

SUMMARY
Humoral immunity provides protection from pathogenic infection and is mediated by antibodies following the differentiation of naïve B cells (nB) to antibody-secreting cells (ASC). This process requires substantial epigenetic and transcriptional rewiring to ultimately repress the nB program and replace it with one conducive to ASC physiology and function. Notably, these reprogramming events occur within the framework of cell division. Efforts to understand the relationship of cell division with reprogramming and ASC differentiation in vivo have uncovered the timing and scope of reprogramming, as well as key factors that influence these events. Herein, we discuss the unique physiology of ASC and how nB undergo epigenetic and genome architectural reorganization to acquire the necessary functions to support antibody production. We also discuss the stage-wise manner in which reprogramming occurs across cell divisions and how key molecular determinants can influence B cell fate outcomes.
Keywords: Plasma cells, B cell differentiation, epigenetics, transcriptomics, cell division, heme
1. INTRODUCTION
The humoral arm of the adaptive immune system relies on robust differentiation of naïve B cells (nB) into antibody-secreting cells (ASC) or plasma cells. Upon antigen encounter, nB become activated, rapidly proliferate, and undergo substantial reprogramming events, with a subset differentiating to ASC (Figure 1). Once B cells are activated, interactions with CD4 T cells play a major role in determining the cell fate choices, antibody specificity and isotype class, and ultimately longevity of the response. Stimulation with T cell-independent (TI) antigens, such as lipids and polysaccharides, leads to the formation of short-lived plasma cells (SLPC). TI antigens can be subdivided into two types1,2. TI type I antigens include pathogen-associated molecular patterns that bind pattern recognition receptors, such as toll-like receptor recognition of bacterial cell wall components or DNA. TI type II antigens have highly repetitive, multivalent structures that activate B cells by crosslinking of their B cell receptors (BCR). TI type I antigens elicit large polyclonal responses, whereas TI type II antigens engage the BCR and thus induce antigen-specific B cell response. Stimulation with a protein antigen induces a T cell-dependent (TD) response in which B cells migrate to germinal centers (GC) and undergo somatic hypermutation and affinity maturation, ultimately resulting in the generation of BCR with higher antigen affinity. The final output of the GC reaction is long-term immunological protection provided by the generation of long-lived plasma cells (LLPC) and memory B cells3. Whether derived from TI or TD responses, antibodies secreted by ASC provide protection from pathogens4. Thus, the formation of ASC and antibodies is the cornerstone of adaptive immunity.
Figure 1. B cell differentiation to ASC requires epigenetic reprogramming.
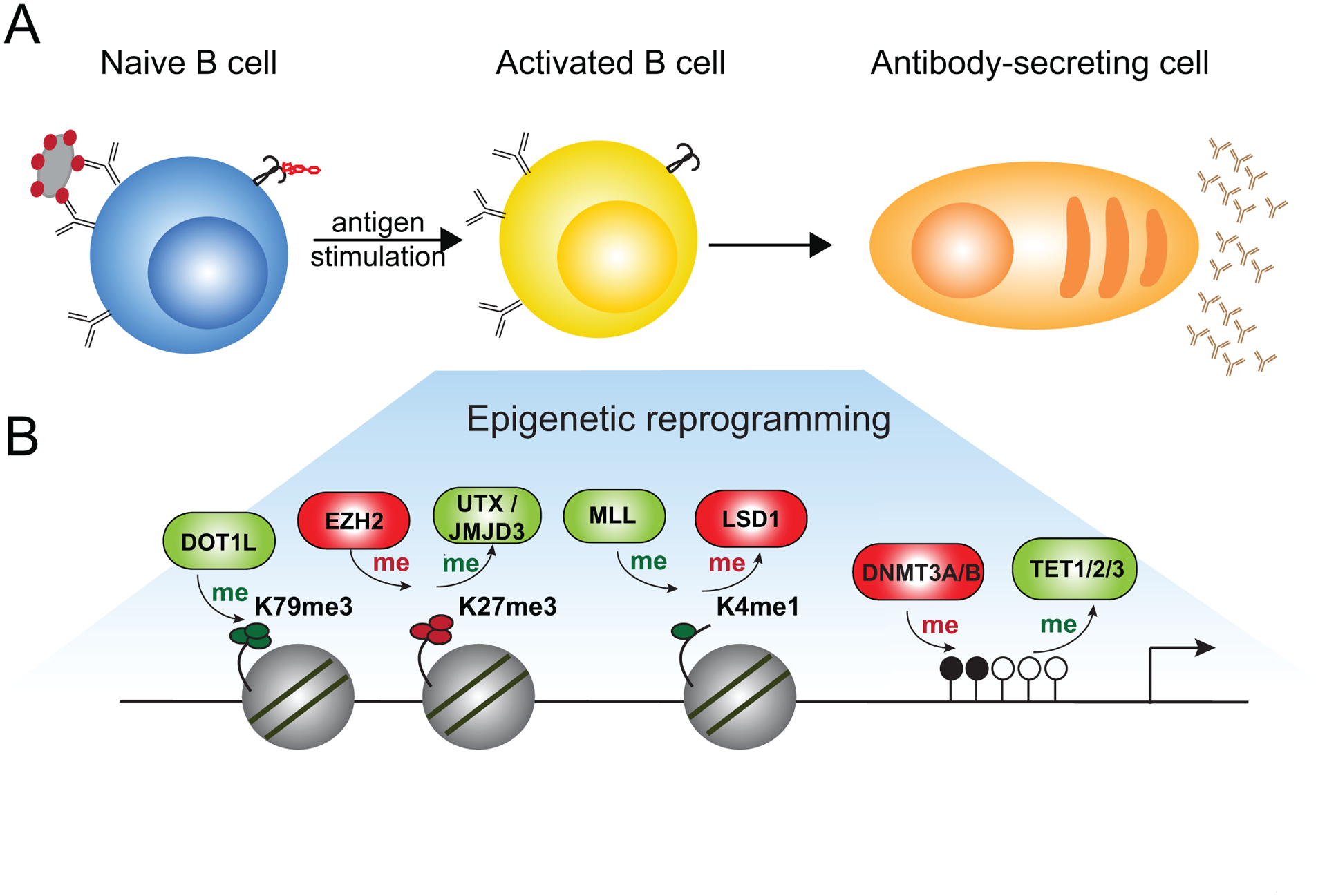
(A) Following stimulation with T-cell independent or T-cell dependent antigen, naïve B cells become activated and differentiate into antibody-secreting cells. (B) The process of naïve B cell reprogramming requires substantial changes to the epigenome. Enzymes that catalyze the addition or removal of DNA methylation and various histone modification are depicted in the shaded box. Enzymes that promote gene expression are colored in green, while enzymes promoting gene repression are colored in red.
2. UNIQUE BIOLOGY OF A PLASMA CELL
2.1. Metabolic adaptations to manufacture and secrete antibodies
The primary function of an ASC is the manufacturing and secretion of antibody molecules. Remarkably, ASC are capable of secreting up to 10,000 antibodies per second5. To support the high rate of antibody secretion, nB undergo significant morphological and bioenergetic changes as they differentiate from a quiescent B cell that does not secrete antibody to an ASC6. This change in metabolism is essential to meet the energy demands required for rapid proliferation of responding B cells and the translational requirements of ASC7,8. Responding activated B cells (actB) utilize both glycolysis and oxidative phosphorylation (OXPHOS), whereas GC B cells also utilize fatty acid oxidation9,10. As actB divide and differentiate towards ASC, they gradually increase their capacity to perform OXPHOS9. This shift towards OXPHOS is in part due to an increase in transcription of more than 100 components of the electron transport chain tricholaric acid cycles9, as well as activity mediated by Protein Kinase C β (PKC-β), which is induced in actB following antigen stimulation11,12. PKC-β was shown to promote mTORC1 signaling to facilitate mitochondrial remodeling and heme biosynthesis, which is necessary for ASC formation. While PKC-β-deficient cells accumulated reactive oxygen species and failed to differentiate, these phenotypic changes were reversed by treatment with hemin suggesting a mechanistic link between PKC-β and heme biosynthesis13. ASC primarily rely on OXPHOS to support antibody secretion, and this metabolic switch is dependent on BLIMP1, one of the master transcriptional regulators of ASC fate. Experimentally, promoting OXPHOS metabolism in responding B cells using dichloroacetate14–16 results in increased ASC formation, suggesting a role for OXPHOS in promoting differentiation9. However, within the ASC lineage there appears to be differences in oxygen consumption. While SLPC consume oxygen nearly at their maximal possible levels, bone marrow LLPC exhibit a high spare respiratory capacity17.
Another metabolic difference specific to LLPC is the high surface level expression of the glucose transporter GLUT117. Interestingly, despite ASC reliance on glucose to support their metabolic demands, genetic deletion of GLUT-1 does not completely block ASC formation, indicating that glucose uptake depends on additional transporters or pathways18,19. Another glucose transporter, GLUT-6, is likely to play a similar role in ASC20,21. Additionally, a large fraction of sugars are shunted to hexosamine biosynthetic pathways in ASC for antibody glycosylation17, which modulates antibody effector functions22. Inhibiting glycosylation using tunicamycin resulted in significant ASC death due to an increase in misfolded proteins and chronic induction of the unfolded protein response (UPR)23. Furthermore, differences in glucose uptake and subsequent pyruvate import into the mitochondria have been attributed to differences in the longevity of SLPC versus LLPC, thus pointing to cross-talk between metabolism and cell survival17. Therefore, while LLPC use glucose for various processes, OXPHOS is still an important source of energy. Additionally, the unique metabolic features of LLPC might allow survival in the hypoxic BM environment24. These studies highlight the unique bioenergetic adaptations of ASC metabolism and how distinct modes may be utilized to promote differentiation and support the demands of antibody generation.
The reliance on OXPHOS is a unique feature of ASC. One of the key drivers of mitochondrial metabolism is the molecule heme. Heme is an iron containing porphyrin ring and is a key part of the cytochrome proteins that function in the electron transport chain and facilitate OXPHOS metabolism25. In addition to its role in the mitochondria, heme can influence B cell fate outcomes through additional mechanisms (Figure 2). One mechanism by which heme promotes ASC formation involves direct binding to the transcription factor BACH2 resulting in its degradation26. BACH2 maintains B cell fate by repressing BLIMP1, thus heme directly promotes ASC formation by degrading BACH2 and allowing for the induction of the ASC transcriptional program. In addition, heme can be degraded through the action of heme oxygenase-1 (HO-1) into carbon monoxide, biliverdin, and iron. Iron is an essential cofactor for many epigenetic erasers, such as histone27 and DNA demethylases28 as well as certain histone deacetylase 8 (HDAC8)29,30. Mice fed an iron-deficient diet exhibited diminished GC B cells and ASC in response to antigen. These findings were further corroborated by ex vivo studies that showed that treatment with an iron chelator impaired B cell proliferation and differentiation31. The impaired ASC response observed following iron depletion was attributed to the failure of H3K9 demethylases to promote the expression of cyclin E1, which regulates entry into S phase of the cell cycle, and resulted in G1/S arrest of iron-deficient B cells31. These data point to heme as a central metabolite with the ability to influence B cell fate through multiple mechanisms.
Figure 2. Heme regulates cell fate outcomes by various mechanisms.
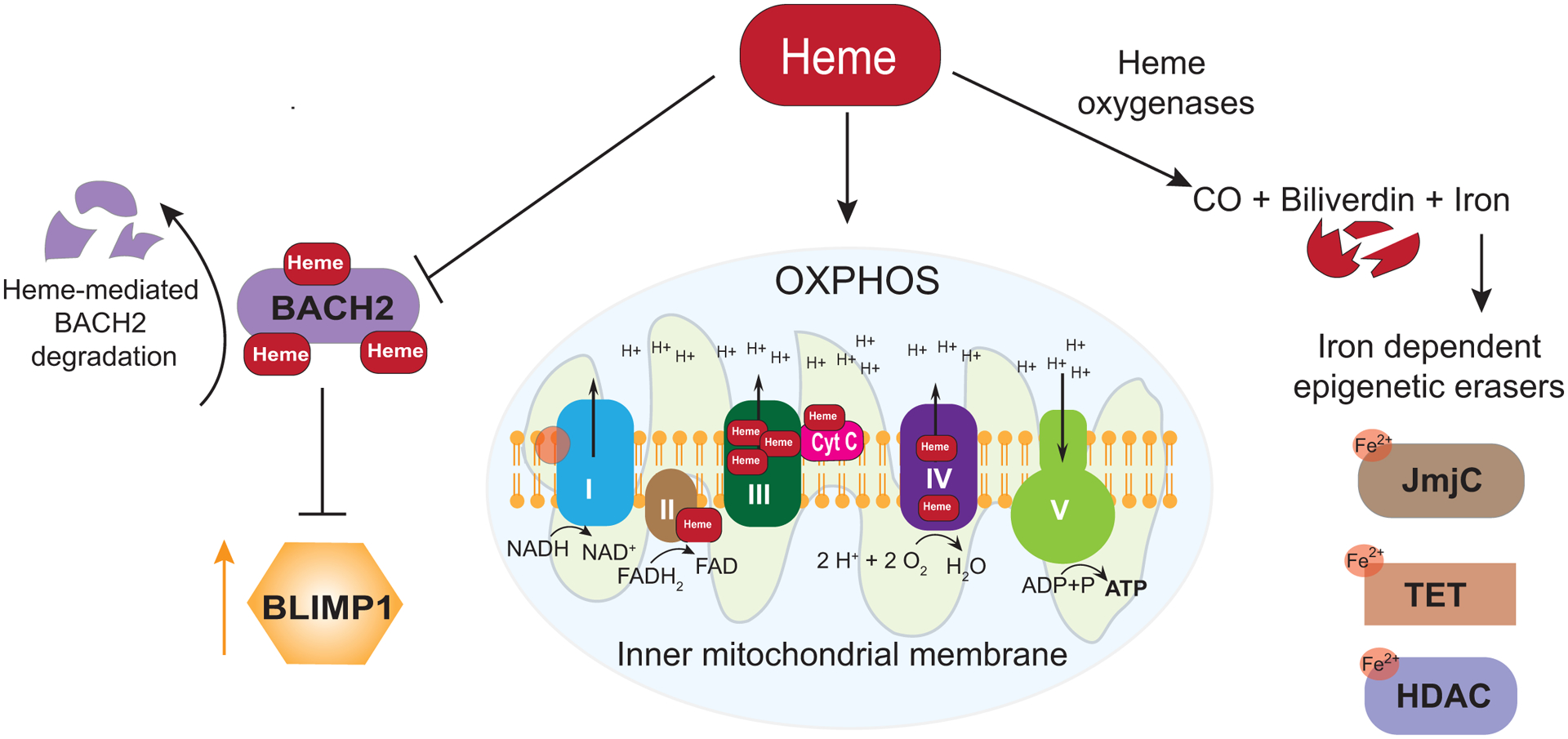
Schematic of the processes regulated by heme that impact ASC formation is shown. Heme directly binds to BACH2 and promotes its degradation, thus relieving BLIMP1 repression (left). Heme is a component of number of cytochrome proteins involved in the electron transport chain of OXPHOS (middle). Heme oxygenase catalyzes heme degradation into biliverdin, carbon monoxide (CO), and free iron (Fe2+). Iron serves as a cofactor for some enzymes that remove epigenetic modifications. Examples are depicted (right).
Memory B cells are able to rapidly differentiate into ASC compared to antigen-inexperienced naïve B cells; however, the mechanisms that facilitate these enhanced secondary responses are not fully understood. Memory B cells were found to harbor unique epigenetic and transcriptional features with primed accessible chromatin surrounding genes important in ASC differentiation, including BLIMP132. Compared to naïve B cells, memory B cells upregulated their iron homeostasis metabolic pathway and possessed greater basal levels of heme, as determined using the expression of HO-132. Treatment of mouse and human naïve and memory B cells with hemin promoted ASC formation and led to an increase in both basal and maximal respiratory capacity32. Although, heme can act through multiple pathways to promote ASC formation (Figure 2), the results mirrored the increase in ASC observed following dichloroacetate treatment to enhance OXPHOS metabolism. Thus, recent studies have demonstrated the critical role of iron and heme in regulating cell fate decisions. More work is necessary to elucidate the precise mechanism(s) and factors regulated by heme/iron (BACH2, OXPHOS, demethylases) and how they impact ASC formation. Additionally, these data highlight the need to understand the interplay and crosstalk between metabolic byproducts and their impact on reprogramming through chromatin modifying enzymes that are dependent on these metabolites33.
In addition to upregulating metabolism to support antibody secretion, ASC also support their energy needs by autophagy, a process that allows cells to degrade proteins and reuse the resulting metabolic intermediates34. This process involves engulfing cytoplasmic contents in a double-membraned vesicle that is delivered to the lysosome for degradation and recycling. When evaluating autophagy in ASC differentiation from murine B cells, significant autophagic induction was observed in both in vitro and in vivo generated ASC, independent of the initial stimulation35. The importance of this pathway is illustrated by the fact that deletion of Atg5, an essential component of the autophagy pathway, resulted in increased ER stress, reduced cellular ATP, and a decrease in ASC survival36. In contrast, an increase in antibody secretion on a per cell basis was observed, suggesting that autophagy may be necessary to limit antibody secretion, thus reducing the ATP expenditure and promoting cell survival.
The high rate of antibody production also requires substantial adaptations of the secretory apparatus and UPR, which is normally induced during stress and initiated by the accumulation of misfolded proteins in the endoplasmic reticulum (ER)37. The UPR consists of three highly conserved signal transductions pathways triggered by three main UPR-inducing ER stress sensors: 1) PKR-like endoplasmic reticulum kinase (PERK), 2) activating transcription factor 6 (ATF6), and 3) inositol-requiring enzyme 1α (IRE1α)38. The PERK pathway leads to an overall suppression of protein translation39,40; however, very limited PERK activation in ASC is observed in vitro41. In fact, genetic ablation of PERK or CHOP (C/EBP homologous protein), a downstream component of the PERK pathway, has no impact on plasma cell survival in vitro42,43. ER stress that exceeds the capacity of adaptive UPR leads to activation of the apoptotic program partially induced by CHOP44. However, it has been reported that LLPC are less sensitive to ER stress associated apoptosis45, although the underlying mechanism remains to be elucidated. The ATF6α pathway can augment ER quality control processes and drive ER expansion, but deletion of ATF6α did not impact antibody secretion or survival in vitro and in vivo46.
The response initiated by IRE1α is best understood in ASC. IRE1α is a ribonuclease and splicing factor that promotes the splicing of X-box protein 1 (XBP1) mRNA to a more stable Xbp1 isoform47. Once generated, XBP1 promotes the expression of genes necessary for the expansion of the secretory apparatus48,49. The importance of XBP1 is highlighted by in vivo studies, where deletion of Xbp1 impeded the ability of ASC to secrete antibodies. XBP1-deficient ASC exhibited normal protein folding but altered glycosylation and lipid synthesis, leading to the failure to mount a proper UPR50. These findings indicate that XBP1 is not required for ASC formation but rather for antibody secretion49,51. While XBP1 is typically thought of as the master transcription factor regulating the UPR response initiated due to the high antibody secretion, a recent study demonstrated that actB upregulate UPR-related genes prior to becoming an ASC52. Upregulation of the actB UPR program was regulated by mTORC1 signaling and the adaptor protein Raptor and occurred prior to XBP1 activity52. This important finding indicates that part of the actB program is to prepare for subsequent antibody synthesis by initiating and building the transcriptional networks to deal with the stress of protein production and secretion.
2.2. Antibody-secreting cell survival and homing
ASC are able to survive in multiple niches in mammals. Both SLPC and LLPC can reside in secondary lymphoid organs, such as the spleen, bone marrow (BM)53,54, and in tissues such as the gut54–56. Since ASC are generated in secondary lymphoid organs, migration of ASC to their survival niche is facilitated by the expression of chemokine receptors. This process is initiated by upregulation of S1PR1, which allows for entry into the bloodstream57, followed by expression of specific chemokine receptors that direct ASC to their niche. Expression of CXCR4 promotes homing to the BM58. Blocking the CXCR4-CXCL12 axis diminished ASC homing to and retention in the BM59, resulting in obvious accumulation of ASCs in the spleen and a concordant decrease in BM ASC60. CXCR3 promotes homing to inflamed tissues61, while CCR10 and CCR9 promote the migration of IgA+ ASC to the gut, with CCR9 being restricted to the small intestine62. Other factors that have been implicated in ASC maturation or rention in the BM include: very late antigen 4 (VLA4)63, CD4464, CD2865, and CD9366 on PCs, as well as ZBTB2067,68 and Aiolos69.
Lasting humoral immunity relies on the long-term survival of LLPC. However, not all ASC are created equally. TI responses predominantly generate SLPC that die within a few days. On the other hand, ASC formed following stimulation with TD antigens are able to home to the BM and persist for decades57,70,71. Thus, understanding the factors regulating ASC survival is crucial to the improvement and development of successful vaccines. The BM is the primary niche of LLPC owning in part to the presence bone marrow stromal cells and cytokines, such as APRIL and BAFF, which promote the expression of the anti-apoptotic gene Mcl1 and thus promoting ASC survival72,73. A recent study identified two novel factors, fibronectin and YWHAZ, that were secreted by stromal cells that promote survival of human LLPC in vitro74. Treatment with antibodies targeting these proteins reduced PC survival in the culture. Furthermore, the combination of mesenchymal stromal cells secretome (including fibronectin and YWHAZ), APRIL, and hypoxic conditions improved the long-term survival of LLPC in vitro and likely in vivo74. However, even within the BM, there is heterogeneity in the lifespan of ASC suggesting that other factors, including cell intrinsic differences are at play.
3. B CELL DIFFERENTIATION IS A COORDINATED MULTISTEP PROCESS
3.1. Initiation of the antibody-secreting cell transcriptional program
B cells and ASC express mutually exclusive gene expression programs75. As a result, B cell differentiation into ASC requires significant transcriptional rewiring that is coordinated by transcription factors (TF)75. For example, Pax576,77, Bach278–80, and Ebf181 are important for establishing or maintaining the nB program, while Bcl682–86 and Irf887,88 regulate actB fate. B cell activation leads to a progressive upregulation of IRF4, which in turn promotes the expression of the master ASC regulator, BLIMP189,90. BLIMP191 (encoded by Prdm1) extinguishes the B cell program and orchestrates the ASC program. BLIMP1 alone coordinates many distinct phenotypes associated with ASC, including: 1) division cessation through repression of C-myc92; 2) loss of MHC class II antigen presentation by repressing expression of the class II transactivator (CIITA)93; and 3) establishment of the ASC program via repression of PAX594,95, a critical TF that maintains B cell fate programs and represses Prdm1. While recent work has demonstrated that repression of Pax5 is not essential for formation of bona fide ASC, normal ASC gene expression programs are significantly dysregulated when PAX5 remains expressed in ASC96. The activity of these factors coordinates programming essential for each cellular stage and have been reviewed extensively70,75
Although there are differences in the cell types that emerge and timing of TI and TD B cell responses, few differences in SLPC or LLPC have been identified, suggesting that the processes that lead to ASC formation are likely similar7,97,98. Indeed, SLPC contribute to early protective antibodies to influenza99, and more recent work demonstrated that extrafollicular responses correlated with early neutralizing antibodies in critically ill COVID-19 patients100. Thus, SLPC contribute to early protection from infection and can be induced during both TD and TI antigen responses.
3.2. Cell division is an essential process during B cell differentiation
Inhibition of proliferation after stimulation of peripheral blood mononuclear cells with pokeweed mitogen prevented the generation of ASC, implicating cell division as essential for B cell differentiation to ASC101. The development of carboxyfluorescein succinimidyl ester (CFSE)102 or CellTrace (CT) dyes allowed for the relationship of cell division and ASC formation to be assessed. These dyes covalently bind free amines on the surface and inside of cells and become diluted through cell proliferation. Application of these dyes has led to the appreciation that cell division is intimately linked to reprogramming events leading to ASC formation103–107.
To investigate the relationships between cellular division and differentiation in vivo, a model system was employed where CT-labeled splenic nB cells were transferred into a B cell-deficient μMT108 hosts followed by inoculation with LPS to stimulate differentiation105. Fine temporal mapping of ASC differentiation revealed that B cells began dividing rapidly 24–36 hr after LPS inoculation and differentiation required a minimum of 8 cell divisions105 that first appeared at 60 hr107. Challenging host mice with a 10,000 fold range of LPS only impacted the frequency of cells that entered the response but did not change the 8 division requirement for ASC formation107. These results are consistent with previous reports indicating ex vivo LPS inoculation results in a quantal all-or-none stimulation paradigm109. Intriguingly, when CT-labeled nB were transferred to wild-type (WT) hosts, the majority of ASC were observed in division 8 but could also be detected as early as division 5 (Figure 3). However, when MYD88-deficient110 mice are used to prevent host cells from responding to LPS, the requirement of 8 cell divisions before ASC formation was restored107. These data indicated that currently unknown cell extrinsic effects in WT mice can impact the cell division kinetics of B cell differentiation. We speculate that other LPS-responding cells present in WT hosts, such as macrophages and dendritic cells111, can influence B cell differentiation. Further supporting this hypothesis comes from experiments in which the TI II antigen 4-hydroxy-3-nitrophenylacetyl (NP)-Ficoll was used to stimulate B cells via BCR. Here, a similar requirement for 8 divisions was observed irrespective of whether the hosts were WT or μMT107. Collectively, these data support the concept that there is a conserved differentiation path to an ASC and that a minimum number of cellular divisions are required before differentiation can occur. This raises the question as to what happens during each of those divisions. As described below, chromatin, epigenetic, and transcription changes occur to allow for the engagement of metabolic pathways necessary for high levels of antibody secretion.
Figure 3. B cell differentiation is coupled to cell division and regulated by both epigenetic and transcription factors.
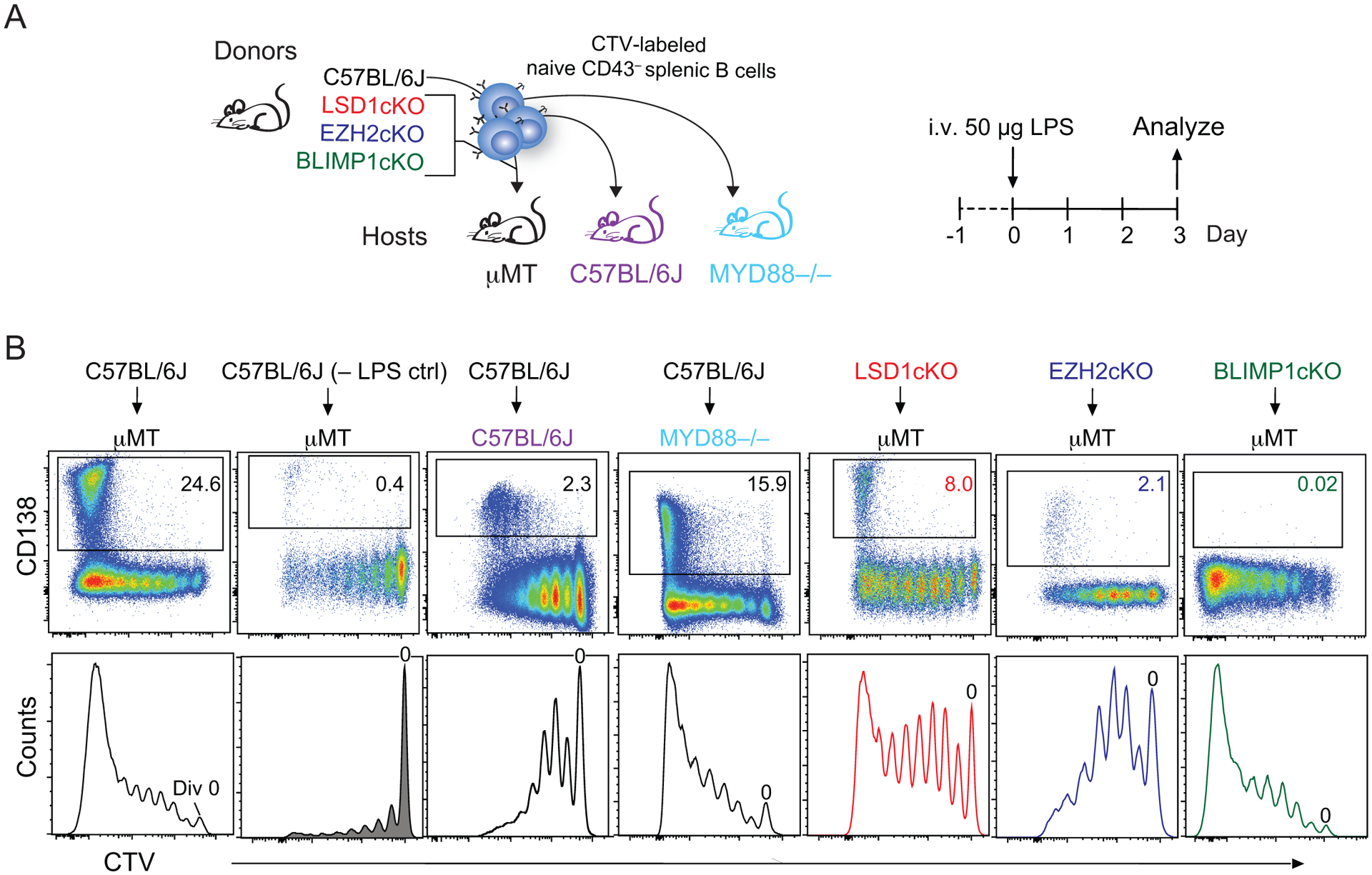
(A) Schematic of an adoptive transfer experimental design. Naïve B cells from the indicated mice were isolated, stained with CellTrace Violet (CTV), and transferred into B-cell deficient μMT, MYD88−/−, or C57BL/6J hosts. After one day, host mice are stimulated with LPS or PBS (– LPS ctrl) and sacrificed three days later for analysis via flow cytometry. Transferred cells are recovered from host spleens. (B) Representative flow cytometry plots depicting CD138 expression versus CTV (top) and histograms of CTV (bottom) with division 0 indicated. The donor:host mouse combination is indicated. Representative examples were reformatted from previously performed and published experiments: C57BL/6J into μMT (from Figure 1, reference107), C57BL/6J into C57BL/6J or MYD88−/− (from Figure 2, reference107), LSD1cKO into μMT (from Figure 5 reference157), EZH2cKO into μMT (from Figure 7 reference152), and BLIMP1 cKO into μMT (from Figure 3 reference107). cKO, conditional knockout.
3.3. Chromatin accessibility changes indicate epigenetic control of B cell differentiation
Epigenetic mechanisms, such as DNA methylation and histone modifications, function to prevent or promote accessibility of DNA to TF112. Therefore, identifying the regions that change accessibility during B cell differentiation can reveal critical regulatory elements and factors that control B cell reprogramming to ASC. The development of the assay for transposase accessible chromatin-sequencing (ATAC-seq)113,114 has revolutionized the study of accessible chromatin landscapes. Using the in vivo model system described above, ATAC-seq was applied to discrete divisions during B cell differentiation to better define the cis-regulatory changes that occur during ASC formation106. These included actB cells in divisions 0, 1, 3, 5, and CD138+ASC115,116 in division 8. Differentially accessible regions (DAR) were determined for all samples compared back to division 0 and revealed that progressive changes in chromatin accessibility occurred as the cells divided, with the majority of chromatin accessibility changes occurring in ASC. TF essential for ASC formation were enriched in ASC DAR including NF-κB, AP1:IRF heterodimers (AICE; high affinity IRF site)117, and IRF:IRF homodimers (ISRE; low affinity IRF site)118,119. Accessibility surrounding NF-κB and AICE sites increased progressively as cells divided, whereas accessibility surrounding ISRE was unique to ASC. These data are consistent with the concentration-dependent activity of IRF4 and high levels of IRF4 expression promoting ISRE binding in ASC90,118–120. Furthermore, these data described a hierarchy of TF activity that is linked to cell division during B cell differentiation. Strikingly, comparing undivided cells to ASC revealed a subset of promoters that was accessible in division 0, but not expressed until ASC formation in division 8, suggesting that epigenetic mechanisms play a role in regulating this “primed” set of genes. These primed genes showed strong enrichment for the gene ontology terms “cell cycle” and “DNA replication” indicating that they might allow the cells to rapidly respond after stimulation106. Indeed, a subset of these primed promoters were enriched for the repressive histone modification H3K27me3, and pharmacological inhibition of the enzyme responsible for H3K27me3 deposition (EZH2) resulted in increased expression of primed genes, such as Prdm1. These data indicate that proper control of H3K27me3 is critical for appropriate expression of ASC-inducing genes and point towards the role of the epigenome to control the timing and magnitude of gene expression during B cell differentiation.
4. B CELL FATE PROGRAMMING AND HETEROGENEITY
B cells encountering an immune challenge respond asynchronously, with cells distributed in all cell divisions and a predictable fraction of ASC forming104,107. Ex vivo stimulation of B cells has led to a stochastic model of differentiation to describe the population-level immune response while accounting for heterogeneity in cell fates121–124. Tracking individual cell fates (whether the cells will divide, die, or differentiate) over time revealed that unrelated cells in the same division have variable outcomes122,123. In contrast, sibling cells in the same division often have the same fate122,123. These data suggest that cell division alone does not determine the fate of a cell and that each division is actually a heterogeneous mix of cells that have adopted distinct fate outcomes.
Recent advances in single cell (sc) RNA-seq now allow for a molecular characterization of heterogeneity within immune populations during an immune response125,126. This can be leveraged to gain a better understanding of cellular transitions, or “trajectories”, that may provide insight into the processes that govern B cell fate decisions. scRNA-seq was applied to the in vivo adoptive transfer system described above to better understand the transcriptional programming that drives B cell heterogeneity107. Trajectory analysis ordered cells by cell division leading to ASC formation and revealed a bifurcation event in actB that occurred around divisions 3–5 during B cell differentiation, with one branch leading to ASC. These data indicate that B cell fates were instructed during the earliest stages of B cell activation and that a subset of B cells are destined to become ASC. ActB that followed this branch were designated “ASC-destined”, while cells that followed the alternative branch were denoted “non-ASC”. ASC-destined actB upregulated critical gene sets for differentiation including MYC-target genes127,128 and OXPHOS9 compared to non-ASC actB in the same division. IRF4, in tandem with BATF at AICE motifs, enforced early reprogramming events that drove cells down the ASC-destined branch. Importantly, ASC-destined cells could be distinguished by loss of CD62L (L-selectin) and may be leveraged in future work to identify cells along each branch. In contrast, non-ASC actB exhibited transcriptional signatures indicating they were responding to inflammatory stimuli and may have a role in shaping the overall immune environment. In this branch, MHC-II expression was maintained. There are likely additional molecular determinants that precede or follow the IRF4-dependent bifurcation event. Future work is also needed to better understand if the programming of cells along each path is fixed or plastic. Ultimately, understanding the full scope of factors that control the path to an ASC has significant therapeutic potential because it may aid in our ability to control desired immune outcomes.
5. EPIGENETIC CONTROL OF ANTIBODY-SECRETING CELL DIFFERENTIATION
As described above, the transcription factor networks regulating plasma cell fate have been well established. In addition, there is also a growing body of evidence suggesting the crucial role of epigenetics in fine-tuning the magnitude of the immune response129,130. Epigenetics is the study of heritable mechanisms that alter gene expression without changes in the DNA sequence. Although multiple epigenetic modifications exist112,131, here we will discuss the consequences of deletion of some epigenetic modifiers that control DNA methylation and histone modification.
5.1. DNA methylation
The methylation of cytosine at CpG dinucleotides is a central epigenetic modification that controls gene expression by recruiting proteins involved in gene repression or by inhibiting the binding of TF to DNA132. DNA methylation localized to promoters or enhancers results in gene repression. Alternatively, methylation across the gene body coupled with loss of methylation at promoters or enhancers supports gene expression133. DNA methylation is mediated by one of three DNA methyltransferases (DNMT). DNMT3A and DNMT3B promote de novo DNA methylation134, while DNMT1 maintains DNA methylation during cell division135. Loss of DNA methylation can occur either via passive loss of methylation during cell division or an active process mediated by Ten-eleven translocation methylcytosine dioxygenases (TET1, TET2, TET3). TET enzymes promote DNA demethylation in a step wise manner by conversion of mCpG into 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), which leads to unmethylated DNA136.
Using the same in vivo model system described above, DNA methylation and transcription were examined in discrete divisions leading to ASC formation105. These included cells in division 0, 1, 3, 5, and 8, with cells in division 8 further delineated based on CD138 status to separate ASC (CD138+) from non-ASC (CD138–)115,116. This study revealed that B cell differentiation is coupled to cell division and is associated with a progressive loss of DNA methylation, with only a small number of loci gaining de novo DNA methylation during ASC formation105. Differentiation of human B cells observed similar trends and indicated the reconfiguration of the DNA methylome was connected to the cell cycle137. Importantly, loss of DNA methylation occurred at many key genes essential for ASC formation, including Prdm1, Irf4, Xbp1, and was enriched at B cell specific enhancer regions105,137. TFs critical for the differentiation of ASC were enriched within demethylated enhancer regions and included IRF family members, BATF (AP-1 family member), NF-κB, and E2A (reviewed in130). Selective demethylation at regulatory regions critical for ASC differentiation suggests that these regions are targeted. Deletion of the DNA demethylase genes encoding TET2 or TET2 and TET3 in hematopoietic stem cells or class-switched B cells led to a reduction in ASC and antibody titers with a corresponding expansion of GC B cells due to a failure to induce the transcriptional program necessary for GC exit138,139. While deletion of Tet2 and Tet3 using inducible Cre-ERT2 followed by ex vivo stimulation did not alter the frequency of ASC generated, there was a significant defect in class switch recombination due to a failure to demethylate the Aicda (AID) locus140. Furthermore, inhibition of DNA methylation105 or enhancement of TET enzymes via ascorbic acid (vitamin C)141 led to an increase in ASC, thus providing functional evidence for the essential role of DNA demethylation in ASC formation.
Despite the fact that de novo DNA methylation occurs only at a small number of loci, those changes are necessary for restraining the commitment to the ASC fate. B cell conditional deletion of Dnmt3a and Dnmt3b led to an increase in GC B cells, as well as ASC with an aberrant transcriptional profile. DNMT3A/B-deficient ASC upregulated genes associated with lysosome function, transcription, as well as various metabolic pathways142. However, despite the well-established role of DNA methylation in ASC formation, it is yet to be determined how these enzymes are recruited to the specific loci. Additionally, more work is needed to uncouple passive and active demethylation events and determine the timing in which these processes occur.
5.2. Enhancer of zeste homolog 2 (EZH2)
Enhancer of zeste homolog 2 (EZH2) is the catalytic component of the Polycomb Complex 2 that mediates histone 3 lysine 27 trimethylation (H3K27me3), which is associated with gene silencing143–145. The active loss of H3K27me3 is mediated by two demethylases: ubiquitously transcribed tetratricopeptide repeat, X chromosome (UTX) and JmjC Domain-Containing Protein 3 (JMJD3)146,147. Mutations in EZH2 are frequently found in Diffuse Large B-cell Lymphoma and Follicular lymphoma, which led to great interest in understanding its role in B cells. In fact, EZH2 was shown to function at multiple stages of B cell development and differentiation148–150. EZH2 is necessary for GC formation and was shown to cooperate with BCL6 and the CBX8-BCOR complex to repress genes associated with ASC fate, as well as cell cycle inhibitors, such as CDKN1A, to allow for the rapid proliferation of GC B cells148. Furthermore, gain of function EZH2 mutant proteins contribute to follicular lymphoma by allowing the mutant B cell to persist and expand in the light zone of the GC in a manner independent of T cell help151. In addition, EZH2 has been shown to directly interact with BLIMP1 to mediate gene repression95. Consistent with these findings, deletion of EZH2 led to an upregulation of Blimp1 target genes and failure to repress the B cell transcriptome. Phenotypically, EZH2-deficiency led to a decrease in ASC formation in vivo, and a profound proliferation defect, with EZH2-deficient B cells accumulating in cell divisions 3–5152 (Figure 3). Furthermore, deletion of Ezh2 led to a failure to upregulate genes associated with the UPR, glycolysis, and OXPHOS, which correlated with reduced ability of the cells to secrete antibodies and perform glycolysis, as well as lower basal respiration rate. Thus, EZH2-dependent gene repression is necessary to repress the B cell program and initiate metabolic and secretory reprogramming during ASC formation.
5.3. Lysine-specific histone demethylase 1A (LSD1)
Histone H3K4 methylation is associated with gene expression, with monomethylation (H3K4me1) associated with enhancers, dimethylation (H3K4me2) with enhancers and gene bodies, and trimethylation (H3K4me3) at RNA polymerase engaged promoters153. Demethylation of H3K4me1/2 (as well as H3K9me1/2) is mediated by the monoamine oxidase Lysine-specific histone demethylase 1A (LSD1)154,155, which has been shown to physically interact with BLIMP1156 and regulate ASC formation157. Deletion of LSD1 led to a failure to decommission nB enhancers, resulting in reduced differentiation of LSD1-deficient B cells into ASC. Furthermore, LSD1-deficiency led to downregulation of genes associated with cell cycle and proliferation, including E2F- and MYC target genes, which corresponded with impaired proliferation157 (Figure 3). In the context of the GC, LSD1 was shown to physically interact with BCL6 to repress enhancers of BCL6 target genes, including nB enhancers that are either lost or poised in GC B cells154. Interestingly, the function of LSD1 in the GC is independent of its catalytic activity, suggesting that it is part of a larger complex with other actions. Pharmacological inhibition of the catalytic domain did not impede GC formation seen in the genetic knockout154. In fact, the Tower domain of LSD1, which interacts with the corepressor complex CoREST, was necessary for survival of lymphoma cell lines154. While deletion of LSD1 leads to a reduction in GC B cells, the opposite is true for the H3K4 methyltransferase (KMT2D)158. The increase in GC B cells was attributed to increased/enhanced proliferation of KMT2D-deficient follicular B cells, as the phenotype was not observed when KMT2D was conditionally deleted in GC B cells158. The role of H3K4 methyltransferases in ASC formation remains to be elucidated. However, evaluating the role of the specific H3K4 methyltransferases is complicated by the existence of four partially redundant H3K4 methyltransferases153.
Conditional deletion of Lsd1 during B cell development resulted in a significant decrease in marginal zone B cells (MZB) but not on follicular B cells (FoB)159. MZB represent a small subset of splenic B cells that predominately respond to TI antigens160,161. Loss of MZB was in fact due to the reprogramming of cells towards the FoB compartment, as the top 200 FoB genes were upregulated in LSD1-deficient MZB. Moreover, chromatin accessibility data indicated that NF-κB like motifs were affected. Further analyses showed that the non-canonical NF-κB family member p52 interacted with LSD1, and that LSD1’s activity through the BAFF signaling pathways during transitional B cell development to MZB, was diminished in LSD1-deficient cells159.
5.4. Disruptor of telomeric silencing 1-like (DOT1L)
Disruptor of telomeric silencing 1-like (DOT1L) facilitates methylation of H3K79, which is associated with gene expression and has been of interest in B cells due to its role in leukemias characterized by translocation of the mixed lineage leukemia (MLL) gene. In the tumor setting, MLL frequently forms fusion complexes with DOT1L-interacting proteins such as AF10, AF4, and ENL. As a result, DOT1L is recruited by MLL fusion complexes to promote malignant gene expression and therefore is a target for drug development162. In the context of B cell differentiation, DOT1L deficiency impaired GC and ASC formation in response to TI and TD antigens in vivo162–164. The failure to mount a robust response following stimulation in vivo was attributed to a failure of DOT1L-deficient B cells to upregulate genes associated with cell movement and migration, resulting in aberrant cell localization within secondary lymphoid organs164. Interestingly, differentiation of DOT1L-deficient B cells or inhibition of DOT1L ex vivo resulted in an increase in ASC. However, while DOT1L-deficient B cells expressed key ASC genes (CD138 and BLIMP1), these ex vivo generated ASC failed to fully downregulated B220 or CD19 suggesting incomplete differentiation. Additionally, ex vivo cultured DOT1L-deficient B cells failed to upregulate the expression of BACH2, MYC, MYC-target genes, and EZH2. Consistent with downregulation of EZH2, DOT1L-deficient B cell upregulated EZH2-target genes, including Cdkn1a, suggesting a mechanistic link between the two epigenetic enzymes163.
6. 3D ARCHITECTURE DURING B CELL DIFFERENTIATION
In addition to changes in transcription factor networks, DNA methylation, and histone modifications, B cell differentiation is also associated with substantial 3D reorganization of the genome165. The advent of chromatin confirmation capture techniques allowed for a closer examination of DNA architecture, which coupled with whole genome sequencing (Hi-C), revealed the presence of topologically associated domains (TAD). TAD are regions of the genome that tend to frequently interact compared with regions outside of TAD166,167. Although this field is young and rapidly progressing, there have been important observations regarding the stage-specific reorganization of the 3D genome and the key enhancer-promoter interactions that mediate B cell differentiation and function.
A comparison of the genomic profiles of nB, actB, or ASC revealed substantial differences in their genomic architecture. B cell differentiation leads to a significant increase in the number of DNA loops and a shift from long-range to mostly short-range interactions168,169. These changes were also associated with alterations in the epigenetic landscape, such as gains in the active histone modification H3K27ac at regions surrounding key genes critical for ASC, including Prdm1, Atf4, Ell2169. Fine scale mapping of chromatin architecture changes by time and cell division ex vivo revealed that the first wave of chromatin reorganization occurred just prior to the first cell division165, a process that is likely driven by MYC168. Genome organization remained largely unchanged during dividing actB until ASC formation, at which a second wave of changes were observed165. The essential role of rewiring the 3D architecture and DNA loop formation is further exemplified by studies investigating the role of Smc3, the catalytic component of the cohesion complex, in GC formation and B cell differentiation170. Genetic deletion of Smc3 impeded GC formation; however, haploinsufficiency of Smc3 resulted in expanded GC and a block in ASC formation. This indicates that the final step of ASC differentiation requires major reorganization of the genome architecture compared to other differentiation stage and is sensitive to changes in SMC3 levels.
Furthermore, the study of 3D architecture revealed the presence of multi-enhancer genes and multi-gene enhancers in actB. The multi-enhancer genes were enriched for gene ontology pathways, such as MHC-II antigen processing and ER associated degradation, while the latter group included genes associated with metabolism and DNA replication171. The important role of such multi-gene enhancers in regulating cell fate decisions was illustrated by another study which evaluated the 3D architecture in GC B cells. This work revealed the presence of an enhancer that functioned as a locus of region control (LRC; a GC-specific enhancer cluster) by interacting with many GC signature genes. Deletion of this region using CRISPR led to abolished GC formation172.
In addition to the global genome architecture studies mentioned above, local enhancer-promoter interactions are also important and dynamic during B cell differentiation. Analysis of the MHC-II in mice and humans has revealed the complex interplay between TF, cis-regulatory elements, and genetic variation in the regulation of antigen presentation and potentially adaptive immunity173–175. The entire MHC-II locus contains a set of loops that are orchestrated by the CCCTC-binding factor (CTCF), which is known to regulate 3D interactions176 and the cohesion complex177–179. CTCF was required for maximal MHC-II gene expression174, and in murine cells, the MHC-II locus is reorganized into distinct compartments when MHC-II gene expression is repressed in ASC180. In the era of genomics, a new set of super enhancers enriched for multiple transcription factor binding sites, active histone modifications, the Mediator complex, and cover large domains greater than 10kb in size have been described181. A super enhancer located between the HLA-DRB1 and HLA-DQA1 genes (termed DR/DQ-SE ) is one of the most acetylated regions of the B cell genome and contains the highest density of genetic polymorphisms in the human genome175. Deletion of the DR/DQ-SE led to significant changes in 3D loops, enrichment of histone modifications and ultimately lower levels of MHC-II gene expression that impaired the ability of B cells to stimulate allogenic T cell proliferation in a mixed lymphocyte reaction assay175. These studies highlight how genetic variation within a super enhancer can impact the 3D architecture of a single locus and ultimately result in variation in MHCII gene expression across a population. This indicates that non-coding genetic variation may be as important as HLA allelic variation within a population. Further work will help delineate the factors controlling MHCII enhancer-promoter interactions and how variation impacts the adaptive immune response.
7. CONCLUDING REMARKS AND OUTLOOK
B cell differentiation to ASC is the cornerstone of humoral immunity. The transition from a quiescent nB to an ASC is a remarkable biological feat that requires substantial physiological alterations, including a shift in metabolism, ER stress pathways, and autophagy to sustain immunoglobulin production. Significant advances have been made in our understanding of the transcriptional and epigenetic events that coordinate these changes, and it has become clear that cell division and epigenetic reprogramming are intimately intertwined, with discrete divisions representing distinct stages of B differentiation105–107.
Following initial activation in vivo to a TI antigen, nB cells begin to proliferate between 24 and 36 hr post-stimulation107 In cell divisions 1–2, accessibility changes at promoters and distal elements are observed and transcriptional amplification is initiated for essential ASC gene sets, including Myc target genes and OXPHOS (Figure 4).9,105. By 48 hr post-stimulation, cells that are able to continue proliferating have progressed as far as division 5107. In divisions 3–5, DNA hypomethylation occurs around enhancer regions containing NF-κB and AP-1 motifs, and cells continue to upregulate / repress pathways initiated in the initial divisions105,106. Additionally, a critical decision point facilitated by IRF4 designates a portion of actB to follow a reprogramming path to an ASC during these cell divisions. Cells that do not follow this path contain transcriptional signatures indicating they are responding to inflammatory stimuli and may ultimately contribute to the immune response. By 60 hr, cells that continue to divide are observed in division 8, with a subset differentiating to ASC (Div 8+)107. During these final divisions before ASC formation is observed, massive epigenetic and transcriptional changes occur. Cell division defects following the deletion of many factors, such as LSD1 and EZH2, that contribute to actB reprogramming are often observed in these divisions152,157 (Figure 3). We speculate that the coupling of actB reprogramming with cell division and cell-cycle promoting genes may have evolved to provide a fail-safe when reprogramming goes awry. Remarkably, substantial differences in epigenetic and transcription reprogramming exist in actB and ASC in division 8. This includes >50,000 demethylated loci and >1,500 differentially expressed genes105. BLIMP1 is one factor essential for the transcriptional changes observed in division 8 ASC, as deletion of BLIMP1 had minimal impact on gene expression in earlier divisions. Additionally, low affinity IRF:IRF (ISRE) and E2A motifs are uniquely enriched in accessible regions in division 8 ASC and positively correlate with predicted target gene expression, implicating these as a major contributor to the final reprogramming events. Collectively, studying chromatin accessibility and gene expression changes in discrete divisions during B cell differentiation, combined with genetic dissection of epigenetic enzymes and transcription factors through genetic deletion, has revealed the step-wise reprogramming events that occur during B cell differentiation. These data can be exploited to understand the timing, mechanism, and scope of reprogramming by factors that impact ASC differentiation.
Figure 4. Cell divisions represent distinct stages during in vivo B cell differentiation in response to LPS.
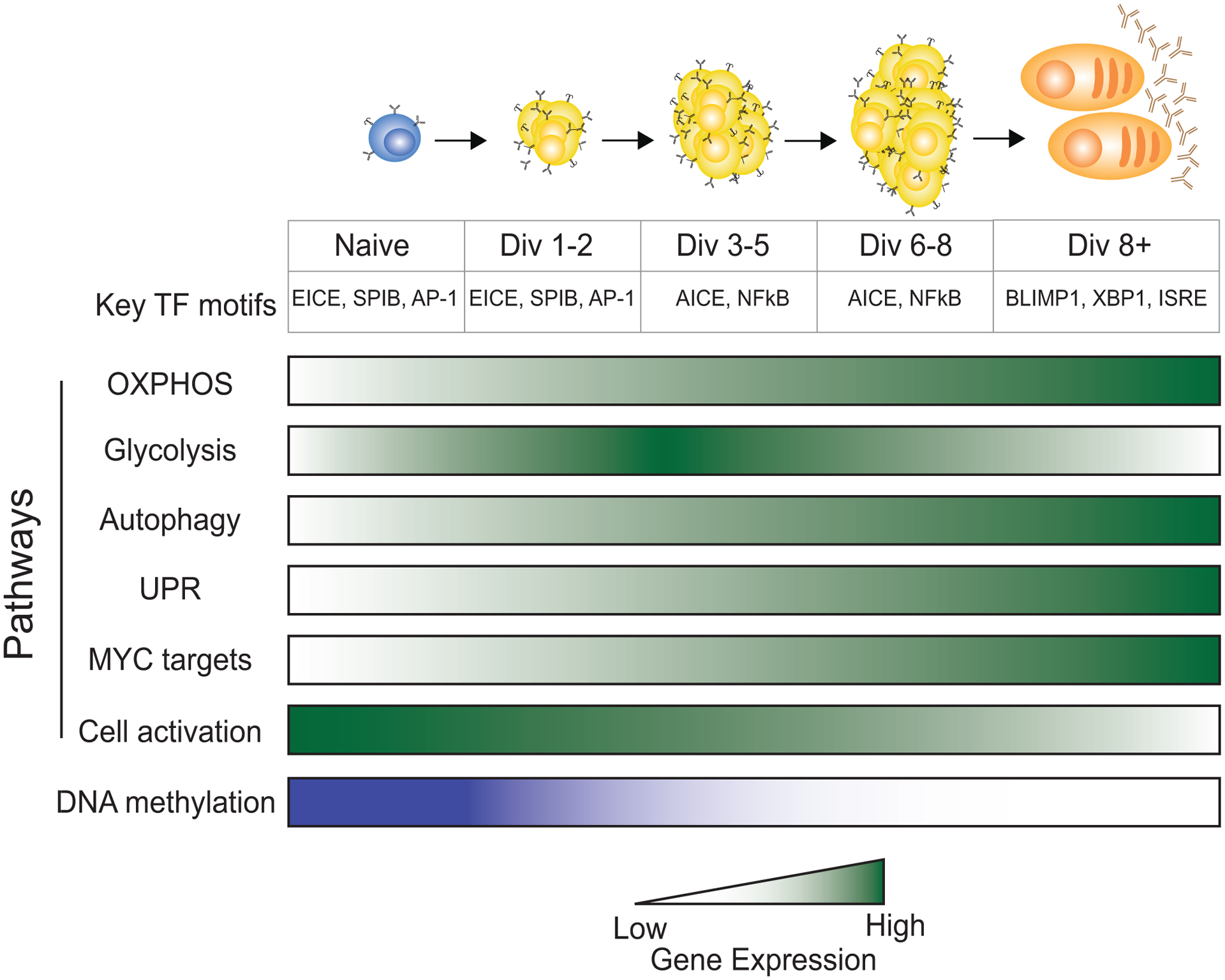
Genome wide accessibility at discrete divisions revealed a hierarchy of transcription factor activity106. Transcription factor motifs enriched at each cell division stage are indicated. RNA-seq of cells in precise divisions revealed a progressive change of gene sets throughout differentiation105. Shaded boxes represent the expression level of genes in the indicated pathways that change as the cells divide during ASC differentiation. Darker color represents higher expression. DNA methylation levels are represented by color with the darker color indicating more methylation.
While considerable progress has been made in understanding the reprogramming events required during B cell differentiation and the timing in which they are initiated, additional efforts are needed to better understand the molecular determinants that influence early cell fate decisions. The advent of scRNA-seq technologies can be leveraged to be more readily determine such factors. While IRF4 is one known component that contributes to such decisions, additional factors that act upstream / downstream remain to be determined. Furthermore, it remains largely unknown whether the same reprogramming events occur following memory B cell reactivation and differentiation of ASC. Although the use of LPS is a useful model to study the events leading to ASC formation, it will be critical to understand the reprogramming events that occur following TD stimulation, which results in a myriad of long-lived cell fates (memory B cells and LLPC). Such an analysis could uncover the timing of cell fate decisions and factors that influence cell outcomes, which may be exploited to influence a desired immune outcome.
ACKNOWLEDGMENTS
We thank the laboratory for helpful discussions on this work. This work was supported by grants to JMB (AI123733, AI125180, AI110483, AI153102-01, ACE, and GM008490) and to CDS AI148471.
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts to report.
REFERENCES
- 1.Allman D, Wilmore JR, Gaudette BT. The continuing story of T-cell independent antibodies. Immunol Rev. 2019;288(1):128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mond JJ, Lees A, Snapper CM. T cell-independent antigens type 2. Annu Rev Immunol. 1995;13:655–692. [DOI] [PubMed] [Google Scholar]
- 3.McHeyzer-Williams LJ, McHeyzer-Williams MG. Antigen-specific memory B cell development. Annu Rev Immunol. 2005;23:487–513. [DOI] [PubMed] [Google Scholar]
- 4.Forthal DN. Functions of Antibodies. Microbiol Spectr. 2014;2(4):AID-0019–2014. [PMC free article] [PubMed] [Google Scholar]
- 5.Vose BM, Bonnard GD. Limiting dilution analysis of the frequency of human T cells and large granular lymphocytes proliferating in response to interleukin 2. I. The effect of lectin on the proliferative frequency and cytotoxic activity of cultured lymphoid cells. J Immunol. 1983;130(2):687–693. [PubMed] [Google Scholar]
- 6.Boothby M, Rickert RC. Metabolic Regulation of the Immune Humoral Response. Immunity. 2017;46(5):743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aronov M, Tirosh B. Metabolic Control of Plasma Cell Differentiation- What We Know and What We Don’t Know. J Clin Immunol. 2016;36Suppl 1:12–17. [DOI] [PubMed] [Google Scholar]
- 8.Dufort FJ, Bleiman BF, Gumina MR, et al. Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via Stat6-dependent regulation of glycolytic metabolism. J Immunol. 2007;179(8):4953–4957. [DOI] [PubMed] [Google Scholar]
- 9.Price MJ, Patterson DG, Scharer CD, Boss JM. Progressive Upregulation of Oxidative Metabolism Facilitates Plasmablast Differentiation to a T-Independent Antigen. Cell Rep. 2018;23(11):3152–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weisel FJ, Mullett SJ, Elsner RA, et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol. 2020;21(3):331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saijo K, Mecklenbrauker I, Santana A, Leitger M, Schmedt C, Tarakhovsky A. Protein kinase C beta controls nuclear factor kappaB activation in B cells through selective regulation of the IkappaB kinase alpha. J Exp Med. 2002;195(12):1647–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su TT, Guo B, Kawakami Y, et al. PKC-beta controls I kappa B kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. 2002;3(8):780–786. [DOI] [PubMed] [Google Scholar]
- 13.Tsui C, Martinez-Martin N, Gaya M, et al. Protein Kinase C-beta Dictates B Cell Fate by Regulating Mitochondrial Remodeling, Metabolic Reprogramming, and Heme Biosynthesis. Immunity. 2018;48(6):1144–1159 e1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez WY, McGee SL, Connor T, et al. Dichloroacetate inhibits aerobic glycolysis in multiple myeloma cells and increases sensitivity to bortezomib. Br J Cancer. 2013;108(8):1624–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen YC, Ou DL, Hsu C, et al. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108(1):72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stockwin LH, Yu SX, Borgel S, et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010;127(11):2510–2519. [DOI] [PubMed] [Google Scholar]
- 17.Lam WY, Becker AM, Kennerly KM, et al. Mitochondrial Pyruvate Import Promotes Long-Term Survival of Antibody-Secreting Plasma Cells. Immunity. 2016;45(1):60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caro-Maldonado A, Wang R, Nichols AG, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192(8):3626–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McBrayer SK, Cheng JC, Singhal S, Krett NL, Rosen ST, Shanmugam M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood. 2012;119(20):4686–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lam WY, Jash A, Yao CH, et al. Metabolic and Transcriptional Modules Independently Diversify Plasma Cell Lifespan and Function. Cell Rep. 2018;24(9):2479–2492 e2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrne FL, Olzomer EM, Brink R, Hoehn KL. Knockout of glucose transporter GLUT6 has minimal effects on whole body metabolic physiology in mice. American journal of physiology Endocrinology and metabolism. 2018;315(2):E286–e293. [DOI] [PubMed] [Google Scholar]
- 22.Jennewein MF, Alter G. The Immunoregulatory Roles of Antibody Glycosylation. Trends Immunol. 2017;38(5):358–372. [DOI] [PubMed] [Google Scholar]
- 23.Hickman S, Kulczycki A, Jr., Lynch RG, Kornfeld S. Studies of the mechanism of tunicamycin in hibition of IgA and IgE secretion by plasma cells. The Journal of biological chemistry. 1977;252(12):4402–4408. [PubMed] [Google Scholar]
- 24.Spencer JA, Ferraro F, Roussakis E, et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature. 2014;508(7495):269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol. 2014;5:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe-Matsui M, Muto A, Matsui T, et al. Heme regulates B-cell differentiation, antibody class switch, and heme oxygenase-1 expression in B cells as a ligand of Bach2. Blood. 2011;117(20):5438–5448. [DOI] [PubMed] [Google Scholar]
- 27.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715–727. [DOI] [PubMed] [Google Scholar]
- 28.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14(6):341–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dowling DP, Gattis SG, Fierke CA, Christianson DW. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry. 2010;49(24):5048–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee P, Murphy B, Miller R, et al. Mechanisms and clinical significance of histone deacetylase inhibitors: epigenetic glioblastoma therapy. Anticancer Res. 2015;35(2):615–625. [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Y, Li C, Wu Q, et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat Commun. 2019;10(1):2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price MJ, Scharer CD, Kania AK, Randall TD, Boss JM. Conserved Epigenetic Programming and Enhanced Heme Metabolism Drive Memory B Cell Reactivation. J Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reid MA, Dai Z, Locasale JW. The impact of cellular metabolism on chromatin dynamics and epigenetics. Nat Cell Biol. 2017;19(11):1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sil P, Muse G, Martinez J. A ravenous defense: canonical and non-canonical autophagy in immunity. Curr Opin Immunol. 2018;50:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cenci S, Autophagy, a new determinant of plasma cell differentiation and antibody responses. Mol Immunol. 2014;62(2):289–295. [DOI] [PubMed] [Google Scholar]
- 36.Pengo N, Scolari M, Oliva L, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol. 2013;14(3):298–305. [DOI] [PubMed] [Google Scholar]
- 37.Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. [DOI] [PubMed] [Google Scholar]
- 39.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. [DOI] [PubMed] [Google Scholar]
- 40.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell. 2000;6(5):1099–1108. [DOI] [PubMed] [Google Scholar]
- 41.Ma Y, Shimizu Y, Mann MJ, Jin Y, Hendershot LM. Plasma cell differentiation initiates a limited ER stress response by specifically suppressing the PERK-dependent branch of the unfolded protein response. Cell Stress Chaperones. 2010;15(3):281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gass JN, Jiang HY, Wek RC, Brewer JW. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol Immunol. 2008;45(4):1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masciarelli S, Fra AM, Pengo N, et al. CHOP-independent apoptosis and pathway-selective induction of the UPR in developing plasma cells. Mol Immunol. 2010;47(6):1356–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. [DOI] [PubMed] [Google Scholar]
- 46.Aragon IV, Barrington RA, Jackowski S, Mori K, Brewer JW. The specialized unfolded protein response of B lymphocytes: ATF6alpha-independent development of antibody-secreting B cells. Mol Immunol. 2012;51(3–4):347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. [DOI] [PubMed] [Google Scholar]
- 48.Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167(1):35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21(1):81–93. [DOI] [PubMed] [Google Scholar]
- 50.McGehee AM, Dougan SK, Klemm EJ, et al. XBP-1-deficient plasmablasts show normal protein folding but altered glycosylation and lipid synthesis. J Immunol. 2009;183(6):3690–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taubenheim N, Tarlinton DM, Crawford S, Corcoran LM, Hodgkin PD, Nutt SL. High rate of antibody secretion is not integral to plasma cell differentiation as revealed by XBP-1 deficiency. J Immunol. 2012;189(7):3328–3338. [DOI] [PubMed] [Google Scholar]
- 52.Gaudette BT, Jones DD, Bortnick A, Argon Y, Allman D. mTORC1 coordinates an immediate unfolded protein response-related transcriptome in activated B cells preceding antibody secretion. Nat Commun. 2020;11(1):723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benner R, Hijmans W, Haaijman JJ. The bone marrow: the major source of serum immunoglobulins, but still a neglected site of antibody formation. Clin Exp Immunol. 1981;46(1):1–8. [PMC free article] [PubMed] [Google Scholar]
- 54.Ellyard JI, Avery DT, Phan TG, Hare NJ, Hodgkin PD, Tangye SG. Antigen-selected, immunoglobulin-secreting cells persist in human spleen and bone marrow. Blood. 2004;103(10):3805–3812. [DOI] [PubMed] [Google Scholar]
- 55.Chernova I, Jones DD, Wilmore JR, et al. Lasting antibody responses are mediated by a combination of newly formed and established bone marrow plasma cells drawn from clonally distinct precursors. J Immunol. 2014;193(10):4971–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ho F, Lortan JE, MacLennan IC, Khan M. Distinct short-lived and long-lived antibody-producing cell populations. Eur J Immunol. 1986;16(10):1297–1301. [DOI] [PubMed] [Google Scholar]
- 57.Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell. 2019;177(3):524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hargreaves DC, Hyman PL, Lu TT, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194(1):45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng Q, Khodadadi L, Taddeo A, et al. CXCR4-CXCL12 interaction is important for plasma cell homing and survival in NZB/W mice. Eur J Immunol. 2018;48(6):1020–1029. [DOI] [PubMed] [Google Scholar]
- 60.Erickson LD, Lin LL, Duan B, Morel L, Noelle RJ. A genetic lesion that arrests plasma cell homing to the bone marrow. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(22):12905–12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hauser AE, Debes GF, Arce S, et al. Chemotactic responsiveness toward ligands for CXCR3 and CXCR4 is regulated on plasma blasts during the time course of a memory immune response. J Immunol. 2002;169(3):1277–1282. [DOI] [PubMed] [Google Scholar]
- 62.Kunkel EJ, Kim CH, Lazarus NH, et al. CCR10 expression is a common feature of circulating and mucosal epithelial tissue IgA Ab-secreting cells. J Clin Invest. 2003;111(7):1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;169(8):4213–4221. [DOI] [PubMed] [Google Scholar]
- 64.Cassese G, Arce S, Hauser AE, et al. Plasma cell survival is mediated by synergistic effects of cytokines and adhesion-dependent signals. J Immunol. 2003;171(4):1684–1690. [DOI] [PubMed] [Google Scholar]
- 65.Rozanski CH, Arens R, Carlson LM, et al. Sustained antibody responses depend on CD28 function in bone marrow-resident plasma cells. J Exp Med. 2011;208(7):1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chevrier S, Genton C, Kallies A, et al. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(10):3895–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Y, Bhattacharya D. Adjuvant-specific regulation of long-term antibody responses by ZBTB20. J Exp Med. 2014;211(5):841–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chevrier S, Emslie D, Shi W, et al. The BTB-ZF transcription factor Zbtb20 is driven by Irf4 to promote plasma cell differentiation and longevity. J Exp Med. 2014;211(5):827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cortés M, Georgopoulos K. Aiolos is required for the generation of high affinity bone marrow plasma cells responsible for long-term immunity. J Exp Med. 2004;199(2):209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. 2015;15(3):160–171. [DOI] [PubMed] [Google Scholar]
- 71.Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Engl J Med. 2007;357(19):1903–1915. [DOI] [PubMed] [Google Scholar]
- 72.Belnoue E, Pihlgren M, McGaha TL, et al. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood. 2008;111(5):2755–2764. [DOI] [PubMed] [Google Scholar]
- 73.Benson MJ, Dillon SR, Castigli E, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol. 2008;180(6):3655–3659. [DOI] [PubMed] [Google Scholar]
- 74.Nguyen DC, Garimalla S, Xiao H, et al. Factors of the bone marrow microniche that support human plasma cell survival and immunoglobulin secretion. Nat Commun. 2018;9(1):3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. 2011;23(5):341–349. [DOI] [PubMed] [Google Scholar]
- 76.Schebesta A, McManus S, Salvagiotto G, Delogu A, Busslinger GA, Busslinger M. Transcription factor Pax5 activates the chromatin of key genes involved in B cell signaling, adhesion, migration, and immune function. Immunity. 2007;27(1):49–63. [DOI] [PubMed] [Google Scholar]
- 77.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401(6753):556–562. [DOI] [PubMed] [Google Scholar]
- 78.Muto A, Ochiai K, Kimura Y, et al. Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J. 2010;29(23):4048–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muto A, Tashiro S, Nakajima O, et al. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature. 2004;429(6991):566–571. [DOI] [PubMed] [Google Scholar]
- 80.Ochiai K, Katoh Y, Ikura T, et al. Plasmacytic transcription factor Blimp-1 is repressed by Bach2 in B cells. The Journal of biological chemistry. 2006;281(50):38226–38234. [DOI] [PubMed] [Google Scholar]
- 81.Vilagos B, Hoffmann M, Souabni A, et al. Essential role of EBF1 in the generation and function of distinct mature B cell types. J Exp Med. 2012;209(4):775–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fukuda T, Yoshida T, Okada S, et al. Disruption of the Bcl6 gene results in an impaired germinal center formation. J Exp Med. 1997;186(3):439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shaffer AL, Yu X, He Y, Boldrick J, Chan EP, Staudt LM. BCL-6 represses genes that function in lymphocyte differentiation, inflammation, and cell cycle control. Immunity. 2000;13(2):199–212. [DOI] [PubMed] [Google Scholar]
- 84.Tunyaplin C, Shaffer AL, Angelin-Duclos CD, Yu X, Staudt LM, Calame KL. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol. 2004;173(2):1158–1165. [DOI] [PubMed] [Google Scholar]
- 85.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276(5312):589–592. [DOI] [PubMed] [Google Scholar]
- 86.Huang C, Geng H, Boss I, Wang L, Melnick A. Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood. 2014;123(7):1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu H, Chaudhri VK, Wu Z, et al. Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat Immunol. 2015;16(12):1274–1281. [DOI] [PubMed] [Google Scholar]
- 88.Carotta S, Willis SN, Hasbold J, et al. The transcription factors IRF8 and PU.1 negatively regulate plasma cell differentiation. J Exp Med. 2014;211(11):2169–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Klein U, Casola S, Cattoretti G, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. 2006;7(7):773–782. [DOI] [PubMed] [Google Scholar]
- 90.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25(2):225–236. [DOI] [PubMed] [Google Scholar]
- 91.Shaffer AL, Lin KI, Kuo TC, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity. 2002;17(1):51–62. [DOI] [PubMed] [Google Scholar]
- 92.Lin Y, Wong K, Calame K. Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science. 1997;276(5312):596–599. [DOI] [PubMed] [Google Scholar]
- 93.Piskurich JF, Lin KI, Lin Y, Wang Y, Ting JP, Calame K. BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat Immunol. 2000;1(6):526–532. [DOI] [PubMed] [Google Scholar]
- 94.Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. 2002;22(13):4771–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Minnich M, Tagoh H, Bonelt P, et al. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol. 2016;17(3):331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu GJ, Jaritz M, Wohner M, et al. Repression of the B cell identity factor Pax5 is not required for plasma cell development. J Exp Med. 2020;217(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shi W, Liao Y, Willis SN, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol. 2015;16(6):663–673. [DOI] [PubMed] [Google Scholar]
- 98.Wiggins KJ, Scharer CD. Roadmap to a plasma cell: Epigenetic and transcriptional cues that guide B cell differentiation. Immunol Rev. 2021;300(1):54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Waffarn EE, Baumgarth N. Protective B cell responses to flu--no fluke! J Immunol. 2011;186(7):3823–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Woodruff MC, Ramonell RP, Nguyen DC, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. 2020;21(12):1506–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jelinek DF, Lipsky PE. The role of B cell proliferation in the generation of immunoglobulin-secreting cells in man. J Immunol. 1983;130(6):2597–2604. [PubMed] [Google Scholar]
- 102.Quah BJ, Parish CR. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J Immunol Methods. 2012;379(1–2):1–14. [DOI] [PubMed] [Google Scholar]
- 103.Heinzel S, Marchingo JM, Horton MB, Hodgkin PD. The regulation of lymphocyte activation and proliferation. Curr Opin Immunol. 2018;51:32–38. [DOI] [PubMed] [Google Scholar]
- 104.Tangye SG, Hodgkin PD. Divide and conquer: the importance of cell division in regulating B-cell responses. Immunology. 2004;112(4):509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barwick BG, Scharer CD, Bally APR, Boss JM. Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation. Nat Immunol. 2016;17(10):1216–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scharer CD, Barwick BG, Guo M, Bally APR, Boss JM. Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat Commun. 2018;9(1):1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scharer CD, Patterson DG, Mi T, Price MJ, Hicks SL, Boss JM. Antibody-secreting cell destiny emerges during the initial stages of B-cell activation. Nat Commun. 2020;11(1):3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350(6317):423–426. [DOI] [PubMed] [Google Scholar]
- 109.Hawkins ED, Turner ML, Wellard CJ, Zhou JH, Dowling MR, Hodgkin PD. Quantal and graded stimulation of B lymphocytes as alternative strategies for regulating adaptive immune responses. Nat Commun. 2013;4:2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hou B, Reizis B, DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29(2):272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500. [DOI] [PubMed] [Google Scholar]
- 113.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Scharer CD, Blalock EL, Barwick BG, et al. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naive SLE B cells. Sci Rep. 2016;6:27030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Smith KG, Hewitson TD, Nossal GJ, Tarlinton DM. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur J Immunol. 1996;26(2):444–448. [DOI] [PubMed] [Google Scholar]
- 116.Kallies A, Hasbold J, Tarlinton DM, et al. Plasma cell ontogeny defined by quantitative changes in blimp-1 expression. J Exp Med. 2004;200(8):967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Glasmacher E, Agrawal S, Chang AB, et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science. 2012;338(6109):975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Singh H, Glasmacher E, Chang AB, Vander Lugt B. The molecular choreography of IRF4 and IRF8 with immune system partners. Cold Spring Harb Symp Quant Biol. 2013;78:101–104. [DOI] [PubMed] [Google Scholar]
- 119.Ochiai K, Maienschein-Cline M, Simonetti G, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity. 2013;38(5):918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sciammas R, Li Y, Warmflash A, Song Y, Dinner AR, Singh H. An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol Syst Biol. 2011;7:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Taylor JJ, Pape KA, Steach HR, Jenkins MK. Humoral immunity. Apoptosis and antigen affinity limit effector cell differentiation of a single naive B cell. Science. 2015;347(6223):784–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Duffy KR, Wellard CJ, Markham JF, et al. Activation-induced B cell fates are selected by intracellular stochastic competition. Science. 2012;335(6066):338–341. [DOI] [PubMed] [Google Scholar]
- 123.Zhou JHS, Markham JF, Duffy KR, Hodgkin PD. Stochastically Timed Competition Between Division and Differentiation Fates Regulates the Transition From B Lymphoblast to Plasma Cell. Front Immunol. 2018;9:2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hasbold J, Corcoran LM, Tarlinton DM, Tangye SG, Hodgkin PD. Evidence from the generation of immunoglobulin G-secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol. 2004;5(1):55–63. [DOI] [PubMed] [Google Scholar]
- 125.Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2018;18(1):35–45. [DOI] [PubMed] [Google Scholar]
- 126.Kunz DJ, Gomes T, James KR. Immune Cell Dynamics Unfolded by Single-Cell Technologies. Front Immunol. 2018;9:1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Heinzel S, Binh Giang T, Kan A, et al. A Myc-dependent division timer complements a cell-death timer to regulate T cell and B cell responses. Nat Immunol. 2017;18(1):96–103. [DOI] [PubMed] [Google Scholar]
- 128.Perez-Olivares M, Trento A, Rodriguez-Acebes S, et al. Functional interplay between c-Myc and Max in B lymphocyte differentiation. EMBO Rep. 2018;19(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang Y, Good-Jacobson KL. Epigenetic regulation of B cell fate and function during an immune response. Immunol Rev. 2019;288(1):75–84. [DOI] [PubMed] [Google Scholar]
- 130.Willis SN, Nutt SL. New players in the gene regulatory network controlling late B cell differentiation. Curr Opin Immunol. 2019;58:68–74. [DOI] [PubMed] [Google Scholar]
- 131.Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489–499. [DOI] [PubMed] [Google Scholar]
- 132.Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget. 2012;3(4):462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. [DOI] [PubMed] [Google Scholar]
- 135.Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. The Journal of biological chemistry. 2004;279(46):48350–48359. [DOI] [PubMed] [Google Scholar]
- 136.Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20(10):590–607. [DOI] [PubMed] [Google Scholar]
- 137.Caron G, Hussein M, Kulis M, et al. Cell-Cycle-Dependent Reconfiguration of the DNA Methylome during Terminal Differentiation of Human B Cells into Plasma Cells. Cell Rep. 2015;13(5):1059–1071. [DOI] [PubMed] [Google Scholar]
- 138.Dominguez PM, Ghamlouch H, Rosikiewicz W, et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov. 2018;8(12):1632–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schoeler K, Aufschnaiter A, Messner S, et al. TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. FEBS J. 2019;286(18):3566–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lio CJ, Shukla V, Samaniego-Castruita D, et al. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci Immunol. 2019;4(34). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Qi T, Sun M, Zhang C, Chen P, Xiao C, Chang X. Ascorbic Acid Promotes Plasma Cell Differentiation through Enhancing TET2/3-Mediated DNA Demethylation. Cell Rep. 2020;33(9):108452. [DOI] [PubMed] [Google Scholar]
- 142.Barwick BG, Scharer CD, Martinez RJ, et al. B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation. Nat Commun. 2018;9(1):1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039–1043. [DOI] [PubMed] [Google Scholar]
- 144.Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111(2):185–196. [DOI] [PubMed] [Google Scholar]
- 145.Muller J, Hart CM, Francis NJ, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111(2):197–208. [DOI] [PubMed] [Google Scholar]
- 146.Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(47):18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lan F, Bayliss PE, Rinn JL, et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449(7163):689–694. [DOI] [PubMed] [Google Scholar]
- 148.Beguelin W, Popovic R, Teater M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer cell. 2013;23(5):677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Caganova M, Carrisi C, Varano G, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013;123(12):5009–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Su IH, Basavaraj A, Krutchinsky AN, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4(2):124–131. [DOI] [PubMed] [Google Scholar]
- 151.Beguelin W, Teater M, Meydan C, et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer cell. 2020;37(5):655–673 e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Guo M, Price MJ, Patterson DG, et al. EZH2 Represses the B Cell Transcriptional Program and Regulates Antibody-Secreting Cell Metabolism and Antibody Production. J Immunol. 2018;200(3):1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hyun K, Jeon J, Park K, Kim J. Writing, erasing and reading histone lysine methylations. Exp Mol Med. 2017;49(4):e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. [DOI] [PubMed] [Google Scholar]
- 155.Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. [DOI] [PubMed] [Google Scholar]
- 156.Su ST, Ying HY, Chiu YK, Lin FR, Chen MY, Lin KI. Involvement of histone demethylase LSD1 in Blimp-1-mediated gene repression during plasma cell differentiation. Mol Cell Biol. 2009;29(6):1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Haines RR, Barwick BG, Scharer CD, Majumder P, Randall TD, Boss JM. The Histone Demethylase LSD1 Regulates B Cell Proliferation and Plasmablast Differentiation. J Immunol. 2018;201(9):2799–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang J, Dominguez-Sola D, Hussein S, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21(10):1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Haines RR, Scharer CD, Lobby JL, Boss JM. LSD1 Cooperates with Noncanonical NF-kappaB Signaling to Regulate Marginal Zone B Cell Development. J Immunol. 2019;203(7):1867–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. 2013;13(2):118–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. 2009;9(11):767–777. [DOI] [PubMed] [Google Scholar]
- 162.Barry ER, Corry GN, Rasmussen TP. Targeting DOT1L action and interactions in leukemia: the role of DOT1L in transformation and development. Expert Opin Ther Targets. 2010;14(4):405–418. [DOI] [PubMed] [Google Scholar]
- 163.Aslam MA, Alemdehy MF, Kwesi-Maliepaard EM, et al. Histone methyltransferase DOT1L controls state-specific identity during B cell differentiation. EMBO Rep. 2021:e51184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kealy L, Di Pietro A, Hailes L, et al. The Histone Methyltransferase DOT1L Is Essential for Humoral Immune Responses. Cell Rep. 2020;33(11):108504. [DOI] [PubMed] [Google Scholar]
- 165.Chan WF, Johanson TM, Allan RS. Three-dimensional genome rewiring during the development of antibody-secreting cells. Biochem Soc Trans. 2020;48(3):1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schoenfelder S, Fraser P. Long-range enhancer-promoter contacts in gene expression control. Nat Rev Genet. 2019;20(8):437–455. [DOI] [PubMed] [Google Scholar]
- 167.Sati S, Cavalli G. Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma. 2017;126(1):33–44. [DOI] [PubMed] [Google Scholar]
- 168.Kieffer-Kwon KR, Nimura K, Rao SSP, et al. Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Molecular cell. 2017;67(4):566–578 e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bortnick A, He Z, Aubrey M, et al. Plasma Cell Fate Is Orchestrated by Elaborate Changes in Genome Compartmentalization and Inter-chromosomal Hubs. Cell Rep. 2020;31(13):107876. [DOI] [PubMed] [Google Scholar]
- 170.Rivas MA, Meydan C, Chin CR, et al. Smc3 dosage regulates B cell transit through germinal centers and restricts their malignant transformation. Nat Immunol. 2021;22(2):240–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chaudhri VK, Dienger-Stambaugh K, Wu Z, Shrestha M, Singh H. Charting the cis-regulome of activated B cells by coupling structural and functional genomics. Nat Immunol. 2020;21(2):210–220. [DOI] [PubMed] [Google Scholar]
- 172.Bunting KL, Soong TD, Singh R, et al. Multi-tiered Reorganization of the Genome during B Cell Affinity Maturation Anchored by a Germinal Center-Specific Locus Control Region. Immunity. 2016;45(3):497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Choi NM, Majumder P, Boss JM. Regulation of major histocompatibility complex class II genes. Curr Opin Immunol. 2011;23(1):81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Majumder P, Gomez JA, Chadwick BP, Boss JM. The insulator factor CTCF controls MHC class II gene expression and is required for the formation of long-distance chromatin interactions. J Exp Med. 2008;205(4):785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Majumder P, Lee JT, Rahmberg AR, et al. A super enhancer controls expression and chromatin architecture within the MHC class II locus. J Exp Med. 2020;217(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rowley MJ, Corces VG. Organizational principles of 3D genome architecture. Nat Rev Genet. 2018;19(12):789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Majumder P, Gomez, J.A., Boss, J.M. The human major histocompatibility complex class II HLA-DRB1 and HLA-DQA1 genes are separated by a CTCF binding enhnacer-blocking element. Journal of Biological Chemistry. 2006;281(27):18435–18443. [DOI] [PubMed] [Google Scholar]
- 178.Majumder P, Boss JM. CTCF controls expression and chromatin architecture of the human major histocompatibility complex class II locus. Mol Cell Biol. 2010;30(17):4211–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Majumder P, Boss JM. Cohesin regulates MHC class II genes through interactions with MHC class II insulators. J Immunol. 2011;187(8):4236–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Majumder P, Scharer CD, Choi NM, Boss JM. B cell differentiation is associated with reprogramming the CCCTC binding factor-dependent chromatin architecture of the murine MHC class II locus. J Immunol. 2014;192(8):3925–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]