

ABSTRACT
The Atg3 protein is highly homologous from yeast to human. Atg3 functions as an E2-like enzyme promoting conjugation of Atg8-family proteins to phosphatidylethanolamine (PE), a lipid molecule embedded in the growing phagophore membrane during stress-induced autophagy. Over the last decade, Atg3 became one of the most explored autophagy proteins, resulting in observations that provided specific insights into the structural mechanisms of its function. In this article, we describe a recent study by Ye et al. that reveals, using the human ATG3, how the membrane binding capability of the enzyme is tightly linked to its conjugation activity. We summarize the current knowledge on important mechanisms that involve protein-protein or protein-membrane interactions of Atg3 and that ultimately lead to efficient Atg8–PE conjugation.
Abbreviations: AH: amphipathic helix; FR: flexible region; HR: handle region; NMR: nuclear magnetic resonance
KEYWORDS: Circular dichroism, LC3B lipidation, liposomes, NMR spectroscopy, protein intrinsic disorder
Covalent attachment of Atg8-family proteins to the lipid phosphatidylethanolamine (PE) is an indispensable step in growth of the phagophore, a double-membrane, cup-shaped precursor of the organelle named the autophagosome, which is built de novo during stress-induced macroautophagy (hereafter autophagy). The Atg8–PE conjugation is executed by a cascade of ubiquitin-like reactions involving several proteins of the core autophagy machinery [1,2]. Briefly, this cascade is initiated by the cysteine protease Atg4 (yeast)/ATG4 (more complex eukaryotes) that cleaves arginine, or a few residues, to expose glycine at the C terminus of Atg8/LC3/GABARAP ubiquitin-like proteins. Afterward, Atg7/ATG7, acting as an E1-like enzyme, activates the Atg8-family proteins as well as Atg12/ATG12, another ubiquitin-like substrate, and delivers each to a corresponding E2-like conjugating enzyme, Atg3/ATG3 or Atg10/ATG10. The Atg10/ATG10 enzyme conjugates the C terminal glycine of Atg12/ATG12 to a single conserved lysine of Atg5/ATG5 in an E3-independent manner. The Atg12−Atg5/ATG12−ATG5 conjugate noncovalently binds Atg16/ATG16L1 and, together, the complex functions as an E3-like enzyme in the conjugation of Atg8-family proteins to PE. Specifically, Atg12/ATG12 in the E3-like complex binds Atg3/ATG3, which promotes transfer of the C terminal glycine in Atg8-family proteins from the catalytic cysteine in Atg3/ATG3 to the amino group of PE on the phagophore membrane [3]. The catalytic cysteine in Atg3/ATG3 is located in the protein core from which several intrinsically disordered domains protrude [4]. These flexible regions are enriched in many negatively charged residues, which together yield the acidic and dynamic enzyme [5–7]. A long flexible region (FR) or loop of approximately 80 amino acids functions in cooperation with the handle region (HR) as an allosteric regulator of Atg3 activity [8]. A short disordered region at the very N terminus of Atg3/ATG3 is foldable into an amphipathic helix that targets the protein to membranes, and makes the conjugation activity membrane curvature dependent [9,10]. How the conjugation activity of Atg3/ATG3 is linked to curvature-dependent membrane binding was so far elusive. The study by Ye et al. [11], described in this article, provides a better understanding of this mechanism.
The authors of this study combined several biophysical and biochemical approaches to show that Atg3/ATG3 proteins from several organisms, including yeast and humans, carry a conserved region at the N terminus that couples their membrane binding capability with conjugation activity. This conserved region of ten amino acids with a consensus motif EYLTP is preceded by a variable region that binds to membranes. Using circular dichroism, Ye et al. showed that intrinsically disordered peptides corresponding to the variable region in human ATG3 or yeast Atg3 fold into an amphipathic helix in the presence of highly curved (sonicated) liposomes, but stay disordered when mixed with extruded liposomes of 50 nm.
A single residue substitution (P21A) in the conserved region of human ATG3 along with chimeric human ATG3 proteins, created by replacing the human variable, conserved, or entire N terminal ATG3 region with those of yeast Atg3, revealed in the in vitro conjugation assays that the conserved region is essential for ATG3 function. Because all these mutants interact with liposomes and create a normal covalent intermediate with LC3B as does the wild-type protein, membrane binding of human ATG3 is necessary but not sufficient for promoting LC3B–PE/LC3-II conjugation on liposomes in vitro. Probing the ATG3P21A variant in atg3−/− mouse embryonic fibroblasts shows a significant defect in LC3B lipidation and autophagic flux in vivo, further supporting the in vitro data.
To visually understand how the N-terminal conserved region in Atg3/ATG3 proteins can affect their conjugation activity executed near the C terminus, Ye et al. applied NMR spectroscopy on human ATG3. They used a mutant carrying a deletion in the 90–190 region, which corresponds to the FR loop that is essential for interaction with ATG7. A “double-edged sword” often unavoidable in studies of proteins containing disordered regions is manifested in ATG3[∆90-190] by improved spectral quality but a significantly reduced LC3B–PE conjugation activity and inefficient formation of the ATG3–LC3B covalent intermediate, as compared to the full-length protein. Fortunately, the conjugation activity of ATG3[∆90-190] can be improved by increasing the concentration of ATG7 in the reaction buffer. Furthermore, the P21A mutation in the 90–190 deletion construct shows decreased conjugation activity as it does in the full-length protein. Testing ATG3 in the in vitro conjugation assays using various membrane mimetics (micelles, bicelles, or nanodiscs), to replace liposomes in the NMR experiments, revealed that only bicelles yield LC3B–PE conjugation activity comparable to that with liposomes. Therefore, bicelles were used for NMR structural analysis of ATG3[∆90-190] and ATG3[∆90-190]P21A.
An overlay of TROSY spectra obtained for ATG3[∆90-190] with and without bicelles revealed significant spectral changes, with approximately 80% of ATG3 residues assigned in the bicelles-bound form. 15N and 1H chemical shift perturbations upon binding to bicelles are observed not only in the N-terminal region, as expected due to membrane binding, but also in the C-terminal region. Although the residues around the active site and catalytic cysteine C264 cannot be assigned, the result of this NMR analysis showed that structural changes in human ATG3 upon membrane binding go beyond rearrangements associated with formation of the N-terminal amphipathic helix.
To better understand these changes, Ye et al. measured TROSY spectra for ATG3[∆90-190]P21A, a mutant disrupted in the conserved N-terminal region of ATG3. 15N and 1H chemical shift differences between ATG3[∆90-190] and ATG3[∆90-190]P21A plotted against amino acid sequence show a good overlap of the spectra for the wild-type and mutant protein in aqueous solution, indicating only minor local differences in their structures. In contrast, chemical shift differences between ATG3[∆90-190] and ATG3[∆90-190]P21A in bicelles are pronounced in both the N- and C-terminal regions, showing that membrane-bound ATG3[∆90-190] adopts a conformation at the N and C terminus different from the structure of membrane-bound ATG3[∆90-190]P21A. This finding can be interpreted to indicate that the P21A mutant is inactive on the membrane because its disrupted N-terminal conserved region cannot mediate the same structural rearrangements that occur near the active site in the wild type and that are essential for LC3B–PE conjugation activity. This further implies that the short conserved region with the consensus motif EYLTP is an important structural element in the Atg3/ATG3 molecule that couples and somehow communicates structural changes at the N terminus upon membrane binding to rearrangements of the active site. How the active site of the Atg3–Atg8/ATG3–LC3 intermediate on the membrane is arranged and the detailed mechanism leading to this architecture remain to be investigated. Ye et al. presume that the structural changes induced upon membrane binding and communicated to the active site of Atg3/ATG3 are distinct from the structural rearrangements induced by binding of the FR loop to Atg12/ATG12 in the E3-like complex. The latter mechanism involves a switch element in the FR [8] that functions as an allosteric activator of the E2-like enzyme upon its binding to Atg7/ATG7 or Atg12/ATG12.
We combined the conclusions by Ye et al. with the outcomes of a number of previous structural studies [3,5–10,12–17], and created a schematic model (Figure 1) that summarizes important mechanisms currently recognized to take place in Atg3/ATG3. The proteins and amino acid residues in this model are denoted using the yeast nomenclature and numbering, but the mechanisms are considered to be very similar in humans. The mechanisms are as follows: (1) The Atg3 enzyme free of any protein is in an autoinhibited conformation that is allosterically controlled by binding of a short helical element in the FR loop on the surface of the protein. Autoinhibition is manifested by having the catalytic cysteine C234 on the catalytic loop reside in a protective pocket of the catalytic domain. This specific position is determined by an α-helix in the handle region (HR) of Atg3. (2) Upon binding of Atg3 to dimeric Atg7, the FR region flips and binds via its helical element to the N-terminal region of Atg7. This movement functions as an allosteric switch communicated throughout the Atg3 molecule to the HR. The helix in the HR gains an additional turn and, thereby, repositions the catalytic loop along with the catalytic cysteine of Atg3 to a new position outside of the protective pocket. Atg3 is activated in the complex with Atg7, when C234 is ready to make a thioester bond with the C terminal glycine (Gly116) in Atg8. This glycine is engaged in a covalent bond with the catalytic cysteine (Cys507) located in a hydrophobic cavity of the Atg7 activation domain. Note that the N terminus and activation domain from the opposite Atg7 protomers are involved with a single copy of Atg3. (3) Once Atg8 is transferred from Atg7 to Atg3, the covalent Atg3–Atg8 intermediate is formed and Atg7 is disengaged. The Atg7-free helical element in the FR presumably reengages the Atg3 surface, the HR releases the additional turn in its helix and, thereby, the C234–G116 bond can reposition into the protective cavity of Atg3. (4) When the cytosolic Atg3–Atg8 intermediate comes into proximity of the phagophore membrane decorated with the Atg12–Atg5-Atg16 complex (E3-like enzyme), two events, presumably separate, take place. One such event is formation of an amphipathic helix at the N terminus of Atg3, which attaches the protein to the lipid bilayer. This event is coupled, via the conserved N-terminal domain, to a structural change around the active site with C234. The other event has not been visualized in the context of full-length proteins, but is assumed to be similar to that in the Atg3–Atg7 interaction. Specifically, the FR of Atg3 can flip so that its helical element binds to the surface of Atg12. This rearrangement can function again as an allosteric switch communicated to the HR, where formation of the additional helical turn in the helix can reposition the Atg3 catalytic loop with the C234–G116 bond out of the protective pocket. Both events just described facilitate the transfer of G116 from C234 to PE on the phagophore membrane. (5) After release of Atg3 and Atg12–Atg5-Atg16 from the membrane, Atg8 is stably conjugated to PE. Whether attaching of Atg3 on the membrane precedes or follows the Atg3–Atg12 interaction remains to be elucidated. A detailed structural insight into the architecture of the Atg3 active site with and without the membrane as well as with and without the Atg12–Atg5-Atg16 complex need to be acquired in order to provide a complete molecular mechanism for Atg3; this means, at some point, obtaining a structure of the Atg3–Atg8 conjugate in the complex with Atg12–Atg5-Atg16 when bound to membranes. Visualization of this intricate assembly is not trivial, and it may not be feasible with purified proteins, but rather becomes a subject for electron cryotomography in the future.
Figure 1.
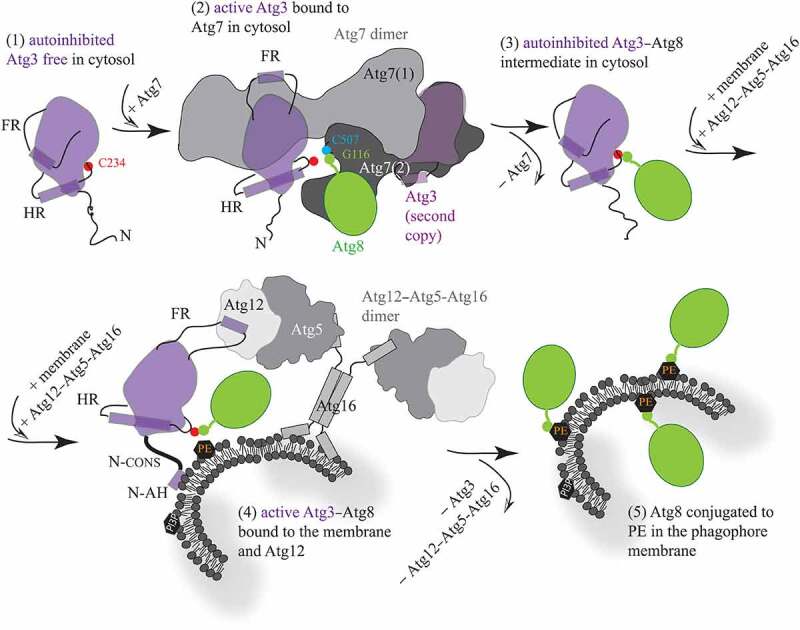
Schematic model for molecular mechanisms in Atg3. (1) Free, autoinhibited Atg3 becomes active upon the interaction of a short helical element in the flexible loop (FR) with the N-terminal domain of Atg7. (2) The flexible FR allosterically regulates enzyme activation that is manifested by helical extension in the handle region (HR) of Atg3 and, thereby, repositioning of the catalytic cysteine (C234). These movements allow for formation of a thioester bond between C234 and the C-terminal glycine (G116) in Atg8 that, after adenylation, resides in the activation domain of Atg7, and is covalently linked to the Atg7 catalytic cysteine (C507). (3) After the transfer of Atg8 from Atg7 to Atg3, the Atg7 dimer is released. The FR presumably reengages the Atg3 surface, causing autoinhibition of the covalent Atg3–Atg8 intermediate. (4) The cytosolic Atg3 molecule attaches to the membrane by embedding the N-terminal amphipathic helix (N-AH) into the lipid bilayer. This event is communicated via the N-terminal conserved region (N-CONS) to a structural rearrangement around C234. Membrane binding of Atg3 is either preceded or followed by binding of a short structural element in the FR to Atg12 in the Atg12–Atg5-Atg16 complex. The Atg3 FR–Atg12 interaction can reposition C234 via an allosteric mechanism, presumably similar to that in the Atg3–Atg7 complex. Structural changes induced by the membrane and Atg12 near Atg3 C234 facilitate the transfer of Atg8 G116 to PE. (5) After Atg3 and Atg12–Atg5-Atg16 detach from the assembly, Atg8 is lipidated on the phagophore membrane
Funding Statement
This work was supported by the National Institute of General Medical Sciences (GM131919) and the Protein Folding Disease FastForward Initiative, University of Michigan.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. [DOI] [PubMed] [Google Scholar]
- [2].Mizushima N.The ATG conjugation systems in autophagy. Curr Opin Cell Biol. 2020;63:1–10. [DOI] [PubMed] [Google Scholar]
- [3].Sakoh-Nakatogawa M, Matoba K, Asai E, et al. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol. 2013;20:433-+. [DOI] [PubMed] [Google Scholar]
- [4].Popelka H, Uversky VN, Klionsky DJ. Identification of Atg3 as an intrinsically disordered polypeptide yields insights into the molecular dynamics of autophagy-related proteins in yeast. Autophagy. 2014;10:1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yamada Y, Suzuki NN, Hanada T, et al. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J Biol Chem. 2007;282:8036–8043. [DOI] [PubMed] [Google Scholar]
- [6].Taherbhoy AM, Tait SW, Kaiser SE, et al. Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol Cell. 2011;44:451–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kaiser SE, Mao K, Taherbhoy AM, et al. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7-Atg3 and Atg7-Atg10 structures. Nat Struct Mol Biol. 2012;19:1242-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zheng Y, Qiu Y, Grace CRR, et al. A switch element in the autophagy E2 Atg3 mediates allosteric regulation across the lipidation cascade. Nat Commun. 2019;10:3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hanada T, Satomi Y, Takao T, et al. The amino-terminal region of Atg3 is essential for association with phosphatidylethanolamine in Atg8 lipidation. Febs Lett. 2009;583:1078–1083. [DOI] [PubMed] [Google Scholar]
- [10].Nath S, Dancourt J, Shteyn V, et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol. 2014;16:415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ye YS, Tyndall ER, Bui V, et al. An N-terminal conserved region in human Atg3 couples membrane curvature sensitivity to conjugase activity during autophagy. Nat Commun. 2021;12. DOI: 10.1038/s41467-020-20607-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fujioka Y, Noda NN, Nakatogawa H, et al. Dimeric coiled-coil structure of saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem. 2010;285:1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Noda NN, Satoo K, Fujioka Y, et al. Structural basis of atg8 activation by a homodimeric E1, Atg7. Mol Cell. 2011;44:462–475. [DOI] [PubMed] [Google Scholar]
- [14].Noda NN, Fujioka Y, Hanada T, et al. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013;14:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hong SB, Kim BW, Lee KE, et al. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol. 2011;18:1323–U1332. [DOI] [PubMed] [Google Scholar]
- [16].Metlagel Z, Otomo C, Takaesu G, et al. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc Natl Acad Sci U S A. 2013;110:18844–18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Popelka H, Reinhart EF, Metur SP, et al. Membrane Binding and homodimerization of Atg16 via two distinct protein regions is essential for autophagy in yeast. J Mol Biol. 2021;433:166809. [DOI] [PMC free article] [PubMed] [Google Scholar]