

Abstract
Schizophrenia is a highly heritable, polygenic disorder. A growing list of common genetic variants have been associated with schizophrenia; there is a clear need to understand the role of these risk factors in the etiology of disease. The majority of these variants occur in non-coding regions of the genome, and are thought to regulate the expression of one or more genes in a cell type specific fashion. Recent advances in stem cell biology and molecular genetics have resulted in two invaluable advances: hiPSC technology makes possible the generation of donor-specific disease-relevant neural cell types, while CRISPR-based techniques can be applied to manipulate individual variants and/or their gene targets. New multiplexed gene manipulation and CRISPR screening techniques show great promise towards dissecting the complex interactions between the myriad disease-associated variants. This review outlines key advances in hiPSC and CRISPR technology, describing their applications and future potential in the field of schizophrenia research.
INTRODUCTION
Schizophrenia is a common yet severe neuropsychiatric disorder impacting approximately 0.3% of the global population1. Twin-based studies have estimated its heritability to be around 80–85%2,3. The polygenic genetic architecture of schizophrenia includes rare genetic variants of large impact (e.g. copy number variants (CNVs)), as well as hundreds of common variants of small effect (e.g. single nucleotide polymorphisms (SNPs))4–6. Although in isolation each SNP confers little disease risk, in aggregate a high polygenic risk score (PRS) substantially increases the risk of schizophrenia7. Most SNPs occur in non-coding portions of the genome8, and half (48.1%) colocalize variants linked to the expression of proximal gene targets in the brain (expression quantitative trait loci (eQTLs))9, although they frequently are thought to also regulate distal genes through trans mechanisms (such as the expression of diffusible factors)10 or cis mechanisms involving chromatin folding.11
There is a critical need to understand the impact of the hundreds of variants linked to schizophrenia, towards uncovering novel pathways and mechanisms underlying schizophrenia risk, and thus potential targets for therapeutic interventions. Here we discuss the most recent developments in stem cell-based technology as a model for studying the mechanisms and pathology underlying schizophrenia, with a particular focus on the potential for unravelling the common genetic variation linked to this disease using these models. We outline the current capabilities of hiPSC technology and further detail the state of the CRISPR field. We discuss the existing limitations and future potential of methods to engineer the genome in a donor-specific fashion across the major cell types of the brain. Overall, we consider impact of functional genomic studies attempting to capture the breadth and complexity of the genetic risk architecture linked to schizophrenia.
STEM CELLS AS A TOOL FOR STUDYING SCHIZOPHRENIA RISK
The advent of hiPSC technology resulted in a patient-specific platform to study development and disease in human cells of interest. hiPSC-derived brain cells can be used to identify disease-specific cellular phenotypes in patient derived lines12, as a platform for drug screening13 and to uncover the impact of genes and variants linked to disease risk, onset, progression, and treatment response14. New large collections pf patient-derived hiPSCs makes it possible to explore disease mechanisms in a cell-type-specific and donor-dependent manner.
It is now possible to generate all of the major cell types of the brain using hiPSCs, with most widely used protocols following one of two major approaches: i) recapitulation of the development signals that specify cell patterning in the developing human brain using morphogens and/or small molecules15,16, and ii) forced overexpression of one or more transcription factors critical to cell fate patterning17,18. These approaches permit the generation of many of the cell types implicated in schizophrenia pathophysiology, including glutamatergic15,17, GABAergic18,19 and dopaminergic neurons20,21, astrocytes22,23, and microglia24 (SEE FIGURE 1), with yields that vary across methodologies but that in some cases can exceed 95%17,25. Nonetheless, both cell type composition and functional maturity frequently vary between donors, and even hiPSC lines derived from the same donor, due to genetic background and stochastic differences between differentiations26. This inter- and intra-donor variability should be considered in the design and analysis of hiPSC-based studies. It is also worth noting that neurons derived from hiPSC lines typically adopt a cellular identity and gene expression consistent with neurons in fetal tissue27, and so are often best suited to the study of the genetic risk during neurodevelopment.
FIGURE 1:
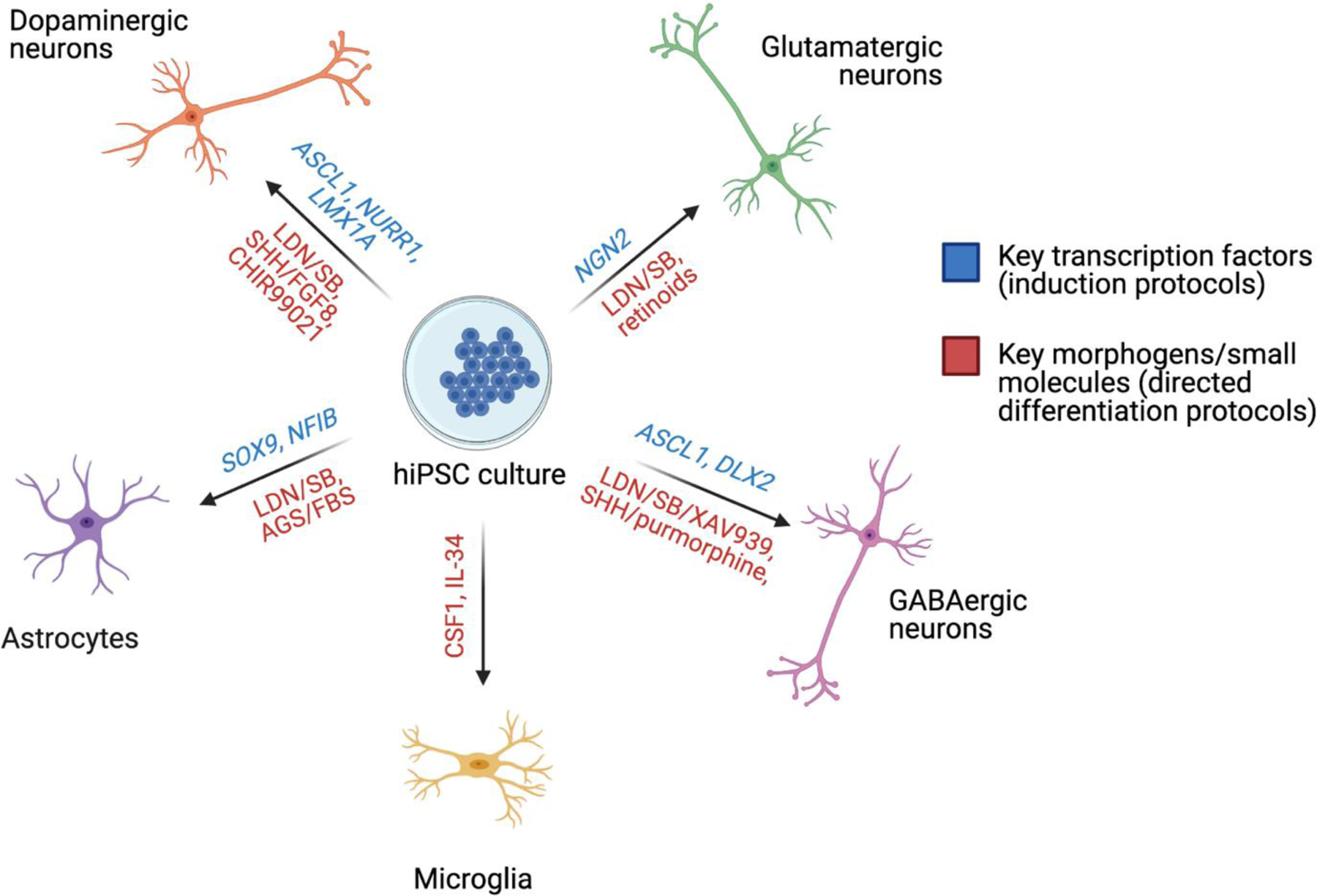
Key directed differentiation and reprogramming techniques for the generation of neural cell types relevant to schizophrenia disease modeling. AGS = astrocyte growth supplement; FBS = fetal bovine serum; LDN = LDN193189; SB = SB431542; SHH = sonic hedgehog. Info from refs16,18–25.
hiPSC lines generated from schizophrenia cases possess all of genetic variants sufficient to result in a disease state, even if these are not yet understood. The impact of these genetic backgrounds can be seen in a number of schizophrenia-relevant cellular phenotypes. Neurons derived from schizophrenia patient hiPSC lines exhibit reduced neuronal connectivity and expression of synaptic proteins, decreases in neurite number, alterations in gene expression, deficits in mitochondrial function and increased oxidative stress12,28–31. These findings are consistent with many of the cellular phenotypes seen in previous animal-based models of schizophrenia and neuropathological studies of postmortem brain tissue from schizophrenia patients. In the former, both genetic and non-genetic mouse models of schizophrenia have found reductions in dendritic spine density and reductions in dendritic arbor size in the cortex (reviewed in detail in32) as well as increases in measures of oxidative stress in cortical interneurons (reviewed in33). In the latter, pyramidal neurons in key regions of the frontal and temporal cortices exhibit reduced dendritic spine density and dendritic field size (reviewed in detail in32,34,35). The transcriptional signatures seen in NPCs and neurons generated from schizophrenia patient hiPSC lines converge with those seen in postmortem cortical tissue from schizophrenia patients26.
Despite methods to ensure reproducibility of results, such as double blinding, being common in hiPSC-based studies28,31,36, many utilise distinct patient cohorts, differentiation protocols, and methods of cellular phenotyping, limiting the generalizability of findings from one study to another. That being said, decreased levels of the synaptic protein PSD95 found in the first hiPSC-based study of schizophrenia were subsequently replicated using hiPSC neurons from independent cases and controls12,37; moreover, decreased synaptic puncta density have been reported in idiopathic schizophrenia hiPSC lines and those from patients with mutations in the schizophrenia-linked gene DISC137,38,40,41. Increased oxidative stress was also broadly indicated across multiple studies27,29,39.
Nonetheless, the large genetic variation between individuals means that case and control cohort designs are underpowered to detect subtle phenotypes such as genome-wide differential gene expression and the molecular mechanisms underpinning cellular phenotypes26. The statistical power of hiPSC-based studies can be maximized by increasing the number of potential donors rather than clonal lines per donor, reducing intra-donor variation through the optimization of neuronal differentiation protocols, and reducing inter-donor heterogeneity by focusing on individuals with specific genetic variants42. This latter point can be stratified into approaches that focus on rare genetic variants with highly penetrant effects on cellular phenotypes, or those that focus on multiple common variants with low individual impact but high impact when combined (i.e. high polygenic risk)42. Both can be studied through patient-derived cell lines, though recent developments in genetic engineering have provided a means of directly inducing or modelling these variants without the requirement of a particular donor genetic background. Towards this, recent hiPSC-based studies apply genetic engineering to dissect individual and combinatorial disease risk factors in a single (isogenic) donor background, providing the opportunity to definitively resolve the causal impact of variants and genes on cellular phenotypes. Genetic engineering has rapidly progressed from early work using zinc finger nucleases (ZFNs) and TALENs43 to CRISPR-Cas9 based systems and their derivatives44, offering myriad strategies to study variants and genes with a high degree of experimental flexibility.
CRISPR-BASED TECHNOLOGIES FACILITATE PRECISE ISOGENIC COMPARISONS OF CAUSAL FUNCTION
The rise of CRISPR-based platforms has been transformative for the study of disease-linked genetic variants in vitro. CRISPR-Cas9 can be targeted to a given sequence with great specificity, offering a higher degree of efficiency in editing, and capable of impacting multiple genes or loci simultaneously.45
The applications of CRISPR have grown from early guideRNA-targeted Cas9 nuclease-based gene knockouts (KO) of disease-relevant genes to newer methods that use Cas nickases or nuclease null mutants fused to effector domains to edit individual base pairs46, perform epigenetic modifications47, manipulate chromatin interactions48, directly activate or repress expression of target genes49 and cleave RNA50 (SEE FIGURE 2). The discovery of Cas9 orthologues with different PAM requirements permits greater flexibility in target sites for manipulation45, while systems such as RNA-targeting CasRX are capable of processing guideRNA arrays via a dedicated RNase domain50–52. Altogether, these many methods permit the high throughput assessment of gene and loci function53, critical for evaluating the sizable list of variants associated with complex polygenic disorders like schizophrenia. We discuss below two CRISPR-based platforms that offer great potential for studying common variants: direct editing of SNPs using CRISPR DNA editors to generate isogenic hiPSC lines, and perturbation of gene expression through CRISPR activation or inhibition (CRISPRa/i).
FIGURE 2:
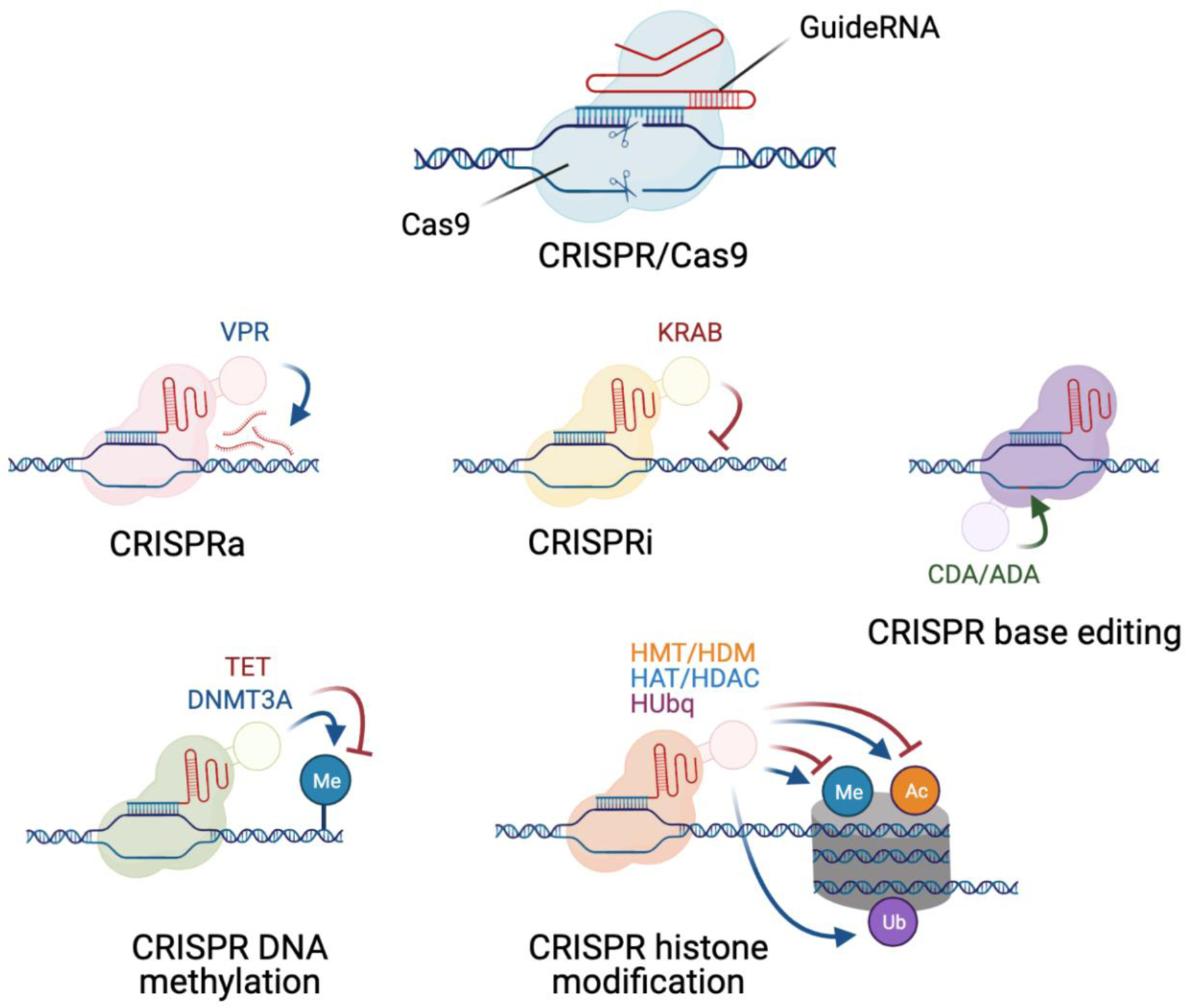
Examples of key CRISPR effectors useful for probing schizophrenia-relevant mechanisms in hiPSC-derived neural cultures: the initial CRISPR/Cas9 targeted DNA cleavage system, CRISPRa, CRISPRi, CRISPR DNA editing systems, CRISPR DNA methylation and CRISPR histone modification. VPR = VP64-p65-Rta; KRAB = Krüppel associated box; CDA = cytidine deaminase; ADA = adenosine deaminase; TET = ten-eleven translocation methylcytosine dioxygenase; DNMT3A = DNA methyltransferase 3A; HMT = histone methyltransferase; HDM = histone demethylase; HAT = histone acetyltransferase; HDAC = histone deacetylase; HUbq = histone ubiquitinase. Info from refs46–49,64.
i). DISSECTING COMMON VARIANTS USING DNA EDITING
CRISPR-Cas9-based genome editing offers a useful means of producing isogenic lines with specific genes and sequences removed. In the context of schizophrenia, this is useful for the study of large copy number variants and the function of genes associated with disease in an experimental system36. However, this form of genome editing lacks the precision or efficiency required to model the myriad common genetic variants at the level of individual base pairs. Towards this, CRISPR DNA editors offer a means to efficiently investigate the role of genetic variation in the etiology of disease at the resolution of individual SNPs.
CRISPR DNA editing is highly precise and offers the benefit of directly editing in the risk alleles for a given disease-associated locus, or conversely editing in non-risk alleles in hiPSC lines from patients with risk SNPs. It has previously been employed to determine the functional impact of common schizophrenia-associated variants: isogenic hiPSC lines have been generated for the schizophrenia-associated variant at rs4702, finding that even a single SNP can influence neuronal activity and morphology54. However, the use of edited hiPSC lines as a means of modelling all known schizophrenia-associated common variants is hindered by the time required to precisely target each individual SNP for editing. DNA editing also requires the knowledge of the specific SNP driving the association signal at the locus, and fine mapping data is only rarely sufficient to establish a single candidate for editing55. These limitations can be partially addressed by prioritizing candidate SNPs which have strong evidence of being the causal variant and editing them in donor lines with varying degrees of polygenic risk to determine epistatic effects, focusing on SNPs which directly alter protein structure and/or highly penetrant de novo variants.
Overall, DNA editing techniques are highly useful for studying the precise impact of key variants linked to schizophrenia risk, but a faster and more scalable approach is often to simply manipulate the impact of known schizophrenia risk variants at the level of their gene targets.
ii). EVALUATING GENE TARGETS USING CRISPRA/I
When a single causal SNP is not after fine-mapping analysis of GWAS data, the identification of candidates for DNA editing experiments problematic. In schizophrenia, the GWAS signal appears to be largely driven by the eQTL signal56,57, and thus experimental models that assess the impact of altering the expression of proximal target genes may offer a valuable insight into the biological consequences of schizophrenia-associated genetic variation.
CRISPRa, CRISPRi, and CRISPR-based RNA cleavage can all be applied to model the impact of a given variant on the expression of specific genes in disease-relevant cell types. Studying gene targets directly bypasses the need for time-consuming generation of each DNA edit in a given hiPSC line individually. While schizophrenia associated genes were once investigated using siRNA-based knockdown58, CRISPR-based methods for manipulation of gene expression have higher specificity and adaptability. However, neither approach well models the impact of variants that alter complex gene splicing patterns rather than expression (splice QTLs). Furthermore, CRISPRa and CRISPRi-based methods lack the ability to precisely control the degree of perturbation in expression levels, in order to match the impact of the eQTL observed in vivo.
Moving forward, pooled CRISPR screens make it increasingly straightforward to manipulate large numbers of genes in parallel, alone and in combination. A broad selection of genes can be targeted in arrayed (“sets of genes”) or pooled (“CRISPR library”) formats, and then analyzed at the population levels or at single cell resolution, respectively.
COMBINATORIAL PERTURBATION OF COMPLEX GENETIC RISK
The study of the interactions between genetic is critical towards understanding the etiology of schizophrenia. A “sets of genes” approach, whereby a selection of disease-associated genes, potentially with a shared biological function, are analyzed in combination can reveal unexpected outcomes of combinatorial perturbations. Towards this, we recently queried the impact of four schizophrenia risk genes individually and in combination, in order to assess additive interactions between the genes54 (SEE FIGURE 3). Perturbing schizophrenia genes together resulted in a synergistic impact on expression of genes relating to synaptic function and others linked to rare and common variant genes for psychiatric disorders like schizophrenia and bipolar disorder. This finding suggests that common and rare genetic variants convey biological effects that converge on specific pathways and genes, while raising the question as to the extent that these findings are generalizable beyond the set of four genes chosen for perturbation.
FIGURE 3:
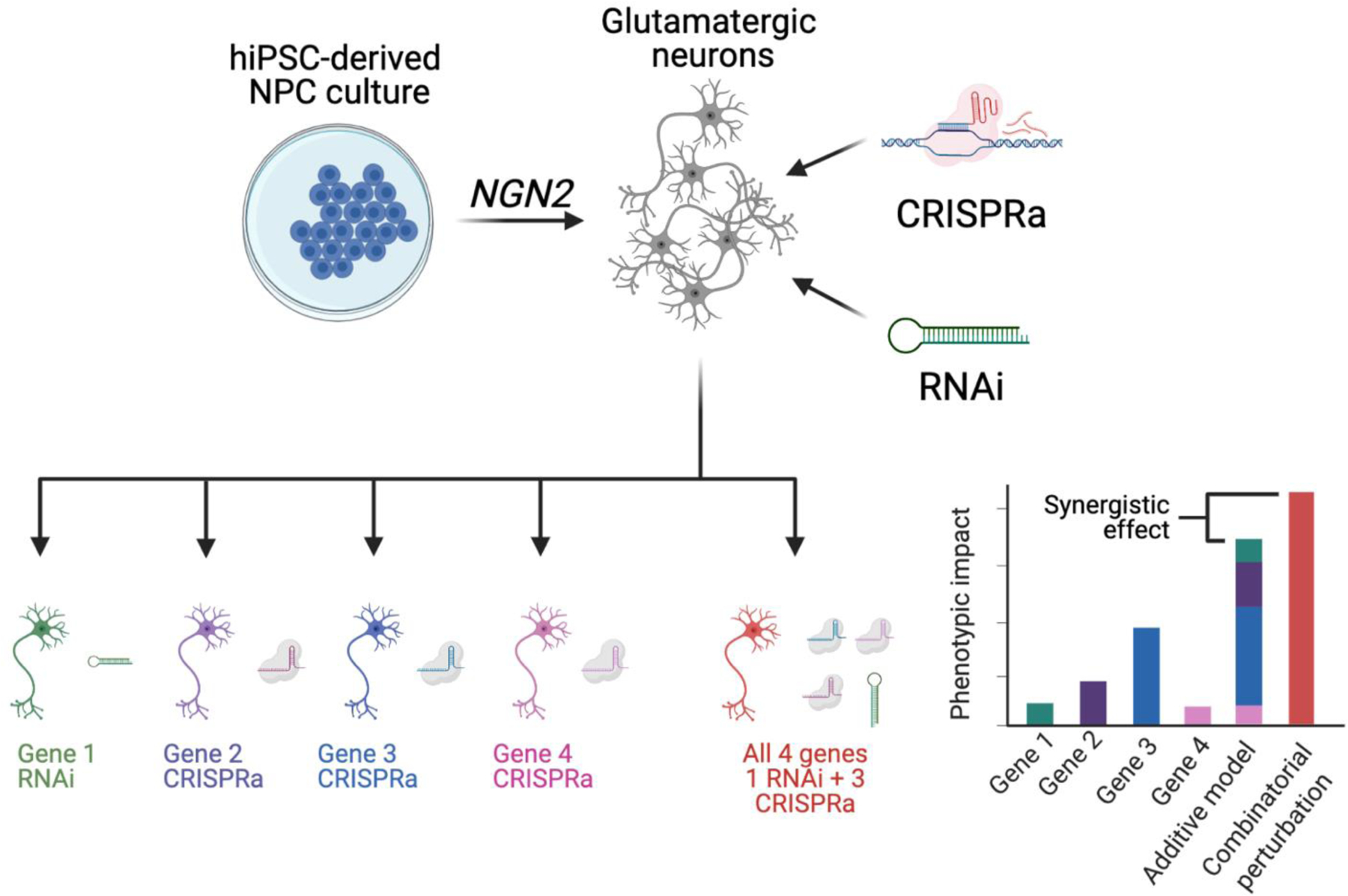
Example of “sets of genes” experimental approach for studying eQTL function and synergy following combinatorial CRISPRa/RNAi in glutamatergic neurons. The synergistic effect for a given combination of perturbations is calculated by summing the impacts of individual perturbations on a particular phenotype and comparing them to the actual measured impact of combinatorial perturbation. Figure partially adapted from ref54.
This approach is useful for dissecting phenotypes arising from specific subsets of eQTLs in detail. This method can be adapted with ease to a wide variety of phenotyping techniques, including morphology, synaptic density, neuronal activity and bulk RNAseq, due to perturbations occurring population-wide within cell cultures rather than on the level of individual cells. Nonetheless, this approach is constrained by the number of guideRNA and shRNA vectors that can be simultaneously delivered into a target cell. Towards this, new Cas12 and CasRX systems have guideRNA array cleavage capabilities, reducing the size of multiplexed guideRNA scaffolds50,52, and so could potentially increase the number of simultaneous gene perturbations possible in a given cell. Moreover, novel CRISPRa and CRISPRi systems utilise different guideRNA scaffold sequences, making possible new bi-directional, combinatorial perturbations of genes using CRISPRa and CRISPRi in the same cell.
Future extensions of this approach could be applied across pathways and cell types, further deciphering the additive impact of risk variants in schizophrenia pathophysiology, and potentially informing improved calculations of PRS. Moving forward, CRISPR screens offer a means of bypassing the time constraints associated with “set of genes” experimental design, scaling up the number of genes that can be simultaneously perturbed.
HIGH THROUGHPUT EVALUATION OF COMPLEX GENETIC RISK USING CRISPR SCREENS
Pooled CRISPR screens can greatly expand the number of variants or genes that can feasibly be examined. These approaches use pooled guideRNA libraries to transduce a population of cells, typically with a low multiplicity of infection (MOI) such that each cell receives one guideRNA only. Each cell undergoes a perturbation at a different locus, and the impact of this perturbation on one or more phenotypes can be evaluated by determining the relative prevalence of each guideRNA among bulk populations of cells using next generation sequencing. Phenotyping methods relying on assessments of guideRNA prevalence were originally limited to simple readouts based on cell frequencies such as proliferation, survival, and reporter gene expression (so-called “grow or glow”)59. However, new single cell RNA sequencing strategies can identify the guideRNA within a given cell, together with its transcriptome, by capturing individual cells in droplets containing barcoded beads that recognize scaffolding sequences on the guideRNA transcript60–62.
One of these techniques, ECCITE-seq, further combines direct guideRNA capture and transcriptome information with cell surface protein detection63. Antibody-mediated cell hashing permits sequencing of cells from multiple samples simultaneously, expanding the number of samples that can be analysed, but the detection of cell surface markers also offers a means to dissect subpopulations of cells from mixed cultures (SEE FIGURE 4). Moreover, the use of direct guideRNA capture allows multiple guideRNAs to be detected within a single cell in protocols with an increased MOI, thus providing a means of dissecting combinatorial perturbations.
FIGURE 4:
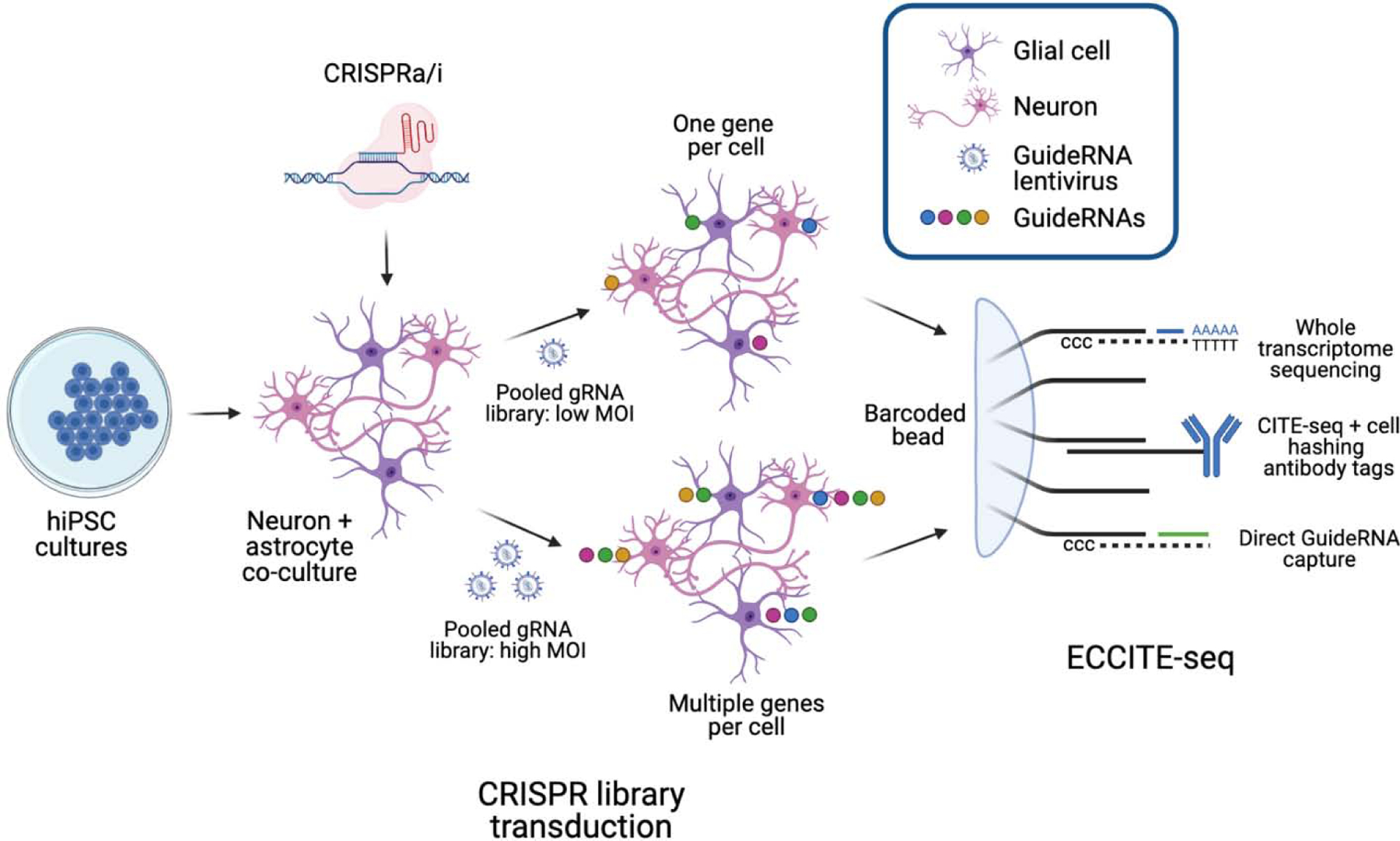
Two potential methods for using ECCITE-seq to determine eQTL function. Mixed neural cultures constitutively expressing CRISPR effectors are transduced with lentiviral guideRNA libraries targeting known eQTL genes. ECCITE-seq can be subsequently used to concatenate global gene expression data, cell identity data and guideRNA expression data in individual cells. Cultures infected with guideRNA libraries at a low MOI can be used to determine the impact of individual perturbations, while high MOI cultures can be used to investigate combinatorial perturbations and synergistic effects. ECCITE-seq schematic adapted from ref63.
Applied to hiPSC-derived neurons and glia, ECCITE-seq can dissect the impact of genetic variants at scale. ECCITE-seq and other CRISPR screens are for the future study of complex polygenic disorders owing to their scalability and the expanding number of cellular phenotyping applications available.
FUTURE APPLICATIONS
The two approaches outlined above offer diverging strategies for studying the common genetic variation underlying schizophrenia risk. The “sets of genes” approach is best suited for detailed dissection of the interactions between risk genes, particularly their convergent pathways. This represents a platform to query all the known variants linked to a particular polygenic disorder simultaneously, serving as an important platform for drug screening and/or personalized medicine. CRISPR screening technologies also make it possible to study of the breadth of genetic variation linked to schizophrenia at scale. Moreover, new applications increase the flexibility of high throughput CRISPR screens to study the impact of variants and genes on transcriptomics, epigenomics, and cellular phenotypes in parallel, uncovering biologically-relevant phenotypes and pathways underlying schizophrenia.
CONCLUSIONS
The recent explosion in CRISPR-based techniques for the manipulation of the genome has provided numerous valuable tools for the study of common genetic variants in the context of schizophrenia, and the continual diversification and improvement of hiPSC-derived neural differentiation protocols has offered an ideal platform to deploy them in. DNA editing is a valuable technique for establishing isogenic lines for the study of key SNPs, while CRISPRa and CRISPRi are a flexible method for modeling the impact and interactions of disease linked eQTLs. Exciting new CRISPR screening techniques such as ECCITE-seq provide a scalable means of studying these eQTLs in bulk and may provide the way forward in capturing the true diversity of genetic variation linked to schizophrenia and the pathophysiology arising from it. These techniques may also be key in future studies of other psychiatric and neurological disorders with complex genetic etiologies.
ACKNOWLEDGEMENTS
This work was partially supported by National Institute of Health (NIH) grants R56 MH101454 (K.J.B), R01 MH106056 (K.J.B.), and R01 MH109897 (K.J.B.).
All figures in this review were created with BioRender.com
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING FINANCIAL INTEREST STATEMENT
The authors declare no conflicts of interest.
References
- 1.Charlson FJ et al. Global Epidemiology and Burden of Schizophrenia : Findings From the Global Burden of Disease Study 2016. 44, 1195–1203 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sullivan PF, Kendler KS & Neale MC Schizophrenia as a Complex Trait: Evidence from a Meta-analysis of Twin Studies. Arch. Gen. Psychiatry 60, 1187–1192 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Hilker R et al. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol. Psychiatry 83, (2018). [DOI] [PubMed] [Google Scholar]
- 4.Gandal MJ, Leppa V, Won H, Parikshak NN & Geschwind DH The road to precision psychiatry: translating genetics into disease mechanisms. Nat. Neurosci 19, 1397–1407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purcell SM et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergen SE et al. Joint contributions of rare CNVs and common SNPs to risk for schizophrenia. Am J Psychiatry 176, 29–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors model the joint impact of PRS and CNVs on schizophrenia risk, finding that CNVs act additively in conjunction with PRS to convey risk, with the degree of polygenic risk required for schizophrenia decreasing with the increasing effect size of each CNV.
- 7.Ripke S, Walters JT & O’Donovan MC Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv (2020). [Google Scholar]; ** Preprint results of the schizophrenia PGC3, the largest schizophrenia GWAS to date. Common variant associations were reported at 270 loci.
- 8.Fromer M et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci 19, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaffe AE et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat. Neurosci 21, 1117–1125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Albert FW & Kruglyak L The role of regulatory variation in complex traits and disease. Nat. Rev. Genet 16, 197–212 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Hu M & Shen Y Gene regulation in the 3D genome. Hum. Mol. Genet 27, R228–R233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brennand KJ et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elitt MS, Barbar L & Tesar PJ Drug screening for human genetic diseases using iPSC models. Hum. Mol. Genet 27, R89–R98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vadodaria KC et al. Serotonin-induced hyperactivity in SSRI-resistant major depressive disorder patient-derived neurons. Mol. Psychiatry 24, 795–807 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Shi Y, Kirwan P & Livesey FJ Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc 7, 1836–1846 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Zhang P, Xia N & Reijo Pera RA Directed dopaminergic neuron differentiation from human pluripotent stem cells. J. Vis. Exp 1–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang N et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 14, 621–628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maroof AM et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–72 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theka I et al. Rapid Generation of Functional Dopaminergic Neurons From Human Induced Pluripotent Stem Cells Through a Single-Step Procedure Using Cell Lineage Transcription Factors. Stem Cells Transl. Med 2, 473–479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriks S et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Canals I et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 15, 693–696 (2018). [DOI] [PubMed] [Google Scholar]
- 23.TCW J et al. An Efficient Platform for Astrocyte Differentiation from Human Induced Pluripotent Stem Cells. Stem Cell Reports 9, 600–614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muffat J et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med 22, 1358–1367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi Y, Kirwan P & Livesey FJ Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc 7, 1836–1846 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Hoffman GE et al. Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brennand K et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 20, 361–368 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ni P et al. iPSC-derived homogeneous populations of developing schizophrenia cortical interneurons have compromised mitochondrial function. Mol. Psychiatry 25, 2873–2888 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robicsek O et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol. Psychiatry 18, 1067–76 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Yu DX et al. Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem cell reports 2, 295–310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kathuria A et al. Synaptic deficits in iPSC-derived cortical interneurons in schizophrenia are mediated by NLGN2 and rescued by N-acetylcysteine. Transl. Psychiatry 9, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaaro-Peled H, Ayhan Y, Pletnikov MV & Sawa A Review of pathological hallmarks of schizophrenia: Comparison of genetic models with patients and nongenetic models. Schizophr. Bull 36, 301–313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steullet P et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol. Psychiatry 22, 936–943 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harrison PJ The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain 122 (Pt 4, 593–624 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Glausier JR & Lewis DA Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srikanth P et al. Genomic DISC1 Disruption in hiPSCs Alters Wnt Signaling and Neural Cell Fate. Cell Rep. 12, 1414–1429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robicsek O et al. Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol. Psychiatry 18, 1067–1076 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Wen Z et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paulsen BDS, Cardoso SC, Stelling MP, Cadilhe DV & Rehen SK Valproate reverts zinc and potassium imbalance in schizophrenia-derived reprogrammed cells. Schizophr. Res 154, 30–5 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Kim NS et al. Pharmacological rescue in patient iPSC and mouse models with a rare DISC1 mutation. Nat. Commun 12, 1–11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X et al. Structural interaction between DISC1 and ATF4 underlying transcriptional and synaptic dysregulation in an iPSC model of mental disorders. Mol. Psychiatry 1, 1346–1360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman GE, Schrode N, Flaherty E & Brennand KJ New considerations for hiPSC-based models of neuropsychiatric disorders. Mol. Psychiatry 24, 49–66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen Z et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pak CH et al. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 17, 316–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaudelli NM et al. Programmable base editing of T to G C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thakore PI, Black JB, Hilton IB & Gersbach CA Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat. Methods 13, 127–137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu P, Chen M, Liu Y, Qi LS & Ding S CRISPR-Based Chromatin Remodeling of the Endogenous Oct4 or Sox2 Locus Enables Reprogramming to Pluripotency. Cell Stem Cell 22, 252–261.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Ho SM et al. Evaluating Synthetic Activation and Repression of Neuropsychiatric-Related Genes in hiPSC-Derived NPCs, Neurons, and Astrocytes. Stem Cell Reports 9, 615–628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konermann S et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors demonstrate knockdown of gene expression using CasRX, a compact RNA-guided Cas RNA cleavage system with the ability to process guideRNA arrays for multiplexed gene perturbation. This system can additionally manipulate alternate splicing, raising the possibility of modelling spliceQTLs in neuronal models.
- 51.Zhang C et al. Structural Basis for the RNA-Guided Ribonuclease Activity of CRISPR-Cas13d. Cell 175, 212–223.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tak YE et al. Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors. Nat. Methods 14, 1163–1166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanson KR et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun 9, 1–15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schrode N et al. Synergistic effects of common schizophrenia risk variants. Nat. Genet 51, 1475–1485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; ** In this study the authors combine DNA editing and combinatorial gene perturbation to investigate the impact of common schizophrenia risk variants in hiPSC-derived neural cells. They find evidence of converging effects on gene expression pathways linked to psychiatric disease and those involved in synaptic function.
- 55.Benner C et al. FINEMAP: Efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huckins LM et al. Gene expression imputation across multiple brain regions provides insights into schizophrenia risk. Nat. Genet 51, 659–674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors combine the largest eQTL reference panel for the postmortem dorsolateral prefrontal cortex with prior schizophrenia GWAS variant data, generating a list of 67 schizophrenia eQTL genes. This eQTL list offers potential biological pathways for further mechanistic evaluation using the hiPSC platforms and CRISPR-based technologies outlined in this review.
- 57.Dobbyn A et al. Landscape of Conditional eQTL in Dorsolateral Prefrontal Cortex and Colocalization with Schizophrenia GWAS. Am. J. Hum. Genet 102, 1169–1184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deans PJM et al. Psychosis risk candidate ZNF804A localizes to synapses and regulates neurite formation and dendritic spine structure. Biol. Psychiatry 82, 49–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian R et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 104, 239–255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dixit A et al. Perturb-Seq : Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Resource Perturb-Seq : Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1857.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Publ. Gr 14, 297–306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kang HM et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36, 89–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mimitou EP et al. Multiplexed detection of proteins , transcriptomes , clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; ** The authors describe a new multimodal experimental platform combining detection of CRISPR perturbations, transcriptomics, cell hashing and cell surface proteins in a single cell format (ECCITE-seq). This technique could potentially be used in combination with hiPSC lines to investigate common genetic variants in the context of polygenic disorders.
- 64.Pulecio J, Verma N, Mejía-Ramírez E, Huangfu D & Raya A CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell 21, 431–447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]