

ABSTRACT
Induction of macroautophagy (hereafter termed autophagy) is a strategy to improve the outcome of antineoplastic therapies by facilitating the induction of immunogenic cancer cell death and the consequent immune recognition of malignant cells. We analyzed 65,000 distinct compounds by means of a phenotypic discovery platform for autophagy induction and identified the IGF1R (insulin like growth factor 1 receptor) inhibitor picropodophyllin (PPP) as a potent inducer of autophagic flux. We found that PPP acts on-target, as an inhibitor of the tyrosine kinase activity of IGF1R and enhances the release of adenosine triphosphate, ATP, from stressed and dying cancer cells in vitro, thereby improving the therapeutic efficacy of chemoimmunotherapy in cancer-bearing mice. This PPP effect was phenocopied by another IGF1R inhibitor, linsitinib. Moreover, in human triple-negative breast cancer, phosphorylation of IGF1R correlates with reduced autophagy, an unfavorable local immune profile and poor prognosis. In summary, IGF1R inhibition may constitute a novel strategy for the treatment of cancer in the context of chemoimmunotherapy.
KEYWORDS: Anticancer immunity, biosensor, drug discovery, high-content screening, immunogenic cell death
The reinstatement of anticancer immunity is the sole therapeutic avenue that holds a promise for achieving remission and recovery from malignancy and (occasionally) definitive cure from the disease. In the setting of chemotherapy, curative effects beyond treatment discontinuation usually rely on the induction of immunogenic cell death (ICD) and the concomitant emission of danger/damage-associated molecular patterns (DAMPs) from stressed and dying malignant cells that elicit innate anticancer immune responses and eventually prime T lymphocyte-mediated tumor immunity. The release of ATP marks a hallmark in the course of ICD as it ligates purinergic receptors expressed by myeloid cells including dendritic cells and thus facilitates their chemoattraction into the tumor bed for igniting tumor antigen presentation.
The ICD-associated release of ATP depends on the induction of premortem autophagy and is absent in cancer cells that lack a functional autophagic machinery. So-called “caloric restriction mimetic” agents such as 3,4-dimethoxychalcone, hydroxycitrate, resveratrol, spermidine and thiostreptone have the ability to induce autophagy and to enhance the efficacy of immunogenic chemotherapy because they stimulate ATP release from stressed and dying malignant cells, thereby boosting local immune responses and improving the immunological and therapeutic outcome of chemotherapy with anthracyclines and oxaliplatin in preclinical models.
Intrigued by these premises, we launched a phenotypic screening campaign employing large compound collections to identify novel autophagy inducers. More than 65,000 chemical agents were tested for their ability to trigger the formation of autophagic puncta in biosensor cell lines expressing green fluorescent protein (GFP), fused to MAP1LC3B/LC3 (microtubule associated protein 1 light chain 3 beta) and primary screening data were further confirmed by means of autophagic flux reporter assays including monomeric red fluorescent protein (mRFP)-GFP-LC3 tandem biosensors and cells expressing an inducible variant of the autophagic substrate Q74-GFP. This approach led to the discovery of the IGF1R inhibitor picropodophyllin (PPP) as a potent, nontoxic, on-target inducer of autophagic flux, that loses its autophagy-stimulatory activity in cells that either lack IGF1R or express a constitutively active mutant of downstream AKT/protein kinase B. Mechanistically, PPP induces the translocation of the transcription factors TFEB and TFE3 from the cytoplasm to the nucleus. In TFEB TFE3 double-knockout cells, PPP loses the ability to trigger the formation of GFP-LC3 puncta, indicating that both transcription factors are essential for PPP-induced autophagy. Furthermore, the capacity of PPP to boost ATP release from cancer cells responding to oxaliplatin-based chemotherapy is reduced in cells lacking the ER stress sensor EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3) or its substrate EIF2A (eukaryotic translation initiation factor 2A), as well as in cells lacking the essential autophagy protein ATG5 [1].
When administered systemically to mice, PPP induces autophagy in heart, liver and brain tissues. Moreover, PPP improves the therapeutic efficacy of chemoimmunotherapy with a combination of immunogenic cytotoxicants such as oxaliplatin and PDCD1/PD-1 blockade in immunocompetent mice bearing fibrosarcomas, lung adenocarcinomas or triple-negative breast cancers. Similarly, the alternative IGF1R inhibitor linsitinib amplifies the antineoplastic effects of oxaliplatin and PDCD1 immune checkpoint blockade in vivo. Of note, improved tumor growth control by IGF1R inhibition is absent when cancer cells lack ATG5 or express either the constitutively active AKT mutant (T308D/S473D) or the ecto-ATPase ENTPD1/CD39, confirming the importance of autophagy-mediated ATP release for anticancer immune responses (Figure 1).
Figure 1.
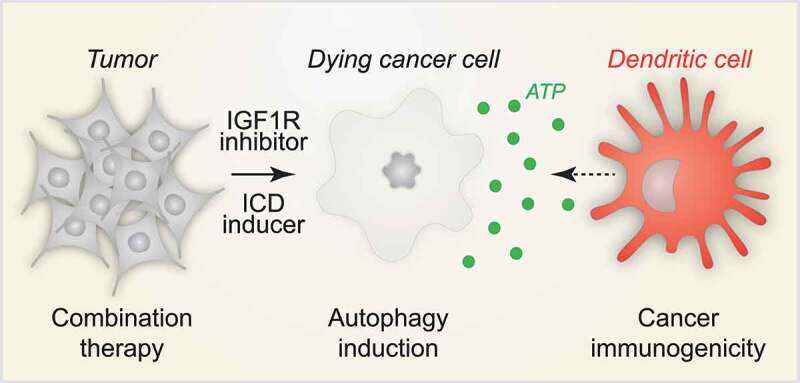
IGF1R inhibition improves immunogenic chemotherapy. Large-scale compound screening led to the discovery of IGF1R inhibition as a strategy for the induction of autophagy that enhances chemotherapy-induced immunogenic cell death (ICD) by boosting the release of the danger/damage-associated molecular pattern (DAMP) molecule ATP. In conclusion, IGF1R inhibition may constitute a novel and druggable therapeutic target for the treatment of cancer that has in settings of chemoimmunotherapy the potential to reinstate anticancer immunity
In combination with chemoimmunotherapy, IGF1R inhibition enhances the infiltration of tumors by cytotoxic T lymphocytes and reduces the abundance of regulatory T cells, thus improving anticancer immunosurveillance in preclinical models. Moreover, in human triple-negative breast cancer patients, the activating phosphorylation of IGF1R detectable by immunohistochemistry correlates with low levels of autophagy, and an immunosuppressive local immune profile, as well as with poor therapeutic outcome. This clinical observation suggests that autophagy induction by IGF1R inhibition might constitute a valid strategy for improving the efficacy of anticancer chemoimmunotherapies.
Funding Statement
OK receives funding by the DIM ELICIT initiative of the Ile de France; GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association “Le Cancer du Sein, Parlons-en!”; Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology (ANR-18-IDEX-0001); the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
Disclosure statement
GK and OK are cofounders of Samsara Therapeutics. GK is a cofounder of Therafast Bio.
Reference
- [1].Wu Q, Tian A, Li B, et al. IGF1 receptor inhibition amplifies the effects of cancer drugs by autophagy and immune-dependent mechanisms. J Immunother Cancer. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]