

Abstract
Epigenetic regulation plays an important role in controlling gene expression during complex processes, such as development of the human brain. Mutations in genes encoding chromatin modifying proteins and in the non-protein coding sequences of the genome can potentially alter transcription factor binding or chromatin accessibility. Such mutations can frequently cause neurodevelopmental disorders, therefore understanding how epigenetic regulation shapes brain development is of particular interest. While epigenetic regulation of neural development has been extensively studied in murine models, significant species-specific differences in both the genome sequence and in brain development necessitate human models. However, access to human fetal material is limited and these tissues cannot be grown or experimentally manipulated ex vivo. Therefore, models that recapitulate particular aspects of human fetal brain development, such as the in vitro differentiation of human pluripotent stem cells (hPSCs), are instrumental for studying the epigenetic regulation of human neural development. Here, we examine recent studies that have defined changes in the epigenomic landscape during fetal brain development. We compare these studies with analogous data derived by in vitro differentiation of hPSCs into specific neuronal cell types or as three-dimensional cerebral organoids. Such comparisons can be informative regarding which aspects of fetal brain development are faithfully recapitulated by in vitro differentiation models and provide a foundation for using experimentally tractable in vitro models of human brain development to study neural gene regulation and the basis of its disruption to cause neurodevelopmental disorders.
Keywords: Human brain development, Pluripotent stem cells, Epigenetic regulation, Chromatin, Neuron, Organoid
1. Introduction
Epigenetic regulation plays a central role in brain development, enabling dynamic control of gene expression in developmental time and space. During embryonic development, large-scale changes in gene expression and chromatin structure occur, upregulating lineage-specific genes and downregulating pluripotency genes and genes expressed in other lineages. Epigenetic regulation alters gene expression by modification of histone tails (for example, adding acetylation or methylation), by altering chromatin accessibility, or by methylating DNA. Human brain development is a protracted process, encompassing much of gestation and producing a diversity of cell types. Accordingly, many different epigenetic regulatory mechanisms shape developmental transitions to generate distinct neuronal and glial cell types (Zhao and Bhattacharyya, 2018). Epigenetic regulation of neural development is of particular interest because mutations in genes encoding proteins that modify or remodel chromatin are highly over-represented as contributors to the etiology of multiple neurodevelopmental disorders (NDDs), including autism spectrum disorder (ASD) (Lewis and Kroll, 2018; Loke et al., 2015). Moreover, many NDD risk variants are located in non-protein coding sequences in the genome, where variation may impact gene regulation by altering transcription factor (TF) binding, chromatin accessibility, or other regulatory mechanisms (Zhou et al., 2019).
Although epigenetic regulation of neural development has been studied extensively in murine models, there are substantial differences between human and murine brain development. These differences include a greater diversity of neural cell types and brain structures in humans, a longer neurodevelopmental timeline, human-specific disease phenotypes not seen in mouse models, and substantial differences in the genome sequence, particularly in non-protein coding regions of the genome (Clowry, 2015; DeFelipe, 2011; Jones, 2009; Letinic et al., 2002; Zhao and Bhattacharyya, 2018). Therefore, the use of human models is necessary to understand neurodevelopment and its disruption to cause NDDs.
Currently, human neural development can be studied through embryonic and fetal cells/tissues or using models mimicking particular aspects of this process (Fig. 1). However, the use of fetal and embryonic material is challenged by limited access and the inability to grow or genetically manipulate these tissues ex vivo, making the study of human brain development difficult. Despite such challenges, some large-scale efforts, including the Encyclopedia of DNA Elements (ENCODE) and the NIH Roadmap Epigenomics projects, have transcriptionally and epigenetically profiled many cell and tissue types from the developing human fetus, including the brain (ENCODE Project Consortium, 2012; Kundaje et al., 2015). In vitro models, which mimic developmental processes by differentiation of human pluripotent stem cells (hPSCs), can also be used to study aspects of brain development without the limitations of using fetal tissue. Here, we discuss transitions in the epigenetic landscape that occur during fetal brain development in vivo and compare these with data obtained for analogous cell or tissue types during the directed differentiation of neurons from hPSCs in vitro.
Fig. 1.
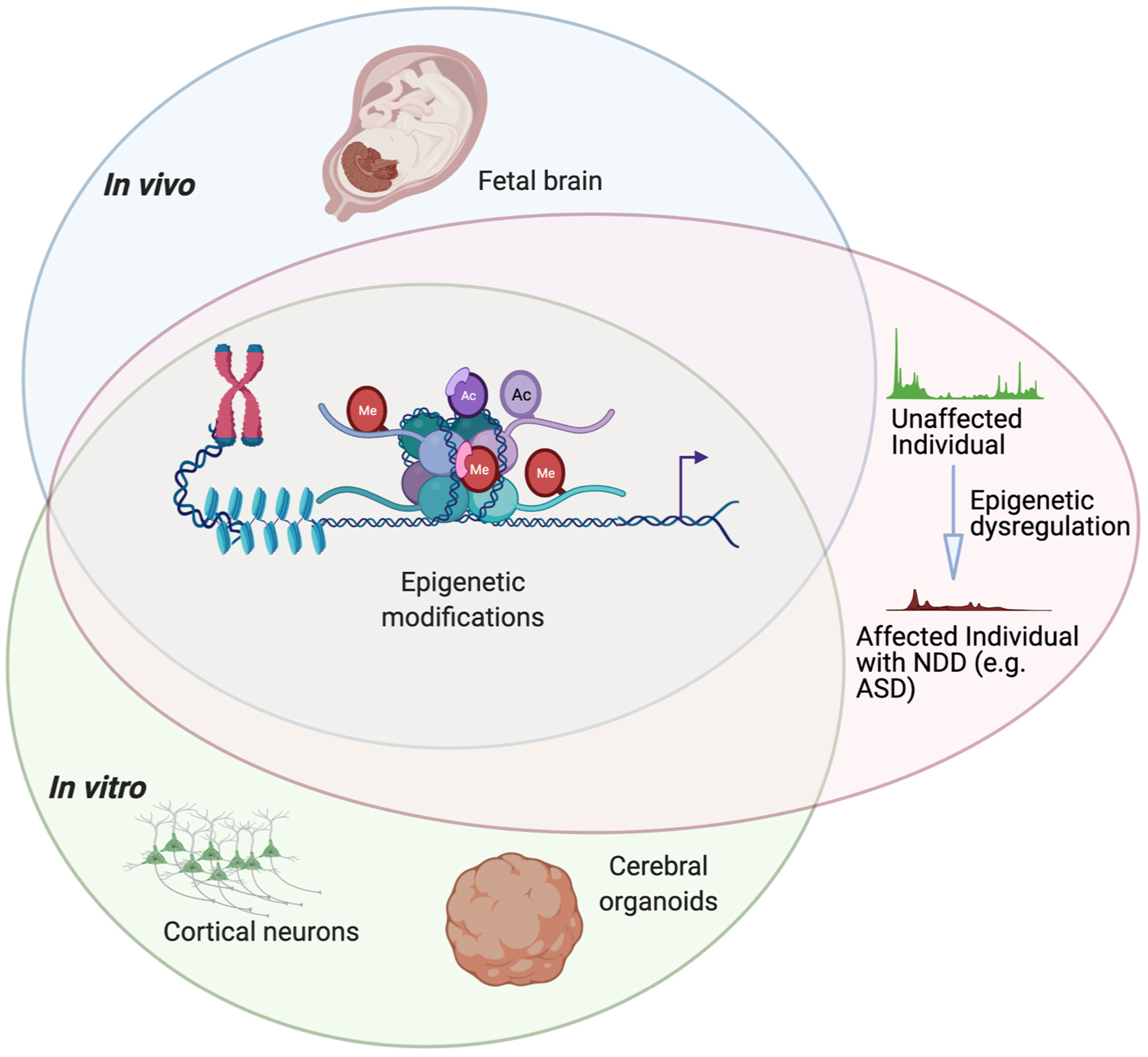
Three models for studying epigenetic regulation of human brain development. High-throughput sequencing approaches such as ATAC-seq or ChIP-seq have been used to interrogate changes in the epigenetic landscape throughout human cortical development. Disruption of epigenetic regulation during embryonic development can contribute to neurodevelopmental disorders such as autism spectrum disorder (ASD). Human brain cells or tissues for use in these assays can be harvested from the developing fetus or produced by in vitro differentiation of human pluripotent stem cells into cortical neurons or cerebral organoids. Figure created using BioRender.com.
2. Epigenetic regulation of gene expression
The major classes of proteins or protein complexes which modulate the epigenome include those that read, write, or erase histone protein tail modifications, remodel the histone-DNA structure, and methylate DNA (Hyun et al., 2017; Sadakierska-Chudy and Filip, 2015). These regulators influence chromatin accessibility, creating euchromatin, which is transcriptionally accessible, or heterochromatin, which is transcriptionally inactive. Although DNA methylation is an important mechanism of epigenetic regulation, many excellent reviews have already been published on this topic (Greenberg and Bourc’his, 2019; Hirabayashi and Gotoh, 2010; Jang et al., 2017). Instead, we focus predominantly on epigenetic regulation through histone modification and chromatin remodeling, as well as the associated changes in chromatin state. Chromatin consists of nucleosomes comprised of a histone octamer, around which DNA is wrapped. The ‘tails’ of histone proteins can acquire different post-translational modifications, influencing overall chromatin structure (Bowman and Poirier, 2015). Histone modifications that are particularly important for developmental gene regulation include tri-methylation of histone 3 lysine 27 (H3K27me3), a repressive modification; H3K4me3, a modification associated with active gene expression; and H3K27ac, a modification associated with active CREs (e.g. enhancers). Often, H3K27me3 and H3K4me3 are present together near gene promoters, creating a ‘bivalent state’ where genes are ‘poised’ for activation (by the removal of H3K27me3) or repression (by the removal of H3K4me3) during development (Burney et al., 2013). As development is a dynamic process during which cells must respond rapidly to cues related to their location and identity, modulating gene expression in this manner during developmental transitions is particularly important.
Several assays are commonly used to study chromatin state, each of which has strengths and limitations. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) uses an antibody directed against a protein of interest (e.g. a particular histone modification) to enrich for the chromatin regions bound by the protein. These protein-bound regions are then identified by sequencing. Although ChIP-seq is a powerful tool, it often results in a high background of non-selective sequence immunoprecipitation. Additionally, it requires a large number of cells and an antibody that can recognize a crosslinked epitope. To assess the general accessibility of chromatin, the Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq) is commonly used, as it requires fewer cells, with single-cell analysis now feasible (Buenrostro et al., 2015). Prior to the development of ATAC-seq in 2015, DNase-seq (also known as DNase hypersensitivity) and MNase-seq were also commonly utilized to assess chromatin accessibility (Klein and Hainer, 2020; Tsompana and Buck, 2014). Like ChIP-seq, DNase/MNase-seq assays require larger cell numbers (tens to hundreds of millions of cells) by comparison with ATAC-seq (Wj, 2014). Nonetheless, ChIP-, DNase-, and MNase-seq have been used to identify key information about chromatin state changes during human brain development and are incorporated into this review. The advantages and disadvantages of using these assays to assess chromatin state in the context of each model (i.e. in fetal tissue, during in vitro differentiation of hPSCs, or in cerebral organoids) are discussed in each section below, especially with respect to limitations pertaining to the numbers or homogeneity of cells assessed in each model. Below, we describe extant data that contribute to our understanding of epigenetic regulation of human brain development, as well as outline knowledge gaps that remain unresolved, particularly related to understanding regulation of chromatin state at neuronal cell type-specific resolution.
3. Epigenetic regulation in the fetal brain
3.1. Background
Human brain development begins in the third gestational week (GW) with the formation of the neural plate (Fig. 2) and extends beyond birth (Stiles and Jernigan, 2010). Using human embryonic or fetal cells or tissues to study epigenetic regulation of human brain development poses several challenges (Moradi et al., 2019). These challenges involve both ethical considerations and practical limitations related to access to human fetal material; specifically, very limited materials can be obtained from each embryo and these materials cannot be expanded or experimentally manipulated ex vivo. The consequences of these limitations are that sample size, and therefore biological replicate numbers, for these studies are often small, encompassing cells or tissues derived from only a few (or sometimes only one) unique embryo(s) or fetus (es) (Supplementary Table 1). Additionally, materials for study are obtained from a small, developing embryo with limited numbers of cells which are not expandable, limiting the number of different chromatin features that can be evaluated. This limited material availability is particularly problematic for methods like ChIP-seq, where assessing enrichment for each histone modification or chromatin-bound protein requires a large number of cells (more than one million per experimental replicate). Consequently, different histone modifications must usually be profiled using material from different embryos, increasing variability and making cross-comparisons more challenging. Given these limitations related to material availability, studies typically focus on a small number of histone modifications, or solely on chromatin accessibility, to increase the number of biological replicate experiments that can be conducted (Table 1; Supplementary Table 1). Finally, since embryonic development is a dynamic process involving constant changes in cell identity and physical location, profiling neural development with spatial and cell type-specific resolution is challenging. Several studies have performed epigenetic profiling on dissected tissues, which provides some spatial resolution but further reduces cell numbers available for any particular assessment (Supplementary Table 1).
Fig. 2.

In vivo-derived human fetal cells and tissues. (A) Studies have performed epigenetic assays such as ChIP-seq, ATAC-seq, RNA-seq, and DNA methylation by using materials derived from human fetuses from 7 to 38 post-conceptional weeks (PCW). The data shown and other related information can be retrieved using accession numbers from the NCBI-GEO and PsychENCODE databases, shown in pink text and described in more detail in Supplementary Table 1. (B) Bulk brain tissue, dissected regions, or single cells have been assayed. The cerebral cortex is a particular focus of this review. Figure created using BioRender.com.
Table 1.
An overview of papers using three models to study the epigenetic regulation of human brain development. (A) Studies using fetal cells and tissues derived from different developmental stages. Post-conceptional weeks = PCW. Gestational weeks = GW. (B) Studies using in vitro differentiation of hPSCs into cortical excitatory neurons, including differentiation method(s) used. (C) Studies using cerebral organoids, including differentiation method (s) used. hCOs = human cortical organoids. hMEGOs = human medial ganglionic eminence organoids.
| (A) FETAL CELLS AND TISSUES | ||||
|---|---|---|---|---|
| Fetal Stage(s) | Bulk/Single-cell | Chromatin Assay(s) | Details | Reference |
| 15–17 PCW (n =3) |
Bulk (dissected coronal sections) |
ATAC-seq | de la Torre Ubieta et al. 2018 | |
| 12 GW (n =2 tech. reps) |
Bulk (whole brain) |
ChIP-seq | H3K27ac, H3K4me1, H3K4me3, H3K27me3, and H3K27me3/H3K4me3 | Yan et al. 2015 |
| Mid-gestation (n =6) |
Single-cell (mix of dissected and whole forebrain) |
ATAC-seq | Ziffra et al. 2020 | |
| 17–38 PCW (n =4) |
Bulk (dissected brain regions) |
ChIP-seq | H3K4me3, H3K27me3, and H3K27ac | Li et al. 2018 |
| 7–12 PCW (n =2) |
Bulk (dissected cortex and separated lobes for 12 PCW) |
ChIP-seq | H3K27ac and H3K4me2 | Reilly et al. 2015 |
| 14–22 GW (n =2–4) |
Bulk (dissected telencephalon and prefrontal cortex) |
ATAC- and ChIP-seq | H3K27ac, H2K27me3, H3K9me3, and H4K20me3 | Markenscoff-Papadimitriou et al. 2020 |
| (B) IN VITRO CELLULAR MODELS | ||||
| Timepoint(s) | Differentiation Method(s) | Chromatin Assay(s) | Details | Reference |
| 3–72 hours | Dual-SMAD | ATAC- and ChIP-seq | H3K27ac and H3K27me3 | Inoue et al. 2019 |
| 12–220 days | Dual-SMAD | ChIP-seq | H3K4me1, H3K4me3, H3K27ac, and H3K27me3 | Ziller et al. 2015 |
| 0–16 days | Dual-SMAD | ATAC-seq | Shang et al. 2018 | |
| 21 days | NGN2 overexpression | ATAC-seq | van der Raadt et al. 2019 | |
| 30 days | Dual-SMAD | ATAC-seq | Forrest et al. 2017 | |
| 16 days and neurons | Dual-SMAD and NGN2 overexpression | ATAC-seq | Schafer et al. 2019 | |
| (C) CEREBRAL ORGANOIDS | ||||
| Timepoint(s) | Differentiation Method(s) | Chromatin Assay(s) | Details | Reference |
| 0–30 days | Single-SMAD w/ Noggin | ChIP-seq | H3K4me3, H3K27ac, and H3K27me3 | Amiri et al. 2018 |
| 20–600 days | Dual-SMAD | ATAC-seq | Trevino et al. 2020 | |
| 0–120 days | Lancaster | ATAC-seq | Bulk and single-cell | Kanton et al. 2019 |
| 0–72 days | Dual-SMAD (hCOs) and SHH agonist (hMGEOs) | ATAC-seq | Xiang et al. 2017 | |
| 0–35 days | Dual-SMAD | ATAC-seq | Single-cell | Ziffra et al. 2020 |
As ATAC-seq requires far fewer cells than ChIP-seq, it is more amenable to studies using fetal tissue; however, it does not provide information regarding changes in histone modification state that centrally regulate development, such as the transition from bivalent to active chromatin (Buenrostro et al., 2015). Other strategies include combining transcriptional and epigenetic profiling (either using dissected tissues or single cells), enabling cell type to be inferred from gene expression (Supplementary Table 1). However, analysis of such data is predicated upon understanding the transcriptional identity of diverse cell types at each developmental stage; this information is often unavailable due to our inability to trace and genetically manipulate human fetal cells. The advent of single-cell sequencing technologies has improved our ability to obtain transcriptional and epigenetic data with spatial and cell type-specific resolution (Armand et al., 2021; Kundaje et al., 2015; Rotem et al., 2015). Below we survey epigenetic studies of human fetal brain development, focusing on profiling of histone modification state and chromatin accessibility using both bulk and single-cell methods.
3.2. ChIP-seq: histone modifications
Despite the challenges described above, several studies have assessed key histone modifications in the developing human brain, including H3K4me3, H3K27me3, and H3K27ac (Table 1; Supplementary Table 1). Initial work to profile these modifications and others was performed as part of the Human Reference Epigenome Mapping Project and is described with other data from this project below. In addition to these studies, Yan et al. profiled the epigenomic landscape of human fetal brain, heart, and liver (Yan et al., 2016). Using two technical replicates from 12 GW embryos, they used ChIP-seq to assess H3K4me1, H3K27ac, H3K4me3, and H3K27me3, and performed sequential ChIP-seq for H3K27me3/H3K4me3 to identify bivalent domains. As expected, many putative CREs identified by H3K27ac enrichment exhibited tissue- and developmental stage-specific enrichment by comparison of peaks present in fetal brain, heart, and liver, and with other H3K27ac peak datasets from corresponding adult tissues and from hESCs. Significantly, both putative fetal brain-specific enhancers and bivalent domains were predicted to regulate genes important for neural development. The authors also identified several so-called ‘super-enhancers,’ defined as large (20–50 kb) regions enriched for H3K27ac signal. These ‘super--enhancers’ usually correspond with and contribute to regulating tissue-and stage-specific gene expression; in this study, they were predicted to regulate genes known to regulate neuronal development (Yan et al., 2016). Although this work was limited in terms of the biological replicates utilized and only analyzed one developmental time point, these data from Yan et al. provide a useful resource and excellent example of the types of analyses and comparisons that can be performed with ChIP-seq and RNA-seq data generated for the developing human brain.
Epigenetic profiling of human fetal cortical development over time was also performed in 2018 by Li et al., in work supported by the PsychENCODE and Brainspan Consortia (Li et al., 2018). This large, collaborative effort assessed the transcriptomic and epigenomic landscape across human brain regions and cell types through both pre- and postnatal development. Comprehensive details of the samples, assays, and time points analyzed are provided in Supplementary Table 1. In short, up to 16 brain regions were dissected in more than 60 postmortem human brains, ranging from five post-conceptional weeks (PCW) to 64 years of age, and assays including RNA-seq, single-cell RNA-seq (scRNA-seq) and ChIP-seq for several histone modifications (H3K27ac, H3K4me3, and H3K27me3) were performed. Although we focus here only on findings from the prenatal samples, further comparisons to data from the postnatal brain can serve as a useful tool to identify regulatory mechanisms that are unique to the developing human brain. For the prenatal samples, the study incorporated histone ChIP-seq data from 17, 19, 21, and 38 PCW embryos with RNA-seq and other data generated from the developing human brain (such as DNA methylation) (Li et al., 2018). This data can be used to define which developmental stages and brain regions in vitro cellular models of cortical development most closely resemble, based upon histone modification state.
3.3. ATAC-seq
Requiring far fewer cells than ChIP-seq and offering a comprehensive landscape of chromatin state, ATAC-seq is a useful tool to study in vivo human brain development (Table 1; Supplementary Table 1). In 2018, de la Torre-Ubieta et al. used ATAC-seq to extensively assess chromatin accessibility and to compare accessibility with changes in gene expression in the developing human cortex (de la Torre-Ubieta et al., 2018). They dissected tissue in the ventral germinal zone (GZ; a neural progenitor/radial glia-rich layer below the cortical plate (CP)) and the dorsal CP (containing differentiating neurons) from the developing forebrain of three female embryos between 15 and 17 PCW. They identified a significant number of differentially accessible regions between the CP and GZ and demonstrated a high correlation between increased chromatin accessibility and expression of the associated genes. Importantly, they showed significant overlap between their data and chromatin accessibility data generated for similar sample types by the NIH Roadmap Epigenomics Project. They also used previously published Hi-C data, which analyzed genome-wide chromatin organization in matched stages and tissue types (Won et al., 2016), to demonstrate that their identified distal CREs were associated with key genes specific to human cortical neurogenesis, including human-specific enhancers. They were also able to test the functional significance of two of their predicted enhancers, which regulated the FGFR2 and EOMES genes, by experimentally manipulating their sequences (de la Torre-Ubieta et al., 2018).
Markenscoff-Papadimitriou et al. recently expanded on this chromatin accessibility work from de la Torre-Ubieta et al., assessing chromatin accessibility in the developing human cortex with increased temporal and anatomical specificity (Markenscoff-Papadimitriou et al., 2020). They dissected nine regions of the mid-gestation (14–19 GW) telencephalon, including six cortical regions and the medial, lateral, and caudal ganglionic eminences. They identified predicted regulatory elements with differential accessibility between the regions and time points studied and correlated these with gene expression. The functional significance of these regions was tested in several ways, including by luciferase assay in neuroblastoma cells and using CRISPR-mediated gene activation (CRISPRa) in a mouse model. Interestingly, two separate sequence variants were identified in the same regulatory element associated with SLC6A1, a gene that causes neurodevelopmental disorders when mutated. When tested by luciferase assay, the two SLC6A1-associated regulatory element variants reduced reporter expression (Markenscoff-Papadimitriou et al., 2020). The chromatin accessibility and gene expression data presented by Markenscoff-Papadimitriou et al. provide a valuable resource, both for comparisons with future studies of chromatin accessibility in developing human brain and for integration with other fetal brain data. Such data may include ChIP-seq for histone modifications or TF binding and transcriptional profiling by single-cell or bulk RNA-seq.
The utility of integrating different types of genome-wide data was already demonstrated in a study comparing bulk tissue ATAC-seq data (de la Torre-Ubieta et al., 2018) with scRNA-seq profiling of 40,000 cells from the developing human brain, using four female embryos between 17 and 18 PCW (Polioudakis et al., 2019). Combining these high-resolution transcriptome data with information about chromatin accessibility allowed them to predict binding and co-expression of TFs previously associated with specific cell types and to identify new regulators of gene expression in a variety of neuronal cell types. This data integration also enabled associations to be drawn between genetic variants that affect both risk for neuropsychiatric disorders and regulation of neuronal cell type-specific gene expression. For example, variants associated with schizophrenia (SZ) risk were associated with gene regulation in multiple neural cell types, including neural progenitors, interneurons, and glutamatergic neurons (Polioudakis et al., 2019). This study by Polioudakis et al. demonstrates the utility of integrating transcriptional and epigenetic data to gain a deeper understanding of human cortical development and to associate these findings with genetic data related to risk for neurodevelopmental or neuropsychiatric disorders.
In addition to scRNA-seq, scATAC-seq can provide valuable information about chromatin accessibility in specific cell types in the developing brain, which bulk analysis cannot resolve. In a recent manuscript, currently available only as a pre-print, Ziffra et al. profiled chromatin accessibility by scATAC-seq for cells from distinct regions of the developing human forebrain (Ziffra et al., 2019). They profiled >75,000 cells from six mid-gestation embryos, preserving information about anatomical region of origin (eight distinct areas) for three embryos, while also analyzing bulk tissue from three embryos. Significantly, they could identify differentially accessible peaks between different cell types and regions, while also observing significant overlap between their data and enhancers identified in another study of bulk cortex tissue, defined by H3K27ac enrichment (Reilly et al., 2015), as well as human-specific forebrain enhancers available through the VISTA Enhancer Browser (Visel et al., 2007). They also analyzed enriched TF binding motifs in the sequences underlying neuronal cell type-specific H3K27ac peaks and found enrichment for binding motifs for TFs known to act in that cell type, such as enrichment for binding motifs for NKX2–1 in medial ganglionic eminence (MGE) progenitors. Although the functional significance of these predicted enhancers cannot be tested in human embryos, Ziffra et al. employed the recently developed activity-by-contact (ABC) model, which predicts enhancers by integrating H3K27ac ChIP-seq, gene expression, chromatin accessibility, and Hi-C datasets (Fulco et al., 2019). This powerful tool allowed them to identify sets of high-confidence putative enhancers for each cell type in the human fetal brain and to predict their target genes. As discussed below, this study by Ziffra et al. also performed scATAC-seq in cortical organoids, identifying a number of shared peaks between the fetal cerebral cortex and cortical organoids, but also showing that a significant portion of peaks identified in primary fetal cells were missing in organoids (Ziffra et al., 2019). While the data from this work are not yet publicly available, they should serve as a resource to understand cell type- and region-specific regulation of human cortical development and will also be useful to compare to other in vitro models of human brain development.
3.4. Human accelerated regions
Although ~99% of the DNA sequence is identical in both the human and chimp genome, about 5% of human DNA sequences have been rearranged, copied, or deleted by comparison to their chimp counterparts (Kehrer-Sawatzki and Cooper, 2007; Watanabe et al., 2004). About 5–10% of the human genome is under positive evolutionary selection, remaining invariant across mammals and thus likely functional. However, some evolutionarily conserved regions - termed human accelerated regions (HARs) - exhibit significantly accelerated nucleotide substitution rates, specific only in the human (Pollard et al., 2006) and ape lineages (del Rosario et al., 2014; Kostka et al., 2018). Other studies have characterized HARs that arose after the divergence of Homo sapiens from other hominid species (Meyer et al., 2012; Prüfer et al., 2014). The function of most HARs have not been elucidated, but many are intergenic and located near developmental genes (Capra et al., 2013); thus they may act as developmental enhancers or long noncoding RNAs (Franchini and Pollard, 2017; Levchenko et al., 2017; Li et al., 2019). Mutated HARs have also been implicated in neurodevelopmental disorders, such as ASD. Doan et al. discovered that individuals with ASD had a 6.5-fold enrichment for rare de novo copy number variants in HARs than sibling-matched controls (Doan et al., 2016). Furthermore, ASD probands have an excess of rare biallelic point mutations in HARs; such mutations in HARs that are active in neurons can contribute ASD risk to approximately 5% of individuals in a family.
In 2015, Prescott et al. epigenetically profiled cranial neural crest cells (CNCCs), using pluripotent-stem-cell-based in vitro differentiation models in both human and chimp (Prescott et al., 2015). The authors studied histone modifications associated with active regulatory elements (H3K4me1, H3K4me3, and H3K27ac) and performed ATAC-seq in both models. Approximately 6% of human CNCC enhancers showed increased or decreased enhancer activity in chimpanzees. Interestingly, human CNCC enhancers overlapped with 163 HARs, while functional testing of 20 of these enhancers demonstrated species-biased activity (Prescott et al., 2015). Kanton et al. also compared potential gene regulatory mechanisms in human and chimpanzee stem cell-derived cerebral organoids using bulk and single-cell accessible chromatin profiling by ATAC-seq (Kanton et al., 2019). Differentially accessible peaks had significant numbers of single nucleotide changes (SNCs) that are conserved in humans and unique versus other primates. Some SNCs are thought to create new or interfere with current TF binding sites. The authors further annotated accessible regions, defining species-specific potential regulatory regions near differentially expressed genes that have human-conserved SNCs or that are HARs, identifying 62 HARs that intersected with these differentially accessible peaks. Differentially expressed genes like cadherin 7 (CDH7), a gene expressed in human but not chimp neurons, were close to some of these HARs, and were perhaps linked to their activity (Kanton et al., 2019). In 2015, Reilly et al. also profiled H3K27ac and H3K4me2 to compare active enhancers and promoters during human, rhesus macaque, and mouse corticogenesis, pinpointing increases (gains) in their activity in humans. Eight thousand nine hundred and ninety-six nonoverlapping enhancers and 2855 promoters in total exhibited gains of these enhancer-associated histone modifications in humans. Whether such increases resulted from human-specific sequence changes was also analyzed. While 48 HARs showed increased H3K27ac or H3K4me2 in the human cortex in general, these gains were not associated with significant human-only accelerated sequence variation (Reilly et al., 2015). Nonetheless, these enhancer-associated histone modification gains were consistently associated with genes important for cortical development and similar increases in H3K27ac enrichment also occur in regions with ancestral regulatory activity, such as promoters or enhancers that are also H3K27ac enriched in rhesus and mouse cortices. Any human-specific gains not enriched in rhesus or mouse may indicate de novo regulatory mechanisms developed through human evolution.
The evolution of human-specific gene regulatory features during neural development might further be driven by transposable elements (Trizzino et al., 2017), which can either act as enhancers or interfere with transcription (Shapiro, 2017). In 2015, Notwell et al. reported that a specific family of transposable elements (MER130) was enriched in active enhancers associated with neocortex development in the mouse (Notwell et al., 2015). Of particular recent interest are Krüppel-associated box (KRAB) domain-containing zinc-finger proteins (KZFPs), a rapidly evolving family of TFs that can bind transposable elements (Ecco et al., 2016; Imbeault et al., 2017). KZFPs transcriptionally silence transposable elements by recruiting other chromatin binding proteins, such as SETDB1 (Pontis et al., 2019). An evolutionary “arms race” between transposable elements and KZFPs in turn may contribute to the evolution of human-specific features of gene regulatory networks, which are of interest for understanding human neural development and disease.
3.5. Other fetal epigenetic data obtained from consortium-based projects
In addition to the previously described PsychENCODE and Brainspan consortia (Li et al., 2018), fetal brain epigenetic data has been collected by other consortia-supported efforts, including the NIH Roadmap Epigenomics Program and the ENCODE project (ENCODE Project Consortium, 2012; Kundaje et al., 2015). Many of these consortia-acquired datasets are outlined in Supplementary Table 2. Some of these data are also included in other published analyses, for example, for comparison to epigenomes of other fetal cell types (Kundaje et al., 2015). Since this work is not specific to fetal brain development, we focused here on highlighting and summarizing the available fetal brain data and creating a curated database (Supplementary Table 2), which may serve as a resource for readers seeking extant fetal brain epigenetic datasets for their analyses. The fetal brain epigenetic data were collected by researchers at the University of Washington, The Broad Institute, and a joint University of California San Francisco-University of British Columbia (UCSF-UBC) team. Most data gathered by the University of Washington team focused on DNase hypersensitivity, analyzing male and female embryos from gestational day 85–142 (~12–20 GW). The research team at the Broad Institute also used material from these embryos to profile histone modifications in conjunction with DNase hypersensitivity profiling. The most complete datasets were generated by the UCSF-UBC team by using twin donors, HuFNSC01 and HuFNSC02 (Shapiro, 2017). The fetal brain epigenetic datasets include whole brain samples, as well as data derived from cortex and GE neurosphere cultures, which were collected from either the cortex or the GE at 17 GW and cultured as neurospheres for approximately three passages in NeuroCult + EGF + FGF (“Genboree Discovery System - Project: XML Submissions/UCSF-UBC/SAMPLE/EDACC.5485, no date (n.d.)”). Although it is not clear whether a specific structure from the GE, such as the MGE was used, this strategy should decrease the heterogeneity of cell types assessed by comparison with whole brain-derived samples, and therefore provides datasets with more tissue-restricted resolution to use for comparisons to data from in vitro-derived neurons. Histone marks profiled included H3K27me3, H3K4me3, H3K36me3, H3K9me3, and H3K4me1, and limited data was also obtained for H3K9ac and H3K27ac. Together with the data discussed previously, this fetal brain epigenetic data from the consortia provide a snapshot of changes in the epigenetic landscape during human cortical development. However, these data are understandably limited, as they lack either tissue- or cell type-specific resolution, and include a limited range of histone modifications, developmental timepoints, and donors. In our conclusions section of this review, below, we describe additional data that would be highly useful, both to understand fetal cortical development and to draw comparisons with in vitro neural differentiation-based studies.
4. In vitro cellular models
4.1. Background
In vitro models that derive different types of human neurons from PSCs circumvent many limitations of using fetal material. For example, in vitro models can generate a large number of human neuronal progenitors and neurons of particular types, which can be expanded and experimentally manipulated. However, some aspects of fetal development may not be fully recapitulated in vitro. Therefore, it is essential to compare data derived from in vitro models with analogous data from the fetal brain, such as gene expression changes and epigenetic regulation. A significant advantage of in vitro models is the ability to use either or both proband-derived iPSCs and genetically engineered PSCs with introduction of a pathogenic NDD-associated mutation, to study the contribution of a specific mutation versus the genetic background to NDD etiology. These approaches involving specific mutations are particularly well-suited to studying NDDs, many of which involve mutations in either chromatin modifying proteins and/or putative CREs (Lewis and Kroll, 2018; Loke et al., 2015). The following sections describe studies that assess changes in histone modification state and chromatin accessibility during in vitro neuronal differentiation in several models. By comparison with data from fetal samples, substantially more histone modifications can be assayed within the same set of experiments, more timepoints during differentiation can be interrogated, and results from multiple types of assays, including RNA-seq, ChIP-seq, and ATAC-seq, can be readily integrated. These rich data provide a more complete picture of the multiple layers of regulatory interactions that drive specification and differentiation of different types of neurons, and the value of these data is increased by comparison with comparable data from the analogous developing neuronal cell types acquired from the fetal brain.
Many strategies have been developed to produce different neuronal cell types from PSCs in vitro, most of which involve either treatment of PSCs with small molecule agonists and antagonists of growth factor signaling or overexpression of neurogenic transcription factors (Fig. 3). Neuronal cell types that can presently be derived from hPSCs include cortical excitatory neurons (cExNs), which correspond to neurons found in different layers of the developing neocortex (Shi et al., 2012); inhibitory cortical interneurons (cINs) (Liu et al., 2013; Maroof et al., 2013; Meganathan et al., 2017; Nicholas et al., 2013); dopaminergic neurons (Kriks et al., 2011); and microglia (Muffat et al., 2016). Here, we focus predominantly on summarizing what is known regarding epigenetic regulation of the development of the two major types of cortical neuron, cExNs and cINs, as dysregulation of these developmental programs is strongly implicated in the etiology of NDDs (Zikopoulos and Barbas, 2013). In addition to the directed differentiation of specific neuronal cell types, cerebral organoids (COs) also provide a promising tool for modeling human forebrain development (Amiri et al., 2018; Kanton et al., 2019; Lancaster et al., 2013; Luo et al., 2016; Marton et al., 2019; Qian et al., 2019). Studies examining epigenetic regulation in COs will be discussed below. By comparison with both tissues derived from the fetal brain and COs, schemes which promote directed differentiation into a specific neuronal cell type can provide a more homogeneous platform for in parallel assessment of different types of phenotypic, transcriptomic, and epigenetic data, especially when using bulk analysis approaches like ChIP-seq, yielding data with stage-and neuronal cell type-specific resolution.
Fig. 3.
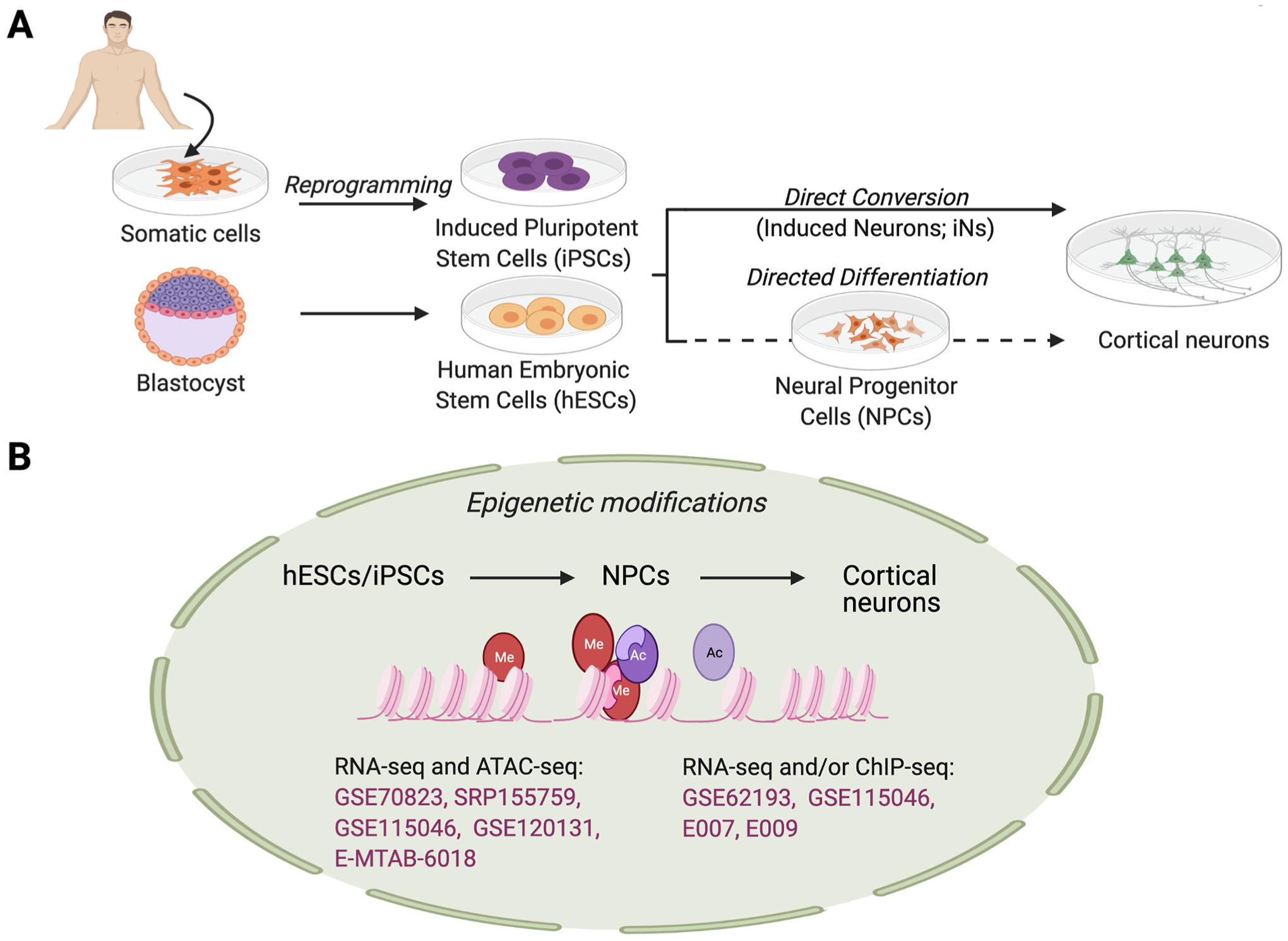
In vitro-derived cortical neurons from human pluripotent stem cells. Human pluripotent stem cells (hPSCs) can be derived from reprogramming of somatic cells (induced pluripotent stem cells; iPSCs) or from the inner cell mass of a blastocyst-stage embryo (human embryonic stem cells; hESCs). Directed differentiation with small molecules or recombinant proteins (via an intermediate neural progenitor cell (NPC) stage) or direct conversion via transcription factor overexpression can be used to produce cortical neurons. (B) Epigenetic assays have been performed on cells at varying stages of differentiation; these can be utilized to analyze epigenetic modifications occurring during directed differentiation or conversion of hESCs/iPSCs to NPCs and NPCs to cortical neurons. Dataset accession numbers are shown in pink text, with additional information for each dataset in Supplementary Table 1. Figure created using BioRender.com.
In general, cortical neurons are produced by patterning PSCs into telencephalic neural progenitors (NPCs) which are then matured into neurons. These differentiation strategies aim to mimic the normal developmental signaling pathways that occur in vivo to produce specific cell fates in vitro. Generation of telencephalic NPCs by directed differentiation is either conducted in monolayer culture or by formation of embryoid bodies (EBs), three-dimensional spheres of cells which enhance cell-to-cell contacts and often improve differentiation efficiency (Zhang et al., 2001). A common approach for specifying telencephalic NPCs involves dual-SMAD inhibition in either monolayer or EB culture (Chambers et al., 2009; Ying et al., 2003). These NPCs can then be patterned towards a dorsal (cExN) or ventral (cIN) telencephalic neuron fate. Formation of dorsal progenitors is promoted in some schemes by adding the Sonic Hedgehog (SHH) signaling inhibitor, Cyclopamine (Li et al., 2009; Liu et al., 2013; Mak et al., 2012; Maroof et al., 2013; Meganathan et al., 2017; Nicholas et al., 2013), while ventral fate can be specified using SHH agonists (Li et al., 2009; Liu et al., 2013; Mak et al., 2012; Maroof et al., 2013; Meganathan et al., 2017; Nicholas et al., 2013). Subsequently, PAX6- and EMX1/2-expressing dorsal telencephalic precursors are differentiated into cExNs (Eiraku et al., 2008; Shi et al., 2012) while NKX2–1-expressing ventral telencephalic precursors are differentiated into cINs (Liu et al., 2013; Meganathan et al., 2017).
As an alternative to the approach described above, forced expression of several neurogenic transcription factors has also been utilized to differentiate hPSCs into cortical neurons. Overexpression of NEUROD1 or NEUROG2 in PSCs can rapidly convert these cells into cExNs (Vierbuchen et al., 2010; Zhang et al., 2013), whereas overexpression of ASCL1 and DLX2 can produce cINs (Yang et al., 2017). Somatic cells, such as fibroblasts can also be trans-differentiated into induced neurons by introduction of transcription factors, such as PTF1A (Xiao et al., 2018). It is important to document and note distinctions between methods used to produce neurons in vitro, as these can complicate comparisons of epigenetic data between studies. This is due to differences both in the identity of the starting cell population (hPSC lines or somatic cells with different genetic backgrounds), as well as variation in how each method recapitulates the epigenetic changes that occur during neural development in vivo and which type of neuronal identity is acquired (Schafer et al., 2019).
4.2. ChIP-seq: histone modifications
Pluripotent stem cell-based differentiation methods provide a valuable platform to study changes in the epigenetic landscape across cortical neural development, as cells can be collected at specific timepoints for a more granular assessment of this regulatory process (Table 1; Supplementary Table 1). Although many studies have defined the epigenetic changes that occur during neural induction of murine ESCs via ChIP-seq, fewer studies have done so during neuronal differentiation of human PSCs. However, several groups have recently begun to elucidate the epigenetic regulation of human neural differentiation, including Inoue et al., who used a dual-SMAD-based differentiation method (Inoue et al., 2019). At six different time points through 72 h of differentiation of the H1 ESC line (male), ChIP-seq for H3K27ac and H3K27me3 was conducted to identify putative enhancers associated with neural differentiation. Regions where H3K27ac and H3K27me3 peaks overlapped were presumed to be inactive and were removed from analysis. Since this paper also integrated this data with chromatin accessibility, determined by ATAC-seq, the main findings are discussed further in a following section of this review. Similarly, Ziller et al. performed epigenomic and transcriptional analysis of six NPC stages derived from human ESCs, also by using a dual-SMAD inhibition protocol (Ziller et al., 2015). Assays were performed on ESCs, neuroepithelial (NE) cells, early radial glial (ERG) cells, mid radial glial (MRG) cells, late radial glial (LRG) cells, and long-term neural progenitors (LNP). These assays included both RNA-seq and ChIP-seq for H3K4me1, H3K4me3, H3K27ac, and H3K27me3, to identify stage-specific regulation of neural specification. Significant changes in histone modification state were observed during the initial specification of NE cell identity, while they also identified an increase in the repressive H3K27me3 mark at the MRG stage; this increase was associated with repression of non-neural gene expression and with activation of many key regulators of neural development and forebrain specification such as PAX6, OTX2, and FOXG1, as well as multiple SOX transcription factors (Ziller et al., 2015). These histone modification state datasets are especially useful for assessing changes in the epigenomic landscape during the early stages of excitatory neural progenitor specification and differentiation.
The NIH Roadmap Epigenomics Project also includes ChIP-seq data for a large number of histone modifications, as well as DNase hypersensitivity and RNA-seq data in H1 ESC-derived NPCs. Together, these data can be utilized to further correlate histone modification status with the regulation of gene expression during neural cell specification (Inoue et al., 2019). A smaller number of histone marks have also been profiled during specification of the H9 ESC line (female) as NPCs (“Roadmap Epigenomics Project - Data, no date (n.d.)”). A summary of these Roadmap Epigenomics datasets is provided in Supplementary Table 1. These data do not include mature cExNs or cINs, nor do they focus on inhibitory NPCs. In order to fill such gaps, additional efforts of this nature will be needed.
4.3. ATAC-seq
As with studies using fetal tissues, chromatin accessibility has also been more widely assessed in in vitro models of neurodevelopment, as it provides a general snapshot of the epigenetic state of cells and can also be effectively used to determine how genetic perturbations affect chromatin state, such as in models using NDD proband-derived PSCs. Several studies have used varying time courses and differentiation strategies to assess chromatin accessibility during directed differentiation of hPSCs into neurons (Table 1; Supplementary Table 1). For example, Shang et al. performed ATAC-seq combined with scRNA-seq analysis at various stages of NPC specification from iPSCs, both to study cellular heterogeneity and to delineate unexpected features of neurogenesis (Shang et al., 2018). They analyzed human iPSCs, EBs, early rosettes (Ros-E, three days after rosette formation), late rosettes (Ros-L, five days after rosette formation), NPCs, and the original somatic fibroblasts from which the iPSCs were derived. Chromatin accessibility changes at each cell stage were assessed by tracing novel ATAC-seq peaks that appeared at each stage of differentiation. Peaks that were conserved across stages were presumed to be associated with housekeeping genes, while stage-specific peaks were regarded as potential CREs that could regulate some aspect of the new cell state or transition to this state. In this work, ~10–50% of the peaks observed at each stage were novel and specific to that stage, while NPC specification corresponded with a large increase in novel peaks and reduction of numbers of pre-existing peaks. After integration with ATAC-seq data, genes expressed in a cell stage-specific manner included those encoding TFs and signaling molecules that regulate cell growth, proliferation, and morphology (Shang et al., 2018). Thus, this study provides both transcriptomic and epigenetic data related to human neural cell specification and differentiation and indicates some regulatory mechanisms underlying acquisition of neuronal fate. In another study, van der Raadt et al. attempted to identify TFs that might influence chromatin accessibility to promote neural cell fate when overexpressed in somatic cells (van der Raadt et al., 2019). They performed ATAC-seq to define differences in chromatin accessibility profiles of human fibroblasts and induced neurons (iNeurons) produced by overexpressing NEUROG2. Among differentially accessible peaks between these cell types (accessible in iNeurons versus fibroblasts), they identified a ONECUT TF binding motif. Accordingly, ONECUT1, ONECUT2, or ONECUT3 overexpression induced chromatin remodeling in fibroblasts within two days and accelerated neuronal differentiation (van der Raadt et al., 2019). This paper demonstrates how epigenetic information can be leveraged to define activities that can regulate neural development.
The temporal interplay between epigenetic and transcriptional regulatory processes during early neural induction is another important topic of study. As introduced above, Inoue et al. assessed transcriptome changes that accompanied neural fate acquisition of hESCs at six time points between 3 and 72 h (Inoue et al., 2019). As expected, expression of neural genes increased, with concomitant decreases in pluripotency gene expression. To identify putative enhancers involved in neural differentiation, the ATAC-seq analysis described above was accompanied by ChIP-seq for activating (H3K27ac) and repressive (H3K27me3) histone modifications at all time points. Changes in these epigenetic modifications over time corresponded with gene expression changes. A set of putative enhancers predicted using these epigenetic modification data were subsequently assessed for activity by massively parallel array analysis. Active enhancers were enriched for binding sites for five TFs, BARHL1, IRX3, LHX5, OTX1, and OTX2, predicting that these TFs regulate neural fate acquisition. Overexpression and knockdown of these TFs respectively up- and down-regulated expression of neural genes (Inoue et al., 2019). A similar approach could be applied to define transcriptional regulators that regulate the development of other cortical cell types and/or act at other temporal stages.
In addition to studying neurodevelopmental processes, several studies have assessed chromatin accessibility differences between affected and unaffected individuals with NDDs, including ASD and SZ, to define neurodevelopmental alterations that may contribute to disease etiology. Schafer et al. performed a time-course analysis in human idiopathic ASD subject-derived iPSCs during neural specification as NPCs (16 days) and during their differentiation into mature neurons (during 2–14 days of NPC differentiation) (Schafer et al., 2019). They also directly differentiated iPSCs into induced neurons (iPSC-iN) by NGN2 overexpression. RNA- and ATAC-seq was performed in both models to identify alterations in chromatin remodeling during neural development in the affected individuals. They observed that pathological dysregulation of chromatin remodeling and transcription prior to the NPC stage was sufficient to induce ASD-associated morphological and functional changes during later neurodevelopment. Interestingly, they also found that epigenetic regulatory processes that normally occur during neural specification and differentiation are not recapitulated during direct conversion of iPSCs into iPSC-iNs (Schafer et al., 2019). Forrest et al. also assessed chromatin state by ATAC-seq in iPSC-derived neurons from individuals with SZ at 27, 33, and 41 days after neural induction (Forrest et al., 2017). They proposed that non-coding SZ risk variants in open chromatin regions (OCRs) were likely to affect neural development, particularly if variants were close to, or within, predicted TF-binding sites within regulatory elements. Significantly, they could correct a risk allele within the putative MIR137 SZ risk locus, partially restoring gene expression and reversing the neurodevelopmental anomalies observed in this model (Forrest et al., 2017). This work relating to NDDs demonstrates the importance of identifying non-coding regulatory regions that control neural development and highlights the role that epigenomic analysis can play in defining variants in the non-coding space that contribute to disease risk.
Intriguingly, another study of chromatin accessibility during iPSC-derived neural differentiation also identified allelic imbalance of gene expression (AIE) related to NDDs, a phenomenon in which the two alleles of a given gene are expressed at different levels in a given cell, due to either genetic variation in regulatory regions or epigenetic inactivation of one allele (Zhang et al., 2020). Allele-specific open chromatin regions (ASoC) may contribute to AIE. Zhang et al. mapped these ASoC regions using ATAC- and RNA-seq, identifying common non-coding risk variants associated with neurodevelopmental disorders. They observed that ASoC and AIE are cell-type specific, with iPSC-derived neurons exhibiting higher levels of ASoC and AIE than iPSCs. They identified heterozygous single nucleotide polymorphisms (SNPs) that exhibited ASoC in each cell type and defined two SNPs of interest: one in the 5′UTR of CHRNA5 and one in the promoter of VPS45. The proteins encoded by both of these genes are involved in synaptic transmission and have pathogenic variants that are strongly associated with NDDs (Zhang et al., 2020). This study demonstrates a powerful method for predicting functional non-coding variants associated with NDD risk.
5. Cerebral organoids
5.1. Background
By comparison with other in vitro differentiation methods, differentiation of multiple neuronal cell types in structured three-dimensional COs more closely recapitulates the tissue structure and organization of the developing brain. This organoid-based approach enables differentiation over very long timelines (months to years), also enabling more mature neurons to be generated. Cerebral organoids can therefore serve as an informative tool to study human neural development (Luo et al., 2016; Qian et al., 2019). Many advantages of non-organoid-based in vitro models of cortical development are also applicable to COs, including the ability to generate large numbers of cells, perform genetic manipulations, and perform assays at multiple timepoints. However, the structure and composition of COs are generally not highly reproducible, which, combined with the heterogeneity of cell types present in each organoid, poses challenges for producing epigenetic data sets from this model.
Initial efforts to construct models of differentiated neurons in three-dimensions date back to the 1990s. Notably, the development in 1992 of methods for culturing various types of neurons as three-dimensional neurospheres was shown to facilitate their culture and differentiation (Reynolds and Weiss, 1992). Cerebral organoids, however, are distinct from neurospheres in that they benefit from a capacity for self-organization of multiple neuronal cell types and therefore more closely imitate in vivo brain development (Qian et al., 2019). Recently, several techniques have emerged to produce COs, stemming from the method described by Lancaster et al. in 2013 (Lancaster et al., 2013). Production of COs generally entails aggregation of PSCs as three-dimensional spheres and their subsequent specification into neural progenitors and neurons with the desired brain regional identity. Current methods for deriving COs include both guided and unguided differentiation approaches. With unguided approaches, the PSCs are formed into spheres, which are subsequently implanted into extracellular matrix and allowed to spontaneously differentiate within a spinning bioreactor (Fig. 4) (Lancaster et al., 2013). As this method does not direct specification of a particular regional neural identity, it produces a heterogenous population of neuronal cell types from multiple brain regions, and results in organoid-to-organoid variability within and across experiments. While this heterogeneity and variability can enable studies of interactions between brain regions or neuronal cell types, it also substantially complicates quantitative analyses (Qian et al., 2019). Guided approaches to producing COs instead utilize specialized media and signaling agonist/antagonists to specify a particular regional identity, enabling derivation of spheroids corresponding with a particular brain region or of a specific neuron type (Fig. 4) (Gordon et al., 2021; Marton et al., 2019; Qian et al., 2019).
Fig. 4.

In vitro-derived cerebral organoids from human pluripotent stem cells. Human pluripotent stem cells (hPSCs) can be differentiated into cerebral organoids using unguided methods that allow for self-organization or into region-specific (e.g. cortex or medial ganglionic eminence; MGE) organoids using guided methods with patterning factors. hPSCs are formed into embryoid bodies (EBs) and are induced into neurons prior to being embedded in Matrigel to provide a scaffold, and then matured on a shaker. (B) Epigenetic assays have been performed on organoids at varying stages of neural induction and maturation, from day (D) 0 to 600. Dataset accession numbers are shown in pink, with additional information for each dataset in Supplementary Table 1. Figure created using BioRender.com.
5.2. ChIP-seq: histone modifications
A few studies have characterized histone modification state in COs, although most extant work focuses primarily on the DNA methylome (Luo et al., 2016), transcriptome (Amiri et al., 2018; Luo et al., 2016; Xiang et al., 2017), or chromatin accessibility (Trevino et al., 2020; Xiang et al., 2017; Ziffra et al., 2019) changes that accompany development (Table 1; Supplementary Table 1). Amiri et al. recently defined transcriptomic changes associated with specific histone modifications (H3K4me3, H3K27ac, and H3K27me3) in developing COs (Amiri et al., 2018). They found that differential activity of enhancers is linked to differential gene expression in the developing organoids, with increased enhancer activity correlating directly with increased gene expression. This correlation was demonstrated through a clustering analysis of differential enhancer activity, which closely followed transcriptomic differences (Amiri et al., 2018). Consistent with other findings, it was observed that the largest change in differential enhancer activity occurred during the specification of stem cells as NPCs; this work suggests that these enhancers regulate gene expression during early aspects of brain development, the developmental timeframe that organoids most closely model. More work is needed in this area to further characterize the relationship between histone modifications and CO development, as well as to determine how this in vitro model compares to its in vivo counterpart.
5.3. ATAC-seq
ATAC-seq is especially valuable for assessing heterogeneous cell populations like COs as, unlike ChIP-seq, it can be performed both in bulk and at a single-cell resolution (Table 1; Supplementary Table 1). In 2017, Xiang et al. employed bulk ATAC-seq to assess epigenomic regulatory mechanisms in regionally restricted brain organoids modeling the MGE and the cerebral cortex (Xiang et al., 2017). Unique open chromatin regions corresponding to genes crucial for development of the MGE or the cortex were observed, suggesting that changes in chromatin accessibility actively contribute to transcriptional regulation during development of those regionally patterned organoids. However, this work did not draw direct comparisons to human fetal data from analogous tissues (Xiang et al., 2017).
A recent study by Trevino et al. likewise collected 117 bulk ATAC-seq samples encompassing both COs of dorsal or ventral character generated from iPSC lines (Trevino et al., 2020). Corresponding human fetal samples were obtained from the dorsal and ventral forebrain at 20 and 21 PCW for comparison. Differences in chromatin accessibility between these CO types were assessed, with consideration of temporal changes. They observed that gene expression correlated more closely with the average accessibility of distal enhancers versus proximal promoters. To assess the relevance of these findings to brain development in vivo, they compared these findings to the human fetal samples, demonstrating that COs cultured for 188–230 days most closely resembled human fetal forebrain at 20–21 PCW. Further comparisons with PSYCHENCODE epigenetic data demonstrated that the COs displayed progressive transformation of their chromatin landscape, which resembled the changes in chromatin remodeling observed in developing fetal tissue (Trevino et al., 2020). This study suggests that COs may accurately recapitulate many aspects of epigenetic regulation in the developing forebrain in vivo and established their relevance for modeling neurodevelopmental pathologies.
While bulk ATAC-seq has provided data on chromatin accessibility, scATAC-seq had not yet been used to characterize COs. However, scATAC-seq was used in the aforementioned pre-print by Ziffra et al., which compares chromatin accessibility in COs and human fetal tissue (Ziffra et al., 2019). These scATAC-seq assays corroborate conclusions drawn in previous studies using bulk ATAC-seq, demonstrating that changes in enhancer chromatin accessibility predict changes in gene expression. Based upon assessment of excitatory neuron lineages over time, epigenomic differences were defined between progenitor cells in different cortical areas, while changes in accessibility corresponded with neuronal differentiation. Interestingly, the highly dynamic changes in chromatin accessibility observed here challenge the conventional model of differentiation as a gradual, progressive process. Furthermore, although COs appear to faithfully mimic many aspects of fetal development, a limitation of this model observed here is an absence of increased accessibility during CO neuronal differentiation at thousands of distal regulatory elements that become active and accessible during fetal brain development (Ziffra et al., 2019). Therefore, this work illustrates both the strengths and limitations of using COs to model aspects of fetal brain development.
6. Conclusions and future perspectives
Although our understanding of epigenetic regulation of human brain development has advanced considerably in the past 10 years, there remain many substantial gaps in knowledge. In many cases, these gaps involve the lack of neuronal cell-type specific resolution of extant data, which limits our understanding of differences in epigenetic regulation across neuronal cell types (Table 1; Supplementary Table 1). Gathering data from additional donors, samples, and timepoints will also enable us to identify both common regulatory processes and those that are specific to a certain timepoint, neuronal cell type, sex, or individual. The sex of most samples (whether fetal brains or cell lines) is noted in most studies, but data from male and female samples are frequently aggregated; this can be problematic, since there are significant differences in developmental gene regulation related to sex (Manoli and Tollkuhn, 2018). Some limitations will be overcome by technological advancements, for example, the single-cell analysis performed by Ziffra et al. (2019), which substantially enhances the resolution of analyses that are possible. Although single-cell sequencing often sacrifices read depth, hampering identification of rare cell types or low-to-moderate gene expression, this tool can be particularly helpful when combined with tissue dissection. Combining tools enables both single-cell-level and bulk-level analysis, while ensuring greater homogeneity of each sample, relative to analysis of bulk cortex or whole fetal brain. Future studies that combine analysis of scATAC-seq and scRNA-seq data will further enable us to define regulatory networks that control the specification and differentiation of different neuronal cell types. Finally, assay innovation will enable collection of data that was previously unattainable due to technical limitations. For example, CUT&RUN (Meers et al., 2019; Skene and Henikoff, 2017) and CUT&Tag (Kaya-Okur et al., 2019) enable low-input, high-resolution, profiling of chromatin state, down to a single-cell level, using MNase- or Tn5 transposase-based methods, respectively. These new approaches will be particularly helpful for fetal samples, allowing histone modification and TF binding profiles to be obtained for a greater number of samples and cell types.
Further studies of these data will also inform our understanding of how epigenetic dysregulation contributes to neurodevelopmental disorders. Although genome-wide association studies (GWAS) have pinpointed hundreds of NDD-associated SNPs, it is often still unclear which risk variants contribute to disease pathology. More generally, more than 90% of disease-associated SNPs are located in DNA regions that do not encode proteins (Maurano et al., 2012). These non-coding risk variants likely affect gene expression by regulating chromatin accessibility for TF binding. Functional genomic assays such as ATAC-seq and ChIP-seq have identified such candidate CREs (cCREs) in the genome. Integrating SNP coordinates with these epigenetic datasets can reveal NDD-associated variants that overlap putative regulatory elements (An et al., 2018; Markenscoff-Papadimitriou et al., 2020). For example, since noncoding CREs often coincide with accessible chromatin regions, Forrest et al. profiled global open chromatin regions in human iPSC models and used these data to define functional noncoding risk variants for SZ (Forrest et al., 2017). Recently, mutations in the noncoding space have also been linked to ASD (Zhou et al., 2019). Machine learning was used to analyze whole genome sequences for 1790 individuals with ASD and their families. Upon comparing de novo variants in the probands to biochemical data illustrating interactions between DNA- and RNA-binding proteins (DBPs and RPBs, respectively) and their targets, Zhou et al. assessed the potential quantitative impact of each mutation on particular transcriptional and post-transcriptional regulatory features, such as histone modification state (Zhou et al., 2019). Indeed, many mutations in ASD probands were found to disrupt binding of DBPs and RBPs and transcriptional regulation, providing support for the functional role of de novo noncoding variants in ASD.
It will also be important to assess the functional significance of identified regulatory elements. While comparison to chromatin-state datasets can help prioritize SNPs that may lie in a regulatory element, this data alone cannot define functional variants. Epigenomic annotations may be incongruous even within the same cell type and thus, variants must be tested to truly define any regulatory significance (Benton et al., 2019). Human accelerated regions may also represent CREs that regulate gene expression in a conserved manner across most vertebrates, including primates (e.g. chimpanzees), but that are significantly different in humans (Capra et al., 2013; Pollard et al., 2006). The presence of these human-specific regulatory elements, as well as additional differences in genome sequence and brain development between other animals and humans indicate that unique regulatory processes underlie human neural development, many of which remain to be elucidated. Massively Parallel Reporter Assays (MPRAs) are a high-throughput method used to assess functional activity of such predicted regulatory elements and changes in activity related to sequence variants. Using this assay, thousands of noncoding sequences can be screened in a single experiment. In an MPRA, candidate regulatory sequences are inserted before a minimal promoter in a vector with a unique DNA barcode that can be transcribed (Ashuach et al., 2019). After constructs are introduced into cells, both DNA construct counts and RNA transcript counts are used to approximate the transcription rate of every barcode. Thus, MPRA testing can provide quantitative measurements of regulatory activity and help supplement disease predictions associated with cCREs. Massively parallel reporter assays have been used to functionally assess cCREs regulating several aspects of neurodevelopment (Grossman et al., 2017; Inoue et al., 2019; Mulvey et al., 2020; Shen et al., 2016). Another approach for functional testing involves epigenome editing to silence or activate cCREs in situ in their genomic context (Chavez et al., 2015; Konermann et al., 2015; Li et al., 2020).
Modeling epigenetic regulation of human fetal brain development increases both our understanding of and our ability to experimentally manipulate these developmental regulatory programs. Recent studies have generated epigenomic data from neuronal cell and tissue types in the developing human brain and have compared these datasets to the analogous in vitro-derived neuronal cell types, providing valuable insights into mechanisms of human brain development and disease etiology. Determining which aspects of human brain development are or are not recapitulated with in vitro models indicates where protocol refinements are needed, and these ongoing refinements continue to enhance the utility of in vitro hPSC differentiation approaches. Together, the increasing amount of epigenetic data available for human brain development is valuable for elucidating epigenetic mechanisms and regulatory networks that control these developmental processes, and for determining how these are disrupted by pathogenic mutations in both TFs and chromatin modifiers and in the non-coding genome to cause neurodevelopmental disorders.
Supplementary Material
Acknowledgements of funding sources
This work was supported by the National Institutes of Health [U01 HG007530 (subaward), U54 HD087011-S1, R01NS114551, and R01MH124808 to KLK, and T32 GM 7067–43 to EMAL]; the M-CM Network to KLK; the Jakob Gene Fund to KLK; the WU Center of Regenerative Medicine (CRM) to KLK; the WU Children’s Discovery Institute (CDI) to KLK; the WU Institute for Clinical and Translational Sciences (ICTS) to KLK; the WU Precision Medicine Pathway to EMAL; the WU Irving Biome Graduate Student Fellowship to EMAL; the WU CDI and Maximizing Access to Research Careers (MARC)-Undergraduate Student Training in Academic Research (U-STAR) program to IA; the WU Departments of Developmental Biology and Nephrology to SD; and the WU Institute for Informatics to SD.
Abbreviations
- hPSC
Human pluripotent stem cell
- TF
Transcription factor
- NDD
Neurodevelopmental disorder
- ASD
Autism spectrum disorder
- HAR
Human accelerated region
- CRE
Cis-regulatory element
- ENCODE
Encyclopedia of DNA elements
- ESC
Embryonic stem cell
- iPSC
Induced pluripotent stem cell
- cExN
Cortical excitatory neuron
- cIN
Cortical interneuron
- CO
Cerebral organoid
- ChIP-seq
Chromatin immunoprecipitation followed by sequencing
- ATAC-seq
Assay for transposase-accessible chromatin with high-throughput sequencing
- GW
Gestational week
- sc
Single-cell
- GZ
Germinal zone
- CP
Cortical plate
- CRISPRa
CRISPR-mediated gene activation
- SZ
Schizophrenia
- MGE
Medial ganglionic eminence
- ABC
Activity-by-contact
- CNCC
Cranial neural crest cell
- SNC
Single nucleotide change
- KZFP
Krüppel-associated box (KRAB) domain-containing zinc-finger protein
- UCSF-UBC
University of California San Francisco-University of British Columbia
- NPC
Neural progenitor cell
- EB
Embryoid body
- SHH
Sonic hedgehog
- NE
Neuroepithelial
- ERG
Early radial glial
- MRG
Mid radial glial
- LRG
Late radial glial
- LNP
Long-term neural progenitors
- Ros-E
Early rosettes
- Ros-L
Late rosettes
- iNeurons or iN
Induced neurons
- OCR
Open chromatin region
- AIE
Allelic imbalance of gene expression
- ASoC
Allele-specific open chromatin region
- SNP
Single nucleotide polymorphism
- GWAS
Genome-wide association study
- DBP
DNA-binding protein
- RBP
RNA-binding protein
- MPRA
Massively parallel reporter assay
Footnotes
Declaration of competing interests
Declarations of interest: none.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neuint.2021.105039.
References
- Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, Pochareddy S, Shin Y, Safi A, Song L, Zhu Y, Sousa AMM, Gerstein M, Crawford GE, Sestan N, Abyzov A, Vaccarino FM, 2018. Transcriptome and epigenome landscape of human cortical development modeled in brain organoids. Science 362. 10.1126/science.aat6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, Currall BB, Dastmalchi C, Dea J, Duhn C, Gilson MC, Klei L, Liang L, Markenscoff-Papadimitriou E, Pochareddy S, Ahituv N, Buxbaum JD, Coon H, Daly MJ, Kim YS, Marth GT, Neale BM, Quinlan AR, Rubenstein JL, Sestan N, State MW, Willsey AJ, Talkowski ME, Devlin B, Roeder K, Sanders SJ, 2018. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362. 10.1126/science.aat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armand EJ, Li J, Xie F, Luo C, Mukamel EA, 2021. Single-cell sequencing of brain cell transcriptomes and epigenomes. Neuron 109, 11–26. 10.1016/j.neuron.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashuach T, Fischer DS, Kreimer A, Ahituv N, Theis FJ, Yosef N, 2019. MPRAnalyze: statistical framework for massively parallel reporter assays. Genome Biol. 20, 183. 10.1186/s13059-019-1787-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton ML, Talipineni SC, Kostka D, Capra JA, 2019. Genome-wide enhancer annotations differ significantly in genomic distribution, evolution, and function. BMC Genom. 20, 511. 10.1186/s12864-019-5779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman GD, Poirier MG, 2015. Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev 115, 2274–2295. 10.1021/cr500350x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J, Wu B, Chang H, Greenleaf W, 2015. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 109, 21–29. 10.1002/0471142727.mb2129s109, 1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burney MJ, Johnston C, Wong K-Y, Teng S-W, Beglopoulos V, Stanton LW, Williams BP, Bithell A, Buckley NJ, 2013. An epigenetic signature of developmental potential in neural stem cells and early neurons. Stem Cell. 31, 1868–1880. 10.1002/stem.1431. [DOI] [PubMed] [Google Scholar]
- Capra JA, Erwin GD, McKinsey G, Rubenstein JLR, Pollard KS, 2013. Many human accelerated regions are developmental enhancers. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20130025. 10.1098/rstb.2013.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L, 2009. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27, 275–280. 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, P R Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, Ter-Ovanesyan D, Braff JL, Davidsohn N, Housden BE, Perrimon N, Weiss R, Aach J, Collins JJ, Church GM, 2015. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328. 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clowry GJ, 2015. An enhanced role and expanded developmental origins for gamma-aminobutyric acidergic interneurons in the human cerebral cortex. J. Anat 227, 384–393. 10.1111/joa.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, Geschwind DH, 2018. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell 172, 289–304. 10.1016/j.cell.2017.12.014 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, 2011. The evolution of the brain, the human nature of cortical circuits, and intellectual creativity. Front. Neuroanat 5 10.3389/fnana.2011.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Rosario RCH, Rayan NA, Prabhakar S, 2014. Noncoding origins of anthropoid traits and a new null model of transposon functionalization. Genome Res 24, 1469–1484. 10.1101/gr.168963.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan RN, Bae B-I, Cubelos B, Chang C, Hossain AA, Al-Saad S, Mukaddes NM, Oner O, Al-Saffar M, Balkhy S, Gascon GG, Homozygosity Mapping Consortium for Autism, Nieto M, Walsh CA, 2016. Mutations in human accelerated regions disrupt cognition and social behavior. Cell 167, 341–354. 10.1016/j.cell.2016.08.071 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecco G, Cassano M, Kauzlaric A, Duc J, Coluccio A, Offner S, Imbeault M, Rowe HM, Turelli P, Trono D, 2016. Transposable elements and their KRAB-ZFP controllers regulate gene expression in adult tissues. Dev. Cell 36, 611–623. 10.1016/j.devcel.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y, 2008. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532. 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Encode Project Consortium, 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Zhang H, Moy W, McGowan H, Leites C, Dionisio LE, Xu Z, Shi J, Sanders AR, Greenleaf WJ, Cowan CA, Pang ZP, Gejman PV, Penzes P, Duan J, 2017. Open chromatin profiling in hiPSC-derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell 21, 305–318. 10.1016/j.stem.2017.07.008 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchini LF, Pollard KS, 2017. Human evolution: the non-coding revolution. BMC Biol 15, 89. 10.1186/s12915-017-0428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, Nguyen TH, Kane M, Perez EM, Durand NC, Lareau CA, Stamenova EK, Aiden EL, Lander ES, Engreitz JM, 2019. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669. 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genboree Discovery System - Project: XML Submissions/UCSF-UBC/SAMPLE/EDACC.5485 [WWW Document], n.d. URL http://genboree.org/java-bin/project.jsp?projectName=XML%20Submissions%2FUCSF-UBC%2FSAMPLE%2FEDACC.5485 (accessed 9.6.20).
- Gordon A, Yoon S-J, Tran SS, Makinson CD, Park JY, Andersen J, Valencia AM, Horvath S, Xiao X, Huguenard JR, Pașca SP, Geschwind DH, 2021. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci 1–12. 10.1038/s41593-021-00802-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MVC, Bourc’his D, 2019. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol 20, 590–607. 10.1038/s41580-019-0159-6. [DOI] [PubMed] [Google Scholar]
- Grossman SR, Zhang X, Wang L, Engreitz J, Melnikov A, Rogov P, Tewhey R, Isakova A, Deplancke B, Bernstein BE, Mikkelsen TS, Lander ES, 2017. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc. Natl. Acad. Sci. U.S.A 114, E1291–E1300. 10.1073/pnas.1621150114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Gotoh Y, 2010. Epigenetic control of neural precursor cell fate during development. Nat. Rev. Neurosci 11, 377–388. 10.1038/nrn2810. [DOI] [PubMed] [Google Scholar]
- Hyun K, Jeon J, Park K, Kim J, 2017. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 49 10.1038/emm.2017.11e324–e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbeault M, Helleboid P-Y, Trono D, 2017. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554. 10.1038/nature21683. [DOI] [PubMed] [Google Scholar]
- Inoue F, Kreimer A, Ashuach T, Ahituv N, Yosef N, 2019. Identification and massively parallel characterization of regulatory elements driving neural induction. Cell Stem Cell 25, 713–727. 10.1016/j.stem.2019.09.010 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang HS, Shin WJ, Lee JE, Do JT, 2017. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes 8. 10.3390/genes8060148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG, 2009. The origins of cortical interneurons: mouse versus monkey and human. Cerebr. Cortex 19, 1953–1956. 10.1093/cercor/bhp088. [DOI] [PubMed] [Google Scholar]
- Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, Guijarro P, Sidow L, Fleck JS, Han D, Qian Z, Heide M, Huttner WB, Khaitovich P, Pääbo S, Treutlein B, Camp JG, 2019. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 574, 418–422. 10.1038/s41586-019-1654-9. [DOI] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, Henikoff S, 2019. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930. 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer-Sawatzki H, Cooper DN, 2007. Structural divergence between the human and chimpanzee genomes. Hum. Genet 120, 759–778. 10.1007/s00439-006-0270-6. [DOI] [PubMed] [Google Scholar]
- Klein DC, Hainer SJ, 2020. Genomic methods in profiling DNA accessibility and factor localization. Chromosome Res 28, 69–85. 10.1007/s10577-019-09619-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F, 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostka D, Holloway AK, Pollard KS, 2018. Developmental loci harbor clusters of accelerated regions that evolved independently in ape lineages. Mol. Biol. Evol 35, 2034–2045. 10.1093/molbev/msy109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim J-W, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L, 2011. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Yaping Liu, Coarfa C, Alan Harris R, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, David Hawkins R, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Scott Hansen R, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh K-H, Feizi S, Karlic R, Kim A-R, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, Jager PLD, Farnham PJ, Fisher SJ, Haussler D, Jones SJM, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai L-H, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, 2015. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, 2013. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letinic K, Zoncu R, Rakic P, 2002. Origin of GABAergic neurons in the human neocortex. Nature 417, 645–649. 10.1038/nature00779. [DOI] [PubMed] [Google Scholar]
- Levchenko A, Kanapin A, Samsonova A, Gainetdinov RR, 2017. Human accelerated regions and other human-specific sequence variations in the context of evolution and their relevance for brain development. Genome Biol Evol 10, 166–188. 10.1093/gbe/evx240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EMA, Kroll KL, 2018. Development and disease in a dish: the epigenetics of neurodevelopmental disorders. Epigenomics 10, 219–231. 10.2217/epi-2017-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Liu Y, Cao H, Zhang Y, Gu Z, Liu X, Yu A, Kaphle P, Dickerson KE, Ni M, Xu J, 2020. Interrogation of enhancer function by enhancer-targeting CRISPR epigenetic editing. Nat. Commun 11 10.1038/s41467-020-14362-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhuang Y, Zhao X, Li X, 2019. Long non-coding RNA in neuronal development and neurological disorders. Front. Genet 9 10.3389/fgene.2018.00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Santpere G, Kawasawa YI, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, Sousa AMM, Werling DM, Kitchen RR, Kang HJ, Pletikos M, Choi J, Muchnik S, Xu X, Wang D, Lorente-Galdos B, Liu S, Giusti-Rodríguez P, Won H, de Leeuw CA, Pardiñas AF, Hu M, Jin F, Li Y, Owen MJ, O’Donovan MC, Walters JTR, Posthuma D, Reimers MA, Levitt P, Weinberger DR, Hyde TM, Kleinman JE, Geschwind DH, Hawrylycz MJ, State MW, Sanders SJ, Sullivan PF, Gerstein MB, Lein ES, Knowles JA, Sestan N, 2018. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362. 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-J, Zhang X, Johnson MA, Wang Z-B, Lavaute T, Zhang S-C, 2009. Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development 136, 4055–4063. 10.1242/dev.036624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu H, Sauvey C, Yao L, Zarnowska ED, Zhang S-C, 2013. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat. Protoc 8, 1670–1679. 10.1038/nprot.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loke YJ, Hannan AJ, Craig JM, 2015. The role of epigenetic change in autism spectrum disorders. Front. Neurol 6 10.3389/fneur.2015.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Lancaster MA, Castanon R, Nery JR, Knoblich JA, Ecker JR, 2016. Cerebral organoids recapitulate epigenomic signatures of the human fetal brain. Cell Rep 17, 3369–3384. 10.1016/j.celrep.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, Huang YA, Iranmanesh S, Vangipuram M, Sundararajan R, Nguyen L, Langston JW, Schüle B, 2012. Small molecules greatly improve conversion of human-induced pluripotent stem cells to the neuronal lineage. Stem Cells Int 2012 140427. 10.1155/2012/140427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli DS, Tollkuhn J, 2018. Gene regulatory mechanisms underlying sex differences in brain development and psychiatric disease. Ann. N. Y. Acad. Sci 1420, 26–45. 10.1111/nyas.13564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markenscoff-Papadimitriou E, Whalen S, Przytycki P, Thomas R, Binyameen F, Nowakowski TJ, Kriegstein AR, Sanders SJ, State MW, Pollard KS, Rubenstein JL, 2020. A chromatin accessibility atlas of the developing human telencephalon. Cell 182, 754–769. 10.1016/j.cell.2020.06.002 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying S-W, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG, Widmer HR, Eggan K, Goldstein PA, Anderson SA, Studer L, 2013. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton RM, Miura Y, Sloan SA, Li Q, Revah O, Levy RJ, Huguenard JR, Pașca SP, 2019. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci 22, 484–491. 10.1038/s41593-018-0316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA, 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers MP, Bryson TD, Henikoff JG, Henikoff S, 2019. Improved CUT&RUN chromatin profiling tools. eLife 8, e46314. 10.7554/eLife.46314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meganathan K, Lewis EMA, Gontarz P, Liu S, Stanley EG, Elefanty AG, Huettner JE, Zhang B, Kroll KL, 2017. Regulatory networks specifying cortical interneurons from human embryonic stem cells reveal roles for CHD2 in interneuron development. Proc. Natl. Acad. Sci. Unit. States Am 114, E11180–E11189. 10.1073/pnas.1712365115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, Sudmant PH, Alkan C, Fu Q, Do R, Rohland N, Tandon A, Siebauer M, Green RE, Bryc K, Briggs AW, Stenzel U, Dabney J, Shendure J, Kitzman J, Hammer MF, Shunkov MV, Derevianko AP, Patterson N, Andrés AM, Eichler EE, Slatkin M, Reich D, Kelso J, Pääbo S, 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226. 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi S, Mahdizadeh H, Šarić T, Kim J, Harati J, Shahsavarani H, Greber B, Moore JB, 2019. Research and therapy with induced pluripotent stem cells (iPSCs): social, legal, and ethical considerations. Stem Cell Res. Ther 10, 341. 10.1186/s13287-019-1455-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai L-H, Aubourg P, Ransohoff RM, Jaenisch R, 2016. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med 22, 1358–1367. 10.1038/nm.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvey B, Lagunas T, Dougherty JD, 2020. Massively parallel reporter assays: defining functional psychiatric genetic variants across biological contexts. Biol. Psychiatr 10.1016/j.biopsych.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, Arnold CM, Chen Y-JJ, Stanley EG, Elefanty AG, Sasai Y, Alvarez-Buylla A, Rubenstein JLR, Kriegstein AR, 2013. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notwell JH, Chung T, Heavner W, Bejerano G, 2015. A family of transposable elements co-opted into developmental enhancers in the mouse neocortex. Nat. Commun 6, 6644. 10.1038/ncomms7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, Vuong CK, Nichterwitz S, Gevorgian M, Opland CK, Lu D, Connell W, Ruzzo EK, Lowe JK, Hadzic T, Hinz FI, Sabri S, Lowry WE, Gerstein MB, Plath K, Geschwind DH, 2019. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801. 10.1016/j.neuron.2019.06.011 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, Rosenbloom KR, Kent J, Haussler D, 2006. Forces shaping the fastest evolving regions in the human genome. PLoS Genet 2, e168. 10.1371/journal.pgen.0020168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontis J, Planet E, Offner S, Turelli P, Duc J, Coudray A, Theunissen TW, Jaenisch R, Trono D, 2019. Hominoid-specific transposable elements and KZFPs facilitate human embryonic genome activation and control transcription in naive human ESCs. Cell Stem Cell 24, 724–735. 10.1016/j.stem.2019.03.012 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SL, Srinivasan R, Marchetto MC, Grishina I, Narvaiza I, Selleri L, Gage FH, Swigut T, Wysocka J, 2015. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163, 68–83. 10.1016/j.cell.2015.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, Li H, Mallick S, Dannemann M, Fu Q, Kircher M, Kuhlwilm M, Lachmann M, Meyer M, Ongyerth M, Siebauer M, Theunert C, Tandon A, Moorjani P, Pickrell J, Mullikin JC, Vohr SH, Green RE, Hellmann I, Johnson PLF, Blanche H, Cann H, Kitzman JO, Shendure J, Eichler EE, Lein ES, Bakken TE, Golovanova LV, Doronichev VB, Shunkov MV, Derevianko AP, Viola B, Slatkin M, Reich D, Kelso J, Pääbo S, 2014. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Song H, Ming G, 2019. Brain organoids: advances, applications and challenges. Development 146, dev166074. 10.1242/dev.166074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SK, Yin J, Ayoub AE, Emera D, Leng J, Cotney J, Sarro R, Rakic P, Noonan JP, 2015. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159. 10.1126/science.1260943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S, 1992. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710. 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Project - Data [WWW Document], n.d. URL. http://www.roadmapepigenomics.org/data/tables/stemcells. accessed 9.6.20.
- Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, Bernstein BE, 2015. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172. 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakierska-Chudy A, Filip M, 2015. A comprehensive view of the epigenetic landscape. Part II: histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox. Res 27, 172–197. 10.1007/s12640-014-9508-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, Kuret TJM, Liyanage M, Mansour AA, Jaeger BN, Marchetto MC, Glass CK, Mertens J, Gage FH, 2019. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat. Neurosci 22, 243–255. 10.1038/s41593-018-0295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Z, Chen D, Wang Q, Wang Shengpeng, Deng Q, Wu L, Liu C, Ding X, Wang Shiyou, Zhong J, Zhang D, Cai X, Zhu S, Yang H, Liu L, Fink JL, Chen F, Liu X, Gao Z, Xu X, 2018. Single-cell RNA-seq reveals dynamic transcriptome profiling in human early neural differentiation. GigaScience 7. 10.1093/gigascience/giy117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JA, 2017. Exploring the read-write genome: mobile DNA and mammalian adaptation. Crit. Rev. Biochem. Mol. Biol 52, 1–17. 10.1080/10409238.2016.1226748. [DOI] [PubMed] [Google Scholar]
- Shen SQ, Myers CA, Hughes AEO, Byrne LC, Flannery JG, Corbo JC, 2016. Massively parallel cis-regulatory analysis in the mammalian central nervous system. Genome Res 26, 238–255. 10.1101/gr.193789.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HPC, Livesey FJ, 2012. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci 15, 477–486. 10.1038/nn.3041. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S, 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. 10.7554/eLife.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL, 2010. The basics of brain development. Neuropsychol. Rev 20, 327–348. 10.1007/s11065-010-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon S-J, Huber N, Pritchard JK, Chang HY, Greenleaf WJ, Pașca SP, 2020. Chromatin accessibility dynamics in a model of human forebrain development. Science 367. 10.1126/science.aay1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trizzino M, Park Y, Holsbach-Beltrame M, Aracena K, Mika K, Caliskan M, Perry GH, Lynch VJ, Brown CD, 2017. Transposable elements are the primary source of novelty in primate gene regulation. Genome Res 27, 1623–1633. 10.1101/gr.218149.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsompana M, Buck MJ, 2014. Chromatin accessibility: a window into the genome. Epigenet. Chromatin 7, 33. 10.1186/1756-8935-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Raadt J, van Gestel SHC, Nadif Kasri N, Albers CA, 2019. ONECUT transcription factors induce neuronal characteristics and remodel chromatin accessibility. Nucleic Acids Res 47, 5587–5602. 10.1093/nar/gkz273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M, 2010. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA, 2007. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res 35, D88–D92. 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Fujiyama A, Hattori M, Taylor TD, Toyoda A, Kuroki Y, Noguchi H, BenKahla A, Lehrach H, Sudbrak R, Kube M, Taenzer S, Galgoczy P, Platzer M, Scharfe M, Nordsiek G, Blöcker H, Hellmann I, Khaitovich P, Pääbo S, Reinhardt R, Zheng H-J, Zhang X-L, Zhu G-F, Wang B-F, Fu G, Ren S-X, Zhao G-P, Chen Z, Lee Y-S, Cheong J-E, Choi S-H, Wu K-M, Liu T-T, Hsiao K-J, Tsai S-F, Kim C-G, OOta S, Kitano T, Kohara Y, Saitou N, Park H-S, Wang S-Y, Yaspo M-L, Sakaki Y, The International Chimpanzee Chromosome 22 Consortium, 2004. DNA sequence and comparative analysis of chimpanzee chromosome 22. Nature 429, 382–388. 10.1038/nature02564. [DOI] [PubMed] [Google Scholar]
- Wj G, 2014. Assaying the epigenome in limited numbers of cells. Methods 72, 51–56. 10.1016/j.ymeth.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, Lee C, Eskin E, Voineagu I, Ernst J, Geschwind DH, 2016. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Tanaka Y, Patterson B, Kang Y-J, Govindaiah G, Roselaar N, Cakir B, Kim K-Y, Lombroso AP, Hwang S-M, Zhong M, Stanley EG, Elefanty AG, Naegele JR, Lee S-H, Weissman SM, Park I-H, 2017. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21, 383–398. 10.1016/j.stem.2017.07.007 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Liu X, Zhang M, Zou M, Deng Q, Sun D, Bian X, Cai Y, Guo Y, Liu S, Li S, Shiang E, Zhong H, Cheng L, Xu H, Jin K, Xiang M, 2018. Direct reprogramming of fibroblasts into neural stem cells by single non-neural progenitor transcription factor Ptf1a. Nat. Commun 9, 2865. 10.1038/s41467-018-05209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Guo H, Hu B, Li R, Yong J, Zhao Y, Zhi X, Fan X, Guo F, Wang X, Wang W, Wei Y, Wang Y, Wen L, Qiao J, Tang F, 2016. Epigenomic landscape of human fetal brain, heart, and liver. J. Biol. Chem 291, 4386–4398. 10.1074/jbc.M115.672931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Chanda S, Marro S, Ng Y-H, Janas JA, Haag D, Ang CE, Tang Y, Flores Q, Mall M, Wapinski O, Li M, Ahlenius H, Rubenstein JL, Chang HY, Buylla AA, Südhof TC, Wernig M, 2017. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 14, 621–628. 10.1038/nmeth.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Q-L, Stavridis M, Griffiths D, Li M, Smith A, 2003. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat. Biotechnol 21, 183–186. 10.1038/nbt780. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhang H, Zhou Y, Qiao M, Zhao S, Kozlova A, Shi J, Sanders AR, Wang G, Luo K, Sengupta S, West S, Qian S, Streit M, Avramopoulos D, Cowan CA, Chen M, Pang ZP, Gejman PV, He X, Duan J, 2020. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science 369, 561–565. 10.1126/science.aay3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S-C, Wernig M, Duncan ID, Brüstle O, Thomson JA, 2001. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol 19, 1129–1133. 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, Xu W, Yang N, Danko T, Chen L, Wernig M, Südhof TC, 2013. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Bhattacharyya A, 2018. Human models are needed for studying human neurodevelopmental disorders. Am. J. Hum. Genet 103, 829–857. 10.1016/j.ajhg.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, Fak JJ, Funk J, Yao K, Tajima Y, Packer A, Darnell RB, Troyanskaya OG, 2019. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet 51, 973–980. 10.1038/s41588-019-0420-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziffra RS, Kim CN, Wilfert A, Turner TN, Haeussler M, Casella AM, Przytycki PF, Kreimer A, Pollard KS, Ament SA, Eichler EE, Ahituv N, Nowakowski TJ, 2019. Single cell epigenomic atlas of the developing human brain and organoids (preprint). Dev. Biol 10.1101/2019.12.30.891549. [DOI] [Google Scholar]
- Zikopoulos B, Barbas H, 2013. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci 7 10.3389/fnhum.2013.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Edri R, Yaffe Y, Donaghey J, Pop R, Mallard W, Issner R, Gifford CA, Goren A, Xing J, Gu H, Cachiarelli D, Tsankov A, Epstein C, Rinn JR, Mikkelsen TS, Kohlbacher O, Gnirke A, Bernstein BE, Elkabetz Y, Meissner A, 2015. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature 518, 355–359. 10.1038/nature13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.