

ABSTRACT
Oro-facial-digital syndrome is a group of rare heterogeneous hereditary disorders characterized by abnormalities of the oral cavity, face and digits, along with varying degrees of mental retardation. Currently, Oro-facial-digital syndrome has been classified into 14 types and two additional unclassified variants have been proposed. Amongst the various variants described, Oro-facial-digital syndrome type I is the most common. We report an interesting subclinical sporadic case of Oro-facial-digital syndrome type I in a 21-year-old female patient. Interestingly, our patient presented with a few novel hitherto unreported clinical findings like midline pits in the philtrum area and a hamartomatous proliferation of tissue in the anterior maxillary alveolar gingival region. This case report highlights the importance of prudent histopathological-clinical correlation, which can direct the flow of clinical investigations leading to the detection and diagnosis of unsuspected conditions as learned in this case. We would also like to emphasize that comprehensive examination of new born for structural abnormalities of the orofacial region is crucial to early diagnosis of syndromes and subsequent referral for further evaluation and management.
Keywords : Mutation, Hamartoma, Cleft Palate, Ciliopathies
INTRODUCTION
Oro-facial-digital syndrome (OFDS) is a group of rare heterogeneous hereditary disorders characterized by morphogenetic impairment of the oral cavity, face and digits, along with varying degrees of mental retardation, almost limited to the female gender. Currently, OFDS have been classified into 14 types and two additional unclassified variants have been proposed. Amongst the various variants described, OFDS type I is the most commonly presented syndrome and yet is quite rare.1 , 2
The first description of OFDS syndrome was given by Mohr in 1941, where he reported a family with significant abnormalities of the oral cavity, face and digits. Oro-facial-digital syndrome type I was first reported in 1954, by Papillon-League and Psaume, hence it is also known as Papillon-League-Psaume syndrome. In 1964, Gorlin & Pindborg coined the term ‘Orodigitofacial dysostosis’. However, due to reports of multi-organ involvement the term ‘Oro-facial-digital syndrome’ is preferred.3
OFDS I is inherited as an X-linked dominant condition, which is lethal to with variable degree of expression within the same family. The gene responsible for this disorder is found on the short arm of the X chromosome (Xp22.3-p22.2). In a study by Ferrante et al.4 mutations in the CXORF5 gene were detected, which was later termed OFD1 gene (MIM# 311200).4
It has been reported that, approximately, 75% of the OFDS I cases are sporadic. Sometimes a female proband with OFDS I may have the disorder as a result of de novo pathogenic variant.5 , 6 The incidence of OFDS I is 1:50000 to 1:250000 live births and the prevalence is estimated to be between 1 out of 25,000 to 1 out of 150,000 live births.5 , 6 , 7
Syndromes show variable expressivity, necessitating recognition and differential diagnosis of the clinical presenting signs and symptoms. A case of oro-facial-digital syndrome type I, with special clinical aspects is presented, highlighting the importance of interpreting histopathological features in the detection and unmasking of unsuspected conditions including syndromes.
CASE REPORT
An excisional biopsy of an anterior maxillary gingival growth was received for routine histopathological examination from a 21-year-old female patient presenting for treatment of mal-aligned anterior teeth. The provisional diagnosis was a fibroma.
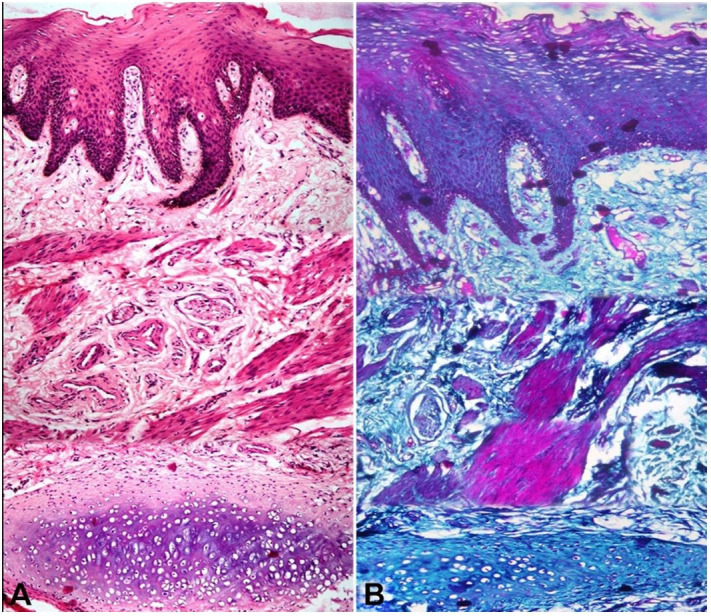
The histopathological evaluation of the Hematoxylin and eosin-stained sections of the biopsied tissue showed stratified squamous parakeratinizing epithelium overlying a fibro cellular stroma. The stroma consisted of loosely arranged collagen fibers, loosely arranged bundles of differentiated smooth muscle fibers, nerve fascicles, thick-walled blood vessels and ectopic cartilaginous tissue (Figure 1A). The Masson Trichrome special stain was used to delineate the different connective tissue components (Figure 1B).
Figure 1. Photomicrographs of the biopsied soft tissue lesion: A – stratified squamous para-keratinized epithelium overlying a fibro-cellular connective tissue stroma predominantly comprising of blood vessels, neural tissue, smooth muscle bundles and forming hyaline cartilage in deeper stroma (H&E, 10X); B – Masson trichrome stain used to differentiate the smooth muscle cells (stained pink) from dense collagen fibres (stained blue) (10X).
As the histopathological findings were suggestive of a hamartoma, a comprehensive clinical anamnesis with radiographic investigations was requisitioned.
Clinical examination revealed a normal-statured, well-oriented female in apparent good health. Extra-orally, micrognathia, pseudo-clefting of the lower lip, two midline pits in the labial philtrum and low set ears were evident (Figure 2A-C). The skin of patient was dry with thin scanty hair, crops of milia were noted on the nose, along the nasolabial folds and chin (Figure 2A, B, D).
Figure 2. Clinical examination: A – showing soft tissue swelling over the alveolar mucosa (arrow head), abnormal frenal attachment, midline diastema, median alveolar cleft, mesio-labial rotation of the right central incisor, pseudo cleft of the lower lip (yellow dotted line), milia (arrows); B – Philtrum pits (arrowhead), milia (arrows); C – Low set ears; D – Thin scanty hair.
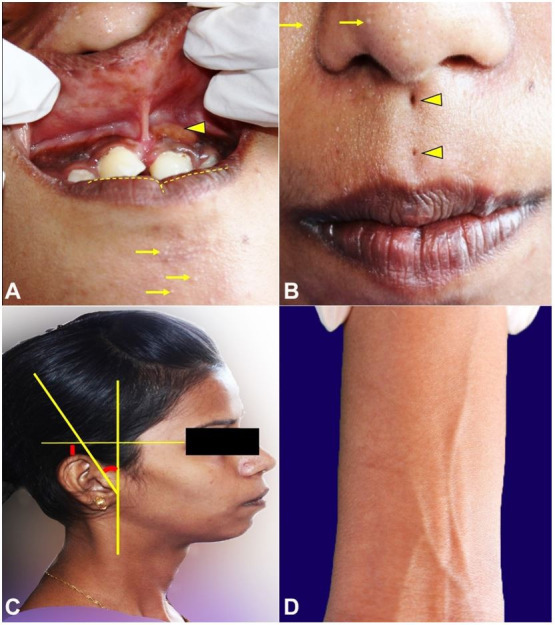
Intra-oral examination of the patient revealed mesio-labial rotation of the right maxillary central incisor, a midline diastema associated with a median alveolar cleft, high labial frenal attachment and an additional small soft tissue gingival swelling (measuring approximately 1x1.5 cm in dimension) in relation to the left maxillary central incisor (Figure 2A) .
There was no evidence of malformation of hands and feet and her medical history was unremarkable. Given this constellation of signs, the patient’s mother was interviewed. The mother confirmed that she had a non-consanguineous marriage, the patient was delivered as a premature baby with low birth weight (exact weight not known) and had learning difficulties. She also mentioned that the patient has a completely normal younger male sibling.
The patient was advised an orthopantamograph (OPG), lateral cephalogram, cone beam computed tomography (CBCT) of the jaw bones and an abdominal ultrasound to rule out polycystic kidney disease. The CBCT (Figure 3A, B) and the OPG confirmed the presence of a median alveolar cleft of the maxilla, while the abdominal ultrasound was unremarkable.
Figure 3. Tomographic examination of maxilla showing median alveolar cleft. A – 3D reconstruction; B – Axial view.

A karyotyping test was conducted. The test revealed an apparently normal karyotype as assessed by conventional cytogenetic analysis (CCA). A review of literature suggested that a normal karyotype has been reported in patients with clinical diagnosis of Oro-facial-digital syndrome type I, as not all genetic mutations are identifiable by CCA and requires use of advanced molecular genetic testing methods to ascertain the clinical diagnosis. In the present case a clinical diagnosis of Oral-facial-digital syndrome type I was concluded upon based on the clinic-radiographic features. In this case advanced molecular genetic tests were not conducted due to financial reasons and thus remains to be a limitation of this case report.
Clinical management of such cases is multidisciplinary and depends on the severity of phenotypic expression of the mutated OFD I gene. Since our patient was not aware of her medical condition and did not present with major anatomical defects, she was informed and counseled about the same and advised to keep in touch for a regular follow up.
DISCUSSION
Oro-facial-digital syndrome type I is a rare, X-linked dominant male lethal ciliopathy with variable clinical presentation owing to varied mutations within the OFDI gene (CXORF5). The OFD I protein is localized to the basal body of the primary cilia.8
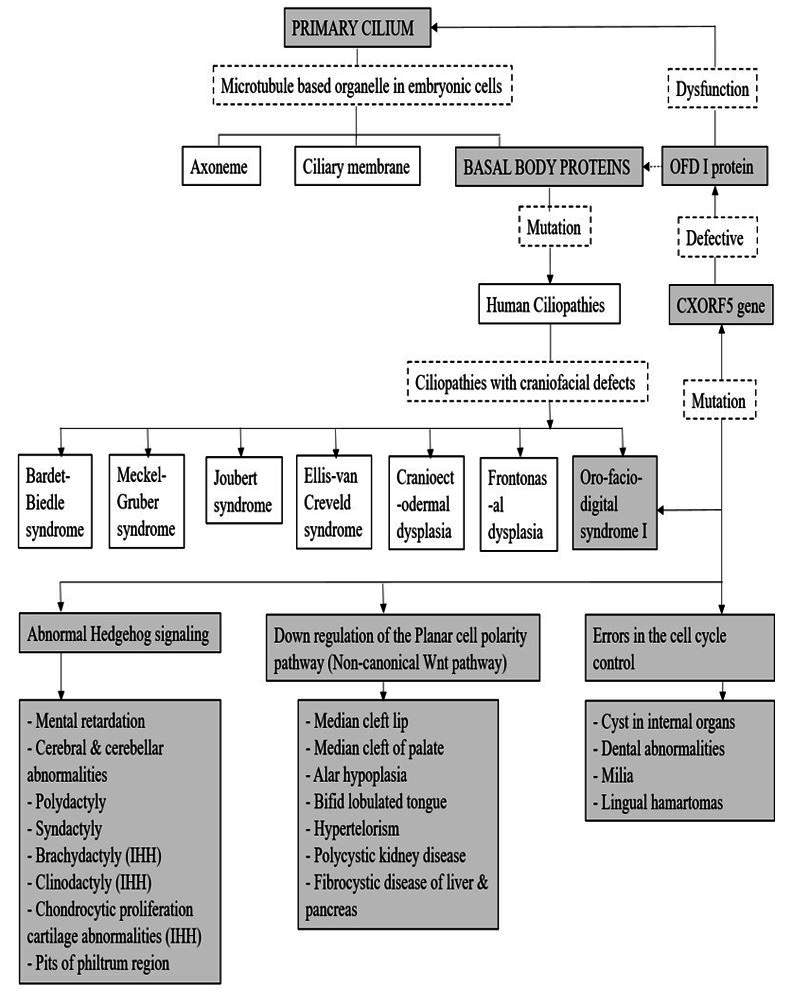
The term ‘primary cilium’ was coined by Sergei Sorokin, to describe an organelle that emanates from the cell surface of most mammalian cell types during growth arrest. The primary cilium provides a means of sequestering the centriole, thus majority of the cells that have primary cilia are non-cycling differentiated cells or stem cells in G0 phase.9 , 10 The primary cilia are found on different cell types in the human body, including the stem, epithelial, endothelial, muscle, connective tissue and the neuronal cells. Increasing evidence suggests that the primary cilium is the key coordinator during development and in tissue homeostasis. Primary cilia also plays a vital role in modulating cell signaling pathways. Experimental studies have shown that, various receptors, ion channels, transporter proteins, downstream effector molecules, are localized to the basal body. Thus, primary cilium helps orchestrate key developmental processes like cell migration, cell differentiation, cell cycle control, plane of cell division and apoptosis. The signaling pathways modulated at the level of the basal body of the primary cilium are diverse and depend on the cell type.9 Genetic mutations in any of the proteins associated with the basal body of the primary cilium can result in various human diseases or syndromes, which are collectively known as ‘Human ciliopathies’. The OFDI protein is one of the proteins associated with the basal body of the primary cilium, when defective results in the clinical manifestations of the OFD I syndrome. The molecular pathogenesis of OFDS type I has been presented in a simplified format using a flow chart (Figure 4).11
Figure 4. Pathogenesis of Oro-facial-digital syndrome type I, according to AlKattan et al., 201511.
Through this case report, we aim to highlight a subclinical sporadic case of OFDS type I, which lacked the easily observable phenotypic features of the syndrome and presented with few novel hitherto unreported clinical findings. To the best of our knowledge, we report the first patient of OFDS type I with midline pits in the philtrum area and a hamartomatous proliferation of tissue in the anterior maxillary alveolar gingival region showing exuberant proliferation of smooth muscle cells, blood vessels, neural tissue and cartilaginous tissue.
The planar cell polarity (PCP) pathway is known to orchestrate proper orientation, migration and intercalation of the tissue cells and the Indian hedgehog pathway (IHH) is associated with chondrocyte proliferation. Thus, as described in Figure 3, down regulation of the PCP pathway and abnormal functioning of the IHH pathway coupled with abnormal cycle control, may have led to the philtrum pits and hamartoma formation in our patient.11 The cartilaginous tissue could have arisen from abnormal proliferation of the remnants of embryonic cartilage precursors from nasal and septal development in the anterior part of the maxilla.12
While our patient had a limited expression of the conventional phenotypic features, she presented with philtrum pits and hamartomatous proliferation of soft tissues of the anterior maxillary gingiva, thus representing yet another facet in the varying phenotypic spectrum of OFDS type I.
In order to ease the clinical evaluation and diagnosis of the varied spectrum of Oro-facial-digital syndromes and the syndromes showing features overlapping with OFDS type I, the authors performed a thorough review of literature and tabulated their clinical features (Table 1) and genetic aberrations (Table 2) for a quick easy review.
Table 1. Comparative analysis of the clinical features evident in Oro-facial-digital syndrome type I and other syndromes constituting its differential diagnosis (EVC = Ellis-van Creveld syndrome; JS = Joubert syndrome; MGS = Meckel-Gruber syndrome; PHS = Pallister-Hall syndrome; SLOS = Smith-Lemli-Opitz syndrome).
| Clinical features/ differential diagnosis | I | II | III | IV | V | VI | VII | VIII | IX | X | XI | XII | XIII | XIV | UI | U II | EVC | JS | MGS | PHS | SLOS | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Types of OFDS | |||||||||||||||||||||||
| Extra oral features | |||||||||||||||||||||||
| Stature [1,5] (Short +, Normal -) | + | - | - | + | - | - | - | - | + | + | - | - | - | - | - | - | + | - | - | - | + | ||
| Eye | Hypertelorism1 | + | + | + | - | - | + | + | + | + | - | + | + | - | - | - | - | - | + | + | - | - | |
| Blepharophimosis1 | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | ||
| Coloboma1 | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | + | + | - | - | ||
| Exophthalmos2 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Seesaw winking2 , 5 | - | - | + | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Epicanthus fold1 | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Telecanthus1 | - | - | - | - | - | - | - | + | - | - | - | - | - | + | - | - | - | - | - | - | - | ||
| Synophrys, Microphthalmia1 , 5 | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Retinal abnormalities1 , 5 | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Epicanthus fold1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | ||
| Ptosis13 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | + | ||
| Nystagmus13 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | ||
| Oculomotor apraxia13 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | ||
| Hypoplastic optic nerve14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Strabismus13 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + | - | - | - | ||
| Congenital cataracts15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | + | ||
| Blepharosis16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | ||
| Microphthalmia14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Nose | Broad bifid tip17 | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Broad nasal root17 | - | + | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Bulbous nose1 , 5 | - | - | + | + | - | - | - | + | - | - | - | - | - | - | - | - | - | - | + | + | - | ||
| Flat nasal root1 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Hypoplasia of the alae11 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Hypoplastic nasal septum14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Short nose upturned nostrils18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + | ||
| Broad or flat nasal bridge18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Nostrils turned forward16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | ||
| Ear | Hearing defects17 | + | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Low set ears1 , 5 | + | - | + | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Abnormal inner ear1 | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Auricular pits & Deafness1 | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | ||
| Malformed ear14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Small ears rotated backwards18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Large external ears14 , 16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | ||
| Intra oral region: | |||||||||||||||||||||||
| Palate | Cleft palate1 , 17 | + | + | + | + | - | + | + | + | + | + | + | - | + | + | + | - | - | + | + | - | + | |
| Lip | Median cleft lip17 | + | + | + | + | + | + | - | + | + | - | - | - | - | - | + | - | - | - | - | + | + | |
| Cleft lip1 | - | - | - | - | - | + | + | - | - | - | - | - | + | - | - | - | - | + | + | - | - | ||
| Short upper lip15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Midline long vertical groove in upper lip18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Long inverted V shape upper lip16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | ||
| Tongue | Cleft1 | - | + | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | |
| Lobulated tongue1 | + | + | + | + | - | + | - | + | + | - | - | - | - | + | + | - | - | + | + | + | - | ||
| Bifid or Trifid1 | + | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | + | - | - | - | ||
| Lingual hamartomas1 | + | + | + | + | - | + | + | + | + | + | - | - | + | + | - | + | - | - | + | + | - | ||
| Ankyloglossia19 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Bifid uvula1 | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Cleft epiglottis14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Microglossia18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Cleft or fissure in the larynx18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Epiglottis hypoplasia1 , 5 | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Bifid epiglottis18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Inflexible epiglottis1 , 2 | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Gingiva | Gingival Frenulae1 | + | + | - | + | + | + | + | + | + | + | + | + | - | + | + | - | + | + | - | + | - | |
| Labiogingival adherence15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Submucosal clefts15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Labial vestibule obliteration15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Buccal frenula18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Abnormal gums16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | ||
| Dentition | Missing teeth17 | + | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Supernumerary teeth1 | + | - | + | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | + | - | - | ||
| Diastema15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Conical teeth15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Natal teeth18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Neonatal teeth14 , 15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | - | - | ||
| Hypodontia15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Enamel dysplasia11 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Enamel hypoplasia1 | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | - | - | ||
| Tooth malformations2 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Premature eruption15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Premature exfoliation15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Mandible | Hypoplastic mandible17 | + | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | |
| Micrognathia14 | + | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | - | - | ||
| Retrognathia1 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | + | - | - | ||
| Short mandible16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | ||
| Jaw winking2 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||||
| Digits [Hands &Feet] | Brachydactyly1 | + | + | - | + | - | + | - | - | + | - | - | - | + | - | - | - | - | - | - | - | - | |
| Clinodactyly1 | + | + | - | + | - | + | + | - | + | - | - | - | + | - | - | - | - | - | + | - | - | ||
| Polydactyly1 | + | + | + | + | + | + | - | + | + | + | + | + | - | + | + | + | + | + | + | + | + | ||
| Syndactyly1 | + | - | - | - | - | + | - | - | - | - | - | - | + | - | - | - | - | - | + | + | + | ||
| Oligodactyly1 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Others systems | |||||||||||||||||||||||
| Skin | Milia5 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Hair | Thick hair1 | - | + | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | |
| Thin dry hair11 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Alopecia11 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |||
| Photosensitivity16 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | ||
| CNS | Mental retardation1 , 17 | + | + | + | + | + | + | - | + | - | - | - | + | - | + | + | - | - | + | - | - | + | |
| Epilepsy2 , 5 , 16 , 18 | - | - | + | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | + | + | ||
| Intellectual disability1 , 5 | - | - | + | - | - | - | + | + | - | - | + | - | - | + | + | - | - | - | - | - | - | ||
| Psychomotor retardation2 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Macrocephaly1 | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | ||
| Microcephaly14 , 16 , 18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + | + | ||
| Neuropsychiatric troubles1 | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | ||
| Encephalocele13 , 14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + | - | - | ||
| Hydrocephaly14 , 16 , 18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + | + | ||
| Anencephaly14 , 16 , 18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + | + | ||
| Cerebellar vermis agenesis14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | ||
| Malformed hypothalamus18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| CVS | Coarctation of the aorta1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | |
| Single atrium15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Defects of the mitral and tricuspid valves15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Patent ductus arteriosus15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | - | - | ||
| Septum hypertrophy1 | - | + | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | ||
| Valve dysplasia1 | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | ||
| Ventricular septal defect1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | - | + | - | - | ||
| Atrial septal defect14 , 15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | + | - | - | ||
| Hypoplastic left heart syndrome15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Congenital heart defects16 , 18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | + | ||
| Kidney | Kidney absent5 , 13 | + | - | + | + | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Polycystic kidney disease1 | + | - | + | + | - | - | + | + | - | - | - | - | - | - | - | - | - | + | + | + | - | ||
| Renal dysplasia1 , 5 | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | + | + | - | - | ||
| Renal failure1 | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | ||
| Fused kidneys1 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | ||
| Agenesis of kidney18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
| Liver | Macrocysts5 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Fibrosis2 , 14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Pancreas | Macrocysts5 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Ovary | Macrocysts5 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Skeletal system | Y-shaped metacarpal1 | - | + | - | - | - | + | - | - | - | - | - | + | - | - | - | + | - | - | - | - | - | |
| Tibia abnormalities1 | - | - | - | + | - | - | - | + | - | - | - | + | - | - | - | - | - | - | - | - | - | ||
| Radius hypoplasia1 | - | - | - | - | - | - | - | + | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Fibular agenesis1 | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | ||
| Vertebral abnormalities1 | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | ||
| Shortening of the middle and distal phalanges15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Deformity of the knees or lumbar lordosis15 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | ||
| Bowing of the long bones of limbs14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | ||
| Talipes equinovarus14 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||
| Abnormally short arms and/or legs and/or dislocated hips18 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | ||
Table 2. Genotypic variation seen in Oro-facial-digital syndrome type I and other syndromes constituting its differential diagnosis (EVC = Ellis-van Creveld syndrome; JS = Joubert syndrome; MGS = Meckel-Gruber syndrome; PHS = Pallister-Hall syndrome; SLOS = Smith-Lemli-Opitz syndrome).
| Type | Phenotype MIM# Number | Inheritance Pattern | Gene | Cytogenetic location |
|---|---|---|---|---|
| Type I20 | 311200 | X linked dominant | CXORF5 | Xp22.3-p22.2 |
| Type II20 | - | Autosomal recessive | Unidentified gene | - |
| Type III20 | 258850 | Autosomal recessive | TMEM231 | 16q23.1 |
| Type IV20 | 258860 | Autosomal recessive | TCTN3 | 10q24.1 |
| Type V20 | 174300 | Autosomal recessive | DDX59 | 1q32.1 |
| Type VI20 | 277170 | Autosomal recessive | C5ORF42 | 5p13.2 |
| Type VII20 | 608518 | X-linked dominant | - | - |
| Type VIII20 | 300484 | X-linked recessive | - | - |
| Type IX20 | 258865 | Autosomal recessive | TBC1D32 | 6q22.31 |
| Type X20 | - | Sporadic | - | - |
| Type XI20 | - | Sporadic | - | - |
| Type XII20 | - | Sporadic | - | - |
| Type XIII20 | - | Sporadic | - | - |
| Type XIV20 | 615948 | Autosomal recessive | C2CD3 | 11q13.4 |
| Unclassified OFD20 | 613580 | Autosomal recessive | WDPCP | 2p15 |
| Unclassified OFD20 | 617563 | Autosomal recessive | TMEM107 | 17p13.1 |
| EVC15 | 225500 | Autosomal recessive | EVC and EVC2 | 4p16.2 |
| JS 1013 | 300804 | Autosomal recessive | OFD1 | Xp22.2 |
| MGS14 | 614209 | Autosomal recessive | B9D1 | 17p11.2 |
| 614175 | B9D2 | 19q13.2 | ||
| 612284 | CC2D2A | 4p15.32 | ||
| 611134 | CEP290 | 12q21.32 | ||
| 249000 | MKS1 | 17q22 | ||
| 611561 | RPGRIP1L | 16q12.2 | ||
| 613885 | TCTN2 | 12q24.31 | ||
| 258860 | TCTN3 | 10q24.1 | ||
| 607361 | TMEM67 | 8q22.1 | ||
| 617562 | TMEM107 | 17p13.1 | ||
| 603194 | TMEM216 | 11q12.2 | ||
| 615397 | TMEM231 | 16q23.1 | ||
| 614424 | TMEM237 | 2q33.1 | ||
| PHS18 | 607324 | Autosomal Dominant | GLI3 | 7p13.1 |
| SLOS16 | 270400 | Autosomal recessive | DHCR7 | 11q13.4 |
CONCLUSION
We contribute to the existing literature, previously unreported features of OFDS I and propose the inclusion of philtrum pits and hamartoma involving any of the oral mucosal tissue (not limited to the tongue) in the clinical presentation of OFDS type I.
Through this case the authors would like to highlight the significance of noting unusual histopathological findings in routine specimens with reappraisal of clinical data which may be crucial to diagnosis of such cases, which are rare and have shown subclinical presentation. The patients diagnosed with OFDS type I are at a risk of developing polycystic kidney disease, hence need to be kept under observation with careful morphological assessment and biochemical monitoring. We would also like to emphasize that comprehensive examination of new born for structural abnormalities of the orofacial region is crucial to early diagnosis and subsequent referral for further evaluation.
Footnotes
How to cite: Syed S, Sawant PR, Spadigam A, Dhupar A. Oro-facial-digital syndrome type I: a case report with novel features. Autops Case Rep [Internet]. 2021;11:e2021315. https://doi.org/10.4322/acr.2021.315
This study was carried out at Goa Dental College and Hospital
Ethics Statement: Patient consent has been obtained.
Financial Support: The authors declare that no financial support was received.
REFERENCES
- 1.Franco B, Thauvin-Robinet C. Update on oral-facial-digital syndromes (OFDS) Cilia. 2016;5(1):12. doi: 10.1186/s13630-016-0034-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.National Organization for Rare Disorders (NORD) Oral-Facial-Digital Syndrome. NORD; 2007. [cited 2020 October 17]. Available from: https://rarediseases.org/rare-diseases/oral-facial-digital-syndrome/ [Google Scholar]
- 3.Dodge JA, Kernohan DC. Oral-facial-digital syndrome. Arch Dis Child. 1967;42(222):214–219. doi: 10.1136/adc.42.222.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrante MI, Giorgio G, Feather SA, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68(3):569–576. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toriello HV, Franco B, Bruel AL, et al. GeneReviews® Oral-facial-digital syndrome type I. Seattle: University of Washington; 2002. [cited 2020 October 17]. Available from: https://www.ncbi.nlm.nih.gov/books/ [Google Scholar]
- 6.Salinas CF, Pai GS, Vera CL, et al. Variability of expression of the orofaciodigital syndrome type I in black females: six cases. Am J Med Genet. 1991;38(4):574–582. doi: 10.1002/ajmg.1320380416. [DOI] [PubMed] [Google Scholar]
- 7.Thauvin-Robinet C, Cossee M, Cormier-Daire V, et al. Clinical, molecular, and genotype–phenotype correlation studies from 25 cases of oral–facial–digital syndrome type I: a French and Belgian collaborative study. J Med Genet. 2006;43(1):54–61. doi: 10.1136/jmg.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma S, Kalish JM, Goldberg EM, Reynoso FJ, Pradhan M. An atypical presentation of a male with oral-facial-digital syndrome type I related ciliopathy. Case Rep Nephrol. 2016;2016:3181676. doi: 10.1155/2016/3181676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123(Pt4):499–503. doi: 10.1242/jcs.050377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorokin SP. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci. 1968;3(2):207–230. doi: 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- 11.AlKattan WM, Al-Qattan MM, Bafaqeeh SA. The pathogenesis of the clinical features of oral-facial-digital syndrome type I. Saudi Med J. 2015;36(11):1277–1284. doi: 10.15537/smj.2015.11.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahajan AM, Ganvir SM, Hazarey VK, Mahajan MC. Chondrosarcoma of the maxilla: a case report and review of literature. J Oral Maxillofac Pathol. 2013;17(2):269–273. doi: 10.4103/0973-029X.119759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.National Organization for Rare Disorders (NORD) Joubert Syndrome. NORD; 2011. [cited 2020 October 17]. Available from: https://rarediseases.org/rare-diseases/joubert-syndrome/ [Google Scholar]
- 14.National Organization for Rare Disorders (NORD) Meckel Syndrome. NORD; 2020. [cited 2020 October 17]. Available from: https://rarediseases.org/rare-diseases/meckel-syndrome/ [Google Scholar]
- 15.Kamal R, Dahiya P, Kaur S, Bhardwaj R, Chaudhary K. Ellis-van Creveld syndrome: a rare clinical entity. J Oral Maxillofac Pathol. 2013;17(1):132–135. doi: 10.4103/0973-029X.110716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.National Organization for Rare Disorders (NORD) Smith Lemli Opitz Syndrome. NORD; 2007. [cited 2020 October 17]. Available from: https://rarediseases.org/rare-diseases/smith-lemli-opitz-syndrome/ [Google Scholar]
- 17.Dave KV, Patel SC, Dudhia BB, Panja P. Orofacial digital syndrome. Indian J Dent Res. 2013;24(1):132–135. doi: 10.4103/0970-9290.114920. [DOI] [PubMed] [Google Scholar]
- 18.National Organization for Rare Disorders (NORD) Pallister-Hall Syndrome. NORD; 2019. [cited 2020 October 17]. Available from: https://rarediseases.org/rare-diseases/pallister-hall-syndrome/ [Google Scholar]
- 19.Dhull KS, Acharya S, Mohanty M, Dhull RS, Panda S. Oro-facial-digital syndrome type I: a case report. J Indian Soc Pedod Prev Dent. 2014;32(2):152–155. doi: 10.4103/0970-4388.130980. [DOI] [PubMed] [Google Scholar]
- 20.Online Mendelian Inheritance in Man® (OMIM®) Orofaciodigital syndrome VI; OFD6. Baltimore: Johns Hopkins University; 2017. [cited 2020 October 17]. Available from: https://www.omim.org/entry/277170. [Google Scholar]