

Abstract
The study of autophagy in the nervous system has predominantly centered on degeneration; however, evidence is now cementing crucial roles for autophagy in neuronal development and growth, especially in axonal and presynaptic compartments. A picture is emerging that autophagy typically promotes the growth of axons and reduces presynaptic stability. Nonetheless, these are not rigid principles, and it remains unclear why autophagy does not always display these relationships during axonal and presynaptic development. Recent progress has identified mechanisms underlying spatiotemporal control of autophagy in neurons and begun to unravel how autophagy is integrated with other cellular processes such as proteasomal degradation and axon guidance. Ultimately, understanding how autophagy is regulated and its role in the developing nervous system is key to comprehending how the nervous system assembles its stereotyped yet plastic configuration. It is also likely to inform how we think about neurodevelopmental disorders and neurodegenerative diseases.
Autophagy in the nervous system, an overview
Macroautophagy, which we will refer to simply as autophagy, is a core catabolic process that is conserved across eukaryotes and is used to recycle organelles and cellular contents. Autophagy involves a complex multistep process with membrane engulfment of organelles, proteins, RNA, and lipids within an autophagosome [1,2]. Autophagosomes fuse with lysosomes to create autolysosomes where enzymatic degradation of contents occurs [2,3]. Autophagy allows cells to remove damaged or unwanted components and provides fresh building blocks for new cellular material. This helps maintain cellular health and supports the appropriate abundance of organelles and proteins within a given subcellular compartment. Autophagy is important during starvation, stress, and aging, and deregulation of autophagy figures prominently in neurodegenerative disease, autoimmune disease, and cancer [4–6]. While much autophagy research has focused on these disease contexts, there is clear evidence autophagy is also important during normal cell growth and development [7,8].
When one considers autophagy in the nervous system, it is essential to begin by noting that neurons have unique autophagic needs [9,10]. This principally stems from several characteristics. Neurons have unusual anatomy with subcellular compartments that are extremely far from the soma, which is particularly notable for axons. Neurons are post-mitotic cells that need to manage highly organized structures, such as growth cones and synapses, which require complex structural reorganization and have high metabolic demands, as post-mitotic cell neurons must manage the turnover of organelles, proteins, and RNA over a lifetime — a daunting but critical task. Finally, neurons must regulate the autophagic response to starvation much differently than other cell types because the nervous system must continue to operate as long as possible during times of starvation. Collectively, these challenges place unusually high demands on autophagy in the nervous system and are likely why altered autophagy is a hallmark of numerous neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and Huntington’s disease [4,10]. Along with this prominent role in neurodegeneration, a significant body of work has shown that autophagy is also essential for the development and growth of the nervous system with emerging roles in neurodevelopmental disorders [9,11,12]. Because this is somewhat counterintuitive (given the catabolic nature of the autophagic process), the role of autophagy in nervous system development has not been as widely recognized as its more heavily studied role in degeneration.
In this review, we discuss how autophagy affects axonal and presynaptic development and delve into recently discovered regulators of autophagy in these compartments. Importantly, new evidence indicates that regulation of autophagy is likely to be a notable difference between neurons and other cell types. These recent findings suggest that the keys to understanding how autophagy can be harnessed to treat neurodegenerative disease may lie within the developing nervous system.
Autophagy affects axon development
Axons are the most dramatically elongated compartment of any cell and require an intense capacity for regulated growth [13,14]. The growth cone facilitates this remarkable growth and sensing ability at the tip of an axon [15–17]. Presynaptic connections must be established with correct postsynaptic target cells, and axon growth must be terminated in an accurate and timely manner in order to ensure correct, efficient nervous system construction. Finally, synaptic contacts and axon integrity must be maintained over time. All these steps in nervous system construction are likely to require autophagic recycling of cellular contents that become damaged or are no longer needed. Autophagy in distal portions of the axon is likely to be important for repurposing materials for structural changes and growth far from the soma. As a result, specialized regulatory mechanisms would be required to control autophagy in neurons, and in particular in axonal and presynaptic compartments.
Early evidence showed autophagosomes form in axon tips [18], indicating the initiation of autophagy constitutively occurs in distal axon regions. These axonal autophagosomes traffic retrogradely to the soma, where they fuse with lysosomes [19–23] (Figure 1). Retrograde trafficking of axonal autophagosomes is also influenced by synaptic activity [24]. Importantly, studies from Drosophila and Caenorhabditis elegans have shown that axonal autophagosomes and retrograde trafficking are evolutionarily conserved and occur in neurons in vivo [25–27]. In the intact Drosophila brain, autophagosomes have been detected at axon termination sites of photoreceptors prior to synapse formation [28]. Work in worms recently provided further in vivo evidence that axonal autophagosomes mature in the soma [29].
Figure 1. Regulation of autophagy in the developing axon.
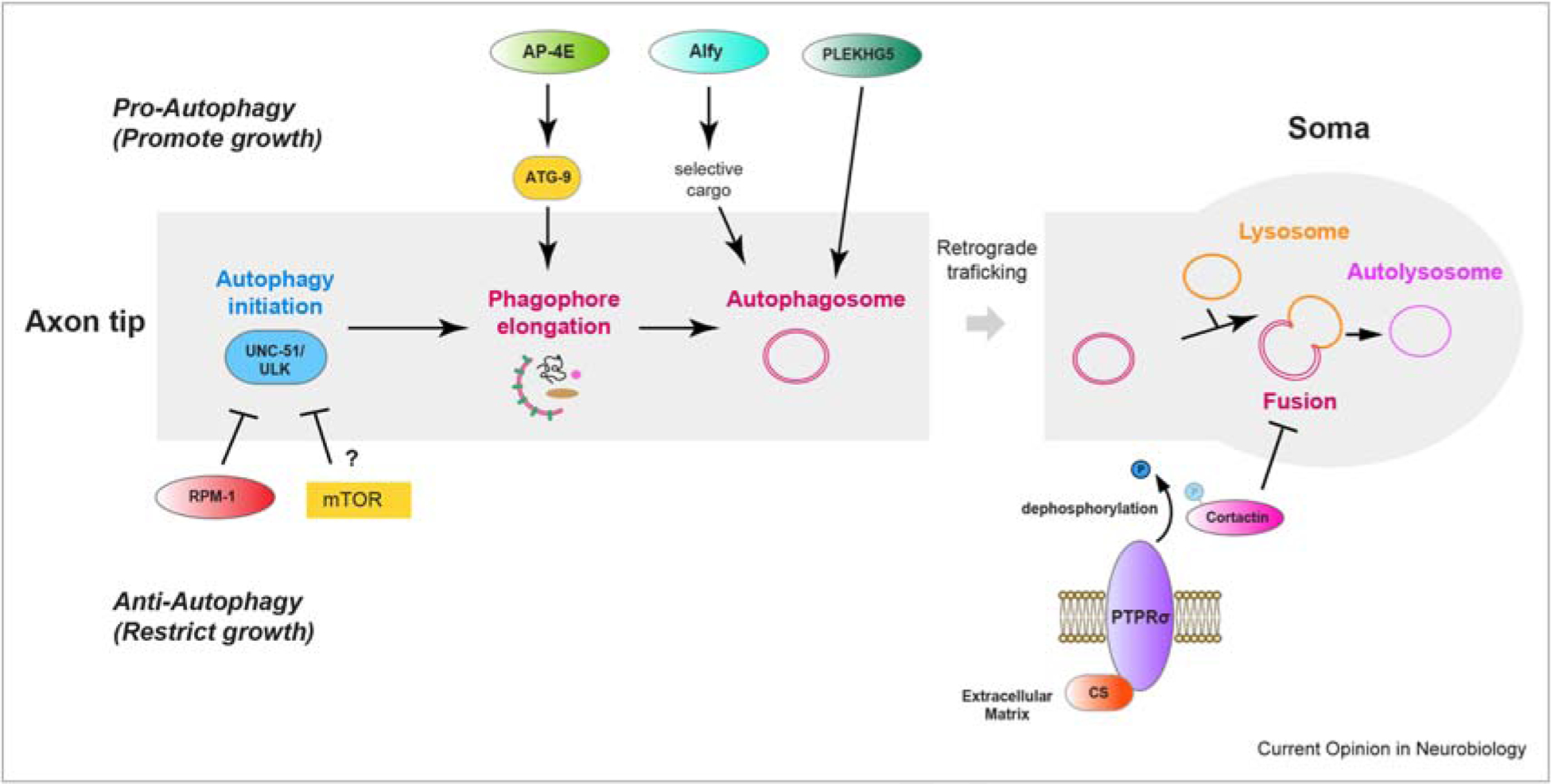
Schematic showing positive (upper) and negative (lower) regulators of axonal autophagy. Diagram integrates findings from a range of different model systems. Recently identified players include (1) Positive regulators AP-4E (adaptor protein complex component), Alfy/WDFY3 (WD40 repeat and FYVE domain protein), and PLEKHG5 (Rab GEF). (2) Negative regulators RPM-1 (E3 ubiquitin ligase that degrades UNC-51/ULK autophagy initiating kinase) and Chondroitin Sulfate (CS)/PTPRs/cortactin pathway. mTOR inhibition of autophagy is noted with a question mark because its role in regulating autophagy in the nervous system generally and axon development remains disputed.
The constitutive initiation of autophagy and autophagosome biogenesis in growing, as well as mature axons, indicate that the recycling of proteins and organelles is important for axonal growth and maintenance. This idea is consistent with studies showing that loss of the core autophagy proteins Atg7 and UNC-51/ULK affect axon extension and morphology in vivo [30–34]. It should be noted that UNC-51/ULK displays autophagy-independent functions in neurons, including effects on axon guidance receptor trafficking [35–39]. Thus, ULK likely represents a key signal intersection in the regulation of axon development. Other early studies primarily observed axon degeneration in mice lacking the core autophagy genes Atg7, Atg5, and FIP200 [40–43]. These studies noted prominent axon degeneration and behavioral/motor deficits as early as postnatal day 14–19. Thus, axon degeneration in such young animals could be considered neurodevelopmental in nature.
Subsequent studies showed that impairing autophagy leads to abnormal axon development and nervous system disease in humans. This includes loss of function mutations in EPG5 [44–47], TECPR2 [48–51], and AP4 [52–55]. In all three cases, agenesis of the corpus callosum, the largest axon tract in the human brain, was observed. Defects in corpus callosum development have been replicated in mice lacking Epg5 [45]. Thus, genetic changes that affect autophagy result in major abnormalities in axon development in the human and mouse brain.
Now, let us turn to the most recent observations made in different systems that indicate autophagy influences axon development. We draw the reader’s attention to an important, growing body of work showing that regulators and mediators of autophagy have a pronounced, evolutionarily conserved effect on axon development (Figure 1). We will start with cell-based systems and then discuss in vivo whole-animal models such as rodents and C. elegans.
Several studies with cell-based neuronal assays have reinforced the concept that altered autophagy leads to changes in axon growth. Cultured motor neurons with reduced Plekhg5, the GEF for the Rab26 GTPase that regulates autophagosome formation, have reduced axon outgrowth and impaired autophagy of synaptic vesicles [56]. In DRG neurons, chondroitin sulfate interacts with the protein tyrosine phosphatase receptor PTPRσ to regulate autophagosome-lysosome fusion in growth cones, and impairing autophagic flux prevents axon outgrowth [57]. Pharmacologically manipulating autophagy in cultured neurons also alters axon outgrowth. Peptides or low concentrations of drugs that increase autophagy stimulate neurite outgrowth and promote regrowth of certain types of axons following injury [58]. Similarly, increasing autophagy with modest starvation or drugs increased axon outgrowth in cultured hippocampal neurons [59]. In contrast, pharmacological inhibition of autophagy [60] or genetic loss of VAMP7 [59] (which promotes membrane fusion during autophagosome formation) results in reduced neurite outgrowth.
Recent studies in mice have identified regulators and cargo mediators of autophagy that affect axon development in vivo. The first is Alfy/WDFY3, a scaffold protein that mediates cargo-selective macroautophagy [61]. Mice lacking Alfy display severe defects in the formation of all the major axon tracts of the brain and spinal cord, including dramatically reduced corpus callosum. Experiments with cortical explants showed that Alfy loss of function inhibits axon outgrowth in response to the trophic effects of Netrin-1 without impacting levels of the netrin receptor DCC. Thus, Alfy regulates autophagy to influence trophic-stimulated axon outgrowth without affecting axon guidance. An investigation into Mir505–3p, a micro RNA that affects axon tract thickness in mice, showed that the autophagy component ATG12 is a central Mir505–3p target [62]. Mir505–3p reduces Atg12 levels to increase axon growth in cultured cortical neurons and callosal axons in vivo. Two independent studies found the opposite phenotype — thinner corpus callosum and failed axon extension — in mice lacking AP-4E [63,64], a positive regulator of autophagosome formation and trafficking [55]. Accompanying these defects was reduced trafficking and soma accumulation of Atg9, which is required for phagosome membrane extension. Consistent with evidence that AP-4E affects axon development via transport of Atg9, a recent study showed that brain specific-knockout of Atg9a results in embryonic dysgenesis of the corpus callosum and impaired axon outgrowth in cultured cortical neurons [65]. Collectively, these findings have solidified the role autophagy plays in axon development and provided important insight into mechanisms that regulate autophagy in the nervous system.
The invertebrate model system C. elegans has also proven extremely valuable in revealing the important, conserved role autophagy plays in axon development in vivo. One key advantage of the C. elegans system is its panoply of autophagy mutants that affect all core steps in the autophagic process [66]. This autophagic toolkit was initially leveraged to show that axon length is increased in the PVD sensory neurons of several autophagy mutants [27].
More recently, proteomic and genetic studies revealed that an E3 ubiquitin ligase, RPM-1/MYCBP2, inhibits autophagy in the nervous system [67]. RPM-1 restricts autophagy by ubiquitinating and degrading the autophagy initiating kinase UNC-51/ULK1 in a subcellular compartment at the tip of the distal axon. RPM-1 inhibition of UNC-51/ULK and autophagy promotes axon termination and restricts axon growth in mechanosensory neurons. Whether RPM-1/MYCBP2 is a conserved inhibitor of autophagy awaits further investigation, but cell-based experiments indicate that human MYCBP2 (also called PAM) can trigger ubiquitination and proteasome-mediated degradation of human ULK [67].
mTOR is the principal inhibitor of ULK and autophagy initiation in non-neuronal cells. Whether mTOR inhibits initiation of autophagy in the nervous system remains disputed and is based almost entirely upon pharmacological approaches [9,68–70]. The discovery that the RPM-1 ubiquitin ligase inhibits initiation of autophagy via UNC-51/ULK now provides a possible explanation for the inconsistent effects of mTOR signaling in the nervous system: neurons potentially deploy two mechanisms to restrict initiation of autophagy, RPM-1/MYCBP2 and mTOR. It is also worth noting that there is evidence for further signaling links between MYCBP2 and mTOR in human cells and mice. MYCBP2 (Phr1 in mice) can ubiquitinate and inhibit the TSC complex that negatively regulates mTOR [71–73]. Recent findings using Hela cells indicate that a lysosomal ATPase associated with Parkinson’s disease, ATP13A2/PARK9, can influence MYCBP2 inhibition of TSC and mTOR to impact autophagy [74]. Thus, RPM-1/MYCBP2 may regulate autophagy directly via UNC-51/ULK and indirectly via the TSC complex. Whether these layers of signaling inhibit the initiation of autophagy in the nervous system remains unknown but is an intriguing possibility.
Other work from the worm model has implicated the L-type calcium channel EGL-19 in the regulation of axon termination and autophagy [75]. EGL-19 functions via Alfy/WdFY3, indicating an important, evolutionarily conserved function for Alfy in axon development. Thus, findings from C. elegans have accelerated the field’s understanding of how autophagy is regulated during axon development and revealed a molecular interface between the ubiquitin-proteasome system and autophagy.
Taken as a whole, work from several model organisms and different types of neurons highlights several general principles regarding the role of autophagy in axon development. Autophagosomes are present in axons, and their biogenesis occurs at and is regulated in distal axon tips. Both regulators of autophagy and core autophagic machinery affect axon development. Finally, and perhaps somewhat surprisingly, more severe phenotypes in axon development generally arise from disruption of autophagy regulators rather than loss of core autophagy components. Why this is the case remains unclear at present.
Differential effects of autophagy on axon growth
Whether autophagy primarily promotes or inhibits the growth of neuronal structures remains an open question. A growing body of work using cell-based and in vivo models has revealed differing effects of autophagy on axon development. We discuss this literature and emphasize that evidence across systems, in general, suggests that autophagy is typically required for axon growth and must be reduced to facilitate axon termination. However, it is important to note that the type of neuron and the time at which axon development is examined could be among the factors influencing whether autophagy functions in a pro or anti-growth capacity.
First, let us begin with the evidence that autophagy is required for axon growth/extension. Many studies have shown that deleting or impairing the autophagic machinery results in shorter axon length both in vivo and in cultured neurons [30,60,65]. Consistent with these findings, impairing positive regulators of autophagy (e.g. AP-4E, Alfy, Plekhg5, VAMP7, or PTPRs) results in loss of axon tracts in vivo and reduced axon outgrowth in cultured neurons [56,57,59,61,63,64]. Finally, removing an inhibitor of autophagy, RPM-1, leads to the opposite phenotype e failed axon termination and excess axon growth [67]. Further support for this relationship between autophagy and axon growth has been observed during axon regeneration [58,76].
A smaller but significant number of studies have shown the opposite relationship: Loss or reduction in autophagy increases axon length [27,31]. Consistent with this, an excess of the autophagy inhibitor Mir-505p results in excess axon growth in vivo and in vitro [62]. Interestingly, a single study noted contrasting effects on neurite extension from knocking out the core autophagy component Atg5 compared to knocking out VAMP7 that is a positive regulator of early steps in autophagosome formation [59].
This apparent paradox in the field might have several explanations. (1) The most straightforward is that different types of neurons might have different needs for autophagy during axon development. For example, autophagy components can influence microtubule stability [77] and axon polarity [59], and different cell types display varying microtubule orientation and polarity arrangements. Together, these effects could help account for the variable results of autophagy on axon growth in different neurons. (2) It is plausible that axonal autophagy must be tightly balanced to facilitate axon development. As a result, impaired or excess autophagy might lead to failure in the same developmental steps. Alternatively, disturbing different components of the pathway might differentially affect such a precise autophagic balance. (3) Finally, and perhaps most interestingly, it is possible that manipulations affecting the regulation of autophagy in specific subcellular locations might yield different outcomes compared to the gross disruption of autophagy in the entire cell. Resolving these hypotheses now awaits a much greater understanding of spatiotemporal regulation of autophagy in the nervous system.
Presynaptic development is regulated by autophagy
The development, maintenance, and refinement of synapses are also influenced by autophagy, although the evidence is less extensive than for axonal development [11,69,70,78]. Indeed, a discussion of autophagy in the axon would not be complete without considering the effects of autophagy on presynaptic terminals, which are important, specialized subcellular compartments within the axon. A series of excellent studies in Drosophila and mice have shown that autophagy influences presynaptic transmission and responds to changes in metabolism and neuronal activity [25,79–83]. Here, we will discuss the role of autophagy in presynaptic development. However, it is important to note that autophagy is regulated at presynaptic terminals by largely different mechanisms during reductions in metabolic activity versus development. Nonetheless, it is clear that precise regulation and fine-tuning of autophagy are required for both correct presynaptic development and function.
Seminal genetic studies in Drosophila were the first to show that autophagy promotes presynaptic terminal growth using the fly neuromuscular junction (NMJ) as a model [84]. Two subsequent studies focused on the mammalian central nervous system showed that striatal dopaminergic neurons lacking the autophagy protein Atg7 have enlarged axon terminals and increased synaptic transmission [82,85]. Importantly, presynaptic enlargement was apparent in dopaminergic neurons of 2-week-old mice, indicating a developmental role for autophagy [85].
The first study to examine the effects of autophagy on presynaptic development in C. elegans evaluated a striking collection of 18 autophagy genes and showed that loss of any step in autophagy disrupts presynaptic terminal formation in AIY interneurons in vivo [27]. It was also revealed that autophagy is initiated near presynaptic terminals with ATG-9 and the KIF1A kinesin controlling autophagosome transport (Figure 2). More recently, work using C. elegans demonstrated that developmental maintenance of glutamatergic presynaptic connections in mechanosensory neurons requires inhibition of autophagy initiation by the RPM-1/MYCBP2 ubiquitin ligase [67] (Figure 2). Presynaptic defects caused by loss of RPM-1 were prevented by removing the autophagy initiating kinase UNC-51/ULK, as well as other autophagy genes. These findings were confirmed using a UNC-51 dominant-negative (DN) construct that circumvents UNC-51 effects on axon guidance. Importantly, behavioral habituation defects caused by loss of presynaptic connections in rpm-1 mutants [86,87] were suppressed by UNC-51 DN [67]. Thus, RPM-1 inhibition of UNC-51 and initiation of autophagy affects presynaptic maintenance and related behavioral outcomes.
Figure 2. Regulation of autophagy during presynaptic development.
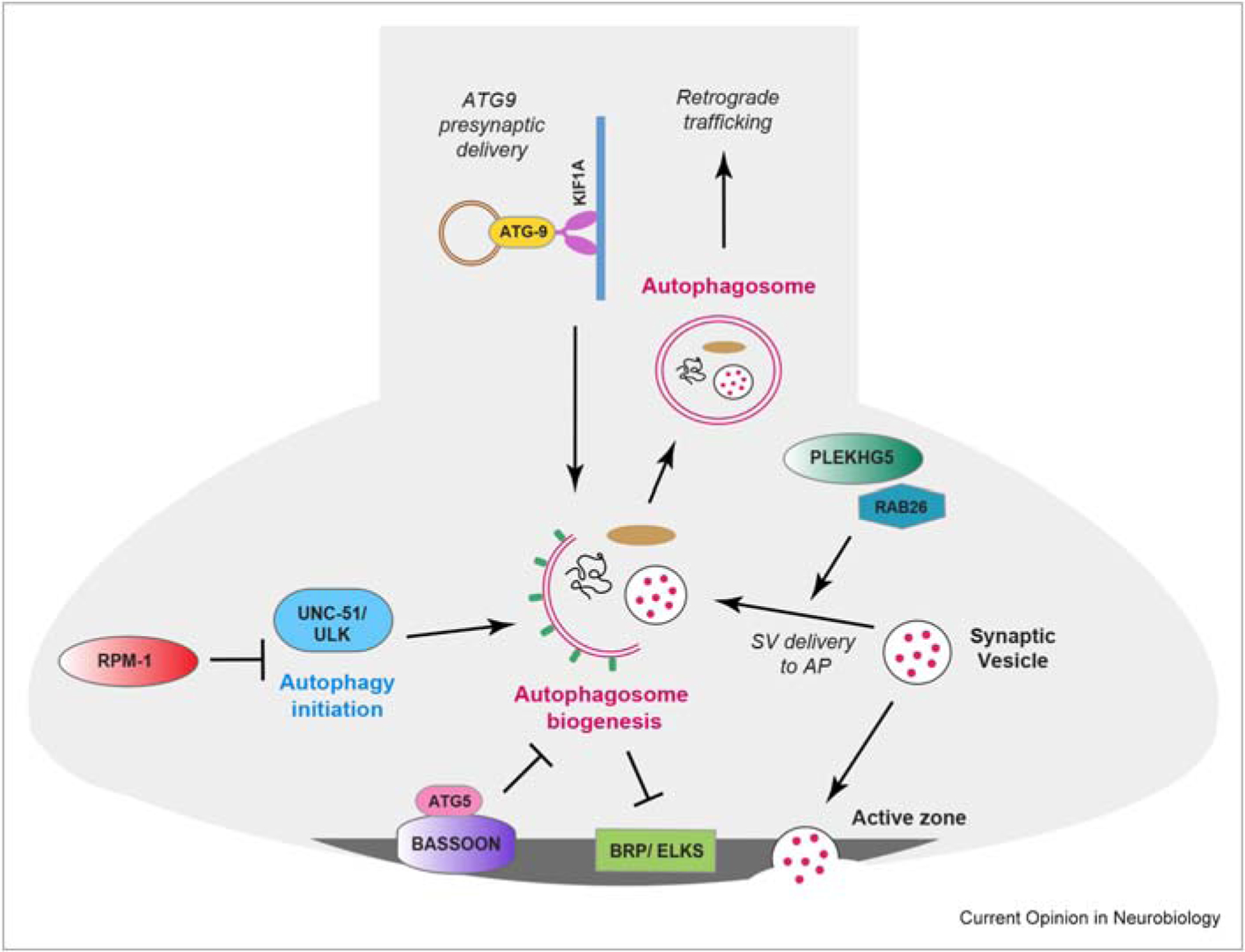
Schematic showing how different molecular players regulate autophagy during presynaptic development. Shown are integrated findings from different model systems and cultured neurons. Positive regulators are the Kif1A kinesin that affects Atg9 presynaptic delivery and PLEKHG5, which is a GEF for Rab26. Negative regulators of presynaptic autophagy include the active zone scaffold Bassoon and the RPM-1 ubiquitin ligase. Also noted is the inhibitory role autophagy plays in regulating levels of BRP and active zone development.
RPM-1 is well known to localize at presynaptic terminals suggesting that it acts locally to regulate presynaptic maintenance and autophagy [88,89]. Notably, observations in flies indicate that the RPM-1 ortholog, Highwire, is degraded by autophagy [84,90]. Since RPM-1 directly inhibits initiation of autophagy, these observations in flies could reflect autophagy feeding back on Highwire. Whether fly Highwire or mammalian orthologs (mouse Phr1 and human MYCBP2/PAM) inhibit autophagy during presynaptic development remains unknown.
Synaptic phenotypes in the mechanosensory neurons of rpm-1 mutants examined by Crawley et al. arise primarily from failed presynaptic maintenance during development [67,88]. Consistent with this, work on mouse auditory cochlear synapses has also shown that pharmacologically increasing autophagy reduces presynaptic connections after initial synapse formation [91]. These findings are consistent with results in young mice, which showed that impaired autophagy leads to increased presynaptic size and increased presynaptic transmission in dopaminergic and glutamatergic neurons [82,83,85].
Extremely interesting links have recently emerged between the presynaptic active zone and autophagy (Figure 2). Both knockdown and knockout approaches in cultured hippocampal neurons showed that Bassoon, an important scaffold in the active zone, inhibits presynaptic autophagy by binding to Atg5, a ubiquitin ligase-like molecule that promotes protein conjugation to growing phagophore membranes [92]. Loss of Bassoon elevated autophagy at presynaptic boutons while strongly reducing synapse numbers. Consistent with these findings, pharmacologically reducing autophagy with wortmannin rescued increased autophagosome formation that occurred with loss of Bassoon. Independent studies confirmed that autophagosomes are present at presynaptic terminals [93] and increased in Bassoon knockout (KO) neurons [94]. Bassoon KO neurons also displayed increased autophagy of synaptic vesicles. Bassoon inhibition of presynaptic autophagy requires the Parkin E3 ubiquitin ligase, which is consistent with proteomic results showing that Bassoon KO neurons have increased ubiquitination of numerous presynaptic proteins [94]. These studies highlight another important functional relationship between autophagy and the ubiquitin-proteasome system in presynaptic development.
Recent findings demonstrate that autophagy has an evolutionarily conserved role in the development of the active zone in the fly brain in vivo. Knocking down Atg9 and Atg5 core autophagy components in mushroom body neurons leads to increased levels of Bruchpilot (Brp)/ELKS, an active zone scaffold [95]. Electron and super-resolution microscopy revealed that impairing autophagy results in enlarged active zones. Altered active zone size was accompanied by impaired memory, consistent with the mushroom body’s role in learning and memory [95]. Interestingly, attenuating autophagy in mushroom body neurons affected Brp levels across the brain. How these noncell autonomous effects occur remains unknown, but further evidence from Drosophila and C. elegans indicates that autophagy can have noncell autonomous effects on neurons [96,97]. In the Drosophila eye, mutants for several autophagy components also resulted in abnormal presynaptic development [98]. Evaluation of Brp showed that Atg7, Atg6, and Atg18 mutants displayed greater numbers of presynaptic terminals in R1–6 and R7 photoreceptors. Presynaptic abnormalities resulted in increased photoreceptor neurotransmission and altered behavioral outcomes with increased visual attention. Impairing autophagy also caused R7 axon terminals to form ectopic synapses with incorrect postsynaptic neurons at inappropriate layers of the medulla. Elegant live imaging with intact fly brain indicated that excess synapse formation arises from developmental alterations in synaptic filopodia dynamics with autophagy promoting more dynamic filopodia, which allows more limited, correct postsynaptic partner choice [98].
Overall, these results point toward autophagy as a key process required for appropriate presynaptic terminal formation and maintenance. Loss of autophagy promotes stabilization of synaptic contacts and enlarged presynaptic morphology in both flies and mammalian neurons. Local inhibition of autophagy by the active zone scaffold Bassoon and the ubiquitin ligase RPM-1 provides spatiotemporal regulation of autophagy required for presynaptic maintenance. Importantly, there is evidence indicating that either defective or excess autophagy can cause incorrect presynaptic development depending on neuronal context. This suggests that a tight balance of presynaptic autophagy is essential for the precise configuration and maintenance of synaptic connections.
Conclusion
Results from a diverse group of experimental systems, including invertebrate models, rodents, and cultured neurons, have substantially driven forward our understanding of how autophagy affects axonal and presynaptic development. Recent findings have also begun to reveal how autophagy is regulated in subcellular axonal compartments to influence nervous system development. A perhaps surprising picture is emerging in which autophagy often promotes growth and overall length of axons, whereas, for presynaptic terminals, the pathway instead functions to limit the size and stability of contacts. This apparent contrasting relationship is logical given the developmental trajectory of an axon. Initially, an axon must continue growing and remain dynamic until correct target cells and the termination location are reached. Growth of the primary axon then needs to be halted while appropriate synaptic contacts (via en passant boutons or collateral branches) are consolidated and maintained. In moving forward, several important questions remain unanswered. (1) How are more specific forms of autophagy, such as mitophagy or cargo-specific autophagy, regulated in the nervous system and in axons? (2) How do different autophagy regulators work together to influence axonal autophagy and presynaptic development? (3) How are several types of degradation/recycling machinery (i.e. autophagy, the ubiquitin-proteasome system, endo-lysosomal system) integrated and controlled to facilitate axon and synapse development? Addressing these questions will undoubtedly deepen our understanding of nervous system development and could provide valuable insight into how we think about and treat neurodevelopmental disorders and neurodegenerative diseases.
Acknowledgement
Brock Grill was supported by National Institutes of Health Grant R01 NS072129. Oliver Crawley was supported by the Severo Ochoa Post-doctoral Program and a Marie Sklodowska-Curie Actions Individual Fellowship.
Footnotes
Conflict of interest statement
The authors have no competing financial interests.
References
- 1.Mizushima N, Yoshimori T, Ohsumi Y: The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011, 27: 107–132. [DOI] [PubMed] [Google Scholar]
- 2.Abildgaard MH, Brynjolfsdottir SH, Frankel LB: The autophagy-RNA interplay: degradation and beyond. Trends Biochem Sci 2020, 45:845–857. [DOI] [PubMed] [Google Scholar]
- 3.Yu L, Chen Y, Tooze SA: Autophagy pathway: cellular and molecular mechanisms. Autophagy 2018, 14:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, et al. : Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93:1015–1034. [DOI] [PubMed] [Google Scholar]
- 5.Levine B, Kroemer G: Biological functions of autophagy genes: a disease perspective. Cell 2019, 176:11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dikic I, Elazar Z: Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 2018, 19: 349–364. [DOI] [PubMed] [Google Scholar]
- 7.Levine B, Klionsky DJ: Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004, 6:463–477. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N, Levine B: Autophagy in mammalian development and differentiation. Nat Cell Biol 2010, 12:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stavoe AKH, Holzbaur ELF: Autophagy in neurons. Annu Rev Cell Dev Biol 2019, 35:477–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto A, Yue Z: Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci 2014, 37:55–78. [DOI] [PubMed] [Google Scholar]
- 11.Birdsall V, Waites CL: Autophagy at the synapse. Neurosci Lett 2019, 697:24–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsh D, Dragich JM: Autophagy in mammalian neurodevelopment and implications for childhood neuorological disorders. Neurosci Lett 2018, 697:29–33. [DOI] [PubMed] [Google Scholar]
- 13.Huber AB, Kolodkin AL, Ginty DD, Cloutier JF: Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci 2003, 26: 509–563. [DOI] [PubMed] [Google Scholar]
- 14.Kolodkin AL, Tessier-Lavigne M: Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb Perspect Biol 2011, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez TM, Letourneau PC: Actin dynamics in growth cone motility and navigation. J Neurochem 2014, 129:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vitriol EA, Zheng JQ: Growth cone travel in space and time: the cellular ensemble of cytoskeleton, adhesion, and membrane. Neuron 2012, 73:1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradke F, Fawcett JW, Spira ME: Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci 2012, 13:183–193. [DOI] [PubMed] [Google Scholar]
- 18.Hollenbeck PJ: Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 1993, 121:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill SE, Colon-Ramos DA: The journey of the synaptic autophagosome: a cell biological perspective. Neuron 2020, 105: 961–973. [DOI] [PubMed] [Google Scholar]
- 20.Maday S, Wallace KE, Holzbaur EL: Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 2012, 196:407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maday S, Holzbaur EL: Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 2014, 30:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S, Sato Y, Nixon RA: Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 2011, 31:7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng XT, Zhou B, Lin MY, Cai Q, Sheng ZH: Axonal autophagosomes use the ride-on service for retrograde transport toward the soma. Autophagy 2015, 11:1434–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T, Martin S, Papadopulos A, Harper CB, Mavlyutov TA, Niranjan D, Glass NR, Cooper-White JJ, Sibarita JB, Choquet D, et al. : Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type a. J Neurosci 2015, 35:6179–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soukup SF, Kuenen S, Vanhauwaert R, Manetsberger J, Hernandez-Diaz S, Swerts J, Schoovaerts N, Vilain S, Gounko NV, Vints K, et al. : A LRRK2-dependent EndophilinA phosphoswitch is critical for macroautophagy at presynaptic terminals. Neuron 2016, 92:829–844. [DOI] [PubMed] [Google Scholar]
- 26.Neisch AL, Neufeld TP, Hays TS: A STRIPAK complex mediates axonal transport of autophagosomes and dense core vesicles through PP2A regulation. J Cell Biol 2017, 216:441–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.••.Stavoe AK, Hill SE, Hall DH, Colon-Ramos DA: KIF1A/UNC-104 transports ATG-9 to regulate neurodevelopment and autophagy at synapses. Dev Cell 2016, 38:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]; Study showed how autophagy affected the presynaptic terminal formation and axon development, and showed autophagosomes forming at presynaptic terminals in vivo, and demonstrated that presynaptic autophagosomes are regulated by the KIF1A kinesin.
- 28.Jin EJ, Kiral FR, Ozel MN, Burchardt LS, Osterland M, Epstein D, Wolfenberg H, Prohaska S, Hiesinger PR: Live observation of two parallel membrane degradation pathways at axon terminals. Curr Biol 2018, 28:1027–1038e1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.••.Hill SE, Kauffman KJ, Krout M, Richmond JE, Melia TJ, Colon-Ramos DA: Maturation and clearance of autophagosomes in neurons depends on a specific cysteine protease isoform, ATG-4.2. Dev Cell 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]; Showed that autophagosomes undergo retrograde trafficking and mature in the cell soma in vivo. The maturation of axonal autophagosomes is regulated by protease activity.
- 30.Coupe B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG: Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metabol 2012, 15:247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ban BK, Jun MH, Ryu HH, Jang DJ, Ahmad ST, Lee JA: Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol 2013, 33:3907–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogura K, Wicky C, Magnenat L, Tobler H, Mori I, Muller F, Ohshima Y: Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev 1994, 8:2389–2400. [DOI] [PubMed] [Google Scholar]
- 33.Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME: A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron 1999, 24:833–846. [DOI] [PubMed] [Google Scholar]
- 34.Mochizuki H, Toda H, Ando M, Kurusu M, Tomoda T, Furukubo-Tokunaga K: Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PloS One 2011, 6, e19632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang B, Kundu M: Canonical and noncanonical functions of ULK/Atg1. Curr Opin Cell Biol 2017, 45:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai T, Garriga G: The conserved kinase UNC-51 acts with VAB-8 and UNC-14 to regulate axon outgrowth in C. elegans. Development 2004, 131:5991–6000. [DOI] [PubMed] [Google Scholar]
- 37.Ogura K, Goshima Y: The autophagy-related kinase UNC-51 and its binding partner UNC-14 regulate the subcellular localization of the Netrin receptor UNC-5 in Caenorhabditis elegans. Development 2006, 133:3441–3450. [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Iyengar R, Li-Harms X, Joo JH, Wright C, Lavado A, Horner L, Yang M, Guan JL, Frase S, et al. : The autophagy-inducing kinases, ULK1 and ULK2, regulate axon guidance in the developing mouse forebrain via a noncanonical pathway. Autophagy 2018, 14:796–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joo JH, Wang B, Frankel E, Ge L, Xu L, Iyengar R, Li-Harms X, Wright C, Shaw TI, Lindsten T, et al. : The noncanonical role of ULK/ATG1 in ER-to-golgi trafficking is essential for cellular homeostasis. Mol Cell 2016, 62:491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. : Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441:880–884. [DOI] [PubMed] [Google Scholar]
- 41.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. : Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441: 885–889. [DOI] [PubMed] [Google Scholar]
- 42.Komatsu M, Wang QJ, Holstein GR, Friedrich VL Jr, Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z: Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 2007, 104:14489–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang CC, Wang C, Peng X, Gan B, Guan JL: Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 2010, 285:3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cullup T, Kho AL, Dionisi-Vici C, Brandmeier B, Smith F, Urry Z, Simpson MA, Yau S, Bertini E, McClelland V, et al. : Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet 2013, 45:83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao YG, Zhao H, Sun H, Zhang H: Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy 2013, 9: 1258–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Byrne S, Jansen L, UK-Im JM, Siddiqui A, Lidov HG, Bodi I, Smith L, Mein R, Cullup T, Dionisi-Vici C, et al. : EPG5-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain 2016, 139:765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hori I, Otomo T, Nakashima M, Miya F, Negishi Y, Shiraishi H, Nonoda Y, Magara S, Tohyama J, Okamoto N, et al. : Defects in autophagosome-lysosome fusion underlie Vici syndrome, a neurodevelopmental disorder with multisystem involvement. Sci Rep 2017, 7:3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oz-Levi D, Ben-Zeev B, Ruzzo EK, Hitomi Y, Gelman A, Pelak K, Anikster Y, Reznik-Wolf H, Bar-Joseph I, Olender T, et al. : Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet 2012, 91:1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heimer G, Oz-Levi D, Eyal E, Edvardson S, Nissenkorn A, Ruzzo EK, Szeinberg A, Maayan C, Mai-Zahav M, Efrati O, et al. : TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur J Paediatr Neurol 2016, 20:69–79. [DOI] [PubMed] [Google Scholar]
- 50.Behrends C, Sowa ME, Gygi SP, Harper JW: Network organization of the human autophagy system. Nature 2010, 466: 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fraiberg M, Tamim-Yecheskel BC, Kokabi K, Subic N, Heimer G, Eck F, Nalbach K, Behrends C, Ben-Zeev B, Shatz O, et al. : Lysosomal targeting of autophagosomes by the TECPR domain of TECPR2. Autophagy 2020:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verkerk AJ, Schot R, Dumee B, Schellekens K, Swagemakers S, Bertoli-Avella AM, Lequin MH, Dudink J, Govaert P, van Zwol AL, et al. : Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am J Hum Genet 2009, 85:40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ebrahimi-Fakhari D, Cheng C, Dies K, Diplock A, Pier DB, Ryan CS, Lanpher BC, Hirst J, Chung WK, Sahin M, et al. : Clinical and genetic characterization of AP4B1-associated SPG47. Am J Med Genet 2018, 176:311–318. [DOI] [PubMed] [Google Scholar]
- 54.Davies AK, Itzhak DN, Edgar JR, Archuleta TL, Hirst J, Jackson LP, Robinson MS, Borner GHH: AP-4 vesicles contribute to spatial control of autophagy via RUSC-dependent peripheral delivery of ATG9A. Nat Commun 2018, 9:3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mattera R, Park SY, De Pace R, Guardia CM, Bonifacino JS: AP-4 mediates export of ATG9A from the trans-Golgi network to promote autophagosome formation. Proc Natl Acad Sci U S A 2017, 114:E10697–E10706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.•.Luningschror P, Binotti B, Dombert B, Heimann P, Perez-Lara A, Slotta C, Thau-Habermann N, RvC C, Karl F, Damme M, et al. : Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Nat Commun 2017, 8:678. [DOI] [PMC free article] [PubMed] [Google Scholar]; Authors used cultured motor neurons to show that the Rab26 GEF Plekhg5 regulates axon growth via effects on autophagy and is required for autophagy of synaptic vesicles.
- 57.••.Sakamoto K, Ozaki T, Ko YC, Tsai CF, Gong Y, Morozumi M, Ishikawa Y, Uchimura K, Nadanaka S, Kitagawa H, et al. : Glycan sulfation patterns define autophagy flux at axon tip via PTPRsigma-cortactin axis. Nat Chem Biol 2019, 15:699–709. [DOI] [PubMed] [Google Scholar]; Demonstrated that a chondroitin sulfate/PTPRs/cortactin pathway regulates autophagy to affect axon development in DRG neurons.
- 58.He M, Ding Y, Chu C, Tang J, Xiao Q, Luo ZG: Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. Proc Natl Acad Sci U S A 2016, 113:11324–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wojnacki J, Nola S, Bun P, Cholley B, Filippini F, Presse MT, Lipecka J, Man Lam S, N’Guyen J, Simon A, et al. : Role of VAMP7-dependent secretion of reticulon 3 in neurite growth. Cell Rep 2020, 33:108536. [DOI] [PubMed] [Google Scholar]
- 60.Clarke JP, Mearow K: Autophagy inhibition in endogenous and nutrient-deprived conditions reduces dorsal root ganglia neuron survival and neurite growth in vitro. J Neurosci Res 2016, 94:653–670. [DOI] [PubMed] [Google Scholar]
- 61.••.Dragich JM, Kuwajima T, Hirose-Ikeda M, Yoon MS, Eenjes E, Bosco JR, Fox LM, Lystad AH, Oo TF, Yarygina O, et al. : Autophagy linked FYVE (Alfy/WDFY3) is required for establishing neuronal connectivity in the mammalian brain. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]; Key study that demonstrated Alfy, a cargo-selective autophagy adaptor, is required for axon growth and development in vivo in mice. Alfy mutants display severe defects in axon tract development in the brain, including defects in the corpus callosum.
- 62.Yang K, Yu B, Cheng C, Cheng T, Yuan B, Li K, Xiao J, Qiu Z, Zhou Y: Mir505–3p regulates axonal development via inhibiting the autophagy pathway by targeting Atg12. Autophagy 2017, 13:1679–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.••.De Pace R, Skirzewski M, Damme M, Mattera R, Mercurio J, Foster AM, Cuitino L, Jarnik M, Hoffmann V, Morris HD, et al. : Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet 2018, 14, e1007363. [DOI] [PMC free article] [PubMed] [Google Scholar]; Initial study showed that loss of AP-4E results in the thin corpus callosum, indicating impaired axon development in vivo. AP-4E knockout animals display Atg9 accumulation in the soma of numerous types of neurons, and impaired Atg9 trafficking.
- 64.Ivankovic D, Drew J, Lesept F, White IJ, Lopez Domenech G, Tooze SA, Kittler JT: Axonal autophagosome maturation defect through failure of ATG9A sorting underpins pathology in AP-4 deficiency syndrome. Autophagy 2020, 16:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.••.Yamaguchi J, Suzuki C, Nanao T, Kakuta S, Ozawa K, Tanida I, Saitoh T, Sunabori T, Komatsu M, Tanaka K, et al. : Atg9a deficiency causes axon-specific lesions including neuronal circuit dysgenesis. Autophagy 2018, 14:764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]; Brain-specific knockout of Atg9 results in failed axon development and dysgenesis of the corpus callosum.
- 66.Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al. : C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010, 141:1042–1055. [DOI] [PubMed] [Google Scholar]
- 67.••.Crawley O, Opperman KJ, Desbois M, Adrados I, Borgen MA, Giles AC, Duckett DR, Grill B: Autophagy is inhibited by ubiquitin ligase activity in the nervous system. Nat Commun 2019, 10:5017. [DOI] [PMC free article] [PubMed] [Google Scholar]; Showed the RPM-1 ubiquitin ligase regulates axon termination and synapse maintenance by ubiquitinating and degrading UNC-51/ULK in subcellular axonal compartments. First study to show that the ubiquitin-proteasome system inhibits initiation of autophagy in the nervous system.
- 68.Maday S, Holzbaur EL: Compartment-specific regulation of autophagy in primary neurons. J Neurosci 2016, 36: 5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nikoletopoulou V, Tavernarakis N: Regulation and roles of autophagy at synapses. Trends Cell Biol 2018, 28:646–661. [DOI] [PubMed] [Google Scholar]
- 70.Lieberman OJ, McGuirt AF, Tang G, Sulzer D: Roles for neuronal and glial autophagy in synaptic pruning during development. Neurobiol Dis 2019, 122:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murthy V, Han S, Beauchamp RL, Smith N, Haddad LA, Ito N, Ramesh V: Pam and its ortholog highwire interact with and may negatively regulate the TSC1.TSC2 complex. J Biol Chem 2004, 279:1351–1358. [DOI] [PubMed] [Google Scholar]
- 72.Han S, Witt RM, Santos TM, Polizzano C, Sabatini BL, Ramesh V: Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal 2008, 20:1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han S, Kim S, Bahl S, Li L, Burande CF, Smith N, James M, Beauchamp RL, Bhide P, Diantonio A, et al. : The E3 ubiquitin ligase, protein associated with Myc (Pam) regulates mammalian/mechanistic target of rapamycin complex 1 (mTORC1) signaling in vivo through N- and C-terminal domains. J Biol Chem 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bento CF, Ashkenazi A, Jimenez-Sanchez M, Rubinsztein DC: The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat Commun 2016, 7:11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buddell T, Friedman V, Drozd CJ, Quinn CC: An autism-causing calcium channel variant functions with selective autophagy to alter axon targeting and behavior. PLoS Genet 2019, 15, e1008488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ko SH, Apple EC, Liu Z, Chen L: Age-dependent autophagy induction after injury promotes axon regeneration by limiting NOTCH. Autophagy 2020, 16:2052–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.••.Negrete-Hurtado A, Overhoff M, Bera S, De Bruyckere E, Schatzmuller K, Kye MJ, Qin C, Lammers M, Kondylis V, Neundorf I, et al. : Autophagy lipidation machinery regulates axonal microtubule dynamics but is dispensable for survival of mammalian neurons. Nat Commun 2020, 11:1535. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrated that core autophagy components can affect microtubule dynamics along with their known roles in autophagy.
- 78.Andres-Alonso M, Kreutz MR, Karpova A: Autophagy and the endolysosomal system in presynaptic function. Cell Mol Life Sci 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vijayan V, Verstreken P: Autophagy in the presynaptic compartment in health and disease. J Cell Biol 2017, 216: 1895–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uytterhoeven V, Lauwers E, Maes I, Miskiewicz K, Melo MN, Swerts J, Kuenen S, Wittocx R, Corthout N, Marrink SJ, et al. : Hsc70–4 deforms membranes to promote synaptic protein turnover by endosomal microautophagy. Neuron 2015, 88: 735–748. [DOI] [PubMed] [Google Scholar]
- 81.••.Vanhauwaert R, Kuenen S, Masius R, Bademosi A, Manetsberger J, Schoovaerts N, Bounti L, Gontcharenko S, Swerts J, Vilain S, et al. : The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J 2017, 36:1392–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]; Presynaptic protein Synaptojanin was shown to affect autophagosome formation at presynaptic terminals following starvation.
- 82.Hernandez D, Torres CA, Setlik W, Cebrian C, Mosharov EV, Tang G, Cheng HC, Kholodilov N, Yarygina O, Burke RE, et al. : Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.••.Kuijpers M, Kochlamazashvili G, Stumpf A, Puchkov D, Swaminathan A, Lucht MT, Krause E, Maritzen T, Schmitz D, Haucke V: Neuronal autophagy regulates presynaptic neurotransmission by controlling the axonal endoplasmic reticulum. Neuron 2021, 109:299–313e299. [DOI] [PMC free article] [PubMed] [Google Scholar]; Showed that autophagy at presynaptic terminals is required to inhibit excitatory presynaptic transmission vis effects on calcium release from the ER.
- 84.••.Shen W, Ganetzky B: Autophagy promotes synapse development in Drosophila. J Cell Biol 2009, 187:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]; First study in any organism to show that autophagy is required for synapse formation in vivo. Considered a seminal study on the function of autophagy in the nervous system.
- 85.Inoue K, Rispoli J, Yang L, Macleod D, Beal MF, Klann E, Abeliovich A: Coordinate regulation of mature dopaminergic axon morphology by macroautophagy and the PTEN signaling pathway. PLoS Genet 2013, 9, e1003845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Giles AC, Opperman KJ, Rankin CH, Grill B: Developmental function of the PHR protein RPM-1 is required for learning in Caenorhabditis elegans. 2015. G3 (Bethesda). [DOI] [PMC free article] [PubMed]
- 87.Crawley O, Giles AC, Desbois M, Kashyap S, Birnbaum R, Grill B: A MIG-15/JNK-1 MAP kinase cascade opposes RPM-1 signaling in synapse formation and learning. PLoS Genet 2017, 13, e1007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borgen MA, Giles AC, Wang D, Grill B: Synapse maintenance is impacted by ATAT-2 tubulin acetyltransferase activity and the RPM-1 signaling hub. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Opperman KJ, Grill B: RPM-1 is localized to distinct subcellular compartments and regulates axon length in GABAergic motor neurons. Neural Dev 2014, 9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tian X, Li J, Valakh V, DiAntonio A, Wu C: Drosophila Rae1 controls the abundance of the ubiquitin ligase Highwire in post-mitotic neurons. Nat Neurosci 2011, 14:1267–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xiong W, Wei W, Qi Y, Du Z, Qu T, Liu K, Gong S: Autophagy is required for remodeling in postnatal developing ribbon synapses of cochlear inner hair cells. Neuroscience 2020, 431: 1–16. [DOI] [PubMed] [Google Scholar]
- 92.••.Okerlund ND, Schneider K, Leal-Ortiz S, Montenegro-Venegas C, Kim SA, Garner LC, Waites CL, Gundelfinger ED, Reimer RJ, Garner CC: Bassoon controls presynaptic autophagy through Atg5. Neuron 2017, 93:897–913.e897. [DOI] [PubMed] [Google Scholar]; Discovered that the active zone protein Bassoon inhibits autophagy in a subcellular axonal compartment, the presynaptic terminal. Bassoon inhibition of autophagy is required for proper synapse formation in cultured hippocampal neurons.
- 93.Hoffmann S, Orlando M, Andrzejak E, Bruns C, Trimbuch T, Rosenmund C, Garner CC, Ackermann F: Light-activated ROS production induces synaptic autophagy. J Neurosci 2019, 39: 2163–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.•.Hoffmann-Conaway S, Brockmann MM, Schneider K, Annamneedi A, Rahman KA, Bruns C, Textoris-Taube K, Trimbuch T, Smalla KH, Rosenmund C, et al. : Parkin contributes to synaptic vesicle autophagy in Bassoon-deficient mice. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]; Showed that Bassoon inhibits autophagy of synaptic vesicles, and that this involves the ubiquitin ligase Parkin.
- 95.••.Bhukel A, Beuschel CB, Maglione M, Lehmann M, Juhasz G, Madeo F, Sigrist SJ: Autophagy within the mushroom body protects from synapse aging in a non-cell autonomous manner. Nat Commun 2019, 10:1318. [DOI] [PMC free article] [PubMed] [Google Scholar]; Showed that autophagy functions noncell autonomously to restrict active zone size across the Drosophila brain.
- 96.Johnson AE, Orr BO, Fetter RD, Moughamian AJ, Primeaux LA, Geier EG, Yokoyama JS, Miller BL, Davis GW: SVIP is a molecular determinant of lysosomal dynamic stability, neurodegeneration and lifespan. Nat Commun 2021, 12:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rowland AM, Richmond JE, Olsen JG, Hall DH, Bamber BA: Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci 2006, 26:1711–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.••.Kiral FR, Linneweber GA, Mathejczyk T, Georgiev SV, Wernet MF, Hassan BA, von Kleist M, Hiesinger PR: Autophagy-dependent filopodial kinetics restrict synaptic partner choice during Drosophila brain wiring. Nat Commun 2020, 11: 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]; Showed that autophagy restricts presynaptic terminal formation in developing fly retina and prevents ectopic synapse formation. Live imaging from intact fly brain indicated that autophagy affects synaptic filopodial dynamics to influence synapse formation.