puro_PDCP10-3FLAG | This study |
| 54 | pMXs-puro_3FLAG-GSTK1 | This study; gene cloned from cDNA library |
| 55 | pMXs-puro_GSTK1-3FLAG | This study |
| 56 | pMXs-puro_3FLAG-MmCHMP5 | This study; gene cloned from pCINeo-mCHMP5 (Addgene, 11771; Depositing Lab: Sankar Ghosh) |
| 57 | pMXs-puro_MmCHMP5-3FLAG | This study |
| 58 | pMXs-puro_3FLAG-MYL6B | This study; gene cloned from cDNA library |
| 59 | pMXs-puro_MYL6B-3FLAG | This study |
| 60 | pMXs-puro_3FLAG-TRAPPC2L | This study; gene cloned from cDNA library |
| 61 | pMXs-puro_TRAPPC2L-3FLAG | This study |
| 62 | pMXs-puro_3FLAG-RAB24 | This study; gene cloned from cDNA library |
| 63 | pMXs-puro_VPS4B-3FLAG | This study; gene cloned from cDNA library |
| 64 | pMXs-puro_3FLAG-VPS4B | This study |
| 65 | pMXs-puro_3FLAG-MTMR6 | This study; gene cloned from cDNA library |
| 66 | pMXs-puro_MTMR6-3FLAG | This study |
| 67 | pMXs-puro_3FLAG-EI24 | This study; gene cloned from cDNA library |
| 68 | pMXs-puro_EI24-3FLAG | This study |
| 69 | pMXs-puro_3FLAG-SAR1B | This study; gene cloned from cDNA library |
| 70 | pMXs-puro_SAR1B-3FLAG | This study |
| 71 | pMXs-puro_3FLAG-CSTB | This study; gene cloned from cDNA library |
| 72 | pMXs-puro_CSTB-3FLAG | This study |
| 73 | pMXs-puro_3FLAG-EDC4 | This study; gene cloned from cDNA library |
| 74 | pMXs-puro_EDC4-3FLAG | This study |
| 75 | pMXs-puro_3FLAG-OPTN | Yamano et al. 2020 |
| 76 | pMXs-puro_3FLAG-SNX3 | This study; gene cloned from cDNA library |
| 77 | pMXs-puro_SNX3-3FLAG | This study |
| 78 | pMXs-puro_3FLAG-RIPK1 | This study; gene cloned from cDNA library |
| 79 | pMXs-puro_RIPK1-3FLAG | This study |
| 80 | pMXs-puro_3FLAG-COASY | This study; gene cloned from cDNA library |
| 81 | pMXs-puro_COASY-3FLAG | This study |
| 82 | pMXs-puro_3FLAG-NMT1 | This study; gene cloned from cDNA library |
| 83 | pMXs-puro_NMT1-3FLAG | This study |
| 84 | pMXs-puro_3FLAG-RAB1B | This study; gene cloned from cDNA library |
| 85 | pMXs-puro_3FLAG-EHD3 | This study; gene cloned from IRAL043P19 (RIKEN BioResource Research Center) |
| 86 | pMXs-puro_3FLAG-HGH1 | This study; gene cloned from IRAL002B07 (RIKEN BioResource Research Center) |
| 87 | pMXs-puro_HGH1-3FLAG | This study |
| 88 | pMXs-puro_3FLAG-YBX3 | This study; gene cloned from IRAL012J06 (RIKEN BioResource Research Center) |
| 89 | pMXs-puro_YBX3-3FLAG | This study |
| 90 | pMXs-puro_3FLAG-HDHD5 | This study; gene cloned from IRAK026K16 (RIKEN BioResource Research Center) |
| 91 | pMXs-puro_HDHD5-3FLAG | This study |
| 92 | pMXs-puro_3FLAG-WHAMM | This study; gene cloned from ARiS122O22 (RIKEN BioResource Research Center) |
| 93 | pMXs-puro_WHAMM-3FLAG | This study |
| 94 | pMXs-puro_3FLAG-RAB1A | This study; gene cloned from cDNA library |
| 95 | pMXs-puro_3FLAG-MmATG16L1 | This study; gene cloned from p3xFLAG-CMV10-mApg16L (Addgene, 24302; Depositing Lab: Noboru Mizushima) |
| 96 | pMXs-puro_3FLAG-HYPK | This study; gene cloned from cDNA library |
| 97 | pMXs-puro_HYPK-3FLAG | This study |
| 98 | pMXs-puro_3FLAG-TBK1 | This study; gene cloned from pWZL Neo Myr Flag TBK1 (Addgene, 20648; Depositing Labs: William Hahn, Jean Zhao) |
| 99 | pMXs-puro_3HA-BCAS3H350A | This study |
| 100 | pMXs-puro_3HA-BCAS3D370A | This study |
| 101 | pMXs-puro_3HA-BCAS3L372A | This study |
| 102 | pMXs-puro_3HA-BCAS3H377A | This study |
| 103 | pMXs-puro_3HA-BCAS3H400A | This study |
| 104 | pMXs-puro_3HA-BCAS3R401A | This study |
| 105 | pMXs-puro_3HA-BCAS3R426A | This study |
| 106 | pMXs-puro_3HA-BCAS3T428A | This study |
| 107 | pMXs-puro_3HA-BCAS3H430A | This study |
| 108 | pMXs-puro_3HA-BCAS3 Δ254-312 | This study |
| 109 | pMXs-puro_3HA-BCAS3 Δ437–560 | This study |
| 110 | pcDNA3.1(+)_C16orf70-3HA | This study |
| 111 | pcDNA3.1(+)_C16orf70-3FLAG | This study |
| 112 | pcDNA3.1(+)_3HA-BCAS3 | This study |
| 113 | pcDNA3.1(+)_3HA-BCAS3(1–471) | This study |
| 114 | pcDNA3.1(+)_3HA-BCAS3(472–913) | This study |
| 115 | pcDNA3.1(+)_3HA-BCAS3(553–913) | This study |
| 116 | pcDNA3.1(+)_3HA-BCAS3(645–913) | This study |
| 117 | pcDNA3.1(+)_3HA-BCAS3(472–913 Δ680–719) | This study |
| 118 | pcDNA3.1(+)_3HA-BCAS3(553–913 Δ680-719) | This study |
| 119 | pcDNA3.1(+)_3HA-BCAS3(645–913 Δ680-719) | This study |
| 120 | pMRX-IP_GFP-mULK1 | Kindly gifted from Dr. Mizushima Lab. |
| 121 | pMXs-puro_GFP-mZFYVE1 | Kindly gifted from Dr. Mizushima Lab. |
| 122 | pMXs-puro_GFP-WIPI1 | Kindly gifted from Dr. Mizushima Lab. |
| 123 | pHA-Ash_C16orf70 | This study |
| 124 | phAG-BCAS3 | This study |
| 125 | pC4M-F2E-3HA-BCAS3-2FKBP-HA | This study |
| 126 | phAG-BCAS3(1–644) | This study |
| 127 | phAG-BCAS3(645–913) | This study |
| 128 | phAG-BCAS3(1–471) | This study |
| 129 | phAG-BCAS3(472–913) | This study |
| 130 | phAG-BCAS3(553–913) | This study |
| 131 | phAG-BCAS3 Δ472–559 | This study |
| 132 | phAG-BCAS3 Δ560–599 | This study |
| 133 | phAG-BCAS3 Δ600–639 | This study |
| 134 | phAG-BCAS3 Δ640–679 | This study |
| 135 | phAG-BCAS3 Δ680-719 | This study |
| 136 | phAG-BCAS3 Δ720–759 | This study |
| 137 | phAG-BCAS3 Δ760–799 | This study |
| 138 | phAG-BCAS3 Δ800–839 | This study |
| 139 | phAG-BCAS3 Δ840–879 | This study |
| 140 | phAG-BCAS3 Δ880–913 | This study |
| 141 | phAG-BCAS3(472–913 Δ680-719) | This study |
| 142 | phAG-BCAS3(553–913 Δ680-719) | This study |
| 143 | phAG-BCAS3(645–913 Δ680-719) | This study |
| 144 | phAG-BCAS3H350A | This study |
| 145 | phAG-BCAS3D370A | This study |
| 146 | phAG-BCAS3L372A | This study |
| 147 | phAG-BCAS3H377A | This study |
| 148 | phAG-BCAS3H400A | This study |
| 149 | phAG-BCAS3R401A | This study |
| 150 | phAG-BCAS3R426A | This study |
| 151 | phAG-BCAS3T428A | This study |
| 152 | phAG-BCAS3H430A | This study |
| 153 | phAG-BCAS3 Δ254-312 | This study |
| 154 | phAG-BCAS3 Δ437–560 | This study |
| 155 | pBABE-puro_Untag-PRKN | Yamano et al. 2020 |
| 156 | pCHAC MCS1-IRES-MCS2 | Allele Biotechnology and Pharmaceuticals, ABP-PVL-BICISIRES |
| 157 | pCHAC_C16orf70-3HA-IRES-MCS2 | This study |
| 158 | pCHAC_C16orf70-3HA-IRES-BCAS3 | This study |
| 159 | pCHAC_C16orf70-3HA-IRES-BCAS3R401A | This study |
| 160 | pRetroQ-mt-Keima | Kindly gifted from Dr. Youle Lab. |
| 161 | pMXs-puro_3FLAG-mULK1 | This study |
| 162 | pET21a vector | Novagen, 69740 |
| 163 | pET21a_BCAS3(645-913)-6×His | This study |
| 164 | pT7-7_GFP-His | Kindly gifted from Dr. Iwai Lab. |
| 165 | pcDNA3.1(+)_GFP-6xHis | This study |
| 166 | pcDNA3.1(+)_GFP-WIPI1-6×His | This study |
| 167 | pcDNA3.1(+)_GFP-BCAS3-6×His | This study |
| 168 | pcDNA3.1(+)_GFP-BCAS3R401A-6×His | This study |
| 169 | pcDNA3.1(+)_GFP-C16orf70-6×His | This study |
Plasmid transfection
For transient expression, plasmid complexes with FuGENE6 (Promega, E269A) in OPTI-MEM (Thermo Fisher Scientific, 31985–070) were generated according to the manufacture’s instruction and then transfected into cells for 24 h or more. For protein purification from HEK293T cells, polyethylenimine (Polysciences, 23966; PEI) was used as the transfection reagent. For HEK293T cells seeded in one 10-cm dish, 10–15 µg of plasmids were mixed with PEI at a ratio of 1:8 in OPTI-MEM and then transfected into cells for 48 h.
For stable expression, retroviral plasmids were cotransfected with Gag-pol and VSV-G plasmids into HEK293T cells grown in a 6-well plate using Lipofectamine LTX (Thermo Fisher Scientific, 15338100). After 12 h of transfection, the culture medium was replaced with fresh medium supplemented with HEPES (Gibco, 15630–080) and the cells were cultivated for another 24 h. Supernatants were filtered and infected into target cells using 8 µg/ml polybrene (Sigma-Aldrich, H9268). After infection for 24 h, the media was replaced with fresh media.
RNA interference
siRNAs for a non-target control (Thermo Fisher Scientific, D-001206-13-20; siGENOME Control siRNApool) or PINK1 [74] were transfected into cells at a final concentration of 10 nM using Lipofectamine RNAi MAX (Thermo Fisher Scientific, 13778150). Cells were analyzed 48 h after transfection.
Immunofluorescence microscopy
Cells grown on 35-mm glass bottom dishes were fixed with 4% paraformaldehyde (Wako, 163–20145; PFA) in PBS for 15 min at room temperature. Cells were then permeabilized with 0.15% (v:v) Triton X-100 (MP Biomedicals, 807426) or 50 µg/ml digitonin (Wako, 043–21371; for observing MAP1LC3B) for 18 min at room temperature, blocked with 0.1% (w:v) gelatin (Sigma-Aldrich, G9391) in PBS for 30 min, and incubated with primary antibodies in 0.1% gelatin for 2 h at room temperature. After washing with PBS containing polyoxyethylene sorbitan monolaurate (nacalai tesque, 35624–02; Tween20) (PBS-T), cells were incubated with secondary antibodies conjugated to Alexa Fluor for 1 h. For primary antibodies, anti-FLAG/DDDDK (Medical & Biological Laboratories, M185-3L), anti-FLAG/DDDDK (Medical & Biological Laboratories, PM020), anti-TOMM20 (Santa Cruz Biotechnology, sc-17764), anti-TOMM20 (Santa Cruz Biotechnology, sc-11415), anti-BCAS3 (this study), anti-HA (Medical & Biological Laboratories, M180-3), anti-HA (Roche, 11867423001), anti-PRKN (Sigma-Aldrich, MAB5512), anti-WIPI2 (Sigma-Aldrich, SAB4200400), anti-ATG13 (Cell Signaling Technologies, 13468), anti-ATG16L1 (Medical & Biological Laboratories, PM040), anti-mtDNA (PROGEN Biotechnik, AC-30-10) and anti-MAP1LC3 (Medical & Biological Laboratories, PM036) were used. For secondary antibodies, Alexa Fluor 488-, 568-, or 647-conjugated goat anti-mouse or anti-rabbit IgG antibodies (Life Technologies, A11034, A11036, A21245, A11029, A11031, A21236) were used. 4ʹ,6-diamidino-2-phenylindole (Invitrogen, D3571; DAPI) was used for nuclear staining. The images were acquired using an inverted confocal microscope (Carl Zeiss; LSM780) with a Plan-Apochromat 63×/1.4 oil lens. DAPI fluorescence was obtained using excitation with a 405 nm laser, whereas fluorescence for YFP, GFP and hAG was obtained using excitation with a 488 nm laser. Other fluorescence signals were obtained using filter sets of the appropriate wavelengths. For image analysis, ZEN microscope software (Carl Zeiss) and Photoshop (Adobe) were used. Z-stack images were constructed from 7–10 images captured in multiple focal planes with a step interval of 1.0 µm.
A TCS SP8 laser scanning confocal microscope (Leica Microsystems) with an HC PLAPO CS2 63×/1.4 oil lens was also used for Figure 5I and 5J. Multiple (34–37) confocal images (pixel size: 45 nm × 45 nm) with a 130 nm z-step size were taken and Huygens Professional deconvolution performed followed by maximum projection.
Western blotting and CBB staining
To briefly confirm protein expression, cells grown in a 6-well plate were washed with PBS and lysed with the appropriate amount of SDS-PAGE sample buffer supplemented with 1,4-dithiothreitol (Roche, 10708984001; DTT). Lysates were boiled at 98°C for 5 min and genomic DNA shredded by sonication. To obtain the total cell lysate, cells grown in a 6-well plate or a 6-cm dish were washed with PBS, collected and solubilized with TNE-N+ buffer (20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% [v:v] Nonidet® P40 [nacalai tesque, 25223–75; NP-40] and a protease inhibitor cocktail [Roche, 11873580001]). After removing insoluble debris by centrifugation at 12,000 × g, 4°C for 15 min, the protein concentration of the supernatants was measured using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23227). The supernatants were mixed with SDS-PAGE sample buffer and boiled at 98°C for 5 min.
Proteins were separated on 4–12% or 12% Bis-Tris SDS–PAGE gels (Invitrogen; NuPAGE) in MES or MOPS buffer, and then transferred to PVDF membranes (Merck Millipore, IPVH00010). Membranes were blocked with 2.5% (w:v) skim-milk (BD Biosciences, 232100) in tris buffered saline with Tween20 (TBS-T) for 30 min, then incubated with primary antibodies for 1.5–2 h, and secondary antibodies for 1 h at room temperature. For primary antibodies, anti-FLAG/DDDDK (Medical & Biological Laboratories, M185-3L), anti-FLAG/DDDDK (Medical & Biological Laboratories, PM020), anti-TOMM20 (Santa Cruz Biotechnology, sc-11415), anti-HA (Medical & Biological Laboratories, M180-3), anti-HA (Roche, 11867423001), anti-GFP (abcam, ab6556), anti-MFN2 (abcam, ab56889), anti-MT-CO2 (abcam, ab110258), anti-PDHA1 (abcam, ab110334), anti-BCAS3 (this study), anti-ATG13 (Cell Signaling Technology, 13468), anti-RB1CC1 (Cell Signaling Technology, 12436S; D10D11), anti-ATG16L (Medical & Biological Laboratories, PM040), anti-MAP1LC3B (Sigma-Aldrich, L7453), anti-WIPI2 (Sigma-Aldrich, SAB4200400), anti-azami-green (Medical & Biological Laboratories, PM011M), anti-ACTA1 (Merck Millipore, MAB1501R; C4) or anti-GAPDH (Merck Millipore, MAB374) antibodies were used. Secondary antibody detection was carried out using a BCIP-NBT Solution Kit for Alkaline Phosphatase Stain (nacalai tesque, 03937–60) with alkaline phosphatases conjugated to goat anti-mouse (Santa Cruz Biotechnology, sc-2008) or goat anti-rabbit IgG (Santa Cruz Biotechnology, sc-2007). Secondary antibody detection done with a Western Lightning Plus ECL (PerkinElmer, NEL105001EA) involved horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Promega, W402B), goat anti-rabbit IgG (Jackson Immuno Research Laboratories, 111–035-144), donkey anti-rat IgG (Jackson Immuno Research Laboratories, 712–035-153), or donkey anti-goat IgG (Santa Cruz Biotechnology, sc-2020). Proteins were detected in the linear range of detection using an ImageQuant LAS 4000 instrument (GE Healthcare Life Sciences).
For immunoblot-based detection of protein-protein interactions, cells grown in a 6-well plate or 6-cm dish were washed with PBS, and solubilized on ice for 15 min in IP buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.5% [w:v] sodium deoxycholate [Sigma-Aldrich, D6750], 1% [v:v] Triton X-100, and a protease inhibitor cocktail, 10 µM MLN7243 [Active Biochem, A-1384]). Unsolubilized debris was removed by centrifugation at 16,000 × g, 4°C for 10 min. A fraction of the supernatants were incubated with anti-DDDDK-tag mAb magnetic beads for 1.5–2 h at 4°C under gentle rotation; the remaining (30% against the bound fraction) was collected as input. Beads were washed with IP buffer three times and bound proteins were eluted with SDS-PAGE sample buffer. The supernatant inputs were precipitated with trichloroacetic acid (Wako, 200–08085) and solubilized with SDS-PAGE sample buffer.
For Coomassie Brilliant Blue (CBB) staining, proteins were separated on 4–12% Bis-Tris SDS–PAGE gels in MES buffer, and gels were incubated in a destaining solution (30% methanol, 10% acetic acid) for 10 min at room temperature. After four 10 min water washes, the proteins were detected using CBB Stain One (nakalai tesque, 04543–51).
LC-MS/MS-based identification of ZFYVE-1 or WIPI1-associated proteins
Cells stably expressing FLAG-tagged proteins grown in a 10-cm dish were treated with or without 10 µM valinomycin for 3 h, and then treated with 0.1% PFA for 10 min at room temperature. After PFA fixation was quenched with 300 mM glycine, cells were washed with PBS, and solubilized on ice for 10 min in RIPA buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% [w:v] SDS, 0.5% [w:v] sodium deoxycholate, 1% [v:v] Triton X-100, and a protease inhibitor cocktail). After centrifugation at 16,000 × g, 4°C for 10 min, the supernatants were incubated with anti-DDDDK-tag mAb magnetic beads (Medical & Biological Laboratories, M185-11) for 1.5 h at 4°C under gentle rotation. The beads were collected using a magnetic stand, washed three times with RIPA buffer, and then another three times with 50 mM ammonium bicarbonate. Proteins bound to the beads were digested with a 200 ng trypsin/Lys-C mix (Promega, V5072) for 16 h at 37°C. The digests were acidified with trifluoroacetic acid (Wako, 206–10731; TFA) and desalted using GL-Tip SDB (GL Sciences, 7820–11200). The eluates were evaporated in a SpeedVac concentrator and dissolved in 0.1% TFA.
LC-MS/MS analysis of the resultant peptides was performed using an EASY-nLC 1200 UHPLC connected to a Q Exactive Plus mass spectrometer equipped with a nanoelectrospray ion source (Thermo Fisher Scientific). The peptides were separated on a 75-µm inner diameter × 150-mm C18 reversed-phase column (Nikkyo Technos, 75-3-155) using a linear 4–28% acetonitrile (ACN) gradient for 0–100 min followed by an increase to 80% ACN for 10 min. The mass spectrometer was operated in a data-dependent acquisition mode with a top 10 MS/MS method. MS1 spectra were measured at a resolution of 70,000, an automatic gain control (AGC) target of 1 × 106 and a mass range from 350 to 1,500 m/z. HCD MS/MS spectra were acquired at a resolution of 17,500, an AGC target of 5 × 104, an isolation window of 2.0 m/z, a maximum injection time of 60 ms and a normalized collision energy of 27. Dynamic exclusion was set to 10 s. Raw data were directly analyzed against the SwissProt database restricted to H. sapiens using Proteome Discoverer version 2.2 (Thermo Fisher Scientific) with Mascot search engine version 2.5 (Matrix Science) used for identification and label-free precursor ion quantification. The search parameters were as follows: (i) trypsin as an enzyme with up to two missed cleavages; (ii) precursor mass tolerance of 10 ppm; (iii) fragment mass tolerance of 0.02 Da; and (iv) acetylation of the protein N-terminus and oxidation of methionine as variable modifications. Peptides and proteins were filtered at a false discovery rate (FDR) of 1% using the percolator node and the protein FDR validator node, respectively. Normalization was performed such that the total sum of abundance values for each sample over all peptides was the same.
Generation of knockout (KO) cell lines
BCAS3-/- and C16orf70-/- HCT116 cell lines were established using a CRISPR-Cas9-based genome editing method with an antibiotic-selection strategy described previously [75]. The guide-RNAs (gRNA) targeted exon 2 in the BCAS3 gene (for line #1: ACTCCACCAGTACAACGACTGGG, for line #2: AGAAGACCCAGTCGTTGTACTGG and for line #3: TGGAGTTGTGGTTCGCCCCCAGG) or exon 1 in the C16orf70 gene (for line #1: GTAGTGCCCGAACGCTCTCTGGG and for line #2: GGTAGTGCCCGAACGCTCTCTGG). The gRNAs were designed using an online CRISPR design tool: Guide Design Resources (https://zlab.bio/guide-design-resources). Three pairs of DNA oligonucleotides: BCAS3-ex2-1-CRISPR-F/R (5ʹ-TGT ATG AGA CCA CAC TCC ACC AGT ACA ACG ACT-3ʹ, 5ʹ-AAA CAG TCG TTG TAC TGG TGG AGT GTG GTC TCA-3ʹ), BCAS3-ex2-2-CRISPR-F/R (5ʹ-TGT ATG AGA CCA CAG AAG ACC CAG TCG TTG TAC-3ʹ, 5ʹ-AAA CGT ACA ACG ACT GGG TCT TCT GTG GTC TCA-3ʹ), and BCAS3-ex2-3-CRISPR-F/R (5ʹ-TGT ATG AGA CCA CTG GAG TTG TGG TTC GCC CCC-3ʹ, 5ʹ-AAA CGG GGG CGA ACC ACA ACT CCA GTG GTC TCA-3ʹ) for the BCAS3 KO, and two pairs of DNA oligonucleotides: C16orf70-ex1-1-CRISPR-F/R (5ʹ-TGT ATG AGA CCA CGT AGT GCC CGA ACG CTC TCT-3ʹ, 5ʹ-AAA CAG AGA GCG TTC GGG CAC TAC GTG GTC TCA −3ʹ), and C16orf70-ex1-2-CRISPR-F/R (5ʹ-TGT ATG AGA CCA CGG TAG TGC CCG AAC GCT CTC-3ʹ, 5ʹ-AAA CGA GAG CGT TCG GGC ACT ACC GTG GTC TCA-3ʹ) for the C16orf70 KO were annealed and inserted into a linearized pEF1-hspCas9-H1-gRNA vector (System Biosciences, CAS700A-1) according to the manufacturer’s instructions.
To construct donor plasmids, 500 bp sequences, which include 247 bp of the 5ʹ and 3ʹ of the gRNA targeting regions as homologous arms and a BamHI site in the middle instead of the gRNA sequence were synthesized in a pUC57-Amp vector (GENEWIZ). The neomycin-resistant gene (NeoR) and the hygromycin-resistant gene (HygroR), including the loxP sites along with the appropriate promoter and terminator [75] were inserted into the BamHI site of the pUC57-Amp plasmids. The resultant pEF1-hspCas9-H1-gRNA plasmids and pUC57-Amp donor plasmids containing NeoR or HygroR were transfected into HCT116 cells using FuGENE HD (Promega, E2311). After 48 h transfection, cells were grown in McCoy’s 5A media containing 700 µg/ml G418 (Sigma-Aldrich, G8168) and 100 µg/ml hygromycin B (Invitrogen, 10687–10). Single colonies were isolated into a 24-well plate. To verify neomycin-resistant and hygromycin-resistant gene insertion, genomic DNA was extracted using a Microprep Kit Quick-gDNA (Zymo Research, D3021) and PCR amplified with primer pairs for BCAS3; BACS3-ex2-check-F2/R2 (5ʹ-TCC CAT CCC ATG ATC TGG TTC-3ʹ, 5ʹ-TCT GAA ATG TGC TTA CGT GAC AG-3ʹ), and for C16orf70; C16orf70-ex1-check-F3/R3 (5ʹ-CGG CAC GCC ACT TTT ACT G-3ʹ, 5ʹ-AGG CAT TTG GGG AGA GCA AG-3ʹ).
BCAS3-/-C16orf70-/- DKO cell lines were established as follows. NeoR and HygroR markers were removed from BCAS3-/- HCT116 (clone #1-24) using Cre Recombinase Gesicles (TaKaRa Bio, 631449). BCAS3-/- HCT116 (clone #1-24) cells grown in a 12-well plate were treated with polybrene and Cre Recombinase Gesicles and centrifuged at 1,200 × g for 30 min. Single colonies were then isolated, and elimination of the NeoR and HygroR markers was confirmed by PCR. CRISPR-Cas9-based genome editing with an antibiotic-selection strategy was likewise used to establish the BCAS3-/-C16orf70-/- HCT116 cell lines.
Preparation of recombinant BCAS3 for antibody production
To purify proteins from E.coli, the human BCAS3 gene (residues 645–913) was amplified and inserted into the EcoRI/XhoI sites of a pET21a vector (Novagen, 69740), which introduced a 6×His C-terminal tag. pET21a/BCAS3(645–913aa) was then introduced into BL21-CodonPlus(DE3)-RIL (Agilent Technologies, 230245). The transformants were cultured in Terrific Broth media at 37°C until the logarithmic growth phase. Protein expression was induced with 1 mM Isopropylβ-D-1-thiogalactopyranoside (IPTG) for 3 h. The cells were collected by centrifugation, lysed with TBS buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 50 µg/ml lysozyme (Wako, 122–02673), 1 µg/ml DNaseI (Worthington Biochemical Corporation, 26P9235), 1 mM DTT, 1 mM MgCl2, a protease inhibitor cocktail) and sonicated. After centrifugation at 8,000 × g, 4°C for 15 min, the pellet was resuspended with Triton X-100 buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 0.1% [v:v] Triton X-100). The insoluble fraction containing recombinant BCAS3 was collected by centrifugation at 8,000 × g, 4°C for 15 min and solubilized with urea buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 6 M urea [GE Healthcare Life Sciences, 17–1319-01]) and incubated with Ni-NTA agarose (QIAGEN, 30250) for 30 min at 4°C under gentle rotation. The agarose was washed three times with urea buffer, and bound proteins were step-wise eluted with urea buffer containing 40, 80, and 200 mM imidazole. Recombinant His-tagged BCAS3 (residues 645–913) was concentrated using an Amicon-Ultra-15 (Merck Millipore, UFC901024), and imidazole was removed using PD midiTrap G-25 (GE Healthcare Life Sciences, 28–9180-08). Anti-BCAS3 antiserum was acquired by immunizing rabbits with the purified BCAS3(645–913aa)-6×His and the anti-BCAS3 antibody was purified using CNBr-activated sepharose 4B coupled to recombinant BCAS3(645–913aa) (Eurofins Genomics).
Preparation of recombinant proteins for liposome flotation assay
To generate purified recombinant GFP, Escherichia coli BL21-CodonPlus(DE3)-RIL (Agilent Technologies) cells harboring pT7-7/GFP-His (a kind gift from Dr. Kazuhiro Iwai, Kyoto university, Japan) were grown in LB medium supplemented with 100 µg/ml ampicillin and 25 µg/ml chloramphenicol at 37°C. GFP-His was expressed at 23°C for 12 h by addition of 500 µM IPTG. The bacterial cell pellets were wash with TBS (50 mM Tris-HCl, pH 7.5, 120 mM NaCl), resuspended in TBS supplemented with lysozyme and a protease inhibitor cocktail, and then stored at −20°C until used. The frozen cell suspension was thawed and sonicated, and insoluble proteins were removed by centrifugation. The resultant supernatant was incubated with equilibrated Ni-NTA agarose resin for 15 min at 4°C. The resin was washed with TBS containing 10 mM imidazole, and the bound proteins were eluted with TBS containing 200 mM imidazole. The buffer was exchanged by dialyzing against TBS containing 10% (v:v) glycerol.
Purified GFP-WIPI1-6× His, GFP-BCAS3-6× His, GFP-BCAS3R401A-6× His and GFP-C16orf70-6× His were similarly generated. Human WIPI1, BCAS3 (WT and R401A) and C16orf70 genes were amplified with primers that included nucleotides encoding a 6× His tag and then inserted into the XhoI site of the pcDNA3.1(+) vector. To generate the N-terminal tag, GFP was amplified and inserted into the BamHI/EcoRI site of the pcDNA3.1(+) constructs.
pcDNA3.1(+)/GFP-WIPI1-6×His, pcDNA3.1(+)/GFP-BCAS3-6×His, pcDNA3.1(+)/GFP-BCAS3R401A-6×His, and pcDNA3.1(+)/GFP-C16orf70-6×His were transfected into HEK293T cells using PEI. After 48 h transfection, cells were harvested from twelve 10-cm dishes, washed with PBS and lysed with lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM DTT, and a protease inhibitor cocktail) for 15 min on ice. After the insoluble proteins were removed by centrifugation at 12,000 × g, 4°C for 10 min, the supernatants were incubated with equilibrated Ni-NTA agarose for 1 h at 4°C under gentle rotation. The resins were washed with TBS containing 1 mM DTT and 20 mM imidazole, and the bound proteins were eluted with TBS containing 500 mM imidazole. Eluted fractions were concentrated, and the buffer composition was gradually exchanged to TBS containing 10% (v:v) glycerol using an Amicon-Ultra-15.
Liposome preparation and liposome flotation assay
To prepare liposomes, the following lipids were used: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (Avanti Polar Lipids, 850725C; PE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, 850457C; PC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein) (Avanti Polar Lipids, 810332P; fluoPE), phosphatidylinositol 3-phosphate diC16 (Echelon Biosciences, P-3016; PtdIns3P), phosphatidylinositol 3,5-bisphosphate diC16 (Echelon Biosciences, P-3516; PtdIns(3,5)P2). PC, PE, and fluoPE were dissolved in chloroform. PtdIns3P and PtdIns(3,5)P2 were dissolved in chloroform:methanol:water (1:2:0.8). Lipids were mixed in a glass tube in the following molar ratios: control liposomes – 70% PC, 28% PE, and 2% fluoPE; 5% PtdIns3P or PtdIns(3,5)P2-containing liposomes – 5% PtdIns3P or PtdIns(3,5)P2, 65% PC, 28% PE, and 2% fluoPE; 15% PtdIns3P or PtdIns(3,5)P2-containing liposomes – 15% PtdIns3P or PtdIns(3,5)P2, 55% PC, 28% PE, and 2% fluoPE. To make lipid films, the organic solvent in the lipid mixtures was removed using a vacuum rotary evaporator. Lipid films were re-hydrated in liposome buffer (20 mM HEPES-KOH, pH 7.5, 150 mM KCl) at a 1 mM lipid concentration. To obtain liposomes, the film suspension was subjected to vigorous vortexing, repeated freeze-thaw cycles, sonication, and finally extrusion through 0.1 µm polycarbonate membranes (Avanti Polar Lipids, 610005–1EA). Each liposome solution was stored at 4°C until used.
Prior to the liposome flotation assay, 1.5-ml microcentrifuge tubes (Eppendorf Himac Technologies, S308892A) were incubated with blocking buffer (5% skim-milk, 1% BSA) for 30 min at room temperature, and washed with liposome buffer. A volume of 30 µl liposome (30 nmol lipids) and purified proteins (~1.5 pmol) were mixed in 50 µl of 10% OptiPrep buffer (20 mM HEPES-KOH, pH 7.5, 150 mM KCl, 10% OptiPrep [Abbott Diagnostics Technologies, 1114542]) in microcentrifuge tubes. After incubating for 45 min at 4°C, the liposome solutions were adjusted to 35% OptiPrep by adding 50 µl of 60% OptiPrep, and then 400 µl of 30% OptiPrep buffer (20 mM HEPES-KOH, pH 7.5, 150 mM KCl, 30% OptiPrep) and 100 µl of liposome buffer were sequentially overlaid (total 600 µl). Samples were centrifuged at 50,000 × g, 4°C for 2 h using a CS150GXII (Hitachi Koki). Liposomes were visually confirmed by fluoPE fluorescence. The 600 µl solution was divided into 3 equivalent fractions (i.e. bottom, middle, and top) by collecting 200 µl aliquots from the bottom of the tube using a micro-syringe (Hamilton). SDS-PAGE sample buffer was added for western blotting.
Quantification of autophagy foci following amino-acid starvation and mitophagy
All images for quantification were acquired under constant excitation strength using a confocal microscope (LSM780) and ZEN software. “The number of BCAS3 foci per cell”, “The relative area of BCAS3 foci per cell” in Figure 6E, “The relative area of foci per cell” in Figure 10E and Fig. S7E, and “The relative mtDNA intensity per cell” in Fig. S6B and S6D were calculated using the Analyze Particles program provided with ImageJ. Non-specific signals were removed by adjusting the lower threshold level. The number of DAPI stained nuclei in the same area were counted and considered as equivalent to the number of cells.
FACS-based mitophagy flux analysis
Cells stably expressing YFP-PRKN and mitochondria-targeted Keima (mt-Keima) were grown in a 6-well plate and treated with or without 4 µM antimycin A, 10 µM oligomycin and 10 µM Q-VD-OPh for 9 h. The cells were then washed with PBS and resuspended with PBS containing 2.5% (v:v) FBS in a polystyrene Round-Bottom Tube (Falcon, 352235). Analysis was performed using FACSDiva software on a BD LSRFortessa X-20 cell sorter (BD Biosciences). mKeima fluorescence was determined based on dual-excitation ratiometric pH measurements using 405-nm (pH 7) and 561-nm (pH 4) lasers with 610/20-nm emission filters. For each sample, 10,000 YFP/mt-Keima double positive cells were collected.
Statistical analysis
Statistical differences between control groups and experimental groups were determined as p-values (P) based on student’s t-test using data obtained from three independent experiments. (n.s.: not significant, *P < 0.05, **P < 0.01, ***P < 0.001)
Comparative modeling of WD40 repeat domain of BCAS3
The structural model of the BCAS3 WD40 repeat domain was constructed based on the K. lactis Hsv2 structure (PDB ID: 4EXV) using the homology model application in MOE 2018.0101 (Chemical Computing Group). The alignment used in the structural model was obtained based on an HHpred [60] prediction with global alignment mode. We used the WD40 repeat domain sequence without the two insertions (residues 254–312 and 437–560) as the query sequence for HHpred.
Supplementary Material
Acknowledgments
We thank Drs. Richard J. Youle and Chunxin Wang for ATG5-/- and ATG13-/- HCT116 cells, pUMVC (Gag-Pol), pCMV-VSV-G (VSV-G), FRB-FIS1, pBMNz-YFP-PRKN, and pRetroQ-mt-Keima plasmids, Dr. Noboru Mizushima for pMRX-IP_GFP-mULK1, pMXs-puro_GFP-mZFYVE1, pMXs-puro_GFP-WIPI1 plasmids, Dr. Kazuhiro Iwai for pT7-7/GFP-His, Ms. Megumi Kawano for LC-MS/MS analysis, Dr. Kosuke Tanegashima for FACS analysis, and Drs. Tetsuya Kotani and Hitoshi Nakatogawa for assistance with the liposome preparation and flotation assay.
Funding Statement
This work was supported by JSPS KAKENHI Grant JP17J03737 (to W.K.); by JSPS KAKENHI Grants JP18H05500 and JP18K06237 (to K.Y.); by JSPS KAKENHI Grants JP18KK0229, JP19H04966 and JP20K06628 (to H.K.); by JSPS KAKENHI Grants JP18H02443 and JP19H05712, the Chieko Iwanaga Fund for Parkinson’s Disease Research, the Takeda Science Foundation and Joint Usage and Joint Research Programs, the Institute of Advanced Medical Sciences, Tokushima University (to N.M.); by JSPS KAKENHI Grant Number JP19H00997 and the Takeda Science Foundation (to K.T.); by JSPS KAKENHI Grants 16K21680, 18K11543 and Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under grant numbers JP19am0101114 (to K.I).
Disclosure statement
No competing interests declared
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Galluzzi L, E H B, Ballabio A.. Molecular definitions of autophagy and related processes. Embo J. 2017;36(13):1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- [3].Feng Y, He D, Yao Z. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang B, Kundu M. Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol. 2017;45:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Noda NN, Fujioka Y. Atg1 family kinases in autophagy initiation. Cell Mol Life Sci. 2015;72(16):3083–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hayashi-Nishino M, Fujita N, Noda T. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433–1437. [DOI] [PubMed] [Google Scholar]
- [7].Axe EL, Walker SA, Manifava M. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Polson HE, de Lartigue J, Rigden DJ. Mammalian atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–522. [DOI] [PubMed] [Google Scholar]
- [9].Proikas-Cezanne T, Takacs Z, Donnes P. WIPI proteins: essential ptdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128(2):207–217. [DOI] [PubMed] [Google Scholar]
- [10].Mizushima N. The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2019;63:1–10. [DOI] [PubMed] [Google Scholar]
- [11].Koyama-Honda I, Tsuboyama K, Mizushima N. ATG conjugation-dependent degradation of the inner autophagosomal membrane is a key step for autophagosome maturation. Autophagy. 2017;13(7):1252–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–174. [DOI] [PubMed] [Google Scholar]
- [13].Thumm M, Egner R, Koch B. Isolation of autophagocytosis mutants of saccharomyces cerevisiae. FEBS Lett. 1994;349(2):275–280. [DOI] [PubMed] [Google Scholar]
- [14].T M H, K A M, S V S. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131(3):591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tian Y, Li Z, Hu W. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141(6):1042–1055. [DOI] [PubMed] [Google Scholar]
- [16].Orvedahl A, Sumpter R Jr., Xiao G. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Morita K, Hama Y, Izume T. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J Cell Biol. 2018;217(11):3817–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Herhaus L, Dikic I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015;16(9):1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol. 2016;26(10):733–744. [DOI] [PubMed] [Google Scholar]
- [20].Yamano K, Matsuda N, Tanaka K. The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016;17(3):300–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in parkinson’s disease. Neuron. 2015;85(2):257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Matsuda N, Sato S, Shiba K. PINK1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Narendra DP, Jin SM, Tanaka A. PINK1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010;8(1):e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kane LA, Lazarou M, Fogel AI. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205(2):143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Koyano F, Okatsu K, Kosako H. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510(7503):162–166. [DOI] [PubMed] [Google Scholar]
- [26].Kazlauskaite A, Kondapalli C, Gourlay R. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at ser65. Biochem J. 2014;460(1):127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ordureau A, Sarraf SA, Duda DM. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell. 2014;56(3):360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sarraf SA, Raman M, Guarani-Pereira V. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vargas JNS, Wang C, Bunker E. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell. 2019;74(2):347–362 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ravenhill BJ, Boyle KB, von Muhlinen N. The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol Cell. 2019;74(2):320–329 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lazarou M, Sliter DA, Kane LA. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yamano K, Kikuchi R, Kojima W. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol. 2020;219(9):e201912144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Itakura E, Kishi-Itakura C, Koyama-Honda I. Structures containing atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of parkin-mediated mitophagy. J Cell Sci. 2012;125(6):1488–1499. [DOI] [PubMed] [Google Scholar]
- [34].Yamano K, Wang C, S A S. Endosomal rab cycles regulate parkin-mediated mitophagy. Elife. 2018;7:e31326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kast DJ, Zajac AL, Holzbaur EL. WHAMM directs the arp2/3 complex to the ER for autophagosome biogenesis through an actin comet tail mechanism. Curr Biol. 2015;25(13):1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Coutts AS. La thangue N B. actin nucleation by WH2 domains at the autophagosome. Nat Commun. 2015;6(1):7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wild P, Farhan H, McEwan DG. Phosphorylation of the autophagy receptor optineurin restricts salmonella growth. Science. 2011;333(6039):228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42):E4439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zoppino FC, Militello RD, Slavin I. Autophagosome formation depends on the small gtPase rab1 and functional ER exit sites. Traffic. 2010;11(9):1246–1261. [DOI] [PubMed] [Google Scholar]
- [40].Winslow AR, Chen CW, Corrochano S. alpha-synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol. 2010;190(6):1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Munafo DB, Colombo MI. Induction of autophagy causes dramatic changes in the subcellular distribution of GFP-rab24. Traffic. 2002;3(7):472–482. [DOI] [PubMed] [Google Scholar]
- [42].Jain M, Bhat GP, Vijayraghavan K. Rudhira/BCAS3 is a cytoskeletal protein that controls cdc42 activation and directional cell migration during angiogenesis. Exp Cell Res. 2012;318(6):753–767. [DOI] [PubMed] [Google Scholar]
- [43].Siva K, Venu P, Mahadevan A. Human BCAS3 expression in embryonic stem cells and vascular precursors suggests a role in human embryogenesis and tumor angiogenesis. PLoS One. 2007;2(11):e1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shetty R, Joshi D, Jain M. Rudhira/BCAS3 is essential for mouse development and cardiovascular patterning. Sci Rep. 2018;8(1):5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rukova B, Staneva R, Hadjidekova S. Whole genome methylation analyses of schizophrenia patients before and after treatment. Biotechnol Biotechnol Equip. 2014;28(3):518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yamano K, Queliconi BB, Koyano F. Site-specific interaction mapping of phosphorylated ubiquitin to uncover parkin activation. J Biol Chem. 2015;290(42):25199–25211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Watanabe T, Seki T, Fukano T. Genetic visualization of protein interactions harnessing liquid phase transitions. Sci Rep. 2017;7(1):46380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lazarou M, Jin SM, Kane LA. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase parkin. Dev Cell. 2012;22(2):320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Narendra D, Tanaka A, Suen DF. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ganley IG, Lam Du H, Wang J. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hosokawa N, Hara T, Kaizuka T. Nutrient-dependent mTORC1 association with the ULK1-atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Valverde DP, Yu S, Boggavarapu V. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol. 2019;218(6):1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Maeda S, Otomo C, Otomo T. The autophagic membrane tether ATG2A transfers lipids between membranes. Elife. 2019;,8:e4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tamura N, Nishimura T, Sakamaki Y. Differential requirement for ATG2A domains for localization to autophagic membranes and lipid droplets. FEBS Lett. 2017;591(23):3819–3830. [DOI] [PubMed] [Google Scholar]
- [55].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–1269. [DOI] [PubMed] [Google Scholar]
- [56].Kishi-Itakura C, Koyama-Honda I, Itakura E. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J Cell Sci. 2014;127(18):4089–4102. [DOI] [PubMed] [Google Scholar]
- [57].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian atg proteins. Autophagy. 2010;6(6):764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jain BP, Pandey S. WD40 repeat proteins: signalling scaffold with diverse functions. Protein J. 2018;37(5):391–406. [DOI] [PubMed] [Google Scholar]
- [59].Stirnimann CU, Petsalaki E, Russell RB. WD40 proteins propel cellular networks. Trends Biochem Sci. 2010;35(10):565–574. [DOI] [PubMed] [Google Scholar]
- [60].Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33(Web Server):W244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Krick R, Tolstrup J, Appelles A. The relevance of the phosphatidylinositolphosphat-binding motif FRRGT of atg18 and atg21 for the cvt pathway and autophagy. FEBS Lett. 2006;580(19):4632–4638. [DOI] [PubMed] [Google Scholar]
- [62].Nair U, Cao Y, Xie Z. Roles of the lipid-binding motifs of atg18 and atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J Biol Chem. 2010;285(15):11476–11488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Baskaran S, Ragusa MJ, Boura E. Two-site recognition of phosphatidylinositol 3-phosphate by PROPPINs in autophagy. Mol Cell. 2012;47(3):339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Krick R, Busse RA, Scacioc A. Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a beta-propeller protein family. Proc Natl Acad Sci U S A. 2012;109(30):E2042–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Katayama H, Kogure T, Mizushima N. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol. 2011;18(8):1042–1052. [DOI] [PubMed] [Google Scholar]
- [66].Gillooly DJ, Morrow IC, Lindsay M. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. Embo J. 2000;19(17):4577–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Filimonenko M, Isakson P, Finley KD. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein alfy. Mol Cell. 2010;38(2):265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhao YG, Sun L, Miao G. The autophagy gene wdr45/wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015;11(6):881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hara T, Nakamura K, Matsui M. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. [DOI] [PubMed] [Google Scholar]
- [70].Komatsu M, Waguri S, Chiba T. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884. [DOI] [PubMed] [Google Scholar]
- [71].Yamada Y, Schaap P. The proppin bcas3 and its interactor kinkyA localize to the early phagophore and regulate autophagy. Autophagy. 2020;1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Joshi D, Inamdar MS. Rudhira/BCAS3 couples microtubules and intermediate filaments to promote cell migration for angiogenic remodeling. Mol Biol Cell. 2019;30(12):1437–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhang Y, Werling U, Edelmann W. Seamless Ligation Cloning Extract (SLiCE) cloning method. Methods Mol Biol. 2014;1116:235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Okatsu K, Uno M, Koyano F. A dimeric PINK1-containing complex on depolarized mitochondria stimulates parkin recruitment. J Biol Chem. 2013;288(51):36372–36384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Koyano F, Yamano K, Kosako H. Parkin-mediated ubiquitylation redistributes MITOL/march5 from mitochondria to peroxisomes. EMBO Rep. 2019;20(12):e47728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Buchan DWA, Jones DT. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res. 2019;47(W1):W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jones DT, Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity. Bioinformatics. 2015;31(6):857–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.