

Abstract
Introduction:
Preclinical data supports antitumor activity of tyrosine kinase inhibitor vandetanib with Ret as the therapeutic target in breast cancer. We investigated the effect of preoperative vandetanib on markers of proliferation and apoptosis in breast cancer.
Methods:
Patients with invasive breast cancer were randomly assigned vandetanib 300 mg or placebo PO daily for two weeks prior to operative resection from 1/2014-6/2017. Pre and post treatment specimens were analyzed by immunohistochemistry (IHC) for Ki-67, TUNEL, and p-ERK with stratification by Ret expression by IHC.
Results:
Ten patients were enrolled. There was no statistically significant difference in ERK activation compared to placebo (p=0.45); however, ERK activation was reduced 74% compared to pretreatment biopsy with vandetinib treatment (p=0.005) without a significant reduction in the placebo group (−29%, p=0.55). Mean change in Ki-67 after vandetanib treatment was +0.3% compared to +2.0% in placebo treated patients, p=0.72. Mean change in TUNEL was +0.48 apoptotic nuclei per high power field in the vandetanib arm compared to +1.02 in the placebo arm, p=0.32. In vandetanib treated patients, Ki-67 was reduced 0.3% in RET-positive tumors compared to increased 1.0% in RET-negative tumors, p=0.43 and TUNEL was increased 0.77 in RET-positive tumors and 0.2 in RET-negative tumors, p=0.21.
Conclusions:
In this pilot study, no statistically significant differences on prespecified markers were seen with vandetanib compared to placebo. In accordance with the investigational hypothesis, there was a non-significant trend with vandetanib treatment of reduction in p-ERK and increased effects in Ret expressing tumors.
Keywords: Breast Cancer, Vandetanib, Ret Proto-oncogene, ERK/MAPK
BACKGROUND
Breast cancer is the most common cancer in women and the second most common cause of cancer death1. The identification of molecular subtypes has allowed for a better understanding of tumor biology, natural history, and specific pathways to target for novel therapeutics2. The majority of breast cancers fall into the luminal subtypes characterized primarily by expression of the estrogen receptor (ER), progesterone receptor (PR), and clinically by sensitivity to antiestrogen treatment.
We previously investigated maintenance of the luminal phenotype and described the transcription factor TFAP2C is a key regulator of genes in the ER-associated cluster3 including the Ret proto-oncogene4. Ret signaling has been implicated in breast cancer growth and resistance to antiestrogen5,6. In luminal breast cancer cell lines disruption of Ret signaling with the tyrosine kinase inhibitor vandetanib leads to decreased phosphorylation of ERK, increased apoptosis, and decreased growth in luminal breast cancer cell lines in vitro7. These findings were supported in a xenograft model, where vandetanib reduced breast cancer tumorigenesis and tumor growth, with greater effects in dual vandetanib and tamoxifen treated animals than either agent alone. In contrast to the cell lines, Ki-67 was not found to be reduced with vandetanib treatment, but apoptotic cells were similarly increased and ERK activation similarly reduced.8 Additionally, treatment of primary breast cancer specimens in vitro with tyrosine kinase inhibitor resulted in decreased ERK activation with augmented response in Ret expressing tumors8,9.
Tyrosine kinase inhibitors have shown efficacy for HER2 amplified breast cancer10,11. In other subtypes, underpowered trails which have not utilized biomarkers to identify patients likely to response have not shown efficacy.12–14 For vandetanib, a phase II trail from May 2002-April 2003 of 46 patients with metastatic breast cancer did not show activity, measured by objective response rate15, but this trial was in patients that had failed at least two lines of therapy and did not report receptor subtypes. A recent phase II trial of the similar TKI, cabozantinib, which has significant anti-Ret activity, showed response of metastatic ER+/HER2− tumors16, supporting a role for this strategy in select patients with ER+/HER2− tumors. In this study we investigated the effect of preoperative treatment with the tyrosine kinase inhibitor vandetanib in patients with breast cancer on markers of proliferation and apoptosis and correlated response with expression of Ret.
METHODS
Trial design and Patients
This investigator-initiated trial examined the effect of a short course preoperative vandetanib treatment on markers of proliferation and apoptosis in breast cancer. The trial was designed as a single center phase 2 randomized placebo-controlled trial. Patients were eligible for enrollment following core needle biopsy demonstrating invasive breast carcinoma. All histologic subtypes were eligible, and patients were excluded for inoperable disease or neoadjuvant systemic therapy. Complete inclusion and exclusion criteria are listed in table 1. The study protocol was approved by the institutional review board at the University of Iowa, the FDA (IND #119274), and registered with clinicaltrials.gov (NCT01934335). Following consent, patients were randomized 1:1 to take vandetanib 300 mg or placebo PO daily for 10-14 days prior to surgery. The dose of 300 mg daily was found to be well tolerated in a phase I clinical trial.17 Placebo was formulated by Astra-Zeneca to appear like the drug. Chemotherapy and radiation were given in the adjuvant setting in patients that received those treatments.
Table 1:
Inclusion and Exclusion Criteria
| Inclusion Criteria | Exclusion Criteria |
|---|---|
| Biopsy proven invasive breast carcinoma | Prolonged QT interval (QTc > 480 milliseconds) |
| Female | Any concomitant medications that are known to be associated with Torsades de Pointes or QT elongation |
| Age ≥ 18 | Hypertension not controlled by medical therapy (systolic BP greater than 160 millimeters of mercury [mmHg] or diastolic blood pressure great than 100 mmHg). |
|
ECOG performance status ≤ 2 |
History of arrhythmia (multifocal premature ventricular contractions, bigeminy, trigeminy, ventricular tachycardia), which is symptomatic or requires treatment (CTCAE Grade 3), symptomatic or uncontrolled atrial fibrillation despite treatment, or asymptomatic sustained ventricular tachycardia. |
|
Life expectancy > 6 months |
Significant cardiac event within 12 weeks, or presence of cardiac disease that in the opinion of the Investigator increases the risk of ventricular arrhythmia. |
|
Ability and willingness to provide consent |
Serum calcium or magnesium outside the institutional range of normal. |
| Serum Potassium < 4.0 mmol/L or above 5.0 mmol/L | |
| Creatinine clearance < 50 ml/min | |
| PT > 12 seconds or PTT > 31 seconds | |
| Platelet count of < 100,000 | |
| Alanine aminotransferase (ALT) > 50 U/L, aspartate aminotransferase (AST) > 65 U/L, or alkaline phosphatase (ALP) > 250 U/L | |
| Any cytotoxic treatments, such as neoadjuvant chemotherapy, planned before subsequent surgical procedure. | |
| Previous exposure to vandetanib | |
| Previous enrollment or randomization in this study | |
| Involvement in the planning and/or conduct of the study (applies to both AstraZeneca staff and staff at UIHC). | |
| Previous or current malignancies of other histologies within the last 5 years, with the exception of in situ carcinoma of the cervix, and adequately treated basal cell or squamous cell carcinoma of the skin. | |
| Patients who have received prior surgical site radiation. | |
| Patients on CYP3A4 inhibitors or inducers | |
| Inability to test core biopsy for study markers | |
| Pregnancy or lactation at the time of study entry |
From 1/2014 to 6/2017 we enrolled 12 patients with histologically proven invasive breast cancer. One patient withdrew from the study after being randomized prior to starting treatment and one patient was excluded from the analysis because there was no residual tumor in the resection specimen, leaving 10 analyzable patients (Figure 1). The trial was closed early due to the acquisition of vandetanib by Genzyme from Astra-Zeneca which resulted in alterations in the consent document for which we decided it was inappropriate to proceed.
FIGURE 1.
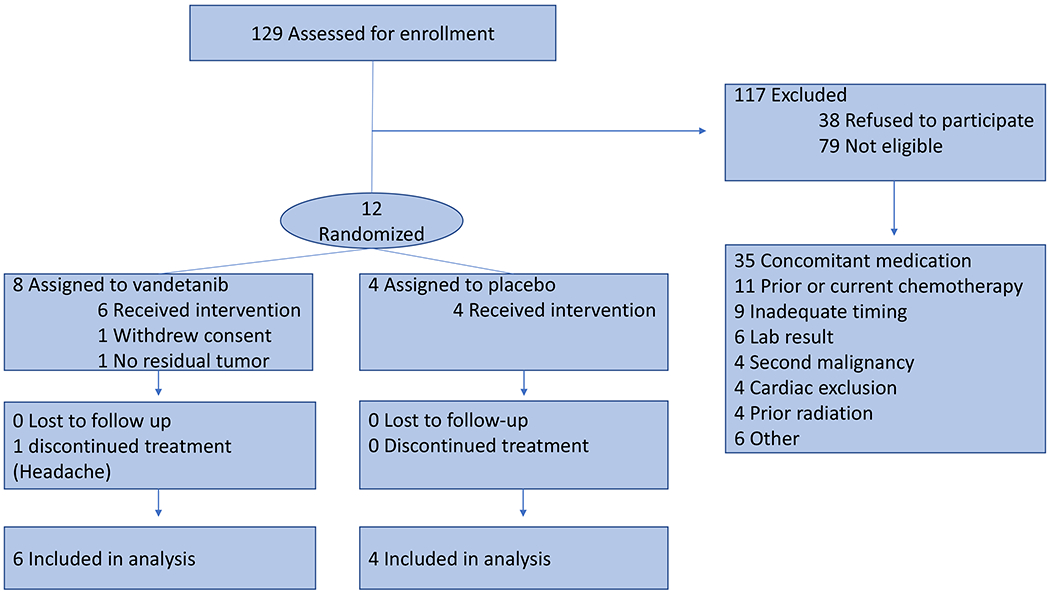
Consort Diagram.
Objectives and endpoints
Our primary endpoint was to determine the change in Ki-67 expression on paired samples obtained before (pretreatment biopsy) and after vandetanib treatment assessed on the surgical resection specimen. Secondary endpoints included determining the change in apoptosis (measured by TUNEL) and ERK activation determined by phosphorylated ERK (p-ERK), stratifying response of the above measures by Ret expression, and surgical complications.
Histologic evaluation
Tumor sections from paraffin-embedded tissue from pretreatment diagnostic biopsies and post treatment resection specimens were selected for immunohistochemical staining. Slides were evaluated and interpreted by a blinded single breast pathologist.
TUNEL Staining
Terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated deoxyuridine-triphosphate (dUTP)-biotin nick-end labelling (TUNEL) staining for apoptosis was performed using the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (#7101; Millipore), as per the manufacturer’s instructions. Briefly, after deparaffinization and rehydration, sections were incubated with proteinase K (#21627; Millipore) for 15 min at room temperature. This was followed by quenching with 3.0% hydrogen peroxide solution in PBS for 5 minutes at room temperature. The slides were rinsed with PBS, blotted, equilibrium buffer was added to the slides followed by the Working TdT Enzyme and the slides were incubated in humidity chamber for 1 hour at room temperature. The stop/wash buffer was added followed by washings with PBS and incubation with Anti-Digoxignenin Conjugate for 30 minutes at room temperature. After PBS washings, the color was developed in Peroxidase Substrate followed by light counterstain with hematoxylin.
Paraffin-embedded sections of normal tonsils were used as positive control. The presence of clear nuclear staining was indicative of apoptotic cells. The number of TUNEL-positive tumor cell nuclei were counted and the apoptotic index was calculated based on the average number of apoptotic nuclei per field in 10 high power fields.
Ki67 Staining
Ki-67 staining was performed using monoclonal mouse anti Ki-67 antigen, clone MIB-1 antibody (1:250, M7240; Dako). Paraffin-embedded sections were deparaffinized and rehydrated. Heat-induced antigen retrieval was performed with citrate buffer, pH 6. After rinsing with Dako 1X Buffer, 3% hydrogen peroxide was used to quench endogenous peroxidase activity at room temperature. Sections were then incubated with Ki-67 antibody for 60 minutes followed by Dako Mouse Envision for 30 minutes and Dako DAB Plus for 5 minutes. This was followed by application of Dako DAB Enhancer and counterstaining with Hematoxylin.
Paraffin-embedded sections of normal tonsils were used as positive control. Ki-67 index was calculated based on the average percentage of cells with nuclear staining. A minimum of 700 cell counts per tumor section was performed.
RET Staining
RET staining was performed using monoclonal rabbit anti- RET (Proto-oncogene tyrosine-protein kinase receptor Ret) antibody (1:50, #3223; Cell Signaling Technology). Paraffin-embedded sections were deparaffinized and rehydrated. Heat-induced antigen retrieval was performed with citrate buffer, pH 6. After rinsing with Dako 1X Buffer, 3% hydrogen peroxide was used to quench endogenous peroxidase activity at room temperature. 10% Goat Serum was then used to block non-specific antibody reactivity (60 minutes). The slides were then incubated with the RET antibody overnight in the refrigerator. After rinsing with Dako 1X Buffer, the secondary biotinylated anti-rabbit 1:500 in 10% Goat/1X Buffer was applied (30 minutes), followed by ABC solution (Vector) , Dako DAB Plus , Dako DAB Enhancer and finally counterstaining with Hematoxylin was performed. Paraffin-embedded sections of a RET- positive medullary thyroid carcinoma case were used as positive control. The percentage of cells with cytoplasmic staining and the intensity of staining (1+, 2+, 3+) were recorded.
Phospho-ERK Staining
Phospho-ERK staining was performed using monoclonal rabbit antibody Phospho p44/42 MAPK (ERK ½), (1:800, #4370S; Cell Signaling Technology). Paraffin-embedded sections were deparaffinized and rehydrated. Heat-induced antigen retrieval was performed with citrate buffer, pH 6. After rinsing with Dako 1X Buffer, 3% hydrogen peroxide was used to quench endogenous peroxidase activity at room temperature. Background Buster (Innovex) was then used to block non-specific antibody reactivity (30 minutes). The slides were then incubated with the pERK 1/2 antibody 1:800 in Dako diluent for 60 minutes. Dako Rabbit Envision System HRP was then applied (30 minutes), followed with Dako DAB Plus, Dako DAB Enhancer and finally counterstaining with Hematoxylin was performed.
P-p44/42 MAPK control slides (#8103S, Cell Signaling Technology) were used. Phospho-ERK scores were calculated based on the summation of the percentages of cells with moderate (2+) and strong (3+) intensity staining. Given the presence of weak patchy (1+) staining in adjacent normal breast epithelium weak (1+) staining was not included in the calculation and only intermediate and strong intensity staining was considered in the calculations. In general, the majority of positive cells in the tumors were strongly positive. The Phospho-ERK scores were calculated based on a minimum of 700 cells counted per tumor section.
CD-31 Staining
CD-31 staining was performed using monoclonal mouse anti-human antibody Clone JC70A (Dako). Paraffin-embedded sections were deparaffinized and rehydrated. Heat-induced antigen retrieval was performed with 1 mmol/L EDTA, pH 9.0. Sections were incubated with CD31 antibody (1:40 dilution) for 30 minutes at room temperature. Paraffin-embedded sections of normal tonsils were used as positive control. CD-31 score was calculated by the average number of CD-31 positive lumen forming structures (microvessels) per high power field in ten high power fields in the most vascular area of the tumor.
Statistical analysis
A sample size of 35 patients per arm was calculated for the primary endpoint of reduction in Ki-67 expression, which based on a standard deviation of 10%, would ensure 90% power to detect a decrease of 5%. Enrollment was planned for 40 patients in each arm (80 total) to account for non-compliance and withdrawal. The Fisher’s exact test for categorical variables and the Student T test for continuous variables were performed. All tests were two-sided p < 5% is considered statistically significant. All analysis was performed using Stata v 15.1 (StataCorp, College Station, Texas, USA).
RESULTS
Patient Characteristics
The clinicopathologic characteristics are listed in table 2. All 10 enrolled patients were positive for the estrogen (ER) and progesterone (PR) receptors and negative for HER2 amplification. All 10 patients were non-Hispanic white. In the vandatanib group, 5 (83%) tumors had ductal histology and 1 (17%) had mucinous carcinoma, whereas in the placebo group all 4 were ductal carcinoma. Adverse events (AEs) occurred in 5 (83%) vandetanib treated patients consisting of grade I diarrhea in 3 patients, hot flashes (2), anorexia (2), rash (1), and grade II headache requiring discontinuation of treatment (1). In the placebo arm 2 patients (50%) had AEs consisting of grade 1 headache and fatigue/fever. One patient in the vandetanib arm had a grade III adverse event with hematoma requiring return to the operating room for evacuation after nipple sparing mastectomy with axillary lymph node dissection and immediate implant reconstruction. No surgical complications were observed in the placebo group.
Table 2:
Clinicopathologic characteristics
| Vandetanib | Placebo | p value | |
|---|---|---|---|
| Number | 6 | 4 | n/a |
| Median age, years, (range) | 57 (50-71) | 65 (45-72) | 0.33 |
| ER+/PR+/HER2− | 6 (100%) | 4 (100%) | 1.0 |
| Ductal Histology | 5 (83%) | 4 (100%) | 1.0 |
| Race — Non-Hispanic White | 6 (100%) | 4 (100%) | 1.0 |
| Stage | |||
| Stage I | 1 (17%) | 2 (50%) | 0.50 |
| Stage II | 5 (83%) | 2 (50%) | |
| Adverse event (any) | 5 (83%) | 2 (50%) | 0.50 |
| Grade 1 | 4 (80%) | 2 (100%) | 1.0 |
| Grade 2 | 1 (20%) | 0 (0%) | 1.0 |
| Operation | |||
| Mastectomy | 2 (33%) | 1 (25%) | 1.0 |
| Lumpectomy | 4 (67%) | 3 (75%) | |
| Complication (any) | 1 (17%) | 0 (0%) | 1.0 |
| Grade 3 | 1 (100%) | n/a | |
| Chemotherapy | 1 (17%) | 0 (0%) | 1.0 |
| Radiation | 4 (67%) | 3 (75%) | 1.0 |
Ki-67
The primary endpoint for the study was a reduction in Ki-67 with vandetanib treatment compared to placebo, figure 2A. Mean pretreatment positivity for Ki-67 was 16% (range 5%-42%). For patients in the vandetanib arm, mean pretreatment Ki-67 was 15.2% and mean posttreatment Ki-67 was 15.5% for an average increase of 0.3%, p=0.92. In the placebo arm mean increase in Ki-67 was 2.0% (p=0.51 pre vs. post treatment), and the comparison to the vandetanib arm was not statistically significant (+0.3% vs. +2.0%, p=0.70).
FIGURE 2. Effects of preoperative vandetanib on markers of proliferation and apoptosis.

Matched specimens from the pretreatment biopsy and posttreatment surgical resection are shown for A) Ki-67, B) TUNEL, C) p-ERK, and D) CD-31 score for microvascular formation. Results are presented as pre and post treatment individual patient scatterplot, waterfall plot of marker change where each bar represents a patient, and graphically as the pre and post-treatment mean in the placebo and vandetanib groups. Error bars represent standard error and p-values compare pre and post treatment samples.
TUNEL
Our preclinical model showed an increase in apoptosis with vandetanib treatment, which we assessed using TUNEL, figure 2B. Mean pretreatment TUNEL apoptotic index in all patients was 1.04 (range 0-4.4). Mean pretreatment TUNEL apoptotic index was 1.22 for patients randomized to vandetanib and mean posttreatment TUNEL apoptotic index was 1.70 for a mean increase of 0.48, p=0.10. The mean increase in TUNEL positivity was 1.02 in patients treated with placebo, p=0.16. The comparison between the change in vandetanib (+0.48) and placebo (+1.02) was not statistically significant (p=0.35).
Phosphorylated ERK
In all patients, mean pretreatment p-ERK score was 112.6 (range 55-240), figure 2C. The comparison of the change in p-ERK between the vandetanib and placebo arms did not achieve statistical significance (mean reduction in p-ERK score 74.7 vs. 36.8, p=0.45). There was a statistically significant reduction in p-ERK in patients treated with vandetanib (102.5 pretreatment vs. 27.8 posttreatment, p=0.005) when analyzed independently. By comparison, there was not a significant reduction in p-ERK in the placebo treated patients (127.8 pretreatment vs. 91 posttreatment, p=0.55).
Microvascular formation
Our previous work in a xenograft model showed reduced microvascular formation in tumors of animals treated with vandetanib8. To assess the effect of preoperative vandetanib on tumor microvasculature we performed CD31 staining and scored the number of CD31 positive lumen forming structures pre and post treatment. The post treatment IHC was unsuccessful in one vandetanib treated patient resulting in 4 analyzable pre-post treatment pairs in the placebo group and 5 pairs in the vandetanib group. In all patients, mean pretreatment CD31 score was 10.4 (range 2-26), figure 2D. Mean post-treatment CD31 score was 10.5 (range 2.4-21.5). There was no significant difference in CD31 score in either the placebo or vandetanib groups. In patients treated with placebo, mean CD31 score was reduced −1.0 compared to mean change in vandetanib treated patients of +0.3, p=0.61.
Treatment Effect by Ret Expression
Utilizing expression of Ret as a biomarker of response was one of the secondary endpoints of this study. Ret was expressed in one patient in the placebo arm so we performed this analysis only using the vandetanib arm, which had three Ret-positive and three Ret-negative patients (figure 3). Mean change in Ki-67 with vandetanib treatment was −0.3% in Ret positive tumors and +1.0% in Ret-negative tumors, p=0.43. Ret positive tumors potentially had a larger increase in TUNEL (0.77) in compared to 0.2 in Ret negative tumors, p=0.21. Finally, Ret-negative tumors had an absolute reduction of p-ERK of 83.7 compared to 65.7 in Ret-positive tumors with vandetanib treatment, p=0.12, although the relative effect was larger in Ret-positive tumors (−88%) than Ret-negative (−59%), p=0.10. Although not reaching statistical significance, Ret-positive tumors demonstrated a trend for augmented response, as hypothesized, by Ki-67 and TUNEL. Both Ret-positive and Ret-negative tumors had a large reduction in ERK activation after vandetanib treatment.
FIGURE 3. Stratification of response by Ret expression.
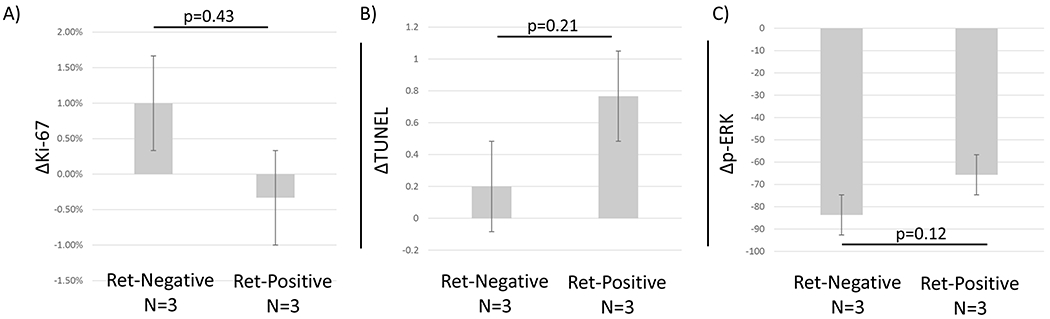
The mean response measured by A)Ki-67, B) TUNEL, and C) p-ERK for patients in vandetanib treated patients was stratified by Ret expression. Ret expressing tumors appear to exhibit an augmented response, although without statistical significance, measured by decreased Ki-67, increased TUNEL, and both Ret positive and Ret negative tumors had decreased p-ERK.
DISCUSSION
In this trial we investigated the effect of preoperative vandetanib treatment in patients with breast cancer on cellular markers of proliferation and apoptosis. The trial closed short of planned accrual due to change in ownership of the investigational drug and none of the trial endpoints met statistical significance, but there are several important trends noted from the data.
Accrual for this trial was quite low. Of 129 patients assessed for enrollment only 50 were eligible for participation. The most common reason patients were not eligible were concomitant medications, which in our patient population was most commonly proton pump inhibitors and H2-antagonists. It is also worth noting that of the 50 patients eligible to participate, 38 (76%) refused to participate. The preoperative window design can be challenging for patients as there are potential side effects from the intervention without potential direct benefit to the participant. As clinicians and scientists we carefully weigh the potential risks to the patient and benefits of knowledge to be gained with oversight from the institutional review board, cancer center protocol review and monitoring committee, and FDA, although this can be difficult to comprehensively convey to the patient. Institutional support for enrollment, as well as industry support for the duration of the trial are essential for successful accrual with statistical power and planned enrollment timeframes need to account for patients declining participation.
An increase in apoptotic cells and a reduction in ERK activation was observed in the placebo group as well as the treatment group. Comments from both our cancer center protocol monitoring committee and the FDA criticized the inclusion of a placebo arm. Without a placebo group the entirety of these changes would have been attributed to the treatment, including a statistically significant reduction in p-ERK. While these findings might be demonstrated with a larger sample size, the importance of the placebo arm to interpret these findings is illustrated. Additionally, the results from the placebo arm inform the expected baseline range, heterogeneity of sampling, and post biopsy changes which are important to understand for future window trials.
Tyrosine kinase inhibitors, such as vandetanib, with activity against VEGFR and EGFR are hypothesized to inhibit wound healing. In this, the first trial of preoperative vandetanib, there was no significant difference in surgical complications. These results demonstrate for the first time the safety of preoperative vandetanib and support the continued investigation of this compound in the preoperative setting.
Although we were not able to reach target enrollment for statistical power, the data in this trial provided a trend that tended to support the investigational hypothesis. Possible reductions in Ki-67 and p-ERK compared to placebo were consistent with preclinical data, although the increase in TUNEL was less in vandetanib treated patients than placebo. Also consistent with the hypothesis was that tumors that express Ret had increased vandetanib effect by Ki-67 and TUNEL, although without statistical significance. A similar, large, reduction in ERK activation was observed in Ret-positive and Ret-negative tumors, which could mean that the primary mechanism of this effect is not mediated through Ret. Vandetanib is a tyrosine kinase inhibitor with VEGFR and EGFR activity and further study is needed to elucidate markers and mechanism of pathway specific responses.
This study showed a decrease in ERK activation in vandetanib treated patients which was not observed in the placebo arm, and due to small sample size was not statistically significant when the two arms were compared. If confirmed, these findings have important potential for clinical relevance in hormone resistant breast cancer, where new therapies are needed. Ret is expressed most commonly in ER-positive breast cancer and our group and others have shown in preclinical models that Ret activation can induce resistance to anti-estrogen therapy, and that inhibition of Ret signaling can augment endocrine sensitivity6,8,18. Further, activation of ERK, which occurs downstream from Ret, has been shown as a mechanism of resistance to endocrine therapy19. This study provides in vivo evidence of a reduction of ERK activation in breast cancer with systemic vandetanib treatment which may also be considered in neoadjuvant trials in combination with anti-estrogen therapy. Our study indicates that vandetanib is well-tolerated and is a potentially promising new therapy to alter hormone response in breast cancer. Further study is needed to determine the effect of vandetanib mediated reduction in ERK signaling, and the role of Ret and other tyrosine kinases, to alter hormone response.
The potential of the strategy of TKI with anti-Ret activity for patients with ER+/HER2− subtype breast cancer was demonstrated in a recent phase II trial of the tyrosine kinase inhibitor Cabozantanib16 which showed patients with hormone receptor positive/HER2 negative breast cancer with bone metastasis had a 38% partial response rate and 50% of patients did not progress. Cabozantanib, similar to vandetanib, is a TKI with activity against MET/RET/VEGFR2, but those levels were not measured or correlated with response. Further study is needed to determine biomarkers of vandetanib and cabozantanib response in endocrine resistant ER+/HER2− breast cancer and define clinical uses in this important and challenging disease.
This study has several limitations. As discussed above accrual was low and enrollment was underpowered. This trial was a window of opportunity design and therefore cannot comment on oncologic outcomes associated with vandetanib treatment. Finally, although all receptor subtypes were eligible for enrollment, only hormone receptor positive, HER2 negative patients were enrolled which prevented a comparison of response by receptor subtype.
CONCLUSION
In combination with the preclinical data, these results support the investigation of vandetanib in breast cancer with expression of Ret as a biomarker of response. Future trials focusing on identifying patients most likely to benefit using biologically relevant markers of response and powered to identify effects of vandetanib on recurrence and survival in patients with breast cancer are warranted.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health grants R01CA109294 (PI: R.J. Weigel), T32CA148062 (PI: R.J. Weigel) and by a generous gift from the Kristen Olewine Milke Breast Cancer Research Fund. PMS, AWL, and ACB were supported by NIH grant T32CA148062. Astra-Zeneca provided vandetanib and placebo for this study, as well as financial support to cover screening and toxicity assessments
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. [DOI] [PubMed] [Google Scholar]
- 2.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woodfield GW, Chen Y, Bair TB, Domann FE, Weigel RJ. Identification of primary gene targets of TFAP2C in hormone responsive breast carcinoma cells. Genes Chromosomes Cancer. 2010;49(10):948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spanheimer PM, Woodfield GW, Cyr AR, et al. Expression of the RET proto-oncogene is regulated by TFAP2C in breast cancer independent of the estrogen receptor. Ann Surg Oncol. 2013;20(7):2204–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gattelli A, Hynes NE, Schor IE, Vallone SA. Ret Receptor Has Distinct Alterations and Functions in Breast Cancer. J Mammary Gland Biol Neoplasia. 2020;25(1):13–26. [DOI] [PubMed] [Google Scholar]
- 6.Morandi A, Martin LA, Gao Q, et al. GDNF-RET signaling in ER-positive breast cancers is a key determinant of response and resistance to aromatase inhibitors. Cancer Res. 2013;73(12):3783–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spanheimer PM, Cyr AR, Gillum MP, Woodfield GW, Askeland RW, Weigel RJ. Distinct pathways regulated by RET and estrogen receptor in luminal breast cancer demonstrate the biological basis for combination therapy. Ann Surg. 2014;259(4):793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spanheimer PM, Park JM, Askeland RW, et al. Inhibition of RET increases the efficacy of antiestrogen and is a novel treatment strategy for luminal breast cancer. Clin Cancer Res. 2014;20(8):2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spanheimer PM, Lorenzen AW, De Andrade JP, et al. Receptor Tyrosine Kinase Expression Predicts Response to Sunitinib in Breast Cancer. Ann Surg Oncol. 2015;22(13):4287–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freedman RA, Gelman RS, Anders CK, et al. TBCRC 022: A Phase II Trial of Neratinib and Capecitabine for Patients With Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer and Brain Metastases. J Clin Oncol. 2019;37(13):1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rimawi MF, Niravath P, Wang T, et al. TBCRC023: A Randomized Phase II Neoadjuvant Trial of Lapatinib Plus Trastuzumab Without Chemotherapy for 12 versus 24 Weeks in Patients with HER2-Positive Breast Cancer. Clin Cancer Res. 2020;26(4):821–827. [DOI] [PubMed] [Google Scholar]
- 12.Yardley DA, Dees EC, Myers SD, et al. Phase II open-label study of sunitinib in patients with advanced breast cancer. Breast Cancer Res Treat. 2012;136(3):759–767. [DOI] [PubMed] [Google Scholar]
- 13.Mayer EL, Isakoff SJ, Klement G, et al. Combination antiangiogenic therapy in advanced breast cancer: a phase 1 trial of vandetanib, a VEGFR inhibitor, and metronomic chemotherapy, with correlative platelet proteomics. Breast Cancer Res Treat. 2012;136(1):169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boer K, Lang I, Llombart-Cussac A, et al. Vandetanib with docetaxel as second-line treatment for advanced breast cancer: a double-blind, placebo-controlled, randomized Phase II study. Invest New Drugs. 2012;30(2):681–687. [DOI] [PubMed] [Google Scholar]
- 15.Miller KD, Trigo JM, Wheeler C, et al. A multicenter phase II trial of ZD6474, a vascular endothelial growth factor receptor-2 and epidermal growth factor receptor tyrosine kinase inhibitor, in patients with previously treated metastatic breast cancer. Clin Cancer Res. 2005;11(9):3369–3376. [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Higgins MJ, Tolaney SM, et al. A Phase II Trial of Cabozantinib in Hormone Receptor-Positive Breast Cancer with Bone Metastases. Oncologist. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holden SN, Eckhardt SG, Basser R, et al. Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol. 2005;16(8):1391–1397. [DOI] [PubMed] [Google Scholar]
- 18.Plaza-Menacho I, Morandi A, Robertson D, et al. Targeting the receptor tyrosine kinase RET sensitizes breast cancer cells to tamoxifen treatment and reveals a role for RET in endocrine resistance. Oncogene. 2010;29(33):4648–4657. [DOI] [PubMed] [Google Scholar]
- 19.Razavi P, Chang MT, Xu G, et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell. 2018;34(3):427–438 e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.