

Abstract
Iron is an essential cofactor for several metabolic processes, including the generation of ATP in mitochondria, which is required for axonal function and regeneration. However, it is not known how mitochondria in long axons, such as those in sciatic nerves, acquire iron in vivo. Because of their close proximity to axons, Schwann cells are a likely source of iron for axonal mitochondria in the PNS. Here we demonstrate the critical role of iron in promoting neurite growth in vitro using iron chelation. We also show that Schwann cells express the molecular machinery to release iron, namely, the iron exporter, ferroportin (Fpn) and the ferroxidase ceruloplasmin (Cp). In Cp KO mice, Schwann cells accumulate iron because Fpn requires to partner with Cp to export iron. Axons and Schwann cells also express the iron importer transferrin receptor 1 (TfR1), indicating their ability for iron uptake. In teased nerve fibers, Fpn and TfR1 are predominantly localized at the nodes of Ranvier and Schmidt-Lanterman incisures, axonal sites that are in close contact with Schwann cell cytoplasm. We also show that lack of iron export from Schwann cells in Cp KO mice reduces mitochondrial iron in axons as detected by reduction in mitochondrial ferritin, affects localization of axonal mitochondria at the nodes of Ranvier and Schmidt-Lanterman incisures, and impairs axonal regeneration following sciatic nerve injury. These finding suggest that Schwann cells contribute to the delivery of iron to axonal mitochondria, required for proper nerve repair.
SIGNIFICANCE STATEMENT This work addresses how and where mitochondria in long axons in peripheral nerves acquire iron. We show that Schwann cells are a likely source as they express the molecular machinery to import iron (transferrin receptor 1), and to export iron (ferroportin and ceruloplasmin [Cp]) to the axonal compartment at the nodes of Ranvier and Schmidt-Lanterman incisures. Cp KO mice, which cannot export iron from Schwann cells, show reduced iron content in axonal mitochondria, along with increased localization of axonal mitochondria at Schmidt-Lanterman incisures and nodes of Ranvier, and impaired sciatic nerve regeneration. Iron chelation in vitro also drastically reduces neurite growth. These data suggest that Schwann cells are likely to contribute iron to axonal mitochondria needed for axon growth and regeneration.
Keywords: axon regeneration, iron, mitochondria, nodes of Ranvier, Schmidt-Lanterman incisures, Schwann cell
Introduction
Mitochondria play a central role in supplying energy required for axon growth and regeneration (Han et al., 2016; Vaarmann et al., 2016; Misgeld and Schwarz, 2017; Mandal and Drerup, 2019). Because of the high-energy demand at distal synapses and growth cones, maintaining energy homeostasis is especially important in neurons with long axons, such as those in peripheral nerves innervating the lower limbs (Berthold et al., 1993). A constant energy supply by mitochondria is therefore key to ensure proper functioning of these axons. Iron is essential for mitochondrial function because the biogenesis of iron-sulfur (Fe-S) cluster and heme, two abundant cofactors of various mitochondrial respiratory chain enzymes, occurs in mitochondria (Hentze et al., 2004; Rouault and Tong, 2005). Iron enters mitochondria via three potential routes that include transient direct contact with endosomes carrying TfR1-Tf-2Fe3+; from the labile iron pool; or non-Tf bound iron bypassing the labile iron pool. Iron exits mitochondria as Fe-S clusters or heme, and excess iron in mitochondria is stored in mitochondrial ferritin (Gao et al., 2021). Fe-S clusters and heme, which are abundant in mitochondrial respiratory chain enzymes, not only play an important role in electron transfer reactions needed for the generation of ATP (Rouault and Tong, 2005), but are also exported out of mitochondria into the cytosol where they are incorporated into other enzymes and proteins (Rouault and Tong, 2005). ATP generated in mitochondria within axons fuels energy-demanding axonal processes, such as the movement of organelles during bidirectional axonal transport, conduction of action potentials, and synaptic function (Li et al., 2004; Misgeld and Schwarz, 2017; Mandal and Drerup, 2019). In addition, mitochondria are needed at the growth cone for axonal regeneration. In vivo imaging of mitochondria in the mouse sciatic nerve over a 6 min period showed that ∼22% of axonal mitochondria are motile and move in both anterograde and retrograde directions (Magrane et al., 2014). It is possible that mitochondria that are considered stable may also move over longer periods of time. Stable mitochondria are generally located in regions of high demand for ATP (Spillane et al., 2013). How and where axonal mitochondria acquire the iron needed for Fe-S and heme biogenesis, and thus ATP generation, are unknown. One possibility is that mitochondria acquire iron in the neuronal cell body, migrate down the axon to sites of high-energy demand, and then return to the cell body to be replenished. Alternatively, axonal mitochondria may obtain iron from Schwann cells along the axon while in transit, potentially acquiring iron where they come in close proximity to Schwann cell cytoplasm, that is, near the paranodal loop regions at the nodes of Ranvier and/or at the Schmidt-Lanterman incisures. The latter are Schwann cell cytoplasmic loops that spiral through the compact myelin and are sites of metabolic activity and where metabolic transfer could occur between Schwann cells and axons.
In the present study, we first investigated whether iron is required for axonal growth by analyzing neurite growth initiation and elongation in DRG cultures treated with an iron chelator. To examine whether Schwann cells may be involved in providing axons with iron, we first assessed whether Schwann cells express the proteins needed to efflux iron. We have previously shown that the iron efflux (export) transporter, ferroportin (Fpn), and the membrane-bound ferroxidase ceruloplasmin (Cp) partner to efflux iron from astrocytes (Jeong and David, 2003). We found that these molecules are indeed expressed by Schwann cells, indicating that they can export iron. We therefore used Cp KO mice to assess whether iron accumulation occurs in Schwann cells in the sciatic nerve, and assessed the localization and number of axonal mitochondria at sites of close apposition between axonal and Schwann cell membranes (Schmidt-Lanterman incisures and nodes of Ranvier). We also assessed whether iron deprivation is detected in axonal mitochondria in Cp KO mice, and the effects of axon regeneration after sciatic nerve injury. Our findings suggest that Schwann cells provide iron to axonal mitochondria, needed for effective axonal regeneration.
Materials and Methods
DRG cultures
Neuronal cultures were prepared from postnatal mouse DRGs (P1-P2) as follows. DRGs were dissected and incubated in 0.25% trypsin (Invitrogen) for 30 min at 37°C. They were then mechanically dissociated by triturating 5-10 times with a 22½ G needle followed by a 25½ G needle. The resulting cell suspension was passed through a 70 μm mesh and centrifuged at 500 × g for 10 min. Cells were resuspended in DMEM containing F12 (DMEM/F12, Invitrogen) with 10% FBS (Invitrogen) and 100 ng/ml NGF (Sigma-Aldrich) and plated onto glass coverslips coated with laminin (5 μg/ml, Sigma-Aldrich). Cells were treated either 5 or 10 h after plating with the iron chelator salicylaldehyde isonicotinoyl hydrazone (SIH, 100 μm) or vehicle and incubated overnight. SIH is an analog of pyridoxal isonicotinoyl hydrazone, which shows high affinity and selectivity for iron, similar to deferoxamine (Richardson and Ponka, 1998) and has been used in vitro and in vivo (Klimtová et al., 2003; Rathore et al., 2008; Jeong et al., 2009).
Schwann cell cultures
Schwann cells were isolated from postnatal mouse sciatic nerve (P5-P7) as follows. Sciatic nerves were taken out and digested first with 1% collagenase at 37°C for 45 min and then with 0.25% trypsin at 37°C for 15 min. Nerves were mechanically dissociated by triturating them first with a 21G needle and then with a 23G needle. The cell suspension was passed through a 70 μm mesh and centrifuged for 10 min at 500 × g. Cells were resuspended in DMEM/F12 with 10% FBS and plated onto poly-L-lysine-coated glass coverslips.
Sholl analysis
To quantify neurite growth, a semi-automated Sholl analysis (Sholl, 1953) was conducted using ImageJ software with Sholl analysis plugin on cultures stained with an anti-neurofilament antibody (see Immunofluorescence). First, the cell bodies and neurites were outlined to exclude adjacent cells. In order to remove background labeling and to better identify neurites, densitometric thresholds were set for each neuron. Using ImageJ, templates of concentric circles increasing in radius by 20 μm were overlaid onto the center of the neuronal cell body. The total number of neurites intersecting each circle was counted using the automated Sholl analysis plugin. The maximal ring with an intersecting process (maximal neurite length) and the sum of the number of intersections (branching complexity) for all rings were generated for each cell. The analysis was conducted for 15 neurons per coverslip (2 coverslips per trial, three experimental trials; n = 3). Data are shown as mean ± SEM. The statistical significance between groups was determined by one-way ANOVA followed by Tukey post hoc analysis.
Sciatic nerve injury
Female Cp−/− (Cp KO) and Cp+/+ (WT) mice on a C57BL/6 background at 8-10 weeks of age were used for sciatic nerve crush injuries. Cp KO mice were previously generated in our laboratory (Patel et al., 2002). Mice were anesthetized with ketamine:xylazine:acepromazine (100:10:3 mg/kg), and the right sciatic nerve was exposed and crushed at the mid-thigh with fine forceps (Dumont #5) for 30 s. The crush site was tagged with a 10-0 suture placed through the epineurium being careful not to go into the nerve. All procedures were approved by the McGill University Animal Care committee and followed the guidelines of the Canadian Council on Animal Care.
Sciatic nerve functional index (SFI)
Before lesion and at 10, 17, and 28 d following sciatic nerve crush injury, mice were allowed to walk down a 40-cm-long track after inking their hind paws. The sciatic nerve functional index (SFI) was calculated from the footprints using a formula developed by de Medinaceli et al. (1982) and modified for mice by Inserra et al. (1998). The analysis was conducted blind. Data are plotted as mean ± SEM (n = 18 WT; 15 Cp KO). Two-way ANOVA followed by Bonferroni post hoc analysis was used to determine statistical significance between groups.
Quantification of GAP-43 labeling
Axonal regeneration after sciatic nerve crush injury was assessed 4 d after crush injury by counting the numbers of GAP-43+ and S100β– fibers (see Immunofluorescence labeling) at 3, 4, and 5 mm distal to the crush site (three sections per animal; n = 3 mice per group). The diameter of the nerve fiber was measured at the respective sites of the axonal counts, and the number of axons was divided by the nerve fiber diameter. Data are mean ± SEM. Two-way ANOVA followed by Bonferroni post hoc analysis was used to determine statistical significance between groups.
Iron histochemistry
Animals were transcardially perfused with 4% PFA in 0.1 m PB, pH 7.4. Modified enhanced Perl's histochemistry was performed on 14-μm-thick cryostat sections of the sciatic nerve to detect iron accumulation as described previously (Rathore et al., 2008). Briefly, sections were incubated for 10 min with 4% potassium ferrocyanide only, followed by incubation with 4% HCl and 4% potassium ferrocyanide (1:1) for 50 min. The reaction product was enhanced with DAB. Sections were counterstained with methyl green and mounted with Entellan (all from Sigma-Aldrich).
RNA isolation and qPCR
Total RNA was isolated from sciatic nerves distal to the crush injury site at 1, 7, and 14 d after injury and from uninjured nerves using RNeasy Mini Kit (QIAGEN). This was followed by reverse transcription into cDNA, using Quantinova kit (QIAGEN, catalog #205411). The qRT-PCR was performed using ABI OneStep cycler (Applied Biosystems) and SYBR PCR master mix (Applied Biosystem) with specific primer sets (Table 1). GAPDH was considered as an internal control gene, and results were analyzed using the ΔΔCT method. The statistical difference between the groups was determined by Mann–Whitney nonparametric test.
Table 1.
Primer sequences used for qPCR
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| Armcx1 | ATCAAAACGCCATCCGTGAG | ACCATGGTATCGTCACACACT |
| Rhot1 | TCCACCTCAAGCCTTCACTT | GTGTCACGTGCAAAGCAGTT |
| Rhot2 | AAGTGGATCCCGCTTGTGAA | TCCTCAAGTGCTTGGCTGAA |
| MfN1 | TTTGGCTGATCGGTGTACCA | CGAACACAGCTTGTGGCAAT |
| MfN2 | AGCAGGAATTGTCTGGGACAT | TGTCCAACCAGCCAGCTTTA |
| Syntaphillin | ATTGACAAGGACAAGGGGCT | TCACTCAGCTTGGTGTAGGT |
| Kif5b | AACCACCAGAAGAAACGAGC | CTGTTTGCAGCGTTTCACCA |
| TfR1 | AAACTGGCTGAAACGGAGGAGACA | GCTGCTTGATGGTGTCAGCAAACT |
| Dmt1 | TGAATCGGGCGAATAAGCAGGA | TCAGCAAAGACGGCAACGACAA |
| Fpn | AACCAGAGTCACTGTCATCAGCCA | TCGGCCCAAGTCAGTGAAGGTAAA |
| Cp | ACATTGCTGCTGAGGAGGTCATCT | TGTTCCTCATCAGGGCCTCTTTGT |
| GAPDH | GGAGAAACCTGCCAAGTATGA | TCCTCAGTGTAGCCCAAGA |
Immunofluorescence
Animals were transcardially perfused with 4% PFA in 0.1 m PB, pH 7.4. The segment of the sciatic nerve containing the lesion site and distal to the lesion was taken out and prepared for cryostat sectioning. Longitudinal sections (10 μm) were incubated in 0.3% Triton-X 100, 5% normal goat/donkey serum (Jackson ImmunoResearch Laboratories), and 2% ovalbumin in PBS for 4-5 h to block nonspecific binding. The tissue sections were then incubated overnight at 4°C with the specific primary antibodies: mouse anti-S100β (for Schwann cells; 1:100; Thermo Fisher Scientific), rabbit anti-ferritin (1:100; Sigma-Aldrich), rabbit anti-GAP-43 (for growth-associated protein 43; 1:500; Chemicon), and rabbit anti-mitochondrial ferritin (1:100; Abcam). Primary antibodies were visualized by incubation with fluorescent-conjugated secondary antibodies: anti-rabbit AlexaFluor-568, anti-rabbit AlexaFluor-488, and streptavidin AlexaFluor-488 (1:500; Invitrogen) for 2 h at room temperature. For labeling F-actin at Schmidt-Lanterman incisures and nodes of Ranvier, rhodamine-conjugated phalloidin (1:500) was used. Sections were coverslipped with ProLong gold antifade mounting media containing DAPI (Invitrogen) and viewed using a confocal laser scanning microscope (FluoView FV1000, Olympus).
For staining of cell cultures, cultures of DRG neurons on coverslips were fixed in 4% PFA, permeabilized in acetic acid-alcohol at 4°C for 20 min, and then incubated with monoclonal mouse SMI32 and SMI312 (both 1:200; Covance) in MEM/HEPES containing 5% normal goat serum and 2% ovalbumin for 30 min. Schwann cell cultures on coverslips were first incubated with IgG-purified polyclonal rabbit anti-Fpn antibody (1:200, Alpha Diagnostics) or rabbit anti-Cp antibody (1:400, Sigma-Aldrich) for 30 min at room temperature, then permeabilized and incubated with monoclonal mouse anti-S100β antibody (1:200, Sigma-Aldrich). Primary antibodies were visualized with rhodamine-conjugated goat anti-mouse IgG (1:400, Jackson ImmunoResearch Laboratories) and Alexa-488-conjugated goat anti-rabbit IgG (1:400; Invitrogen) for 30 min at room temperature. Cells were coverslipped with DAPI-containing mounting medium (Vector Laboratories) and viewed with a Carl Zeiss Axioskop 2 plus microscope.
Quantification of ferritin+ Schwann cells and mitochondrial ferritin
Sciatic nerve tissue sections stained for S100β and ferritin were viewed using confocal laser scanning microscope (FluoView FV1000, Olympus), and micrographs were taken with the FV10-ASW 3.0 software (Olympus). For comparing between the groups, the same setting of brightness and contrast was applied in all the immunostained images. Using ImageJ software, double-labeled ferritin+/S100β+ cells were quantified in both the groups (n = 4/group), and the percentage of double-labeled cells in the total S100β+ population was calculated. Data are mean ± SEM. For statistical significance, Mann–Whitney nonparametric test was used to compare between two groups.
For mitochondrial ferritin quantification, micrographs were taken with the same software as mentioned above. Using ImageJ software, the images were binarized and in the internodal region along the axon outlined, and the percentage of mitochondrial ferritin staining in the occupied area was measured. Results are presented as mean ± SEM, and the Mann–Whitney nonparametric test was used for statistical significance.
Teased nerve preparation and quantification of mitochondria
Teased fiber preparation and immunofluorescence technique were performed as described previously (Catenaccio and Court, 2018). Briefly, uninjured and 7 d postcrush injured sciatic nerves were harvested from animals, drop-fixed in 4% PFA for 1 h, followed by removal of the perineurium/epineurium in a 35 mm culture dish containing 1× PBS. Then, nerve segments were cut into 0.5-cm-long pieces, placed on top of a TESPA-coated slide in a drop of 1× PBS, and the bundle of fibers was teased apart using a pair of fine forceps (Dumont #5, #91150-20; Fine Science Tools). For immunolabeling, teased nerves on slides were incubated in prechilled (−20°C) acetone for 20 min, then incubated in blocking solution (0.1% Triton X-100 and 5% cold fish gelatin in PBS) for 1 h, followed by overnight incubation (4°C) with rabbit anti-porin polyclonal antibody diluted in blocking solution (1:100, ab15895, Abcam). Then, slides were incubated for 2 h at room temperature in blocking solution containing Alexa-488-conjugated goat anti-rabbit IgG (1:400; Invitrogen) and rhodamine-conjugated phalloidin (1:500), which strongly labels F-actin at Schmidt-Lanterman incisures and nodes of Ranvier. In axons, the F-actin cytoskeleton is found in the cortical axoplasm, while in Schwann cells, the cytoplasm contains an F-actin meshwork in regions of noncompact myelin, namely, the nodes of Ranvier and Schmidt-Lanterman incisures (Kun et al., 2012). Slides were coverslipped with DAPI-containing mounting medium (Vector Laboratories) and viewed in a confocal laser scanning microscope (FluoView FV1000, Olympus). Micrographs were taken as single-plane images with the FV10-ASW 3.0 software (Olympus). Mitochondrial distribution and quantification along individual teased fibers were analyzed by placing a predefined ROI (15 µm2) at Schmidt-Lanterman incisures, nodes of Ranvier, and internodes, using the ImageJ software. Images were binarized, and mitochondrial counts were evaluated as percentage of labeling (occupied area) within the ROI at the different fiber regions along the axons. Schmidt-Lanterman incisures and nodes of Ranvier in teased fibers were distinguishable from each other based on their characteristic appearance in rhodamine-conjugated phalloidin labeling, which stains F-actin in Schmidt-Lanterman incisures as funnel-shaped profiles and nodes of Ranvier as narrowed constricted rings, as previously described (Catenaccio and Court, 2018). For the uninjured-teased nerve measurements, we analyzed ∼300 Schmidt-Lanterman, ∼200 internodes, and ∼29 nodes of Ranvier from 4 WT and 4 Cp KO mice. For injured teased nerves, we measured ∼180 Schmidt-Lanterman, ∼120 internodes, and ∼30 nodes of Ranvier from 3 WT and 3 Cp KO animals. Data are mean ± SEM. Unpaired t test was used to determine statistical significance. As we were interested in comparing differences in the localization of mitochondria to the nodes of Ranvier and Schmidt-Lanterman incisures in uninjured and injured nerves, we selected fibers that have Schmidt-Lanterman incisures and nodes based on the rhodamine-phalloidin stain.
Epon embedding and quantification of axon diameter
Animals were perfused with 4% PFA and 2% glutaraldehyde in sodium cacodylate buffer (0.1 m; pH 7.4). Uninjured WT and Cp KO sciatic nerve segments were postfixed in 1% osmium tetroxide plus 0.8% potassium ferrocyanide for 2 h. Afterward, nerve segments were dehydrated and embedded in Epon resin. Morphometric analysis was used in five sampled areas from the semithin cross sections (1 µm) of the nerve, stained with 1% toluidine blue, at 1000× magnification. Measurements of axon diameter were calculated using the software ImageJ from 3 WT and 3 Cp KO mice, and data are presented as the mean ± SEM. Unpaired t test was used to determine statistical significance.
Statistical analysis
All experimental data were analyzed using GraphPad Prism 5.0 and represented as mean ± SEM. Statistical comparisons between groups were determined by one-way or two-way ANOVA, followed by Tukey or Bonferroni post hoc test as indicated in each of the subsections above. Unpaired t test was used to determine statistical significance for mitochondrial quantification in teased nerve fibers. The Mann–Whitney nonparametric test was used to assess qPCR results, for quantifying mitochondrial ferritin, and double-label ferritin+/S100β+ cells. A value of p ≤ 0.05 was considered significant. All analyses were conducted blinded to the experimental or control groups.
Results
Iron is required for initiation and elongation of neurite growth
We first assessed whether iron plays a role in the initiation of neurite outgrowth using DRG cultures. The objective of these experiments was to assess the role of iron in neurite growth, not to dissect out the role of Schwann cells. Postnatal mouse DRG neurons were plated onto laminin-coated coverslips in the presence of NGF and allowed to attach to the substrate. Five hours after plating, DRG neurons had attached to the substrate but had not initiated neurite outgrowth yet (Fig. 1Ai,Aii). To render cells iron-deficient, the iron chelator SIH was added to the cultures 5 h after plating and left overnight. SIH has lipophilic properties, with a high affinity for iron (Klimtová et al., 2003; Cheah et al., 2006), and has been effectively used to chelate iron in vitro and in vivo (Rathore et al., 2008; Jeong et al., 2009). Immunofluorescence labeling for neurofilament revealed impaired neurite outgrowth in the presence of the iron chelator (Fig. 1Ai,Aiv) compared with vehicle-treated controls (Fig. 1Ai,Aiii). Sholl analysis showed a drastic reduction in neurite outgrowth (Fig. 1Avi; Control vs Vehicle: p < 0.001; Control vs SIH: p = 0.03; Vehicle vs SIH: p < 0.001; n = 3 per group, F(2,6) = 64.04, p < 0.001, one-way ANOVA with Tukey's multiple comparison test) and branching complexity (Fig. 1Avii; Control vs Vehicle: p = 0.004; Vehicle vs SIH: p = 0.006; n = 3 per group, F(2,6) = 17.52, p = 0.003, one-way ANOVA with Tukey's multiple comparison test) in the iron-chelated group compared with vehicle-treated controls, indicating that iron is required for the initiation of neurite outgrowth from DRG neurons.
Figure 1.
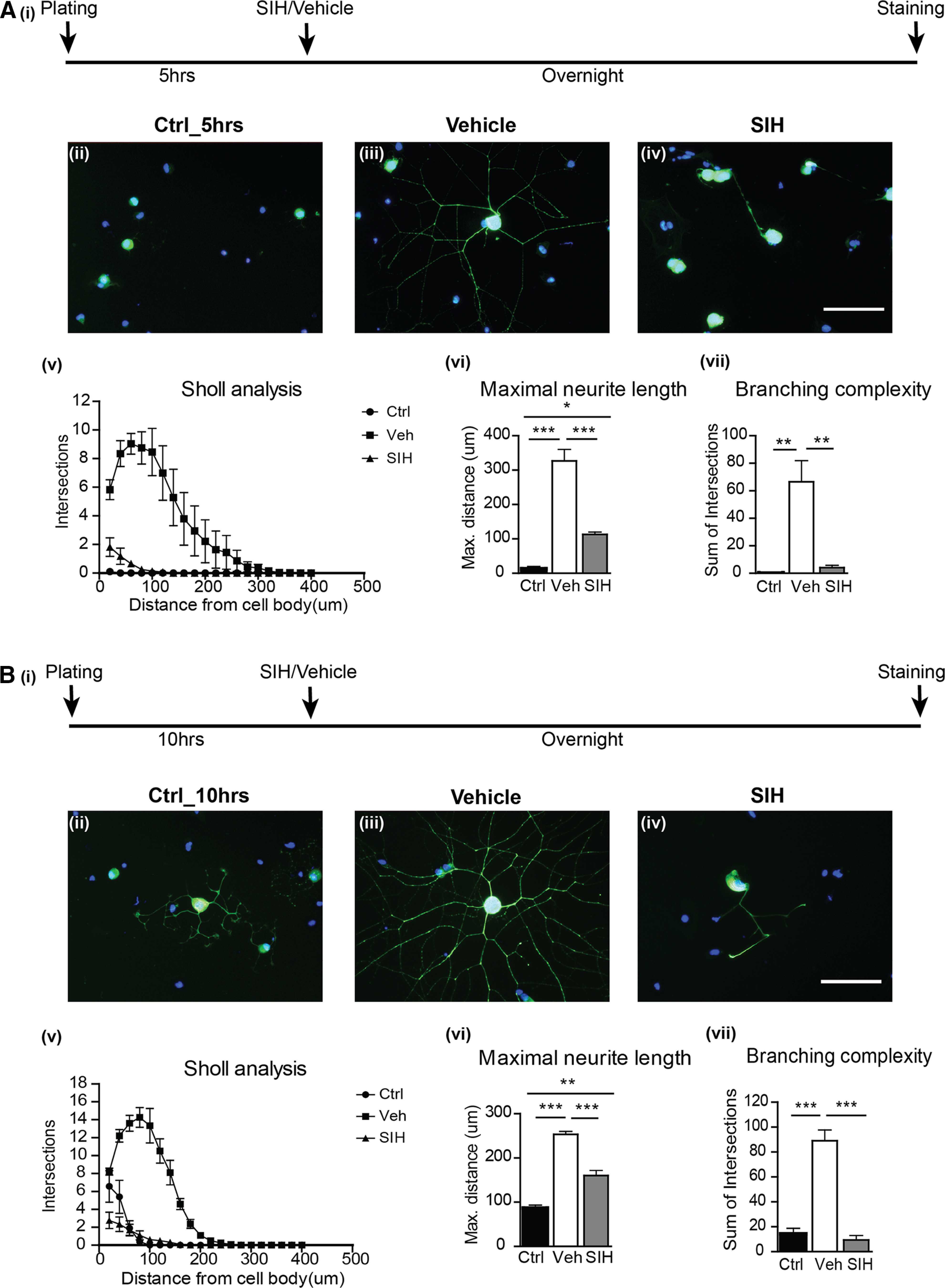
A, Initiation of neurite outgrowth is impaired in DRG neurons under iron-deficient conditions. Postnatal (P1-P2) mouse DRG neurons were plated onto laminin-coated coverslips for 5 h to allow cells to attach, and then treated overnight with the iron chelator SIH or vehicle. Ai, The experimental timeline. Aii, Immunofluorescence labeling with the anti-neurofilament antibodies (SMI32/SMI312) showed no neurite outgrowth in the first 5 h after plating. After overnight incubation, vehicle-treated control cultures demonstrated robust neurite growth (Aiii). In contrast, the initiation of neurite outgrowth was markedly impaired in the SIH-treated group (Aiv). Scale bar: Aiv, 100 μm. Sholl analysis revealed a significant reduction in neurite branching complexity in the SIH-treated group versus the vehicle-treated group (Av,Avii) as well as in the maximal neurite length (Av,Avi). Data are mean ± SEM (n = 3). *p < 0.05. **p < 0.01. ***p < 0.001. B, Extension of neurite outgrowth is impaired in DRG neurons under iron-deficient conditions. DRG neuronal cultures were plated onto laminin-coated coverslips for 10 h to allow neurite outgrowth to start, and then treated overnight with the iron chelator SIH or vehicle. Bi, The experimental timeline. Bii, Immunofluorescence labeling with the anti-neurofilament antibodies (SMI32/SMI312) showed that neurite outgrowth had already started in the first 10 h after plating. Biii, After overnight incubation, vehicle-treated control cultures demonstrated extensive neurite growth. Biv, In contrast, SIH-treated cultures showed marked impairment of neurite extension. Scale bar: Biv, 100 μm. Sholl analysis revealed a significant reduction in neurite branching complexity (Bv,Bvii) and maximal neurite length (Bv,Bvi) in the SIH-treated group versus the vehicle-treated group. Data are mean ± SEM (n = 3). **p < 0.01. ***p < 0.001.
To assess whether iron is also required for the elongation of neurites once growth has already been initiated, DRG neurons were plated for 10 h before the iron chelator was added (Fig. 1Bi). At this point, DRG neurons had already started to extend short neurites (Fig. 1Bii). Cells were then treated with the iron chelator or vehicle overnight. Immunofluorescence labeling for neurofilaments revealed a marked reduction in the elongation of neurites in the iron chelator-treated group (Fig. 1Biv) compared with vehicle-treated controls (Fig. 1Biii). Sholl analysis showed significant reduction in the elongation of neurites (Fig. 1Bvi; Control vs Vehicle: p < 0.001; Control vs SIH: p = 0.002; Vehicle vs SIH: p < 0.001; n = 3 per group, F(2,6) = 94.20, p < 0.001, one-way ANOVA with Tukey's multiple comparison test) and branching complexity (Fig. 1Bvii; Control vs Vehicle: p < 0.001; Vehicle vs SIH: p < 0.001; n = 3 per group, F(2,6) = 56.13, p < 0.001, one-way ANOVA with Tukey's multiple comparison test), suggesting that iron is also required for the elongation of neurites from DRG neurons.
Neurite growth requires ATP, which is generated in mitochondria (Vaarmann et al., 2016; Sheng, 2017) with the help of respiratory chain enzymes that are rich in Fe-S clusters (Complex I, II, and III) and Fe-containing heme (Complex IV) required for electron transfer. ATP is also produced in the tricarboxylic acid cycle, which contains enzymes harboring Fe-S clusters (aconitase and SDH). Iron is therefore essential to maintain proper mitochondrial function. In peripheral nerves, such as sciatic nerves, which have very long axons, an important question is how and where mitochondria in these axons acquire iron. It is possible that axonal mitochondria acquire iron while they travel (motile) or reside for periods of time (stable) along axons. Because of their close association with axons, Schwann cells are ideally located to supply axons with iron. We therefore assessed whether Schwann cells possess iron export proteins required to release iron. The only known iron exporter is Fpn, which needs to partner with a ferroxidase, such as Cp to export iron (Jeong and David, 2003; Levi and Taveggia, 2014).
Schwann cells express proteins required for iron efflux
Double immunofluorescence labeling of Schwann cells isolated from postnatal mouse sciatic nerve revealed that Schwann cells express Cp (Fig. 2i-iii) and Fpn (Fig. 2iv-vi) on the cell surface. Interestingly, immunofluorescence labeling of teased nerve preparations of the adult mouse sciatic nerve showed that Fpn appears to be localized to Schwann cells at the nodes of Ranvier and Schmidt-Lanterman incisures (Fig. 3), suggesting that these regions might be sites of iron efflux/export from Schwann cells in vivo. In addition, the iron uptake protein transferrin receptor 1 (TfR1) is expressed in the cell body of Schwann cells (Fig. 4iv-vi), and in axons in regions near the nodes of Ranvier and Schmidt-Lanterman incisures (Fig. 4i-iii). These findings suggest regional differences in iron uptake/efflux in Schwann cells–iron uptake is likely to occur in the region of the cell body via TfR1 (Fig. 4iv-vi), and efflux via Fpn in regions near the nodes of Ranvier and Schmidt-Lanterman incisures. To assess the possibility that Schwann cells efflux or export iron via Fpn/Cp, we used Cp KO mice. We have previously shown that astrocytes, which normally express Cp are unable to efflux iron in Cp KO mice (Jeong and David, 2003), resulting in iron accumulation in these cells (Patel et al., 2002; Jeong and David, 2006). The iron efflux transporter Fpn appears to be unable to function on its own but requires the help of Cp to export iron from astrocytes (Jeong and David, 2003). We therefore investigated whether impairment of iron efflux proteins, as seen in Cp KO mice, also leads to iron accumulation in Schwann cells in the sciatic nerve of these mice.
Figure 2.

Cell surface expression of Fpn and Cp in Schwann cells. Schwann cells were isolated from postnatal mouse (P5-P7 days old) sciatic nerve. Double immunofluorescence labeling with anti-Cp (i) or anti-Fpn (iv) and the Schwann cell marker anti-S100β (ii,v) antibodies demonstrated that Schwann cells express Cp and Fpn. iii, vi, Merged images, including DAPI staining, to visualize cell nuclei. Scale bar, 50 μm.
Figure 3.

Localization of the iron exporter Fpn in normal axons. Teased nerve fiber preparations of uninjured sciatic nerve from adult WT mice stained for (i) Fpn (green) and (ii) rhodamine-conjugated phalloidin (red), which strongly labeled F-actin at Schmidt-Lanterman incisures and nodes of Ranvier. The node of Ranvier (arrow) and the Schmidt-Lanterman incisure (arrowhead) can be distinguished by their characteristic morphology (constricted ring and funnel shape, respectively). Staining for Fpn was mainly localized to these two sites (iii; merged image), being slightly more intense at paranodal loop regions of the nodes of Ranvier than at the Schmidt-Lanterman incisures. Scale bar, 15 µm.
Figure 4.

Localization of the iron importer TfR1 in normal axons. Teased nerve fiber preparations of uninjured sciatic nerve from adult WT mice stained for TfR1 (green) and rhodamine-conjugated phalloidin (red) to label F-actin localized at nodes of Ranvier (arrow) and Schmidt-Lanterman incisures (arrowhead). TfR1 staining (i) was mainly localized along the axon (not the surrounding Schwann cell or myelin), with increased staining at the region near the node of Ranvier (arrows) (ii,iii). In some preparations, in which the Schwann cell bodies were visible (iv-vi), TfR1 staining was prominent at the cell body (arrows) as detected by the presence of DAPI-labeled Schwann cell nucleus (v) and the TfR1 labeling, which appeared to wrap around the axon (v,vi). Scale bars: iii, 15 µm; vi, 10 µm.
Iron export is disrupted in Schwann cells in Cp KO mice
As expected, iron histochemistry revealed iron deposition in cells that have a Schwann cell morphology in the sciatic nerve of Cp KO mice (Fig. 5A). To confirm that these cells are Schwann cells, we conducted double immunofluorescence labeling for ferritin and the Schwann cell marker S100β (Fig. 5Bi and ii). Ferritin is a very good surrogate marker for iron, as each ferritin molecule can sequester up to 4500 atoms of iron. Therefore, an increase in ferritin expression is a good indicator of iron accumulation in cells. We found that 63.1 ± 4.2% of S100β+ Schwann cells were double-labeled for ferritin in the sciatic nerve of Cp KO mice compared with 4 ± 1.1% double-labeled in WT mice (Fig. 5Biii; WT vs Cp KO: p < 0.0001, n = 4 per group, Mann–Whitney test). These results indicate that Schwann cells are unable to efflux/export iron effectively in Cp KO mice. In addition, sciatic nerves from Cp KO mice also showed a 2- to 3-fold increase in expression of the iron import proteins TfR1 (Fig. 5Ci; WT vs Cp KO: p = 0.05, n = 3 or 4 per group, Mann–Whitney test) and divalent metal transporter 1 (Dmt1) (Fig. 5Cii; WT vs Cp KO: p = 0.028, n = 3 or 4 per group, Mann–Whitney test) compared with sciatic nerves from WT mice. Therefore, the lack of iron export because of the absence of Cp, along with the increase in iron importers, is likely to account for the increased iron accumulation seen in Schwann cells in Cp KO mice. We next examined whether impaired iron efflux from Schwann cells in Cp KO mice has an effect on the localization of mitochondria along sciatic nerve axons.
Figure 5.

Iron accumulation in Schwann cells in Cp KO mice. Ai-Aiv, Iron histochemistry shows iron accumulation in cells that have a Schwann cell morphology in longitudinal sections of the sciatic nerve of Cp KO mice. Bi, Double immunofluorescence labeling of WT nerve for S100β (green) and ferrtin (red) and nuclear stain DAPI. Very few S100β+ Schwann cells express ferritin (arrowhead) in WT nerve. Single S100β+ cells (arrows). Bii, Cp KO nerve shows many S100β+ Schwann cells double-labeled for ferritin (arrowheads). Single S100β+ cells (arrows). Biii, Significant increase in the percentage of S100β+ Schwann cells double-labeled with ferritin in Cp KO nerves. n = 4 per group. ****p < 0.0001. Scale bars: Aiv, 20 μm; Bi, 15 μm. Ci, Cii, qPCR analysis of uninjured nerves from WT and Cp KO mice shows statistically significant increase in mRNA expression of iron importers TfR1 and Dmt1. n = 3 or 4 per group, each with 3 pooled nerves. *p = 0.03.
Selective distribution of mitochondria along axons
If iron released by Schwann cells is taken up by axons for utilization in axonal mitochondria, we might expect such transfer to occur at sites where Schwann cell cytoplasm comes in close proximity with axonal membranes, such as the paranodal loops at the nodes of Ranvier and Schmidt-Lanterman incisures. Mitochondrial staining in teased sciatic nerve preparations of WT mice (8–10 weeks old) showed ∼2.5-fold more mitochondria at the nodes of Ranvier than at Schmidt-Lanterman incisures and internodal regions (Fig. 6i; Table 2), possibly reflecting the greater energy demands at the nodes of Ranvier associated with the conduction of action potentials and other metabolic needs. However, iron export proteins are expressed at the nodes of Ranvier and Schmidt-Lanterman incisures, suggesting that both regions may be sites of iron transfer, with the nodes of Ranvier being more important under normal conditions (Fig. 3). We found a similarly high number of mitochondria at the nodes of Ranvier in Cp KO mice, in which iron release from Schwann cells is impaired, compared with WT mice (Fig. 6vi; Table 2). Surprisingly, the number of mitochondria at the Schmidt-Lanterman incisures was twice as high in these mice compared with WT mice (Fig. 6v; WT vs Cp KO: p < 0.0001, unpaired t test). The selective increase in mitochondria at Schmidt-Lanterman incisures and not at the nodes of Ranvier in Cp KO mice suggests that, under conditions of decreased iron efflux from Schwann cells (i.e., in Cp KO mice), the Schmidt-Lanterman incisures may serve as an additional site for iron acquisition by axonal mitochondria. Increase in mitochondria is also seen in the internodal regions in Cp KO mice (Fig. 6vii; Table 2; WT vs Cp KO: p < 0.0001, unpaired t test). Interestingly, mRNA expression of Miro1 (WT: 1 ± 0.2; Cp KO: 3.34 ± 0.5; WT vs Cp KO: p = 0.028, n = 3 or 4 per group, Mann–Whitney test), Kif5b (WT: 1 ± 0.2; Cp KO: 2.2 ± 0.15; WT vs Cp KO: p = 0.05, n = 3 or 4 per group, Mann–Whitney test), and Armcx1 (WT: 1 ± 0.2; Cp KO: 3.9 ± 0.75; WT vs Cp KO: p =0.028, n = 3 or 4 per group, Mann–Whitney test), three molecules that enhance mitochondrial trafficking, was increased in sciatic nerves of Cp KO mice.
Figure 6.
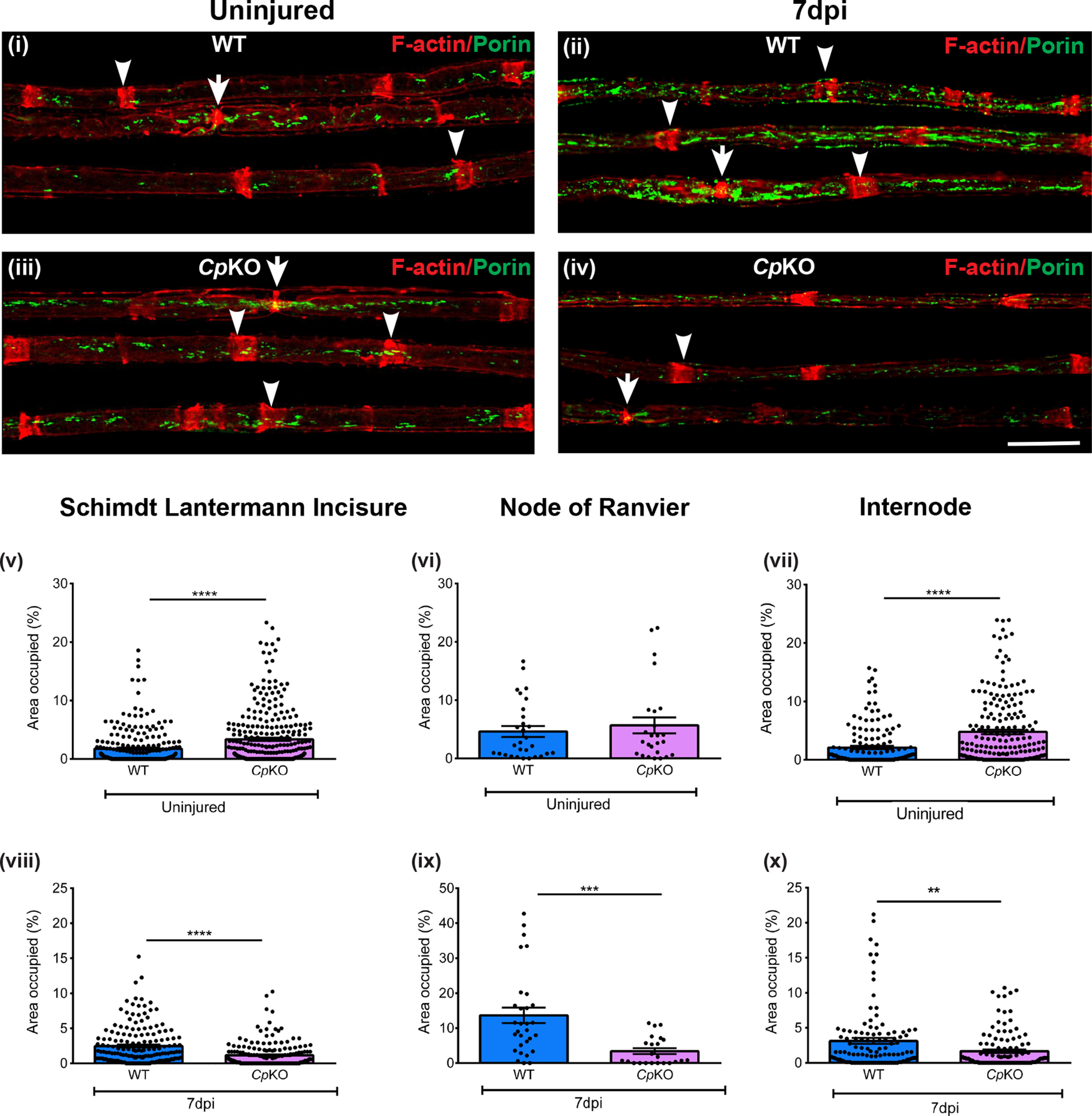
Changes in mitochondria in axons in Cp KO mice and effects of crush injury. Teased nerve fiber preparation of uninjured (i) and injured (ii) nerve fibers of WT mice. Note the marked increase in mitochondria in regenerating nerves of WT mice 7 d after crush injury (ii). Bottom panels, Uninjured (iii) and injured (iv) nerves of Cp KO mice. Note the marked reduction in mitochondria in regenerating nerves of Cp KO mice (iv) compared with WT mice (ii) 7 d after crush injury. Green represents mitochondrial profiles (porin staining). Red represents Schmidt-Lanterman incisures (arrowheads) and nodes of Ranvier (arrows) (rhodamine-phalloidin staining). v-x, Graphs represent area (%) occupied by mitochondria at the Schimdt-Lanterman incisures, nodes of Ranvier, and internode in teased axons from uninjured (v-vii) and injured (viii-x) sciatic nerves of WT and Cp KO mice. Uninjured teased fibers: Schmidt-Lanterman incisures, WT, n = 242, Cp KO mice, n = 301; nodes of Ranvier, WT mice, n = 25, Cp KO mice, n = 29; internode, WT, n = 158, Cp KO mice, n = 205 from 4 different animals of each genotype. Injured teased fibers: Schmidt-Lanterman incisures, WT mice, n = 174, Cp KO mice, n = 182; nodes of Ranvier, WT mice, n = 23, Cp KO mice, n = 30; internode, WT mice, n = 113, Cp KO mice, n = 120 from 3 different animals of each genotype. **p < 0.01. ***p < 0.001. ****p <0.0001. Scale bar, 20 µm.
Table 2.
Mitochondrial quantification in distinct axonal regions from WT and Cp KO mice
| Schmidt-Lanterman incisures (%) | Nodes of Ranvier (%) | Internode (%) | |
|---|---|---|---|
| WT uninjured | 1.7 ± 0.2 | 4.6 ± 0.9 | 2 ± 0.2 |
| Cp KO uninjured | 3.4 ± 0.2 | 5.7 ± 1.3 | 4.8 ± 0.2 |
| WT injured | 2.5 ± 0.2 | 13.6 ± 2.2 | 3.1 ± 0.4 |
| Cp KO injured | 1.1 ± 0.1 | 3.4 ± 0.8 | 1.7 ± 0.2 |
Numbers indicate the percent area occupied by mitochondria at different regions along individual axons from teased nerve fibers of uninjured and 7 d postcrush nerves from WT and Cp KO mice. Data are mean ± SEM.
Reduction in mitochondrial ferritin in axons in Cp KO mice
If axonal mitochondria get iron from Schwann cells, we would expect to see a reduction in mitochondrial iron in axonal mitochondria in Cp KO mice. As ferritin is a good surrogate marker for iron, we assessed the expression of mitochondrial ferritin in sciatic nerve axons of WT and Cp KO mice. Mitochondrial ferritin differs from cytosolic ferritin (Arosio and Levi, 2010), and antibodies are available that selectively bind only to mitochondrial ferritin (Jeong et al., 2009; Zarruk et al., 2015). We stained longitudinal cryostat sections of sciatic nerves from adult WT and Cp KO mice with rabbit anti-mitochondrial ferritin antibody and with rhodamine-phalloidin to visualize the nodes of Ranvier and Schmidt-Lanterman incisures. The latter staining allowed us to outline the internodal regions in axons and assess the extent of mitochondrial ferritin staining within these regions using ImageJ. Staining for mitochondrial ferritin was abundant in WT axons but sparce in Cp KO animals, that is, 3-fold greater in WT than Cp KO nerves (Fig. 7; WT vs Cp KO: p = 0.0043, n = 4 per group, Mann–Whitney test), suggesting iron depletion in axonal mitochondria in Cp KO mice. F-actin staining was used as a guide to select only the inner portion of the internodal region to avoid including Schwann cells in the counts. These results show that, in the absence of iron efflux from Schwann cells in Cp KO mice, there is less iron in axons, and provide further support for the hypothesis that Schwann cells provide iron to axons via the Cp-Fpn mechanism.
Figure 7.

Reduction of mitochondrial ferritin in Cp KO mice. i, Staining for mitochondrial ferritin in uninjured WT sciatic nerve. ii, In comparison, there is marked reduction in mitochondrial ferritin staining in Cp KO nerves. iii, Quantification of the area in internodal regions occupied by mitochondrial ferritin staining. Note the significant reduction in Cp KO nerves. n = 4 per group. **p = 0.0043. Scale bar, 30 µm.
Impaired axonal regeneration and functional recovery after sciatic nerve crush injury in Cp KO mice
Axonal regeneration and motor recovery following sciatic nerve crush injury were assessed in adult Cp KO and WT mice. The rate of motor recovery after sciatic nerve crush was determined using the SFI, defined as the ratio of the length of the foot (tip of big toe to the tip of the heel) versus the width of the foot (the distance between the first and fifth toes). Following sciatic nerve injury, the SFI dropped to a value of ∼80, indicating a marked loss of motor control of the injured hind paw in WT and Cp KO mice (Fig. 8A). The recovery of the motor disability toward a normal SFI value closer to 0 reflects axonal regeneration and reinnervation of the target muscles in the foot. Cp KO mice exhibited significantly lower SFI values at 10 and 17 d after injury compared with WT mice, indicating impaired motor recovery (Fig. 8A; day 10 WT vs Cp KO: p = 0.02; day 17 WT vs Cp KO: p = 0.006; n = 15-18 per group, F(2,93) = 51.60, p < 0.0001, two-way ANOVA with Bonferroni's multiple comparison test).
Figure 8.

Motor recovery and axonal regeneration after sciatic nerve injury are impaired in Cp KO mice. A, Recovery of motor function was determined by analyzing the SFI, conducted in Cp KO and WT mice at 10, 17, and 28 d following sciatic nerve crush injury. Note the significant reduction in the SFI values in Cp KO mice at 10 and 17 d after nerve crush as compared to WT mice. The value at 28 d was also lower in Cp KO mice but did not reach significance. These data indicate that functional recovery after nerve crush injury, as assessed by SFI, was reduced in Cp KO versus WT mice. Data are mean ± SEM (n = 18 WT; 15 Cp KO). *p = 0.02. **p = 0.006. B, Quantification of the numbers of GAP43+ axons 3, 4, and 5 mm distal to the lesion site revealed a significant decrease of regenerating axons in Cp KO versus WT mice. Data are mean ± SEM (n = 3). *p < 0.05. **p < 0.01. C, Immunofluorescence analysis of longitudinal sections of crushed sciatic nerves using anti-GAP43 antibody, a marker for regenerating axons, showed less regenerating axons at 4 mm distal to the lesion in Cp KO (Ci,Cii) versus WT (Ciii,Civ) mice 4 d after injury. Cii, Civ, Images with DAPI staining to label cell nuclei. Scale bar, 200 μm.
We further investigated directly whether axonal regeneration after sciatic nerve injury is impaired in Cp KO mice by immunofluorescence labeling for GAP-43, a protein expressed in growing axons (Skene, 1989; Kawasaki et al., 2018). Four days after nerve crush, GAP-43 immunoreactivity was reduced distal to the lesion site in Cp KO mice compared with WT mice (Fig. 8B,C). Quantification of the numbers of GAP-43+/S100β– axonal profiles showed significant reduction of regenerating axons 3 and 4 mm distal to the crush site in Cp KO versus WT mice (Fig. 8B; 3 mm from lesion site WT vs Cp KO: p = 0.0089; 4 mm from lesion site WT vs Cp KO: p = 0.035; n = 3 per group, F(2,12) = 13.94, p = 0.0007, two-way ANOVA with Bonferroni's multiple comparison test), indicating impaired axonal regeneration in Cp KO mice. We next assessed localization of axonal mitochondria after sciatic nerve crush injury in WT and Cp KO mice.
Alterations in the distribution of axonal mitochondria after nerve injury in WT and Cp KO mice
Axonal regeneration is associated with increased energy demands (Han et al., 2016; Vaarmann et al., 2016). To determine whether a potential lack of axonal iron supply in Cp KO mice has an effect on the localization of axonal mitochondria in a situation of high-energy demand, we examined mitochondrial distribution in WT and Cp KO mice after sciatic nerve crush injury. As we were interested in comparing differences in the localization of mitochondria to the nodes of Ranvier and Schmidt-Lanterman incisures in uninjured and injured nerves, we selected fibers that have Schmidt-Lanterman incisures and nodes of Ranvier based on the rhodamine-phalloidin stain. The presence of myelinated fibers 7 d after sciatic nerve crush injury has also been reported (Stettner et al., 2018). The diameter of injured regenerating nerve fibers is significantly smaller compared with uninjured nerves (uninjured WT: 5.3 ± 0.1; injured WT: 3.2 ± 0.1; uninjured WT vs injured WT: p < 0.0001; uninjured Cp KO: 5.2 ± 0.1; injured Cp KO: 3.2 ± 0.08; uninjured Cp KO vs injured Cp KO: p < 0.0001; n = 3 or 4 animals per group, F(3,289) = 114.1, p < 0.0001, one-way ANOVA with Tukey's multiple comparison test). Consistent with earlier reports, nerve injury in WT mice resulted in an increase of mitochondria in all three regions analyzed 7 d after injury (Fig. 6i,ii; Table 2), with an increase of ∼50% at the Schmidt-Lanterman incisures and ∼3-fold at the nodes of Ranvier (Table 2). In contrast, in Cp KO mice, the number of mitochondria was markedly reduced in all three regions 7 d after injury compared with uninjured nerves (Fig. 6iii,iv; Table 2). Comparing mitochondrial numbers after injury in both genotypes revealed a 4-fold reduction at the nodes of Ranvier and a 2.2-fold reduction at the Schmidt-Lanterman incisures in Cp KO versus WT mice (Fig. 6viii; WT vs Cp KO: p < 0.0001, unpaired t test; Fig. 6ix; WT vs Cp KO: p = 0.0002, unpaired t test; Fig. 6x; WT vs Cp KO: p = 0.0012, unpaired t test). Thus, while nerves of Cp KO mice appear to compensate for the lack of iron export from Schwann cells by increasing the number of mitochondria under normal conditions, they seem to be unable to cope with the marked increase in energy demands required for regeneration following injury. This striking reduction in mitochondria in injured Cp KO mice could be because of the lack of iron needed for mitochondrial motility or biogenesis.
Expression of molecules involved in mitochondrial fusion and motilitiy
To understand the mechanisms that may underlie the drastic reduction in mitochondria in injured axons in Cp KO mice, we assessed the mRNA expression of molecules involved in mitochondrial biogenesis and motility. Interestingly, the mRNA expression of mitofusin 1 and 2 (Mfn1 and Mfn2), which are outer mitochondrial membrane proteins involved in mitochondrial fusion, was significantly lower in injured sciatic nerves of Cp KO mice compared with WT mice (Fig. 9i; 14 dpi WT vs Cp KO: p = 0.01; Fig. 9ii; 7 dpi WT vs Cp KO: p = 0.03; 14 dpi WT vs Cp KO: p = 0.01, n = 3 or 4 per group, Mann–Whitney test). In nerves of WT mice, Mfn2 mRNA was increased ∼2-fold at 7 and 14 d after injury, and Mfn1 was increased ∼2-fold at 14 d after injury (Fig. 9i; Naive WT vs 14 dpi WT: p = 0.03; Fig. 9ii; Naive WT vs 7 dpi WT: p = 0.03; Naive WT vs 14 dpi WT: p = 0.03, n = 3 or 4 per group, Mann–Whitney test). In contrast, the expression of Mfn1 and Mfn2 remained unchanged from baseline levels at both time points after injury in Cp KO mice (Fig. 9i,ii). In addition to its role in mitochondrial fusion, Mfn2 has also been shown to be involved in mitochondrial motility by partnering with the outer mitochondrial membrane proteins Miro1 and Miro2, linking mitochondria to kinesin. The mRNA expression of Miro1 and Miro2 was also significantly lower in injured nerves from Cp KO mice compared with WT mice, with Miro1 being reduced at 7 and 14 d after injury and Miro2 at 14 d after injury (Fig. 9iii; 7 dpi WT vs Cp KO: p = 0.05; 14 dpi WT vs Cp KO: p = 0.03; Fig. 9iv; 14 dpi WT vs Cp KO: p = 0.03, n = 3 or 4 per group, Mann–Whitney test). Miro1 expression was transiently elevated at 1 d after injury in both genotypes (Fig. 9iii; Naive WT vs 1 dpi WT: p = 0.04; Naive Cp KO vs 1 dpi Cp KO: p = 0.01; Fig. 9iv; Naive Cp KO vs 1 dpi Cp KO: p = 0.05; n = 3 or 4 per group, Mann–Whitney test).). These data suggest that mitochondrial motility after sciatic nerve injury might be impaired in Cp KO mice. Interestingly, mRNA expression of Armadillo Repeat Containing, X-Linked 1 (Armcx1), a mitochondrial protein that increases mitochondrial transport and axonal regeneration (Cartoni et al., 2016), was elevated over 2-fold in injured versus uninjured nerves of WT mice (Fig. 9v; Naive WT vs 14 dpi WT: p = 0.05; Naive Cp KO vs 14 dpi Cp KO: p = 0.03; n = 3 or 4 per group, Mann–Whitney test). In contrast, it was significantly reduced below control levels in injured nerves of Cp KO mice at 14 d after injury (Fig. 9v; 14 dpi WT vs Cp KO: p = 0.03; n = 3 or 4 per group, Mann–Whitney test) in line with the impaired axonal regeneration seen in Cp KO mice. The mRNA expression of two other molecules involved in mitochondrial motility, namely, syntaphilin (snph) and kinesin-1, was also altered following sciatic nerve injury. SNPH is an axonal mitochondria-specific protein that anchors and immobilizes mitochondria by bridging it to microtubules (Chen and Sheng, 2013). Knockdown of snph has previously been shown to increase mitochondrial motility (Kang et al., 2008; Chen and Sheng, 2013) and axonal regeneration (Zhou et al., 2016). We found that mRNA expression of snph in injured nerves of WT mice increased ∼5-fold at 1 d after injury and transiently returned to control uninjured levels at 7 d after injury, before increasing to 2-fold at 14 d after injury (Fig. 9vi; Naive WT vs 1 dpi WT: p = 0.05; Naive WT vs 14 dpi WT: p = 0.03; n = 3 or 4 per group, Mann–Whitney test). In contrast, snph levels in injured nerves of Cp KO mice were lower than in injured nerves of WT mice at 1 and 14 d, reaching significance at 14 d after injury (Fig. 9vi; 14 dpi WT vs Cp KO: p = 0.01; n = 3 or 4 per group, Mann–Whitney test). The lower levels of snph expression after nerve injury in Cp KO compared with WT mice might reflect an attempt to increase mitochondrial motility to compensate for the drastic reduction in mitochondrial numbers in injured Cp KO nerves (Table 2). The mRNA expression of Kif5B (kinesin-1), a protein required for fast anterograde axonal transport of mitochondria (Pilling et al., 2006; Sheng, 2017), was also significantly lower in injured nerves of Cp KO compared with WT mice (Fig. 9vii; 1 dpi WT vs Cp KO: p = 0.03; 14 dpi WT vs Cp KO: p = 0.01; n = 3 or 4 per group, Mann–Whitney test), potentially contributing to the lower overall number of mitochondria in injured nerves of Cp KO mice.
Figure 9.

Expression of genes involved in mitochondrial fusion and transport. qPCR analysis of mitochondrial genes at 1, 7, and 14 d after sciatic nerve crush injury in WT and Cp KO mice. Values were compared with uninjured nerves indicated by the horizontal dotted line at 1. Expression of Mfn1 (i) and Mfn2 (ii) was increased in nerves of WT mice 14 d after injury, whereas no change in expression was detected in injured nerves of Cp KO mice. The same was noted for the mitochondrial transport genes Rhot1 (iii) and Rhot2 (iv), Armcx1 (v), Snph (vi), and Kif5b (vii) at 14 d after injury (except Armcx1, which was significantly reduced in Cp KO mice). At earlier time points, increases were detected in Mfn2, Rhot1, snph, and Kif5b in nerves of WT mice. Only Rhot2 was significantly higher in Cp KO than WT mice at 1 d after injury. Data are mean ± SEM (n = 4 per group, each with 3 pooled nerves). *p ≤ 0.05 versus naive. #p ≤ 0.05 versus WT.
Discussion
Iron plays an important role in maintaining energy metabolism in neurons. As part of Fe-S clusters and heme, iron is required for the function of enzymes of the mitochondrial respiratory chain and the tricarboxylic acid cycle, and thus for the production of ATP. While Fe-S clusters are found in Complex I (NADH dehydrogenase), Complex II (succinate dehydrogenase), Complex III, and cytochrome bc1 complex, iron-containing heme is present in Complex IV (cytochrome c oxidase) of the respiratory chain (Rouault and Tong, 2005; Stehling and Lill, 2013). These iron-containing enzymes are involved in electron transfer reactions that ultimately lead to generation of ATP via oxidative phosphorylation. The ATP produced by axonal mitochondria is needed for various energy-demanding processes, such as membrane repolarization, bidirectional transport of organelles, and maintenance of synaptic function, including recycling of synaptic vesicles (Hollenbeck and Saxton, 2005; Misgeld and Schwarz, 2017; Sheng, 2017; Mandal and Drerup, 2019). Additionally, iron is essential in Schwann cells for myelination, as it is a cofactor for enzymes involved in cholesterol and lipid synthesis (Connor and Menzies, 1996). After axonal injury, ATP generated in axonal mitochondria is also required for growth cone motility and axonal regeneration (Han et al., 2016; Mandal and Drerup, 2019). Despite the especially high-energy demands of axons, it is unknown how axonal mitochondria acquire iron.
We first confirmed the role of iron in neurite growth by using iron chelation in DRG cell culture, which completely blocked neurite growth. Unlike glial cells or other non-neuronal cells that have small cell bodies in which mitochondria do not need to be mobilized far, some neurons have very long axons which require mitochondria to travel for long distances to meet the energy demands at various regions along the axon. For example, axons within peripheral nerves innervating the limbs, such as sciatic nerves, can extend several feet long in humans, requiring mitochondria to be mobilized to axonal sites far away from the cell body. However, where and how mitochondria in these long-projecting axons acquire iron for their metabolic needs have not been studied.
Because of their close proximity to axons, Schwann cells are the most likely source of iron for axonal mitochondria located far away from the cell body. Previous work has shown that Schwann cells express a GPI-anchored cell surface form of the ferroxidase Cp (Salzer et al., 1998), similar to astrocytes in the CNS (Patel and David, 1997; Patel et al., 2000). We confirm the expression and membrane localization of Cp in Schwann cells. We also demonstrate here that Schwann cells express the iron efflux transporter Fpn, and thus contain the proteins needed to efflux iron. Fpn has previously been shown to partner with Cp to efflux iron from astrocytes (Jeong and David, 2003). Astrocytes that lack Cp are unable to efflux iron, resulting in iron accumulation in astrocytes in the CNS of Cp KO mice (Patel et al., 2002; Jeong and David, 2003, 2006) and in patients with mutations in the Cp gene (Gitlin, 1998). Our findings show that Cp-deficient Schwann cells accumulate iron in Cp KO mice, supporting the role of Cp in iron efflux from Schwann cells. Excess iron in Schwann cells was detected by iron histochemistry and increased ferritin labeling. Intracellular iron levels regulate ferritin synthesis via binding of iron response proteins to iron response element, which are stem loop structures in the 5′ untranslated region of ferritin mRNA. We hypothesized that transfer of iron from Schwann cells to axons is likely to occur at regions where Schwann cell cytoplasm comes into close contact with axons, namely, the paranodal loops at the nodes of Ranvier and the Schmidt-Lanterman incisures. In support of this hypothesis, we detected expression of Fpn at the nodes of Ranvier and Schmidt-Lanterman incisures in teased nerve preparations. Additionally, the iron uptake protein TfR1 was localized at the Schwann cell body, and to axonal membrane at the nodes of Ranvier and Schmidt-Lanterman incisures, likely mediating iron uptake into Schwann cells and into axons, respectively. Neurons and Schwann cells possess iron importers (TfR1) and exporters (Fpn), but the localization of these varies in different regions (along the axon or different parts of Schwann cells) depending on whether there is a need for iron uptake or release. The aggregation of organelles along axons generally indicates a reduced rate of organelle transit. Interestingly, the numbers of axonal mitochondria in the sciatic nerve of WT mice were 2- to 3-fold greater at the nodes of Ranvier than at the Schmidt-Lanterman incisures or internodal regions, in line with the high metabolic demands of conduction of action potentials at the nodes of Ranvier. Interestingly, in Cp KO mice, in which iron efflux from Schwann cells is impaired, mitochondrial numbers at the Schmidt-Lanterman incisures were increased by 2-fold, while numbers at the nodes of Ranvier were similar compared with WT mice. This selective increase of mitochondrial numbers at the Schmidt-Lanterman incisures and not at the nodes of Ranvier suggests that this is specific to Schmidt-Lanterman incisures and not a general accumulation. Although mitochondrial numbers at the Schmidt-Lanterman incisures were higher in Cp KO than WT mice, the values were nevertheless ∼60% lower than at the nodes of Ranvier, suggesting that, under conditions of reduced iron availability, Schmidt-Lanterman incisures could serve as additional sites of iron uptake to complement possibly reduced uptake at the nodes of Ranvier. We suggest that, in Cp KO mice, mitochondria might linger longer at the Schmidt-Lanterman incisures to acquire iron via other mechanisms that may be less efficient, resulting in the buildup of mitochondria at these sites.
Long projecting axons develop and are maintained in adult Cp KO mice, and assessment of axonal diameter showed only a small shift from large-diameter to small-diameter myelinated axons in Cp KO mice (Fig. 10). This suggests that there are likely to be other mechanisms in addition to the Cp-Fpn efflux mechanism that provide iron to axonal mitochondria. There is evidence that bone marrow-derived macrophages can release ferritin-bound iron via the multivesicular body (MVB)-exosome pathway (Truman-Rosentsvit et al., 2018). MVBs are also found in neurons, being 50 times more abundant in cell bodies than in axons (Altick et al., 2009). Nevertheless, MVBs are detected in axons and may be transported in anterograde or retrograde transport (Altick et al., 2009; Von Bartheld and Altick, 2011; Ye et al., 2018). There is also in vitro and in vivo EM evidence that ferritin can be taken up by spinal ganglion neurons and become localized in MVBs (Rosenbluth and Wissig, 1964). It is therefore possible that ferritin-bound iron packaged in MVBs traveling anterogradely from the cell body or retrogradely from the synaptic targets may provide iron for axonal mitochondria. In addition, Schwann cells or fibroblasts may also provide iron via ferritin-bound iron released in MVB-exosome transport. Nevertheless, our data also suggest that the Cp-Fpn pathway also contribute iron from Schwann cells to axonal mitochondria.
Figure 10.
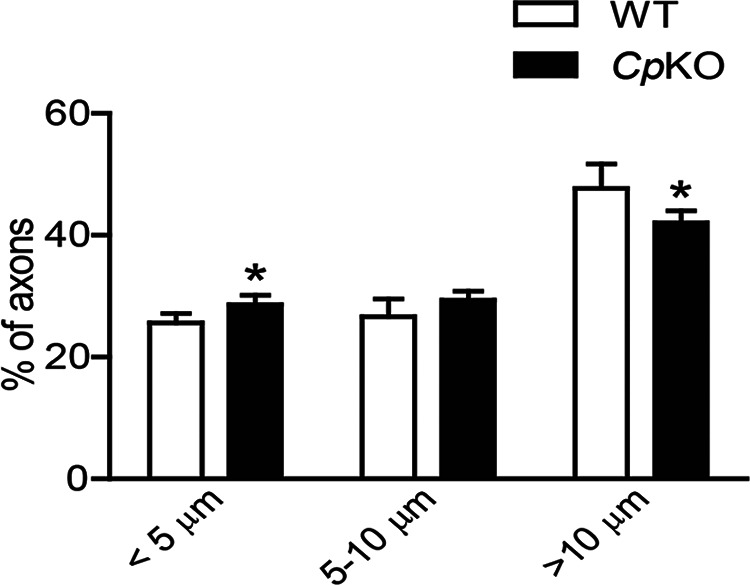
Quantification of myelinated fiber diameters was done on Epon-embedded semithin sections stained with toluidine blue. The percent distribution is shown in three ranges. Note the slight shift from large- to small-diameter axons in Cp KO mice compared with WT mice. n = 3 per group. *p < 0.05.
Considering the high metabolic demand associated with axonal regeneration (Mandal and Drerup, 2019), it is not surprising that sciatic nerve crush injury in WT mice resulted in an >2-fold increase in the overall number of mitochondria at 7 d after injury. The role of iron in this energy-demanding process was supported by our neurite outgrowth assay in cell culture, demonstrating impaired initiation of neurite outgrowth and extension of neurites in the presence of an iron chelator. In vivo, axonal regeneration following sciatic nerve injury was also found to be impaired in Cp KO mice compared with WT mice. Interestingly, there was a 3-fold reduction in the number of mitochondria in injured regenerating nerves of Cp KO versus WT mice. Thus, the reduction of mitochondria seen in injured axons of Cp KO mice may result from potential axonal iron deficiency because of impaired iron release from Schwann cells in these mice. This is supported by our findings of reduced mitochondrial ferritin (a surrogate marker for iron) in axons of Cp KO nerves compared with WT nerves. We hypothesized that the potential axonal iron deficiency in Cp KO mice may lead to reduced mitochondrial biogenesis and motility (Rensvold et al., 2016; Bastian et al., 2019), two energy- and thus iron-dependent processes, and subsequently, a decrease in the number of axonal mitochondria. This is supported by recent evidence that iron deficiency reduces mitochondrial motility in hippocampal dendrites, affecting anterograde transport in these unmyelinated structures (Bastian et al., 2019). Interestingly, sciatic nerve injury in WT mice was associated with increased expression of mitofusins, particularly Mfn2. Since mitofusins are mitochondrial outer membrane GTPases essential for mitochondrial fusion (Filadi et al., 2018), the increased Mfn expression might indicate greater mitochondrial fusion, a process associated with increased ATP generation (Filadi et al., 2018). Importantly, there was no increase in the expression of Mfn1 and Mfn2 in injured sciatic nerves of Cp KO mice, suggesting that mitochondrial fusion and ATP production after injury might be lower in these mice compared with WT mice. Such reduced ATP production could possibly explain the impaired regeneration seen in Cp KO mice. Mitofusins also regulate mitochondrial dynamics, with Mfn2 being required for mitochondrial motility in axons. A study using cultured neurons from Mfn2 KO mice demonstrated that lack of Mfn2 results in slower anterograde and retrograde transport of mitochondria, along with reduced fusion (Misko et al., 2010). The reduced expression of Mfn2 in injured sciatic nerves of Cp KO mice may therefore contribute to the overall reduction in axonal mitochondria in the injured nerves of Cp KO mice. Mfn1 and Mfn2 also interact with the Rho GTPases Miro1 and Miro2, which form a complex with other molecules that connect to kinesin for mitochondrial motility (Misko et al., 2010). We found a significant increase in the expression of Miro1 and Miro2 in injured nerves of WT mice, suggesting increased mitochondrial transport. However, no increase was detected in injured nerves of Cp KO mice, which correlates with the observed reduction in mitochondrial numbers and impaired axonal regeneration. We also found a significant reduction in the expression of Kif5b (kinesin-1) in injured nerves of Cp KO compared with WT mice. Considering that the kinesin-1 family is the main anterograde motor for mitochondrial transport in neurons (Sheng, 2017), reduction in Kif5b in injured nerves of Cp KO mice could contribute to the reduction in the number of mitochondria and impaired axonal regeneration seen in these mice. Armcx1 is another mitochondrial protein shown to be upregulated after nerve injury (Cartoni et al., 2016). Its overexpression has been associated with enhanced axonal mitochondrial transport and axonal regeneration (Cartoni et al., 2016). We also observed an increased expression of Armcx1 in regenerating sciatic nerves of WT mice but detected reduced expression in injured nerves of Cp KO mice, in which mitochondrial transport and axonal regeneration are impaired. It is noteworthy that previous work has shown that enhanced mitochondrial transport and ATP production promote axonal regeneration (Zhou et al., 2016). Our work points to a deficit in axonal mitochondria and reduced axonal regeneration in sciatic nerves of Cp KO mice in which iron export from Schwann cells is disrupted.
In conclusion, this study addresses an important question about how and where axonal mitochondria acquire iron needed for the activity of respiratory chain enzymes and, consequently, the production of ATP. Our findings support the hypothesis that axonal mitochondria are likely to acquire iron from Schwann cells, particularly at the paranodal loops at the nodes of Ranvier and the Schmidt-Lanterman incisures. Schwann cells express the ferroxidase Cp and the iron exporter Fpn, and therefore possess the molecular machinery to efflux iron. Iron accumulation in Schwann cells of Cp KO mice further confirmed the role of Cp in iron efflux from Schwann cells. We used these mice to assess the potential role of Schwann cells in providing axonal mitochondria with iron. The impaired axonal regeneration, the substantial changes in axonal mitochondrial numbers and its localization, as well as the reduced expression of molecules involved in mitochondrial fusion and motility in injured nerves of Cp KO mice suggest that Schwann cells may play a role in supplying axonal mitochondria with iron.
Footnotes
This work was supported by Canadian Institutes of Health Research Grant MOP-142231 to S.D. P.J. and K.S. received postdoctoral fellowship and studentship, respectively, from the MS Society of Canada. B.S.M. received CAPES/CNPq studentship award and International Society for Neurochemistry travel grant support. We thank Laura Curran and Ourania Tsatas for technical help.
The authors declare no competing financial interests.
References
- Altick AL, Baryshnikova LM, Vu TQ, von Bartheld CS (2009) Quantitative analysis of multivesicular bodies (MVBs) in the hypoglossal nerve: evidence that neurotrophic factors do not use MVBs for retrograde axonal transport. J Comp Neurol 514:641–657. 10.1002/cne.22047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arosio P, Levi S (2010) Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 1800:783–792. 10.1016/j.bbagen.2010.02.005 [DOI] [PubMed] [Google Scholar]
- Bastian TW, von Hohenberg WC, Georgieff MK, Lanier LM (2019) Chronic energy depletion due to iron deficiency impairs dendritic mitochondrial motility during hippocampal neuron development. J Neurosci 39:802–813. 10.1523/JNEUROSCI.1504-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold CH, Fabricius C, Rydmark M, Andersen B (1993) Axoplasmic organelles at nodes of Ranvier: I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol 22:925–940. 10.1007/BF01218351 [DOI] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, He Z (2016) The mammalian-specific protein Armcx1 regulates mitochondrial transport during axon regeneration. Neuron 92:1294–1307. 10.1016/j.neuron.2016.10.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catenaccio A, Court FA (2018) Teased fiber preparation of myelinated nerve fibers from peripheral nerves for vital dye staining and immunofluorescence analysis. Methods Mol Biol 1739:329–337. 10.1007/978-1-4939-7649-2_21 [DOI] [PubMed] [Google Scholar]
- Cheah JH, Kim SF, Hester LD, Clancy KW, Patterson SE 3rd, Papadopoulos V, Snyder SH (2006) NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 51:431–440. 10.1016/j.neuron.2006.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Sheng ZH (2013) Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol 202:351–364. 10.1083/jcb.201302040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Menzies SL (1996) Relationship of iron to oligodendrocytes and myelination. Glia 17:83–93. [DOI] [PubMed] [Google Scholar]
- de Medinaceli L, Freed WJ, Wyatt RJ (1982) An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol 77:634–643. 10.1016/0014-4886(82)90234-5 [DOI] [PubMed] [Google Scholar]
- Filadi R, Pendin D, Pizzo P (2018) Mitofusin 2: from functions to disease. Cell Death Dis 9:330. 10.1038/s41419-017-0023-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Zhou Q, Wu D, Chen L (2021) Mitochondrial iron metabolism and its role in diseases. Clin Chim Acta 513:6–12. 10.1016/j.cca.2020.12.005 [DOI] [PubMed] [Google Scholar]
- Gitlin JD (1998) Aceruloplasminemia. Pediatr Res 44:271–276. 10.1203/00006450-199809000-00001 [DOI] [PubMed] [Google Scholar]
- Han SM, Baig HS, Hammarlund M (2016) Mitochondria localize to injured axons to support regeneration. Neuron 92:1308–1323. 10.1016/j.neuron.2016.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–297. 10.1016/S0092-8674(04)00343-5 [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM (2005) The axonal transport of mitochondria. J Cell Sci 118:5411–5419. 10.1242/jcs.02745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inserra MM, Bloch DA, Terris DJ (1998) Functional indices for sciatic, peroneal, and posterior tibial nerve lesions in the mouse. Microsurgery 18:119–124. [DOI] [PubMed] [Google Scholar]
- Jeong SY, David S (2003) Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J Biol Chem 278:27144–27148. 10.1074/jbc.M301988200 [DOI] [PubMed] [Google Scholar]
- Jeong SY, David S (2006) Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J Neurosci 26:9810–9819. 10.1523/JNEUROSCI.2922-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S (2009) Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J Neurosci 29:610–619. 10.1523/JNEUROSCI.5443-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH (2008) Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132:137–148. 10.1016/j.cell.2007.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki A, Okada M, Tamada A, Okuda S, Nozumi M, Ito Y, Kobayashi D, Yamasaki T, Yokoyama R, Shibata T, Nishina H, Yoshida Y, Fujii Y, Takeuchi K, Igarashi M (2018) Growth cone phosphoproteomics reveals that GAP-43 phosphorylated by JNK is a marker of axon growth and regeneration. iScience 4:190–203. 10.1016/j.isci.2018.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimtová I, Šimůnek T, Mazurová Y, Kaplanová J, Štěrba M, Hrdina R, Geršl V, Adamcová M, Poňka P (2003) A study of potential toxic effects after repeated 10-week administration of a new iron chelator–salicylaldehyde isonicotinoyl hydrazone (SIH) to rabbits. Acta Med (Hradec Kralove) 46:163–170. 10.14712/18059694.2019.27 [DOI] [PubMed] [Google Scholar]
- Kun A, Canclini L, Rosso G, Bresque M, Romeo C, Hanusz A, Cal K, Calliari A, Sotelo Silveira J, Sotelo JR (2012) F-actin distribution at nodes of Ranvier and Schmidt-Lanterman incisures in mammalian sciatic nerves. Cytoskeleton (Hoboken) 69:486–495. 10.1002/cm.21011 [DOI] [PubMed] [Google Scholar]
- Levi S, Taveggia C (2014) Iron homeostasis in peripheral nervous system, still a black box? Antioxid Redox Signal 21:634–648. 10.1089/ars.2013.5813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Zhai XY, Ohsato K, Futamata H, Shimada O, Atsumi S (2004) Mitochondrial accumulation in the distal part of the initial segment of chicken spinal motoneurons. Brain Res 1026:235–243. 10.1016/j.brainres.2004.08.016 [DOI] [PubMed] [Google Scholar]
- Magrane J, Cortez C, Gan WB, Manfredi G (2014) Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet 23:1413–1424. 10.1093/hmg/ddt528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Drerup CM (2019) Axonal transport and mitochondrial function in neurons. Front Cell Neurosci 13:373. 10.3389/fncel.2019.00373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Schwarz TL (2017) Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96:651–666. 10.1016/j.neuron.2017.09.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH (2010) Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30:4232–4240. 10.1523/JNEUROSCI.6248-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel BN, David S (1997) A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem 272:20185–20190. 10.1074/jbc.272.32.20185 [DOI] [PubMed] [Google Scholar]
- Patel BN, Dunn RJ, David S (2000) Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem 275:4305–4310. 10.1074/jbc.275.6.4305 [DOI] [PubMed] [Google Scholar]
- Patel BN, Dunn RJ, Jeong SY, Zhu Q, Julien JP, David S (2002) Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J Neurosci 22:6578–6586. 10.1523/JNEUROSCI.22-15-06578.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM (2006) Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell 17:2057–2068. 10.1091/mbc.e05-06-0526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathore KI, Kerr BJ, Redensek A, Lopez-Vales R, Jeong SY, Ponka P, David S (2008) Ceruloplasmin protects injured spinal cord from iron-mediated oxidative damage. J Neurosci 28:12736–12747. 10.1523/JNEUROSCI.3649-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensvold JW, Krautkramer KA, Dowell JA, Denu JM, Pagliarini DJ (2016) Iron deprivation induces transcriptional regulation of mitochondrial biogenesis. J Biol Chem 291:20827–20837. 10.1074/jbc.M116.727701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DR, Ponka P (1998) Pyridoxal isonicotinoyl hydrazone and its analogs: potential orally effective iron-chelating agents for the treatment of iron overload disease. J Lab Clin Med 131:306–315. 10.1016/S0022-2143(98)90180-9 [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Wissig SL (1964) The distribution of exogenous ferritin in toad spinal ganglia and the mechanism of its uptake by neurons. J Cell Biol 23:307–325. 10.1083/jcb.23.2.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault TA, Tong WH (2005) Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol 6:345–351. 10.1038/nrm1620 [DOI] [PubMed] [Google Scholar]
- Salzer JL, Lovejoy L, Linder MC, Rosen C (1998) Ran-2, a glial lineage marker, is a GPI-anchored form of ceruloplasmin. J Neurosci Res 54:147–157. [DOI] [PubMed] [Google Scholar]
- Sheng ZH (2017) The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol 27:403–416. 10.1016/j.tcb.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl DA (1953) Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat 87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Skene JH (1989) Axonal growth-associated proteins. Annu Rev Neurosci 12:127–156. 10.1146/annurev.ne.12.030189.001015 [DOI] [PubMed] [Google Scholar]
- Spillane M, Ketschek A, Merianda TT, Twiss JL, Gallo G (2013) Mitochondria coordinate sites of axon branching through localized intra-axonal protein synthesis. Cell Rep 5:1564–1575. 10.1016/j.celrep.2013.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehling O, Lill R (2013) The role of mitochondria in cellular iron-sulfur protein biogenesis: mechanisms, connected processes, and diseases. Cold Spring Harb Perspect Biol 5:a011312. 10.1101/cshperspect.a011312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettner M, Zenker J, Klingler F, Szepanowski F, Hartung HP, Mausberg AK, Kleinschnitz C, Chrast R, Kieseier BC (2018) The role of peripheral myelin protein 2 in remyelination. Cell Mol Neurobiol 38:487–496. 10.1007/s10571-017-0494-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman-Rosentsvit M, Berenbaum D, Spektor L, Cohen LA, Belizowsky-Moshe S, Lifshitz L, Ma J, Li W, Kesselman E, Abutbul-Ionita I, Danino D, Gutierrez L, Li H, Li K, Lou H, Regoni M, Poli M, Glaser F, Rouault TA, Meyron-Holtz EG (2018) Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 131:342–352. 10.1182/blood-2017-02-768580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaarmann A, Mandel M, Zeb A, Wareski P, Liiv J, Kuum M, Antsov E, Liiv M, Cagalinec M, Choubey V, Kaasik A (2016) Mitochondrial biogenesis is required for axonal growth. Development 143:1981–1992. 10.1242/dev.128926 [DOI] [PubMed] [Google Scholar]
- Von Bartheld CS, Altick AL (2011) Multivesicular bodies in neurons: distribution, protein content, and trafficking functions. Prog Neurobiol 93:313–340. 10.1016/j.pneurobio.2011.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye M, Lehigh KM, Ginty DD (2018) Multivesicular bodies mediate long-range retrograde NGF-TrkA signaling. Elife 7:e33012. 10.7554/eLife.33012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarruk JG, Berard JL, Passos dos Santos R, Kroner A, Lee J, Arosio P, David S (2015) Expression of iron homeostasis proteins in the spinal cord in experimental autoimmune encephalomyelitis and their implications for iron accumulation. Neurobiol Dis 81:93–107. 10.1016/j.nbd.2015.02.001 [DOI] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH (2016) Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol 214:103–119. 10.1083/jcb.201605101 [DOI] [PMC free article] [PubMed] [Google Scholar]