

Abstract
Comorbid anxiety and depressive symptoms in chronic pain are a common health problem, but the underlying mechanisms remain unclear. Previously, we have demonstrated that sensitization of the CeA neurons via decreased GABAergic inhibition contributes to anxiety-like behaviors in neuropathic pain rats. In this study, by using male Sprague Dawley rats, we reported that the CeA plays a key role in processing both sensory and negative emotional-affective components of neuropathic pain. Bilateral electrolytic lesions of CeA, but not lateral/basolateral nucleus of the amygdala (LA/BLA), abrogated both pain hypersensitivity and aversive and depressive symptoms of neuropathic rats induced by spinal nerve ligation (SNL). Moreover, SNL rats showed structural and functional neuroplasticity manifested as reduced dendritic spines on the CeA neurons and enhanced LTD at the LA/BLA-CeA synapse. Disruption of GluA2-containing AMPAR trafficking and endocytosis from synapses using synthetic peptides, either pep2-EVKI or Tat-GluA2(3Y), restored the enhanced LTD at the LA/BLA-CeA synapse, and alleviated the mechanical allodynia and comorbid aversive and depressive symptoms in neuropathic rats, indicating that the endocytosis of GluA2-containing AMPARs from synapses is probably involved in the LTD at the LA/BLA-CeA synapse and the comorbid aversive and depressive symptoms in neuropathic pain in SNL-operated rats. These data provide a novel mechanism for elucidating comorbid aversive and depressive symptoms in neuropathic pain and highlight that structural and functional neuroplasticity in the amygdala may be important as a promising therapeutic target for comorbid negative emotional-affective disorders in chronic pain.
SIGNIFICANCE STATEMENT Several studies have demonstrated the high comorbidity of negative affective disorders in patients with chronic pain. Understanding the affective aspects related to chronic pain may facilitate the development of novel therapies for more effective management. Here, we unravel that the CeA plays a key role in processing both sensory and negative emotional-affective components of neuropathic pain, and LTD at the amygdaloid LA/BLA-CeA synapse mediated by GluA2-containing AMPAR endocytosis underlies the comorbid aversive and depressive symptoms in neuropathic pain. This study provides a novel mechanism for elucidating comorbid aversive and depressive symptoms in neuropathic pain and highlights that structural and functional neuroplasticity in the amygdala may be important as a promising therapeutic target for comorbid negative emotional-affective disorders in chronic pain.
Keywords: AMPAR, amygdala, LTD, neuropathic pain, neuroplasticity, pain-related negative emotion
Introduction
Pain associates both sensory and emotional aversive components, and often leads to anxiety and depression when it becomes chronic. Comorbid anxiety and depressive symptoms in chronic pain are a common health problem, but the underlying mechanisms remain unclear. Previously, we have demonstrated that sensitization of neurons in the CeA contributes to anxiety-like behaviors in neuropathic pain rats (Jiang et al., 2014). In this study, we sought to investigate the neuronal mechanisms underlying the comorbid aversive and depressive symptoms in neuropathic pain. Because persistent pain is frequently associated with psychological and emotional dysfunction (Baliki and Apkarian, 2015), understanding the neuronal and molecular mechanisms involved in the affective component of pain may have considerable clinical importance for the treatment of chronic pain.
Several studies have shown that the persistent pain arising from peripheral nerve injury induces maladaptive changes in the corticolimbic brain areas (Taylor, 2018), in which the amygdala plays a key role in emotional processing, neuropsychiatric disorders, and the emotional-affective dimension of pain (Neugebauer, 2015). In particular, the lateral amygdala (LA) and basolateral amygdala (BLA) and the central amygdala have been implicated in the affective components and consequences of neuropathic pain (Yalcin et al., 2014; Neugebauer et al., 2020). Using in vivo neural calcium imaging in freely behaving mice, Corder et al. (2019) have identified in the BLA a critical neural ensemble target that mediates aversive chronic pain unpleasantness. In a separate study, the CeA has been identified as a pivotal relay station in a specific serotonergic pathway from the dorsal raphe nucleus to the lateral habenula in governing comorbid depressive symptoms in chronic pain (Zhou et al., 2019). The LA/BLA is conceptualized as the main input of the amygdala receiving polymodal sensory, including nociceptive inputs from the thalamus and the cortex (Neugebauer, 2015). Through associative processing, the LA/BLA attaches emotional-affective content to the sensory inputs and transmits highly integrated information to the CeA, the main output station of the amygdala, for modulating the emotional components of pain (Veinante et al., 2013).
Neuroplasticity in the amygdala has emerged as an important contributor to emotional-affective aspects of pain (Thompson and Neugebauer, 2017). Enhanced neurotransmission in the BLA-CeA pathway and increased excitability of CeA neurons have been shown in rodent models of inflammatory pain and neuropathic pain (Neugebauer and Li, 2003; Neugebauer et al., 2003; Li et al., 2017). Rats with spared nerve injury display signs of depressive-like behavior accompanied by increased amygdala volume, which is associated with an increased generation of new neurons in the CeA as well as in the BLA (Goncalves et al., 2008). These neuroplastic changes might contribute to the development of depressive-like symptoms frequently present in prolonged pain syndromes (Thompson and Neugebauer, 2017). LTP and LTD, the two major forms of synaptic plasticity which are thought to be the cellular basis for learning and memory (Abraham et al., 2019), are observed in most brain pathologies, including chronic pain and affective disorders (Doan et al., 2015; Zhuo, 2019). Importance of AMPA-type glutamate receptors (AMPARs) has emerged notably in LTP and LTD (Diering and Huganir, 2018). AMPARs are homotetramers or heterotetramers assembled from GluA1-A4 subunits, and the vast majority are GluA1/2 and GluA2/3 heteromers (Traynelis et al., 2010). Phosphorylation of either GluA1 serine 831 (GluA1Ser831) or 845 (GluA1Ser845) residues regulates GluA1-containing, Ca2+-permeable AMPAR (CP-AMPAR) insertion into the postsynaptic membrane and facilitates LTP induction, whereas phosphorylation of GluA2 serine 880 (GluA2Ser880) promotes GluA2-containing, Ca2+-impermeable AMPAR (CI-AMPAR) endocytosis and leads to LTD (Diering and Huganir, 2018; Park, 2018).
From these lines of evidence, we hypothesized that the AMPAR-mediated long-term synaptic plasticity in the amygdala may be involved in the negative affective components of chronic pain. We uncovered that the AMPAR-mediated LTD in the LA/BLA-CeA pathway, resulting from the endocytosis of GluA2-containing AMPARs involved in the comorbid aversive and depressive symptoms in rats with neuropathic pain.
Materials and Methods
Chemicals, reagents, and antibodies
Pentobarbital sodium (P3761), D(-)-2-amino-5-phospho-nopentanoic acid (D-AP5, A8054), bicuculline (B7686), and phenethylamine (128945) were purchased from Sigma-Aldrich. 1-Naphthyl-acetyl-spermine-trihydrochloride (NASPM, 2766) was obtained from Tocris Bioscience. TTX (AF3015) and DAPI (ab228549), respectively, were purchased from YEASEN Bio Technologies and Abcam. All these chemicals were dissolved in 0.9% sterile saline with stock concentration of 1 M, stored in −20°C, and diluted to desired concentrations just before experiments. Pep2-EVKI peptide (YNVYGIEEVKI) and nonfunctional peptide pep2-SVKE (YNVYGIESVKE) were purchased from Tocris Bioscience. The active Tat-GluA2(3Y) peptide (869YKEGYNVYG877) and the inactive control peptide Tat-GluA2(3A) (869AKEGYNVYG877) were synthesized by R&D Systems. All the peptides were dissolved in the ACSF at desired concentrations. Primary antibodies, including mouse anti-rat GluA1 (ab1504), rabbit anti-rat GluA2 (ab20673), mouse anti-pGluA2Ser880 (ab2834726), and rabbit anti-GFP (ab7260), were purchased from Abcam. Rabbit anti-pGluA1Sser831 (04-823), rabbit anti-pGluA1Ser845 (04-1073), and mouse anti-NeuN (MAB377) were purchased from Merck Millipore. Mouse anti-GFAP (GA5, 3670S) and rabbit anti-Iba1 (019-19741) were purchased from Cell Signaling Technology and Wako Chemicals, respectively. Primary antibody mouse anti-GAPDH (TA-08) and secondary antibodies, including HRP-labeled goat anti-rabbit IgG (ZDR-5306), HRP-labeled goat anti-mouse IgG (ZD-2305), and FITC-labeled goat anti-rabbit IgG (ZF-0511), were obtained from ZSGB-Bio. HRP-labeled AlexaFluor-647 goat anti-mouse IgG (ab150116) was purchased from Abcam. Unless otherwise specified, all other chemicals and reagents were purchased from Sigma-Aldrich.
Animals
Male Sprague Dawley rats weighing 150-200 g at the beginning of the experiment were provided by the Department of Experimental Animal Sciences, Peking University Health Science Center, and were randomly assigned to experimental groups. The following factors are considered for the reason to use male animals rather than female animals: many neural and behavioral functions are affected by estrogens, including mood, cognitive function, and pain (Marrocco and McEwen, 2016; McEwen and Milner, 2017). A substantial body of evidence suggests that the sex hormones, particularly estradiol and progesterone, play an important role in pain perception (Vincent and Tracey, 2010). Strong links between fluctuations in mood and sex hormones (especially the endogenous estrogen) also have been reported (Bäckström et al., 1983; Schmidt and Rubinow, 2009; Wharton et al., 2012; Hernández-Hernández et al., 2019; Frokjaer, 2020). In addition, the menstrual (or estrous) cycle has been reported to alter pain perception (Hellström and Anderberg, 2003; Ibironke and Aji, 2011; Kaur et al., 2018), depression (Stuart and Redman, 1991; Jenkins et al., 2001; D'Souza and Sadananda, 2017; Dan et al., 2019), and even BLA neuronal activity (Blume et al., 2017). Therefore, to rule out the influence of sex hormones and estrous cycle on pain and depression, only male animals were selected to be used in the present study.
The rats were housed in separated cages with free access to food and water, and maintained in a room temperature kept at 24 ± 1°C under natural light–dark cycle. All animal experimental procedures were conducted in accordance with the guidelines of the International Association for the Study of Pain (Zimmermann, 1983) and were approved by the Animal Care and Use Committee of Peking University.
Spinal nerve ligation (SNL) surgery
Under general anesthesia via intraperitoneal injection (i.p.) of pentobarbital sodium (50 mg/kg), the left lumbar 5 (L5) spinal nerves distal to the DRGs were tightly ligated with 4–0 silk sutures as described previously (Kim and Chung, 1992; Jiang et al., 2014; Ding et al., 2015). In control animals, sham surgery with identical procedure, except for ligation of the L5 spinal nerves, was received. Any rats exhibiting motor deficiency or lack of tactile allodynia were excluded from the study.
Behavioral studies
All behavioral experiments thereafter were performed in a blinded fashion; that is, the behavioral experimenters were kept blind from the groupings of the rats.
Assessment of mechanical allodynia
Mechanical allodynia, as a behavioral sign of neuropathic pain, was assessed by measuring 50% paw withdrawal threshold (PWT) as described in our previous reports (Jiang et al., 2014; Ding et al., 2015). The 50% PWT in response to a series of von Frey filaments (Stoelting) was determined by the Up and Down method as described previously (Chaplan et al., 1994). In brief, the rat was placed on a metal mesh floor covered with an inverted clear plastic cage (18 × 8 × 8 cm) and allowed a 20 min period for habituation. Eight von Frey filaments with approximately equal logarithmic incremental (0.224) bending forces were chosen (0.41, 0.70, 1.20, 2.00, 3.63, 5.50, 8.50, and 15.10 g). Each trial started with a von Frey force of 2.00 g delivered perpendicularly to the plantar surface of the left hindpaw for ∼2-3 s. An abrupt withdrawal of the foot during stimulation or immediately after the removal of the filament was recorded as a positive response. Whenever there was a positive or negative response, the next weaker or stronger filament was applied, respectively. This procedure was done until 6 stimuli after the first change in response had been observed. The 50% PWT was calculated using the following formula: 50% PWT (g) = 10[Xf + κδ], where Xf is the value of the final von Frey filament used (in log units), κ is a value measured from the pattern of positive/negative responses, and δ = 0.224, which is the average interval (in log units) between the von Frey filaments (Dixon, 1980). An allodynic rat is defined as that the 50% PWT is <4.0 g (i.e., withdrawal in response to non-noxious tactile stimulus) (Zimmermann, 2001).
Conditioned place avoidance (CPA) test
The nociceptive mechanical stimulation-induced CPA (the mCPA) was conducted as previously described (Watanabe et al., 2002) with slight modification. The place conditioning apparatus consists of compartments (30/21/30 cm, width/length/height) with distinctive visual color and floor texture and a neutral compartment (30/10/30 cm, width/length/height). One of the two equal-sized compartments was black with a smooth floor; the other was white with a textured floor. The apparatus was enclosed by a sound-attenuated and light-attenuated box under conditions of dim illumination (40 lux) and masking white noise. To reduce contamination by olfactory cues during measurement and conditioning, the apparatus was thoroughly cleaned with 10% ethanol every time it was used. The experimental process consisted of four distinct sessions, including a habituation session, a preconditioning session, a conditioning session, and the test session. On days 11 and 12 after surgery, the rats were individually placed on the neutral compartment and allowed to climb down and freely explore the three compartments for 30 min, so that they habituated themselves to the apparatus. On days 13 and 14 after surgery, the same trial was performed, and the time spent in each compartment over 15 min was measured. On days 15-20 after surgery, place conditioning was performed as follows: in the evening (6:00 P.M. to 9:00 P.M.), each rat was confined to the black compartment and given a 300 g von Frey stimulus every 30 s for 15 min; in the morning (6:00 A.M. to 9:00 A.M.), each rat was confined to the white compartment for 15 min without any stimulus. On day 21 after surgery, as the test session, the rats were individually placed on the neutral platform and allowed to freely explore the two compartments without any stimulus. The time spent in each compartment over 15 min was then measured. The CPA scores represent the time spent in the pain-paired compartment in preconditioning session minus the time spent in the same compartment in the test session, and are expressed as mean ± SEM.
The predator odor stress-induced CPA (the oCPA) was performed as described previously (Ferrero et al., 2011; Edwards et al., 2013; Schreiber et al., 2017) with slight modification. In brief, rats were exposed to predator odor (2-phenylethylamine odor [PEA]) versus no odor in two separate contexts that differed on both visual (wall patterns) and tactile (floor grids) cues, and underwent a 4 d CPA procedure. On the first day, rats were allowed 15 min to freely explore two conditioning chambers with distinct tactile and visual same as described in the pain-related CPA procedure. The time that rats spent in the two conditioning chambers was recorded as the preconditioning baseline. On the second day, rats were placed in one chamber without odor (neutral environment) for 30 min. On the third day, rats were exposed in the opposing context for 30 min with a PEA-soaked sponge (10 ml diluted in 100 ml of water) placed under the corner of the chamber (predator odor environment), or no odor for control animals. On the fourth day, rats were again allowed to explore the two conditioning chambers without PEA in a 15 min video recording. The predator oCPA scores were calculated as a difference score between preconditioning time (the first day) spent in odor-paired context and postconditioning time (the fourth day) spent in odor-paired context, and are expressed as mean ± SEM.
Sucrose preference test (SPT)
SPT was performed as previously described (Chen et al., 2018). Briefly, rats were habituated to 1% sucrose solution for 48 h, and then were deprived of water for 12 h, followed by the SPT, in which each rat had free access to two bottles that contained 1% sucrose or tap water at the same time. The position of the two bottles was changed every half an hour. The volumes of water and sucrose consumed were measured at 48 h later. The volume of the sucrose solution consumed (i.e., sucrose preference), expressed as a percentage of the total liquid ingested during a 48 h tested period, was used as a measure of the rats' sensitivity to reward.
Forced swim test (FST)
FST was conducted according to a previously described method (Detke et al., 1995). Briefly, a 60 cm high behavioral test cylinder (20 cm in diameter) was maintained at 18°C-22°C and filled with 30 cm height of water, which ensured that the rats could not support themselves by touching the bottom with their paws or tail. Rats were forced to swim for 6 min, and behaviors were monitored by video camera for subsequent analysis. The rats were considered “immobile” when they ceased struggling and remained floating motionless in the water, with only movements necessary to maintain their heads above water. The duration of immobility was recorded in the last 4 min of the 6 min testing period because most animals are very active at the beginning of the FST, and the potential effects of the treatment can be obscured during the first 2 min (Bogdanova et al., 2013).
Assessment of locomotor function
Inclined-plate test and rotarod test were used for the assessment of animal's locomotor function. For inclined-plate test, rats were placed crosswise to the long axis of an inclined plate. The initial angle of the inclined plate was 50 degrees. The angle was adjusted in 5 degree increments. The maximum angle of the plate was determined on which the rat maintained its body position for 5 s without falling (Rivlin and Tator, 1977; Chen et al., 2018). The rotarod test was performed according to the method described previously (Rozas et al., 1997). Briefly, rats first underwent a training procedure, in which the animals were placed on a rotating drum with the constant speed of 4 rpm, forcing them to walk forward to avoid falling. Training sessions were conducted 1 and 2 d before the experiments, with three trials per day, with a minimum of 30 min intertrial rest intervals. In the testing procedure, the rod was accelerated from 4-40 rpm over 5 min. Animals' latencies (in seconds) to their first fall were recorded with a cutoff time of 300 s. The rod was cleaned with 75% alcohol before the next animal was tested. Animals displayed motor dysfunction after surgery or drug administration were excluded from the study.
Electrolytic lesions of bilateral LA/BLA and CeA
After 4 d of SNL surgery, rats were anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and mounted on a stereotaxic apparatus (Stoelting). The scalp was incised, and small burr holes were made on the skull for electrode insertion. For electrolytic lesions, stainless-steel electrodes (0.3 mm in diameter) insulated with epoxy, except for 0.5 mm at the tip, were lowered bilaterally into the LA/BLA (2.5 mm anterior, 5.1 mm lateral, 8.8 mm ventral to the bregma) or the CeA (2.1 mm anterior, 4.2 mm lateral, 7.0 mm ventral to the bregma). Coordinates were adapted from the rat brain atlas of Paxinos and Watson (2014). The lesion was made by passing anodal current (DC current, 2.0 mA, 10 s for CeA, and 2.0 mA, 20 s for LA/BLA) at each site through a Lesion-Making Device (53500, Ugo Basile). The sham-operated rats were treated in the same manner, except that no current was passed. After surgery, animals were allowed to recover for at least 1 week and acclimated to daily handling before commencement of behavioral procedures.
Cannula implantation and intra-CeA microinjection
Rats were anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and placed in a stereotaxic apparatus (Stoelting) with flat-skull position for bilateral implantation of stainless-steel guide cannulae (22-gauge, Plastics One) to 0.5 mm above the CeA (2.1 mm anterior, 4.2 mm lateral, 7.0 mm ventral to the bregma). Coordinates were adapted from the rat brain atlas of Paxinos and Watson (2014). Rats were allowed a 7 d recovery before the experiments began. For the microinjection studies, rats received a bilateral infusion of 0.5 μl peptides into the CeA. Both microinjectors (28 gauge) were attached by polyethylene tubing to a 1 μl Hamilton syringe with infusions being driven by a microinjection pump (Harvard Apparatus). The microinjectors remained in position 60 s after the completion of the microinjection to ensure the entire dose had infused into the brain region. The working concentration of peptides used in the in vivo experiments was as follows: pep2-EVKI and pep2-SVKE, 10 μg/μl, Tat-GluA2(3Y) and Tat-GluA2(3A), 75 pmol.
Histologic verification
Following the completion of the experiments, rats were deeply anesthetized with overdose pentobarbital sodium (375 mg/ml in 2 ml/kg, i.p.) and underwent intracardiac perfusion with 50 ml of 0.9% saline followed by 50 ml of 10% formalin. Bains were extracted, postfixed in 10% formalin and potassium ferrocyanide solution for 7 d to be used for LA/BLA or CeA electrolytic lesion verification. After submerged in 20% sucrose, tissues were sliced into 30-μm-thick coronal sections using a cryostat, and subjected to Neutral Red staining to confirm the lesion locations in the LA/BLA and the CeA via microscopic inspection. Sites of lesions were verified histologically and plotted on standard diagrams adapted from Paxinos and Watson (2014). Only data from rats with correct lesion locations were further analyzed.
To verify the cannula placement in the intra-CeA microinjection experiments, 0.5 μl of black India ink was injected into the CeA immediately before the rat was killed at the end of each experiment, and the ink diffusion into the CeA was histologically evaluated. In brief, deeply anesthetized rats were perfused transcardially with 0.9% saline followed by 4% PFA. After postfixed in PFA at 4°C overnight, the brains were transferred to a 20% sucrose solution in saline for cryoprotection. Coronal sections of 30 μm were cut on a microtome, mounted on charged slides, and stained with cresyl violet staining solution (Beyotime Biotechnology) following the Nissl staining method. The sections were examined under a light microscope (Leica Microsystems, DMI 3000B), and determination of the location of infusion needle tips within the CeA was made according to the standardized atlas plates of Paxinos and Watson (2014) by an observer blind to drug treatment condition. Only data from rats with correct cannula placements were included in the statistical analysis.
Construction of lentiviral vectors expressing pep2-EVKI, pep2-SVKE, Tat-GluA2(3Y), or Tat-GluA2(3A)
Construction and production of recombinant lentivirus-expressing pep2-EVKI (LV-pep2-EVKI), pep2-SVKE (LV-pep2-SVKE), Tat-GluA2(3Y) (LV-Tat-GluA2(3Y)), or Tat-GluA2(3A) (LV-Tat-GluA2(3A)) linked with EGFP tag were completed by Likeli Technologies using pLVX-mCMV vector encoding EGFP. In brief, the pLVX-mCMV-EGFP vector was digested with the restriction enzyme NheI and AgeI (Likeli Technologies), and was then combined with PCR fragments encoding pep2-EVKI, pep2-SVKE, Tat-GluA2(3Y), and Tat-GluA2(3A), respectively. Next, by using Lipofectamine 2000 (Invitrogen), the total vector system composed of pLVX-mCMV-EGFP, pHelper1.0 vector, and pHelper2.0 vector was cotransfected into HEK-293T cells. Recombinant lentiviral vectors expressing pep2-EVKI, pep2-SVKE, Tat-GluA2(3Y), or Tat-GluA2(3A) linked with EGFP tag were obtained from supernatant liquid at 48 h after transfection, and the final virus titer of 4.0 × 108 TU/ml was used in this study.
Intra-CeA injections of lentiviral vectors
All surgical procedures were performed under pentobarbital sodium anesthesia (50 mg/kg, i.p.). Rats were placed in a stereotaxic frame, and two 2.5 μl injections of LV-pep2-EVKI, LV-pep2-SVKE, LV-Tat-GluA2(3Y), or LV-Tat-GluA2(3A) were made into bilateral CeA at coordinates (2.1 mm anterior, 4.2 mm lateral, 7.0 mm ventral to the bregma). A Hamilton syringe fitted with a glass capillary needle (Hamilton Gas Tight syringe 80000, 26 s/2 inch needle; Hamilton; coated in SigmaCote) was used for injection. The needle was lowered to the site, and vector injection began immediately at a rate of 0.5 μl/minute and remained in place after the injection for an additional 5 min before retraction. Histologic identification of lentiviral vectors infection was performed using immunofluorescence staining of GFP with NeuN, GFAP, and Iba-1, respectively, and counterstained with the nuclear marker DAPI at the end of experiments.
Immunofluorescence staining
The rats were anesthetized with pentobarbital sodium (60 mg/kg, i.p.) and then transcardially perfused with 150 ml of 0.01 M PBS, pH 7.4, followed by 500 ml of 4% PFA in 0.1 M PB, pH 7.4. After perfusion, the brains were postfixed in 4% PFA for 3-4 h, and were kept at 4°C overnight in a 0.1 M PB containing 30% sucrose until the brains completely sunk to the bottom of the container. Coronal 30-μm-thick serial sections of the brain were cut with a freezing microtome (CM1950, Leica Microsystems).
For immunostaining, the tissues were washed 3 times in PBS for 5 min each and blocked in 10% goat serum (in 0.1 M PBS) with 0.3% Triton-X 100 for 1 h at room temperature. Then, tissues were incubated with the respective primary antibody in PBS at 4°C overnight, which includes rabbit anti-GFP (1:100), mouse anti-NeuN (1:200), mouse anti-GFAP (1:500), and rabbit anti-Iba1 (1:500). Then, after 3 washes in PBS, tissues were incubated with appropriate secondary antibodies, including FITC-labeled goat anti-rabbit IgG (1:200) or AlexaFluor-647 goat anti-mouse IgG (1:200) at room temperature for 1 h. Sections were counterstained with the nuclear marker DAPI (100 ng/ml) carrying blue fluorescence for 10 min at room temperature, and were mounted on slides and coverslipped with glycerin-gelatin. Negative controls were obtained by repeating the same procedure with the omission of the primary or secondary antibodies. Visualization of fluorescence signal was performed by confocal microscopy at excitation wavelengths of 488 nm (green), 543 nm (red) as well as 405 nm and 647 nm (blue). At least three sections per animal and four fields per section were analyzed to establish reproducibility.
Golgi-Cox staining and Sholl analysis
Staining was performed using the method as previously described (Han et al., 2019) with slight modification. In brief, rats were anesthetized with pentobarbital sodium (50 mg/kg, i.p.) and killed by decapitation. The brain was removed quickly from the skull, rinsed with 0.1 M PBS (4°C), and immediately immersed in Golgi-Cox solution (5% potassium dichromate; 5% mercuric chloride; 5% potassium chromate, in distilled water) in the dark for 14 d at room temperature. The impregnation solution was changed every 3 d during this period. After impregnation, brains were dehydrated in 30% sucrose solution at 4°C. Using a cryostat microtome, the brain was cut into 100 μm sections and mounted into gelatin-coated slides. Mounting slides were dried at 37°C overnight, and were ready for the following staining: 1 min in distilled water, 30 min in ammonia solution (in the dark), and 1 min in distilled water (in the dark); then, 30 min in Kodak developer (in the dark) and 1 min in distilled water; next, in a graded series of ethanol (1 min in 50%, 1 min in 70%, 1 min in 96%, and 5 min in 100% ethanol twice) and CXA solution (chloroform, xylene, ethanol with equal volume mix). After putting coverslip on the stained slides with neutral resin-based medium, the covered slides were placed in the room temperature for several days drying and then ready for further imaging.
Neurons were traced using Neurolucida (MicroBrightFields Bioscience) at 40× magnification. Only a neuron that was completely filled was used for further tracing; thus, secondary and tertiary dendrites with spines were visible. Analysis of neuronal tracings was performed by Neurolucida Explorer software (MicroBrightFields Bioscience). To minimize bias, the experimenter remained blind to the treatment conditions throughout the procedure. For each animal, 2 or 3 well-stained LA/BLA or CeA neurons were traced for evaluation. Sholl analysis was used to assess the morphologic structure of dendrites, in which the radius interval of each section was set to 20 μm, starting from 40 μm and ending at a 200 μm distance from the soma. The length of dendrites was measured by Neurolucida 360 (MicroBrightFields Bioscience), and the density of dendritic spines was calculated. The spine density was expressed as the number of spines per 100 μm dendrite.
Western blot analysis
Synaptosomal fraction preparation
Synaptosomal fraction was extracted using modified methods as described in previous studies (Dunah and Standaert, 2001; Li et al., 2017). Briefly, the CeA tissues were homogenized in ice-chilled lysis buffer containing 0.32 M sucrose, 10 mM HEPES, 2 mM EDTA, and 1 mM PMSF. The extract was centrifuged at 1000 × g for 10 min to remove nuclei and large debris (P1). The supernatant (S1) was centrifuged at 10,000 × g for 15 min to obtain the synaptosomal fraction (P2). The P2 pellet was solubilized with 1% SDS in TEVP buffer containing 10 mM Tri-HCl, pH 7.4, 5 mM NaF, 1 mM EDTA, 1 mM EGTA, 2 mM PMSF, and 1% SDS at 4°C for 1 h, and centrifuged at 15,000 × g for 5 min to get the supernatant. The concentration of protein was determined using a BCA assay kit (Pierce).
Western blotting
Equal amounts of protein samples (30 μg) were denatured and then separated through SDS-PAGE using 8% running gels and transferred to a PVDF membrane (Bio-Rad). After blocking with 5% nonfat milk in TBS and Tween (TBST, 20 mm Tris-HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween-20) for 60 min at room temperature, the membrane was incubated with the following primary antibodies at 4°C overnight, respectively: mouse anti-rat GluA1 (1:1000, Abcam), rabbit anti-rat GluA2 (1:1000, Abcam), rabbit anti-pGluA1Sser831 (1:4000, Millipore), rabbit anti-pGluA1Ser845 (1:5000, Millipore), mouse anti-pGluA2Ser880 (1:1000, Abcam), and mouse anti-GAPDH (1:1000, ZSGB-Bio). The blots were washed in TBST and then were incubated in HRP-labeled goat anti-rabbit IgG (1:2000, ZSGB-Bio) or HRP-labeled goat anti-mouse IgG (1:2000, ZSGB-Bio) secondary antibodies as needed. Protein bands were visualized using an enhanced chemiluminescence detection kit (Pierce) followed by autoradiography using Hyperfilm MP (Santa Cruz Biotechnology).
Electrophysiological studies
Amygdaloid slice preparation
After decapitation, the rat brain was quickly dissected in oxygenated cutting solution (in mM as follows: 110 choline chloride, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 1.3 NaH2PO4, 25 NaHCO3, 1.3 ascorbate sodium, 0.6 sodium pyruvate, and 25 glucose, 290-300 mOsm, equilibrated to pH ∼7.4 with a mixture of 95% O2 and 5% CO2). Coronal brain slices (300 μm thick) containing the amygdale were prepared using a vibrating microtome (Leica Microsystems, VT 1000S). After incubation in oxygenated modified ACSF (in mM as follows: 125 NaCl, 2.5 KCl, 2.0 CaCl2, 1.3 MgCl2, 1.3 NaH2PO4, 25 NaHCO3, 25 glucose, 1.3 ascorbate sodium, and 0.6 sodium pyruvate, 300-310 mOsm, equilibrated to pH ∼7.4 with a mixture of 95% O2 and 5% CO2) at 34°C for 1 h, a single brain slice was transferred to the recording chamber and perfused continuously with the oxygenated ACSF at ∼2 ml/min. Only one or two brain slices per animal were used, one neuron was recorded in each slice, and a fresh slice was used for each new experimental protocol. Numbers in the manuscript refer to the number of neurons tested for each parameter.
Field potential recording and LTP induction
Field EPSPs (fEPSPs) were elicited by stimulating LA/BLA (cell bodies, including some fibers of passage) with concentric bipolar tungsten electrodes (FHC) supplied by a stimulator (STG4002, Multi-Channel Systems) and recorded in the CeA with glass Ag/AgCl microelectrodes filled with 3 M NaCl (1–3 MΩ). Electrical stimuli (100 μs square-wave pulses) were delivered at frequencies 0.033 Hz. Input-output relationships were obtained by increasing the stimulus intensity from 0.1 to 1.0 mA in 0.1 mA increments. For LTP induction, fEPSP amplitudes were adjusted to ∼30% of maximal response and recorded for 20 min as baseline. Then, a conditioning high-frequency stimulation (HFS) with the following protocols was delivered as indicated, respectively: 1 train, 1× baseline intensity, 100 Hz for 1 s, at 30 s intervals; 2 trains, 1× baseline intensity, 100 Hz for 1 s, at 30 s intervals; 5 trains, 1× baseline intensity, 100 Hz for 1 s, at 30 s intervals; 5 trains, 2.5× baseline intensity, 100 Hz for 1 s, at 30 s intervals. Subsequent fEPSPs in response to the test stimulation were recorded for at least 60 min, and their amplitude quantified as a percentage change with respect to baseline. Bicuculline (Sigma, 10 μM) was present in the perfusion solution to block inhibitory synaptic transmission.
Whole-cell patch-clamp recording
Whole-cell patch-clamp recording was performed on CeA neurons at room temperature using a Multiclamp 700B amplifier and Digidata 1440A digitizer with Axon pCLAMP 11 software (Molecular Devices). Neurons of the CeA were visually identified by using an upright infrared microscope fitted with a water immersion lens (Axioskop 2 FS plus, Carl Zeiss). The recording pipette had a resistance of 5-8 MΩ when filled with an internal solution containing the following (in mM): 128 CsCl, 10 HEPES, 10 EGTA, 10 TEACl, 1 QX314, 2 MgATP, 1 CaCl2, 2 NaCl, 280-300 mOsm, pH adjusted to 7.2-7.4 with KOH. Voltage-clamp recordings were made to measure EPSCs in CeA neurons at a holding potential of −70 mV, with both pipette and membrane capacitance cancellation, filtered at 2 kHz, and digitized at 10 kHz. Membrane resistance and series resistance (Rs) were monitored continuously during all experiments, with >20% change in Rs resulting in the omission of that experiment.
Evoked EPSCs (eEPSCs) at the LA/BLA-CeA synapse were elicited by electrical stimuli (100 μs duration, 0.1-1.0 mA amplitude) of LA/BLA at 0.05 Hz with concentric bipolar tungsten electrodes (FHC), in oxygenated recording ACSF containing GABAA receptor antagonist, bicuculline (10 μM). AMPAR-mediated eEPSCs (AMPAR currents) were isolated by adding NMDAR antagonist, D-AP5 (50 μM) in bicuculline (10 μM) containing ACSF bath, and the amplitude of AMPAR-mediated eEPSC was taken at the peak of synaptic response recorded at –70 mV. Current-voltage (I–V) relationship of AMPAR-mediated eEPSCs was measured at different holding potentials ranging from –70 to 30 mV in 20 mV increments, and then plotted using eEPSC amplitude measured at each membrane potential (normalized to averaged eEPSC amplitude at –70 mV). LTD was induced by low-frequency stimulation (LFS, 480 pulses at 1 Hz) with postsynaptic cell depolarized to −50 mV for 8 min. After LFS, cells were held back to −70 mV. For amplitude comparison between responses, eEPSCs from 10 min before the LTD induction protocol were averaged and compared with the averaged responses recorded in 40 min after LFS using a two-tailed paired t test.
Paired-pulse ratio (PPR) of eEPSCs was examined by delivering a pair of electrical pulses (100 μs duration) with interpulse intervals (IPIs) of 25, 50, 100, or 200 ms to the LA/BLA-CeA pathway, given every 30 s. Stimulation currents were titrated to achieve responses that were ∼80% of the maximum eEPSC amplitudes, which was on average 350-450 μA. The PPR was defined as the ratio of the average amplitude of the second eEPSC (eEPSC2) to that of the first eEPSC (eEPSC1) (eEPSC2/eEPSC1) at the indicated time point.
For miniature EPSC (mEPSC) recording, TTX (1 μM) was added to the ACSF to block action potential-dependent synaptic transmission in the presence of bicuculline (10 μM). A fixed length of traces (5 min) was analyzed for frequency, amplitude, decay time, and rise time of mEPSCs using MiniAnalysis program 6.0 (Synaptosoft). The root mean square of the background noise was computed for each set of data. The detection threshold for an event was set to 3-4 times the root mean square value. The peaks were detected automatically, but each detected event was then visually inspected to prevent the inclusion of false data.
Experimental design and statistical analysis
The experimental design is illustrated in the figures and described in figure legends. Images were carefully selected to show the average effect obtained for each experimental condition. Statistical analyses were performed with GraphPad Prism 8.0 for Windows (GraphPad Software). All experiments were performed at least three independent experiments, and all quantitative biochemical data and histologic staining are representative of at least three independent biological replicates. Shapiro–Wilk tests were used to assess normality in the distribution (Gaussian distribution) for each group, and only the data were normally distributed and variances were similar between groups to be compared were subjected to parametric statistical tests; otherwise, specific nonparametric tests were applied as indicated in the text. Two-tailed unpaired Student's t test was used for the comparison of the mean values between two groups. One-way analysis of variance (ANOVA) with Tukey's post hoc test or two-way ANOVA (treatment and time factors) with Sidak's post hoc test was used for multiple comparison. All data are reported as mean ± SEM, and differences with p < 0.05 were considered statistically significant.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
Results
The amygdala critically contributes to the comorbid aversive and depressive symptoms in neuropathic pain
Previously, we have revealed that the amygdala (especially the CeA) plays an important role in neuropathic pain-related anxiety-like behaviors in rats (Jiang et al., 2014). To determine whether the amygdala is also a contributor to comorbid aversive and depressive symptoms in neuropathic pain, we first identified the pain-related aversion and depressive-like behaviors in a rat model of neuropathic pain induced by SNL (Kim and Chung, 1992). In line with our previous reports (Geng et al., 2010; Jiang et al., 2014; Ding et al., 2015), ligation of L5 spinal nerve in rats produced stable mechanical allodynia, a behavioral sign of neuropathic pain characterized by decreased PWT in response to a series of von Frey filament stimuli (Jensen and Finnerup, 2014). The decreased PWT emerged on day 1 after surgery (5.01 ± 0.48 g SNL vs 13.42 ± 0.63 g, p < 0.0001), lasted at least 5 weeks after surgery at the end of our observation (3.65 ± 0.43 g SNL vs 12.57 ± 0.76 g, p < 0.0001) (n = 10, F(7,126) = 20.87; Fig. 1A). Apart from comorbid anxiety-like behaviors in SNL rats with pain allodynia observed in our previous study (Jiang et al., 2014), we currently found that aversive emotion and depressive-like symptoms also emerged in neuropathic pain rats following SNL surgery (Fig. 1B-E). The nociceptive mCPA, a behavioral phenotype reflecting pain-related aversive emotion (i.e., the pain-related CPA) (Johansen et al., 2001), was acquired in rats with SNL surgery. The CPA score, represented by the time spent in the pain-paired compartment in preconditioning session minus the time spent in the same compartment in the test session (Watanabe et al., 2002), was significantly increased in SNL-operated rats (93.00 ± 18.28, n = 14) compared with sham-operated rats (−48.67 ± 33.17, n = 12) (p = 0.0007, t(24) = 3.89; Fig. 1B) at 3 weeks after surgery, indicating that the SNL rats spent less time in the pain-paired compartment on the postconditioning day than that on the preconditioning day, and acquired pain-related aversion. Also, the predator oCPA was performed to assess whether the general CPA not related to pain is altered in the SNL animals. The results showed that, although both SNL- and sham-SNL rats exhibited evident avoidance of the predator-paired chamber after 4 d exposure of predator odor, no significant difference in the oCPA score was observed between the two groups (67.66 ± 5.22 SNL vs 65.58 ± 6.15 sham, n = 10, p = 0.7998, t(18) = 0.26; Fig. 1C) at 3 weeks after surgery.
Figure 1.
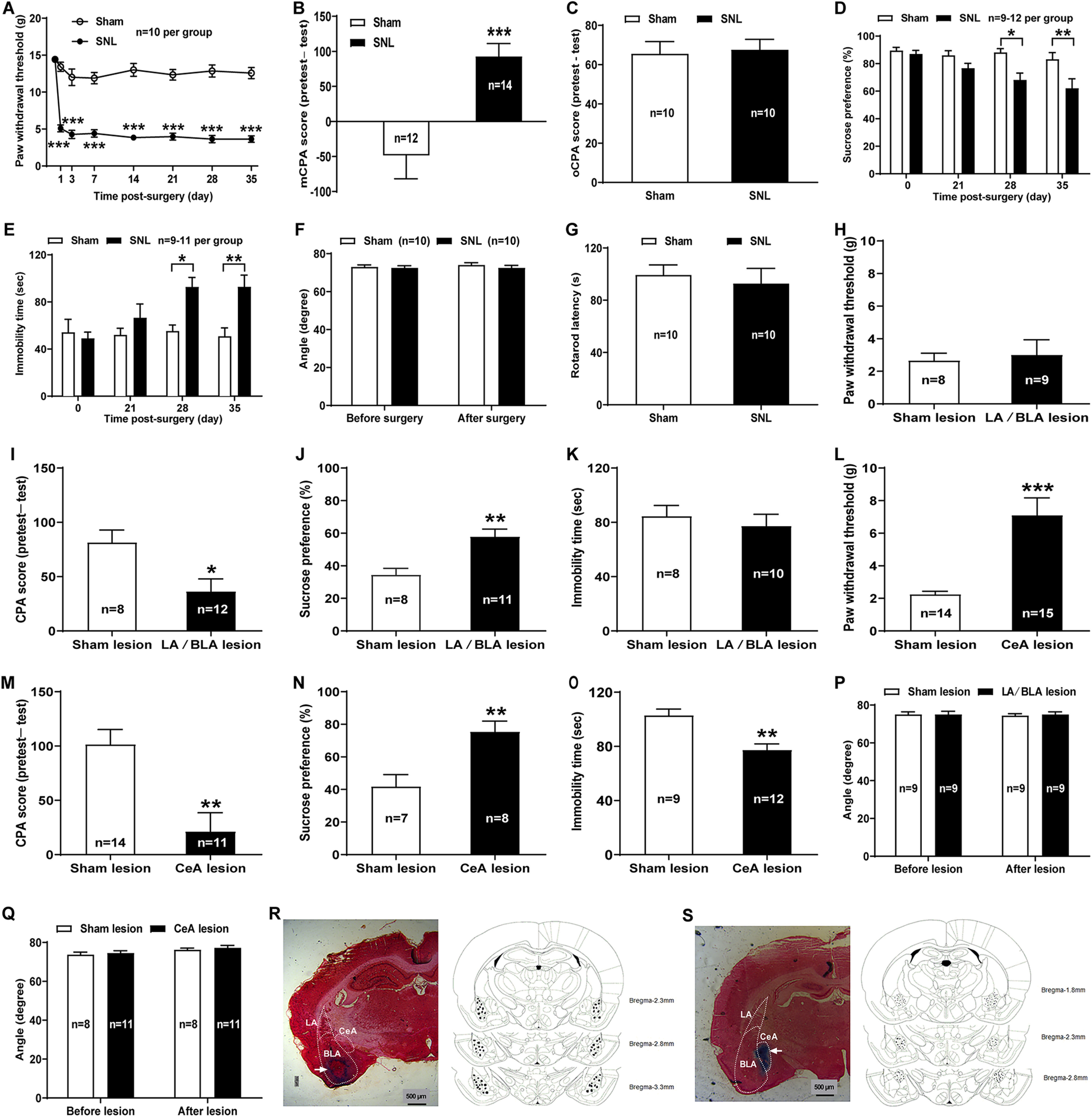
Distinct roles of LA/BLA and CeA in mediating sensory and negative emotional components of neuropathic pain in rats. A–E, Comorbid aversive and depressive symptoms in neuropathic pain in rats receiving SNL surgery. A, PWT in response to von Frey filament stimuli in SNL and sham SNL groups (n = 10 rats per group, F(7,126) = 20.87, p < 0.0001 vs sham, two-way repeated-measures ANOVA with Sidak's post hoc test). B, The nociceptive mCPA in SNL and sham SNL groups (n = 12 rats in sham and n = 14 rats in SNL, t(24) = 3.89, p = 0.0007 vs sham on day 21 after surgery, two-tailed unpaired Student's t test). C, The predator oCPA in SNL and sham SNL groups (n = 10 rats per group, t(18) = 0.26, p = 0.7998 vs sham on day 21 after surgery, two-tailed unpaired Student's t test). D, SPT in SNL and sham SNL groups (n = 9-12 rats per group, F(3,76) = 1.98, p = 0.0141 and p = 0.0024 vs sham on days 28 and 35 after surgery, two-way repeated-measures ANOVA with Sidak's post hoc test). E, FST in SNL and sham SNL groups (n = 9-11 rats per group, F(3,67) = 2.87, p = 0.0297 and p = 0.0077 vs sham on days 28 and 35 after surgery, two-way repeated-measures ANOVA with Sidak's post hoc test). The same animals were tested repeatedly on different days for A, D, and E. F, G, Inclined-plate test (F) and rotarod test (G) in SNL and sham SNL rats (n = 10 rats per group, F(1,36) = 0.17, p = 0.9462 vs sham at 5 weeks after surgery, two-way ANOVA with Sidak's post hoc test for F; t(18) = 0.47, p = 0.6446 vs sham at 5 weeks after surgery, two-tailed unpaired Student's t test for G). H–K, Effects of bilateral LA/BLA electrolytic lesions on the pain sensitivity as well as the aversive and depressive-like symptoms of SNL-operated rats. L–O, Effects of bilateral CeA electrolytic lesions on the pain sensitivity as well as the aversive and depressive-like symptoms of SNL-operated rats. H, L, PTW in response to von Frey filament stimuli measured on day 35 after surgery in lesion and sham lesion groups. I, M, The mCPA measured on day 21 after surgery in lesion and sham lesion groups. J, N, SPT measured on day 35 after surgery in lesion and sham lesion groups. K, O, FST measured on day 35 after surgery in lesion and sham lesion groups. *p < 0.05, **p < 0.01, ***p < 0.001, versus sham (two-tailed unpaired Student's t test for H–O). P, Q, Inclined-plate test measured on day 35 after surgery in lesion and sham lesion groups. n = 9, F(1,32) = 0.04, p = 0.9999 for P; n = 8-11, F(1,34) = 0.01, p = 0.9923 for Q, versus sham on day 35 after surgery (two-way ANOVA with Sidak's post hoc test). R, S, Histologically verified electrolytic lesion sites in LA/BLA (R) and CeA (S). Left, Photomicrograph of a histologic section stained with neutral red to illustrate the electrolytic lesion sites in BLA and CeA (arrow). Right, Representative three coronal sections through the rat amygdala are shown in sequence from anterior to posterior. The numbers in the right margin indicate millimeters posterior to the bregma. Filled circles in bilateral hemispheres represent the approximate positions of the electrolytic lesion sites corresponding to some representative rats in bilateral LA/BLA and CeA electrolytic lesions groups, respectively.
To date, research in rodents has been dominated by two types of assays designed to test for depressive-like behavior: behavioral despair tests, such as the FST, and measures of anhedonia, such as the SPT (Hales et al., 2014; Weinstock, 2017). We performed SPT and FST at 3, 4, and 5 weeks after surgery, and the results revealed that, compared with sham-operated rats, the SNL-operated rats had a significantly lower preference for sucrose solution (on day 35 after surgery: 62.03 ± 6.92% SNL vs 83.13 ± 4.90% sham, n = 12, p = 0.0024, F(3,76) = 1.98; Fig. 1D), and exhibited more time of immobility (on day 35 after surgery: 92.89 ± 9.91 s SNL vs 50.89 ± 7.09 s sham, n = 9, p = 0.0077, F(3,67) = 2.87; Fig. 1E) at 4 and 5 weeks but not 3 weeks after surgery, indicating increased anhedonia and despair behaviors in neuropathic pain rats. In addition, the inclined-plate test and rotarod test were performed to assess whether the animal's locomotor function was impaired after SNL surgery. Similar to previous reports (Kim and Chung, 1992; Jiang et al., 2014), no significant impairment of locomotor activity was found in rats, as assessed by both inclined-plate test (72.5 ± 1.34° SNL vs 74.00 ± 1.25° sham, n = 10, p = 0.9462, F(1,36) = 0.17; Fig. 1F) and rotarod test (92.83 ± 11.53 s SNL vs 99.34 ± 7.73 s sham, n = 10, p = 0.6446, t(18) = 0.47; Fig. 1G) at 5 weeks after SNL surgery. Together, these data suggest a comorbidity of aversive and depressive symptoms with neuropathic pain in SNL-operated rats.
To further identify the contribution of the amygdala (especially the two main functional subnuclei, the LA/BLA and the CeA), to comorbid aversive and depressive symptoms in neuropathic pain, we investigated the effects of bilateral LA/BLA or CeA electrolytic lesions on the components of pain sensory (mechanical allodynia) and pain emotion (pain-related aversion and depressive-like symptoms) in SNL-operated rats. The results showed that bilateral LA/BLA lesions did not affect the mechanical allodynia of SNL-operated rats as indicated by the PWT in response to von Frey filament stimuli (3.00 ± 0.93 g LA/BLA lesion vs 2.66 ± 0.45 g sham lesion, n = 9 LA/BLA lesion and n = 8 sham lesion, p = 0.7520, t(15) = 0.32; Fig. 1H). However, bilateral lesions of these subnuclei attenuated the SNL-induced aversive emotion indicated by decreased CPA score (36.29 ± 11.60 LA/BLA lesion vs 81.48 ± 11.43 sham lesion, n = 12 LA/BLA lesion and n = 9 sham lesion, p = 0.0139, t(19) = 2.71; Fig. 1I), and partially rescued the depressive-like behaviors of neuropathic pain rats assessed in SPT (57.77 ± 4.76% LA/BLA lesion vs 34.50 ± 3.97% sham lesion, n = 11 LA/BLA lesion and n = 8 sham lesion, p = 0.0024, t(17) = 3.56; Fig. 1J), although the lesions of bilateral LA/BLA had no significant effect on the immobility time of SNL-operated rats assessed in FST (p = 0.5559, t(16) = 0.60; Fig. 1K). In agreement with previous findings (Corder et al., 2019), these results raise the possibility that the LA/BLA, which serves as the main input of the amygdala receiving polymodal sensory information from the thalamus and cortex (Neugebauer, 2015), probably mainly contributes to the negative emotional components of chronic pain.
Unlike the LA/BLA, bilateral CeA lesions affected both sensory and negative emotional components of neuropathic pain (Fig. 1L–O). For instance, the SNL-induced reduction of ipsilateral PWT was effectively restored in rats receiving bilateral CeA lesions (7.11 ± 1.07 g, n = 15) compared with sham lesions (2.24 ± 0.20 g, n = 14) (p = 0.0002, t(27) = 4.34; Fig. 1L), supporting the key role of the CeA in persistent pain processing (Neugebauer et al., 2004; Kato et al., 2018). In addition, lesions of bilateral CeA to SNL rats not only reduced the CPA score in CPA test (21.18 ± 17.37 CeA lesion vs 101.4 ± 13.77 sham lesion, n = 11 CeA lesion and n = 14 sham lesion, p = 0.0013, t(23) = 3.67; Fig. 1M), but also rescued the decreased preference for sucrose solution in SPT (75.40 ± 6.59% CeA lesion vs 41.81 ± 7.34% sham lesion, n = 8 CeA lesion and n = 7 sham lesion, p = 0.0046, t(13) = 3.42; Fig. 1N) and the increased immobility time in FST (77.33 ± 4.52 s CeA lesion vs 102.9 ± 14.09 s sham lesion, n = 12 CeA lesion and n = 9 sham lesion, p = 0.0011, t(19) = 3.86; Fig. 1O) in SNL rats, indicating the contribution of the CeA to the negative emotional components of neuropathic pain.
Moreover, the results from inclined-plate test revealed that the animal's locomotor function was not impaired either after bilateral LA/BLA electrolytic lesions (at 5 weeks after surgery: 75.00 ± 1.44° lesion vs 74.44 ± 1.02° sham lesion, n = 9, p = 0.9999, F(1,32) = 0.04; Fig. 1P) or CeA electrolytic lesions (at 5 weeks after surgery: 77.27 ± 1.24° lesion vs 76.25 ± 0.82° sham lesion, n = 11 lesion and n = 8 sham lesion, p = 0.9923, F(1,34) = 0.01; Fig. 1Q). Sites of lesions were verified histologically and plotted on standard diagrams adapted from Paxinos and Watson (2014). Representative images of histologic verification of electrolytic lesions were presented in Figure 1R (LA/BLA lesion) and Figure 1S (CeA lesion), respectively.
Collectively, these data suggest that the CeA, which receives the highly integrated sensory and emotional information from the LA/BLA, plays a critical role in mediating both pain hypersensitivity and pain-related negative emotion, including aversive and depressive-like symptoms of neuropathic pain.
Enhancement of LTD at the LA/BLA-CeA synapses in neuropathic pain rats with comorbid aversive and depressive-like symptoms
Maladaptive neuroplastic changes are a brain mechanism of chronic pain (Kuner and Flor, 2016). To determine the involvement of amygdala plasticity in comorbid aversive and depressive-like symptoms in neuropathic pain, we first examined the morphologic structure changes in neuronal somata and dendrites of both CeA neurons and LA/BLA neurons by performing Golgi-Cox staining for the brain tissues of SNL-operated rats at 4 weeks when they emerged in pain hypersensitivity and pain-related negative emotion (Figs. 2 and 3). The results showed that the SNL-operated rats had significantly less dendritic spine density on the CeA neurons compared with the sham-operated rats (in numbers/100 μm dendrite: 16.93 ± 2.34 SNL vs 30.22 ± 2.62 sham, n = 13 SNL and n = 15 sham, p = 0.0009, t(26) = 3.74; Fig. 2A,B), whereas no differences were found in the total and average dendrite length, the total numbers of dendrite nodes and dendrite ends, as well as the dendrite length of order 1-4 dendrite branches between the two groups (Fig. 3A–L).
Figure 2.
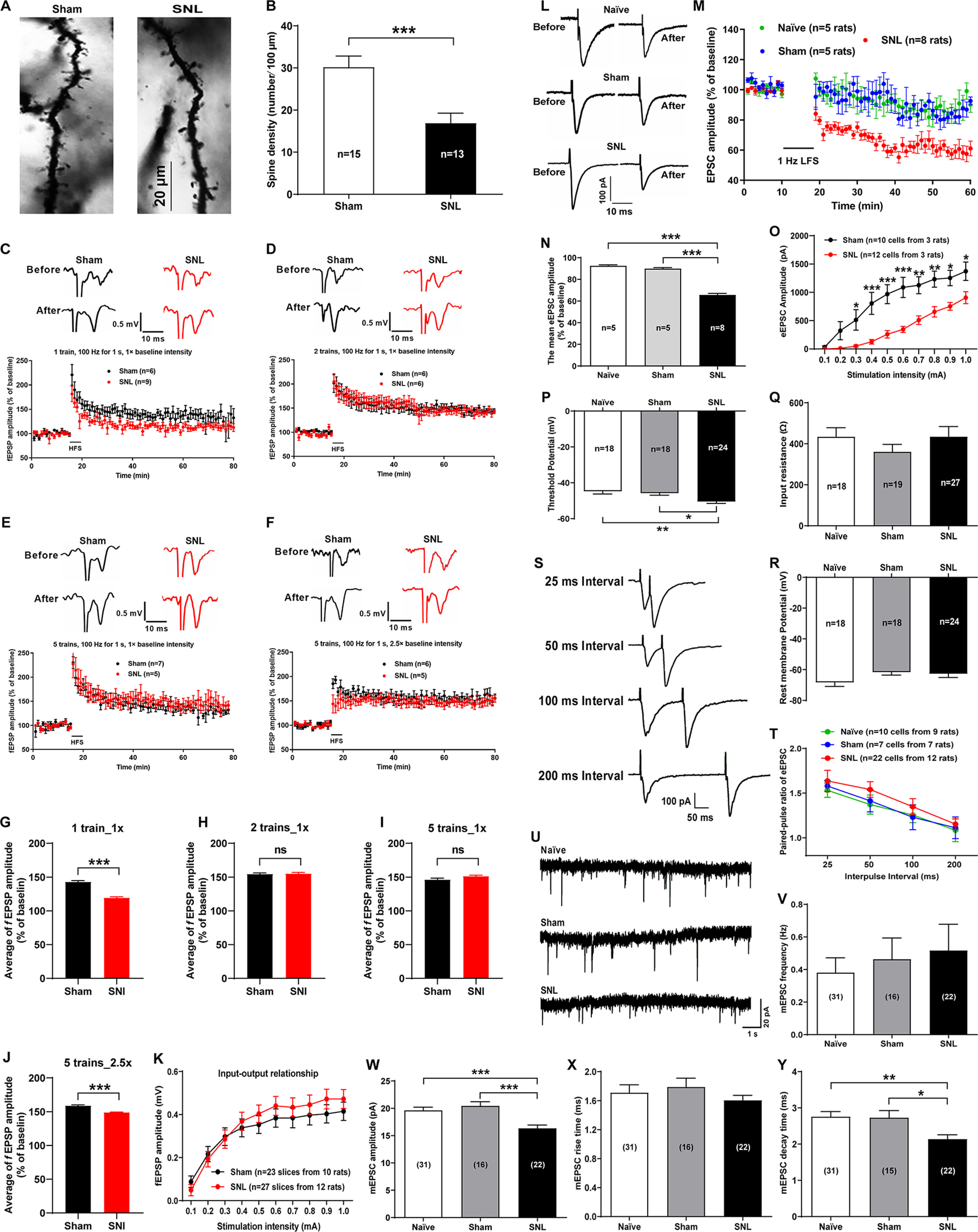
Alterations of spine density, basal synaptic properties of CeA neurons, and LTD at the LA/BLA-CeA synapse of SNL-operated rats. A, B, Morphologic plasticity of the CeA neurons in sham- and SNL-operated rats. A, Representative images of Golgi-Cox–stained dendritic spines in the CeA neurons of sham- (left) and SNL-operated (right) rats. Scale bar, 20 μm. B, Summary plot of dendritic spine density expressed as the spine numbers per 100 μm dendrite length (n = 13-15 neurons from 5 rats per group, t(26) = 3.74, p = 0.009 vs sham, two-tailed unpaired Student's t test). C–J, LTP of fEPSPs at the LA/BLA-CeA synapse recorded from the amygdaloid slices of sham- and SNL-operated rats. C–F, LTP induced by different protocols of a conditioning HFS, including 1 train, 1× baseline intensity, 100 Hz for 1 s, at 30 s intervals (C); 2 trains, 1× baseline intensity, 100 Hz for 1 s, at 30 s intervals (D); 5 trains, 1× baseline intensity, 100 Hz for 1 s, at 30 s intervals (E); and 5 trains, 2.5× baseline intensity, 100 Hz for 1 s, at 30 s intervals (F). Histograms represent the mean time course of fEPSPs before and after HFS in sham and SNL groups. Top, Traces are representative fEPSPs recorded at 10 min before and 60 min after HFS of the LA/BLA. Calibration: 0.5 mV, 10 ms. G–J, Bar graph represents the mean amplitude of fEPSPs (% of baseline) during the 20-80 min following HFS. ***p < 0.001; ns, no statistical difference, compared between the SNL and the sham SNL groups (two-tailed unpaired Student's t test). K, Input-output relationship of fEPSP amplitude between the SNL and the sham SNL groups measured by increasing the stimulus intensity from 0.1 to 1.0 mA in 0.1 mA increment (n = 23-27 slices from 10-12 rats, F(9,480) = 0.43, p > 0.05 compared between the SNL and the sham SNL groups, two-way ANOVA with Sidak's post hoc test). L–N, LTD of eEPSCs at the LA/BLA-CeA synapse in sham- and SNL-operated rats. L, Representative traces of eEPSCs before and after LFS. Calibration: 100 pA, 10 ms. M, Average eEPSC amplitudes (normalized to baseline) over the time course of the LFS protocol (480 pulses at 1 Hz with postsynaptic cell depolarized to −50 mV). N, Average eEPSC amplitudes (normalized to baseline) are plotted during the 20-60 min following LFS (n = 5-8 rats per group, F(2,161) = 177.8, p < 0.0001 vs naïve and sham, one-way ANOVA with Tukey's post hoc test). O, Input-output relationship of eEPSCs at the LA/BAL-CeA synapse in sham- and SNL-operated rats. *p < 0.05, **p < 0.01, ***p < 0.001, SNL versus sham, n = 10-12 cells, F(9,200) = 1.83 (two-way ANOVA with Sidak's post hoc test). P–R, The intrinsic properties of CeA neurons, including threshold potential (P), input resistance (Q), and resting membrane potentials (R) in naïve, sham-, and SNL-operated rats. *p < 0.05, **p < 0.01, versus naïve and sham (one-way ANOVA with Tukey's post hoc test). S, T, PPR of eEPSCs evoked by a pair of electrical pulses (100 μs duration) with IPIs of 25, 50, 100, or 200 ms to the LA/BLA-CeA pathway, given every 30 s. S, Representative paired-pulse traces of eEPSCs recorded from a CeA pyramidal cell in response to paired-pulse stimuli at different IPIs as indicated. Calibration: 100 pA, 50 ms. T, Summary plot for PPR of eEPSCs against IPIs in naïve, sham-, and SNL-operated rats (n = 7–22 cells from 7–12 rats per group, F(6,138) = 0.06, p > 0.05 versus sham, two-way ANOVA with Sidak's post hoc test). U–Y, mEPSCs recorded in the CeA neurons in naïve, sham-, and SNL-operated rats. U, Representative traces of mEPSCs in naïve, sham-, and SNL-operated rats. Calibration: 20 pA, 1 s. V–Y, Summary plots for mean frequency (V), amplitude (W), rise time (X), and decay time (Y) of mEPSCs measured in naïve, sham-, and SNL-operated rats, respectively (n = 15-31 cells from 12–15 rats per group). *p < 0.05, **p < 0.01, ***p < 0.001, versus naïve and sham (one-way ANOVA with Tukey's post hoc test).
Figure 3.
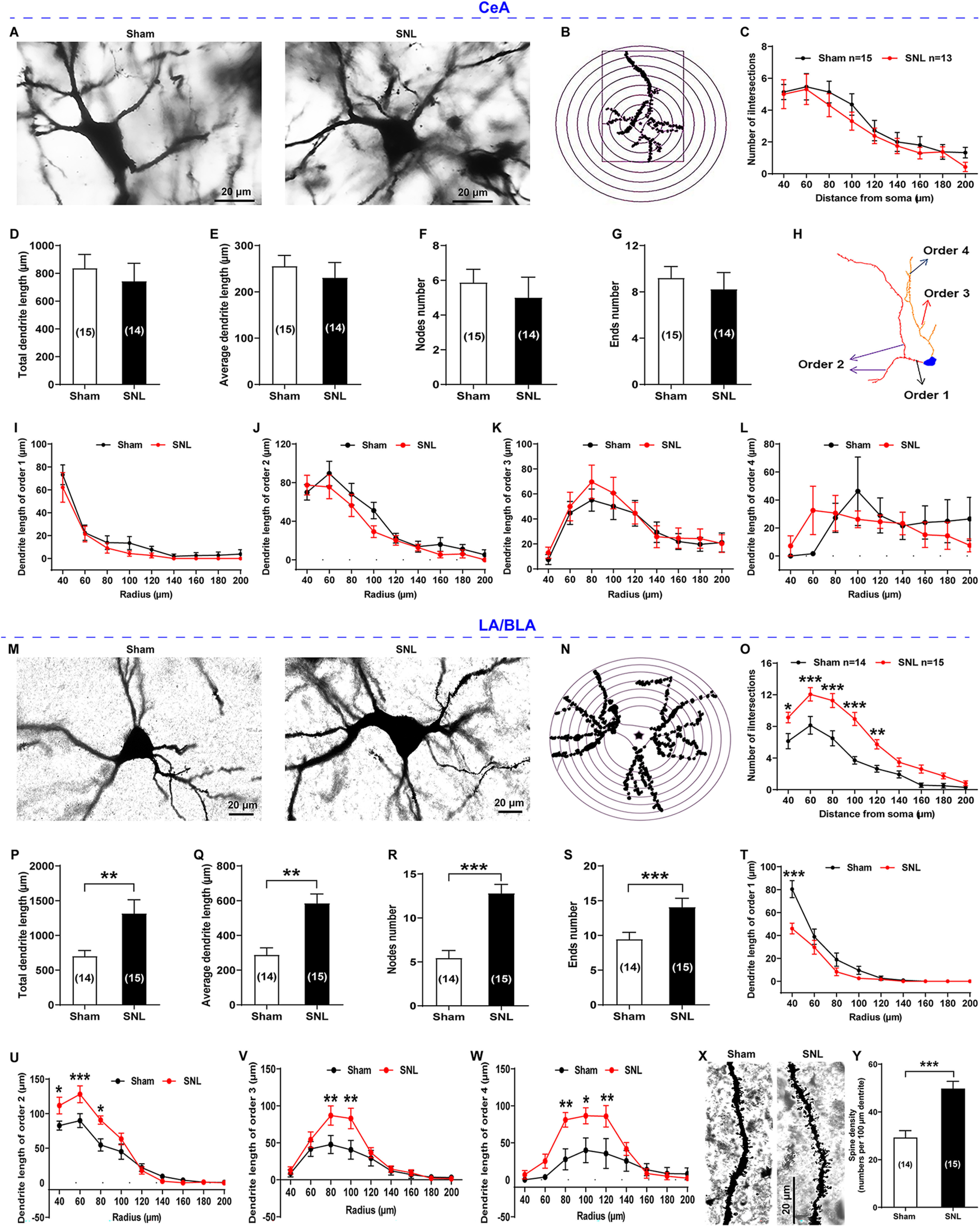
Golgi-Cox staining and Sholl analysis of CeA neurons and LA/BLA neurons in sham- and SNL-operated rats. A–L, CeA neurons. M-Y, LA/BLA neurons. A, M, Representative images of Golgi-Cox–stained CeA neurons (A) and LA/BLA neurons (M) obtained from sham- (left) and SNL-operated (right) rats. Scale bar, 20 μm. B, N, Representative CeA neuron (B) and LA/BLA neuron (N) with concentric circles used for the Sholl analysis. The radius interval between the circles was set to 20 μm per step, ranging from 40 to 200 μm from the center of the neuronal soma to the end of the dendrites. C, O, The numbers of dendritic intersections per circle were quantified (n = 13-15 neurons from 3 rats, F(8,194) = 0.15, p > 0.05 for C; n = 14 or 15 neurons from 3 rats, F(8,243) = 3.08, *p < 0.05, **p < 0.01, ***p < 0.001 for O, compared between sham and SNL groups, two-way ANOVA with Sidak's post hoc test). D–G, P–S, Comparisons of total dendrite length (t(27) = 0.58, p = 0.5685 for B; t(27) = 2.80, p = 0.0093 for P), average dendrite length (t(27) = 0.63, p = 0.5347 for C; t(27) = 4.37, p = 0.0002 for Q), numbers of dendrite nodes (t(27) = 0.63, p = 0.5360 for D; t(27) = 5.50, p < 0.0001 for R), and dendrite ends (t(27) = 0.57, p = 0.5734 for E; t(31) = 2.81, p = 0.0085 for S) of CeA neurons and LA/BLA neurons between sham-operated and SNL-operated rats (n = 14 or 15 neurons from 3 rats, compared between sham and SNL groups, two-tailed unpaired Student's t test). H, Neuronal reconstruction of representative Golgi-Cox–stained CeA neurons used for dendrite branch order analysis. I–L, T–W, Comparisons of the dendrite length of order 1-4 dendrite branches of CeA neurons and LA/BLA neurons between sham-operated and SNL-operated rats (n = 12-15 neurons from 3 rats). *p < 0.05, **p < 0.01, ***p < 0.001, compared between sham and SNL groups (two-way ANOVA with Sidak's post hoc test). X, Representative images of Golgi-Cox–stained dendritic spines in the LA/BLA neurons of sham- (left) and SNL-operated (right) rats. Scale bar, 20 μm. Y, Summary plot of dendritic spine density expressed as the spine numbers per 100 μm dendrite length (n = 14 or 15 neurons from 5 rats per group, t(27) = 4.92, p < 0.0001 vs sham, two-tailed unpaired Student's t test).
In contrast, the dendritic spine density of LA/BLA neurons was significantly increased in the SNL-operated rats compared with the sham-operated rats (in numbers/100 μm dendrite: 49.80 ± 3.02 SNL vs 29.29 ± 2.86 sham, n = 15 SNL and n = 14 sham, p < 0.0001, t(27) = 4.92; Fig. 3X,Y). Also, a significant increase in the total and average dendrite length, the total numbers of dendrite nodes and dendrite ends, as well as the dendrite length of order 1–4 dendrite branches was found in the LA/BLA neurons in SNL-operated rats compared with the sham-operated rats (Fig. 3M–W).
Since dendritic spines usually serve as postsynaptic partners mediating synaptic transmission, we then examined the functional consequence of this structure change by using in vitro electrophysiological recording from the amygdaloid slices at 4 weeks after surgery when SNL-operated rats emerged comorbid aversive and depressive symptoms in neuropathic pain. We first recorded baseline fEPSPs in the LA/BLA-CeA pathway at a frequency of 0.033 Hz and at a stimulation intensity adjusted to give an fEPSP amplitude of ∼30% maximum response. Then we applied the following four different protocols of HFS for inducing LTP, respectively: 1 train of 100 Hz stimulation for 1 s duration at an interval of 30 s with onefold baseline intensity (1 train, 100 Hz for 1 s, 1× baseline intensity)-HFS; 2 trains, 100 Hz for 1 s, 1× baseline intensity-HFS; 5 trains, 100 Hz for 1 s, 1× baseline intensity-HFS; and 5 trains, 100 Hz for 1 s, 2.5× baseline intensity-HFS. The results revealed that a reliable LTP of fEPSPs at the LA/BLA-CeA synapse could be induced by each protocol of HFS at both SNL-operated rats and sham-operated rats. However, no significant difference in LTP amplitude was observed between the two groups, except for a slight decrease in mean amplitude of fEPSPs after HFS by the protocol of 1 train, 1× baseline intensity and 5 trains, 2.5× baseline intensity, in SNL-operated rats compared with sham-operated rats (Fig. 2C–J). Also, no significant difference in the input-output relationship of fEPSP amplitude, as measured by increasing the stimulus intensity from 0.1 to 1.0 mA in 0.1 mA increment, was found between the SNL and the sham SNL groups (n = 23-27 slices from 10-12 rats, p > 0.05, F(9,480) = 0.43; Fig. 2K).
Furthermore, using whole-cell patch-clamp recording of the eEPSC at the LA/BLA-CeA synapse in the presence of GABAA receptor antagonist bicuculline (10 μM), we examined LTD of eEPSC at the LA/BLA-CeA synapse induced by LFS consisting of 1 Hz, 480 pulses combining postsynaptic depolarization to −50 mV for 8 min. We found that SNL surgery induced a significant enhancement of LTD, as indicated by a decreased mean eEPSC amplitude, at the LA/BLA-CeA synapse compared with naïve and sham-operated rats (Fig. 2L–N). When compared with the baseline responses (averaged at 0-10 min before LFS), the mean eEPSC amplitude after LFS was decreased from 92.56 ± 0.92% of the naïve group and 89.97 ± 0.92% of the sham-operated group to 65.74 ± 1.38% of the SNL-operated group (n = 5-8 rats per group, p < 0.0001, F(2,161) = 177.8; Fig. 2N), suggesting an enhanced LTD at the LA/BLA-CeA synapses in neuropathic pain rats with comorbid aversive and depressive-like symptoms. Additionally, the input-output relationship of eEPSCs and the intrinsic properties of CeA neurons were examined. The input-output relationship of eEPSCs revealed that the eEPSC was significantly decreased in SNL rats compared with sham SNL rats almost at all stimulation intensity. The eEPSC amplitude was decreased from the stimulation intensity of 0.3 mA (51.58 ± 19.93 pA SNL vs 512.61 ± 180.78 pA sham, p = 0.0378) to the maximal stimulation intensity of 1.0 mA (906.07 ± 105.52 pA SNL vs 1372.78 ± 162.24 pA sham, p = 0.0339) used in this study (n = 12 cells SNL and n = 10 cells sham, F(9,200) = 1.83; Fig. 2O), which might reflect the loss of spines/synapses in SNL rats. With respect to the intrinsic properties of CeA neurons, the threshold potential was more hyperpolarized in SNL rats (−50.45 ± 1.08 mV) than that in naïve (−44.73 ± 1.55 mV) and sham SNL (−45.77 ± 1.16 mV) rats (n = 18-24 cells, p = 0.0051 SNL vs naïve and p = 0.0255 SNL vs sham, F(2,57) = 6.36; Fig. 2P). However, there was no significant difference in the input resistance (in Ω: 434.7 ± 49.30 SNL vs 434.4 ± 44.26 naïve and 360.5 ± 36.67 sham, n = 18-27 cells, p > 0.9999 SNL vs naïve and p = 0.4606 SNL vs sham, F(2,63) = 0.86; Fig. 2Q) and the resting membrane potentials (in mV: −62.70 ± 2.38 SNL vs −68.32 ± 2.58 naïve and −79.00 ± 1.91 sham, n = 18-24 cells, p = 0.2154 SNL vs naïve and p = 0.9445 SNL vs sham, F(2,65) = 2.07; Fig. 2R) among the naïve, sham SNL, and SNL groups.
Then, we examined PPR of eEPSCs (McKernan and Shinnick-Gallagher, 1997) and mEPSCs (Wyllie et al., 1994), both of them may reflect basal synaptic properties in the CNS. Two orthodromic synaptic stimuli of equal intensity were applied at varying intervals, and the resulting EPSCs were recorded in whole-cell voltage clamp for PPR analysis. Peak amplitudes of the initial eEPSC (eEPSC1) and the second eEPSC (eEPSC2) were measured as the difference between the current level before the stimulus artifact and the peak of the eEPSC and PPR was calculated as the ratio of eEPSC2 over eEPSC1. It is well known that when two action potentials are elicited in close succession in the presynaptic cell, the second action potential triggers the release of more vesicles than the first, because the release probability remains transiently elevated because of the residual Ca2+ that remains in the nerve terminal after the first action potential (Debanne et al., 1996). In the present study, stimulation at different IPI, including 25, 50, 100, and 200 ms were applied to the LA/BLA in the presence of bicuculline (10 μM), for inducing paired eEPSCs at the LA/BLA-CeA synapse. The results showed that, in either SNL- or sham-operated rats, the second eEPSC was typically larger than the first one at each IPI (Fig. 2S), indicating a paired-pulse facilitation of eEPSC at the LA/BLA-CeA synapse. However, no significant difference in the PPR of eEPSC was observed at each IPI among naïve, sham-operated, and SNL-operated rats (n = 7-22 cells from 7-12 rats per group, p = 0.9989, F(6,138) = 0.06; Fig. 2T). These results indicate that, despite the spine loss in the CeA neurons, the probability of release at the LA/BLA-CeA synapse is probably not impaired in SNL-operated rats.
The mEPSC results from the spontaneous fusion of neurotransmitter-containing vesicles to the presynaptic terminal membrane, and various factors affect the mEPSC frequency and amplitude. Presynaptic changes at the transmitter release site and changes in synapse number can affect mEPSC frequency (Han and Stevens, 2009), whereas changes at the postsynaptic membrane would alter mEPSC amplitude (quantal size) (Wyllie et al., 1994; Han et al., 2005). In this study, we analyzed the mEPSCs of CeA neurons in the presence of TTX (1 μM) and bicuculline (10 μM) (to block action potential-mediated synaptic activity and GABAA receptors, respectively). Our data revealed that the amplitude and decay time of mEPSCs were significantly decreased in the CeA neurons of SNL-operated rats compared with naïve and sham-operated rats (n = 15-31 cells, amplitude: 16.35 ± 0.59 mV SNL vs 19.63 ± 0.58 mV naïve and 20.45 ± 0.74 mV sham, p = 0.0006 SNL vs naïve and p = 0.0003 SNL vs sham, F(2,66) = 10.92; decay time: 2.14 ± 0.12 ms SNL vs 2.76 ± 0.14 ms naïve and 2.73 ± 0.19 ms sham, p = 0.0064 SNL vs naïve and p = 0.0368 SNL vs sham, F(2,65) = 5.67) (Fig. 2U,W,Y). In contrast, no difference on the frequency and rise time of mEPSCs was found among the three groups (n = 16-31 cells, frequency: p = 0.6979 SNL vs naïve and p = 0.9602 SNL vs sham, F(2,66) = 0.34; rise time: p = 0.7353 SNL vs naïve and p = 0.5097 SNL vs sham, F(2,65) = 0.64) (Fig. 2U,V,X).
Phosphorylation of postsynaptic AMPARs and reduction of GluA2-containing AMPARs-mediated currents in the CeA neurons of SNL-operated rats
Changes in the properties and postsynaptic abundance of AMPARs are major mechanisms underlying various forms of synaptic plasticity, including LTP and LTD (Diering and Huganir, 2018). In particular, phosphorylation of AMPARs is well known to play a pivotal role in the expression of synaptic plasticity (Park, 2018). We hence examined changes in protein abundance and phosphorylation levels of postsynaptic AMPARs by using Western blotting from CeA synaptosomal fraction extracted from SNL-operated rats at 4 weeks after surgery when they emerged comorbid aversive and depressive symptoms in neuropathic pain (Fig. 4). The results showed that the total protein abundance of both GluA1 and GluA2 in the CeA synaptosomal fraction was increased in SNL-operated rats compared with naïve and sham-operated rats (GluA1: n = 8, p = 0.0073 SNL vs naïve and p = 0.0007 SNL vs sham, F(2,21) = 10.68; GluA2: n = 6, p = 0.0235 SNL vs naïve and p = 0.0109 SNL vs sham, F(2,15) = 6.82) (Fig. 4A,C,E,G,I,K). However, the phosphorylation level of GluA1 at serine 845 residue (pGluA1Ser845) was decreased (n = 5, p = 0.0066 SNL vs naïve and p = 0.0131 SNL vs sham, F(2,12) = 8.75), whereas the phosphorylated GluA2 at serine 880 residue (pGluA2Ser880) was increased (n = 6 rats per group, p < 0.0001 SNL vs naïve and sham, F(2,15) = 35.73) in SNL-operated rats compared with naïve and sham-operated rats (Fig. 4F,H,J,L). By contrast, no significant difference in phosphorylated GluA1 at serine 831 residue (pGluA1Ser831) was observed among the three groups (n = 5, p = 0.9190 SNL vs naïve and p = 0.8071 SNL vs sham, F(2,12) = 0.20) (Fig. 4B,D). Phosphorylation of GluA1Ser831 and GluA1Ser845 is implicated in regulating GluA1-containing, CP-AMPAR insertion into the postsynaptic membrane and facilitating LTP induction, whereas phosphorylation of GluA2Ser880 is critical for promoting GluA2-containing, CI-AMPAR endocytosis and leads to LTD (Diering and Huganir, 2018; Park, 2018). Using whole-cell patch-clamp recording from amygdaloid slices during 3-4 weeks after surgery when comorbid aversive and depressive symptoms in neuropathic pain emerged in SNL-operated rats, the AMPAR-mediated eEPSCs (AMPAR currents) evoked at the LA/BLA-CeA synapse were recorded in the presence of D-AP5 (NMDAR antagonist, 50 μM) and bicuculline (GABA receptor antagonist, 10 μM). Here, we applied D-AP5 and bicuculline to block both NMDARs and GABA receptors activities, and subsequently recording the uncontaminated AMPAR currents. Our results revealed that the AMPAR currents were significantly decreased in SNL-operated rats (2.00 ± 0.30 pA/pF) compared with naïve (3.99 ± 0.51 pA/pF) and sham-operated rats (4.10 ± 0.42 pA/pF) (n = 16-26 cells, p = 0.0013 SNL vs naïve and p = 0.0020 SNL vs sham, F(2,51) = 10.09; Fig. 5A), indicating the reduction of functional AMPARs on the membrane surface of CeA neurons in neuropathic rats. Based on the fact that GluA2-lacking AMPARs are Ca2+-permeable and show an inward rectification I–V relationship, whereas GluA2-containing AMPARs are Ca2+-impermeable and have a linear I–V relationship (Greger et al., 2017), we further identified the subunit components of the reduced AMPARs on the membrane surface of CeA neurons in SNL rats by examining the I–V relationship of AMPAR-mediated eEPSC at the LA/BLA-CeA synapse. Interestingly, a consistent, linear I–V relationship curve emerged among naïve, sham-operated, and SNL-operated rats, and no significant difference on the I–V relationship curve was found among the three groups (n = 9-17 cells, p = 0.9997, F(10,183) = 0.11; Fig. 5B). Moreover, we applied a selective CP-AMPAR antagonist, NASPM (50 μM), to block GluA2-lacking AMPARs (Gerace et al., 2014), and found that NASPM was not able to inhibit the AMPAR-mediated eEPSC at the LA/BLA-CeA synapse either in SNL-operated rats or in naïve or sham-operated rats (n = 5 or 6 rats per group, p > 0.9999 SNL vs naïve p > 0.9999 SNL vs sham, and p = 0.9998 sham vs naïve F(2,12) = 0.0001; Fig. 5C,D). These results suggest that, the GluA2-containing, CI-AMPARs are probably partly responsible for the reduction of AMPAR-mediated eEPSCs (AMPAR currents) evoked at the LA/BLA-CeA synapse in SNL rats.
Figure 4.

Reduction of phosphorylated GluA1Ser845 and elevation of phosphorylated GluA2Ser880 in the CeA synaptosomal fraction of SNL-operated rats. A–D, Western blot of GluA1 and phosphorylated GluA1 at serine 831 residue (pGluA1Ser831) expression in the CeA synaptosomal fraction of naïve, sham-, and SNL-operated rats. A, Representative of Western blot bands, GAPDH is used as an internal control. B–D, Summary plots for the relative intensity of pGluA1Ser831 and GluA1, and ratio of pGluA1Ser831/GluA1 (n = 5-8 rats per group). **p < 0.01, ***p < 0.001, versus naïve and sham (one-way ANOVA with Tukey's post hoc test). E–H, Western blot of GluA1 and phosphorylated GluA1 at serine 845 residue (pGluA1Ser845) expression in the CeA synaptosomal fraction of naïve, sham-, and SNL-operated rats. E, Representative of Western blot bands. F–H, Summary plots for the relative intensity of pGluA1Ser845 and GluA1, and ratio of pGluA1Ser845/GluA1 (n = 5-8 rats per group). *p < 0.05, **p < 0.01, ***p < 0.001, versus naïve and sham (one-way ANOVA with Tukey's post hoc test). I–L, Western blot of GluA2 and phosphorylated GluA2 at serine 880 residue (pGluA2Ser880) expression in the CeA synaptosomal fraction of naïve, sham-, and SNL-operated rats. I, Representative of Western blot bands. J–L, Summary plots for the relative intensity of pGluA2Ser880 and GluA2, and ratio of pGluA2Ser880/GluA2 (n = 5 or 6 rats per group). *p < 0.05, **p < 0.01, ***p < 0.001, versus naïve and sham (one-way ANOVA with Tukey's post hoc test).
Figure 5.
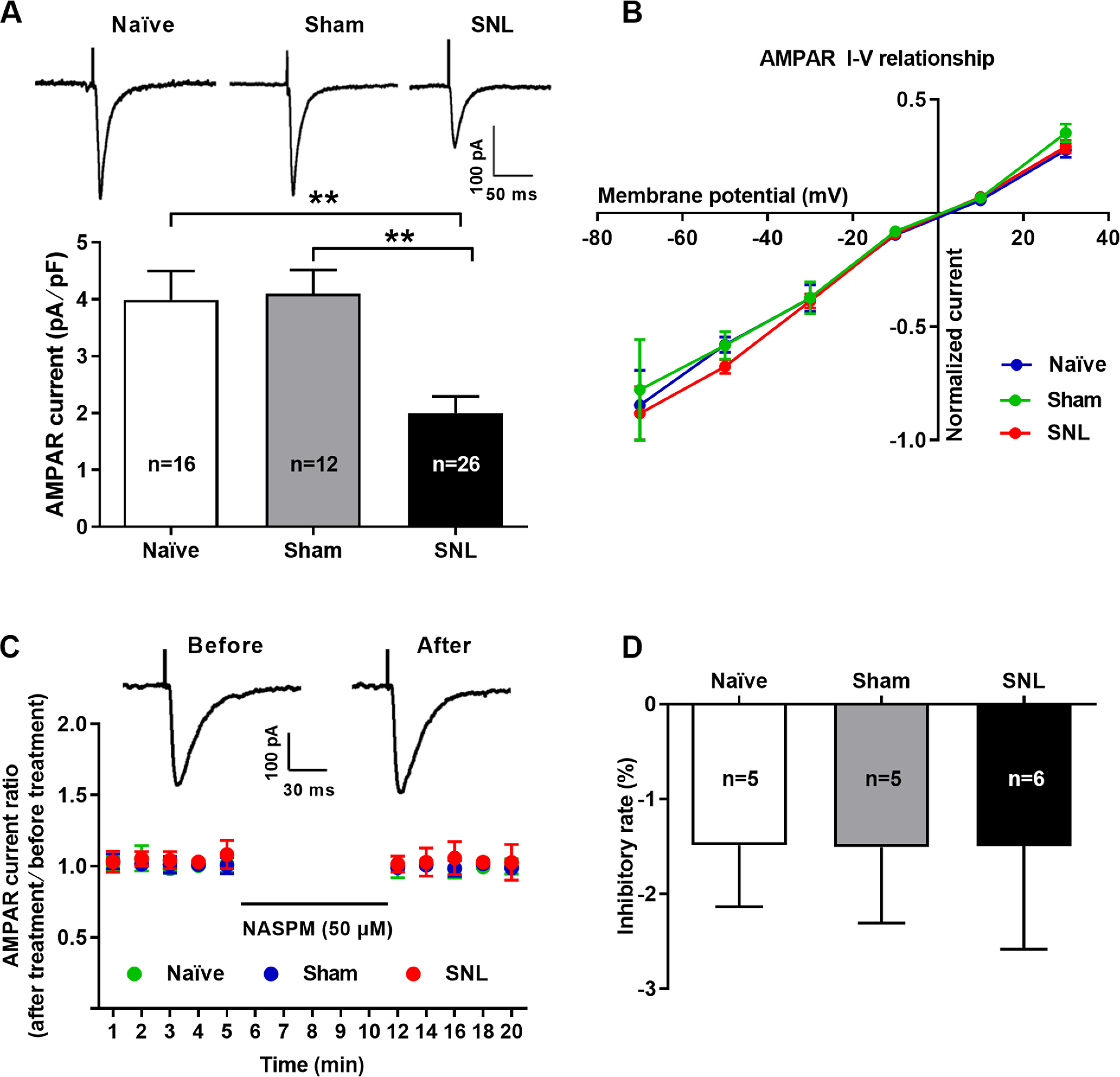
Reduction of GluA2-containing AMPAR-mediated EPSCs at the LA/BLA-CeA synapse in SNL-operated rats. A, Whole-cell voltage-clamp recording of AMPAR-mediated EPSCs evoked by stimulating the LA/BLA-CeA synapses in the presence of D-AP5 (50 μM) in bicuculline (10 μM) containing ACSF bath (to block both NMDAR and GABA receptor activities, and subsequently recording the uncontaminated AMPAR currents). Top, Traces of AMPAR-mediated eEPSCs (AMPAR currents) in naïve, sham-, and SNL-operated rats. Calibration: 100 pA, 50 ms. Bottom, Summary plot for the amplitude of AMPAR currents (normalized to membrane capacitance, in pA/pF) in naïve, sham-, and SNL-operated rats (n = 16 cells/3 naïve rats, 12 cells/3 sham rats, and 26 cells/4 SNL rats, F(2,51) = 10.09, p = 0.0013 vs naïve and p = 0.002 vs sham, one-way ANOVA with Tukey's post hoc test). B, I–V relationship of averaged AMPAR-mediated eEPSCs at different holding potentials ranging from –70 to 30 mV in 20 mV increments (normalized to averaged eEPSC amplitude at –70 mV), and plot of pooled data were linear in naïve, sham-, and SNL-operated rats (n = 9-17 cells from 5-7 rats per group, F(10,183) = 0.11, p > 0.05, SNL vs naïve, SNL vs sham, and naïve vs sham, two-way ANOVA with Sidak's post hoc test). C, D, Bath application of GluA2-lacking AMPAR antagonist NASPM (50 μM) did not affect the AMPAR-mediated eEPSCs (AMPAR currents) in all groups. C, Dot plots for the effects of NASPM (50 μM) application on normalized AMPAR currents (ratio of after treatment to before treatment) over the time course. Inset, Traces of AMPAR-mediated eEPSCs obtained from the recording before (left) and after (right) NASPM application. Calibration: 100 pA, 30 ms. D, Summary histogram illustrating the inhibitory rate of NASPM on AMPAR-mediated eEPSCs in naïve, sham-, and SNL-operated rats (n = 5 or 6 rats per group, F(2,12) = 0.0001, p > 0.9999 vs naïve and sham, one-way ANOVA with Tukey's post hoc test).
GluA2-containing AMPAR endocytosis contributes to LTD at the LA/BLA-CeA synapse and comorbid aversive and depressive symptoms in neuropathic pain in SNL-operated rats
To further determine whether the GluA2-containing AMPAR endocytosis is required for LTD at the LA/BLA-CeA synapse and comorbid aversive and depressive symptoms in neuropathic pain in rats following peripheral nerve injury, two synthetic peptides, pep2-EVKI and Tat-GluA2(3Y), were administered to prevent the GluA2-containing AMPARs trafficking and endocytosis from synapses, and the effects on LTD at the LA/BLA-CeA synapse as well as the effects on pain hypersensitivity and pain-related aversive and depressive symptoms were examined in amygdaloid slices of neuropathic rats in vitro and in SNL-operated rats in vivo, respectively (Fig. 6). Pep2-EVKI, which mimics C terminus residues of GluA2, disrupts the interactions between GluA2 and protein interacting with C-kinase (PICK1) (Daw et al., 2000), and subsequently prevents GluA2-containing AMPARs trafficking to and from the plasma membrane (Famous et al., 2008), whereas Tat-GluA2(3Y), a synthetic peptide containing a short C-terminal sequence of GluA2, has been proven to prevent GluA2-containing AMPAR endocytosis from synapses (Brebner et al., 2005; Benneyworth et al., 2019). In the in vitro experiments, we found that either preincubated amygdaloid slices with pep2-EVKI (100 μM) to prevent GluA2-containing AMPARs trafficking to and from the plasma membrane (Fig. 6A–C), or with Tat-GluA2(3Y) (10 μM) to block GluA2-containing AMPAR endocytosis from synapses (Fig. 6I–K), abrogated the SNL-induced enhancement of LTD at the LA/BLA-CeA synapse compared with their corresponding control peptide (inactive or nonfunctional peptide), pep2-SVKE (100 μM), and Tat-GluA2(3A) (10 μM). Compared with the baseline responses (averaged at 0-10 min pre-LFS), the mean eEPSC amplitude after LFS (20-60 min post-LFS) was 72.05 ± 1.51 of pep2-EVKI (functional peptide) versus 38.01 ± 1.60 of pep2-SVKE (control peptide) (n = 7–10 rats per group, p < 0.0001, t(131) = 15.07; Fig. 6C), and 106.0 ± 1.01 of Tat-GluA2(3Y) (active peptide) versus 38.78 ± 1.82 of Tat-GluA2(3A) (inactive peptide) (n = 4 or 5 rats per group, p < 0.0001, t(104) = 31.26; Fig. 6K), respectively. Consistently, results from the in vivo experiments revealed that bilateral microinjection of either pep2-EVKI (10 μg/μl, 0.5 μl per side) or Tat-GluA2(3Y) (75 pmol, 0.5 μl per side) into the CeA at 1 h before behavioral test could effectively restore the SNL-induced mechanical allodynia (Pep2-EVKI: n = 9-14 rats per group, p < 0.0001 post-treatment: Pep2-EVKI vs post-treatment: Pep2-SVKE, F(2,66) = 17.84, Fig. 6D; Tat-GluA2(3Y): n = 9-17 rats per group, p < 0.0001 post-treatment: Tat-GluA2(3Y) vs post-treatment: Tat-GluA2(3A), F(2,75) = 51.09, Fig. 6L) and comorbid aversive and depressive symptoms in SNL-operated rats, as indicated in CPA test (Pep2-EVKI: n = 7-10 rats per group, p = 0.0014 post-treatment: Pep2-EVKI vs post-treatment: Pep2-SVKE, F(2,48) = 5.69, Fig. 6E; Tat-GluA2(3Y): n = 9-14 rats per group, p = 0.0002 post-treatment: Tat-GluA2(3Y) vs post-treatment: Tat-GluA2(3A), F(2,64) = 8.41, Fig. 6M), SPT (Pep2-EVKI: n = 8 or 9 rats per group, p = 0.0092 post-treatment: Pep2-EVKI vs post-treatment: Pep2-SVKE, F(1,30) = 4.33, Fig. 6F; Tat-GluA2(3Y): n = 10-12 rats per group, p = 0.0029 post-treatment: Tat-GluA2(3Y) vs post-treatment: Tat-GluA2(3A), F(1,40) = 5.28, Fig. 6N), and FST (Pep2-EVKI: n = 9 or 10 rats per group, p = 0.0132 post-treatment: Pep2-EVKI vs post-treatment: Pep2-SVKE, F(1,33) = 4.88, Fig. 6G; Tat-GluA2(3Y): n = 9 rats per group, p = 0.0349 post-treatment: Tat-GluA2(3Y) vs post-treatment: Tat-GluA2(3A), F(1,32) = 4.81, Fig. 6O), respectively. Moreover, the results from inclined-plate test revealed that the animal's locomotor function was not impaired either after pep2-EVKI/pep2-SVKE or Tat-GluA2(3Y)/Tat-GluA2(3A) microinjection (Fig. 6H,P).
Figure 6.
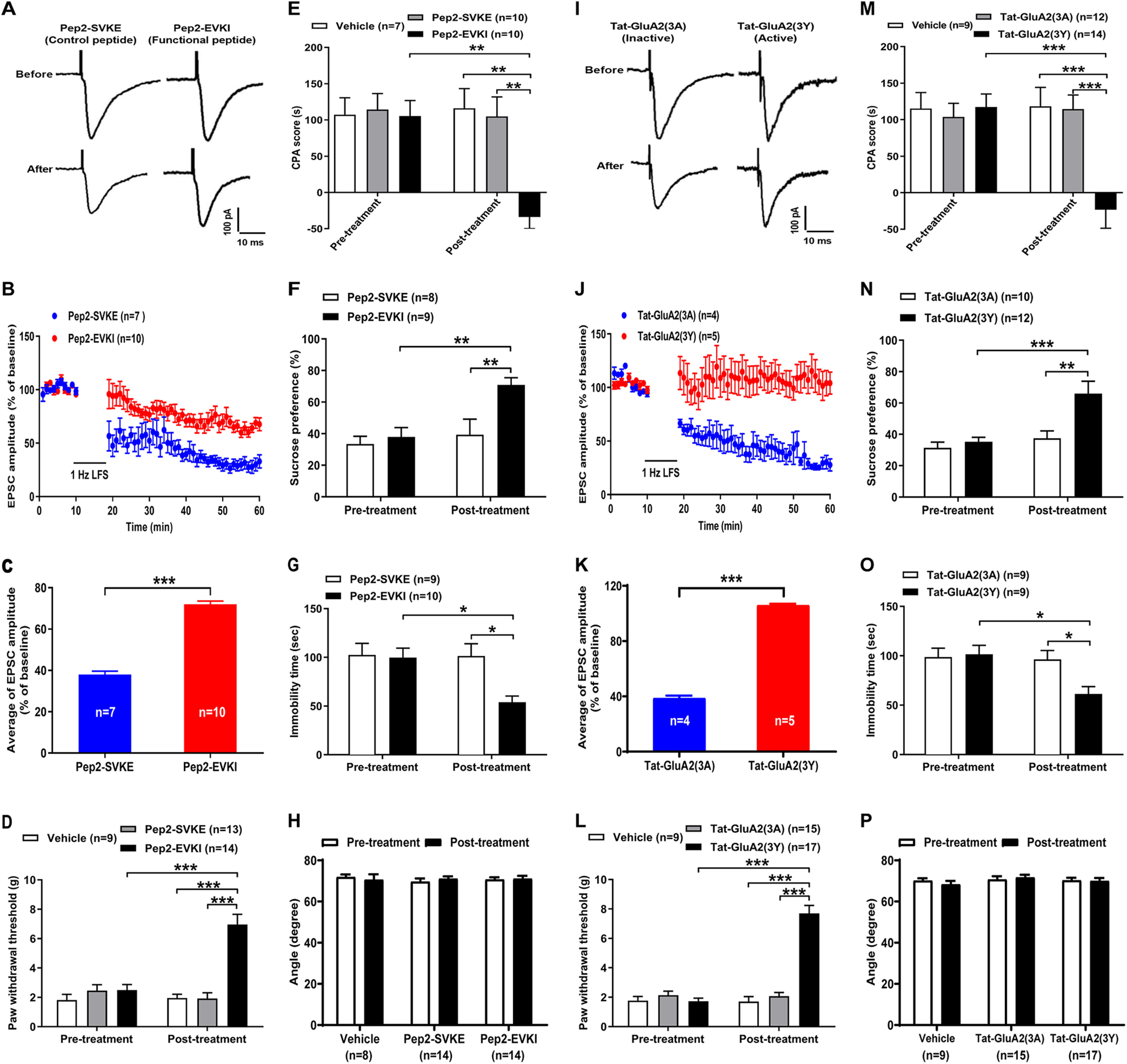
Disruption of GluA2-containing AMPAR endocytosis by the peptides abrogates the enhanced LTD at the LA/BLA-CeA synapse and comorbid aversive and depressive symptoms in neuropathic pain in SNL-operated rats. A–C, Preventing GluA2-containing AMPARs (GluA2-AMPARs) trafficking to and from the plasma membrane by pep2-EVKI peptide inhibits the enhancement of LTD at the LA/BLA-CeA synapse in SNL-operated rats. A, Representative traces of AMPAR-mediated eEPSCs at the LA/BLA-CeA synapse before and after LFS (480 pulses at 1 Hz with postsynaptic cell depolarized to −50 mV) in the presence of either pep2-EVKI peptide (100 μM) or control peptide pep2-SVKE (100 μM) in bicuculline (10 μM) and D-AP5 (50 μM) containing ACSF bath (to block both NMDARs and GABA receptor activities, and subsequently recording the uncontaminated AMPAR currents). Calibration: 100 pA, 10 ms. B, Average eEPSC amplitudes (normalized to baseline) over the time course of the LFS protocol in pep2-EVKI and pep2-SVKE groups. C, Average eEPSC amplitudes (normalized to baseline) are plotted during the 20-60 min following LFS in pep2-EVKI and pep2-SVKE groups (n = 7–10 rats, t(131) = 15.07, p < 0.0001 compared between pep2-EVKI and pep2-SVKE groups, two-tailed unpaired Student's t test). D–G, Bilateral microinjection of pep2-EVKI (10 μg/μl, 0.5 μl per side) in the CeA attenuates comorbid aversive and depressive-like symptoms in neuropathic pain in SNL-operated rats. D, PWT in response to von Frey filament stimuli in pep2-EVKI and pep2-SVKE groups (n = 9–14 rats per group, F(2,66) = 17.84, p < 0.0001 vs pep2-SVKE post-treatment, two-way ANOVA with Sidak's post hoc test). E, CPA test (n = 7-10 rats per group, F(2,48) = 5.69, p = 0.0014 vs pep2-SVKE post-treatment, two-way ANOVA with Sidak's post hoc test). F, SPT (n = 8 rats pep2-SVKE and 9 rats pep2-EVKI, F(1,30) = 4.33, p = 0.0092 vs pep2-SVKE post-treatment, two-way ANOVA with Sidak's post hoc test). G, FST (n = 9 or 10 rats per group, F(1,33) = 4.88, p = 0.0123 vs pep2-SVKE post-treatment, two-way ANOVA with Sidak's post hoc test). I-O, Preventing GluA2-AMPAR endocytosis by Tat-GluA2(3Y) peptide inhibits the enhancement of LTD at the LA/BLA-CeA synapse in SNL-operated rats. I, Representative traces of AMPAR-mediated eEPSCs at the LA/BLA-CeA synapse before and after LFS in the presence of either Tat-GluA2(3Y) peptide (10 μM) or control peptide Tat-GluA2(3A) (10 μM) in bicuculline (10 μM) and D-AP5 (50 μM) containing ACSF bath. Calibration: 100 pA, 10 ms. J, Average eEPSC amplitudes (normalized to baseline) over the time course of the LFS protocol in Tat-GluA2(3Y) and Tat-GluA2(3A) groups. K, Average eEPSC amplitudes (normalized to baseline) are plotted during the 20-60 min following LFS in Tat-GluA2(3Y) and Tat-GluA2(3A) groups (n = 4 rats Tat-GluA2(3A) and n = 5 rats Tat-GluA2(3YA), t(104) = 31.26, p < 0.0001 compared between Tat-GluA2(3Y) and Tat-GluA2(3A) groups, two-tailed unpaired Student's t test). L-O, Bilateral microinjection of GluA2(3Y) (75 pmol, 0.5 μl per side) in the CeA attenuates comorbid aversive and depressive-like symptoms in neuropathic pain in SNL-operated rats. L, PWT in response to von Frey filament stimuli in Tat-GluA2(3Y) and Tat-GluA2(3A) groups (n = 9-17 rats per group, F(2,75) = 51.09, p < 0.0001 vs Tat-GluA2(3A) post-treatment, two-way ANOVA with Sidak's post hoc test). M, CPA test (n = 9-14 rats per group, F(2,26) = 8.41, p = 0.0002 vs Tat-GluA2(3A) post-treatment, two-way ANOVA with Sidak's post hoc test). N, SPT (n = 10-12 rats per group, F(1,40) = 5.28, p = 0.0029 vs Tat-GluA2(3A) post-treatment, two-way ANOVA with Sidak's post hoc test). O, FST (n = 9 rats per group, F(1,32) = 4.81, p = 0.0349 vs Tat-GluA2(3A) post-treatment, two-way ANOVA with Sidak's post hoc test). H, P, Inclined-plate test measured before and after peptide microinjection in pep2-EVKI/pep2-SVKE (H) and Tat-GluA2(3Y)/Tat-GluA2(3A) (P) groups.
Additionally, the effects of these peptides on sham SNL rats were examined to test the specificity of the peptides (Fig. 7). The results from patch-clamp recording of amygdaloid slices showed that either preincubated amygdaloid slices with pep2-EVKI (100 μM) or with Tat-GluA2(3Y) (10 μM) had no significant effect on the LTD at the LA/BLA-CeA synapse compared with their corresponding control peptide (inactive or nonfunctional peptide), pep2-SVKE (100 μM), and Tat-GluA2(3A) (10 μM) (Fig. 7A,B,G,H). Likewise, results from the in vivo experiments revealed that, compared with their corresponding control peptide (pep2-SVKE or Tat-GluA2(3A)), bilateral microinjection of either pep2-EVKI (10 μg/μl, 0.5 μl per side) or Tat-GluA2(3Y) (75 pmol, 0.5 μl per side) into the CeA at 1 h before behavioral test, had no significant effect on the PWT (Fig. 7C,I), the CPA score (Fig. 7D,J), as well as on the sucrose preference (Fig. 7E,K) and the immobility time in the FST (Fig. 7F,L). Representative images of histologic verification of microinjection sites are presented in Figure 7M, N.
Figure 7.
Effects of microinjection of the peptides (pep2-EVKI/pep2-SVKE or Tat-GluA2(3Y)/Tat-GluA2(3A)) in bilateral CeA on LTD, pain sensitivity, and pain-related negative emotion in sham-operated rats. A–F, pep2-EVKI/pep2-SVKE. G-L, Tat-GluA2(3Y)/Tat-GluA2(3A). A, B, G, H, Effects of microinjection of the peptides on LTD of eEPSCs at the LA/BLA synapse. A, G, Representative traces of eEPSCs before and after LFS in pep2-EVKI/pep2-SVKE (A) and Tat-GluA2(3Y)/Tat-GluA2(3A) (G) groups. Calibration: 100 pA, 10 ms. B, H, Average eEPSC amplitudes (normalized to baseline) over the time course of the LFS protocol (480 pulses at 1 Hz with postsynaptic cell depolarized to −50 mV). C-F, I-L, Effects of microinjection of the peptides on pain sensitivity and pain-related negative emotion indicated by the PWT (C,I), CPA score (D,J), SPT (E,K), and immobility time in FST (F,L) in sham-operated rats (n = 6 rats per group). p > 0.05 (two-way ANOVA with Sidak's post hoc test). M, N, Histologically verified microinjection sites in the CeA. M, Examples of the Nissl-stained coronal section to illustrate the track of the microinjection cannula and the tip placement (as arrow shows) in left and right CeA. N, Representative four coronal sections through the rat amygdala are shown in sequence from anterior to posterior. Numbers in the right margin indicate millimeters posterior to the bregma. Filled circles in bilateral hemispheres represent the approximate positions of the cannula tips corresponding to some representative rats in the intra-CeA microinjection group. Diagrams are adapted from Paxinos and Watson (2014) and show coronal sections through bilateral hemispheres at different levels posterior to the bregma. Scale bar, 500 μm.
Furthermore, to determine the long-lasting effects of both pep2-EVKI and Tat-GluA2(3Y) on comorbid aversive and depressive symptoms in neuropathic pain, we performed additional experiments to examine the actions of intra-CeA overexpressing pep2-EVKI or Tat-GluA2(3Y) by infecting lentivirus-expressing pep2-EVKI or Tat-GluA2(3Y) linked with EGFP tag (abbreviated as LV-pep2-EVKI and LV-Tat-GluA2(3Y), respectively) to bilateral CeA neurons before SNL surgery to rats. Using immunofluorescence staining of GFP with NeuN (the neuronal marker), GFAP (the astrocyte marker), and Ibal-1 (the microglial cells marker) in the CeA tissues from the lentivirus injected rats, we observed a prominent GFP-positive expression with NeuN, as well as a few GFP-positive expressions with GFAP or Ibal-1 (Fig. 8) in the CeA. As expected, overexpression of either pep2-EVKI or Tat-GluA2(3Y) in bilateral CeA neurons of SNL rats could significantly prevent the decrease in spine density of CeA neurons, abrogate the enhanced LTD at the LA/BLA-CeA synapse of SNL rats, and attenuate the SNL-induced neuropathic pain and pain-related aversive and depressive-like behaviors last at least for 5 weeks after surgery until experiment termination (Fig. 9). Compared with their control peptides (pep2-SVKE, Tat-GluA2(3A)), overexpression of either pep2-EVKI or Tat-GluA2(3Y) produced a significant increase in dendritic spine density of CeA neurons in SNL rats (Fig. 9A–D) (in numbers/100 μm dendrite: 35.21 ± 2.80 pep2-EVKI vs 17.15 ± 1.51 pep2-SVKE, n = 14 pep2-EVKI and n = 13 pep2-SVKE, p < 0.0001, t(25) = 5.54, Fig. 9B; 32.54 ± 2.74 Tat-GluA2(3Y) vs 17.50 ± 1.43 Tat-GluA2(3A), n = 14 Tat-GluA2(3Y) and n = 13 Tat-GluA2(3A), p < 0.0001, t(25) = 4.98, Fig. 9D). Moreover, overexpression of either pep2-EVKI or Tat-GluA2(3Y) could significantly inhibit the enhanced LTD at the LA/BLA-CeA synapse in SNL rats (Fig. 9E–G,M–O). Compared with the baseline responses (averaged at 0-10 min pre-LFS), the mean eEPSC amplitude after LFS (20-60 min post-LFS) was 91.25 ± 0.93 of pep2-EVKI OE (functional peptide) versus 47.45 ± 0.81 of pep2-SVKE OE (control peptide) (n = 5 rats per group, p < 0.0001, t(78) = 35.48; Fig. 9G), and 94.38 ± 1.05 of Tat-GluA2(3Y) OE (active peptide) versus 58.11 ± 0.88 of Tat-GluA2(3A) OE (inactive peptide) (n = 5 rats per group, p < 0.0001, t(78) = 26.58; Fig. 9O), respectively. The behavioral results showed that overexpression of either pep2-EVKI or Tat-GluA2(3Y) could abrogate the decreased PWT of SNL-operated rats on day 35 after surgery (Pep2-EVK: n = 9-11 rats per group, p < 0.0001, Pep2-EVKI OE vs Pep2-SVKE OE, F(1,36) = 82.03, Fig. 9H; Tat-GluA2(3Y): n = 9 rats per group, p < 0.0001, Tat-GluA2(3Y) OE vs Tat-GluA2(3A) OE, F(1,32) = 35.27, Fig. 9P). Similarly, the enhanced CPA score (Pep2-EVK: n = 9-11 rats per group, p = 0.0033 Pep2-EVKI OE vs Pep2-SVKE OE, F(1,36) = 7.40, Fig. 9I; Tat-GluA2(3Y): n = 9 rats per group, p = 0.0001, Tat-GluA2(3Y) OE vs Tat-GluA2(3A) OE, F(1,36) = 12.91, Fig. 9Q), the reduced sucrose preference (Pep2-EVK: n = 9-11 rats per group, p < 0.0001, Pep2-EVKI OE vs Pep2-SVKE OE, F(1,36) = 23.45, Fig. 9J; Tat-GluA2(3Y): n = 9 rats per group, p < 0.0001, Tat-GluA2(3Y) OE vs Tat-GluA2(3A) OE, F(1,32) = 31.71, Fig. 9R), and the increased immobility time in the FST (Pep2-EVK: n = 9-11 rats per group, p = 0.0048, Pep2-EVKI OE vs Pep2-SVKE OE, F(1,36) = 8.94, Fig. 9K; Tat-GluA2(3Y): n = 9 rats per group, p = 0.0027, Tat-GluA2(3Y) OE vs Tat-GluA2(3A) OE, F(1,32) = 8.47, Fig. 9S) in SNL-operated rats were rescued by intra-CeA overexpressing pep2-EVKI or Tat-GluA2(3Y) compared with their control peptides (pep2-SVKE, Tat-GluA2(3A)). Also, the results from inclined-plate test revealed that the animal's locomotor function was not impaired after overexpressing either pep2-EVKI/pep2-SVKE or Tat-GluA2(3Y)/Tat-GluA2(3A) in the CeA in SNL rats (Fig. 9L,T). Together, these data present solid evidence supporting our hypothesis that, the endocytosis of GluA2-containing AMPARs contributes to the LTD at the LA/BLA-CeA synapse and the comorbid aversive and depressive symptoms in neuropathic pain in SNL-operated rats.
Figure 8.

Histologic identification of lentiviral expression and infected target cells using immunofluorescence staining with GFP and NeuN, GFAP, or Iba-1 in the CeA. Staining was performed on day 35 post-SNL/sham SNL surgery. Shown are representative images for the immunofluorescence staining of GFP (green) with NeuN (a neuronal marker), GFAP (an astrocyte marker), Iba-1 (a microglial marker), and DAPI (the nuclear marker) in the CeA tissue in sham-operated or SNL-operated rats, which received microinjection of lentivirus-expressing pep2-EVKI, pep2-SVKE, Tat-GluA2(3Y), and Tat-GluA2(3A), respectively. Scale bar, 25 μm. n = 3 biological replicates.
Figure 9.
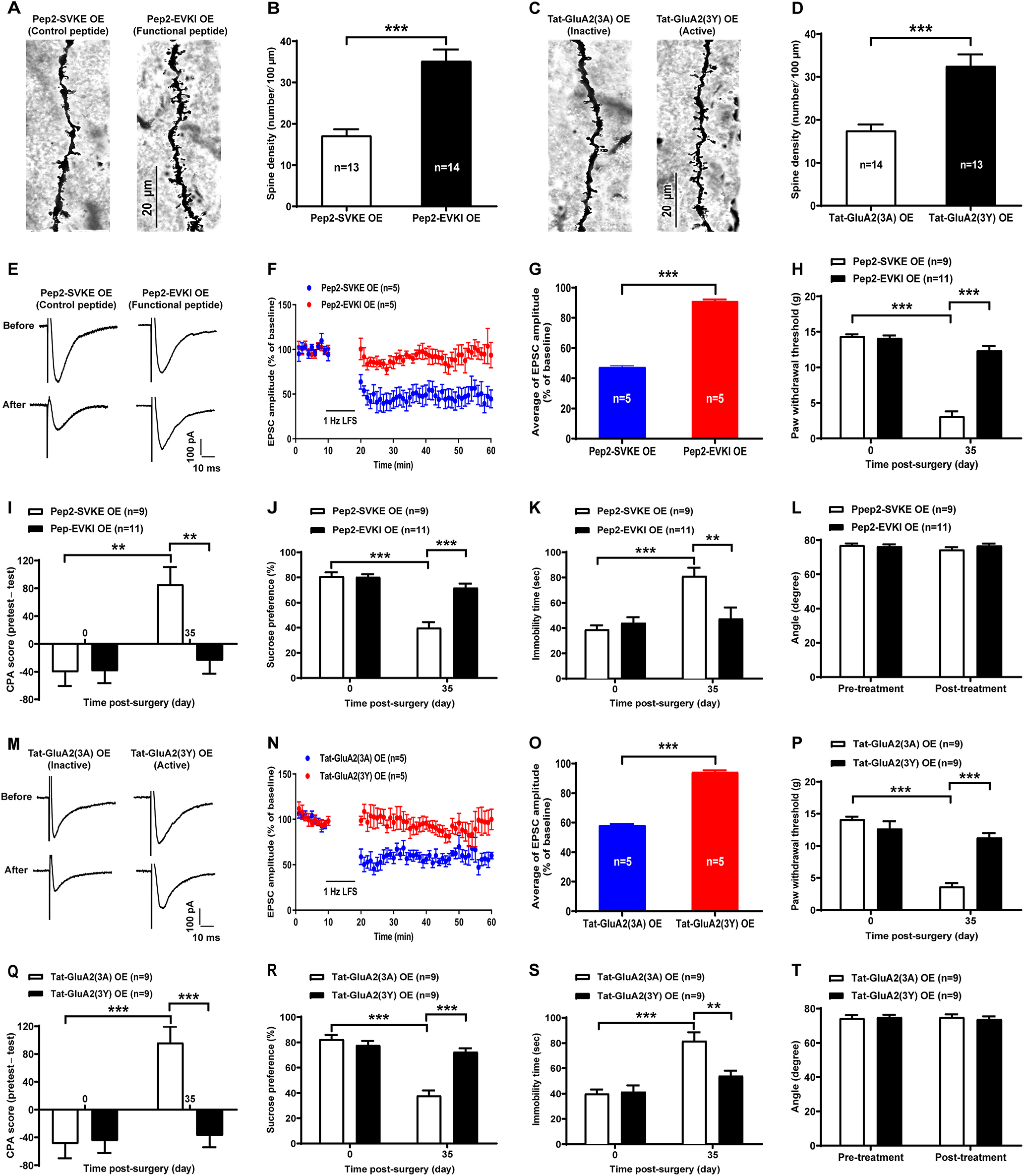
Effects of overexpressing pep2-EVKI or Tat-GluA2(3Y) in bilateral CeA neurons on spine density, LTD, as well as pain sensitivity and pain-related negative emotion in SNL-operated rats. A-D, Spine density of CeA neurons. A, C, Representative images of Golgi-Cox–stained dendritic spines in the CeA neurons overexpressed pep2-EVKI/pep2-SVKE (A) or Tat-GluA2(3Y)/Tat-GluA2(3A) (C). B, D, Summary plot of dendritic spine density expressed as the spine numbers per 100 μM dendrite length (n = 13 or 14 neurons from 5 rats per group, t(25) = 5.54 for B, t(25) = 4.50 for D). p < 0.0001 vs control peptide (two-tailed unpaired Student's t test). E–G, M–O, LTD of eEPSCs at the LA/BLA synapse. E, M, Representative traces of AMPAR-mediated eEPSCs at the LA/BLA-CeA synapse before and after LFS (480 pulses at 1 Hz with postsynaptic cell depolarized to −50 mV) in bicuculline (10 μM) and D-AP5 (50 μM) containing ACSF bath in SNL rats overexpressed pep2-EVKI/pep2-SVKE (E) or Tat-GluA2(3Y)/Tat-GluA2(3A) (M). Calibration: 100 pA, 10 ms. F, N, Average eEPSC amplitudes (normalized to baseline) over the time course of the LFS protocol in pep2-EVKI/pep2-SVKE (F) and Tat-GluA2(3Y)/Tat-GluA2(3A) (N) groups. G, O, Average eEPSC amplitudes (normalized to baseline) are plotted during the 20-60 min following LFS in pep2-EVKI/pep2-SVKE (G) and Tat-GluA2(3Y)/Tat-GluA2(3A) (O) groups (n = 5 rats, t(78) = 35.48 for G, t(78) = 26.58 for O). p < 0.0001 versus control peptide (two-tailed unpaired Student's t test). H, P, PWT in response to von Frey filament stimuli in SNL rats overexpressing (OE) pep2-EVKI or Tat-GluA2(3Y) compared with their control peptides (n = 9–11 per group, F(1,36) = 82.03 for H, F(1,32) = 35.27 for P). ***p < 0.001 (two-way ANOVA with Sidak's post hoc test). I, Q, CPA test (n = 9–11 per group, F(1,36) = 7.40 for I, F(1,36) = 12.91 for Q). **p < 0.01, ***p < 0.001 (two-way ANOVA with Sidak's post hoc test). J, R, SPT (n = 9–11 per group, F(1,36) = 23.45 for J, F(1,32) = 37.71 for R). ***p < 0.001 (two-way ANOVA with Sidak's post hoc test). K, S, FST (n = 9-11 per group, F(1,36) = 8.94 for K, F(1,32) = 8.47 for S). **p < 0.01, ***p < 0.001 (two-way ANOVA with Sidak's post hoc test). L, T, Inclined-plate test measured before and after lentiviral expressing in pep2-EVKI/pep2-SVKE OE (L) and Tat-GluA2(3Y)/Tat-GluA2(3A) OE (T) groups.
Discussion
A recent study has shown that the CeA is a pivotal relay station in a specific serotonergic pathway from the dorsal raphe nucleus to the lateral habenula in governing comorbid depressive symptoms in chronic pain (Zhou et al., 2019), whereas Corder et al. (2019) have identified in the BLA a critical neural ensemble target that mediates aversive chronic pain unpleasantness. To determine the key subregions of the amygdala that are involved in sensory and negative emotional components of neuropathic pain, we performed bilateral LA/BLA or CeA electrolytic lesions to SNL-operated rats before surgery. In line with Corder et al. (2019) reports showing that, silencing the BLA nociceptive ensemble alleviates aversive pain unpleasantness without altering the sensory detection of noxious stimuli or withdrawal reflexes, we found that bilateral LA/BLA lesions only attenuated aversive emotion and partly inhibited depressive-like behaviors, but did not affect pain sensitivity itself in SNL-operated rats, implying the main contribution of the LA/BLA in the emotional-affective dimension of pain processing. In contrast, bilateral lesions of CeA not only alleviated pain hypersensitivity but also abrogated aversive and depressive symptoms of neuropathic rats, suggesting the key role of CeA in processing both sensory and negative emotional-affective components of chronic pain. It has been demonstrated that the latero-capsular part of the CeA, which is termed nociceptive amygdala, receives nociceptive-specific information directly from the spinal cord and brainstem through the spino-parabrachio-amygdaloid pain pathway, whereas the LA/BLA, which receives polymodal sensory information, including emotional-affective information from cortical and thalamic areas, is relayed to the CeA. The CeA integrates polysensory and nociceptive-specific information, attaching emotional significance to painful stimuli (Thompson and Neugebauer, 2017). These findings may partly explain that, although the LA/BLA-CeA synaptic transmission is important, the lesion of LA/BLA only affects the emotional components but not sensory components of pain, whereas the lesion of CeA affects both sensory and emotional components of pain.
Structural and functional neuroplasticity in the amygdala is implicated in stress response, emotional processing, neuropsychiatric disorders, and the emotional-affective dimension of pain (Qiao et al., 2016; Thompson and Neugebauer, 2017). Chronic stress causes an increase in dendritic arborization and spine density on pyramidal neurons of the BLA (Mitra et al., 2005; Qin et al., 2011; Padival et al., 2013) but not the CeA (Vyas et al., 2003; Cui et al., 2008); and these neuroplastic changes are highly related to stress-induced anxiety (Patel et al., 2018; Zhang et al., 2019) and depressive-like behaviors (Yi et al., 2017; Yang et al., 2020). Our present data showed that the SNL-operated rats had less dendritic spine density on the CeA neurons at 4 weeks after surgery when they emerged pain hypersensitivity and pain-related negative emotion, supporting the understanding that plastic changes in the CeA are also important for comorbid aversive and depressive-like symptoms in neuropathic pain. We speculated that these discrepant findings are probably because of the differences in experimental model used, in days after surgery, or in different neuronal populations examined between our present study and previous studies. Indeed, in line with previous reports, we found a significant increase in dendritic spine density, as well as in total and average dendrite length, and in dendritic arborization of LA/BLA neurons in SNL-operated rats. Also, a decrease in neuronal dendritic spine density in the CeA and medial nucleus of amygdala emerges in anxiety-like rats (Moonat et al., 2011; You et al., 2014; Stefaniuk et al., 2017), and in rats with emotional instability or depression (Matsuo et al., 2017). During synaptic plasticity, the number and shapes of dendritic spines undergo radical reorganizations. LTP induction promotes spine head enlargement as well as the formation and stabilization of new spines, whereas LTD results in their shrinkage and retraction. Ample evidence exists supporting that LTP and LTD are associated with bidirectional modifications in spine number and/or morphology. LTP is accompanied by enlargement of existing spines and/or formation of new spines, whereas LTD is linked to spine shrinkage and/or elimination (Asrar and Jia, 2013; Chidambaram et al., 2019). In line with this understanding, we indeed found that the functional synaptic plasticity, such as LTP and LTD in the LA/BLA-CeA pathway, could be induced in both SNL-operated rats and sham-operated rats, in which no significant difference in LTP amplitude and input-output relationship of fEPSP amplitude was observed between the two groups, whereas an enhanced LTD at the LA/BLA-CeA synapse emerged in SNL-induced neuropathic rats, suggesting that LTD at the LA/BLA-CeA synapse, associated with the loss of dendritic spine on the CeA neurons, likely underlies the pathogenesis of neuropathic pain and pain-related negative emotion, including aversive and depressive-like symptoms.
In addition, we have examined mEPSCs and PPR of eEPSC at the LA/BLA-CeA synapse in naïve, sham-operated, and SNL-operated rats, both of mEPSCs and PPR may reflect the basal synaptic properties. We found a significant decrease in amplitude, but not in frequency, of mEPSCs in SNL-operated rats compared with control rats. Also, no difference in PPR of eEPSC at the LA/BLA-CeA synapse was observed at each IPI among naïve, sham-operated, and SNL-operated rats. In this study, we observed an unexpected result; that is, in SNL-operated rats, there was a significant decrease in dendritic spine density of CeA neurons and a large decrease in eEPSC amplitude (the input-output relationship of eEPSCs), but no desired decrease in mEPSCs frequency was found. Similar results were also observed in a previous study, in which they found that, in 8 DIV cultured neurons silenced for 24 h, there was a decrease in synapse number, but a statistically insignificant decrease in mEPSC frequency (Han and Stevens, 2009). We do not know the exact reasons for this discrepancy because various factors affect the mEPSC frequency and amplitude. For example, mEPSC frequency depends on at least three factors: synapse number, size of the readily releasable pool (RRP) of synaptic vesicles, and release rate per vesicle (Han and Stevens, 2009). It is possible that an increase in silent synapses (Isaac et al., 1996; Nakayama et al., 2005) or any changes in the release rate per vesicle (Han and Stevens, 2009) could offset the loss of synapses in the CeA neurons of SNL rats. In addition, evidence has been documented that the population of morphologically docked synaptic vesicles, called the RRP, corresponds to the release-ready neurotransmitter quanta (Schikorski and Stevens, 2001). An increase in RRP of each synapse would predict an increase in release probability, and this could offset the reduction of mEPSC frequency in neurons with spines lost (Ultanir et al., 2007). Additionally, several studies in cultured hippocampal slices and cortical neurons have observed an increase in the number of shaft synapses while dendritic spines are lost (Fishbein and Segal, 2007; Mateos et al., 2007). These shaft synapses could also compensate for the loss of dendritic spines and subsequently offset the desired decrease in mEPSC frequency (Segal, 2010). Also, dendritic spine morphology plays a pivotal role in higher brain functions, such as learning and memory (Sekino et al., 2007). Generally, there are four types of dendritic spines: filopodia, thin, stubby, and mushroom, of which the first two are immature and the last two are stable and mature (Tanokashira et al., 2012). Some studies have demonstrated that the number of mature spines is correlated with synaptic plasticity (Kreple et al., 2014; Francis et al., 2017), whereas immature synapses comprised lots of filopodia spines which may release neurotransmitters in higher level (Andreae et al., 2012; Burk et al., 2018). Therefore, the dendritic spine morphology associated with its dynamic change could also affect mEPSC frequency (Sekino et al., 2007). An increase in elongated dendritic filopodia, which recruits (or contains) more excitatory synapse number on dendritic shafts (Fishbein and Segal, 2007; Segal, 2010; Vessey et al., 2010), could offset the loss of dendritic spines and subsequently abrogate the reduction of mEPSC frequency in the CeA neurons of SNL rats. Of course, the above lines of evidence just provide some possible explanations for the discrepancy of unchanged mEPSC frequency with the loss of dendritic spines in the CeA neurons of SNL rats. The exact causes for this unexpected result need further investigation.
Phosphorylation of postsynaptic AMPARs is critical for regulating synaptic plasticity (Park, 2018). For example, phosphorylation of GluA1Ser831 and GluA1Ser845 is implicated in regulating GluA1-containing, CP-AMPAR insertion into the postsynaptic membrane, and facilitating LTP induction, whereas phosphorylation of GluA2Ser880 is essential for promoting GluA2-containing, CI-AMPAR endocytosis and leads to LTD (Diering and Huganir, 2018; Park, 2018). Our findings of decreased pGluA1Ser845 and increased pGluA2Ser880 in the CeA synaptosomal fraction raise a possibility that SNL surgery may lead to either an impaired LTP at the LA/BLA-CeA synapse due to the reduction of CP-AMPAR insertion into the postsynaptic membrane, or a facilitated LTD resulting from CI-AMPAR endocytosis, or both. Moreover, we found a significant decrease in AMPAR-mediated eEPSCs (AMPAR currents) evoked at the LA/BLA-CeA synapse of SNL-operated rats, further confirming the functional reduction of AMPARs in postsynaptic membrane of the CeA neurons. Also, we found a consistent, linear I–V relationship of AMPAR currents among naïve, sham-operated, and SNL-operated rats, and that application of NASPM, a selective CP-AMPAR antagonist to block GluA2-lacking AMPARs (Gerace et al., 2014), had no effect on these AMPAR-mediated currents. It is well accepted that the GluA2-lacking AMPARs are CP-AMPARs and show inward rectification I–V relationship, whereas the GluA2-containing AMPARs are CI-AMPARs and have a linear I–V relationship (Greger et al., 2017). We thus speculated that, under neuropathic condition, phosphorylation of GluA2Ser880 might result in the internalization of GluA2-containing, CI-AMPARs from postsynaptic membrane, which is considered as a contributor of LTD at the LA/BLA-CeA synapse (Diering and Huganir, 2018; Park, 2018). Indeed, two synthetic peptides, pep2-EVKI and Tat-GluA2(3Y), which have been shown to be able to prevent the GluA2-containing AMPARs (CI-AMPARs) trafficking and endocytosis from synapses, respectively (Brebner et al., 2005; Famous et al., 2008; Benneyworth et al., 2019), could abrogate the SNL-induced enhancement of LTD at the LA/BLA-CeA synapse in vitro, and alleviated the SNL-induced mechanical allodynia and comorbid aversive and depressive symptoms in vivo, suggesting that the endocytosis of GluA2-containing AMPARs is probably related to the LTD at the LA/BLA-CeA synapse and the comorbid aversive and depressive symptoms in neuropathic pain in SNL-operated rats.
In conclusion, this study demonstrates that the CeA plays a key role in processing both sensory and negative emotional-affective components of neuropathic pain. Endocytosis of GluA2-containing AMPARs from synapses owing to the phosphorylation of GluA2Ser880 mediates LTD at the LA/BLA-CeA synapse, which might be associated with the pathogenesis of neuropathic pain and pain-related negative emotion, such as aversive and depressive-like symptoms in SNL-operated rats.
Footnotes
This work was supported by National Natural Science Foundation of China Grants 81974169, 81671085, and 61527815; and National Key R&D Program of China 2019YFC1712104.
The authors declare no competing financial interests.
References
- Abraham WC, Jones OD, Glanzman DL (2019) Is plasticity of synapses the mechanism of long-term memory storage? NPJ Sci Learn 4:9. 10.1038/s41539-019-0048-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreae LC, Fredj NB, Burrone J (2012) Independent vesicle pools underlie different modes of release during neuronal development. J Neurosci 32:1867–1874. 10.1523/JNEUROSCI.5181-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrar S, Jia Z (2013) Molecular mechanisms coordinating functional and morphological plasticity at the synapse: role of GluA2/N-cadherin interaction-mediated actin signaling in mGluR-dependent LTD. Cell Signal 25:397–402. 10.1016/j.cellsig.2012.11.007 [DOI] [PubMed] [Google Scholar]
- Bäckström T, Sanders D, Leask R, Davidson D, Warner P, Bancroft J (1983) Mood, sexuality, hormones, and the menstrual cycle: II. Hormone levels and their relationship to the premenstrual syndrome. Psychosom Med 45:503–507. 10.1097/00006842-198312000-00004 [DOI] [PubMed] [Google Scholar]
- Baliki MN, Apkarian AV (2015) Nociception, pain, negative moods, and behavior selection. Neuron 87:474–491. 10.1016/j.neuron.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benneyworth MA, Hearing MC, Asp AJ, Madayag A, Ingebretson AE, Schmidt CE, Silvis KA, Larson EB, Ebner SR, Thomas MJ (2019) Synaptic depotentiation and mGluR5 activity in the nucleus accumbens drive cocaine-primed reinstatement of place preference. J Neurosci 39:4785–4796. 10.1523/JNEUROSCI.3020-17.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume SR, Freedberg M, Vantrease JE, Chan R, Padival M, Record MJ, DeJoseph MR, Urban JH, Rosenkranz JA (2017) Sex- and estrus-dependent differences in rat basolateral amygdala. J Neurosci 37:10567–10586. 10.1523/JNEUROSCI.0758-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanova OV, Kanekar S, D'Anci KE, Renshaw PF (2013) Factors influencing behavior in the forced swim test. Physiol Behav 118:227–239. 10.1016/j.physbeh.2013.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT (2005) Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310:1340–1343. 10.1126/science.1116894 [DOI] [PubMed] [Google Scholar]
- Burk K, Ramachandran B, Ahmed S, Hurtado-Zavala JI, Awasthi A, Benito E, Faram R, Ahmad H, Swaminathan A, McIlhinney J, Fischer A, Perestenko P, Dean C (2018) Regulation of dendritic spine morphology in hippocampal neurons by copine-6. Cereb Cortex 28:1087–1104. 10.1093/cercor/bhx009 [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chen L, Li S, Cai J, Wei TJ, Liu LY, Zhao HY, Liu BH, Jing HB, Jin ZR, Liu M, Wan Y, Xing GG (2018) Activation of CRF/CRFR1 signaling in the basolateral nucleus of the amygdala contributes to chronic forced swim-induced depressive-like behaviors in rats. Behav Brain Res 338:134–142. 10.1016/j.bbr.2017.10.027 [DOI] [PubMed] [Google Scholar]
- Chidambaram SB, Rathipriya AG, Bolla SR, Bhat A, Ray B, Mahalakshmi AM, Manivasagam T, Thenmozhi AJ, Essa MM, Guillemin GJ, Chandra R, Sakharkar MK (2019) Dendritic spines: revisiting the physiological role. Prog Neuropsychopharmacol Biol Psychiatry 92:161–193. 10.1016/j.pnpbp.2019.01.005 [DOI] [PubMed] [Google Scholar]
- Corder G, Ahanonu B, Grewe BF, Wang D, Schnitzer MJ, Scherrer G (2019) An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363:276–281. 10.1126/science.aap8586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Sakamoto H, Higashi S, Kawata M (2008) Effects of single-prolonged stress on neurons and their afferent inputs in the amygdala. Neuroscience 152:703–712. 10.1016/j.neuroscience.2007.12.028 [DOI] [PubMed] [Google Scholar]
- Dan R, Canetti L, Keadan T, Segman R, Weinstock M, Bonne O, Reuveni I, Goelman G (2019) Sex differences during emotion processing are dependent on the menstrual cycle phase. Psychoneuroendocrinology 100:85–95. 10.1016/j.psyneuen.2018.09.032 [DOI] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT (2000) PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron 28:873–886. 10.1016/S0896-6273(00)00160-4 [DOI] [PubMed] [Google Scholar]
- Debanne D, Guérineau NC, Gähwiler BH, Thompson SM (1996) Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol 491:163–176. 10.1113/jphysiol.1996.sp021204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detke MJ, Rickels M, Lucki I (1995) Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology (Berl) 121:66–72. 10.1007/BF02245592 [DOI] [PubMed] [Google Scholar]
- Diering GH, Huganir RL (2018) The AMPA receptor code of synaptic plasticity. Neuron 100:314–329. 10.1016/j.neuron.2018.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Cai J, Li S, Liu XD, Wan Y, Xing GG (2015) BDNF contributes to the development of neuropathic pain by induction of spinal long-term potentiation via SHP2 associated GluN2B-containing NMDA receptors activation in rats with spinal nerve ligation. Neurobiol Dis 73:428–451. 10.1016/j.nbd.2014.10.025 [DOI] [PubMed] [Google Scholar]
- Dixon WJ (1980) Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20:441–462. 10.1146/annurev.pa.20.040180.002301 [DOI] [PubMed] [Google Scholar]
- Doan L, Manders T, Wang J (2015) Neuroplasticity underlying the comorbidity of pain and depression. Neural Plast 2015:504691. 10.1155/2015/504691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza D, Sadananda M (2017) Estrous cycle phase-dependent changes in anxiety- and depression-like profiles in the late adolescent Wistar-Kyoto rat. Ann Neurosci 24:136–145. 10.1159/000477151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG (2001) Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. J Neurosci 21:5546–5558. 10.1523/JNEUROSCI.21-15-05546.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Baynes BB, Carmichael CY, Zamora-Martinez ER, Barrus M, Koob GF, Gilpin NW (2013) Traumatic stress reactivity promotes excessive alcohol drinking and alters the balance of prefrontal cortex-amygdala activity. Transl Psychiatry 3:e296. 10.1038/tp.2013.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famous KR, Kumaresan V, Sadri-Vakili G, Schmidt HD, Mierke DF, Cha JH, Pierce RC (2008) Phosphorylation-dependent trafficking of GluR2-containing AMPA receptors in the nucleus accumbens plays a critical role in the reinstatement of cocaine seeking. J Neurosci 28:11061–11070. 10.1523/JNEUROSCI.1221-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero DM, Lemon JK, Fluegge D, Pashkovski SL, Korzan WJ, Datta SR, Spehr M, Fendt M, Liberles SD (2011) Detection and avoidance of a carnivore odor by prey. Proc Natl Acad Sci USA 108:11235–11240. 10.1073/pnas.1103317108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein I, Segal M (2007) Miniature synaptic currents become neurotoxic to chronically silenced neurons. Cereb Cortex 17:1292–1306. 10.1093/cercor/bhl037 [DOI] [PubMed] [Google Scholar]
- Francis TC, Chandra R, Gaynor A, Konkalmatt P, Metzbower SR, Evans B, Engeln M, Blanpied TA, Lobo MK (2017) Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol Psychiatry 22:1512–1519. 10.1038/mp.2017.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer VG (2020) Pharmacological sex hormone manipulation as a risk model for depression. J Neurosci Res 98:1283–1292. 10.1002/jnr.24632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng SJ, Liao FF, Dang WH, Ding X, Liu XD, Cai J, Han JS, Wan Y, Xing GG (2010) Contribution of the spinal cord BDNF to the development of neuropathic pain by activation of the NR2B-containing NMDA receptors in rats with spinal nerve ligation. Exp Neurol 222:256–266. 10.1016/j.expneurol.2010.01.003 [DOI] [PubMed] [Google Scholar]
- Gerace E, Masi A, Resta F, Felici R, Landucci E, Mello T, Pellegrini-Giampietro DE, Mannaioni G, Moroni F (2014) PARP-1 activation causes neuronal death in the hippocampal CA1 region by increasing the expression of Ca(2+)-permeable AMPA receptors. Neurobiol Dis 70:43–52. 10.1016/j.nbd.2014.05.023 [DOI] [PubMed] [Google Scholar]
- Goncalves L, Silva R, Pinto-Ribeiro F, Pego JM, Bessa JM, Pertovaara A, Sousa N, Almeida A (2008) Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp Neurol 213:48–56. 10.1016/j.expneurol.2008.04.043 [DOI] [PubMed] [Google Scholar]
- Greger IH, Watson JF, Cull-Candy SG (2017) Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron 94:713–730. 10.1016/j.neuron.2017.04.009 [DOI] [PubMed] [Google Scholar]
- Hales CA, Stuart SA, Anderson MH, Robinson ES (2014) Modelling cognitive affective biases in major depressive disorder using rodents. Br J Pharmacol 171:4524–4538. 10.1111/bph.12603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YY, Chen ZH, Shang YJ, Yan WW, Wu BY, Li CH (2019) Cordycepin improves behavioral-LTP and dendritic structure in hippocampal CA1 area of rats. J Neurochem 151:79–90. 10.1111/jnc.14826 [DOI] [PubMed] [Google Scholar]
- Han JS, Li W, Neugebauer V (2005) Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci 25:10717–10728. 10.1523/JNEUROSCI.4112-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han EB, Stevens CF (2009) Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc Natl Acad Sci USA 106:10817–10822. 10.1073/pnas.0903603106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström B, Anderberg UM (2003) Pain perception across the menstrual cycle phases in women with chronic pain. Percept Mot Skills 96:201–211. 10.2466/pms.2003.96.1.201 [DOI] [PubMed] [Google Scholar]
- Hernández-Hernández OT, Martínez-Mota L, Herrera-Pérez JJ, Jiménez-Rubio G (2019) Role of estradiol in the expression of genes involved in serotonin neurotransmission: implications for female depression. Curr Neuropharmacol 17:459–471. 10.2174/1570159X16666180628165107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibironke GF, Aji KE (2011) Pain threshold variations in female rats as a function of the estrus cycle. Niger J Physiol Sci 26:67–70. [PubMed] [Google Scholar]
- Isaac JT, Oliet SH, Hjelmstad GO, Nicoll RA, Malenka RC (1996) Expression mechanisms of long-term potentiation in the hippocampus. J Physiol Paris 90:299–303. 10.1016/s0928-4257(97)87901-6 [DOI] [PubMed] [Google Scholar]
- Jenkins JA, Williams P, Kramer GL, Davis LL, Petty F (2001) The influence of gender and the estrous cycle on learned helplessness in the rat. Biol Psychol 58:147–158. 10.1016/s0301-0511(01)00111-9 [DOI] [PubMed] [Google Scholar]
- Jensen TS, Finnerup NB (2014) Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol 13:924–935. 10.1016/S1474-4422(14)70102-4 [DOI] [PubMed] [Google Scholar]
- Jiang H, Fang D, Kong LY, Jin ZR, Cai J, Kang XJ, Wan Y, Xing GG (2014) Sensitization of neurons in the central nucleus of the amygdala via the decreased GABAergic inhibition contributes to the development of neuropathic pain-related anxiety-like behaviors in rats. Mol Brain 7:72. 10.1186/s13041-014-0072-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Fields HL, Manning BH (2001) The affective component of pain in rodents: direct evidence for a contribution of the anterior cingulate cortex. Proc Natl Acad Sci USA 98:8077–8082. 10.1073/pnas.141218998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato F, Sugimura YK, Takahashi Y (2018) Pain-associated neural plasticity in the parabrachial to central amygdala circuit: pain changes the brain, and the brain changes the pain. Adv Exp Med Biol 1099:157–166. 10.1007/978-981-13-1756-9_14 [DOI] [PubMed] [Google Scholar]
- Kaur S, Benton WL, Tongkhuya SA, Lopez CM, Uphouse L, Averitt DL (2018) Sex differences and estrous cycle effects of peripheral serotonin-evoked rodent pain behaviors. Neuroscience 384:87–100. 10.1016/j.neuroscience.2018.05.017 [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363. [DOI] [PubMed] [Google Scholar]
- Kreple CJ, Lu Y, Taugher RJ, Schwager-Gutman AL, Du J, Stump M, Wang Y, Ghobbeh A, Fan R, Cosme CV, Sowers LP, Welsh MJ, Radley JJ, LaLumiere RT, Wemmie JA (2014) Acid-sensing ion channels contribute to synaptic transmission and inhibit cocaine-evoked plasticity. Nat Neurosci 17:1083–1091. 10.1038/nn.3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R, Flor H (2016) Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci 18:20–30. 10.1038/nrn.2016.162 [DOI] [PubMed] [Google Scholar]
- Li MJ, Liu LY, Chen L, Cai J, Wan Y, Xing GG (2017) Chronic stress exacerbates neuropathic pain via the integration of stress-affect-related information with nociceptive information in the central nucleus of the amygdala. Pain 158:717–739. 10.1097/j.pain.0000000000000827 [DOI] [PubMed] [Google Scholar]
- Marrocco J, McEwen BS (2016) Sex in the brain: hormones and sex differences. Dialogues Clin Neurosci 18:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos JM, Lüthi A, Savic N, Stierli B, Streit P, Gähwiler BH, McKinney RA (2007) Synaptic modifications at the CA3-CA1 synapse after chronic AMPA receptor blockade in rat hippocampal slices. J Physiol 581:129–138. 10.1113/jphysiol.2006.120550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo S, Matsuda KI, Takanami K, Mori T, Tanaka M, Kawata M, Kitawaki J (2017) Decrease in neuronal spine density in the postpartum period in the amygdala and bed nucleus of the stria terminalis in rat. Neurosci Lett 641:21–25. 10.1016/j.neulet.2017.01.040 [DOI] [PubMed] [Google Scholar]
- McEwen BS, Milner TA (2017) Understanding the broad influence of sex hormones and sex differences in the brain. J Neurosci Res 95:24–39. 10.1002/jnr.23809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P (1997) Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390:607–611. 10.1038/37605 [DOI] [PubMed] [Google Scholar]
- Mitra R, Jadhav S, McEwen BS, Vyas A, Chattarji S (2005) Stress duration modulates the spatiotemporal patterns of spine formation in the basolateral amygdala. Proc Natl Acad Sci USA 102:9371–9376. 10.1073/pnas.0504011102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonat S, Sakharkar AJ, Zhang H, Pandey SC (2011) The role of amygdaloid brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein and dendritic spines in anxiety and alcoholism. Addict Biol 16:238–250. 10.1111/j.1369-1600.2010.00275.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Kiyosue K, Taguchi T (2005) Diminished neuronal activity increases neuron-neuron connectivity underlying silent synapse formation and the rapid conversion of silent to functional synapses. J Neurosci 25:4040–4051. 10.1523/JNEUROSCI.4115-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V (2015) Amygdala pain mechanisms. Handb Exp Pharmacol 227:261–284. 10.1007/978-3-662-46450-2_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W (2003) Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J Neurophysiol 89:716–727. 10.1152/jn.00799.2002 [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G, Gereau RW (2003) Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci 23:52–63. 10.1523/JNEUROSCI.23-01-00052.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS (2004) The amygdala and persistent pain. Neuroscientist 10:221–234. 10.1177/1073858403261077 [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Mazzitelli M, Cragg B, Ji G, Navratilova E, Porreca F (2020) Amygdala, neuropeptides, and chronic pain-related affective behaviors. Neuropharmacology 170:108052. 10.1016/j.neuropharm.2020.108052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padival M, Quinette D, Rosenkranz JA (2013) Effects of repeated stress on excitatory drive of basal amygdala neurons in vivo. Neuropsychopharmacology 38:1748–1762. 10.1038/npp.2013.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J (2018) Phosphorylation of the AMPAR-TARP complex in synaptic plasticity. Proteomes 6:40. 10.3390/proteomes6040040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Anilkumar S, Chattarji S, Buwalda B (2018) Repeated social stress leads to contrasting patterns of structural plasticity in the amygdala and hippocampus. Behav Brain Res 347:314–324. 10.1016/j.bbr.2018.03.034 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (2014) The rat brain in stereotaxic coordinates, Ed 7. San Diego: Academic. [DOI] [PubMed] [Google Scholar]
- Qiao H, Li MX, Xu C, Chen HB, An SC, Ma XM (2016) Dendritic spines in depression: what we learned from animal models. Neural Plast 2016:8056370. 10.1155/2016/8056370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Xia Z, Huang T, Smith CB (2011) Effects of chronic immobilization stress on anxiety-like behavior and basolateral amygdala morphology in Fmr1 knockout mice. Neuroscience 194:282–290. 10.1016/j.neuroscience.2011.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin AS, Tator CH (1977) Objective clinical assessment of motor function after experimental spinal cord injury in the rat. J Neurosurg 47:577–581. 10.3171/jns.1977.47.4.0577 [DOI] [PubMed] [Google Scholar]
- Rozas G, Guerra MJ, Labandeira-García JL (1997) An automated rotarod method for quantitative drug-free evaluation of overall motor deficits in rat models of parkinsonism. Brain Res Brain Res Protoc 2:75–84. 10.1016/s1385-299x(97)00034-2 [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF (2001) Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci 4:391–395. 10.1038/86042 [DOI] [PubMed] [Google Scholar]
- Schmidt PJ, Rubinow DR (2009) Sex hormones and mood in the perimenopause. Ann NY Acad Sci 1179:70–85. 10.1111/j.1749-6632.2009.04982.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber AL, Lu YL, Baynes BB, Richardson HN, Gilpin NW (2017) Corticotropin-releasing factor in ventromedial prefrontal cortex mediates avoidance of a traumatic stress-paired context. Neuropharmacology 113:323–330. 10.1016/j.neuropharm.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M (2010) Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur J Neurosci 31:2178–2184. 10.1111/j.1460-9568.2010.07270.x [DOI] [PubMed] [Google Scholar]
- Sekino Y, Kojima N, Shirao T (2007) Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem Int 51:92–104. 10.1016/j.neuint.2007.04.029 [DOI] [PubMed] [Google Scholar]
- Stefaniuk M, Beroun A, Lebitko T, Markina O, Leski S, Meyza K, Grzywacz A, Samochowiec J, Samochowiec A, Radwanska K, Kaczmarek L (2017) Matrix metalloproteinase-9 and synaptic plasticity in the central amygdala in control of alcohol-seeking behavior. Biol Psychiatry 81:907–917. 10.1016/j.biopsych.2016.12.026 [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Redman SJ (1991) Mechanisms of presynaptic inhibition studied using paired-pulse facilitation. Neurosci Lett 126:179–183. 10.1016/0304-3940(91)90548-8 [DOI] [PubMed] [Google Scholar]
- Tanokashira D, Morita T, Hayashi K, Mayanagi T, Fukumoto K, Kubota Y, Yamashita T, Sobue K (2012) Glucocorticoid suppresses dendritic spine development mediated by down-regulation of caldesmon expression. J Neurosci 32:14583–14591. 10.1523/JNEUROSCI.2380-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM (2018) Corticolimbic circuitry in the modulation of chronic pain and substance abuse. Prog Neuropsychopharmacol Biol Psychiatry 87:263–268. 10.1016/j.pnpbp.2017.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JM, Neugebauer V (2017) Amygdala plasticity and pain. Pain Res Manag 2017:8296501. 10.1155/2017/8296501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62:405–496. 10.1124/pr.109.002451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultanir SK, Kim JE, Hall BJ, Deerinck T, Ellisman M, Ghosh A (2007) Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc Natl Acad Sci USA 104:19553–19558. 10.1073/pnas.0704031104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veinante P, Yalcin I, Barrot M (2013) The amygdala between sensation and affect: a role in pain. J Mol Psychiatry 1:9. 10.1186/2049-9256-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey JP, Schoderboeck L, Gingl E, Luzi E, Riefler J, Di Leva F, Karra D, Thomas S, Kiebler MA, Macchi P (2010) Mammalian Pumilio 2 regulates dendrite morphogenesis and synaptic function. Proc Natl Acad Sci USA 107:3222–3227. 10.1073/pnas.0907128107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent K, Tracey I (2010) Sex hormones and pain: the evidence from functional imaging. Curr Pain Headache Rep 14:396–403. 10.1007/s11916-010-0139-1 [DOI] [PubMed] [Google Scholar]
- Vyas A, Bernal S, Chattarji S (2003) Effects of chronic stress on dendritic arborization in the central and extended amygdala. Brain Res 965:290–294. 10.1016/s0006-8993(02)04162-8 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Nakagawa T, Yamamoto R, Maeda A, Minami M, Satoh M (2002) Involvement of glutamate receptors within the central nucleus of the amygdala in naloxone-precipitated morphine withdrawal-induced conditioned place aversion in rats. Jpn J Pharmacol 88:399–406. 10.1254/jjp.88.399 [DOI] [PubMed] [Google Scholar]
- Weinstock M (2017) Prenatal stressors in rodents: effects on behavior. Neurobiol Stress 6:3–13. 10.1016/j.ynstr.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton W, Gleason CE, Olson SR, Carlsson CM, Asthana S (2012) Neurobiological underpinnings of the estrogen-mood relationship. Curr Psychiatry Rev 8:247–256. 10.2174/157340012800792957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Manabe T, Nicoll RA (1994) A rise in postsynaptic Ca2+ potentiates miniature excitatory postsynaptic currents and AMPA responses in hippocampal neurons. Neuron 12:127–138. 10.1016/0896-6273(94)90158-9 [DOI] [PubMed] [Google Scholar]
- Yalcin I, Barthas F, Barrot M (2014) Emotional consequences of neuropathic pain: insight from preclinical studies. Neurosci Biobehav Rev 47:154–164. 10.1016/j.neubiorev.2014.08.002 [DOI] [PubMed] [Google Scholar]
- Yang M, Luo CH, Zhu YQ, Liu YC, An YJ, Iqbal J, Wang ZZ, Ma XM (2020) 7,8-Dihydroxy-4-methylcoumarin reverses depression model-induced depression-like behaviors and alteration of dendritic spines in the mood circuits. Psychoneuroendocrinology 119:104767. 10.1016/j.psyneuen.2020.104767 [DOI] [PubMed] [Google Scholar]
- Yi ES, Oh S, Lee JK, Leem YH (2017) Chronic stress-induced dendritic reorganization and abundance of synaptosomal PKA-dependent CP-AMPA receptor in the basolateral amygdala in a mouse model of depression. Biochem Biophys Res Commun 486:671–678. 10.1016/j.bbrc.2017.03.093 [DOI] [PubMed] [Google Scholar]
- You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC (2014) Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int J Neuropsychopharmacol 17:313–322. 10.1017/S1461145713001144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Liu TH, He Y, Pan HQ, Zhang WH, Yin XP, Tian XL, Li BM, Wang XD, Holmes A, Yuan TF, Pan BX (2019) Chronic stress remodels synapses in an amygdala circuit-specific manner. Biol Psychiatry 85:189–201. 10.1016/j.biopsych.2018.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Jin Y, Meng Q, Zhu X, Bai T, Tian Y, Mao Y, Wang L, Xie W, Zhong H, Zhang N, Luo MH, Tao W, Wang H, Li J, Li J, Qiu BS, Zhou JN, Li X, Xu H, et al. (2019) A neural circuit for comorbid depressive symptoms in chronic pain. Nat Neurosci 22:1649–1658. 10.1038/s41593-019-0468-2 [DOI] [PubMed] [Google Scholar]
- Zhuo M (2019) Long-term cortical synaptic changes contribute to chronic pain and emotional disorders. Neurosci Lett 702:66–70. 10.1016/j.neulet.2018.11.048 [DOI] [PubMed] [Google Scholar]
- Zimmermann M (1983) Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16:109–110. 10.1016/0304-3959(83)90201-4 [DOI] [PubMed] [Google Scholar]
- Zimmermann M (2001) Pathobiology of neuropathic pain. Eur J Pharmacol 429:23–37. 10.1016/s0014-2999(01)01303-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.