

A cancer ubiquitome landscape identifies metabolic reprogramming as target of Parkin tumor suppression.
Abstract
Changes in metabolism that affect mitochondrial and glycolytic networks are hallmarks of cancer, but their impact in disease is still elusive. Using global proteomics and ubiquitome screens, we now show that Parkin, an E3 ubiquitin ligase and key effector of mitophagy altered in Parkinson’s disease, shuts off mitochondrial dynamics and inhibits the non-oxidative phase of the pentose phosphate pathway. This blocks tumor cell movements, creates metabolic and oxidative stress, and inhibits primary and metastatic tumor growth. Uniformly down-regulated in cancer patients, Parkin tumor suppression requires its E3 ligase function, is reversed by antioxidants, and is independent of mitophagy. These data demonstrate that cancer metabolic networks are potent oncogenes directly targeted by endogenous tumor suppression.
INTRODUCTION
Tumors universally reprogram their metabolism (1) to engender cellular plasticity, promote adaptation to an unfavorable microenvironment, and facilitate disease progression (2). Mechanistically, this involves aberrant utilization of glycolysis even when oxygen is present, the “Warburg effect” (3), as well as exploitation of anabolic and antioxidant mechanisms maintained by the pentose phosphate pathway (PPP) (4). Mitochondria also play an important role in tumor metabolism (5), and their functions in oxidative bioenergetics (6, 7), redox balance (8), and mitochondrial dynamics (9, 10) contribute to aggressive disease traits and metastatic dissemination (11). However, evidence that glycolytic and mitochondrial reprogramming is a genuine, disease-driving oncogene antagonized by endogenous tumor suppression mechanisms has remained elusive.
A regulator of mitochondrial integrity, including in cancer, Parkin (12) is an E3 ubiquitin ligase altered in up to 50% of familial, early-onset, Parkinson’s disease (PD) (13). Parkin neuroprotection has been linked to its ability to remove subpar mitochondria (14) through the autophagy-lysosome machinery (15), i.e., mitophagy, a process that requires phosphorylation of Parkin (as well as nearby ubiquitin) by phosphatase and tensin homolog (PTEN)–induced kinase 1 (PINK1) (16) in response to mitochondrial damage (17). There is evidence that Parkin may have functions outside the central nervous system (18) and potentially antagonize tumor growth (19) via regulation of cyclin levels (20) or suppression of glycolytic reprogramming (21). However, a tumor suppression function of Parkin has not been clearly defined: PD patients have paradoxically lower incidence of malignancy (22), Parkin can promote cancer in certain conditions (23), and the mechanisms of a potential antitumorigenic function have not been firmly established, variously linked to PTEN loss (24), disruption of iron homeostasis (25), or inhibition of serine metabolism (26). In this study, we used global proteomics and ubiquitome screens to unravel a previously unidentified role of Parkin in cancer, independent of mitophagy (12).
RESULTS
A mitophagy-independent role of Parkin in cancer
To study a role of Parkin in cancer, we reintroduced Parkin in Parkin-negative prostate adenocarcinoma PC3 cells (fig. S1A). In the absence of mitophagy-inducing stimuli, recombinant Parkin was recruited to mitochondria of PC3 cells (fig. S1B), together with phosphorylation of ubiquitin on Ser65 (fig. S1C), a hallmark of PINK1 stabilization/activation (16). Under these conditions, reconstitution with Parkin did not affect mitochondrial inner membrane potential in PC3 or neuroblastoma SK-N-SH cells (SKN; fig. S1, D and E). In addition, hallmarks of autophagy, such as expression of p62 or processing of LC3 to its lipidated form, were indistinguishable in control or Parkin-expressing cells (fig. S1F).
Next, we looked at a potential activation of mitophagy in this response. Transient reexpression of Parkin in PC3 cells did not cause loss of mitochondrial outer membrane proteins normally degraded during mitophagy (fig. S2A) (27), and mitochondrial mass was unchanged in control or Parkin-expressing cells (fig. S2B). The colocalization of lysosomes (LAMP1) with mitochondria (TOMM20), a prerequisite of mitophagy, was unaffected in the presence of Parkin (fig. S2, C and D), and overall mitochondrial structure as determined by transmission electron microscopy was indistinguishable in the presence or absence of Parkin, with preservation of cristae length, branching, and tubular network (fig. S2E). Consistent with these data, quantification of mitophagy by mitochondrial Keima-Red fluorescence reporter activity showed no statistically significant differences in vector or Parkin-expressing PC3 and SKN cells (fig. S2F), as determined by ratiometric determination of phycoerythrin (PE)/BV605 fluorescence of 1.2 ± 0.96 (vector) and 1.05 ± 0.77 (Parkin) in PC3 cells and 0.72 ± 0.56 (vector) and 0.54 ± 0.44 (Parkin) in SKN cells (fig. S2G). As control, treatment with the mitochondrial uncoupler and mitophagy inducer carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (16) induced loss of mitochondrial mass (fig. S2B) and increased mitochondrial Keima-Red reporter activity in PC3 (PE/BV605: Parkin, 0.79; Parkin + FCCP, 2.4) and SKN (Parkin, 1.03; Parkin + FCCP, 4.79) cells.
Parkin inhibition of tumor cell motility
On the basis of these data, we next examined potential function(s) of Parkin in cancer independently of mitophagy. Reconstitution of glioblastoma LN229 cells with Parkin reduced the area and length of focal adhesion (FA) (Fig. 1, A and B) and decreased the rate of FA assembly and disassembly (Fig. 1C and fig. S3A), two requirements of cell movements (28). Quantitatively, Parkin suppressed the formation of new (vector, 19.9 ± 12.5%; Parkin, 0%) and decayed (vector, 60.1 ± 29%; Parkin, 35.8 ± 29%) FA while increasing the fraction of stable FA (vector, 14.5 ± 7.4%; Parkin, 64 ± 26.5%). Consistent with these data, Parkin inhibited single-cell motility of tumor types, including PC3 (Fig. 1D), prostate adenocarcinoma DU145, and SKN (fig. S3B) cells, reducing the speed of individual tumor cell movements compared to controls (Fig. 1E). Directional cell migration quantified in a wound closure assay was also inhibited in the presence of Parkin (Fig. 1F). In terms of signaling requirements, reexpression of Parkin in PC3 cells reduced the levels and phosphorylation of FA kinase (FAK; Y397 and Y925) and Src (Y416), which are important for cell movements, whereas Akt was not affected (fig. S3C). Last, Parkin expression blocked the invasion across Matrigel of tumor types, including PC3, DU145, and LN229 cells (Fig. 1G). Despite the profound inhibition of cell motility, Parkin-expressing cells remained responsive to a chemotactic gradient (fig. S4, A and B), indicating that actin cytoskeletal dynamics was not globally disrupted in these settings.
Fig. 1. Parkin regulation of tumor cell motility.
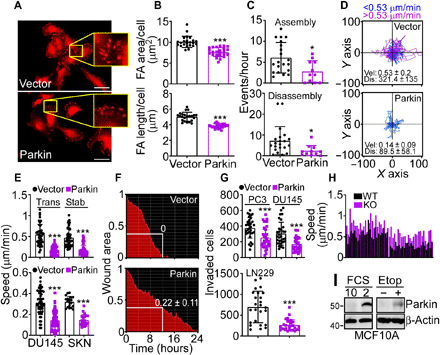
(A and B) LN229 cells transfected with vector or Parkin were labeled for vinculin and analyzed by confocal microscopy [(A), representative images] with quantification of the area and length of focal adhesion (FA) (B). Scale bars, 50 μm. Inset, magnifications of indicated areas. Mean ± SD (N = 3). ***P < 0.0001. (C) Talin-GFP–labeled LN229 cells as in (A) were quantified for the rate (events per hour) of FA assembly (top) or disassembly (bottom) (11 to 14 cells per condition). Mean ± SD (N = 3). *P = 0.03. (D) PC3 cells as in (A) were analyzed for single-cell motility (49 cells per condition). The cutoff velocities for slow-moving (blue) or fast-moving (purple) cells and the mean ± SD speed of cell motility (μm/min) and distance traveled (μm) are indicated. (E) PC3 cells transiently (Trans) or stably (Stab) transfected with vector or Parkin (top) or DU145 or SKN cells transfected with vector or Parkin (bottom) were analyzed for single-cell motility with quantification of speed of cell movements. Mean ± SD (19 to 68 cells analyzed; N = 3). ***P < 0.0001. (F) PC3 cells as in (A) were analyzed in a wound closure assay with quantification of the residual wound area after 12 hours. (G) The indicated tumor cell types transfected with vector or Parkin were analyzed for invasion across Matrigel-coated Transwell inserts. Mean ± SD (N = 3). ***P = 0.0003. (H) Wild-type (WT) or Parkin knockout (KO) MEFs were analyzed for single-cell motility, and the speed of cell movements was quantified (N = 51). P < 0.0001. (I) Breast epithelial MCF10A cells were incubated in 2 or 10% FCS or exposed to etoposide (Etop) for 24 hours and analyzed by Western blotting.
In reciprocal experiments, homozygous deletion of Parkin in Parkin knockout (KO) mouse embryonic fibroblasts (MEFs) increased single-cell motility compared to wild-type (WT) MEF (fig. S4C), resulting in faster speed of cell movements (Fig. 1H).
On the basis of these data, we next looked for other pathophysiologic condition(s) that could stabilize Parkin in the absence of mitochondrial damage or activation of mitophagy. Accordingly, exposure of breast epithelial MCF10A cells, which express endogenous Parkin to stress conditions, such as nutrient deprivation [2% fetal calf serum (FCS)] or the DNA-damaging agent etoposide, resulted in strong up-regulation of endogenous Parkin expression (Fig. 1I). Under these conditions, etoposide treatment was not associated with mitochondrial damage, as organelle inner membrane potential was unchanged in the presence or absence of endogenous or recombinant Parkin (fig. S4, D and E). Similar to the data obtained with KO MEF, silencing of endogenous Parkin in MCF10A cells by small interfering RNA (siRNA) (fig. S4F) significantly increased cell migration, compared to control siRNA transfectants (fig. S4, G and H).
Parkin inhibition of primary and metastatic tumor growth
Next, we examined the effect of Parkin on tumor growth in vivo. We found that Parkin-expressing PC3 cells formed significantly smaller tumors when engrafted subcutaneously in immunocompromised nonobese diabetic (NOD) severe combined immunodeficiency (SCID) γ mice compared to control transfectants (mean ± SD tumor volume at day 26: vector, 883 ± 274 mm3; Parkin, 364 ± 98 mm3; P < 0.0001; Fig. 2A). In these settings, Parkin expression suppressed metastatic dissemination to the lungs, whereas vector-transfected PC3 cells formed extensive lung metastases (Fig. 2, B and C). As an independent model of metastasis, we next injected PC3 cells directly into the spleen of NOD SCID γ mice and looked at liver colonization 11 days after reconstitution. Here, control PC3 cells generated extensive metastatic foci in the liver, whereas expression of Parkin inhibited the number and surface areas of liver metastases (Fig. 2, D and E).
Fig. 2. Parkin inhibition of primary and metastatic tumor growth.
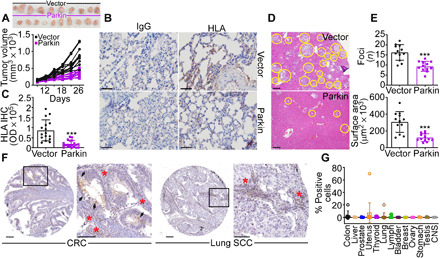
(A) PC3 cells stably transfected with vector or Parkin were injected subcutaneously on the flank of immunocompromised NOD SCID γ mice (two tumors per mouse; 10 tumors per group), and tumor volume was quantified with a caliper at the indicated time intervals. Top: Macroscopic images of tumors harvested from the two animal groups at the end of the experiment. (B and C) The conditions are as in (A), and lungs isolated from the indicated animal groups were stained with immunoglobulin G (IgG) or an antibody to human HLA by immunohistochemistry (IHC) [(B), representative images] and quantified (C). Scale bars, 100 μm. OD, optical density. ***P < 0.0001. (D) PC3 cells as in (A) were injected into the spleen of NOD SCID γ mice (five animals per group), and metastatic foci to the liver (yellow circles, representative images) were examined after 11 days by hematoxylin and eosin staining and light microscopy. Scale bars, 200 μm. (E) The conditions are as in (D), and the number (top) and surface area (bottom) of liver metastases were quantified by morphometry. Mean ± SD (N = 60). ***P < 0.0001. (F and G) A universal cancer TMA (Tissue Microarray) was stained with an antibody to Parkin by IHC [(F), representative images], and the percentage of positive cells in each tumor sample was quantified (G). Each symbol corresponds to an individual tumor sample. Scale bars, 100 μm. Inset, magnification of indicated areas. Arrowheads and red asterisks indicate tumor cells or infiltrating lymphocytes, respectively, positive for Parkin. CNS, central nervous system; CRC, colorectal cancer; SCC, squamous cell carcinoma.
When analyzed by immunohistochemistry (IHC) in a universal cancer microarray, Parkin was undetectable in patient-derived tissue samples, with only modest reactivity in infiltrating lymphocytes (Fig. 2, F and G, and fig. S5A). Parkin mRNA levels were also uniformly reduced in all tumor types in The Cancer Genome Atlas (TCGA) database (fig. S5B), with significant loss of expression in many common malignancies, compared to normal tissues (fig. S5C). Last, all tumor cell lines in the Cancer Cell Line Encyclopedia (CCLE) showed decreased levels of Parkin mRNA (fig. S5D), without significant alterations of copy number (fig. S5E). Consistent with these data, proteomics analysis of the CCLE database (12,755 proteins) demonstrated that only 1 of 42 batches (8 of 378 cell lines) contained detectable Parkin levels (identifier sp|O60260|PRKN2_HUMAN) and with very low peptide detection (fig. S5F), together supporting the notion of Parkin as a tumor suppressor.
Parkin E3 ligase activity regulates tumor cell motility
On the basis of these data, we next asked whether Parkin E3 ligase activity was required for inhibition of tumor responses. For these experiments, we changed the Parkin PINK1 phosphorylation site on Ser65 to Ala (S65A) or the active site at Cys431 to Ser (C431S) to generate E3 ligase loss-of-function mutants (fig. S6A). Consistent with the data above, reconstitution of PC3 cells with WT Parkin suppressed single-cell motility (fig. S6B) and potently inhibited Matrigel invasion (fig. S6, C and D). In contrast, expression of Parkin S65A or C431S mutant did not reduce PC3 single-cell motility (fig. S6B) or Matrigel invasion (fig. S6, C and D).
As an independent approach, we next focused on the Parkin activator PINK1. In preliminary experiments, we found that PC3 cells constitutively expressed a basal level of PINK1, which was strongly up-regulated, i.e., stabilized in the presence of the mitochondrial uncoupler FCCP (fig. S7A), in agreement with previous observations (16). Transfection of PC3 cells with PINK1-directed siRNA efficiently suppressed PINK1 protein expression in PC3 cells (fig. S7A), whether in the presence or absence of Parkin (fig. S7, A and B). Under these conditions, silencing of PINK1 significantly, albeit not completely, restored Matrigel invasion of PC3 cells in the presence of Parkin (fig. S7, C and D). To test the specificity of this response, we next targeted the essential regulators of autophagy, p62 or Atg5, and looked at their involvement in Parkin functions. In these experiments, siRNA silencing of p62 (fig. S7E) had no effect on Parkin suppression of PC3 cell invasion across Matrigel (fig. S7F). Similarly, siRNA knockdown of Atg5 (fig. S7G) did not reverse the inhibition of single-cell motility mediated by Parkin (fig. S7H), and Parkin suppression of the speed of cell movements was indistinguishable in control or Atg5-silenced cells (fig. S7I).
A Parkin proteome and ubiquitome in cancer
To elucidate the function of Parkin in cancer, we next used SILAC (stable isotope labeling by amino acids in cell culture) labeling to map global proteomic changes in vector- or Parkin-expressing PC3 cells in the absence of mitophagy-inducing stimuli. In two independent experiments (Fig. 3A), we detected a total of 5557 proteins (Fig. 3B), with 319 and 222 proteins showing increased or decreased abundance in the presence of Parkin, respectively (Fig. 3C and table S1). By bioinformatics analysis, the cancer Parkin proteome identified here was predicted to affect protein networks of cell movements, including extracellular matrix (FN1 ↑1.8-fold; FBLN1 ↑10.3-fold), EMT (Epithelial Mesenchymal Transition) and metastasis (MYOD1 ↑9.9-fold; KIF14 ↓2.5-fold; CYR61 ↓2.1-fold; STARD10 ↓1.9-fold), suppressed extracellular signal–regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) pathway (DLGAP5 ↓1.8-fold; SBSN ↓8.9-fold), and down-regulated oncogenic signaling (TACSTD2/TROP2 ↓59.4-fold; UBE2T ↓1.9-fold; BICD1 ↓1.8-fold; HMGCS1 ↓1.8-fold) (Fig. 3D). Selective inspection of a mitochondrial proteome (MitoCarta 3.0, N = 741 proteins) in this dataset (Fig. 3E) revealed that Parkin modulated the expression of electron transport chain complex I (MT-ND1 ↑1.6-fold; NDUFA3 ↑1.8-fold; NDUFC2 ↑1.4-fold; NDUFA4 ↑1.4-fold) and complex IV (COX6A1 ↑1.3-fold) subunits, endogenous antioxidant mechanisms (PRDX4 ↓1.6-fold; SOD2 ↓1.4-fold; GFER ↓1.4-fold; SOD1 ↑2.1-fold), and organelle protein translation (MRP63 ↑2.1-fold; MRPL54 ↑1.6-fold; MRPL52 ↑1.4-fold; MRPS15 ↓4.7-fold) (Fig. 3F).
Fig. 3. Landscape of a Parkin proteome and ubiquitome in cancer.
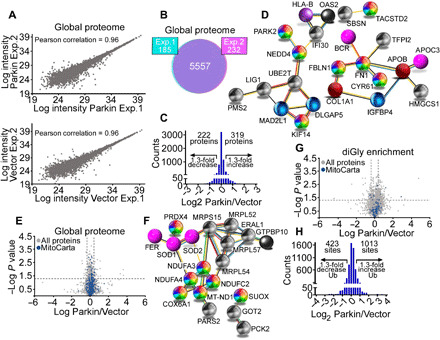
(A) Scatter plots of protein intensities from two independent global proteome SILAC experiments in PC3 cells transfected with vector (light labeled) or Parkin (heavy labeled) in the absence of mitochondrial uncouplers. A Pearson correlation coefficient is indicated. (B) The conditions are as in (A), and concordance in detection of proteins between experiment 1 (Exp. 1, 97%) and experiment 2 (Exp. 2, 96%) is shown. (C) Changes in protein levels in PC3 cells transfected with Parkin. A 1.3-fold difference cutoff was defined as potentially biologically important. (D) STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) analysis of predicted protein networks modulated by Parkin in the global proteome of PC3 cells. (E) Volcano plot of the Parkin global proteome in PC3 cells. The distribution of total proteins (N = 5557) and MitoCarta 3.0–associated proteins (N = 741) is indicated. (F) STRING analysis of predicted mitochondrial protein networks modulated by Parkin in the global proteome. (G) Volcano plot of a Parkin ubiquitome in PC3 cells in the absence of mitochondrial uncouplers. The distribution of total (N = 3796) and MitoCarta 3.0–associated ubiquitination sites (N = 112) is indicated. (H) Changes in Parkin ubiquitome identified using SILAC-based ubiquitin remnant motif enrichment (K-ε-GG), with 1013 and 423 sites showing increased or decreased ubiquitination in the presence of Parkin using a 1.3-fold cutoff.
Next, we used SILAC-based ubiquitin remnant motif enrichment (K-ε-GG) to characterize a Parkin ubiquitome in cancer. In two independent experiments in the absence of mitophagy-inducing stimuli (fig. S8A), we identified 1821 proteins (fig. S8B) with 3796 ubiquitinated sites (fig. S8C), with 112 mitochondria-associated ubiquitination sites (Fig. 3G and table S2) present in both experiments. Using a 1.3-fold cutoff, 1013 and 423 sites showed increased or decreased ubiquitination in the presence of Parkin, respectively (Fig. 3H). Bioinformatics analysis of this dataset identified four main protein networks in the Parkin cancer ubiquitome. These included (i) cell death (MCL1, HK1, and HMGB1), (ii) mitochondrial dynamics (RHOT1, MFN2, and FIS1), (iii) endoplasmic reticulum (CCT7) and mitochondrial (HSPA1A, also known as HSP72) proteostasis, and (iv) glucose metabolism, involving both glycolysis (HK1 and TPI1) and the non-oxidative phase of the PPP (TKT and TALDO1) (Fig. 4A and fig. S8D). Experimentally, the SILAC ratios of proteins and ubiquitinated sites identified in both replicates showed very low variability, with a median coefficient of variation (CV) of 5.6 and 6%, respectively, and >95% of identified proteins and ubiquitination sites with a CV of <30% (fig. S8, E and F).
Fig. 4. Novel Parkin ubiquitome in cancer.
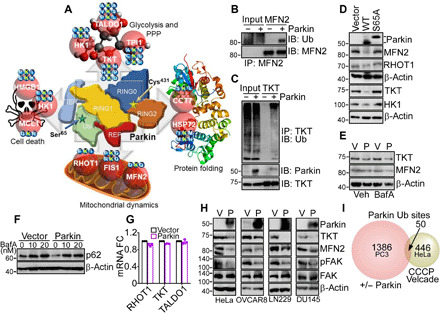
(A) Bioinformatics analysis of predicted protein networks regulated by Parkin ubiquitination in the absence of mitochondrial uncoupling, including cell death (HK1, MCL1, and HMGB1), glucose metabolism involving glycolysis (HK1 and TPI1) and the PPP (TALDO1 and TKT), protein folding (CCT7 and HSPA1A, also known as HSP72), and mitochondrial dynamics (RHOT1, FIS1, and MFN2). Parkin-directed ubiquitination sites (Lys, K) identified by SILAC proteomics in each target protein are indicated. (B) PC3 cells in the presence or absence of Parkin were immunoprecipitated with an antibody to MFN2, and immune complexes were probed with antibodies to ubiquitin (Ub) or MFN2 by Western blotting. IP, immunoprecipitation; IB, immunoblot. (C) The conditions are as in (B) except that TKT immune complexes precipitated from PC3 cells with or without Parkin were probed with antibodies to Parkin, TKT or Ub, by Western blotting. (D) PC3 cells transfected with WT Parkin or E3 ligase–defective Parkin S65A mutant were analyzed by Western blotting. (E) PC3 cells transfected with vector (Veh) or Parkin (P) were treated with bafilomycin A (BafA; 20 nM for 6 hours) and analyzed by Western blotting. (F) PC3 cells transfected with vector or Parkin were treated with the indicated concentrations of BafA and analyzed by Western blotting. (G) PC3 cells transfected with vector or Parkin were analyzed for changes in mRNA expression of RHOT1, TKT, or TALDO1 by reverse transcription PCR. Mean ± SD (N = 3). (H) The indicated tumor cell types transfected as in (E) were analyzed by Western blotting. (I) Comparison between the Parkin ubiquitination sites identified in PC3 cells in the absence of mitochondrial uncoupling (this study) and HeLa cells treated with CCCP plus Velcade (Exp. ID 57) as reported in (27). The number of ubiquitination sites is indicated. Only sites showing significant changes in SILAC ratios as defined in the respective studies were compared.
In validation experiments, MFN2 (Fig. 4B) or TKT (Fig. 4C) immunoprecipitated from PC3 cells reacted with an antibody to ubiquitin in the presence of Parkin. In addition, expression of WT Parkin reduced the levels of its target proteins—MFN2, RHOT1, TKT, and HK1—by Western blotting, whereas a Parkin S65A mutant had no effect (Fig. 4D). Consistent with the data above, treatment of PC3 cells with the autophagy inhibitor bafilomycin A (BafA) did not affect Parkin-induced degradation of MFN2 or TKT (Fig. 4E). Conversely, in control experiments, treatment of PC3 cells with BafA stabilized the levels of p62 in the presence or absence of Parkin (Fig. 4F). In parallel experiments, mRNA levels of RHOT1, TKT, or TALDO1 were unchanged in control or Parkin-expressing PC3 cells (Fig. 4G). We next looked at the generality of this pathway in a panel of genetically unrelated tumor cell lines, including cervical carcinoma HeLa, ovarian cancer OVCAR8, LN229, and DU145 cells. Expression of Parkin in these conditions comparably induced degradation of its targets, TKT and MFN2, in all tumor cell lines tested, concomitantly with down-regulation of phosphorylated FAK (Fig. 4H). Last, the cancer Parkin ubiquitome identified here in the absence of mitochondrial damage showed negligible overlap with the ubiquitome of CCCP (Carbonyl cyanide m-chlorophenyl hydrazone)–treated HeLa cells, with only 50 ubiquitination sites identified in common between the two datasets when sites identified as changing were compared (Fig. 4I).
Parkin regulation of mitochondrial dynamics controls tumor cell motility
In the cancer Parkin ubiquitome, RHOT1 and MFN2 regulate mitochondrial dynamics and have been implicated in tumor cell motility and metastasis (9). Consistent with this, Parkin suppressed mitochondrial dynamics in PC3 and SKN cells (fig. S9A), reducing the rates of mitochondrial fusion and fission events, compared to controls (fig. S9B). As a result, Parkin inhibited the subcellular movements of mitochondria (fig. S9C), with near-complete suppression of individual mitochondrial speed compared to control conditions (fig. S9D). Validating the specificity of these findings, reconstitution of Parkin-expressing cells with MFN2, but not MFN1 (fig. S9E), rescued mitochondrial dynamics (Fig. 5A) and restored the rate of fusion and fission events to levels of control cells (Fig. 5B). In parallel experiments, reconstitution of Parkin-expressing cells with RHOT1 (fig. S9F) normalized mitochondrial motility and increased the speed of mitochondrial movements indistinguishably from controls (Fig. 5C). Consistent with these data, RHOT1 expression restored mitochondrial accumulation at the cortical cytoskeleton of PC3 cells, which was suppressed in the presence of Parkin (Fig. 5D). Together, normalization of mitochondrial dynamics and subcellular mitochondrial trafficking by MFN2 and RHOT1 corrected single-cell motility (Fig. 5E) and the speed of individual cell movements (fig. S9G) in the presence of Parkin. Reconstitution with RHOT1 and MFN2 also restored PC3 cell invasion across Matrigel, comparably to control transfectants (Fig. 5F). These results were specific because expression of a Parkin ubiquitination–resistant RHOT1 K194R mutant escaped degradation in the presence of Parkin (fig. S9F) and was sufficient to increase migration of MCF10A cells compared to controls (Fig. 5G). In contrast, WT RHOT1 did not affect MCF10A cell migration (Fig. 5G).
Fig. 5. Parkin regulation of mitochondrial dynamics and tumor cell motility.
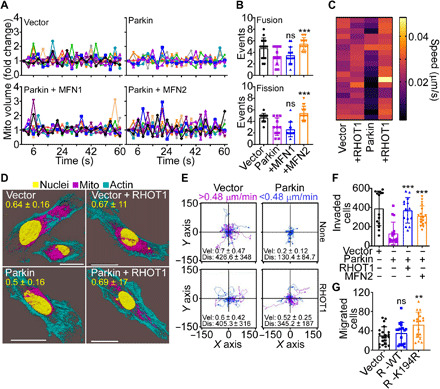
(A and B) PC3 cells transfected with vector or Parkin were reconstituted with MFN1 or MFN2 and analyzed for changes in mitochondrial volume by time-lapse videomicroscopy (A) and mitochondrial fusion (>1.3-fold mitochondrial volume) and fission (<0.7-fold mitochondrial volume) events in a 60-s time interval were quantified (B). Mean ± SD (N = 3). ***P = 0.0002 to 0.0004; ns, not significant. (C) PC3 cells transfected with vector or Parkin were reconstituted with RHOT1, and the speed of individual mitochondrial movements was quantified in a heatmap. Each bar corresponds to an individual mitochondrion (N = 40; P < 0.0001). (D) PC3 cells reconstituted as in (C) were imaged for mitochondrial accumulation at the cortical cytoskeleton by confocal microscopy (representative images). Scale bars, 20 μm. The quantification (mean ± SD; N = 3) of cortical mitochondria is indicated per each condition tested. (E) The conditions are as in (C), and motility of PC3 cells reconstituted with vector or RHOT1 was quantified in single-cell analysis by time-lapse video microscopy. Each tracing corresponds to the movement of an individual cell (representative experiment). The cutoff velocities for slow-moving (blue) or fast-moving (purple) cells and the average (mean ± SD) speed of cell motility [velocity (Vel); μm/min] and distance traveled (Dis; μm) are indicated. (F) PC3 cells expressing vector or Parkin were reconstituted with RHOT1 or MFN2 and analyzed for invasion across Matrigel-coated Transwell inserts. Mean ± SD (N = 3). ***P < 0.0001. (G) Breast epithelial MCF10A cells were transfected with WT RHOT1 or Parkin ubiquitination–resistant K194R RHOT1 mutant and analyzed for cell migration. Mean ± SD (N = 3). **P = 0.002.
Parkin regulation of the PPP controls oxidative stress and tumor cell motility
As shown here, a novel protein network targeted by Parkin in cancer is the PPP, and we next tested the impact of this pathway on tumor responses. Consistent with the results of the ubiquitome screen, Parkin inhibited TKT activity in PC3 cells, compared to vector transfectants (Fig. 6A). This was associated with reduced flux through the PPP compared to glycolysis in [1,2-13C]glucose labeling experiments (Fig. 6B), resulting in decreased incorporation of PPP-derived carbons into metabolites dihydroxyacetone phosphate (DHAP), 3-phosphoglycerate (3PG), phosphoenolpyruvate (PEP), and lactate (Fig. 6C). In addition, Parkin expression inhibited glucose consumption and lactate generation in PC3 cells, two hallmarks of glycolysis (fig. S10A). As a result of impaired bioenergetics, Parkin-expressing PC3 cells exhibited decreased adenosine 5′-triphosphate (ATP) production and heightened phosphorylation of the energy sensor AMPK (fig. S10B), a marker of cellular starvation. These metabolic changes were specific because Parkin did not affect the lipidomic profile of PC3 cells (fig. S10C) and the levels of saturated (SFA), monounsaturated (MUFA), or polyunsaturated (PUFA) fatty acids were unchanged compared to controls (fig. S10, D and E).
Fig. 6. Parkin inhibition of the PPP controls tumor cell motility.
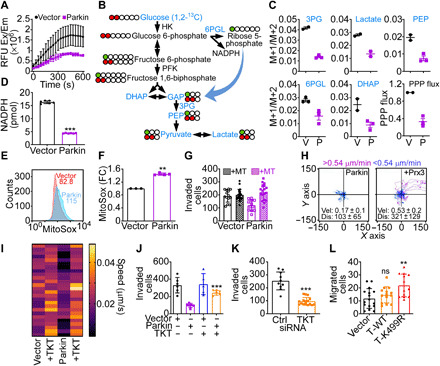
(A) PC3 cells transfected with vector or Parkin were analyzed for TKT activity. Mean ± SD (n = 2). (B) PPP flux analysis. Metabolites quantified by [1,2-13C]glucose labeling are indicated in blue. (C) The conditions are as in (B), and the M+1 (PPP) to M+2 (glycolysis) isotopolog metabolite ratio was quantified (N = 2 to 3). (D) PC3 cells as in (A) were analyzed for NADPH levels. Mean ± SD (N = 3). ***P < 0.0001. (E and F) The conditions are as in (A), and mitochondrial superoxide production was analyzed by flow cytometry [(E), representative tracings] and quantified (F). MFI values are indicated. FC, fold change. Mean ± SD (N = 3). **P = 0.002. (G) PC3 cells as in (A) were treated with vehicle or MitoTempo (MT) and analyzed for Matrigel invasion. Mean ± SD (n = 3). (H) PC3 cells expressing Parkin were reconstituted with vector or Prx3 and analyzed for single-cell motility. The cutoff velocities for slow-moving (blue) or fast-moving (purple) cells and the mean ± SD speed of cell motility (μm/min) and distance traveled (μm) are indicated. For Prx3-transfected cells: speed, 0.53 ± 0.2 μm/min; distance, 321 ± 121.6 μm (N = 3). (I) PC3 cells as in (A) were reconstituted with TKT, and the speed of mitochondrial movements was quantified in a heatmap (N = 28 to 35; P < 0.0001). (J) The conditions are as in (I), and PC3 cells were analyzed for Matrigel invasion. Mean ± SD (n = 3). ***P < 0.0001. (K) PC3 cells transfected with control siRNA (Ctrl) or TKT-directed siRNA were analyzed for Matrigel invasion. Mean ± SD (n = 3). ***P < 0.0001. (L) MCF10A cells expressing vector, WT TKT, or Parkin K499R TKT mutant were analyzed for cell migration. Mean ± SD (n = 3). **P = 0.002.
The PPP has an important antioxidant function (29), and accordingly, Parkin inhibition of TKT lowered NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) levels (Fig. 6D), resulting in heightened production of mitochondrial reactive oxygen species (ROS) (Fig. 6, E and F) in PC3 cells. Consistent with the notion that oxidative stress is a potent inhibitor of mitochondrial trafficking and tumor cell motility (30, 31), ROS scavenging using MitoTempo restored mitochondrial trafficking in Parkin-expressing PC3 cells and normalized the speed of mitochondrial movements to levels of control transfectants (fig. S11A). As a result, MitoTempo fully rescued single-cell movements (fig. S11B), speed of cell motility (fig. S11C), and Matrigel invasion (Fig. 6G) of Parkin-expressing PC3 cells. Reconstitution of Parkin-expressing PC3 cells with antioxidant Prx3 gave similar results, restoring the speed of cell motility and the total distance traveled by individual cells comparably to controls (Fig. 6H).
Last, we examined the specificity of PPP signaling in this response. Reconstitution of Parkin-expressing PC3 cells with TKT (fig. S11D) was sufficient to rescue subcellular mitochondrial movements (fig. S11E) and restore the speed of individual mitochondria to levels of control transfectants (Fig. 6I). This translated in increased PC3 cell invasion across Matrigel (fig. S11F), indistinguishably from controls (Fig. 6J). In reciprocal experiments, siRNA silencing of TKT (fig. S11G) inhibited tumor cell invasion, whereas a control siRNA had no effect (Fig. 6K). Last, expression of a Parkin ubiquitination–resistant TKT K499R mutant escaped degradation by Parkin in PC3 cells (fig. S11D) and promoted MCF10A cell migration, whereas WT TKT had no effect (Fig. 6L). Consistent with these results, TKT K499R mutant immunoprecipitated from Parkin-expressing cells showed considerably reduced reactivity with an antibody to ubiquitin, compared to WT TKT immune complexes precipitated under the same conditions (fig. S11H).
DISCUSSION
In this study, we have shown that Parkin targets glycolytic and mitochondrial networks to antagonize tumor growth. This pathway is independent of mitochondrial damage or activation of mitophagy and requires Parkin E3 ligase activity to degrade effectors of mitochondrial dynamics (MFN2 and RHOT1) and the non-oxidative phase of the PPP (TKT). This creates acute metabolic and oxidative stress, suppresses subcellular mitochondrial trafficking, inhibits phosphorylation of cell motility kinases, FAK and Src, and blocks tumor cell movements, with profound suppression of primary and metastatic tumor growth in vivo (Fig. 7).
Fig. 7. Schematic model for stress-regulated Parkin tumor suppression.
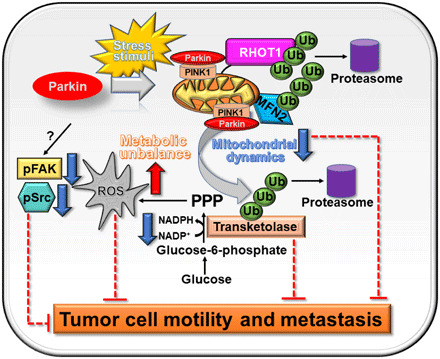
In response to stress stimuli, Parkin is recruited to mitochondria of tumor cells and promotes ubiquitination of protein substrates involved in mitochondrial dynamics and the PPP in a PINK1-dependent manner, affecting ROS production, metabolism and tumor cell motility, invasion, and metastasis.
Parkin neuroprotection in PD has been consistently linked to mitochondrial quality control (32), a process that eliminates damaged, subpar, and potentially dangerous organelles via mitophagy (14). We have shown here that this pathway is not an obligatory requirement in cancer, where Parkin can be recruited to mitochondria with active E3 ligase function in the absence of organelle damage or evidence of mitophagy. Other emerging functions of Parkin in cancer, such as modulation of iron homeostasis (25) or serine metabolism (26), have been equally linked to E3 ligase activity in the absence of mitophagy-inducing stimulation. There is also recent evidence challenging a long-held belief that mitophagy functions in tumor suppression, as activation of this pathway has been linked to progression of KRAS-driven pancreatic cancer (33) and resistance to chemotherapy-induced cell death (34).
Together, we propose the existence of additional mechanisms of Parkin activation in cancer, consistent with our observation that stress stimuli commonly found in a tumor microenvironment, such as transient nutrient deprivation or DNA damage, stabilize Parkin in the absence of mitochondrial damage. In line with this possibility, the Parkin ubiquitome identified here showed negligible overlap with that reported in neurons (35) or in tumor cells after acute mitochondrial damage (27), encompassing new protein targets in metabolism (TKT, TALDO1, and TP1I), damage-associated molecular pattern (HMGB1), chaperone function (CCT7), and unique ubiquitination sites in MCL1 (LGK136R), HK1 (HEK101N and GFK624A), MFN2 (QDK171Q), Fis1 (LPK64G), and Hspa1a (DMK88H) that may differentially affect protein fate.
Functionally, Parkin-induced degradation of RHOT1 and MFN2 suppressed mitochondrial dynamics in tumor cells, inhibiting the subcellular movements of mitochondria and their accumulation at the cortical cytoskeleton. Consistent with an important role of mitochondria in cancer (5), changes in organelle size, shape, and subcellular distribution, i.e., mitochondrial dynamics, have been associated with various traits of advanced disease (10), in particular tumor cell motility, invasion, and metastatic competence (9). Specifically, cortical mitochondria act as regional energy sources (36, 37) fueling membrane lamellipodia dynamics, FA turnover, and Rho guanosine triphosphatase (GTPase) signaling (38) to increase cell movements (39). Consistent with a role of Parkin in mitochondrial dynamics via ubiquitination of MFN proteins (40), MFN2 and RHOT1 have been recognized as important effectors of tumor chemotaxis (41), influencing Myc-driven metastasis (42).
As one of the new targets uncovered here, Parkin induced degradation of the non-oxidative PPP enzyme TKT (4), resulting in glycolytic starvation, loss of ATP production, and generation of mitochondrial ROS in tumor cells. This is consistent with an emerging role of PPP anabolic and antioxidant mechanisms in cancer, exploited for tumor growth (43), therapy resistance (44), and NADPH-dependent antioxidant responses (29). Here, Parkin-induced metabolic and oxidative stress may contribute to the observed inhibition of tumor growth in vivo, whereas ROS have been implicated as potent antagonists of mitochondrial trafficking, tumor cell motility, and invasion (30). The ability of antioxidants (or TKT) to reverse Parkin blockade of mitochondrial trafficking and restore tumor cell movements is consistent with this scenario, reinforcing a critical role of redox balance in tumor progression (45), including metastatic dissemination (8, 46).
In summary, the data presented here identify glycolytic and mitochondrial reprogramming not only as hallmarks of tumor metabolism (5) but also as potent, disease-driving oncogenes targeted by endogenous tumor suppression mechanisms. Uniformly lost in patients with genetically disparate malignancies, Parkin emerged as a critical, stress-regulated effector of this tumor suppression pathway, activating metabolic and oxidative “danger” signals to antagonize malignant cell proliferation and metastatic competence in vivo (Fig. 7). Although novel for Parkin and at variance with its neuroprotective function in PD (47), this model is reminiscent of other cardinal tumor suppressors (48). Like Parkin, stress-regulated stabilization of p53 also suppresses mitochondrial dynamics (49), increases ROS (50), and triggers metabolic unbalance (51) via inhibition of glycolysis (52) and the PPP (53).
METHODS
Patient material
A cancer universal tissue microarray utilized in a previous study (54) was used to investigate the expression of Parkin in different tumor types by IHC. Parkin expression was scored as the percentage of positive tumor cells out of the total number of tumor cells in each core. Normal brain cortex was used as positive control.
Cells and cell culture
Prostate adenocarcinoma PC3 and DU145, neuroblastoma SKN, glioblastoma LN229, ovarian cancer OVCAR8, breast epithelial MCF10A, and cervical carcinoma HeLa cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and maintained in culture according to the supplier’s specifications. MEFs were prepared from WT or Parkin KO mice. Cell passaging was limited to <40 passages from receipt, and cell lines were authenticated by STR (Short Tandem Repeat) profiling with AmpFLSTR Identifiler PCR Amplification Kit (Life Technologies) at the Wistar Institute’s Genomics facility. Mycoplasma-free cultures were confirmed at the beginning of the studies and every 2 months afterward by direct polymerase chain reaction (PCR) of cultures using Bioo Scientific Mycoplasma Primer Sets (catalog no. 375501) and Hot Start polymerase (QIAGEN). Conditioned medium was prepared from exponentially growing cultures of NIH3T3 cells (ATCC) maintained in Dulbecco’s modified Eagle’s medium supplemented with d-glucose (4.5 g/liter), sodium pyruvate, 10 mM Hepes, and 10% fetal bovine serum for 48 hours.
Antibodies and reagents
Antibodies to Parkin, PINK1, MFN1, and MFN2 were acquired from Cell Signaling Technology. The antibody to Parkin used for IHC was from LSBio and used at 1:500 dilution. Antibodies to TKT and vinculin were from Abcam. Antibodies to Src, Tyr416-phosphorylated Src, FAK, and Tyr925- or Tyr397-phosphorylated FAK were from Cell Signaling Technology. Antibodies to β-actin and RHOT1 were from Sigma-Aldrich and Santa Cruz Biotechnology, respectively. MitoTracker Green, Phalloidin Alexa Fluor 488, MitoTracker Deep Red FM, and secondary antibodies used in immunofluorescence studies were from Molecular Probes. FCCP, MitoTempo, and etoposide were purchased from Sigma-Aldrich. BafA was purchased from Cell Signaling Technology. NADPH was determined by PicoProbe NADPH quantification fluorometric assay kit from BioVision. Kits for glucose uptake, lactate production, and TKT activity were purchased from BioVision.
Plasmids and transfections
Human complementary DNA (cDNA) encoding Parkin and RHOT1 was purchased from GeneCopoeia. cDNA plasmids encoding TKT, MFN1, and MFN2 were obtained from OriGene. In some experiments, PC3 or SKN cells were transfected with vector or Parkin cDNA, and stably expressing clones were selected in the presence of neomycin for 14 days and confirmed by Western blotting. E3 ligase loss-of-function Parkin cDNA mutants S65A and C431S were purchased from Addgene. Ubiquitination-defective plasmids for RHOT1 (K194R), MFN2 (K171R), and TKT (K499R) were generated using a Stratagene QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies) and confirmed by DNA sequencing.
For transfection, cells were mixed with 2 μg of the various cDNA constructs plus 4 μl of X-tremeGENE HP (Roche) in complete medium for 24 hours, washed, and processed for individual experiments. Gene knockdown experiments by siRNA were carried out as described (54). The following siRNA sequences were used: control, ON-TARGETplus non-targeting siRNA pool (Dharmacon), Parkin-directed siRNA (Sigma-Aldrich), PINK1-directed siRNA (Dharmacon), and TKT-directed siRNA (Dharmacon). The various tumor cell types were transfected with the individual siRNA pools at 40 nM in Lipofectamine RNAiMAX (Invitrogen) at a 1:1 ratio (vol siRNA 20 μM/vol Lipofectamine RNAiMAX). After 72 hours, transfected cells were validated for target protein knockdown by Western blotting and processed for functional experiments.
Protein analysis
Protein lysates were prepared in radioimmunoprecipitation assay buffer [150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM tris (pH 8.0)] in the presence of EDTA-free Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail (Roche). Equal amounts of protein lysates were separated by SDS gel electrophoresis, transferred to polyvinylidene difluoride membranes, and incubated with primary antibodies of various specificities. Protein bands were visualized by chemiluminescence. For ubiquitination analysis, cells were washed with 10 mM N-ethylmaleimide containing phosphate-buffered saline (PBS). The cell pellet was lysed by sonication in 50 μl of 2% SDS containing 50 mM tris-HCl (pH 7.5) and boiled in a heat block for 10 min at 95°C. A total of 950 μl of 50 mM tris-HCl (pH 7.5) buffer was added to the boiled lysates followed by centrifugation.
Mitochondrial ROS and inner membrane potential
Parkin-expressing PC3 cells were stained with MitoSOX Red mitochondrial superoxide indicator (5 mM; Thermo Fisher Scientific) for 10 min in complete medium. To quantify mitochondrial inner membrane potential, Parkin-expressing cells were incubated in the presence or absence of etoposide (50 μM) for 48 hours and stained with tetramethylrhodamine, ethyl ester (10 μM) for 30 min. Cells were washed in PBS (pH 7.4) and analyzed on a FACSCalibur flow cytometer. Intact cells were gated in the forward scatter/side scatter (FSC/SSC) plot to exclude small debris.
Mitochondrial mass
PC3 cells expressing Parkin or control vector were treated with or without the mitochondrial uncoupler FCCP (20 μM) for 16 hours and stained with MitoTracker Green (5 mM; Thermo Fisher Scientific) for 1 hour in complete medium. After washes in PBS (pH 7.4), cells were analyzed on a FACSCalibur flow cytometer. Intact cells were gated in the FSC/SSC plot to exclude small debris.
SILAC ubiquitome and global proteomic analyses
PC3 cells expressing Parkin (heavy SILAC labeled with 13C615N4 l-arginine, 13C615N2 l-lysine) or vector (light SILAC labeled) were processed in duplicate for ubiquitome analysis using the PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit (Cell Signaling Technology). Briefly, 20 mg of 1:1 mix of heavy- and light-labeled lysates was reduced with dithiothreitol, alkylated with iodoacetamide, and digested in solution with trypsin as described previously (55, 56). Tryptic peptides were desalted using Sep-Pak C18 (Waters), and ubiquitinated peptides were enriched using the K-ε-GG antibody. Samples were subjected to replicate liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis using a 4-hour LC gradient on a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) as described previously (57). For global proteome analysis without immune affinity enrichment, 100 μg of in-solution digested samples was fractionated into 10 fractions using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo Fisher Scientific) and analyzed in replicate by LC-MS/MS.
MS data were analyzed using MaxQuant 1.6.8.0 (58). MS/MS spectra were searched against a UniProt human protein database (October 2019) and a common contaminants database using full tryptic specificity with up to five missed cleavages and static carbamidomethylation of Cys. Variable modifications included Met oxidation, Lys carbamylation, and peptide N-terminal carbamylation. Variable diglycine addition to Lys was also considered in the search of ubiquitome samples. Consensus identification lists were generated with false discovery rates of 1% at protein, peptide, and site levels. Reverse hits, contaminants, and identifications without any H/L (Heavy/Light) ratio were removed from all datasets. Ubiquitinated sites were determined from the GlyGly (K)Sites.txt table. For global proteome analysis, protein identifications were obtained from the proteinGroups.txt table. Mitochondrial proteins were identified from the MitoCarta 3.0 database. For global proteome network analysis, proteins were required to have a consistent minimum fold change of 1.45 and a minimum SILAC ratio count of 2. Mitochondrial proteins were required to have a consistent minimum fold change of 1.3 and a minimum ratio count of 2 for network analysis.
Lipidomics screening
Samples were spiked with EquiSPLASH mix (Avanti Polar Lipids), and lipid species were identified by LipidSearch 4.2 (Thermo Fisher Scientific) from MS/MS spectra with 5 ppm (parts per million) precursor and product ion mass tolerances and were filtered by expected adduct and identification quality. Peak areas were used for quantification and were corrected by EquiSPLASH lipids for represented classes and normalized to protein amount in each sample. Lipid classes were quantified by summing peak areas of all species in a given class. For saturation analysis, fatty acids incorporated into lipids were assigned as SFA, MUFA, or PUFA based on the number of carbon double bonds (0, 1, or >1, respectively). Each lipid species with fatty acid level identification was weighted by its number of fatty acids of each type. SFA, MUFA, and PUFA levels for each class in a sample were determined by summing the weighted peak areas for lipid species in the class.
Total fatty acid quantification
Lipid extracts were saponified with 0.3 M KOH in 90% methanol at 80°C. Samples were acidified with formic acid, and fatty acids were extracted with hexane. LC-MS was performed in negative ion mode on a Thermo Fisher Scientific Q-Exactive HF-X mass spectrometer and a Vanquish Horizon UHPLC system. Gradient LC separation used an Accucore C18 column (2.1 mm × 150 mm; Thermo Fisher Scientific) with water and acetonitrile solvents containing 0.1% acetic acid. Full MS scans were acquired at 120,000 resolution from 180 to 650 m/z (mass/charge ratio). Fatty acids were identified by accurate mass and retention time based on pure standards and quantified by peak area using TraceFinder 4.1 (Thermo Fisher Scientific).
Quantification of PPP flux
Cells were labeled until isotopic steady state with [1,2-13C]glucose to quantify 13C incorporation into intermediates of glycolysis and PPP. Polar metabolites were extracted with 80% methanol and analyzed by LC-MS as described previously (57). Data analysis was performed using TraceFinder 4.1 (Thermo Fisher Scientific). Metabolites were identified by accurate mass and retention time based on pure standards, and all possible carbon isotopologs determined from chemical formulas were quantified by peak area. Data were corrected for natural 13C abundance and isotope tracer purity (99%) using IsoCorrectoR (59). Direct glycolysis produces M+2-labeled metabolites, while PPP produces M+1-labeled metabolites. The ratio of M+1 to M+2 labeling for a given metabolite indicates the ratio of flux going through the PPP versus glycolysis. PPP flux was calculated using the following formula: Glucose consumption rate × (M+1 lactate/(M+1 lactate + M+2 lactate)).
FA dynamics
LN229 cells were transfected with vector or Parkin for 24 hours, plated on high optical quality 35-mm glass bottom plates, and transduced with Talin-GFP (green fluorescent protein) BacMam virus for 18 hours. Time-lapse videomicroscopy was carried out using a Leica TCS SP8 Scanning Laser Confocal Microscope system with an HCX PL APO CS 63× 1.40 numerical aperture (NA) oil ultraviolet objective. Acquisition of live cells using an integrated Leica LAS software was performed every 3 min per frame for a total interval of 2 hours. The time-lapse videomicroscopy movies were analyzed using the Focal Adhesion Analysis Server (FAAS) (https://faas.bme.unc.edu/) with quantification of rates of FA assembly and disassembly. In other experiments, LN229 cells transfected with vector or Parkin cDNA were stained for vinculin (1:100) to label FA followed by quantification of FA length and area using NIS-Elements.
Cortical mitochondria
Mitochondria/F-actin composite images were analyzed in ImageJ as described (60). For quantification of cortical mitochondria, a mask was manually created around the periphery of the cell based on the F-actin channel and subsequently applied to the mitochondrial channel to measure intensity at the cortical region. The intensity was normalized to total mitochondrial intensity per cell and background-subtracted. A minimum of 20 cells was analyzed in each independent experiment to obtain mean values.
Mitochondria time-lapse videomicroscopy
Cells (2 × 104) growing on high optical quality glass bottom 35-mm plates (MatTek Corporation) were incubated with 100 nM MitoTracker Deep Red FM dye for 30 min and imaged on a Leica TCS SP8 X inverted laser scanning confocal microscope using a 63× 1.40 NA oil objective. Short duration time-lapse sequences were carried out using a Tokai Hit incubation chamber equilibrated to 37°C and 5% CO2. Time lapse was performed for 6 min (3 s per frame). Individual 12-bit images were acquired using a white-light supercontinuum laser (0.2% at 645 nm) and HyD detectors at 5× digital zoom with a pixel size of 70 nm × 70 nm. A pinhole setting of 1 Airy units provided a section thickness of 0.896 μm. Each time point was captured with a step size of 0.15 μm. At least seven cells under each condition were collected for analysis. Initial postprocessing of three-dimensional (3D) sequences was carried out with Huygens software to deconvolve the images, and then they were imported into LAS X software to study fission and fusion events. A workflow capable of tracking time-dependent changes in mitochondrial volume was designed on the LAS X software platform. For each cell, the volume of mitochondria over time was analyzed in four different areas (with an average of 10 mitochondria per area) in 3D images. Variations in mitochondrial volume were evaluated by fold change over time: A fold change of >1.3 denoted a fusion event, and a fold change of <0.7 denoted a fission event. The average fission and fusion events in the four different areas were used for each cell analyzed.
Mitophagy assay
PC3 cells stably expressing mitochondrial-targeted Keima-Red (Addgene, catalog no. 56018) were transfected with vector or Parkin, detached with trypsin, washed, and suspended in PBS. FCCP-treated cells (20 μM for 16 hours) were used as positive control. Cells were analyzed on an LSR 18 flow cytometer at 405- and 561-nm lasers and 610/20 filters, and the percentage of PE/Texas Red–positive cells and Brilliant Violet (BV605) cells were calculated. The ratio of PR/BV605-positive cells, representative of mitophagy, was quantified. Intact cells were gated in the FSC/SSC plot to exclude small debris.
Single-cell motility analysis
Experiments were carried out essentially as described using 2D chemotaxis chambers (Ibidi) and a gradient setup with NIH3T3 conditioned medium (42). Videomicroscopy was performed over 10 hours, with a time-lapse interval of 10 min. Stacks were imported into ImageJ, and images were aligned according to subpixel intensity registration with the StackReg plugin for ImageJ43. At least 30 cells were tracked using the Manual Tracking plugin for ImageJ, and the tracking data from four independent time-lapse experiments were pooled and exported into Chemotaxis and Migration Tool v2.0 (Ibidi) for calculation of quantitative parameters of speed of cell movements and total distance traveled by individual cells. In some experiments, a forward migration index and Rayleigh distribution statistics were used to quantify directional compared to random cell movements.
Cell invasion
Experiments were carried out as described (42) using Growth Factor Reduced Matrigel–coated 8-mm PET (Polyethylene terepthalate) Transwell chambers (Corning) and NIH3T3 conditioned medium placed in the lower chamber as chemoattractant. Cells were allowed to invade for 16 to 24 hours, noninvading cells were scraped off the top side of the membranes, and the invaded cells on the Transwell insert were fixed in methanol. Membranes were mounted in medium containing DAPI (4′,6-diamidino-2-phenylindole) (Vector Labs) and analyzed by fluorescence microscopy. Five random fields at ×10 magnification were collected for each membrane. Digital images were batch-imported into ImageJ, thresholded, and analyzed with the Analyze particle function.
Transmission electron microscopy
PC3 cells were transfected with vector or Parkin for 72 hours. Cells were fixed for 16 hours with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer. After washes, samples were post-fixed in 2% osmium tetroxide for 1 hour at 22°C and rinsed in H2O before en bloc staining with 2% uranyl acetate. After dehydration through a graded ethanol series, samples were infiltrated and embedded in EMbed 812 (Electron Microscopy Sciences, Fort Washington, PA). Sections were stained with uranyl acetate and lead citrate and imaged with a JEOL 1010 electron microscope at 50,000×.
Parkin expression analysis
Analysis of TCGA tumor expression data for Parkin mRNA (RNA sequencing values) was performed using the FireBrowse portal (http://firebrowse.org) to compare differential expression in cancer versus the corresponding normal tissues. The CCLE (https://portals.broadinstitute.org/ccle/) portal was used to evaluate Parkin mRNA expression and copy number variation. Individual cancer cell lines were grouped by primary tissue of origin according to the CCLE classification and plotted along with copy number variation. Proteomics data from the CCLE database containing 12,755 proteins from 378 cell lines were downloaded (61) and analyzed for Parkin expression in 42 batches (“ten-plex” experiments). Only 1 of the 42 batches had Parkin detected (identifier sp|O60260|PRKN2_HUMAN) and only with one peptide.
Animal studies
Experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). Protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of The Wistar Institute. Sample size was determined by power analysis. All animals were included in the analysis. Surgical procedures for a liver metastasis model were carried out in isoflurane-anesthetized animals following aseptic technique inside a biosafety cabinet, and a slow-release buprenorphine formulation was administered for pain relief. PC3 cells stably expressing vector or Parkin at 80% confluency were suspended in PBS, and 1 × 106 cells were injected (50 μl) in the spleen of anesthetized 6- to 8-week-old male NOD SCID γ (NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (The Jackson Laboratory). After 24 hours, the spleen was surgically removed, and animals were euthanized 11 days later. Metastatic dissemination to liver was quantified by morphometry, and the number and surface area of metastatic foci were calculated under the various conditions tested. In other experiments, PC3 cells stably transduced with vector or Parkin were engrafted subcutaneously on the flanks of NOD SCID γ mice (10 animals per group), and tumor growth was measured with a caliper throughout a 26-day interval. Lungs collected from the various animal groups were analyzed for metastatic dissemination by reactivity with an antibody to human leukocyte antigen (HLA) by IHC.
Statistical analysis
Data are expressed as mean ± SD of results from a minimum of three independent experiments. Unpaired, two-tailed Student’s t test was used for two-group comparative analyses. In some cases, correction for multiple testing by the Benjamini-Hochberg procedure was obtained. For multiple-group comparisons, analysis of variance (ANOVA) was used. Rayleigh distribution statistics were used for experiments of cellular chemotaxis. All statistical analyses were performed using the GraphPad software package (Prism 9.0) for Windows. P < 0.05 was considered statistically significant.
Acknowledgments
We thank J. Hayden and F. Keeney of the Wistar Imaging Core Facility for assistance with time-lapse videomicroscopy as well as the staff of the Wistar Proteomics and Metabolomics Core Facility for LC-MS/MS analysis. We thank the Electron Microscopy Core at the University of Pennsylvania for assistance with transmission electron microscopy. Funding: This work was supported by NIH grants P01 CA140043 (D.C.A., L.R.L., and D.W.S.), R35 CA220446 (D.C.A.), R50 CA221838 (H.-Y.T.), and R50 CA211199 (A.V.K.). The Thermo Q-Exactive HF-X mass spectrometer was purchased with NIH grant S10 OD023586. Author contributions: E.A. and D.C.A. conceived the project. E.A. performed experiments of mitochondrial dynamics, subcellular mitochondrial trafficking, and tumor cell motility and invasion, including mouse models of metastasis in vivo. J.C.G. performed experiments of tumor metabolic reprogramming. A.V.K. analyzed bioinformatics data. H.-Y.T., A.R.G., and D.W.S. performed proteomics and metabolomics experiments. V.V. analyzed primary patient samples for differential Parkin expression in a universal cancer tissue microarray by immunohistochemistry. E.A., L.R.L., D.W.S., and D.C.A. analyzed data. E.A. and D.C.A. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The raw data files listing all the proteins and ubiquitylation sites have been uploaded to the MassIVE public repository and can be accessed at https://massive.ucsd.edu/ with the accession MSV000087097. Reviewer login credentials are MSV000087097_reviewer (username) and WiS7ar1nS7 (password).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabg7287/DC1
REFERENCES AND NOTES
- 1.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Jia D., Lu M., Jung K. H., Park J. H., Yu L., Onuchic J. N., Kaipparettu B. A., Levine H., Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 116, 3909–3918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potter M., Newport E., Morten K. J., The Warburg effect: 80 years on. Biochem. Soc. Trans. 44, 1499–1505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Payen V. L., Porporato P. E., Baselet B., Sonveaux P., Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway. Cell. Mol. Life Sci. 73, 1333–1348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis R. J., Chandel N. S., Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Reyes I., Cardona L. R., Kong H., Vasan K., McElroy G. S., Werner M., Kihshen H., Reczek C. R., Weinberg S. E., Gao P., Steinert E. M., Piseaux R., Budinger G. R. S., Chandel N. S., Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan A. S., Baty J. W., Dong L. F., Bezawork-Geleta A., Endaya B., Goodwin J., Bajzikova M., Kovarova J., Peterka M., Yan B., Pesdar E. A., Sobol M., Filimonenko A., Stuart S., Vondrusova M., Kluckova K., Sachaphibulkij K., Rohlena J., Hozak P., Truksa J., Eccles D., Haupt L. M., Griffiths L. R., Neuzil J., Berridge M. V., Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 21, 81–94 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Piskounova E., Agathocleous M., Murphy M. M., Hu Z. P., Huddlestun S. E., Zhao Z. Y., Leitch A. M., Johnson T. M., DeBerardinis R. J., Morrison S. J., Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altieri D. C., Mitochondrial dynamics and metastasis. Cell. Mol. Life Sci. 76, 827–835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Senft D., Ronai Z. A., Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 39, 43–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vyas S., Zaganjor E., Haigis M. C., Mitochondria and cancer. Cell 166, 555–566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernardini J. P., Lazarou M., Dewson G., Parkin and mitophagy in cancer. Oncogene 36, 1315–1327 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N., Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Pickles S., Vigie P., Youle R. J., Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Youle R. J., Narendra D. P., Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iguchi M., Kujuro Y., Okatsu K., Koyano F., Kosako H., Kimura M., Suzuki N., Uchiyama S., Tanaka K., Matsuda N., Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 288, 22019–22032 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lazarou M., Sliter D. A., Kane L. A., Sarraf S. A., Wang C., Burman J. L., Sideris D. P., Fogel A. I., Youle R. J., The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veeriah S., Taylor B. S., Meng S., Fang F., Yilmaz E., Vivanco I., Janakiraman M., Schultz N., Hanrahan A. J., Pao W., Ladanyi M., Sander C., Heguy A., Holland E. C., Paty P. B., Mischel P. S., Liau L., Cloughesy T. F., Mellinghoff I. K., Solit D. B., Chan T. A., Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 42, 77–82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulogiannis G., McIntyre R. E., Dimitriadi M., Apps J. R., Wilson C. H., Ichimura K., Luo F., Cantley L. C., Wyllie A. H., Adams D. J., Arends M. J., PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. U.S.A. 107, 15145–15150 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong Y., Zack T. I., Morris L. G., Lin K., Hukkelhoven E., Raheja R., Tan I. L., Turcan S., Veeriah S., Meng S., Viale A., Schumacher S. E., Palmedo P., Beroukhim R., Chan T. A., Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 46, 588–594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C., Lin M., Wu R., Wang X., Yang B., Levine A. J., Hu W., Feng Z., Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. U.S.A. 108, 16259–16264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansson B., Jankovic J., Low cancer rates among patients with Parkinson’s disease. Ann. Neurol. 17, 505–509 (1985). [DOI] [PubMed] [Google Scholar]
- 23.Park K. R., Yun J. S., Park M. H., Jung Y. Y., Yeo I. J., Nam K. T., Kim H. D., Song J. K., Choi D. Y., Park P. H., Han S. B., Yun H. M., Hong J. T., Loss of parkin reduces lung tumor development by blocking p21 degradation. PLOS ONE 14, e0217037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta A., Anjomani-Virmouni S., Koundouros N., Dimitriadi M., Choo-Wing R., Valle A., Zheng Y., Chiu Y. H., Agnihotri S., Zadeh G., Asara J. M., Anastasiou D., Arends M. J., Cantley L. C., Poulogiannis G., PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol. Cell 65, 999–1013.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C., Zhang Y., Cheng X., Yuan H., Zhu S., Liu J., Wen Q., Xie Y., Liu J., Kroemer G., Klionsky D. J., Lotze M. T., Zeh H. J., Kang R., Tang D., PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev. Cell 46, 441–455.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J., Zhang C., Wu H., Sun X. X., Li Y., Huang S., Yue X., Lu S. E., Shen Z., Su X., White E., Haffty B. G., Hu W., Feng Z., Parkin ubiquitinates phosphoglycerate dehydrogenase to suppress serine synthesis and tumor progression. J. Clin. Invest. 130, 3253–3269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarraf S. A., Raman M., Guarani-Pereira V., Sowa M. E., Huttlin E. L., Gygi S. P., Harper J. W., Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roussos E. T., Condeelis J. S., Patsialou A., Chemotaxis in cancer. Nat. Rev. Cancer 11, 573–587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayes J. D., Dinkova-Kostova A. T., Tew K. D., Oxidative stress in cancer. Cancer Cell 38, 167–197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Debattisti V., Gerencser A. A., Saotome M., Das S., Hajnoczky G., ROS control mitochondrial motility through p38 and the motor adaptor miro/trak. Cell Rep. 21, 1667–1680 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghosh J. C., Seo J. H., Agarwal E., Wang Y., Kossenkov A. V., Tang H. Y., Speicher D. W., Altieri D. C., Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene 38, 6926–6939 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouton-Liger F., Jacoupy M., Corvol J. C., Corti O., PINK1/parkin-dependent mitochondrial surveillance: From pleiotropy to Parkinson’s disease. Front. Mol. Neurosci. 10, 120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humpton T. J., Alagesan B., DeNicola G. M., Lu D., Yordanov G. N., Leonhardt C. S., Yao M. A., Alagesan P., Zaatari M. N., Park Y., Skepper J. N., Macleod K. F., Perez-Mancera P. A., Murphy M. P., Evan G. I., Vousden K. H., Tuveson D. A., Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 9, 1268–1287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villa E., Proics E., Rubio-Patino C., Obba S., Zunino B., Bossowski J. P., Rozier R. M., Chiche J., Mondragon L., Riley J. S., Marchetti S., Verhoeyen E., Tait S. W. G., Ricci J. E., Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. 20, 2846–2859 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Ordureau A., Paulo J. A., Zhang W., Ahfeldt T., Zhang J., Cohn E. F., Hou Z., Heo J. M., Rubin L. L., Sidhu S. S., Gygi S. P., Harper J. W., Dynamics of PARKIN-dependent mitochondrial ubiquitylation in induced neurons and model systems revealed by digital snapshot proteomics. Mol. Cell 70, 211–227.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunniff B., McKenzie A. J., Heintz N. H., Howe A. K., AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 27, 2662–2674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desai S. P., Bhatia S. N., Toner M., Irimia D., Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J. 104, 2077–2088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caino M. C., Chae Y. C., Vaira V., Ferrero S., Nosotti M., Martin N. M., Weeraratna A., O'Connell M., Jernigan D., Fatatis A., Languino L. R., Bosari S., Altieri D. C., Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Invest. 123, 2907–2920 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuler M. H., Lewandowska A., Caprio G. D., Skillern W., Upadhyayula S., Kirchhausen T., Shaw J. M., Cunniff B., Miro1-mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol. Biol. Cell 28, 2159–2169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou J., Eldeeb M., Wang X., Beyond deubiquitylation: USP30-mediated regulation of mitochondrial homeostasis. Adv. Exp. Med. Biol. 1038, 133–148 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Zhou W., Hsu A. Y., Wang Y., Syahirah R., Wang T., Jeffries J., Wang X., Mohammad H., Seleem M. N., Umulis D., Deng Q., Mitofusin 2 regulates neutrophil adhesive migration and the actin cytoskeleton. J. Cell Sci. 133, jcs248880 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agarwal E., Altman B. J., Ho Seo J., Bertolini I., Ghosh J. C., Kaur A., Kossenkov A. V., Languino L. R., Gabrilovich D. I., Speicher D. W., Dang C. V., Altieri D. C., Myc regulation of a mitochondrial trafficking network mediates tumor cell invasion and metastasis. Mol. Cell. Biol. 39, e00109-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dasgupta S., Rajapakshe K., Zhu B., Nikolai B. C., Yi P., Putluri N., Choi J. M., Jung S. Y., Coarfa C., Westbrook T. F., Zhang X. H.-F., Foulds C. E., Tsai S. Y., Tsai M.-J., O’Malley B. W., Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nature 556, 249–254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding Y., Gong C., Huang R. C., Sui P., Lin K. H., Liang G., Yuan L., Xiang H., Chen J., Yin T., Alexander P. B., Wang Q.-F., Song E.-W., Li Q.-J., Wood K. C., Wang X.-F., Synthetic lethality between HER2 and transaldolase in intrinsically resistant HER2-positive breast cancers. Nat. Commun. 9, 4274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R. S., Chandel N. S., Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Gal K., Ibrahim M. X., Wiel C., Sayin V. I., Akula M. K., Karlsson C., Dalin M. G., Akyürek L. M., Lindahl P., Nilsson J., Bergo M. O., Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 7, 308re308 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Devine M. J., Plun-Favreau H., Wood N. W., Parkinson’s disease and cancer: Two wars, one front. Nat. Rev. Cancer 11, 812–823 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Levine A. J., p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 20, 471–480 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Moulder D. E., Hatoum D., Tay E., Lin Y., McGowan E. M., The roles of p53 in mitochondrial dynamics and cancer metabolism: The pendulum between survival and death in breast cancer? Cancers 10, 189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maillet A., Pervaiz S., Redox regulation of p53, redox effectors regulated by p53: A subtle balance. Antioxid. Redox Signal. 16, 1285–1294 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Kruiswijk F., Labuschagne C. F., Vousden K. H., p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 16, 393–405 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Schwartzenberg-Bar-Yoseph F., Armoni M., Karnieli E., The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 64, 2627–2633 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Bensaad K., Tsuruta A., Selak M. A., Vidal M. N., Nakano K., Bartrons R., Gottlieb E., Vousden K. H., TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Seo J. H., Rivadeneira D. B., Caino M. C., Chae Y. C., Speicher D. W., Tang H. Y., Vaira V., Bosari S., Palleschi A., Rampini P., Kossenkov A. V., Languino L. R., Altieri D. C., The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLOS Biol. 14, e1002507 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bengsch F., Tu Z., Tang H. Y., Zhu H., Speicher D. W., Zhang R., Comprehensive analysis of the ubiquitinome during oncogene-induced senescence in human fibroblasts. Cell Cycle 14, 1540–1547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu H., Le L., Tang H. Y., Speicher D. W., Zhang R., Detection of the ubiquitinome in cells undergoing oncogene-induced senescence. Methods Mol. Biol. 1534, 127–137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J., Agarwal E., Bertolini I., Seo J. H., Caino M. C., Ghosh J. C., Kossenkov A. V., Liu Q., Tang H. Y., Goldman A. R., Languino L. R., Speicher D. W., Altieri D. C., The mitophagy effector FUNDC1 controls mitochondrial reprogramming and cellular plasticity in cancer cells. Sci. Signal. 13, eaaz8240 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Heinrich P., Kohler C., Ellmann L., Kuerner P., Spang R., Oefner P. J., Dettmer K., Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci. Rep. 8, 17910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caino M. C., Seo J. H., Aguinaldo A., Wait E., Bryant K. G., Kossenkov A. V., Hayden J. E., Vaira V., Morotti A., Ferrero S., Bosari S., Gabrilovich D. I., Languino L. R., Cohen A. R., Altieri D. C., A neuronal network of mitochondrial dynamics regulates metastasis. Nat. Commun. 7, 13730 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nusinow D. P., Szpyt J., Ghandi M., Rose C. M., McDonald E. R. III, Kalocsay M., Jane-Valbuena J., Gelfand E., Schweppe D. K., Jedrychowski M., Golji J., Porter D. A., Rejtar T., Wang Y. K., Kryukov G. V., Stegmeier F., Erickson B. K., Garraway L. A., Sellers W. R., Gygi S. P., Gygi, quantitative proteomics of the cancer cell line encyclopedia. Cell 180, 387–402.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/35/eabg7287/DC1