

ABSTRACT
Coronaviruses are commonly characterized by a unique discontinuous RNA transcriptional synthesis strategy guided by transcription-regulating sequences (TRSs). However, the details of RNA synthesis in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have not been fully elucidated. Here, we present a time-scaled, gene-comparable transcriptome of SARS-CoV-2, demonstrating that ACGAAC functions as a core TRS guiding the discontinuous RNA synthesis of SARS-CoV-2 from a holistic perspective. During infection, viral transcription, rather than genome replication, dominates all viral RNA synthesis activities. The most highly expressed viral gene is the nucleocapsid gene, followed by ORF7 and ORF3 genes, while the envelope gene shows the lowest expression. Host transcription dysregulation keeps exacerbating after viral RNA synthesis reaches a maximum. The most enriched host pathways are metabolism related. Two of them (cholesterol and valine metabolism) affect viral replication in reverse. Furthermore, the activation of numerous cytokines emerges before large-scale viral RNA synthesis.
IMPORTANCE SARS-CoV-2 is responsible for the current severe global health emergency that began at the end of 2019. Although the universal transcriptional strategies of coronaviruses are preliminarily understood, the details of RNA synthesis, especially the time-matched transcription level of each SARS-CoV-2 gene and the principles of subgenomic mRNA synthesis, are not clear. The coterminal subgenomic mRNAs of SARS-CoV-2 present obstacles in identifying the expression of most genes by PCR-based methods, which are exacerbated by the lack of related antibodies. Moreover, SARS-CoV-2-related metabolic imbalance and cytokine storm are receiving increasing attention from both clinical and mechanistic perspectives. Our transcriptomic research provides information on both viral RNA synthesis and host responses, in which the transcription-regulating sequences and transcription levels of viral genes are demonstrated, and the metabolic dysregulation and cytokine levels identified at the host cellular level support the development of novel medical treatment strategies.
KEYWORDS: SARS-CoV-2, transcriptome, cholesterol, valine, TNF, transcription-regulating sequence
INTRODUCTION
The outbreak of coronavirus disease 19 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has brought about a global health panic and led to more than 120 million cases of infection and 2 million deaths (WHO, as of March 26th, 2021) (1). SARS-CoV-2 has a positive-sense, single-stranded RNA genome of approximately 30 kb, similar to that of SARS-CoV (2), and thus belongs to the family Coronaviridae in the order Nidovirales. Coronaviruses (CoVs) carry the largest genomes of all RNA viruses and employ a distinct RNA synthesis mechanism. Upon invading host cells, the first 5′ open reading frame 1ab (ORF1ab) of the viral genomic RNA (gRNA) is translated to generate polyprotein 1a and polyprotein 1ab, which mainly function as the RNA synthetase complex to execute both genome replication and genome transcription, yielding gRNA and subgenomic mRNAs (sgmRNAs), respectively (reviewed in reference 3). Although genome replication is accomplished through a regular continuous process, the sgmRNA used for transcription is synthesized in a discontinuous step guided by RNA signals known as transcription-regulating sequences (TRSs), commonly located in the 5′ untranslated region (UTR) (known as the leader TRS) and in adjacent regions upstream of the 5′ end of each ORF (known as body TRSs) (Fig. 1A) (4). TRSs contain a conserved 6- to 7-nucleotide (nt) core sequence (core TRS) surrounded by variable sequences. During negative-strand synthesis, the viral RNA synthetase pauses when it crosses a body TRS and switches to the leader TRS as a template, which results in a discontinuous leader-to-body fusion transcript (5). Positive-strand sgmRNAs are transcribed from the fused negative-strand intermediates. Finally, from 5′ to 3′, an sgmRNA is composed of a sequence identical to the genomic 5′ UTR with a TRS (referred to as a leader UTR in an sgmRNA), the ORF to be translated, and downstream, nontranslated ORFs with a coterminus. In addition to ORF1ab, the SARS-CoV-2 genome contains 8 to 9 canonical ORFs according to the current annotation (2), including the widely accepted spike (S), envelope (E), membrane (M), and nucleocapsid (N) structural proteins, as well as several accessory proteins (ORF3, ORF6, ORF7, ORF8) that are separated by body TRSs; however, it has been reported that some noncanonical sgmRNAs are also generated (6). As its close relative, SARS-CoV produces 8 sgmRNAs (4) whose synthesis is directed by the conserved core TRS ACGAAC, as demonstrated by preliminary studies (7). Although the same sequences exist in the SARS-CoV-2 genome and are predicted to be the core TRSs (2), the putative TRSs reported in the SARS-2-CoV transcriptome (6, 8) do not correspond well with previous genomic results. Thus, further studies are needed to reveal the detailed mechanism guiding SARS-CoV-2 RNA synthesis, in which a key issue will be the core TRS. Moreover, the homology between gRNAs and coterminal sgmRNAs leads to difficulty in identifying the expression level changes in SARS-CoV-2 genes by conventional real-time quantitative RT-PCR (qRT-PCR), and this issue is exacerbated by the lack of available antibodies. SARS-CoV-2 infection induces substantial transcriptional dysregulation of the host in human cell lines and severe cytokine storms in bronchoalveolar cells in vivo (9–11). Using high-throughput RNA sequencing (RNA-Seq), we obtained time-scaled transcriptomes of both viral and host genes in SARS-CoV-2-infected human lung cells, in which the SARS-CoV-2 transcriptional mechanism was established and the dysregulation of several host pathways was discovered and verified.
FIG 1.

Transcription of SARS-Cov-2. (A) To transcribe the ORFs from S to N (8 ORFs shown in various colors), 8 corresponding sgmRNAs were discontinuously synthesized. In each sgmRNA, 5′ genomic sequences (from the 5′ leader UTR to the leader TRS) and 3′ genomic sequences (from the body TRS to the polyA tail) are fused, which is demonstrated with red dashed-solid lines; that is, each sgmRNA consists of a leader UTR (white rectangle with black frame) with a TRS (red circles), the ORF to be translated (colored rectangles; e.g., pink in S), downstream ORFs with a 3′ UTR (black rectangle), and a poly A tail. The 5′ structure of the S sgmRNA is enlarged (black dashed square). (B) Site-by-site sequencing depth of the SARS-CoV-2 genome from the +20,000 position to the 3′ end at 12 hpi. ORFs from S to N are shown with different colors. The junctions (red dashed circles) between ORF1ab and S, between S and ORF3, between E and M, and between ORF6 and ORF7 are enlarged in 4 inset figures. No shading is applied for the ORF1ab or sites not within the ORF (ORF gaps and 5′/3′ UTR of gRNA), with start sequences of repeated synthesis sites indicated at the junctions.
RESULTS
SARS-CoV-2 RNA synthesis.
The origins of each read at different time points after infection were evaluated by aligning the reads to the genome (Fig. S2A). Viral RNA was nearly undetectable from 0 to 6 hpi and still accounted for only a small percentage of total RNA (8.6%) at 12 hpi. The viral RNA percentage rose to 78% at 24 hpi and then remained relatively steady. Thus, viral genome replication and/or transcription increased sharply from 12 to 24 hpi. Mild cytopathy emerged as early as 24 hpi and then continued to intensify, which resulted in cell detachment and death at 96 hpi (data not shown). To avoid RNA degradation induced by cell death, RNAs were collected no later than 72 hpi. The viral genome copy number also reached a peak at 24 hpi and did not increase significantly thereafter (Fig. S2B). In the mock-treated cells, the percentage of viral RNA remained as low as 0.2%, indicating that the applied algorithm was reliable enough to distinguish between viral RNAs and host RNAs. It is worth noting that viral RNA accounted for as much as 80% of the total RNA present at 72 hpi, demonstrating that the virus comprehensively hijacked the biochemical and biophysical activities of the host cells.
At 12 hpi, the sequencing depth of ORF1ab (266 to 21,290 nt) was generally lower than 5,000 (Fig. S3A), as the corresponding RNA synthesis came exclusively from gRNA replication. The sequencing depth of sites at the downstream ORFs was generally higher than that at the upstream ORFs (Fig. 1B); the reason for this was that discontinuous sgmRNA transcription of coronaviruses (CoVs) leads to repeated synthesis of 3′ sequences. More specifically, each sgmRNA transcription induced a novel repeated synthesis starting around the beginning of the corresponding gene ORF (Fig. 1B, circles in red dotted lines), where the fused body TRSs were located. The repeated synthesis started either with a sequence of “TAAACGAAC…” for major cases (start sites of S, ORF3, M, ORF7, and N) or with a sequence of “ACGAAC…” for minor cases (start site of E, see Fig. S3B). Furthermore, the two sequences above were located around the beginning of each currently considered ORF and therefore were regarded as the core TRSs of SARS-CoV-2 (2). Considering that ORFs (S, ORF3, M, ORF7, and N) of SARS-CoV-2 are located end-to-end (the downstream body TRS is located adjacent to the stop codon sites of the upstream ORF) with TAA as the stop codon, it is highly probable that repeated synthesis of TAA stems from upstream ORFs rather than these sites functioning as true TRSs. For ORFs not located adjacent to the upstream stop codon (E), the repeated synthesis started with “ACGAAC…” rather than “TAAACGAAC…,” indicating that the former is actually the start site of the TRS. Furthermore, a portion of the 5′ UTR that ended with “…ACGAAC” (at the +75 site in the genome) was also found to be repeatedly synthesized (Fig. S3A), where the 5′ coleader of sgmRNAs was expected to occur. Taking this into account, ACGAAC could be the core TRS of SARS-CoV-2, and we provide more evidence for this below.
sgmRNA and TRS identification.
The repeated synthesis of probable TRSs was revealed by the sequencing depth of the genome sites in the above data. Conservatively existing in each sgmRNA, the TRSs of CoVs are short sequences where the leader UTR and the ORF are discontinuously fused. Through site-by-site query using query reads homologous to the 5′ UTR, the downstream return reads were analyzed to answer two questions. (i) Are the return reads able to be (partially) aligned to ORFs? If yes, the last nt of the corresponding query read still belongs to the conservative coleader (leader UTR with leader TRS) of sgmRNAs. If no, the nt is considered located in ORF1. (ii) What are the sequences in return reads that are aligned to the corresponding ORFs, and what are their respective abundances? From our results, the coleader sequence of sgmRNAs was extended as much as possible to a discontinuous fused point where the end of the leader TRS was exactly located (illustrated in Fig. S1). To explain the results in a clear way, the probable ACGAAC TRS upstream of the ORFs was designated on the order of A−6C−5G−4A−3A−2C−1, and the first site of each downstream ORF in the return reads was +1. Thus, the ending sites of the query reads applied were located at −7 to +5.
For each query read whose ending site was located at −7 to −1, the return reads containing all 8 canonical ORFs were observed and each of them revealed a stable abundance (Fig. 2), indicating that the coleader was extended at least to the −1 site in all sgmRNAs. However, discrepancies began to appear beginning at the +1 site. For query reads whose ending sites were located +3 downstream, no return reads existed except the continuous return reads from the gRNA (or ORF1), indicating that the coleader should be located in the upstream genomic region of the +3 site. A point of confusion arose with the results of +1 to +2 sites, which seemed to exist as a part of a specific leader sequences in the sgmRNAs of M, ORF3, and E. However, it is worth noting that the 5′ sequences in the M/ORF3/E ORFs downstream of “…A−6C−5G−4A−3A−2C−1” were identical to the +1 or +1 to +2 sites identified above. Hence, these sites could also be considered to stem from the ORF sequences (though they are displayed as a part of continuous leader in Fig. 2). For most return reads, the fused sites were extended to the −1 site, which led to the absence of 5′ UTR sequences downstream of the “…ACGAAC” in sgmRNAs (truncated at the red dashed line in Fig. 2). Thus, the leader TRS was considered to end at the −1 site. Moreover, for query reads with ending sites located at −7 to −2, the return reads were discontinuous, of which the 5′ sequences were aligned continuously to 5′ UTR and their 3′ sequences were aligned to the corresponding ORF. For the query read whose ending site was located at −1, the return reads were totally aligned to the ORFs (S to N). All of the identified ORFs were totally identical to the predicted ORFs revealed by a previous SARS-CoV-2 genome study (2), which are displayed downstream of the +1 sites in Fig. 2. Combined with the results revealed by repeated synthesis, a sequence of “…ACGAAC” was shown to conservatively exist in all canonical sgmRNAs, followed by the exact ORF sequences that had already been predicted, which indicated that ACGAAC was the core TRS of canonical sgmRNAs and that sgmRNAs were synthesized perfectly from leader to body without any addition, deletion, or mutation following a strict and conservative discontinuous strategy.
FIG 2.

Site-by-site query reveals the discontinuous and continuous RNA synthesis near the core TRS region. The sgmRNAs of 8 canonical genes are composed of a 5′ coleader (coleader UTR to the left of the TRS in the middle), specific ORFs, and polyA tails (right). The final nt of the query reads (−7 to +5 sites) is displayed on the horizontal axis. The site-matched return reads are aligned discontinuously with their 5′ sequences mapped to the coleader UTR and 3′ sequences aligned to probable ORFs (displayed on the vertical, including widely accepted S, ORF6, ORF7, ORF8, N, M, ORF3, and E, and truncated/mutated ORFs, including N1 and ORF7p1 to 3), except for the genomic reads (as gRNA, or ORF1), which are continuously aligned to the genome. The site-matched read counts are indicated by colored circles. The solid red outline indicates the coleader of the sgmRNAs and gRNA, which ends at the red dashed line for the reads of most sgmRNAs. The ORFs revealed by return reads of each sgmRNA are demonstrated downstream of the coleader, with coding sequences in italics. The discontinuous synthesis of N1 is indicated by a wavy line. A GAACttt sequence, underscored with a dashed line, exists upstream of the three truncated ORFs (ORF7p1 to ORF7p.3) and is a minor TRS used in noncanonical sgmRNA synthesis. The localization of their first sites relative to the stop codon of ORF7 is indicated by the superscript number with an underscore.
The homologous 5′ coleader between the sgmRNAs and the gRNA causes great difficulty in analyzing gene expression by qRT-PCR. In contrast, the monitoring of short reads of sgmRNAs containing TRSs is not subject to the same problem, and the read counts of return reads reflect the expression levels of the corresponding sgmRNAs. Moreover, gRNA synthesis and sgmRNA synthesis are both carried out by the RNA synthetase complex in CoVs, which are comparable to some extent. As shown in Fig. 1, N sgmRNA presented the highest read count, followed by the other sgmRNAs in the order ORF7 > ORF3 > M > ORF8 > S ≈ ORF6 > E, although the time factor and host calibrators were not considered in this analysis. Interestingly, the E gene presented the lowest read count, which was even lower than that of gRNAs, although all read counts of other sgmRNAs were much higher than that of gRNA. Its low expression also suggested why the repeated synthesis of E gene start sites were not remarkable (Fig. S3B). Hence, our results indicated that the major activity of the RNA synthetase complex was transcription (discontinuous RNA synthesis) rather than genome replication (continuous RNA synthesis) after viral entry.
In addition to canonical transcripts that produce sgmRNAs composed of a leader UTR with a conservative TRS and a full-length downstream ORF, few noncanonical sequences from the virus pool were discovered. One read for N contained a full-length ORF and could therefore be used to express the standard protein, which was designated “N1” here (Fig. 2). The N1 sgmRNA contained only 2 nt of the core TRS at the 5′ end, which was directly connected to the downstream ORF, and the read count was only ∼5% of the gRNA read count. It is worth noting that the N1 sgmRNA was indicated to be the canonical sgmRNA with the highest expression level among all N sgmRNAs (approximately three times that of the canonical N sgmRNA that we determined) in a previous publication (6). However, according to our results, which should be more accurate in detecting expression levels (see “Expression of noncanonical sgmRNA” below), the read count of N1 sgmRNA accounted for less than 1% of that of the canonical N sgmRNA (Table S5) at different time points after infection. Several sgmRNAs with truncated ORF7 sequences were also identified, including ORF7p1 to ORF7p3, with read counts of approximately one half, one third, and one quarter of the N1 read counts, respectively. Only the sgmRNA of ORF7p1 contained an integral TRS. Interestingly, the leader UTR and downstream sequences of ORF7p1 to ORF7p3 were all linked by a GAACttt sequence (dashed line in Fig. 2, identical to genome sites −4 to +3), which was indicated to be another TRS-related sequence guiding discontinuous RNA synthesis. The GAACttt sequences were connected to variable truncated ORF7 with each of their first sites located near the stop codon of ORF7. In addition to the sgmRNAs described above, no novel ORFs were transcribed into any individual sgmRNA transcripts, including ORF7b and ORF10, which were predicted previously (2, 6).
By merging all read pools at various time points after infection, the expression level of each gene was evaluated according to the obtained read counts, as indicated above. However, the results of the strategy of aligning long reads (30 nt) to sgmRNAs could be affected by the relatively short read length of Illumina sequencing (150 nt), which might lead to incorrect quantification of expression levels. To evaluate the expression levels of 8 genes at different time points, a short specific read (SSR) alignment-based strategy was applied.
Expression of SARS-CoV-2 genes.
The major differences between sgmRNAs and gRNAs are the sequences located near TRSs. The sequences of gRNAs are continuous, while in sgmRNAs, the leader TRS and body TRS are fused, which generates a discontinuous leader UTR-TRS-ORF structure with a highly specific sequence (Fig. 2B). To evaluate the expression of each SARS-CoV-2 gene, we designed minimal specific reads (MSRs) for the 8 canonical sgmRNAs based on the sgmRNA sequences identified above. Manual alignment showed that the MSRs were not homologous to human mRNAs or other SARS-CoV-2 sequences and were able to reflect the expression levels of corresponding genes. However, the results may not be comparable among different genes because the MSRs were of various lengths, and the lengths probably affected the read counts obtained. To make their lengths uniform, the regions homologous to ORF sequences that were less than 6 nt long were elongated to 6 nt, generating SSRs of 18 nt, which were then used to compare the expression levels of SARS-CoV-2 genes at each time point.
In fact, the results indicated that measuring the time-scaled expression levels of SARS-CoV-2 genes by MSR and SSR read counts according to our strategy was reasonable. The evidence included the following. (i) MSRs did not exist in the read pools of most uninfected mock-treated cells, while the read counts of infected cells continued to increase with infection time, which was in accordance with the transcriptional process after infection (see Tables S2 and S3). (ii) The read counts of most SSRs were usually lower, albeit slightly, than those of MSRs, indicating that read length differences between SSRs and MSRs had few effects on the read counts and that the SSRs were also highly specific. (iii) We also measured the counts of three continuous 18-nt reads (referred to as continuous SSRs) located at the beginning of the S, ORF3, and N ORFs. From 5′ to 3′, the counts of the continuous reads increased rapidly (Table S4). At 72 hpi, compared to the corresponding read counts of discontinuous sgmRNAs, the continuous read count for the S region was lower, that of ORF3 was approximately twice as high, and that of N was four times as high, which was in accordance with the read depths indicated by our results above (Fig. 1). Thus, the SSRs used to equilibrate expression were distinguished from continuous sequences that were repeatedly synthesized in various sgmRNAs.
After calibration according to the expression levels of reference host genes, the relative expression levels of 8 SARS-CoV-2 genes were illustrated using read count per fragments per kilobase per million mapped reads (RCPF) values. We classified the genes according to their expression levels, in which the highest RCPF of each gene was used (Fig. S4A). The highly expressed genes (RCPF ≥ 150,000) included N (496,000) and ORF7 (178,000). The moderately expressed genes (150,000 > RCPF ≥ 50,000) were ORF3 (88,000), M (82,000), and ORF8 (52,000). The group with the lowest expression (50,000 > RCPF) included S (45,000), ORF6 (41,000), and E (16,000). The results were very similar to those calculated using the 30-nt reads as described above (Fig. 2). A clearer view of the expression changes was obtained by separating the structural proteins and accessory proteins (Fig. 3). There was an obvious tenfold difference in expression level between the genes with the highest and lowest expression. Interestingly, the expression differences among accessory proteins were much lower than those among structural proteins. More unexpectedly, both the S and E structural genes were expressed at low mRNA levels, and E showed the lowest expression among all of the SARS-CoV-2 genes. Most genes reached their expression peaks at 24 hpi. As the most highly expressed genes, the expression trends of N and ORF7 were divergent (Fig. 3 and Fig. S4B). The expression of N peaked at 24 hpi, while that of ORF7 was higher at 72 hpi. Moreover, the expression curves did not cross each other in most cases (Fig. S4A), indicating that the relative expression levels of various genes were constant over time. Thus, highly expressed genes were clearly identifiable in early infection.
FIG 3.
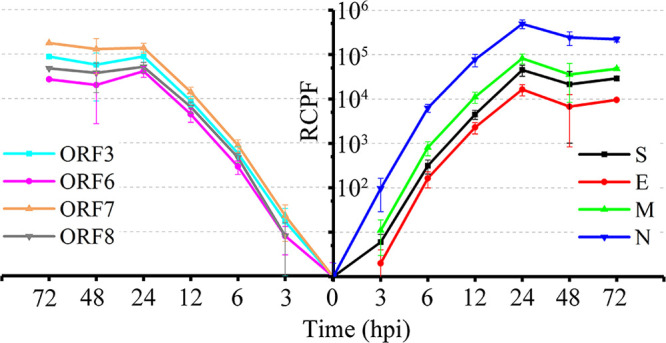
SARS-CoV-2 gene expression at various times after infection, including structural proteins (right) and accessory proteins (left).
As all sgmRNAs and gRNA share homologous coleaders and cotermini, monitoring the expression of a SARS-CoV-2 sgmRNA by qRT-PCR requires carefully designed primers to avoid false priming. The central principle of design for a specific primer pair contains two points. (i) One of the primers (sense or antisense) should be located on the most highly specific region of the template, which should contain as few consecutive matches as possible with unexpected targets; therefore, the primer will be located on the discontinuously fused sites of sgmRNAs and thus will be composed of the core TRS with flanking sequences. (ii) Situations in which the pair of primers falsely prime the same unintended target should be avoided in case a nonspecific product is generated. In our study, the sense primer was designed to be located on the discontinuous region, and thus the antisense primer should be located downstream in the coding sequence (CDS) in order to generate an amplicon homologous to the corresponding sgmRNA. However, as sgmRNAs of CoVs are coterminal, an antisense primer located in the target ORF not only primes the corresponding sgmRNA but also primes sgmRNAs coding upstream ORFs. For example, an antisense primer located in ORF6 also falsely primes the gRNAs and sgmRNAs coding S, ORF3, M, and E (considered unintended templates), but sgmRNAs coding ORF7, ORF8, and N would not be falsely primed. Meanwhile, as the sense primer priming the discontinuously fused sites of a target sgmRNA still contains sequences homologous to those of the unintended templates (including sequences located in the coleader UTR and TRS), it is still possible that nonspecific products will be generated. However, this is avoidable for some sgmRNAs. Primer pairs designed according to our principle for the S and ORF3 genes will falsely prime gRNA and/or S sgmRNA, respectively. The unintended amplicons are at least 4,000 bp in length, which will be extremely difficult to amplify with the short extension time in the qRT-PCR procedure. Therefore, monitoring the expression of S and ORF3 sgmRNAs by qRT-PCR was highly accurate and feasible. We monitored the expression of the ORF3 sgmRNA and found that the expression changes identified by PCR were consistent with those identified by RNA-Seq and could be clearly distinguished from the genome replication changes identified by PCR (Fig. S4C). Most of the mock-treated samples showed no ORF3 signal, and thus the primer pair was specific to the ORF3 sgmRNA. Finally, based on the bidirectional verification between qRT-PCR and RNA-Seq, the expression levels of the sgmRNAs identified by RNA-Seq were considered highly credible.
Expression of noncanonical sgmRNA.
Previous studies have demonstrated that the classical synthesis of CoV sgmRNAs follows discontinuous instructions strictly controlled by TRS sequences, such as the ACGAAC core TRS of SARS-CoV (7), although more-consolidated evidence is also needed. The TRS of SARS-CoV-2, however, was not confirmed before our results. Recent studies suggest that canonical transcription uses variable TRSs to guide discontinuous sgmRNA synthesis and that a TRS is not directly linked to the leader UTR or corresponding ORFs at 5′/3′ adjacent flanking sites (6, 8), which seems inconsistent with classical theories. To examine the exact strategy guiding sgmRNA synthesis, SSRs for several published noncanonical sgmRNAs were designed using the same method applied above, and their time-scaled expression levels were obtained. Of note, these sgmRNAs were considered either highly expressed or canonical in previous studies. As expected, the abundance of most sgmRNAs consistently remained very low (see Table S5), less than 2% of the corresponding canonical sgmRNAs; therefore, they should be regarded as noncanonical sgmRNAs, mutant RNAs, or sequencing errors. More detailed results can be found in Fig. S5 and Table S6, and as the final result, only noncanonical sgmRNAs of the ORF3 and N genes were consolidated (highlighted in Table S6).
Host gene transcriptomic changes.
The RNA-Seq analysis also provided information on large-scale host transcriptome changes after infection. Considering that viral RNA may account for a large proportion of total RNA, which could affect subsequent host transcriptome analysis, deeper sequencing was performed on a cDNA library of infected cells, which produced 9 GB of data in addition to the 6 GB of data obtained from mock-infected cells. We identified 8,954 differentially expressed genes (DEGs) from 0 to 72 hpi, including both upregulated and downregulated genes (Fig. S6A). Although viral transcription and genome replication had already reached their peak levels at 24 hpi, the dysregulation of host cells continued to exacerbate from 24 hpi to 72 hpi, which was reflected by an increase in DEG numbers. Interestingly, most of the DEGs identified during early infection (0 to 12 hpi) did not overlap those identified during late infection (24 to 72 hpi) (Fig. S6B). There were only two exceptions, PTX3 and IFNL2, which were continuously upregulated during both early and late infection. In contrast, the upregulated and downregulated gene sets identified at 24, 48, and 72 hpi presented high overlap with each other, indicating substantial divergence in the host response at different infection time points.
The large number of DEGs demonstrated that a series of normal cell physiological processes were hijacked, and these genes were significantly enriched in several pathways (Fig. S7), including the ribosome-related, oxidative phosphorylation, and tumor necrosis factor (TNF) signaling pathways. With the Ingenuity Pathway Analysis (IPA) database, dysregulation trends (by Z-score) and credibility (by P value) of enriched pathways at 72 hpi were further analyzed (Fig. 4A; also see below).
FIG 4.
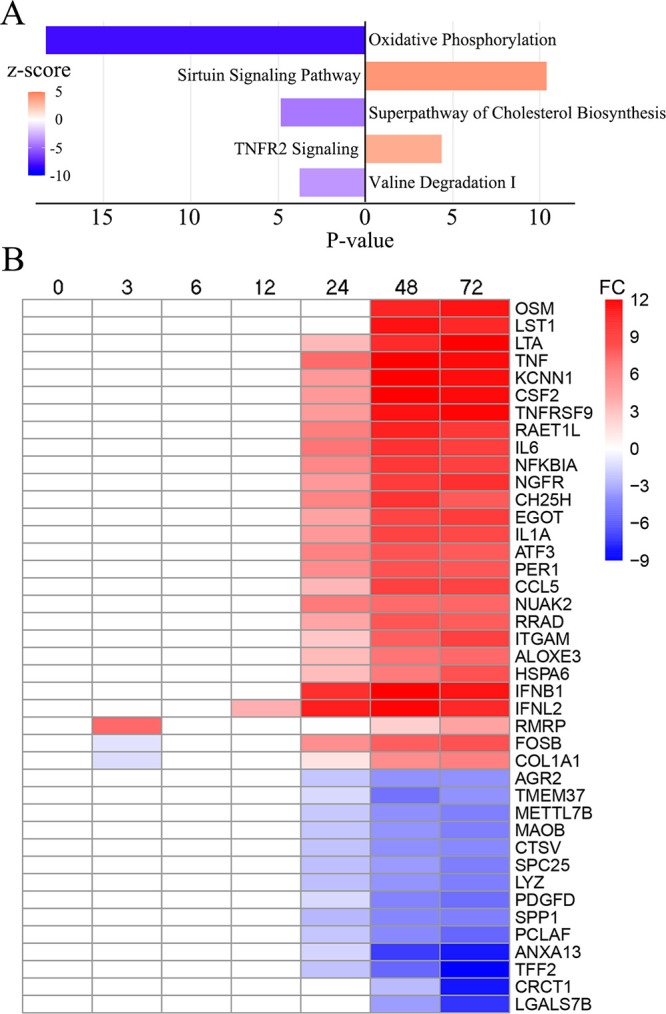
Host transcriptome deviation over time after SARS-CoV-2 infection. (A) Dysregulated pathways enriched according to IPA. Activated and deactivated pathways at 72 hpi are indicated in orange and navy, respectively. Pathways with absolute –log10P values (P values) of less than 3 were removed. (B) Heatmap of the host transcriptome at 0 to 72 hpi. The genes showing the largest log2fold changes (FC) are demonstrated in red and navy.
Oxidative phosphorylation perturbed by infection.
After SARS-CoV-2 infection, the most deviant cellular process identified was oxidative phosphorylation, which controls the majority of energy metabolism in aerobic organisms. The expression of almost all genes in mitochondrial complex 1 through complex 5 showed slight but universal decreases, including the NADH:ubiquinoneoxidoreductase complex (NDUFA/B/S/V, FC = −1.1 to −3.1), the succinate dehydrogenase complex (SDHA/B/C/D, FC = −1.6 to −2.0), ubiquinol-cytochrome c reductase (UQCR, FC = −1.2 to −2.5), cytochrome c oxidases (COX5/7/8/11/15/17, FC = −2.8 to −1.4), the ATP synthetase complex (FC = −1.3 to −2.8), and even the main electron transporter, cytochrome c. The expression changes of two genes, NDUFS6 and ATP5PF, which are subunits of complex 1 and complex 5, respectively, were checked by qRT-PCR (Fig. 5A). The qRT-PCR and RNA-Seq results were highly consistent and indicated that oxidative phosphorylation was inactive from 12 to 72 hpi. The inhibition of oxidative phosphorylation can be linked to several neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease (see Fig. S7), in which a mitochondrial energy deficit is regarded as one of the key cellular phenomena (12), implying that medicines interfering with oxidative phosphorylation could be candidates for treating COVID-19. Similar results on the cellular level were also suggested by proteomic methods (13).
FIG 5.

Mutual influence between host responses and viral reproduction. Gene expression changes (log2fold changes) validated by qRT-PCR, including (A) oxidative phosphorylation-related genes, (B) sirtuin-related genes, (C) cholesterol, valine, and fatty acid metabolism-related genes, (D) TNF-related genes, (E) cytokines, and (F) others. (G) Valine and cholesterol treatment affects the reproduction of SARS-CoV-2. Compared to the expression of uninfected cells in panels A to F (located on the lateral axis) or the untreated negative control (NC in panel G), significant differences are determined using Student’s t test and marked by asterisks; *, P < 0.05 and **, P < 0.01.
Transcriptional dysregulation induced via the sirtuin pathway.
Mammalian sirtuins (SIRTs) are NAD-dependent lysine deacetylases, among which SIRT1, SIRT6, and SIRT7 are primarily nuclear enzymes that regulate transcription factors. Through histone modification functions, SIRTs coordinate gene expression programs and thus direct the cellular metabolic state (14). After infection, we observed the upregulation of SIRT1 (FC = 2.114) along with tens of related downstream genes, including histones, nicotinate phosphoribosyltransferase (NAPRTase), and numerous transcriptional regulators and receptors. High SIRT1-mediated expression was observed for transcription factors, including the NR1H2 and ABCA1 activators involved in cholesterol efflux, which has been regarded as a response to energy usage transformation in acute inflammation (15). Almost all histones and one histone lysine methyltransferase, DOT1, were upregulated (FC = 2.2 to 6.8), which consequently promoted the formation of heterochromatin and thus inhibited the transcription of normal cellular processes (16). Downstream genes regulated by SIRT1, GADD45, FOXO1, and SOD2, which function in oxidative stress, were shown to be highly expressed, and their expression was highly related to pneumonia (17). High expression of several autophagy-related genes (ATGs), including ATG4, ATG14, and ATG101, was demonstrated after infection, which could be promoted by activated SIRT1 in the cytoplasm with the involvement of upregulated NOS3 (18). Considering the probable molecular and clinical relationship between SARS-CoV-2 and hypertension (19, 20), the question of whether NOS gene functions, especially those related to hypertension and/or inflammation, are related to SARS-CoV-2 infection could be of interest. Finally, the upregulation of SIRT1, histone H2B, and FOXO1 was identified by qRT-PCR, indicating that the pathway could be activated no later than 48 hpi (Fig. 5B).
SIRTs regulate the renin–angiotensin system by inhibiting the angiotensin II type I receptor (21). Thus, three related genes were of interest. Lower expression (FC = −1.7) of AGTRAP was observed after infection. More importantly, the expression change in another component of the renin-angiotensin system, angiotensin I converting enzyme 2 (ACE2), which has been regarded as the primary receptor of SARS-CoV-2 (19), was indicated by RNA-Seq. However, neither ACE2 nor its interactor, ACE, showed significant expression changes after infection according to qRT-PCR (Fig. 5F). Interestingly, higher expression of TMPRSS2, another core gene related to the host entry of SARS-CoV-2, was demonstrated after infection by qRT-PCR. The change was observed as early as 24 hpi and continued to increase with infection (Fig. 5B).
Mutual influence between material metabolism and viral replication.
After infection, we observed a diverse set of downregulated genes with functions in material metabolism, enriched mainly in cholesterol synthesis and branched-chain amino acid (BCAA) degradation. As early as 24 hpi, the universal inhibition of the cholesterol synthesis pathway was identified, and this dysregulation continued to increase with the prolongation of viral infection. Downregulation was observed for 5 genes in the mevalonate pathway, which is involved in the first step of cholesterol synthesis, including acetyl-CoA acetyltransferase (ACAT1 and ACAT2), phosphomevalonate kinase, diphosphomevalonate decarboxylase, and isopentenyl-diphosphate d-isomerase (FC = −1.1 to −3.3). Most genes involved in the downstream steps of the cholesterol synthesis pathway were also inhibited (FC = −1.1 to −1.7) except for sterol 14-demethylase. Significant downregulation of ACAT2 and DHCR24 from 24 hpi to 72 hpi was clearly demonstrated by qRT-PCR (Fig. 5C).
l-valine, l-leucine, and l-isoleucine are three BCAAs. The catabolic pathways of BCAAs can be divided into two sequential series of reactions (reviewed in reference 22). The first is the process that catalyzes the conversion of all three BCAAs to their respective acyl-CoA derivatives, which is accomplished by specific transaminases and the universal branched-chain keto acid dehydrogenase complex (BCKDH). The next individual pathways are completely different for the three BCAAs and are comprised of enzymes specific for each amino acid. Nearly all enzymes in the valine degradation pathway remained downregulated from 24 to 72 hpi. The qRT-PCR results indicated that the expression of the beta subunit of BCKDH (BCKDHB) was significantly downregulated during late infection (Fig. 5C). Notably, CoA-related coenzymes, such as hydroxysteroid 17-beta dehydrogenase 4 (HSD17B4) and acyl-CoA dehydrogenase 2 (ACAT2), are involved in both lipid and amino acid degradation. They were verified to be significantly downregulated, especially at 72 hpi. Furthermore, we also observed the inhibition of hydroxyacyl-CoA dehydrogenase (HADH), which functions exclusively in fatty acid oxidation.
To determine whether SARS-CoV-2 production is truly affected by cholesterol and valine metabolism, Calu-3 cells were treated with overnourished medium containing high concentrations of cholesterol and valine, respectively. Compared to untreated cells, both cholesterol- and valine-treated cells produced much higher levels of virus (Fig. 5G), demonstrating that their cellular functions are highly related to SARS-CoV-2 replication.
Cytokine storms and TNFR2 signaling.
Two components of the tumor necrosis factor (TNF) family involved in immune response modulation and the induction of inflammation (TNF-alpha, FC = 11.5 and lymphotoxin, FC = 12.2) were among the most drastically changed gene sets during late infection. Enriched activation of noncanonical TNF receptor 2 (TNFR2) signaling was demonstrated by the significantly higher expression of TNFR2 itself along with several signaling components, including TNF receptor-associated factor 2 (TRAF2), which functions in the nuclear translocation of NFKB and ultimately promotes the expression of downstream genes (23).
Interestingly, another common TNF-induced NFKB inhibitor, TNF-alpha-induced protein 3 (TNFAIP3), was also highly expressed after infection (72 hpi, FC = 11.8), as were five other inhibitors (NFKBIA/B/C/D/E, FC = 2.2 to 9.0). Both of these findings indicated that bidirectional regulation of NFKB induced by the TNF pathway occurred during infection. The qRT-PCR results demonstrated that the upregulation of TNF and lymphotoxin-alpha (LTA) became significant as early as 12 hpi (Fig. 5D), although this result was not indicated by the RNA-Seq. The increase in TNFR2 emerged much later. At the endpoint of the TNFR2 signaling pathway, NFKB showed a gradual increase after 24 hpi, along with the inhibitors TNFAIP3 and NFKBIA. In fact, the most activated genes identified were cytokines (Fig. 4). Though not fully identified by RNA-Seq, IFNB, IFNL, interleukin 1A (IL-1A), and IL-6 were activated as early as 12 hpi (Fig. 5E), indicating that the initial host response emerged before large-scale viral RNA synthesis.
DISCUSSION
On the basis of transcriptome and proteome analyses, several publications have provided insight into gene expression changes in SARS-CoV-2-infected cells; however, most of them have focused on host responses rather than the expression of viral genes (9, 10, 13, 24). Currently, the lack of reliable antibodies against most SARS-CoV-2 proteins limits the examination of SARS-CoV-2 gene expression, and the homology between sgmRNAs and gRNAs also causes universal false priming in qRT-PCR-based methods. Thus, transcriptome analysis based on RNA-Seq presents a huge advantage. The SARS-CoV-2 gene showing the highest expression was the N gene, followed by ORF7, ORF3, M, ORF8, and S ≈ ORF6, with the E gene showing the lowest expression; these findings were in general accordance with a previous study (6). The major difference was that we regarded S as a gene with relatively low mRNA expression and E as the gene with the lowest expression on the basis of relatively long sgmRNA read mapping (Fig. 2) and SSR mapping (Fig. S4A). A recent study exploring protein-comparable antibodies against SARS-CoV-2 proteins in patients (25) also showed good agreement with our data, indicating that most of the highly expressed genes that we identified elicit strong specific antibody responses, while the response elicited by the S protein was mild, although its immunogenicity should be high enough.
In fact, discontinuous RNA synthesis guided by a TRS is a conserved strategy commonly used by all CoVs (5), including SARS-CoV-2 and its precursor, SARS-CoV. Eight sgmRNAs of SARS-CoV have previously been proven to be synthesized (4) and are primarily translated into eight canonical proteins by the host machinery (if frameshifts are not taken into consideration). RT-PCR and Sanger sequencing analyses of the 5′ discontinuous portions of sgmRNAs have indicated that the core TRS of SARS-CoV is ACGAAC (7). However, Sanger sequencing-based analysis can provide only limited information about all discontinuous RNA sequences, which makes it difficult to distinguish whether the results are consistent with common cases of discontinuous RNA synthesis. Considering the high homology between the genomes of SARS-CoV-2 and SARS-CoV, ACGAAC could also function as the core TRS of SARS-CoV-2 (2). However, an RNA-Seq study conducted using Nanopore technology indicated that the TRS of SARS-CoV-2 is flexible and that some discontinuous RNAs that are not guided by a TRS are synthesized, in addition to the canonical sgmRNAs (6). Another Nanopore sequencing study demonstrated that ACGAAC exists in most sgmRNAs; however, the leader UTR and ORFs that are located around TRSs of some sgmRNAs reported are not in line with the transcription strategy commonly employed by CoVs (8). Despite the limitations imposed by the short reads obtained via Illumina sequencing, this technology provides accurate and exhaustive information to distinguish most cases of discontinuous RNA synthesis from rare cases with lower sequencing errors. Our Illumina sequencing results demonstrated that the majority of discontinuous SARS-CoV-2 RNA synthesis near the TRS region followed a highly conserved principle, leading to the transcription of eight canonical sgmRNAs with the same conserved TRS employed by SARS-CoV. The upstream and downstream sequences in the sgmRNAs were also in line with the widely recognized discontinuous RNA synthesis mechanism. Although a proportion of the discontinuous RNAs were demonstrated to not be in accordance with universal CoV transcription strategies, their expression levels were much lower than those of canonical sgmRNAs guided by TRS according to our results. Some sequences with quite low expression should be regarded as sequencing errors. We suppose that the highly variable RNA synthesis strategy of SARS-CoV-2 indicated by previous studies could have been caused by the low accuracy of Nanopore sequencing.
The host transcriptional response to SARS-CoV-2 infection identified by us can be inferred to be divided into two major components: material metabolism and energy utilization, and cytokine-related transcriptional regulation centered on TNF. The application of high levels of cholesterol and valine can promote viral replication. Recently, the knockout of MBTPS2, a protease facilitating the transcription factor-induced activation of fatty acid and cholesterol synthesis, has been reported to inhibit SARS-CoV-2 replication (26), indicating that low levels of cholesterol biosynthesis impede SARS-CoV-2 infection. However, another CRISPR-based screening study demonstrated that increasing cholesterol biosynthesis tended to inhibit SARS-CoV-2 infection (27). Although high cardiovascular cholesterol in vivo has been associated with severe manifestations of SARS-CoV-2 infection (28), additional studies will be required to reveal the detailed relationship between cholesterol (or valine) metabolism and SARS-CoV-2 infection. The observed transcriptome enrichment also indicated a relationship between the host response and TNFR2. In fact, rather than TNFR2 (fragments per kilobase per million mapped reads [FPKM] = 0.3), Calu-3 cells express high levels of TNFR1 (FPKM ≈ 70) before infection. However, TNFR1 exhibits ∼3-fold higher expression and TNFR2 exhibits ∼10-fold higher expression. The former has been reported to induce strong, widespread proinflammatory activities in various cell types, while the latter is considered to be exclusively expressed in T cells (29), indicating that Calu-3 cells could be a good model for TNFR2-related immunological studies. TNF has also been indicated to drive inflammation in peripheral blood mononuclear cells of SARS-CoV-2 patients (30), and it serves as a key cytokine in SARS-CoV-2-infected normal human bronchial epithelial cells (31). However, a previous study indicated that blocking TNFR1 and TNFR2 activity does not affect the mortality associated with a highly pathogenic respiratory virus infection (32). Therefore, the relationship between cytokine storms in other types of infectious pneumonia and TNF-related signaling deserves further study.
MATERIALS AND METHODS
Cells and virus.
Calu-3 cells were purchased from the China Infrastructure of Cell Line Resource and cultured in minimum essential medium (MEM; HyClone, USA) supplemented with 10% fetal bovine serum (FBS; BI, Israel), 4 mM l-glutamine (Thermo, USA), 1 mM sodium pyruvate (Thermo, USA), nonessential amino acids (Thermo, USA), and penicillin-streptomycin at 37°C under 5% CO2. Vero E6 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Corning, USA) supplemented with 10% FBS and penicillin-streptomycin at 37°C under 5% CO2. SARS-CoV-2 (BJ01, accession number MT291831) was passaged in Vero E6 cells. A total of 2 × 106 Calu-3 cells were cultured in T25 flasks 2 days before infection; the cell number increased to 6 × 106 just before infection. High-titer stocks of virus were diluted to 7 × 105 50% tissue culture infective dose (TCID50)/ml with MEM containing 2% FBS. After the medium was removed, the cells were inoculated with 1 mL diluted virus for 1 hour and washed once with cold phosphate-buffered saline (PBS), at which time the zero-timepoint of infection was set. Mock-treated cells were treated with MEM containing 2% FBS for 1 hour. At a series of timepoints (0, 3, 6, 12, 24, 48, and 72 hours postinfection [hpi]), viral RNA copies in the supernatant were quantified by qRT-PCR, and cellular RNAs from both the virus and host were analyzed. All experiments with SARS-CoV-2 were performed in a biosafety level 3 containment laboratory approved by the Institute of Military Veterinary Medicine.
RNA extraction and sequencing.
Cellular RNA was extracted with an RNAsimple extraction kit (Tiangen, China). After the RNA concentration and integrity were checked using a Qubit 2.0 (Thermo, USA) and an Agilent 2100 Bioanalyzer (Agilent, USA), sequencing libraries were generated using the NEBNext Ultra II directional RNA library prep kit for Illumina (NEB, USA) following the manufacturer’s recommendations. Briefly, mRNA was purified from total RNA using oligonucleotide dT attached magnetic beads. Fragmentation was carried out using NEBNext first-strand synthesis reaction buffer. First-strand cDNA was synthesized using random hexamer primers and M-MuLV reverse transcriptase, and second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After the adenylation of the 3′ ends of DNA fragments, NEBNext adaptors with a hairpin loop structure were ligated to the fragments to prepare for hybridization. cDNA fragments150 to 200 bp in length were selected and then purified with AMPure XP beads (Beckman, USA). Then, PCR was performed with Phusion high-fidelity DNA polymerase, universal PCR primers, and an index (X) primer. Finally, the PCR products were purified with AMPure XP beads, and library insert size was assessed on an Agilent 2100 system. The clustering of the index-coded samples was performed on a cBot cluster generation system. After cluster generation, the library was sequenced on the Illumina NovaSeq 6000 platform, and 150-bp paired-end reads were generated.
TRS identification by short read query.
After rRNA was removed by alignment to the Rfam database, reads of infected cells were mapped to the SARS-CoV-2 genome (MT291831) using Hisat2, generating read pools containing viral sequences. Read pools at various time points were combined to avoid the influence of time. To identify the core TRS, 12 continuous genomic sequences of 15 nt in length were selected and referred to as short “query reads” (see Fig. S1). Their first nt were located one by one downstream at a specific region of the 5′ UTR. By querying the combined read pool containing all viral sequences, all sequences of 30 nt in length whose 5′ 15-nt sequences were identical to the query reads were returned and their numbers were counted. The 15-nt sequences downstream of the corresponding query reads were referred to as their “return reads.” Located around the possible TRS, the return reads could be either manually aligned continuously to gRNAs or aligned discontinuously to sgmRNAs with the 5′ partial sequence homologous to the leader UTR (upstream of the leader TRS) and the 3′ sequence homologous to various ORFs (downstream of the body TRS). When a site could be regarded as either 5′ continuous (continuous to upstream query reads) or 3′ continuous (continuous to downstream ORFs), it was designated 5′ continuous, as we intended to identify the probable leader TRS as long as possible.
In parallel, two additional types of 15-nt query reads with sequences homologous to the beginning of the ORFs were used: the first type started at the body TRS (6 nt) and ended at +9 nt of the downstream ORF (referred to as “in-TRS reads”), and the second type was homologous to +1 to +15 nt of the ORFs adjacent to the downstream body TRS (referred to as “after-TRS reads”). For each known ORF, an in-TRS read and an after-TRS read were used. By querying the combined read pool, all sequences of 30 nt whose last 15 nt at the 3′ end were completely identical to the query reads were chosen; their numbers were counted, and only the top 5 were further analyzed. The 15-nt sequences upstream of the corresponding query reads were referred to as their “return reads.”
Transcriptome of SARS-CoV-2.
Minimal specific reads (MSRs) homologous to the discontinuous regions of 8 canonical sgmRNAs were designed. With lengths of 15 to 18 nt, each MSR contained a leader UTR of 6 nt (−12 to −7 nt upstream of the leader TRS), a core TRS of 6 nt, and 3 to 6 nt of the ORF (+1 to +3 to 6 nt downstream of the body TRS). The elaborately selected sequence made it specific and able to reflect the expression levels of the corresponding SARS-CoV-2 genes. The detailed sequences are listed in Table S3. The specificity for host mRNA and other sgmRNAs/gRNAs of SARS-CoV-2 was checked and ensured by manual alignment to human reference RNA and published SARS-CoV-2 transcriptome/genome sequences (2, 6, 8). The sequences of MSRs and elongated short specific reads (SSRs) can be obtained from Table S4. The fragments per kilobase per million mapped reads (FPKM) values of beta-2-microglobulin (B2M) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as double references were applied to calibrate SARS-CoV-2 gene expression. The time-scaled read count per FPKM (RCPF) of each SARS-CoV-2 gene was used to reflect the expression level and was calculated as follows:
Host transcriptome.
Reads that were not mapped to the viral genome were mapped to the reference human genome (GRCh38). Differences in the expression of host RNAs were evaluated on the basis of FPKM values with the software RSEM using the common criteria. Differential expression was determined by comparing virus-infected replicates to time-matched, mock-treated replicates according to the criteria of an absolute log2fold change (log2FC) greater than 1 and a false discovery rate (FDR)-adjusted P value of <0.05 for each time point. Functional analysis and KEGG pathway enrichment were performed using clusterProfiler. The Ingenuity Pathway Analysis (IPA) database was used to determine dysregulated pathways, in which the P value reflected the percentage of genes showing expression changes consistent with the predicted dysregulation, and the Z-score reflected the global extent of dysregulation from the total expression differences. Only pathways with both an absolute log(P value) of >3 and Z-score of >3 were selected.
qRT-PCR.
The synthesis of first-strand cDNA was carried out using oligonucleotide dT primers and a Maxima H Minus first-strand cDNA synthesis kit (Thermo, USA). The qRT-PCR was performed in a Bio-Rad CFX96 system by using SsoAdvanced Universal SYBR green supermix (Bio-Rad). The procedure included initial denaturation for 30 s at 95°C and 45 cycles of amplification (5 s at 95°C and 30 s at 60°C), followed by 10 s at 95°C to evaluate the melting curve. The 2−ΔΔCT method (33) was used to analyze relative gene expression data. Double references, GAPDH (NM_002046.7) and B2M (NM_004048.4), were applied for calibration. The canonical sgmRNA sequence of ORF3 provided by our data was used to design qPCR primers for ORF3. The sense primer was located at the discontinuous fused region of its sgmRNA, and the antisense primer was located at the ORFs. All primers are shown in Table S1.
Viral genome copy number.
Viral RNA in the supernatant was extracted using a magnetic viral DNA/RNA kit (Tiangen, China) following the manufacturer’s instructions. RNA genome copy numbers were identified with a 2019-nCoV nucleic acid detection kit using an RT-PCR fluorescence probe (Puruikang, China).
Cholesterol and valine treatment.
Overnourished medium was prepared by adding 50 mg/100 ml cholesterol and 3 mM valine to normal Calu-3 medium. After treatment with overnourished medium for 24 hours, Calu-3 cells in 24-well plates were washed with normal medium twice and infected with virus. After 1 hour of infection, the supernatant was drained, and the cells were maintained with overnourished medium for 48 hours.
Data availability.
Sequencing data were archived at the NCBI Sequence Read Archive (SRA) under accession number SRP324059.
ACKNOWLEDGMENTS
This work was supported by the National Key Research and Development Program (grant 2016YFD0500203). We thank Chengfeng Qin and Xiaofeng Li for kindly providing the SARS-CoV-2 strain.
X.W. and Y.G. designed the study. X.W., Y.Z., F.Y., T.W., and W.S. performed the experiments. X.W. and Y.Z. wrote the paper. N.F., W.W., H.W., H.H., S.Y., X.X., and Y.G. revised the paper. Y.G. and W.S. acquired the funding.
The authors declare that they have no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Yuwei Gao, Email: yuwei0901@outlook.com.
Kanta Subbarao, The Peter Doherty Institute for Infection and Immunity.
REFERENCES
- 1.World Health Organization. 2020. WHO coronavirus disease (COVID-19) dashboard. https://covid19.who.int/.
- 2.Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, Hu Y, Tao Z-W, Tian J-H, Pei Y-Y, Yuan M-L, Zhang Y-L, Dai F-H, Liu Y, Wang Q-M, Zheng J-J, Xu L, Holmes EC, Zhang Y-Z. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snijder EJ, Decroly E, Ziebuhr J. 2016. Chapter three - the nonstructural proteins directing coronavirus RNA synthesis and processing, p 59–126. In Ziebuhr J (ed), Advances in virus research, vol 96. Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LLM, Guan Y, Rozanov M, Spaan WJM, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J Mol Biol 331:991–1004. 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sola I, Almazán F, Zúñiga S, Enjuanes L. 2015. Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol 2:265–288. 10.1146/annurev-virology-100114-055218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim D, Lee J-Y, Yang J-S, Kim JW, Kim VN, Chang H. 2020. The Architecture of SARS-CoV-2 transcriptome. Cell 181:914–921.e10. 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thiel V, Ivanov KA, Putics Á, Hertzig T, Schelle B, Bayer S, Weißbrich B, Snijder EJ, Rabenau H, Doerr HW, Gorbalenya AE, Ziebuhr J. 2003. Mechanisms and enzymes involved in SARS coronavirus genome expression. J Gen Virol 84:2305–2315. 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 8.Taiaroa G, Rawlinson D, Featherstone L, Pitt M, Caly L, Druce J, Purcell D, Harty L, Tran T, Roberts J, Scott N, Catton M, Williamson D, Coin L, Duchene S. 2020. Direct RNA sequencing and early evolution of SARS-CoV-2. bioRxiv 10.1101/2020.03.05.976167:2020.03.05.976167. [DOI]
- 9.Chua RL, Lukassen S, Trump S, Hennig BP, Wendisch D, Pott F, Debnath O, Thürmann L, Kurth F, Völker MT, Kazmierski J, Timmermann B, Twardziok S, Schneider S, Machleidt F, Müller-Redetzky H, Maier M, Krannich A, Schmidt S, Balzer F, Liebig J, Loske J, Suttorp N, Eils J, Ishaque N, Liebert UG, von Kalle C, Hocke A, Witzenrath M, Goffinet C, Drosten C, Laudi S, Lehmann I, Conrad C, Sander L-E, Eils R. 2020. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat Biotechnol 38:970–979. 10.1038/s41587-020-0602-4. [DOI] [PubMed] [Google Scholar]
- 10.Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, Guo D, Hu W, Yang J, Tang Z, Wu H, Lin Y, Zhang M, Zhang Q, Shi M, Liu Y, Zhou Y, Lan K, Chen Y. 2020. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect 9:761–770. 10.1080/22221751.2020.1747363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stukalov A, Girault V, Grass V, Bergant V, Karayel O, Urban C, Haas DA, Huang Y, Oubraham L, Wang A, Hamad SM, Piras A, Tanzer M, Hansen FM, Enghleitner T, Reinecke M, Lavacca TM, Ehmann R, Wölfel R, Jores J, Kuster B, Protzer U, RR Ziebuhr J, Thiel V, Scaturro P, Mann M, Pichlmair A. 2020. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. bioRxiv 10.1101/2020.06.17.156455:2020.06.17.156455. [DOI] [PubMed]
- 12.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. 2009. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106:14670–14675. 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bojkova D, Klann K, Koch B, Widera M, Krause D, Ciesek S, Cinatl J, Münch C. 2020. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583:469–472. 10.1038/s41586-020-2332-7. [DOI] [PubMed] [Google Scholar]
- 14.Chang H-C, Guarente L. 2014. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab 25:138–145. 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. 2007. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28:91–106. 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 16.Zhang D, Li S, Cruz P, Kone BC. 2009. Sirtuin 1 functionally and physically interacts with disruptor of telomeric silencing-1 to regulate alpha-ENaC transcription in collecting duct. J Biol Chem 284:20917–20926. 10.1074/jbc.M109.020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storz P. 2011. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal 14:593–605. 10.1089/ars.2010.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. 2008. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105:3374–3379. 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280.e8. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, Zhao Y, Li Y, Wang X, Peng Z. 2020. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 323:1061–1069. 10.1001/jama.2020.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyazaki R, Ichiki T, Hashimoto T, Inanaga K, Imayama I, Sadoshima J, Sunagawa K. 2008. SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 28:1263–1269. 10.1161/ATVBAHA.108.166991. [DOI] [PubMed] [Google Scholar]
- 22.Neinast M, Murashige D, Arany Z. 2019. Branched chain amino acids. Annu Rev Physiol 81:139–164. 10.1146/annurev-physiol-020518-114455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker SJ, Reddy EP. 1998. Modulation of life and death by the TNF receptor superfamily. Oncogene 17:3261–3270. 10.1038/sj.onc.1202568. [DOI] [PubMed] [Google Scholar]
- 24.Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Cheng L, Li J, Wang X, Wang F, Liu L, Amit I, Zhang S, Zhang Z. 2020. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 26:842–844. 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 25.Hachim A, Kavian N, Cohen CA, Chin AWH, Chu DKW, Mok CKP, Tsang OTY, Yeung YC, Perera RAPM, Poon LLM, Peiris JSM, Valkenburg SA. 2020. ORF8 and ORF3b antibodies are accurate serological markers of early and late SARS-CoV-2 infection. Nat Immunol 21:1293–1301. 10.1038/s41590-020-0773-7. [DOI] [PubMed] [Google Scholar]
- 26.Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, Carlson-Stevermer J, Zengel JR, Richards CM, Fozouni P, Oki J, Rodriguez L, Joehnk B, Walcott K, Holden K, Sil A, Carette JE, Krogan NJ, Ott M, Puschnik AS. 2020. Genetic screens identify host factors for SARS-CoV-2 and common cold coronaviruses. Cell 184:106–119.e14. 10.1016/j.cell.2020.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daniloski Z, Jordan TX, Wessels H-H, Hoagland DA, Kasela S, Legut M, Maniatis S, Mimitou EP, Lu L, Geller E, Danziger O, Rosenberg BR, Phatnani H, Smibert P, Lappalainen T, tenOever BR, Sanjana NE. 2021. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 184:92–105.e16. 10.1016/j.cell.2020.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radenkovic D, Chawla S, Pirro M, Sahebkar A, Banach M. 2020. Cholesterol in relation to COVID-19: should we care about it? J Clin Med 9:1909. 10.3390/jcm9061909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Medler J, Wajant H. 2019. Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin Ther Targets 23:295–307. 10.1080/14728222.2019.1586886. [DOI] [PubMed] [Google Scholar]
- 30.Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, Choi B, Nam SK, Sa M, Kwon J-S, Jeong SJ, Lee HK, Park SH, Park S-H, Choi JY, Kim S-H, Jung I, Shin E-C. 2020. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol 5:eabd1554. 10.1126/sciimmunol.abd1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–1045.e1039. 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salomon R, Hoffmann E, Webster RG. 2007. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci U S A 104:12479–12481. 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S2 to S5<br>. Download JVI.00600-21-s0001.xlsx, XLSX file, 0.02 MB (24.4KB, xlsx)
Table S6<br>. Download JVI.00600-21-s0002.xlsx, XLSX file, 0.01 MB (15.9KB, xlsx)
Fig. S1 to S7 and Table S1<br>. Download JVI.00600-21-s0003.pdf, PDF file, 4.4 MB (4.4MB, pdf)
Data Availability Statement
Sequencing data were archived at the NCBI Sequence Read Archive (SRA) under accession number SRP324059.