

ABSTRACT
The influenza A virus genome is comprised of eight single-stranded negative-sense viral RNA (vRNA) segments. Each of the eight vRNA segments contains segment-specific nonconserved noncoding regions (NCRs) of similar sequence and length in different influenza A virus strains. However, in the subtype-determinant segments, encoding hemagglutinin (HA) and neuraminidase (NA), the segment-specific noncoding regions are subtype specific, varying significantly in sequence and length at both the 3′ and 5′ termini among different subtypes. The significance of these subtype-specific noncoding regions (ssNCR) in the influenza virus replication cycle is not fully understood. In this study, we show that truncations of the 3′-end H1-subtype-specific noncoding region (H1-ssNCR) resulted in recombinant viruses with decreased HA vRNA replication and attenuated growth phenotype, although the vRNA replication was not affected in single-template RNP reconstitution assays. The attenuated viruses were unstable, and point mutations at nucleotide position 76 or 56 in the adjacent coding region of HA vRNA were found after serial passage. The mutations restored the HA vRNA replication and reversed the attenuated virus growth phenotype. We propose that the terminal noncoding and adjacent coding regions act synergistically to ensure optimal levels of HA vRNA replication in a multisegment environment. These results provide novel insights into the role of the 3′-end nonconserved noncoding regions and adjacent coding regions on template preference in multiple-segmented negative-strand RNA viruses.
IMPORTANCE While most influenza A virus vRNA segments contain segment-specific nonconserved noncoding regions of similar length and sequence, these regions vary considerably both in length and sequence in the segments encoding HA and NA, the two major antigenic determinants of influenza A viruses. In this study, we investigated the function of the 3′-end H1-ssNCR and observed a synergistic effect between the 3′-end H1-ssNCR nucleotides and adjacent coding nucleotide(s) of the HA segment on template preference in a multisegment environment. The results unravel an additional level of complexity in the regulation of RNA replication in multiple-segmented negative-strand RNA viruses.
KEYWORDS: influenza A virus, HA segment-specific noncoding nucleotides, transcription, replication, viral RNA promoter, template preference
INTRODUCTION
Influenza A viruses are a major health threat that cause seasonal epidemics as well as occasional global pandemics (1). The genome of influenza A viruses consists of eight single-stranded negative-sense viral RNA (vRNA) segments, which associate with the viral polymerase and nucleoprotein (NP) to form viral ribonucleoprotein (vRNP) complexes (2). The viral polymerase consists of three subunits, polymerase basic 1 (PB1), polymerase basic 2 (PB2), and polymerase acidic (PA) proteins, and carries out transcription and replication of the vRNA in the nucleus of infected cells. Transcription of the negative-sense vRNA genome produces positive-sense viral mRNAs with a 5′-terminal N7-methylguanosine (m7G) cap and 3′ poly(A) tail (3, 4). During transcription, the viral polymerase makes an essential interaction with the C-terminal domain (CTD) of the large subunit of cellular RNA polymerase II (5). Replication of the negative-sense vRNA genome is a two-step process that requires polymerase dimerization (6–10). In the first step, the viral polymerase copies the vRNA template into a positive-sense cRNA replicative intermediate, which in the second step serves as template for the polymerase to synthesize vRNA. Both cRNA and vRNA assemble into RNP complexes with newly synthesized polymerase and NP (11). Progeny vRNPs are exported from the cell nucleus and are selectively packaged into progeny virions, which are released from the infected cells by budding (12).
Each influenza A virus vRNA segment encodes one or two major open reading frames (ORFs) in the negative sense, which are flanked by 3′ and 5′ noncoding terminal sequences. The first 12 and 13 nucleotides at the 3′ and 5′ ends, respectively, are highly conserved among different vRNA segments and form a promoter structure, which is bound by the viral polymerase (13). The conserved terminal promoter sequences and coding region are separated by nonconserved noncoding regions (NCRs), which are variable in length and nucleotide composition in different vRNA segments but in most instances are highly conserved in the same vRNA segment of different influenza virus strains (14). In the subtype-determinant segments, encoding hemagglutinin (HA) and neuraminidase (NA), the segment-specific NCRs are also subtype specific, varying significantly in length and sequence at both the 3′ and 5′ ends among the different subtypes (15).
The NCRs are known to play multiple roles in the replication cycle of influenza A viruses. These include regulatory roles in transcription and replication of vRNA segments as well as roles in determining splicing and translation efficiencies (16–22). NCRs and the adjacent coding regions have also been proposed to contain signals that are required for selective packaging of the eight vRNA segments into progeny virions (23–37).
In this study, we aimed to characterize the role of the 3′-terminal NCR in the HA vRNA of influenza A virus. These regions differ in length (ranging between 5 to 32 nucleotides) as well as sequence between different influenza A subtypes, suggesting a subtype-specific role (15). Here, we focus on the 3′-terminal NCR of the H1 subtype vRNA that is characterized by a 20-nucleotide-long U-rich sequence. We demonstrate that HA vRNA templates with truncations in the 3′-end H1-subtype-specific noncoding region (3′-end H1-ssNCR) are replicated less efficiently than the wild-type (WT) HA vRNA template in infected cells, leading to viral attenuation. However, point mutations (G76A or C56U/A; numbering starting from the 3′ end) in the coding region of the HA vRNA segment reverses the deficiency in vRNA replication and restores virus replication. These results highlight the importance of sequences beyond the conserved promoter structure for vRNA template utilization.
RESULTS
The 3′-end H1-ssNCR is not required for transcription and replication in single-template RNP reconstitution assays.
To assess the importance of 3′-end H1-ssNCR for the replication of influenza A virus, we first generated six pHW2000 plasmids to express the HA vRNA segment of influenza A/WSN/33 (H1N1) (WSN) virus with truncations in the 3′-end H1-ssNCR. Specifically, we progressively truncated the 3′-end H1-ssNCR by deleting three nucleotides at a time upstream of the HA start codon, producing mutants 3L1, 3L2, 3L3, 3L4, 3L5, and 3L6 with 3-, 6-, 9-, 12-, 15-, and 18-nt deletions, respectively (Fig. 1A).
FIG 1.

The 3′-end H1-ssNCR is not essential for transcription and replication in single-template RNP reconstitution assays. (A) Schematic representation of mutants with serial truncations at the 3′-end H1-ssNCR of the HA segment of WSN virus. The red boxes represent the 3′- and 5′-terminal promoter regions. The orange boxes represent the start codon (AUG) of HA vRNA (UAC in negative sense). The lines represent the remaining nucleotides. The triangles represent the deleted nucleotides. The truncation is three nucleotides at a time. In 3L6, the short line next to the red box at the 3′ end represents two conserved nucleotides CC (in negative sense) next to the 3′-terminal promoter region. (B, C) HEK-293T cells were cotransfected with plasmids expressing WSN (B) or CA04 (C) viral polymerase subunits, NP, and wild-type or mutant HA vRNA (wild type or mutant). A polymerase containing an active site mutation (PB1a, D445A/446A) was used as negative control (NC). Accumulation of RNA 24 h posttransfection was analyzed by primer extension and 6% PAGE. SE, short exposure; LE, long exposure. The graph shows the mean intensity signals of mutant HA RNAs relative to those of wild-type HA RNAs. The data represent the means ± standard error of mean (SEM) of three independent experiments.
To characterize the effect of 3′-end H1-ssNCR truncations on RNA synthesis, RNPs were reconstituted by coexpression of the three polymerase subunits, NP and wild type or 3′-end H1-ssNCR-truncated HA vRNA in human HEK-293T cells, and the steady-state levels of positive- and negative-sense RNAs were analyzed by primer extension. The catalytically inactive polymerase (PB1a) was used as a negative control (38). All 3′-end H1-ssNCR-truncated HA vRNAs were transcribed and replicated by the viral polymerase to levels similar to that of the wild type HA vRNA (Fig. 1B). These results suggest that the conserved 12 nucleotides and the two adjacent HA segment-specific CC nucleotides at the 3′ end of HA vRNA (as present in the 3L6 mutant) are sufficient for viral polymerase to efficiently replicate and transcribe the vRNA in a single-template RNP reconstitution system.
To address whether this result is specific to influenza A/WSN/33 or could be generalized to other influenza A virus strains, we introduced the same 3′-end H1-ssNCR truncations into the HA segment of the pandemic A/California/04/2009 (H1N1) virus (CA04) and examined the effect of the truncations on RNA synthesis in the context of the CA04 RNP reconstitution system. We obtained similar results as with the WSN virus (Fig. 1C). Together, these results show that the 3′-end H1-ssNCR is not required for transcription and replication in the single-template RNP reconstitution assay.
Truncations of the 3′-end H1-ssNCR lead to virus attenuation.
Next, we used reverse genetics to generate WSN and CA04 viruses with 3′-end H1-ssNCR truncations, and titers of the rescued viruses (P0) were examined by plaque assay. In the context of WSN viruses, the 3L1 and 3L2 viruses showed similar titers compared to the wild-type virus, while the 3L3 virus showed about a 10-fold reduction. The 3L4 and 3L5 viruses were severely attenuated, showing a more than 3 log reduction. No plaques were detected for the 3L6 virus (Fig. 2A).
FIG 2.

Truncations of the 3′-end H1-ssNCR lead to reduced virus replication. (A, B) Titers of WSN (A) and CA04 (B) rescued viruses (P0). Wild-type or mutant viruses were rescued in HEK-293T cells by cotransfecting eight pHW2000-PB2, -PB1, -PA, -HA (wild type or mutant), -NP, -NA, -M, and -NS plasmids. The graph shows the mean titers of wild-type and mutant viruses in P0. The data represent the means ± SEM of three independent experiments. One-way analysis of variance (ANOVA) with Dunnett correction for multiple testing; asterisks represent a significant difference from wild-type virus as follows: **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (C) Growth curves of WSN wild-type and mutant viruses (P2). The growth curves of the WSN viruses were determined by plaque assay on MDCK cells at 24 h, 36 h, 48 h, 60 h, and 72 h postinfection. The graph shows the mean titers of wild-type and mutant viruses. The data represent the means ± SEM of three independent experiments. Two-way analysis of variance with Dunnett correction for multiple testing; asterisks represent a significant difference from wild-type virus as follows: *, P < 0.05; **, P < 0.01. (D) Titers of CA04 wild-type and mutant viruses (P6). CA04 WT, 3L5, and 3L6 were passaged in embryonated eggs six times (P6), and the P6 virus titers were detected by TCID50. The data represent the means ± SEM of three independent experiments. One-way analysis of variance with Dunnett correction for multiple testing; asterisks represent a significant difference from wild-type virus as follows: **, P < 0.01; ***, P < 0.001. (E, F) Reverse cDNA sequences of the 3′ terminus of WSN (E) or CA04 (F) HA vRNA of viruses harvested at 48 h postinfection from the growth curve experiment above. The sequencing results are the representatives from three independent biological replicates (n = 3).
In the case of CA04 viruses (P0), all truncation mutants (3L1 to 3L6) could be rescued, and we measured the virus replication efficiency by 50% tissue culture infective dose (TCID50). As shown in Fig. 2B, we observed similar titers for the rescued WT and 3L1 to 3L4 mutant viruses, but the titers of 3L5 and 3L6 mutant viruses were significantly lower than WT. Taken together, these results suggest that 3′-end H1-ssNCR truncations lead to virus attenuation, although the extents vary between different truncations and different viruses.
Attenuated P0 viruses show improved growth upon virus passage.
To further characterize these 3′-end H1-ssNCR-truncated mutant viruses, the rescued WSN virus (P0) was then passaged in MDCK cells. Interestingly, the growth curves of all passaged WSN viruses (P2) in MDCK cells, including the 3L4 and 3L5 mutants, reached maximal viral titers between 1 × 107 and 1 × 108 PFU/ml, similar to the wild type (Fig. 2C). To address the molecular basis of the improved growth, we sequenced the 3′ terminus of HA vRNA of WSN viruses (P2). We found that 3L3, 3L4, and 3L5 mutants were unstable and contained mutations in addition to the truncations introduced. In the 3L3 mutant, we observed a duplication of six nucleotides, resulting in the insertion of UAUUUU (ATAAAA in positive sense) upstream of the HA start codon, extending the 3′-end H1-ssNCR to a length identical to that in 3L1. In the mutant viruses 3L4 and 3L5, a point mutation G76A (C76T in positive sense) was found in the HA open reading frame, which changes the 15th HA amino acid from threonine to isoleucine. We also passaged these mutant WSN viruses in A549 cells and sequenced the virus at P2. We found exactly the same mutations as described above for 3L3, 3L4, and 3L5. Moreover, we repeatedly found an independent synonymous C56U (G56A in positive sense) mutation in the HA open reading frame of mutant virus 3L5 (Fig. 2E). For the highly attenuated 3L5 and 3L6 CA04 mutant viruses, we did not observe any improved cytopathic effect (CPE) after 10 passages in MDCK cells. We then passaged these viruses in embryonated chicken eggs and analyzed virus titers by TCID50. The CA04 3L6 mutant showed significantly higher virus titers than 3L5 at P6 (Fig. 2D). We sequenced the CA04 3L6 (P6) virus and found a synonymous mutation C56A (G56T in positive sense) at exactly the same position as we found in WSN 3L5 virus passaged in A549 cells (Fig. 2F). Together, these results show that the 3L3, 3L4, and 3L5 mutant WSN viruses and the 3L6 CA04 virus with 3′-end H1-ssNCR truncations are unstable, and upon passage, they can accumulate mutations at specific positions in the adjacent HA coding region, which might improve their growth.
The G76A and C56U mutations are responsible for the improved growth of the attenuated viruses.
In order to assess the effect of the G76A mutation on WSN virus growth, we generated recombinant viruses WT-G76A, 3L4-G76A, and 3L5-G76A using reverse genetics, and titers of the rescued viruses (P0) were examined by plaque assay. In contrast to the significant attenuation (3 log reduction) observed for the original 3L4 and 3L5 mutants, the 3L4-G76A and the 3L5-G76A mutants showed similar titers compared to that of the wild-type virus (Fig. 3A). We sequenced the 3′ terminus of the HA vRNA of the 3L4, 3L4-G76A, 3L5, and 3L5-G76A viruses and found that in the 3L4 and 3L5 viruses, both G and A (C and T in positive sense) were present at position 76, indicating that these viruses are unstable and obtain the G76A mutation very quickly. In contrast, no mutation was observed at position 76 or at any other position in the sequenced part of the 3′ terminus of the HA vRNA (position 1 to 151) of the 3L4-G76A and 3L5-G76A viruses (Fig. 3B). To address whether the C56U mutation has the same effect as the G76A mutation, we plaque purified both 3L5-G76A and 3L5-C56U mutant viruses and examined virus growth by plaque assay. The 3L5-C56U mutant showed a similar titer to those of the 3L5-G76A mutant and wild-type virus (Fig. 3C). Together, these results show that the G76A and C56U mutations in the open reading frame of the HA vRNA stabilize the 3L4 and 3L5 viruses with 3′-end H1-ssNCR truncations and reverse the attenuation in virus replication.
FIG 3.

The G76A and C56U mutations improve the growth of 3L4 and 3L5 viruses. (A) Titers of rescued viruses (P0). Wild-type or mutant viruses were rescued in HEK-293T cells, and virus titers were examined by plaque assay on MDCK cells. The graph shows the mean titers of wild-type and mutant viruses. The data represent the means ± SEM of three independent experiments. Two-way analysis of variance (ANOVA) with Dunnett correction for multiple testing; asterisks represent a significant difference from wild-type virus as follows: ****, P < 0.0001. (B) Reverse cDNA sequence traces of the 3′ terminus HA vRNA of 3L4, 3L4-G76A, 3L5, and 3L5-G76A mutant viruses from panel A. The sequence traces are the representatives from three independent biological replicates (n = 3). (C) Titers of WSN wild-type and mutant viruses (P2). WSN WT, 3L5-G76A, and 3L5-C56U were passaged in MDCK cells twice (P2), and the P2 virus titers were detected by plaque assay.
The 3′-end H1-ssNCR-truncated mutant viruses show decreased HA vRNA levels in virions and in cells.
To address which step in the virus life cycle is affected by the truncations in the 3′-end H1-ssNCR and to elucidate how the G76A mutation improves viral growth, we analyzed HA vRNA levels in the rescued WSN virus (P0) as well as in the transfected cells used for the generation of P0 stock of virus using reverse transcription-quantitative PCR (RT-qPCR). Compared to the levels of NA vRNA, the 3′-end H1-ssNCR-truncated mutant WSN viruses 3L2, 3L3, 3L4, and 3L5 showed decreased levels of HA vRNA in both the harvested virions and the transfected cells. However, the G76A mutation in the 3L4-G76A and 3L5-G76A viruses significantly increased the HA vRNA levels in both virions and cells (Fig. 4A).
FIG 4.
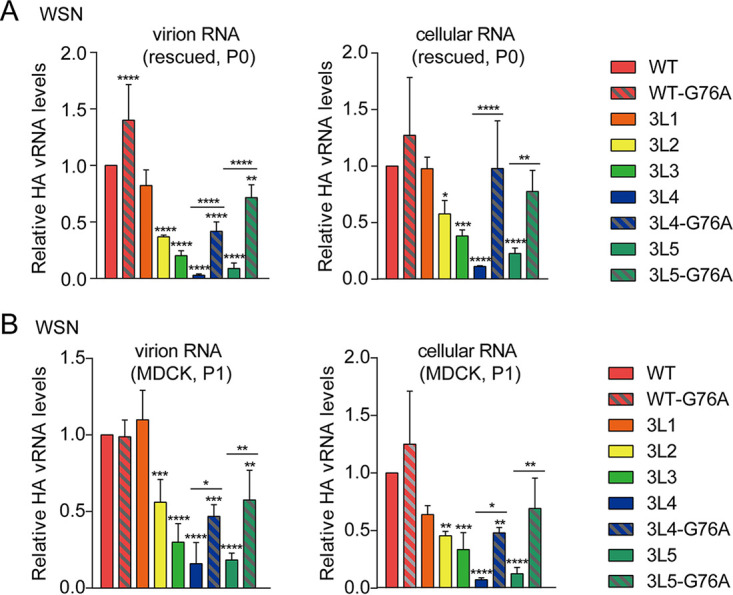
The 3′-end H1-ssNCR-truncated mutant viruses show decreased HA vRNA levels in virions and in cells. (A, B) Wild-type or mutant viruses were rescued in HEK-293T cells and then passaged in MDCK cells once. The vRNA levels of P0(A) and P1(B) virus in virions and in cells were analyzed by RT-qPCR. The graphs show the mean values of HA vRNA relative to those of NA vRNA. The data represent the means ± SEM of three independent experiments. Two-way analysis of variance (ANOVA) with Dunnett correction for multiple testing; asterisks represent a significant difference from wild-type virus as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
To investigate the reduction in HA vRNA levels further, the rescued viruses (P0) were passaged in MDCK cells (P1). At 24 h postinfection (p.i.), RNA was isolated from the passaged viruses and the infected cells, followed by the analysis of HA vRNA by RT-qPCR. In general, the results were consistent with the results observed before passage, showing that 3′-end H1-ssNCR truncations lead to decreased HA vRNA levels in both virions and cells while the G76A mutation increased HA vRNA levels (Fig. 4B). Together, these results show that truncations in the 3′-end H1-ssNCR lead to deficiency in RNA replication, resulting in decreased HA vRNA levels in cells and thus in virions. The G76A mutation increases HA vRNA levels in cells and thus in virions, promoting the improved replication of the 3′-end H1-ssNCR-truncated mutant viruses.
The 3′-end H1-ssNCR-truncated mutant templates are replicated less efficiently than the wild-type HA template in multiple-template RNP reconstitution assays.
The findings above demonstrate that truncations in the 3′-end H1-ssNCR do not significantly affect RNA synthesis in a single-template RNP reconstitution assay (Fig. 1B) but result in decreased HA vRNA levels in virions and in cells during viral rescue and virus passage (Fig. 4). It has been proposed that the gene segments of influenza A viruses could compete for available viral polymerase (39). To address the effect of truncations in 3′-end H1-ssNCR, seven pHW2000 plasmids (PB2, PB1, PA, NP, HA (wild type or mutant), NA, and NS) were cotransfected into human HEK-293T cells, and the accumulation of positive- and negative-sense RNAs was analyzed by primer extension. The pHW2000-M plasmid was excluded to avoid the formation of virus particles. Mutant HA vRNA templates with 3′-end H1-ssNCR truncations showed reduced steady-state levels of mRNA, cRNA, and vRNA compared to those of the wild-type HA segment. However, the 3L4-G76A and 3L5-G76A mutant HA vRNA templates with the additional G76A mutation showed increased RNA levels compared to those without the G76A mutation (Fig. 5A, top). The steady-state levels of mRNA, cRNA, and vRNA derived from the NA segment were not affected (Fig. 5A, bottom). We conducted the same experiment described above for the CA04 3′-end H1-ssNCR truncation mutants and obtained very similar results (Fig. 5B).
FIG 5.

The 3′-end H1-ssNCR-truncated mutant templates are replicated less efficiently than the wild-type HA template in multiple-template RNP reconstitution assays. (A, B) Seven pHW2000-PB2, -PB1, -PA, -HA (wild type or mutant), -NP, -NA, and -NS plasmids derived from WSN (A) or CA04 (B) were transfected into HEK-293T cells. A polymerase containing an active site mutation (PB1a, D445A/446A) was used as negative control (NC). Accumulation of HA and NA RNAs was analyzed by primer extension and 6% PAGE. The graph shows the mean intensity signals of mutant HA and NA RNAs relative to those of wild-type RNAs. The data represent the means ± SEM of three independent experiments. Two-way analysis of variance (ANOVA) with Dunnett correction for multiple testing; asterisks represent a significant difference from wild-type virus as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
The results above indicate that the HA vRNA templates with 3′-end H1-ssNCR truncations are transcribed and replicated less efficiently than the wild-type HA vRNA template. To address this further, we carried out an RNP reconstitution assay in which we coexpressed wild-type HA vRNA and a 3′-end H1-ssNCR-truncated HA vRNA, together with plasmids expressing polymerase and nucleoprotein. We used a transcription-deficient polymerase (PA-D108A) (40) to avoid the generation of mRNA, which produces a signal that overlaps with cRNA, hampering the quantitation of primer extension analyses. In comparison to the steady-state level of the wild-type HA vRNA template, the 3L2 to 3L6 3′-end H1-ssNCR-truncated mutant vRNA templates produced significantly reduced levels of cRNA. However, the 3L4-G76A and 3L5-G76A mutant templates with the additional G76A mutation produced significantly increased levels of cRNA compared to those of 3L4 and 3L5 without the G76A mutation and were not significantly different compared to cRNA levels produced by the wild-type HA vRNA template (Fig. 6A). We also observed similar effects when the C56U mutation was introduced into the 3L4 and 3L5 mutants (Fig. 6B). Together, these results show that the 3L2 to 3L6 3′-end H1-ssNCR-truncated mutants have a disadvantage compared to the wild-type HA segment in RNA synthesis. However, the introduction of the G76A or the C56U mutation in the 3L4 and 3L5 mutants eliminates this disadvantage and restores the deficiencies in RNA synthesis.
FIG 6.

The G76A and C56U mutations increased the replication of the 3′-terminal H1-ssNCR-truncated mutant template in multitemplate RNP recombination assays. HEK-293T cells were cotransfected with pcDNA-PB2, pcDNA-PB1, pcDNA-PA-D108A, pcDNA-NP, pPOL1-HA, and pPOL1-mut-HA with G76A(A) or C56U(B). A polymerase containing an active site mutation (PB1a, D445A/446A) was used as negative control (NC). Accumulation of RNA 24 h posttransfection was analyzed by primer extension. The graph shows the mean intensity signals of mutant HA RNAs relative to those of wild-type HA RNAs. The data represent the means ± SEM of three independent experiments. Two-way analysis of variance (ANOVA) with Dunnett correction for multiple testing; asterisks represent a significant difference from wild-type virus as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
DISCUSSION
In this study, we aimed to characterize the role of the 3′-end H1-ssNCR in the HA vRNA of influenza A viruses. We found that 3′-end H1-ssNCR-truncated mutant templates could be transcribed and replicated as efficiently as the wild-type template in single-template RNP reconstitution assays. However, recombinant viruses (WSN and CA04) with truncations in the 3′-end H1-ssNCR showed decreased HA vRNA levels both in virions and infected cells, and those with the longest deletions were severely attenuated. This was the result of reduced replication of the mutant HA vRNAs in the presence of the other seven vRNA segments in both transfected and infected cells. Interestingly, we found that a point mutation at a specific position located in the adjacent HA coding region of the attenuated viruses could restore the HA vRNA levels and virus growth. We propose that although not essential, the 3′-end H1-ssNCR as well as the adjacent coding region play a role in determining how efficiently the viral polymerase transcribes and replicates a template in the presence of the other seven vRNA templates during influenza A virus replication.
It has been reported that mutations introduced into the 3′-end NCR of a segment led to the reduction of this segment both in infected cells and in virions (16, 17), while other studies using RNP reconstitution assays found no evidence for a significant role of 3′-end NCR in regulating viral RNA synthesis (15, 18). We show here that the conserved 12 nucleotides and the two adjacent HA segment-specific CC nucleotides at the 3′-end of HA vRNA (as present in the 3L6 mutant) are sufficient for the viral polymerase to efficiently replicate and transcribe the vRNA in a single-template RNP reconstitution system. However, in a multisegment environment, such as during reverse genetics and viral infection, we observed a reduced replication of the 3′-end H1-ssNCR mutants, indicating that 3′-end NCRs influence replication and transcription and contribute to the fine-tuning of these processes. This is consistent with a previous report in which it was proposed that influenza virus genome segments could compete for available polymerase, especially at an early stage of infection. The ability of a segment to compete is dependent on its length and sequences both in the coding region and untranslated regions, although the mechanisms involved remain unknown (38). Our study, for the first time, reveals very distinct synthesis capacities of the same 3′-end NCR mutant templates in single-segment and multisegment environments and, as such, may help resolve the inconsistent conclusions on the regulatory role of the nonconserved NCR reported in the literature. It would be interesting to test whether 5′-end H1-ssNCR mutants have similar effects. Our data could be particularly relevant to the vRNA segments encoding HA and NA, which are known to act antagonistically and their activities have to be balanced during viral infection (41–45). The subtype-specific variation in sequence and length of NCRs of HA and NA vRNA could be a mechanism by which the levels of HA and NA are fine-tuned in different influenza A virus subtypes.
Upon viral passage, we identified an insertion in the 3′-end H1-ssNCR and a point mutation in the coding region of HA vRNA that improved viral growth. It remains unclear how a single-nucleotide mutation more than 20 to 40 nucleotides downstream of the mutated 3′-end H1-ssNCR restores vRNA replication. A recent study investigating the global high-resolution structure of the influenza A virus genome showed that vRNA in the context of vRNP is capable of accommodating secondary RNA structures with extensive base pairing (46). Le Sage et al. recently reported that vRNA-vRNA interactions in the context of vRNPs are highly flexible and redundant (47). We speculate that truncations in the segment-specific noncoding region could inhibit the formation of a beneficial secondary structure or promote the formation of an inhibitory secondary structure, and the single-nucleotide mutation could revert this. Alternatively, considering that different reversion mutations were obtained in different hosts (G76A for WSN in MDCK cells, either G76A or C56U for WSN in A549 cells, and C56A for CA04 in eggs), we speculate that the synergistic effect between 3′-terminal noncoding and adjacent coding regions of the H1 vRNA could also be mediated by a host factor. However, the underlying mechanisms that lead to the reversal of viral attenuation as a result of these single nucleotide mutations remain unknown and need further study to clarify. It should also be noted that the effect of 3′-end H1-ssNCR truncations on vRNA replication could be due to defects in the second step of replication, i.e., the replication of cRNA into vRNA, with the mutations affecting the 5′-end of the HA cRNA. In multisegment RNP reconstitution assays, mRNA levels were also affected (Fig. 5). It remains unclear whether this reduction in mRNA is a result of reduced vRNA template levels or if the truncations directly affect transcription as well.
It has been proposed that the 3′ and 5′ NCRs, together with the adjacent terminal coding regions of each segment, serve as segment-specific packaging signals in the selective genome-packaging model (29). However, replication of the mutant HA templates used in previous studies was mainly examined in single-template RNP reconstitution assays. It is unclear whether the reductions in HA vRNA in virions were entirely due to reduced packaging or reduced replication in a multisegment environment. In our study, we cannot exclude the possibility that the packaging efficiency of HA vRNA is also affected in the 3′-end H1-ssNCR-truncated mutant viruses. We speculate that the nucleotides in the ssNCRs of the HA segment might function as cis-acting signals in both vRNA replication and HA vRNA packaging. Furthermore, one could argue that defective particles (DI) could be generated upon reduction of HA vRNA due to a packaging defect (35). However, considering that the segment with the single-nucleotide mutation quickly outcompeted the truncated segment and the HA vRNA levels in virions recovered to wild-type levels after two virus passages, it is unlikely that a lot of DI particles would have been generated that would have interfered with virus growth in passages one and two.
In summary, we demonstrate that 3′-end H1-ssNCR truncations lead to decreased HA-specific RNA synthesis, resulting in virus attenuation. Point mutations found in the adjacent coding region reverse the defect in vRNA replication and restore virus growth. These findings unravel the importance of the NCR and the adjacent coding region in regulating the replication and transcription of vRNA segments during influenza A virus replication.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
Human embryonic kidney 293T (HEK-293T) cells and human lung carcinoma cell line A549 were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). Madin-Darby canine kidney (MDCK) cells were cultured in minimal essential medium (MEM) supplemented with 10% FCS and 2 mM l-glutamine. Cells were maintained at 37°C and 5% CO2. Plasmids pcDNA-PB2, pcDNA-PB1, pcDNA-PA, pcDNA-NP, and pcDNA-PB1a for the RNP reconstitution system of influenza A/WSN/33 (H1N1) virus (38, 48) have been described previously. Recombinant influenza A/WSN/33 (H1N1) virus was generated using the pHW2000 eight-plasmid system (49). The pHW2000 eight-plasmid system and the RNP reconstitution system of influenza A/California/04/2009 (H1N1) virus were generated in the same way as those of the WSN virus. Plasmids pHW2000-HA-3L1 to pHW2000-HA-3L6 were generated from pHW2000-HA by PCR amplification of the HA sequences, with corresponding primers introducing truncations in the 3′-end H1-ssNCR. The PCR products were then ligated into the BsmBI site of the pHW2000 vector. To construct the pHW2000-mut plasmid to express vRNA but not mRNA, the pHW2000 plasmid was modified by removing the truncated immediate early promoter of the human cytomegalovirus (CMV). The plasmids pHW2000-mut-HA-3L1 to pHW2000-mut-HA-3L6 were then generated by ligating the PCR products mentioned above into the pHW2000-mut plasmid. The plasmids with G76A or C56U mutation were generated from the corresponding pHW2000-HA or pHW2000-mut-HA plasmids using site-directed PCR mutagenesis.
Reverse genetics.
Recombinant influenza A/WSN/33 (H1N1) (WSN) virus or influenza A/California/04/2009 (H1N1) (CA04) virus was generated using the pHW2000 eight-plasmid system (49). Approximately 106 HEK-293T cells were transfected with 0.5 μg each of pHW2000-PB2, pHW2000-PB1, pHW2000-PA, pHW2000-HA (wild type or mutant), pHW2000-NP, pHW2000-NA, pHW2000-M, and pHW2000-NS using Lipofectamine 2000 and Opti-MEM according to the manufacturer’s instructions. At 24 h posttransfection, DMEM containing 10% FCS was replaced with DMEM containing 0.5% FCS and 100 U/ml penicillin-streptomycin (Thermo Fisher). The virus supernatant was harvested 48 h after changing medium. For serial passage, A/WSN/33 wild-type and mutant viruses were used to infect MDCK or A549 cells at a multiplicity of infection (MOI) of 0.001. A/California/04/2009 wild type and mutant viruses were diluted 10-fold with Opti-MEM and serial passaged in 9-day-old embryonated eggs (50). The virus titer was examined by plaque assay (A/WSN/33 virus) or TCID50 (A/California/04/2009 virus).
RNA sequencing.
The RNA of virus stock was extracted with TRI LS Reagent (Sigma-Aldrich) and reverse transcribed using the SuperScript III first-strand synthesis system (Invitrogen) with universal influenza A virus reverse transcription (RT) primers (vRNA_3_ga GTTCAGACGTGTGCTCTTCCGATCTAGCGAAAGCAGG; vRNA_3_aa GTTCAGACGTGTGCTCTTCCGATCTAGCAAAAGCAGG) (the nucleotide corresponding to position 4 at the 3′-end of vRNA, which can be either U or C, is underlined). The RT products were then amplified by PCR using HA segment-specific primers (Fwd_5′-GTGCTCTTCCGATCTAGCAAAAGCAGG-3′; Rev_5′-GATGTTACATTTCCCCAATTGTAGTGGGGC-3′ (A/WSN/33 virus); Rev_5′-GGTGTTTCCACAATGTAGGACCATGAGCT-3′ [A/California/04/2009 virus]), followed by sequencing of the PCR product.
Virus growth curve.
MDCK cells were infected with either wild-type WSN or recombinant virus at a multiplicity of infection (MOI) of 0.001. At 24, 36, 48, 60, and 72 h postinfection (p.i.), the supernatants were collected. The virus titers were determined by plaque assay on MDCK cells.
RNP reconstitution assay and primer extension analysis.
For single-template RNP reconstitution assays, vRNPs were reconstituted by transiently transfecting approximately 106 HEK-293T cells with 1 μg each of pcDNA-PB2, pcDNA-PB1/pcDNA-PB1a, pcDNA-PA, pcDNA-NP, and pHW2000-mut-HA (wild type or mutant) using Lipofectamine 2000 and Opti-MEM according to the manufacturer’s instructions. For double-template RNP reconstitution assays with a transcription-deficient but replication-competent polymerase (D108A) (40), approximately 106 HEK-293T cells were transfected as before with 1 μg each of pcDNA-PB2, pcDNA-PB1/pcDNA-PB1a, pcDNA-PA-D108A, pcDNA-NP, and pHW2000-mut-HA (wild type and mutant). For seven-template RNP reconstitution assays, pHW2000-PB2, -PB1, -PA, -HA (wild type or mutant), -NP, -NA, and -NS plasmids were transfected into HEK-293T cells. Plasmids pcDNA-PB1a and pPOLI-PB1 encoding polymerase with an active site mutation (D445A/446A) (38) were used as negative controls. Cells were harvested 24 h posttransfection. Total RNA was extracted using TRI Reagent (Sigma-Aldrich) and dissolved in 20 μl of nuclease-free water. RNA was analyzed by primer extension using 32P-labeled primers specific for negative- or positive-sense viral RNAs as well as 5S rRNA. The primers for segment 4 or 6 negative- or positive-sense RNA of A/WSN/33 virus and 5S rRNA have been described previously (51). 5S rRNA was used as internal loading control. In order to improve the separation of primer extension products from segment 4 negative- and positive-sense RNAs of the 3′-end H1-ssNCR-truncated WSN mutants, primer 5′-TTCGAGCAGGTTAACAGAATG-3′ was used to analyze the positive-sense HA RNAs. For the RNP reconstitution assays using pcDNA-PA-D108A, primer 5′-GCTACAAATGCATATAACAGGACC-3′ or 5′-AGGACCAGTAGTTTTGCC-3′ annealing close to the 5′ terminus of HA cRNA was used to analyze HA cRNA. For A/California/04/2009 virus, primer 5′-CTTCTAGAAGGTTAACAGAGTGTG-3′ was used to detect positive-sense RNA. Primer 5′-ATTGGTACTGGTAGTCTCCC-3′ was used to detect negative-sense RNA. In the RNP reconstitution assays using pcDNA-PA-D108A, primer 5′-GCGGTTGCAAATGTATATAGC-3′ annealing close to the 5′ terminus of HA cRNA was used to analyze HA cRNA. Primer extension products were analyzed by 6% or 12% denaturing PAGE with 7 M urea in Tris-borate-EDTA (TBE) buffer and visualized by phosphor imaging on an FLA-5000 scanner (Fuji). ImageJ was used to analyze the 32P-derived signal (52).
qPCR analysis of virion RNA and cellular RNA.
Wild-type or recombinant A/WSN/33 viruses and transfected or infected cells were harvested after virus rescue or infection. Virus was treated with 5 μl Benzonase (Novagen) at 37°C for 30 min to remove nucleic acids outside of the virions. Then virion RNA was extracted using TRI LS Reagent (Sigma-Aldrich), followed by DNase treatment (Turbo) and cleanup by using the RNA Clean & Concentrator-5 kit (Zymo Research). Cellular RNA was extracted using TRI Reagent (Sigma-Aldrich), followed by the same DNase treatment and cleanup as above. The virion RNA or cellular RNA was dissolved in 15 μl nuclease-free water. Approximately 40 ng virion RNA or cellular RNA was reverse transcribed by SuperScript III (Invitrogen) with universal influenza A virus reverse transcription (RT) primers as above. The RT product was then diluted 80-fold and used as a template for qPCR. One microliter of diluted RT product was used for a TaqMan probe-based qPCR using Brilliant III ultra-fast probe high ROX QPCR master mix (Agilent Technologies). The TaqMan probes and primer sequences are as follows: HA_Fw 5′-GTTTGGTGTTTCTACAATGTAGGACC-3′; HA_Rv 5′-CGAAGACAGACACAACGGGA-3′; HA_Taq [JOE]5′-CTGGAAGCAGTGAGTCGCA-3′[BHQ2]; NA_Fw 5′-CTGTGTATAGCCCACCCACG-3′; NA_Rv 5′-GCAACCAAGGCAGCATTACC-3′; NA_Taq [JOE]5′-TGCTGGGCAGGACTCAACTTCAGT-3′[BHQ1]. The primers named with “_Taq” are TaqMan probes labeled with fluorophores (JOE) and quenchers (BHQ1, BHQ2). A 40-cycle qPCR was carried out according to the manufacturer’s instructions on a StepOnePlus instrument (Applied Biosystems). Two technical replicates were performed for each segment per sample. The relative concentrations of vRNAs were determined on the basis of an analysis of cycle threshold values.
Statistics.
GraphPad Prism software, version 6, was used for statistical analysis. Two-way analysis of variance (ANOVA) with Dunnett correction was used for two-variable comparisons, while one-way analysis of variance (ANOVA) with Dunnett correction was used for one-variable comparisons. P values of <0.05 were considered to be significant.
ACKNOWLEDGMENTS
We thank Erich Hoffmann for the pHW2000 plasmids. We thank Hongjie Zhang (Core Facility for Protein Research, Institute of Crystal Structure of Biophysics, Chinese Academy of Sciences) for technical support with autoradiography.
This work was supported by grants from the National Mega-Project for Infectious Diseases (2018ZX10101001-004), the CAMS Innovation Fund for Medical Sciences (2016-12M-1-014), the UK Medical Research Council (MR/R009945/1), and the China Scholarship Council (201806210420).
Contributor Information
Ervin Fodor, Email: ervin.fodor@path.ox.ac.uk.
Tao Deng, Email: dengt@im.ac.cn.
Stacey Schultz-Cherry, St. Jude Children's Research Hospital.
REFERENCES
- 1.Taubenberger JK, Kash JC. 2010. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 7:440–451. 10.1016/j.chom.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisfeld AJ, Neumann G, Kawaoka Y. 2015. At the centre: influenza A virus ribonucleoproteins. Nat Rev Microbiol 13:28–41. 10.1038/nrmicro3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fodor E, Te Velthuis AJW. 2020. Structure and function of the influenza virus transcription and replication machinery. Cold Spring Harb Perspect Med 10:a038398. 10.1101/cshperspect.a038398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wandzik JM, Kouba T, Cusack S. 27April2020. Structure and function of influenza polymerase. Cold Spring Harb Perspect Med 10.1101/cshperspect.a038372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker AP, Fodor E. 2019. Interplay between influenza virus and the host RNA polymerase II transcriptional machinery. Trends Microbiol 27:398–407. 10.1016/j.tim.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorba N, Coloma R, Ortin J. 2009. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog 5:e1000462. 10.1371/journal.ppat.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan H, Walker AP, Carrique L, Keown JR, Serna Martin I, Karia D, Sharps J, Hengrung N, Pardon E, Steyaert J, Grimes JM, Fodor E. 2019. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 573:287–290. 10.1038/s41586-019-1530-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. 2012. Organization of the influenza virus replication machinery. Science 338:1631–1634. 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang S, Sun D, Liang H, Wang J, Li J, Guo L, Wang X, Guan C, Boruah BM, Yuan L, Feng F, Yang M, Wang L, Wang Y, Wojdyla J, Li L, Wang J, Wang M, Cheng G, Wang HW, Liu Y. 2015. Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 A resolution. Mol Cell 57:925–935. 10.1016/j.molcel.2014.12.031. [DOI] [PubMed] [Google Scholar]
- 10.Carrique L, Fan H, Walker AP, Keown JR, Sharps J, Staller E, Barclay WS, Fodor E, Grimes JM. 2020. Host ANP32A mediates the assembly of the influenza virus replicase. Nature 587:638–643. 10.1038/s41586-020-2927-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.York A, Hengrung N, Vreede FT, Huiskonen JT, Fodor E. 2013. Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc Natl Acad Sci U S A 110:E4238–E4245. 10.1073/pnas.1315068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lakdawala SS, Fodor E, Subbarao K. 2016. Moving on out: transport and packaging of influenza viral RNA into virions. Annu Rev Virol 3:411–427. 10.1146/annurev-virology-110615-042345. [DOI] [PubMed] [Google Scholar]
- 13.Robertson JS. 1979. 5' and 3' terminal nucleotide sequences of the RNA genome segments of influenza virus. Nucleic Acids Res 6:3745–3758. 10.1093/nar/6.12.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desselberger U, Racaniello VR, Zazra JJ, Palese P. 1980. The 3' and 5'-terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene 8:315–328. 10.1016/0378-1119(80)90007-4. [DOI] [PubMed] [Google Scholar]
- 15.Zhao L, Peng Y, Zhou K, Cao M, Wang J, Wang X, Jiang T, Deng T. 2014. New insights into the nonconserved noncoding region of the subtype-determinant hemagglutinin and neuraminidase segments of influenza A viruses. J Virol 88:11493–11503. 10.1128/JVI.01337-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng H, Palese P, Garcia-Sastre A. 1996. Nonconserved nucleotides at the 3' and 5' ends of an influenza A virus RNA play an important role in viral RNA replication. Virology 217:242–251. 10.1006/viro.1996.0111. [DOI] [PubMed] [Google Scholar]
- 17.Bergmann M, Muster T. 1996. Mutations in the nonconserved noncoding sequences of the influenza A virus segments affect viral vRNA formation. Virus Res 44:23–31. 10.1016/0168-1702(96)01335-4. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Peng Y, Zhao L, Cao M, Hung T, Deng T. 2015. Influenza A virus utilizes a suboptimal Kozak sequence to fine-tune virus replication and host response. J Gen Virol 96:756–766. 10.1099/vir.0.000030. [DOI] [PubMed] [Google Scholar]
- 19.Zheng M, Wang P, Song W, Lau SY, Liu S, Huang X, Mok BW, Liu YC, Chen Y, Yuen KY, Chen H. 2015. An A14U substitution in the 3' noncoding region of the M segment of viral RNA supports replication of influenza virus with an NS1 deletion by modulating alternative splicing of M segment mRNAs. J Virol 89:10273–10285. 10.1128/JVI.00919-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma J, Liu K, Xue C, Zhou J, Xu S, Ren Y, Zheng J, Cao Y. 2013. Impact of the segment-specific region of the 3'-untranslated region of the influenza A virus PB1 segment on protein expression. Virus Genes 47:429–438. 10.1007/s11262-013-0969-0. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Lee CW. 2009. Sequencing and mutational analysis of the non-coding regions of influenza A virus. Vet Microbiol 135:239–247. 10.1016/j.vetmic.2008.09.067. [DOI] [PubMed] [Google Scholar]
- 22.Maeda Y, Goto H, Horimoto T, Takada A, Kawaoka Y. 2004. Biological significance of the U residue at the −3 position of the mRNA sequences of influenza A viral segments PB1 and NA. Virus Res 100:153–157. 10.1016/j.virusres.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Fujii K, Fujii Y, Noda T, Muramoto Y, Watanabe T, Takada A, Goto H, Horimoto T, Kawaoka Y. 2005. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J Virol 79:3766–3774. 10.1128/JVI.79.6.3766-3774.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutchinson EC, Curran MD, Read EK, Gog JR, Digard P. 2008. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J Virol 82:11869–11879. 10.1128/JVI.01634-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang Y, Hong Y, Parslow TG. 2005. cis-Acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J Virol 79:10348–10355. 10.1128/JVI.79.16.10348-10355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marsh GA, Hatami R, Palese P. 2007. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J Virol 81:9727–9736. 10.1128/JVI.01144-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchinson EC, Wise HM, Kudryavtseva K, Curran MD, Digard P. 2009. Characterisation of influenza A viruses with mutations in segment 5 packaging signals. Vaccine 27:6270–6275. 10.1016/j.vaccine.2009.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gog JR, Afonso EDS, Dalton RM, Leclercq I, Tiley L, Elton D, von Kirchbach JC, Naffakh N, Escriou N, Digard P. 2007. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res 35:1897–1907. 10.1093/nar/gkm087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. 2003. Selective incorporation of influenza virus RNA segments into virions. Proc Natl Acad Sci U S A 100:2002–2007. 10.1073/pnas.0437772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muramoto Y, Takada A, Fujii K, Noda T, Iwatsuki-Horimoto K, Watanabe S, Horimoto T, Kida H, Kawaoka Y. 2006. Hierarchy among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. J Virol 80:2318–2325. 10.1128/JVI.80.5.2318-2325.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dos Santos Afonso E, Escriou N, Leclercq I, van der Werf S, Naffakh N. 2005. The generation of recombinant influenza A viruses expressing a PB2 fusion protein requires the conservation of a packaging signal overlapping the coding and noncoding regions at the 5' end of the PB2 segment. Virology 341:34–46. 10.1016/j.virol.2005.06.040. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe T, Watanabe S, Noda T, Fujii Y, Kawaoka Y. 2003. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J Virol 77:10575–10583. 10.1128/jvi.77.19.10575-10583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marsh GA, Rabadan R, Levine AJ, Palese P. 2008. Highly conserved regions of influenza A virus polymerase gene segments are critical for efficient viral RNA packaging. J Virol 82:2295–2304. 10.1128/JVI.02267-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozawa M, Maeda J, Iwatsuki-Horimoto K, Watanabe S, Goto H, Horimoto T, Kawaoka Y. 2009. Nucleotide sequence requirements at the 5' end of the influenza A virus M RNA segment for efficient virus replication. J Virol 83:3384–3388. 10.1128/JVI.02513-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang Y, Huang T, Ly H, Parslow TG, Liang Y. 2008. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J Virol 82:229–236. 10.1128/JVI.01541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odagiri T, Tashiro M. 1997. Segment-specific noncoding sequences of the influenza virus genome RNA are involved in the specific competition between defective interfering RNA and its progenitor RNA segment at the virion assembly step. J Virol 71:2138–2145. 10.1128/JVI.71.3.2138-2145.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Li J, Zhao L, Cao M, Deng T. 2017. Dual roles of the hemagglutinin segment-specific noncoding nucleotides in the extended duplex region of the influenza A virus RNA promoter. J Virol 91:e01931-16. 10.1128/JVI.01931-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vreede FT, Jung TE, Brownlee GG. 2004. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J Virol 78:9568–9572. 10.1128/JVI.78.17.9568-9572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Widjaja I, de Vries E, Rottier PJ, de Haan CA. 2012. Competition between influenza A virus genome segments. PLoS One 7:e47529. 10.1371/journal.pone.0047529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hara K, Schmidt FI, Crow M, Brownlee GG. 2006. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding, and virion RNA promoter binding. J Virol 80:7789–7798. 10.1128/JVI.00600-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neverov AD, Kryazhimskiy S, Plotkin JB, Bazykin GA. 2015. Coordinated evolution of influenza A surface proteins. PLoS Genet 11:e1005404. 10.1371/journal.pgen.1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaymard A, Le Briand N, Frobert E, Lina B, Escuret V. 2016. Functional balance between neuraminidase and haemagglutinin in influenza viruses. Clin Microbiol Infect 22:975–983. 10.1016/j.cmi.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 43.Diederich S, Berhane Y, Embury-Hyatt C, Hisanaga T, Handel K, Cottam-Birt C, Ranadheera C, Kobasa D, Pasick J. 2015. Hemagglutinin-neuraminidase balance influences the virulence phenotype of a recombinant H5N3 influenza A virus possessing a polybasic HA0 cleavage site. J Virol 89:10724–10734. 10.1128/JVI.01238-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu R, Zhu X, McBride R, Nycholat CM, Yu W, Paulson JC, Wilson IA. 2012. Functional balance of the hemagglutinin and neuraminidase activities accompanies the emergence of the 2009 H1N1 influenza pandemic. J Virol 86:9221–9232. 10.1128/JVI.00697-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yen HL, Liang CH, Wu CY, Forrest HL, Ferguson A, Choy KT, Jones J, Wong DD, Cheung PP, Hsu CH, Li OT, Yuen KM, Chan RW, Poon LL, Chan MC, Nicholls JM, Krauss S, Wong CH, Guan Y, Webster RG, Webby RJ, Peiris M. 2011. Hemagglutinin-neuraminidase balance confers respiratory-droplet transmissibility of the pandemic H1N1 influenza virus in ferrets. Proc Natl Acad Sci U S A 108:14264–14269. 10.1073/pnas.1111000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dadonaite B, Gilbertson B, Knight ML, Trifkovic S, Rockman S, Laederach A, Brown LE, Fodor E, Bauer DLV. 2019. The structure of the influenza A virus genome. Nat Microbiol 4:1781–1789. 10.1038/s41564-019-0513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le Sage V, Kanarek JP, Snyder DJ, Cooper VS, Lakdawala SS, Lee N. 2020. Mapping of influenza virus RNA-RNA interactions reveals a flexible network. Cell Rep 31:107823. 10.1016/j.celrep.2020.107823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fodor E, Crow M, Mingay LJ, Deng T, Sharps J, Fechter P, Brownlee GG. 2002. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J Virol 76:8989–9001. 10.1128/jvi.76.18.8989-9001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97:6108–6113. 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tannock GA, Paul JA, Barry RD. 1984. Relative immunogenicity of the cold-adapted influenza virus A/Ann Arbor/6/60 (A/AA/6/60-ca), recombinants of A/AA/6/60-ca, and parental strains with similar surface antigens. Infect Immun 43:457–462. 10.1128/iai.43.2.457-462.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robb NC, Smith M, Vreede FT, Fodor E. 2009. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J Gen Virol 90:1398–1407. 10.1099/vir.0.009639-0. [DOI] [PubMed] [Google Scholar]
- 52.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]