

Abstract
Endothelial-to-mesenchymal transition (EndMT) is a dynamic process in which endothelial cells suppress constituent endothelial properties and take on mesenchymal cell behaviors. To begin the process, endothelial cells loosen their cell-cell junctions, degrade the basement membrane and migrate out into the perivascular surroundings. These initial endothelial behaviors reflect a transient modulation of cellular phenotype, i.e., a phenotypic modulation, that is sometimes referred to as partial EndMT. Loosening of endothelial junctions and migration are also seen in inflammatory and angiogenic settings such that endothelial cells initiating EndMT have overlapping behaviors and gene expression with endothelial cells responding to inflammatory signals or sprouting to form new blood vessels. Reduced endothelial junctions increases permeability, which facilitates leukocyte trafficking, while endothelial migration precedes angiogenic sprouting and neovascularization; both endothelial barriers and quiescence are restored as inflammatory and angiogenic stimuli subside. Complete EndMT proceeds beyond phenotypic modulation such that mesenchymal characteristics become prominent and endothelial functions diminish. In pro-adaptive, regenerative settings the new mesenchymal cells produce extracellular matrix and contribute to tissue integrity whereas in mal-adaptive, pathologic settings the new mesenchymal cells become fibrotic, overproducing matrix to cause tissue stiffness, which eventually impacts function. Here we will review what is known about how transforming growth factor beta (TGFβ) influences this continuum from junctional loosening to cellular migration and its relevance to cardiovascular diseases.
Graphical Abstract:
TGFβ1-3 and Wnt signaling initiate EndMT while laminar shear stress-induced ERK5 and KLF2/4 along with FGFR1 and BMPR2 signaling prevent EndMT. Acetyl CoA, by enhancing fatty acid oxidation, also minimizes EndMT. Endothelial cell morphology changes dramatically during EndMT as transcription factors Snail1 and Slug suppress endothelial genes and increase mesenchymal genes.
Introduction
EndMT was first described in the endocardial cushions of the embryonic heart by Markwald and colleagues in the mid-1970s1, 2. For decades this endothelial transition was referred to as epithelial-to-mesenchymal transition (EMT) but in recent years the terms EndMT and EndoMT have become widely used to refer specifically to endothelial versus epithelial transitions. Hallmarks of EndMT are down-regulated adherens junction molecules such as VE-cadherin, increased cellular migration aided by increased matrix metalloproteinases (MMPs), and up-regulated myofibroblastic markers such as α-smooth muscle actin (α-SMA). Correspondingly, EndMT results in diminished endothelial properties such as cytokine-induced leukocyte adhesion and tube formation and increased mesenchymal properties such as collagen deposition and contractility. The transcription factors Snai1, Snai2 (also known as Slug), Twist and nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) orchestrate the changes in gene expression needed to induce or attenuate EndMT. Endothelial cells from different parts of the vasculature show varying propensities to undergo EndMT3, which may be in part due to embryonic origin. Heart valves are lined with a specialized endothelium derived from FLK1+ (a.k.a.VEGFR2+) progenitor cells that originate from cardiac mesoderm4; these valvular endothelial cells (VEC) are among the most readied to undergo EndMT. Yet, heterogeneity exists as some clonally expanded VECs show robust transforming growth factor beta (TGFβ)-mediated EndMT while other VEC clones resist EndMT signals and maintain an endothelial phenotype5-7. EndMT propensity may also be influenced by the location along the leaflet or cusp, as VECs facing onward blood flow (ventricular side the aortic valve; atrial side of the mitral valve) experience different hemodynamic forces compared to the downstream side. Indeed, distinct molecular phenotypes have been described in VECs on either side of the aortic valve8, 9. At present, the molecular basis for the differential susceptibility to EndMT signals among valvular, arterial, venous, microvascular and lymphatic endothelial cells is not fully understood.
TGFβ driven EndMT
TGFβ is well known for its role in maintaining homeostasis among cells within a given environment10yet it can become chronically elevated in disease settings, where it activates fibroblasts to become myofibroblasts, increases angiogenesis and suppresses immune responses11. It is also a major driver of EndMT during development and in pathological settings in adult life, where inflammatory cytokines such as interleukin-1β (IL1β) and mechanical forces imparted by abnormal shear stress contribute as well12. Endothelial TGFβ1 signaling was recently shown to drive cardiac valve regeneration in adult zebrafish by increasing cell-cycle entry and differentiation into new valve cells13. Other TGFβ family members, including bone morphogenic proteins (BMP), modulate TGFβ signaling in intricate ways14. For example, elegant studies by Hiepen and colleagues have shown BMP9-BMPR2 signaling maintains vascular quiescence and blocks EndMT by preventing mixed heterodimeric receptor complexes and TGFβ autostimulation15. Here we will focus on TGFβ and its direct role in EndMT.
Three TGFβ family members, TGFβ1, TGFβ2 and TGFβ3, can each stimulate EndMT in cultured endothelial cells, with TGFβ2 playing a prominent role. Indeed, TGFβ1 and TGFβ3 were shown to increase endogenous TGFβ2 in endothelial cells, including VECs 16, indicating TGFβ1 and TGFβ3 work indirectly by elevating TGFβ2. In vivo TGFβ2 signaling was shown to initiate EndMT in chick endocardial cushion16, 17 and this was further demonstrated in mice18. TGFβ2 stimulates expression of the transcription factor Slug, which then initiates EndMT19. While the direct role of TGFβ3 in EndMT induction was questioned when TGFβ3−/− mice showed no valve defects20, a recent study noted abnormally formed valves among the several morphological defects in TGFβ3−/− mice21. Recent work shows knocking out endothelial TGFβ receptors type I and II (TGFβRI and TGFβRII) reduces vascular permeability22, implicating TGFβ signaling in the earliest steps in EndMT. Endothelial TGFβ signaling can reduce cell-cell contacts and promote angiogenesis23,24; this suggests commonality in TGFβ-stimulated endothelial behaviors in EndMT, permeability and angiogenesis25.
TGFβ binds to a tetrameric complex on the plasma membrane composed of two TGFβRI and two TGFβRII, both dual specificity kinases. In endothelial cells, the prominent type I receptors are the activin receptor-like kinases 1 and 5 (ALK1 and ALK5). ALK1 is activated by bone morphogenetic protein-9/10 (BMP9/10) and usually promotes endothelial quiescence while TGFβ activates ALK5. ALK5 phosphorylates the transcription factors Sma genes and the Drosophila Mad, Mothers against decapentaplegic (SMAD)- 2 and −3 that, once phosphorylated, combine with SMAD4 and translocate to the nucleus to regulate expression of specific target genes. This is referred to as canonical TGFβ signaling pathway. The same TGFβ-bound receptor complex also activates the mitogen activated protein kinase (MAPK) pathway via recruitment and phosphorylation of the Src homology domain-containing protein ShcA, resulting in assembly of ShcA-Grb2-SOS complexes that in turn lead to phosphorylation and activation of ERK1/2. ERK1/2 then moves from the cytoplasm into the nucleus to activate transcription of non-canonical TGFβ target genes. Both canonical and non-canonical TGFβ pathways are functional in EndMT26-29.
TGFβ is also known to induce EndMT via epigenetic reconfiguration. This occurs either through regulation of EndMT transcription factors such as Snail130-35 or via SMAD molecules that directly interact with genome modifiers such as tripartite motif containing 33 (TRIM33)36, forkhead box H1 (FOXH1)37, histone acetyltransferases (HATs) including CREB-binding protein (CBP/P300)38 and P300/CBP-associated factor (P/CAF)39, histone deacetylases (HDACs)40, and SWItch/Sucrose Non-Fermentable (SW1/SNF)41. More recently, FOXM1 was found to drive TGFβ-induced EndMT by binding to the promoter of SNAI1 in human umbilical vein endothelial cells (HUVECs), which increased SNAIL1 expression42.
In addition, miRNA-mediated modulation of TGFβ-induced EndMT has been studied in the context of development and disease. Some examples of inhibitory roles of miRNA on TGFβ-induced EndMT include: miR-23, which was shown to restrict endocardial cushion formation43; miR-126, which was shown to inhibit EndMT in rat bone marrow-derived endothelial progenitor cells44, and miR-148b, which blocked EndMT and angiogenesis in a skin wound healing model45. miRNAs were also found to promote EndMT: examples are miR-49446, miR-2147 and miR-374; the latter works by downregulating MAPK7 (see below) at sites of developing atherosclerosis48. In depth review of the role(s) of miRNAs in (TGFβ-induced) EndMT can be found in these recent publications49, 50.
EndMT and endothelial permeability
The integrity of the endothelial barrier is strongly influenced by laminar shear forces and the molecular signals induced by shear stress. In particular, extracellular signal-related kinase (ERK) 5, also called MAPK7 and big-mitogen kinase-1 (BMK-1), has long been known to promote vascular integrity. ERK5 is increased in response to normal laminar shear stress (15 dynes/ cm2) and this promotes endothelial barrier integrity by increasing expression of two transcription factors - the Kruppel-like factors KLF2 and KLF4 - both of which exert anti-inflammatory and atheroprotective effects on the endothelium. These effects are mediated by the ability of KLF2 and KLF4 to inhibit nuclear factor kappa B (NF-κB), a master transcriptional regulator that orchestrates many aspects of leukocyte trafficking across the endothelium. In addition, KLF2 and KLF4 inhibit TGFβ signaling directly by interfering with SMAD2 and SMAD3 respectively; this action maintains endothelial VE-cadherin and prevents mesenchymal gene transcription51. In contrast, disturbed shear stress favors TGFβ signaling, which promotes EndMT51. Abnormally low shear stress (4 dynes/cm2) was shown to induce EndMT by increasing the transcription factor Snai1, which inhibits expression of VE-cadherin. Diminished VE-cadherin results in loosened adherens junctions, which in turn increases vascular permeability52. As such, inflammatory cytokines promote EndMT by perturbing endothelial barrier function via disassembly of adherens and tight junctions. For example, interleukin-6 (IL-6) was found to increase the permeability of HUVECs by causing a redistribution of tight junction proteins and the actin-cytoskeleton53. Interestingly, IL-6 is strongly increased in BMPR2-deficient endothelial cells, which are predisposed to EndMT15. Tumor necrosis factor alpha (TNFα) combined with interferon gamma (IFNγ) also increased permeability in HUVECs by reducing junctional adhesion molecule54. Chen and colleagues provide further insights on the impact of cytokines on permeability and EndMT55. In summary, dismantling of endothelial cell-cell junctions is a first step in EndMT (see Figure 1) as well as inflammation and angiogenesis.
Figure 1.
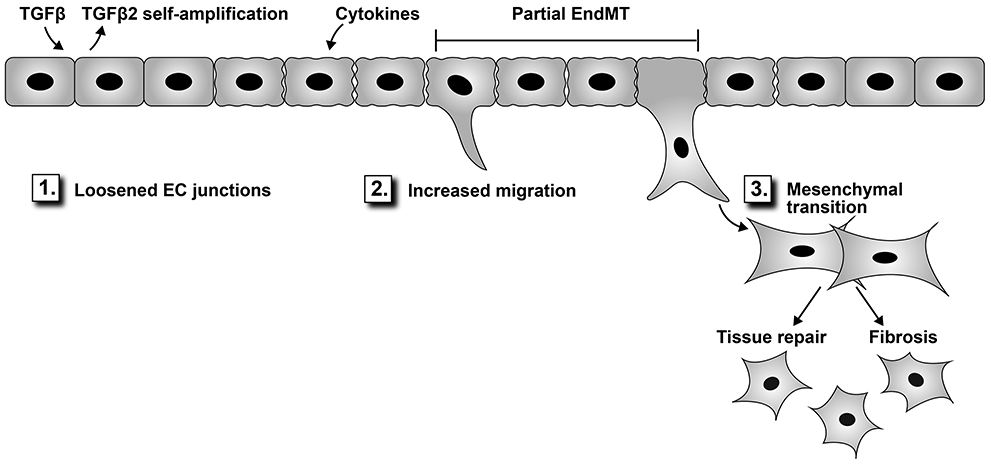
Endothelial cells exposed to TGFβ and cytokines begin EndMT by loosening junctions between endothelial cells (1), which facilitates migration into the perivascular compartment (2). The mesenchymal transition (3) can be partial, wherein cells exhibit both endothelial and mesenchymal characteristics. This state can be transient – recently referred to as endothelial to mesenchymal activation89- as the cells revert to a functional endothelial phenotype. Fully transitioned cells with diminished endothelial and increased mesenchymal functions can contribute to tissue repair but in disease settings often exacerbate fibrosis.
EndMT and endothelial migration
Endothelial cell migration is a hallmark of EndMT, angiogenesis and vascular repair. In EndMT, the cells migrate individually whereas collective migration is seen in sprouting angiogenesis where endothelial tip and stalk cells migrate in a coordinated manner56. In EndMT, loosened cell-cell junctions allow subsets of endothelial cells to migrate as single cells and invade the sub-endothelial mesenchyme. TGFβ increases MMPs and endothelial podosomes, actin-rich microdomains that protrude into the basement membrane; this likely facilitates breach of the basement membrane by focal degradation of matrix proteins and the cellular migration that ensues. The zinc-finger transcription factor SNAI2 (i.e., SLUG), known for its role in cell movement, is consistently increased in cells undergoing TGFβ-mediated EndMT and is required for EndMT in endocardial cushions57. Welch-Reardon and colleagues showed SLUG is also expressed in migrating angiogenic EC and that reducing SLUG decreased membrane type 1 MMP (MT1-MMP) and in turn MMP2 and MMP9, which reduced the ability of endothelial cells to sprout and migrate into various extracellular matrices58. This again underscores overlapping cellular behaviors between angiogenesis and EndMT, and as proposed by these authors, angiogenesis requires a transient adoption of mesenchymal features which they refer to as partial EndMT25. Despite the known requirements for SLUG and MMPs in EndMT, at present we have only a nascent molecular understanding of how the increased migration in EndMT is achieved. One might speculate that EndMT-specific migration involves SLUG-mediated transcription of genes needed for podosome formation and that complex interactions between the podosomes and the extracellular matrix, as well as surrounding cells in that milieu, provide important guideposts for migration59.
Restraints on EndMT
Endothelial phenotype and function are maintained by signaling pathways that counter TGFβ-driven EndMT. For example, fibroblast growth factor-2 (FGF-2, aka basic FGF), a growth factor known to play important roles in vascular integrity60 and endothelial proliferation61, 62, was shown to inhibit TGFβ signaling in endothelial cells in 199062. Subsequent studies showed removal of basic FGF from culture media stimulated EndMT in aortic VECs5 and in HUVECs63. When a variety of growth factors were analyzed for effects on mitral VECs, basic FGF proved to be the strongest negative regulator of TGFβ induced EndMT64. Recent mechanistic studies have shown FGF signaling through endothelial FGF-receptor 1 (FGFR1), as opposed to FGFR3 or FGFR4, inhibits TGFβ signaling and EndMT. It was shown that activated FGFR1 recruits FGFR substrate 2a (FRS2a), an adaptor protein, and this leads to increased levels of the microRNA let-7, which in turn reduces expression of TGFβRI and its signaling65, 66. Inflammatory cytokines IFNγ, TNFα and IL1β, however, diminish FGFR1 signaling65, which could explain how inflammatory cytokines augment EndMT in some cultured endothelial cells. TNFα and IL1β potently activate NF-κB, which leads to rapid induction of leukocyte adhesion molecules (e.g., E-selectin, vascular cell adhesion molecule 1; VCAM1, intercellular adhesion molecule 1; ICAM1). These molecules attract leukocytes to the endothelium at sites of inflammation. In contrast, endothelial cells that have undergone EndMT show diminished cytokine-induced E-selectin, VCAM1 and ICAM1 and concomitantly decreased adherence of leukocytes6, 67. Thus, the loss of an endothelial function that occurs during EndMT would appear to blunt leukocyte trafficking across the endothelium to sites of inflammation. Indeed, TGFβ/Smad and NF-κB are known to antagonize one another in inflammatory responses68.
Bone morphogenic protein-receptor 2 (BMPR2) also prevents EndMT by dampening endothelial TGFβ signaling. BMPR2 loss of function mutations in patients with pulmonary arterial hypertension (PAH) are associated with increased TGFβ signaling and expression of EndMT markers69. When BMP2R is absent, formation of a mixed heteromeric BMPR1/TGFβR type I/TGFβR type II complex is favored, and this leads to increased canonical TGFβ signaling and EndMT15. These authors describe endothelial BMPR2 as a gatekeeper that maintains homeostatic BMP/TGFβ signaling. Reduced BMPR2 was shown to result in increased chromatin factor high mobility group AT-hook 1 (HMGA1), which in turn increased SLUG and initiated EndMT in pulmonary arterial endothelial cells70.
Endothelial phenotype is also maintained by acetyl-CoA levels generated by fatty acid oxidation. Xiong and colleagues showed that acetyl CoA increases SMAD7, an inhibitory SMAD that dampens TGFβ signaling; this inhibits EndMT71. Endothelial-specific knock out of carnitine palmitoyltransferase II (Cpt2), an enzyme required for the shuttle of fatty acids into the mitochondria for oxidation, resulted in thickened mitral valve leaflets and aortic valve cusps. Lineage tracing showed endothelial contribution to the increased valve interstitial cells in the Cpt2-deficient mice, demonstrating that decreased endothelial fatty acid oxidation increased EndMT during valve development. In vitro studies in which human pulmonary microvascular endothelial cells were treated with 10 ng/ml TGFβ1 + 1 ng/ml IL1β for 7 days, conditions used in prior studies72, showed that increases in mesenchymal cell markers could be reversed by supplying acetate into culture medium to replenish acetyl CoA. The acetate increased SMAD7 and decreased phospho-SMAD2 levels, consistent with reduced TGFβ signaling71. In summary, laminar shear stress, FGFR1 and BMPR2 signaling and fatty acid oxidation suppress EndMT –whereas disturbed shear stress, TGFβ and Wnt signaling (see below) stimulate EndMT.
Wnt signaling in EndMT
In contrast to inhibitory pathways, Wnt (wingless-type mammary tumor virus integration site family of proteins) signaling can augment TGFβ signaling. Wnt ligands bind to Frizzled receptors on the plasma membrane, which triggers changes in intracellular β-catenin, a primary effector of canonical Wnt signaling. In quiescent, non-Wnt-stimulated endothelial cells, β-catenin interacts with VE-cadherin at the cytoplasmic face of adherens junctions. Cytosolic levels of β-catenin are kept low by constitutive ubiquitination and proteasomal degradation such that in the absence of Wnt ligands and/or presence of Wnt inhibitors, the canonical Wnt pathway is in an ‘off state’. Wnt ligand-receptor binding releases β-catenin from the adherens junction and reduces its turnover by proteasomal degradation, resulting in cytosolic accumulation that then favors β-catenin translocation into the nucleus where it increases transcription of the lymphocyte enhancer factor/T-cell transcription factor (Lef/TCF). In some settings, Wnt and TGFβ signaling converge in the nucleus where β-catenin interacts with SMAD transcriptional regulators as well as Lef/TCF to orchestrate the transcriptional regulation of shared target genes68.
Canonical Wnt signaling is required for EndMT in endocardial cushions in the developing heart - cushion explants or endothelial cells deficient in β-catenin fail to undergo EndMT73. Wnt’s role in EndMT has also been studied in cell culture models under mechanical stress. Balachandran and colleagues showed that β-catenin plays a role when mitral VECs undergo EndMT under high cyclic mechanical strain74, and indeed EndMT occurs upon mechanical stretch of mitral valve leaflets in a large animal model75. Zhong and colleagues showed that Wnt-β-catenin can drive EndMT in aortic VECs and is influenced by matrix stiffness7. Endothelial Wnt signaling and EndMT have also been analyzed within days after myocardial infarction (MI), produced by ligating the left anterior descending coronary artery in mice76. In this elegant study, lineage tracing of endothelial cells expressing the TOPGAL reporter, a readout for canonical Wnt-β-catenin signaling, showed Wnt signaling in α-SMA-positive cells of endothelial origin, indicating a post-MI Wnt activation coincident with EndMT. Interestingly, activation of Wnt/β-catenin in cultured bovine aortic endothelial cells, without TGFβ or FGF in the culture medium, was sufficient to induce mesenchymal markers and reduce endothelial markers76. The authors proposed that Wnt signaling in endothelial cells produces a migratory CD31+/αSMA+ intermediate phenotype that reflects cells that have partially transitioned to a mesenchymal state. Cells that have undergone partial EndMT can either undergo angiogenesis to produce new blood vessels, reverting to an endothelial phenotype, or proceed to a CD31-/α-SMA+ collagen-producing myofibroblast. The authors concluded that the endothelial cells contributed to cardiac repair after MI by both partial and complete EndMT pathways76. The mechanisms that control whether endothelial cells revert to an endothelial phenotype after partial EndMT or complete a full transition to mesenchymal phenotype will be important to elucidate as this information could be used to both increase angiogenesis and provide matrix-producing cells to enhance tissue repair.
EndMT in different types of endothelial cells
The extent to which the location of endothelial cells within the cardiovascular system influences EndMT and what type of stimulus or stimuli are needed to induce partial or complete EndMT can be gleaned from the literature. First, endothelial cells isolated from heart valves – mitral, aortic or pulmonic - show robust EndMT in vitro when treated with 1-2 ng/ml TGFβ for 4-5 days. In these studies, EndMT is defined as cells: 1) co-expressing endothelial and mesenchymal markers, 2) exhibiting increased cellular migration, 3) showing a loss of endothelial properties such as leukocyte adhesion and tubule formation, and 4) exhibiting increased mesenchymal/myofibroblastic properties such as collagen production and contractility5-7, 16, 77. In vivo studies of the mitral valve in large animal models show EndMT is associated with valve leaflet growth and thickening16, 78-80. The propensity of VECs to undergo EndMT may be enhanced by the inherent plasticity of endocardial endothelial cells4, 81 and possibly expression of the Type III TGFβ-receptor, which was shown to necessary for EndMT in atrioventricular cushions and sufficient to drive EndMT in ventricular endocardium82.
Some arterial ECs can be induced to undergo EndMT. Endothelial lineage tracing provides compelling evidence for arterial EndMT in mouse models of atherosclerotic plaque development83 as well as partial EndMT behaviors in formation of collateral vessels in injured neonatal hearts84. In contrast, some sources of arterial endothelial cells - isolated from the coronary artery, carotid artery or pulmonary artery and analyzed in vitro - showed no increase in EndMT markers in response to TGFβ3, 16, 26. In contrast, primary aortic ECs were shown to undergo robust EndMT in response to TGFβ23. Venular endothelial cells such as HUVECs, treated with TGFβ1, showed increased expression of transgelin (SM22α) and SLUG, and decreased expression of CD31 and VE-cadherin, compared with no treatment, indicating EndMT29. In another study, HUVECs treated with TGFβ2 plus IL1β for 24 hours showed EndMT characteristics that were attributed to p65-dependent NF-κB activation85. Microvascular intestinal endothelial cells were non-responsive to TGFβ unless TNFα, another NF-κB activating inflammatory cytokine, was included72. In another study, human dermal microvascular endothelial cells did not undergo EndMT in response to TGFβ2 alone, whereas in parallel, human pulmonary VECs showed robust EndMT6. In summary, the findings suggest inflammatory signals contribute to changes in endothelial phenotype, perhaps by priming the cells to respond to EndMT inducing stimuli such as TGFβ. Interestingly, lymphatic endothelial cells have been reported to undergo EndMT. For example, Kaposi’s sarcoma herpesvirus (KSHV) infection of lymphatic endothelial cells stimulates EndMT through viral protein activation of the NOTCH pathway and MT1-MMP86. Further, FGF2 helps maintain a differentiated lymphatic endothelial cell identity by opposing TGFβ-activated SMAD2 via Ras/ERK MAPK signaling87. In summary, we have much to learn about the requirements and conditions under which endothelium in different vasculature beds undergoes EndMT.
A limitation of studying EndMT in vitro is that while many types of endothelial cells have been reported to undergo EndMT, the increase in mesenchymal markers is often not robust and functional assays are lacking. An overarching limitation for many in vitro EndMT assays is that the purity of the starting endothelial cultures in not readily apparent. This is a concern because a small number of contaminating cells in primary endothelial cell cultures, such as smooth muscle cells or fibroblasts, can lead to an appearance of EndMT on a western blot or in qPCR assays because TGFβ increases α-SMA, a commonly used marker for EndMT, in fibroblasts, for example. In addition to assays on whole cell lysates, techniques such as flow cytometry and/or immunofluorescence should be employed to 1) assess potential for other cell types in the endothelial cultures and to 2) verify co-localized endothelial and mesenchymal markers to document cells have initiated EndMT (Figure 2). In addition, functional assays that measure contractility (mesenchymal) and cytokine-induced leukocyte adhesion (endothelial) should be conducted to determine the extent to which cells have acquired mesenchymal functions and reduced endothelial functions. Cells that retain endothelial markers (e.g., VE-cadherin) and endothelial functions have undergone partial EndMT whereas cells that have lost endothelial markers and functions, and express mesenchymal markers and gained mesenchymal functions, have undergone complete EndMT.
Figure 2.

Mitral VECs in culture undergoing EndMT. Cells were co-stained with anti-VE-cadherin (green), anti-α-SMA (red) and DAPI (blue) to highlight nuclei. A 72-hour stimulus converted many VE-cadherin-positive VECs within the cobblestone monolayer to α-SMA-positive/VE-cadherin-positive cells (yellow/orange) with mesenchymal morphology, indicating partial EndMT. Scale bar, 50μm.
EndMT in cardiovascular disease
The mechanisms that activate EndMT in settings of injury, inflammation or stress are under intense scrutiny in order to understand the extent to which EndMT is a driver or a consequence of cardiovascular pathology55, 69, 88. The dynamic nature of EndMT is also emerging, particularly from new single cell RNA-seq studies that have revealed a transient EndMT in the non-cardiomyocyte fraction after MI. Lineage tracing in the mice showed that within 3 days after MI, a subset of endothelial cells underwent a transient mesenchymal activation, termed endothelial to mesenchymal activation, that lasted approximately 10 days. HUVECs treated with TGFβ2 mimicked endothelial to mesenchymal activation, which was reversed upon withdrawal of TGFβ289. The authors speculate that the transient mesenchymal state may facilitate angiogenesis, which would be beneficial for the ischemic myocardium, but also raise the possibility that matrix deposition from the temporary mesenchyme may have detrimental effects. These new observations, made possible by state-of-the-art transcriptomics, underscore the necessity for deeper understanding of the EndMT continuum. The role of BMP signaling and how dysregulation of this pathway contributes to cardiovascular diseases are discussed in detail elsewhere90.
Ischemic mitral regurgitation (IMR)
EndMT is required for formation of the four cardiac valves during embryogenesis, but is strongly suppressed in adult valvular endothelium. Yet EndMT capability is not lost. Ovine, porcine and human VECs isolated from adult valves readily undergo EndMT in response to TGFβ5, 6, 75, 91, suggesting EndMT capability is retained, possibly throughout life, in order to replenish valve interstitial cells, major producers of the valve extracellular matrix. This speculation is not supported, however, by lineage tracing studies, which failed to find evidence of EndMT in mitral valves of mice at 4 months of age92. In contrast, recent studies in adult zebrafish indicate some endothelial cells undergo EndMT in uninjured valves13. Large animal studies devised to study IMR, which is the most common form of valvular heart disease in the U.S.93 and is associated with excess mortality after MI94, 95, showed that EndMT can be reactivated in adult valves. Mitral VECs undergoing EndMT were detected and quantified in mitral valve leaflets exposed in vivo to mechanical stretch designed to mimic the tethering of mitral valve leaflets that occurs when the left ventricle enlarges after MI. EndMT coincided with an increase in mitral valve leaflet area and thickness, leading to the concept that the mitral valve leaflets are not passive flaps but can adapt to changing hemodynamics, and that the valvular endothelium is central to this adaptive response75. When mechanical stretch was combined with MI, EndMT was further increased over that seen in mitral valves with mechanical stretch alone, but there was excessive sub-endothelial localized TGFβ on the atrial side, increased collagen deposition and leukocyte infiltration78, all of which was blocked by losartan given over the 2 month post-MI time period79. The rationale for testing losartan therapy in this model was based on its negative effect on TGFβ signaling, in addition to its inhibitory effect on angiotensin II-receptor binding96. These studies suggest that preventing excessive TGFβ-driven EndMT may reduce fibrosis in the leaflets, which develops over several months post-MI, causing IMR.
Significantly increased EndMT was found at 6 months after inferior MI in sheep; this study led to the unexpected discovery of CD45, a protein tyrosine phosphatase, expressed along the MV leaflet endothelium and a corresponding increase in VE-cadherin-positive endothelial cells expressing CD45 and α-SMA, almost 15 times higher in IMI versus in sham animals. The increase was detected by flow cytometric analysis of mitral valve cells isolated immediately after mitral valves were removed from sham and post-MI sheep. In vitro, TGFβ induced expression of CD45, α-SMA, SLUG, collagen type I alpha 2 (COL1A2) and TGFβ2 in mitral VEC, all of which were blocked by inclusion of a CD45 phosphatase inhibitor16. We speculated that CD45 drives a maladaptive, pro-fibrotic form of EndMT in the mitral valve endothelium, possibly through post-translational mechanisms such as dephosphorylation of molecules involved in EndMT. EndMT has been recognized as a contributor to other types of valve diseases such as rheumatic heart disease97 and calcific aortic valve disease (CAVD). In CAVD in vitro models, microRNA-483 was shown to protect against endothelial inflammation and EndMT, which can precede calcification, by downregulating ubiquitin E2 ligase C, resulting in decreased hypoxia inducible factor 1 subunit alpha (HIF1α)98. The authors went on to show a HIF1α inhibitor reduced calcification in a porcine CAVD model. CAVD and the role of EndMT in CAVD have been recently reviewed elsewhere99, 100.
Atherosclerosis –
Studies in murine models of atherosclerosis have revealed EndMT, with confirmatory studies in human atherosclerotic plaques83, 101. Evrard and colleagues employed inducible endothelial lineage tracing and single cell imaging to identify endothelial cells that had undergone partial EndMT and those that had completely transitioned to mesenchymal cells. Three to nine percent of the intimal plaque fibroblast-like cells were endothelial-derived, and thus had undergone complete EndMT; and further, these cells were correlated with plaque instability83. Chen and colleagues discovered that endothelial FGFR1 prevented TGFβ-driven EndMT, neointimal formation, and the progressive development of atherosclerosis101. Moonen et al. showed that disturbed flow decreased the ERK5/MAPK7 signaling that normally protects endothelium from EndMT and led to neointimal hyperplasia and atherogenic differentiation of EC102. Boström et al. found aortic endothelial cells from ApoE−/− mice fed a high fat/high cholesterol ‘western diet’ expressed mesenchymal markers and showed signs of calcification103. Helmke and colleagues reported partial EndMT in aortic arch sections from ApoE−/− mice fed a western diet. The transitioning endothelial cells were identified by expression of the stem cell marker stem cell antigen 1 (Sca-1)104. Lastly, evidence in support of partial EndMT was reported in patients with critical limb ischemia, a hazardous manifestation of atherosclerosis105. In this study, endothelial cells of the patients’ intramuscular arterioles were bulked up and re-oriented such that they narrowed or occluded the arterioles. The endothelial cells maintained CD31 expression and remained on the luminal side of the vessel, however, the mesenchymal markers S100A4 and N-cadherin expression were increased. Quantification showed 76% of arterioles in had at least one endothelial cell positive for nuclear localized SNAI1, and increased nuclear pSMAD2/3 was detected as well, indicating TGFβ signaling. Taken together these murine and human studies strongly implicate partial and complete EndMT in atherosclerotic lesions and point to EndMT as a potential target for limiting the development and progression of unstable plaques. An important question is at what point along the continuum from endothelial to mesenchymal do cells begin to exert deleterious effects on the vessel wall, and what are the molecular mechanisms at each juncture.
In a recent study, single cell RNA-seq analysis of carotid arteries exposed to disturbed blood flow (d-flow) versus stable flow (s-flow) revealed that endothelial cells respond differently at the genomic level to disturbed versus stable flow. Andueza and colleagues identified s-flow- and d-flow-dependent enrichment of specific transcription factor binding sites and cis-regulatory elements106. D-flow reprogrammed the carotid arterial ECs into an array of phenotypes from inflammatory, to mesenchymal (i.e., EndMT), stem/progenitor-like, hematopoietic, and immune cell-like phenotypes. Furthermore, the authors showed that d-flow rapidly induced, whereas s-flow prevented, robust atherosclerotic development within 2 weeks in hypercholesterolemic mice. This fascinating study underscores the complexity of the endothelium and suggests its plasticity may extend beyond EndMT.
Pulmonary arterial hypertension (PAH)
PAH is a fatal disease marked by the cancer-like proliferation of pulmonary vascular smooth muscle and endothelial cells that is strongly linked to germline loss-of-function mutations in BMPR2107, 108. Examination of pulmonary artery plexiform lesions from PAH patients revealed cells co-expressing both endothelial markers and α-SMA, indicating EndMT109. BMPR2 loss of function was shown to increase the EndMT-related transcription factor Twist-1 and the mesenchymal intermediate filament protein vimentin, and repress VE-cadherin and p120-catenin, thus indicating a role for BMPR2 signaling in preventing EndMT in the animal model of severe PAH110. Another study showed that when BMPR2 falls below 50% of its normal level, BMP9 switches from promoting endothelial quiescence to promoting endothelial overgrowth111. Recently, another group of researchers showed that BMP9 induced a partial EndMT in pulmonary microvascular endothelial cells isolated from PAH patients but not in pulmonary microvascular endothelial cells from non-PAH lungs. The authors traced the mechanism to BMP-9 induced expression of IL-6 in the PAH pulmonary microvascular endothelial cells. They showed that neutralizing anti-IL-6 blocked BMP9-induced increases in SNAI1 and SNAI2/SLUG, preserved the endothelial cobblestone morphology, sustained the expression and junctional organization of VE-cadherin and resulting barrier function, and blocked expression of SM22α112. This study identified autocrine IL-6 as an inflammatory mediator that can drive EndMT in the lung microvasculature and thereby enhances our understanding of how inflammatory molecules operate in EndMT.
PAH endothelial cells have also been shown to express stem cell markers. Suzuki and colleagues used endothelial-specific lineage tracing in a mouse model wherein Sugen 5416 and hypoxia were used to induce PAH113. The authors delineated partial EndMT as cells enriched for the stem cell marker CD133/Prom1 and endothelial marker CD34 whereas CD31−CD45−Sca-1+ lung cells were designated complete EndMT. The cells identified as having fully transitioned to a mesenchymal phenotype – the CD31−/CD45−/Sca-1+ cells - showed higher proliferative and migratory capacity compared to non-endothelial CD326+ lung mesenchymal cells. Further, the complete EndMT cells could be induced to differentiate into smooth muscle-like cells when grown in appropriate media. This indicates a de-differentiation followed by redifferentiation pathway in this setting. Normal pulmonary vascular endothelial cells were shown to undergo EndMT, exhibit increased proliferation and express stem cell markers in a model of acute lung injury, yet the cells lacked the features of complete EndMT seen in PAH114. In summary, accumulating evidence suggests that partial and complete EndMT play important roles in initiation and progression of PAH.
Cerebral cavernous malformations (CCM)
EndMT has been suggested to play a driving role in cerebral cavernous malformation-1 (CCM1). CCM1 is a vascular malformation characterized by enlarged capillary-like channels, loss of function mutations in KRIT1 and cerebral hemorrhage. In mouse models of CCM1, upregulation of Erk5, Klf4 and Bmp6 were shown to drive cerebral endothelial cells to undergo EndMT. The role of KLF4 in regulating EndMT appears to be dependent on the cellular context. Cuttano et al. showed that in the absence of Krit1, Klf4 was increased in ECs of different organs but the full upregulation of EndMT markers was detected only in brain ECs suggesting that Klf4 upregulation is critical to promote the EndMT, however, the strength of this effect is context-dependent and specific to the brain vasculature115. In contrast, another study demonstrated a causal role of Klf2/Klf4 in CCM formation but found no evidence of EndMT involvement116. The role of abnormal flow and low fluid shear stress in the malformed CCM vessels may have an impact on the development of abnormal endothelial responses and changes in phenotype, as proposed by Gamble and colleagues117. Taken together, the role of EndMT – either partial or complete - in vascular malformations is not fully defined and requires further investigation. Many other EndMT-mediated human diseases are reviewed and discussed elsewhere118.
Drugs that prevent EndMT
Several TGFβ blocking drugs have been proposed as EndMT inhibitors. Losartan, an angiotensin II type 1 receptor antagonist, impairs EndMT by blocking TGFβ signaling, as shown in vitro in TGFβ-treated mitral VEC26. Bartko and colleagues showed systemic Losartan therapy over 2 months allowed adaptive growth of mitral valve leaflets but prevented thickening, EndMT and fibrotic changes79. Moreover, the effect of Losartan in hypertensive cardiac fibrosis was studied using spontaneous hypertensive rat models where it was shown that Losartan downregulated TGFβ/Smad signaling, increased CD31 expression, and reduced collagen 1 in the left ventricle119. There is little evidence that Losartan inhibits angiogenesis per se and thus it may not affect partial EndMT in angiogenic sprouting. Indeed, Jain and colleagues have shown Losartan increases vascular perfusion in dense tumor stroma by reducing extracellular matrix120, 121 ; from this one might speculate that Losartan might increase vascular perfusion in fibrotic tissue. Other drugs include Kallistatin, which was shown to upregulate endothelial nitric oxide synthase (eNOS) via its active site domain and inhibit miR21, SNAI1 expression, and reactive oxygen species formation through its heparin-binding domain122, Linagliptin, a dipeptidyl peptidase IV inhibitor (DPP-4) that impairs DPP-4 interaction with integrin β1, was shown to block TGFβ2-induced EndMT in cultured human microvascular endothelial cells via upregulation of miR29123. Macitentan, an endothelin-1 receptor inhibitor, was shown to impair EndMT induced by either endothelin-1 or TGFβ1124. Ideally, EndMT-inhibitors would be targeted to specific sites in a temporally controlled manner to avoid systemic inhibition of EndMT that may be needed for regeneration at other sites, as shown recently in regenerating atrioventricular valves in zebrafish13. Up-to-date lists of the drugs with EndMT-inhibiting properties are available88, 125. As the triggers and timing of mal-adaptive EndMT are elucidated, the possibility of targeted prevention of EndMT in cardiovascular disease – both temporal and site-specific - may become feasible using drugs therapies.
Summary
EndMT represents prototypical plasticity of endothelium. The onset and degree of EndMT capability is dictated by inherent features such as embryonic origin and genetics and external factors such as hemodynamics, cytokines and hypoxia78, 126. EndMT relies on loosened cellular junctions, sprouting, and migration to move into the extravascular milieu; these are activities that overlap with leukocyte diapedesis and angiogenesis. Partially transitioned cells have several fates – revert to endothelial phenotype, transition fully to a mesenchymal cell, or pause at an elusive ‘partial EndMT’ state. Molecular drivers of EndMT include TGFβ and Wnt signaling molecules while FGFR1, ERK5, and BMPR2 signaling and as recently shown, fatty acid oxidation, restrain EndMT. Expression levels of different receptors sensitize the cells to specific TGFβ isoforms to fine tune endothelial responses in different cellular contexts127. While lineage tracing strategies using cell-specific constitutive versus inducible Cre drivers have advanced our understanding of EndMT considerably – Zhou and colleagues provide an excellent summary of these tools128 – there is still little known about which molecules control the rate and extent to which partial versus complete EndMT occurs. There is also little to distinguish pro-adaptive versus maladaptive EndMT. The animal models described above for IMR, atherosclerosis and PAH provide essential tools to address these questions and develop strategies to harness the endothelial plasticity to achieve tissue repair and regeneration.
Highlights.
TGFβ, Wnt signaling, and disturbed shear stress promote EndMT.
Normal laminar shear stress, FGFR1 and BMPR2 signaling and fatty acid oxidation suppress EndMT.
Initial steps in EndMT – loosening of endothelial junctions and migration – are similar to initial steps of angiogenesis and leukocyte diapedesis.
EndMT is associated with mitral valve leaflets thickening and mitral regurgitation after myocardial infarction.
EndMT in pulmonary arterial hypertension is driven by loss of BMPR2 and dysregulated BMP-9 signaling.
Sources of Funding
NHLBI of the National Institutes of Health under award number R01HL141917 and T32 HL007917.
List of non-standard abbreviations and acronyms
- ALK
activin receptor-like kinase
- α-SMA
α-smooth muscle actin
- BMP9/10
bone morphogenetic protein-9/10
- BMPR2
bone morphogenic protein-receptor 2
- CAF
CBP-associated factor
- CAVD
calcific aortic valve disease
- CBP
CREB-binding protein
- CCM
cerebral cavernous malformations
- COL
collagen
- Cpt2
carnitine palmitoyltransferase II
- DDP-4
dipeptidyl peptidase IV inhibitor
- d-flow
disturbed blood flow
- EMT
epithelial to mesenchymal transition
- EndMT
endothelial to mesenchymal transition
- eNOS
endothelial nitric oxide synthase
- ERK
extracellular signal-related kinase
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- FGRS
FGFR substrate
- FOXH1
forkhead Box H1
- HIF1a
hypoxia inducible factor 1 subunit alpha
- HMGA
high Mobility Group AT-hook
- HUVECs
human umbilical vein endothelial cells
- IL1β
interleukin-1β
- IMR
Ischemic mitral valve regurgitation
- KLF
Kruppel-like factor
- KSHV
Kaposi’s sarcoma herpesvirus
- Lef
lymphocyte enhancer factor
- MAPK
mitogen activated protein kinase (MAPK)
- MI
myocardial infarction
- MMPs
matrix metalloproteinases
- MT1
membrane type 1
- NFAT1
nuclear factor of activated T-cells, cytoplasmic 1
- NF-κB
nuclear factor kappa B
- PAH
pulmonary arterial hypertension
- PTH
parathyroid hormone
- Sca1
stem cell antigen 1
- Shc
Src homology domain-containing protein
- s-flow
stable flow
- SMAD
Sma genes and the Drosophila Mad, Mothers against decapentaplegic
- TCF
T cell transcription factor
- TGFβ
transforming growth factor beta
- TGFβRI/II
TGFβ receptors type I/II
- TNFα
tumor necrosis factor-α
- TRIM33
tripartite motif containing 33
- VEC
valve endothelial cells
- Wnt
wingless-related integration site
Footnotes
Disclosures- none.
References
- 1.Markwald RR, Fitzharris TP and Manasek FJ. Structural development of endocardial cushions. Am J Anat. 1977;148:85–119. [DOI] [PubMed] [Google Scholar]
- 2.Markwald RR, Fitzharris TP and Smith WN. Structural analysis of endocardial cytodifferentiation. Developmental Biology. 1975;42:160–80. [DOI] [PubMed] [Google Scholar]
- 3.Ursoli Ferreira F, Eduardo Botelho Souza L, Hassibe Thome C, Tomazini Pinto M, Origassa C, Salustiano S, Marcel Faca V, Olsen Camara N, Kashima S and Tadeu Covas D. Endothelial Cells Tissue-Specific Origins Affects Their Responsiveness to TGF-beta2 during Endothelial-to-Mesenchymal Transition. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakano A, Nakano H, Smith KA and Palpant NJ. The developmental origins and lineage contributions of endocardial endothelium. Biochim Biophys Acta. 2016;1863:1937–47. [DOI] [PubMed] [Google Scholar]
- 5.Paranya G, Vineberg S, Dvorin E, Kaushal S, Roth SJ, Rabkin E, Schoen FJ and Bischoff J. Aortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro. Am J Pathol. 2001;159:1335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paruchuri S, Yang JH, Aikawa E, Melero-Martin JM, Khan ZA, Loukogeorgakis S, Schoen FJ and Bischoff J. Human pulmonary valve progenitor cells exhibit endothelial/mesenchymal plasticity in response to vascular endothelial growth factor-A and transforming growth factor-beta2. Circ Res. 2006;99:861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhong A, Mirzaei Z and Simmons CA. The Roles of Matrix Stiffness and ss-Catenin Signaling in Endothelial-to-Mesenchymal Transition of Aortic Valve Endothelial Cells. Cardiovasc Eng Technol. 2018;9:158–167. [DOI] [PubMed] [Google Scholar]
- 8.Simmons CA, Grant GR, Manduchi E and Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. 2005;96:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF and Davies PF. Hypercholesterolemia induces side-specific phenotypic changes and peroxisome proliferator-activated receptor-gamma pathway activation in swine aortic valve endothelium. Arterioscler Thromb Vasc Biol. 2010;30:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huk DJ, Austin BF, Horne TE, Hinton RB, Ray WC, Heistad DD and Lincoln J. Valve Endothelial Cell-Derived Tgfbeta1 Signaling Promotes Nuclear Localization of Sox9 in Interstitial Cells Associated With Attenuated Calcification. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:328–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akhurst RJ. Targeting TGF-beta Signaling for Therapeutic Gain. Cold Spring Harb Perspect Biol. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma J, Sanchez-Duffhues G, Goumans MJ and Ten Dijke P. TGF-beta-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front Cell Dev Biol. 2020;8:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bensimon-Brito A, Ramkumar S, Boezio GLM, Guenther S, Kuenne C, Helker CSM, Sánchez-Iranzo H, Iloska D, Piesker J, Pullamsetti S, Mercader N, Beis D and Stainier DYR. TGF-β Signaling Promotes Tissue Formation during Cardiac Valve Regeneration in Adult Zebrafish. Developmental cell. 2020;52:9–20.e7. [DOI] [PubMed] [Google Scholar]
- 14.Rol N, Kurakula KB, Happé C, Bogaard HJ and Goumans MJ. TGF-β and BMPR2 Signaling in PAH: Two Black Sheep in One Family. International journal of molecular sciences. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiepen C, Jatzlau J, Hildebrandt S, Kampfrath B, Goktas M, Murgai A, Cuellar Camacho JL, Haag R, Ruppert C, Sengle G, Cavalcanti-Adam EA, Blank KG and Knaus P. BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFbeta responses and altered cell mechanics. PLoS Biol. 2019;17:e3000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bischoff J, Casanovas G, Wylie-Sears J, Kim DH, Bartko PE, Guerrero JL, Dal-Bianco JP, Beaudoin J, Garcia ML, Sullivan SM, Seybolt MM, Morris BA, Keegan J, Irvin WS, Aikawa E and Levine RA. CD45 Expression in Mitral Valve Endothelial Cells After Myocardial Infarction. Circ Res. 2016;119:1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabbineni H, Verma A and Somanath PR. Isoform-specific effects of transforming growth factor β on endothelial-to-mesenchymal transition. Journal of cellular physiology. 2018;233:8418–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA and Klewer SE. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Developmental biology. 2002;248:170–81. [DOI] [PubMed] [Google Scholar]
- 19.Romano LA and Runyan RB. Slug is an essential target of TGFbeta2 signaling in the developing chicken heart. Developmental biology. 2000;223:91–102. [DOI] [PubMed] [Google Scholar]
- 20.Azhar M, Schultz Jel J, Grupp I, Dorn GW 2nd, Meneton P, Molin DG, Gittenberger-de Groot AC and Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine & growth factor reviews. 2003;14:391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chakrabarti M, Al-Sammarraie N, Gebere MG, Bhattacharya A, Chopra S, Johnson J, Peña EA, Eberth JF, Poelmann RE, Gittenberger-de Groot AC and Azhar M. Transforming Growth Factor Beta3 is Required for Cardiovascular Development. Journal of cardiovascular development and disease. 2020;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen PY, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, Gujja S, Cilfone NA, Kauffman KJ, Sun L, Sun H, Zhang X, Aryal B, Canfran-Duque A, Liu R, Kusters P, Sehgal A, Jiao Y, Anderson DG, Gulcher J, Fernandez-Hernando C, Lutgens E, Schwartz MA, Pober JS, Chittenden TW, Tellides G and Simons M. Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat Metab. 2019;1:912–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madri JA, Pratt BM and Tucker AM. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. The Journal of cell biology. 1988;106:1375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Budi EH, Mamai O, Hoffman S, Akhurst RJ and Derynck R. Enhanced TGF-beta Signaling Contributes to the Insulin-Induced Angiogenic Responses of Endothelial Cells. iScience. 2019;11:474–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welch-Reardon KM, Wu N and Hughes CC. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arteriosclerosis, thrombosis, and vascular biology. 2015;35:303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wylie-Sears J, Levine RA and Bischoff J. Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-beta-induced phosphorylation of ERK. Biochem Biophys Res Commun. 2014;446:870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dufton NP, Peghaire CR, Osuna-Almagro L, Raimondi C, Kalna V, Chauhan A, Webb G, Yang Y, Birdsey GM, Lalor P, Mason JC, Adams DH and Randi AM. Dynamic regulation of canonical TGFβ signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nature communications. 2017;8:895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pardali E, Sanchez-Duffhues G, Gomez-Puerto MC and Ten Dijke P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. International journal of molecular sciences. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooley BC, Nevado J, Mellad J, Yang D, St Hilaire C, Negro A, Fang F, Chen G, San H, Walts AD, Schwartzbeck RL, Taylor B, Lanzer JD, Wragg A, Elagha A, Beltran LE, Berry C, Feil R, Virmani R, Ladich E, Kovacic JC and Boehm M. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Science translational medicine. 2014;6:227ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jamora C, Lee P, Kocieniewski P, Azhar M, Hosokawa R, Chai Y and Fuchs E. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLoS biology. 2005;3:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Alvarez C, Blanco MJ, Perez R, Rabadan MA, Aparicio M, Resel E, Martinez T and Nieto MA. Snail family members and cell survival in physiological and pathological cleft palates. Developmental biology. 2004;265:207–18. [DOI] [PubMed] [Google Scholar]
- 32.Yanez-Mo M, Lara-Pezzi E, Selgas R, Ramirez-Huesca M, Dominguez-Jimenez C, Jimenez-Heffernan JA, Aguilera A, Sanchez-Tomero JA, Bajo MA, Alvarez V, Castro MA, del Peso G, Cirujeda A, Gamallo C, Sanchez-Madrid F and Lopez-Cabrera M. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. The New England journal of medicine. 2003;348:403–13. [DOI] [PubMed] [Google Scholar]
- 33.Peinado H, Quintanilla M and Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. The Journal of biological chemistry. 2003;278:21113–23. [DOI] [PubMed] [Google Scholar]
- 34.Kaimori A, Potter J, Kaimori JY, Wang C, Mezey E and Koteish A. Transforming growth factor-beta1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. The Journal of biological chemistry. 2007;282:22089–101. [DOI] [PubMed] [Google Scholar]
- 35.Medici D, Potenta S and Kalluri R. Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. The Biochemical journal. 2011;437:515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow-Tsang LF, Liu JX, Kim H, Barlas A, Manova-Todorova K, Kaartinen V, Studer L, Mark W, Patel DJ and Massague J. A poised chromatin platform for TGF-beta access to master regulators. Cell. 2011;147:1511–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aragon E, Wang Q, Zou Y, Morgani SM, Ruiz L, Kaczmarska Z, Su J, Torner C, Tian L, Hu J, Shu W, Agrawal S, Gomes T, Marquez JA, Hadjantonakis AK, Macias MJ and Massague J. Structural basis for distinct roles of SMAD2 and SMAD3 in FOXH1 pioneer-directed TGF-beta signaling. Genes & development. 2019;33:1506–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janknecht R, Wells NJ and Hunter T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes & development. 1998;12:2114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simonsson M, Kanduri M, Gronroos E, Heldin CH and Ericsson J. The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. The Journal of biological chemistry. 2006;281:39870–80. [DOI] [PubMed] [Google Scholar]
- 40.Simonsson M, Heldin CH, Ericsson J and Gronroos E. The balance between acetylation and deacetylation controls Smad7 stability. The Journal of biological chemistry. 2005;280:21797–803. [DOI] [PubMed] [Google Scholar]
- 41.Xi Q, He W, Zhang XH, Le HV and Massague J. Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor beta transcriptional program. The Journal of biological chemistry. 2008;283:1146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song S, Zhang R, Cao W, Fang G, Yu Y, Wan Y, Wang C, Li Y and Wang Q. Foxm1 is a critical driver of TGF-beta-induced EndMT in endothelial cells through Smad2/3 and binds to the Snail promoter. Journal of cellular physiology. 2019;234:9052–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagendijk AK, Goumans MJ, Burkhard SB and Bakkers J. MicroRNA-23 restricts cardiac valve formation by inhibiting Has2 and extracellular hyaluronic acid production. Circ Res. 2011;109:649–57. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Zhang Z, Zhang DY, Zhu J, Zhang T and Wang C. microRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS One. 2013;8:e83294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miscianinov V, Martello A, Rose L, Parish E, Cathcart B, Mitić T, Gray GA, Meloni M, Al Haj Zen A and Caporali A. MicroRNA-148b Targets the TGF-β Pathway to Regulate Angiogenesis and Endothelial-to-Mesenchymal Transition during Skin Wound Healing. Molecular therapy : the journal of the American Society of Gene Therapy. 2018;26:1996–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, Zhu Y, Hu L, Yan F and Chen J. miR-494 induces EndMT and promotes the development of HCC (Hepatocellular Carcinoma) by targeting SIRT3/TGF-β/SMAD signaling pathway. Sci Rep. 2019;9:7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park DH and Thum T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler Thromb Vasc Biol. 2012;32:361–9. [DOI] [PubMed] [Google Scholar]
- 48.Vanchin B, Offringa E, Friedrich J, Brinker MG, Kiers B, Pereira AC, Harmsen MC, Moonen JA and Krenning G. MicroRNA-374b induces endothelial-to-mesenchymal transition and early lesion formation through the inhibition of MAPK7 signaling. The Journal of pathology. 2019;247:456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki HI. MicroRNA Control of TGF-β Signaling. International journal of molecular sciences. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J MicroRNAs as critical regulators of the endothelial to mesenchymal transition in vascular biology. BMB reports. 2018;51:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krenning G, Barauna VG, Krieger JE, Harmsen MC and Moonen JR. Endothelial Plasticity: Shifting Phenotypes through Force Feedback. Stem cells international. 2016;2016:9762959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, Mammoto A, Chen J, Ariaans M, Francis SE, Van der Heiden K, Ridger V and Evans PC. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci Rep. 2017;7:3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desai TR, Leeper NJ, Hynes KL and Gewertz BL. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. The Journal of surgical research. 2002;104:118–23. [DOI] [PubMed] [Google Scholar]
- 54.Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T, Okawa K, Iwamatsu A and Kita T. Cutting edge: combined treatment of TNF-alpha and IFN-gamma causes redistribution of junctional adhesion molecule in human endothelial cells. Journal of immunology (Baltimore, Md : 1950). 1999;163:553–7. [PubMed] [Google Scholar]
- 55.Chen PY, Schwartz MA and Simons M. Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Frontiers in cardiovascular medicine. 2020;7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Michaelis UR. Mechanisms of endothelial cell migration. Cellular and molecular life sciences : CMLS. 2014;71:4131–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Niessen K, Fu Y, Chang L, Hoodless PA, McFadden D and Karsan A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. 2008;182:315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welch-Reardon KM, Ehsan SM, Wang K, Wu N, Newman AC, Romero-Lopez M, Fong AH, George SC, Edwards RA and Hughes CC. Angiogenic sprouting is regulated by endothelial cell expression of Slug. J Cell Sci. 2014;127:2017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamada KM and Sixt M. Mechanisms of 3D cell migration. Nature reviews Molecular cell biology. 2019;20:738–752. [DOI] [PubMed] [Google Scholar]
- 60.De Smet F, Tembuyser B, Lenard A, Claes F, Zhang J, Michielsen C, Van Schepdael A, Herbert JM, Bono F, Affolter M, Dewerchin M and Carmeliet P. Fibroblast growth factor signaling affects vascular outgrowth and is required for the maintenance of blood vessel integrity. Chem Biol. 2014;21:1310–1317. [DOI] [PubMed] [Google Scholar]
- 61.Xiao L and Dudley AC. Fine-tuning vascular fate during endothelial-mesenchymal transition. J Pathol. 2017;241:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fafeur V, Terman BI, Blum J and Bohlen P. Basic FGF treatment of endothelial cells down-regulates the 85-KDa TGF beta receptor subtype and decreases the growth inhibitory response to TGF-beta 1. Growth Factors. 1990;3:237–45. [DOI] [PubMed] [Google Scholar]
- 63.Ishisaki A, Hayashi H, Li AJ and Imamura T. Human umbilical vein endothelium-derived cells retain potential to differentiate into smooth muscle-like cells. The Journal of biological chemistry. 2003;278:1303–9. [DOI] [PubMed] [Google Scholar]
- 64.Wang Z, Calpe B, Zerdani J, Lee Y, Oh J, Bae H, Khademhosseini A and Kim K. High-throughput investigation of endothelial-to-mesenchymal transformation (EndMT) with combinatorial cellular microarrays. Biotechnology and bioengineering. 2016;113:1403–12. [DOI] [PubMed] [Google Scholar]
- 65.Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, Ali R, Medina PP, Yu J, Slack FJ, Anderson DG, Kotelianski V, Wang F, Tellides G and Simons M. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell reports. 2012;2:1684–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen PY, Qin L, Tellides G and Simons M. Fibroblast growth factor receptor 1 is a key inhibitor of TGFbeta signaling in the endothelium. Sci Signal. 2014;7:ra90. [DOI] [PubMed] [Google Scholar]
- 67.Shapero K, Wylie-Sears J, Levine RA, Mayer JE Jr. and Bischoff J. Reciprocal interactions between mitral valve endothelial and interstitial cells reduce endothelial-to-mesenchymal transition and myofibroblastic activation. J Mol Cell Cardiol. 2015;80:175–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo X and Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G and Baker AH. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73:190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L and Rabinovitch M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation. 2016;133:1783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, Fergusson MM, Yu ZX, Rovira II, Brittain EL, Wolfgang MJ, Jurczak MJ, Fessel JP and Finkel T. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol Cell. 2018;69:689–698 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E and Fiocchi C. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S and Dejana E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. The Journal of cell biology. 2004;166:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balachandran K, Alford PW, Wylie-Sears J, Goss JA, Grosberg A, Bischoff J, Aikawa E, Levine RA and Parker KK. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci U S A. 2011;108:19943–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Handschumacher MD, Sullivan S, Johnson B, Titus JS, Iwamoto Y, Wylie-Sears J, Levine RA and Carpentier A. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation. 2009;120:334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I and Hatzopoulos AK. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech. 2011;4:469–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wylie-Sears J, Aikawa E, Levine RA, Yang JH and Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Hjortnaes J, Beaudoin J, Szymanski C, Bartko PE, Seybolt MM, Handschumacher MD, Sullivan S, Garcia ML, Mauskapf A, Titus JS, Wylie-Sears J, Irvin WS, Chaput M, Messas E, Hagege AA, Carpentier A, Levine RA and Leducq Transatlantic Mitral N. Myocardial Infarction Alters Adaptation of the Tethered Mitral Valve. J Am Coll Cardiol. 2016;67:275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bartko PE, Dal-Bianco JP, Guerrero JL, Beaudoin J, Szymanski C, Kim DH, Seybolt MM, Handschumacher MD, Sullivan S, Garcia ML, Titus JS, Wylie-Sears J, Irvin WS, Messas E, Hagege AA, Carpentier A, Aikawa E, Bischoff J and Levine RA. Effect of Losartan on Mitral Valve Changes After Myocardial Infarction. J Am Coll Cardiol. 2017;70:1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beaudoin J, Dal-Bianco JP, Aikawa E, Bischoff J, Guerrero JL, Sullivan S, Bartko PE, Handschumacher MD, Kim DH, Wylie-Sears J, Aaron J and Levine RA. Mitral Leaflet Changes Following Myocardial Infarction: Clinical Evidence for Maladaptive Valvular Remodeling. Circ Cardiovasc Imaging. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang H, Lui KO and Zhou B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ Res. 2018;122:774–789. [DOI] [PubMed] [Google Scholar]
- 82.Brown CB, Boyer AS, Runyan RB and Barnett JV. Requirement of type III TGF-beta receptor for endocardial cell transformation in the heart. Science. 1999;283:2080–2. [DOI] [PubMed] [Google Scholar]
- 83.Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d'Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M and Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Das S, Goldstone AB, Wang H, Farry J, D'Amato G, Paulsen MJ, Eskandari A, Hironaka CE, Phansalkar R, Sharma B, Rhee S, Shamskhou EA, Agalliu D, de Jesus Perez V, Woo YJ and Red-Horse K. A Unique Collateral Artery Development Program Promotes Neonatal Heart Regeneration. Cell. 2019;176:1128–1142 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maleszewska M, Moonen JR, Huijkman N, van de Sluis B, Krenning G and Harmsen MC. IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology. 2013;218:443–54. [DOI] [PubMed] [Google Scholar]
- 86.Cheng F, Pekkonen P, Laurinavicius S, Sugiyama N, Henderson S, Gunther T, Rantanen V, Kaivanto E, Aavikko M, Sarek G, Hautaniemi S, Biberfeld P, Aaltonen L, Grundhoff A, Boshoff C, Alitalo K, Lehti K and Ojala PM. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell Host Microbe. 2011;10:577–90. [DOI] [PubMed] [Google Scholar]
- 87.Ichise T, Yoshida N and Ichise H. FGF2-induced Ras-MAPK signalling maintains lymphatic endothelial cell identity by upregulating endothelial-cell-specific gene expression and suppressing TGFbeta signalling through Smad2. J Cell Sci. 2014;127:845–57. [DOI] [PubMed] [Google Scholar]
- 88.Man S, Sanchez Duffhues G, Ten Dijke P and Baker D. The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis. 2019;22:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tombor LS, John D, Glaser SF, Luxán G, Forte E, Furtado M, Rosenthal N, Baumgarten N, Schulz MH, Wittig J, Rogg EM, Manavski Y, Fischer A, Muhly-Reinholz M, Klee K, Looso M, Selignow C, Acker T, Bibli SI, Fleming I, Patrick R, Harvey RP, Abplanalp WT and Dimmeler S. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun. 2021;12:681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gomez-Puerto MC, Iyengar PV, García de Vinuesa A, Ten Dijke P and Sanchez-Duffhues G. Bone morphogenetic protein receptor signal transduction in human disease. The Journal of pathology. 2019;247:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mahler GJ, Farrar EJ and Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim AJ, Alfieri CM and Yutzey KE. Endothelial Cell Lineage Analysis Does Not Provide Evidence for EMT in Adult Valve Homeostasis and Disease. Anatomical record (Hoboken, NJ : 2007). 2019;302:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG and Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet (London, England). 2006;368:1005–11. [DOI] [PubMed] [Google Scholar]
- 94.Grigioni F, Detaint D, Avierinos JF, Scott C, Tajik J and Enriquez-Sarano M. Contribution of ischemic mitral regurgitation to congestive heart failure after myocardial infarction. Journal of the American College of Cardiology. 2005;45:260–7. [DOI] [PubMed] [Google Scholar]
- 95.Grigioni F, Enriquez-Sarano M, Zehr KJ, Bailey KR and Tajik AJ. Ischemic mitral regurgitation: long-term outcome and prognostic implications with quantitative Doppler assessment. Circulation. 2001;103:1759–64. [DOI] [PubMed] [Google Scholar]
- 96.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP and Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xian S, Chen A, Wu X, Lu C, Wu Y, Huang F and Zeng Z. Activation of activin/Smad2 and 3 signaling pathway and the potential involvement of endothelial-mesenchymal transition in the valvular damage due to rheumatic heart disease. Molecular medicine reports. 2021;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fernandez Esmerats J, Villa-Roel N, Kumar S, Gu L, Salim MT, Ohh M, Taylor WR, Nerem RM, Yoganathan AP and Jo H. Disturbed Flow Increases UBE2C (Ubiquitin E2 Ligase C) via Loss of miR-483-3p, Inducing Aortic Valve Calcification by the pVHL (von Hippel-Lindau Protein) and HIF-1α (Hypoxia-Inducible Factor-1α) Pathway in Endothelial Cells. Arterioscler Thromb Vasc Biol. 2019;39:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ma X, Zhao D, Yuan P, Li J, Yun Y, Cui Y, Zhang T, Ma J, Sun L, Ma H, Zhang Y, Zhang H, Zhang W, Huang J, Zou C and Wang Z. Endothelial-to-Mesenchymal Transition in Calcific Aortic Valve Disease. Acta Cardiologica Sinica. 2020;36:183–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goody PR, Hosen MR, Christmann D, Niepmann ST, Zietzer A, Adam M, Bönner F, Zimmer S, Nickenig G and Jansen F. Aortic Valve Stenosis: From Basic Mechanisms to Novel Therapeutic Targets. Arterioscler Thromb Vasc Biol. 2020;40:885–900. [DOI] [PubMed] [Google Scholar]
- 101.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA and Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. The Journal of clinical investigation. 2015;125:4514–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moonen JR, Lee ES, Schmidt M, Maleszewska M, Koerts JA, Brouwer LA, van Kooten TG, van Luyn MJ, Zeebregts CJ, Krenning G and Harmsen MC. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res. 2015;108:377–86. [DOI] [PubMed] [Google Scholar]
- 103.Boström KI, Yao J, Guihard PJ, Blazquez-Medela AM and Yao Y. Endothelial-mesenchymal transition in atherosclerotic lesion calcification. Atherosclerosis. 2016;253:124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Helmke A, Casper J, Nordlohne J, David S, Haller H, Zeisberg EM and von Vietinghoff S. Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. Faseb j. 2019;33:2278–2289. [DOI] [PubMed] [Google Scholar]
- 105.Chevalier J, Yin H, Arpino JM, O'Neil C, Nong Z, Gilmore KJ, Lee JJ, Prescott E, Hewak M, Rice CL, Dubois L, Power AH, Hamilton DW and Pickering JG. Obstruction of Small Arterioles in Patients with Critical Limb Ischemia due to Partial Endothelial-to-Mesenchymal Transition. iScience. 2020;23:101251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Andueza A, Kumar S, Kim J, Kang DW, Mumme HL, Perez JI, Villa-Roel N and Jo H. Endothelial Reprogramming by Disturbed Flow Revealed by Single-Cell RNA and Chromatin Accessibility Study. Cell reports. 2020;33:108491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Southgate L, Machado RD, Gräf S and Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nat Rev Cardiol. 2020;17:85–95. [DOI] [PubMed] [Google Scholar]
- 108.Hautefort A, Mendes-Ferreira P, Sabourin J, Manaud G, Bertero T, Rucker-Martin C, Riou M, Adão R, Manoury B, Lambert M, Boet A, Lecerf F, Domergue V, Brás-Silva C, Gomez AM, Montani D, Girerd B, Humbert M, Antigny F and Perros F. Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension. Circulation. 2019;139:932–948. [DOI] [PubMed] [Google Scholar]
- 109.Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ and Holmes AM. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am J Pathol. 2015;185:1850–8. [DOI] [PubMed] [Google Scholar]
- 110.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S and Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. 2015;131:1006–18. [DOI] [PubMed] [Google Scholar]
- 111.Theilmann AL, Hawke LG, Hilton LR, Whitford MKM, Cole DV, Mackeil JL, Dunham-Snary KJ, Mewburn J, James PD, Maurice DH, Archer SL and Ormiston ML. Endothelial BMPR2 Loss Drives a Proliferative Response to BMP (Bone Morphogenetic Protein) 9 via Prolonged Canonical Signaling. Arterioscler Thromb Vasc Biol. 2020;40:2605–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Szulcek R, Sanchez-Duffhues G, Rol N, Pan X, Tsonaka R, Dickhoff C, Yung LM, Manz XD, Kurakula K, Kiełbasa SM, Mei H, Timens W, Yu PB, Bogaard HJ and Goumans MJ. Exacerbated inflammatory signaling underlies aberrant response to BMP9 in pulmonary arterial hypertension lung endothelial cells. Angiogenesis. 2020;23:699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Suzuki T, Carrier EJ, Talati MH, Rathinasabapathy A, Chen X, Nishimura R, Tada Y, Tatsumi K and West J. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2018;314:L118–l126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Suzuki T, Tada Y, Nishimura R, Kawasaki T, Sekine A, Urushibara T, Kato F, Kinoshita T, Ikari J, West J and Tatsumi K. Endothelial-to-mesenchymal transition in lipopolysaccharide-induced acute lung injury drives a progenitor cell-like phenotype. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1185–98. [DOI] [PubMed] [Google Scholar]
- 115.Cuttano R, Rudini N, Bravi L, Corada M, Giampietro C, Papa E, Morini MF, Maddaluno L, Baeyens N, Adams RH, Jain MK, Owens GK, Schwartz M, Lampugnani MG and Dejana E. KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol Med. 2016;8:6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou Z, Tang AT, Wong WY, Bamezai S, Goddard LM, Shenkar R, Zhou S, Yang J, Wright AC, Foley M, Arthur JS, Whitehead KJ, Awad IA, Li DY, Zheng X and Kahn ML. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature. 2016;532:122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li J, Zhao Y, Coleman P, Chen J, Ting KK, Choi JP, Zheng X, Vadas MA and Gamble JR. Low fluid shear stress conditions contribute to activation of cerebral cavernous malformation signalling pathways. Biochimica et biophysica acta Molecular basis of disease. 2019;1865:165519. [DOI] [PubMed] [Google Scholar]
- 118.Piera-Velazquez S and Jimenez SA. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiological reviews. 2019;99:1281–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu M, Peng Z, Zu C, Ma J, Lu S, Zhong J and Zhang S. Losartan Attenuates Myocardial Endothelial-To-Mesenchymal Transition in Spontaneous Hypertensive Rats via Inhibiting TGF-beta/Smad Signaling. PLoS One. 2016;11:e0155730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popović Z, Huang P, Bawendi MG, Boucher Y and Jain RK. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhao Y, Cao J, Melamed A, Worley M, Gockley A, Jones D, Nia HT, Zhang Y, Stylianopoulos T, Kumar AS, Mpekris F, Datta M, Sun Y, Wu L, Gao X, Yeku O, Del Carmen MG, Spriggs DR, Jain RK and Xu L. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Natl Acad Sci U S A. 2019;116:2210–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Guo Y, Li P, Bledsoe G, Yang ZR, Chao L and Chao J. Kallistatin inhibits TGF-beta-induced endothelial-mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Experimental cell research. 2015;337:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, Ishigaki Y, Kitada M, Srivastava SP and Koya D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63:2120–31. [DOI] [PubMed] [Google Scholar]
- 124.Cipriani P, Di Benedetto P, Ruscitti P, Capece D, Zazzeroni F, Liakouli V, Pantano I, Berardicurti O, Carubbi F, Pecetti G, Turricchia S, Alesse E, Iglarz M and Giacomelli R. The Endothelial-mesenchymal Transition in Systemic Sclerosis Is Induced by Endothelin-1 and Transforming Growth Factor-beta and May Be Blocked by Macitentan, a Dual Endothelin-1 Receptor Antagonist. The Journal of rheumatology. 2015;42:1808–16. [DOI] [PubMed] [Google Scholar]
- 125.Choi KJ, Nam JK, Kim JH, Choi SH and Lee YJ. Endothelial-to-mesenchymal transition in anticancer therapy and normal tissue damage. Experimental & molecular medicine. 2020;52:781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manavski Y, Lucas T, Glaser SF, Dorsheimer L, Günther S, Braun T, Rieger MA, Zeiher AM, Boon RA and Dimmeler S. Clonal Expansion of Endothelial Cells Contributes to Ischemia-Induced Neovascularization. Circ Res. 2018;122:670–677. [DOI] [PubMed] [Google Scholar]
- 127.van Meeteren LA and ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell and tissue research. 2012;347:177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li Y, Lui KO and Zhou B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. 2018;15:445–456. [DOI] [PubMed] [Google Scholar]