

Abstract
Subcellular compartmentalization of macromolecules increases flux and prevents inhibitory interactions to control biochemical reactions. Inspired by this functionality, we sought to build designer compartments that function as hubs to regulate the flow of information through cellular control systems. We report a synthetic membraneless organelle platform to control endogenous cellular activities through sequestration and insulation of native proteins. We engineer and express a disordered protein scaffold to assemble micron size condensates and recruit endogenous clients via genomic tagging with high-affinity dimerization motifs. By relocalizing up to ninety percent of a targeted enzymes to synthetic condensates, we efficiently control cellular behaviors, including proliferation, division, and cytoskeletal organization. Further, we demonstrate multiple strategies for controlled cargo release from condensates to switch cells between functional states. These synthetic organelles offer a powerful and generalizable approach to modularly control cell decision-making in a variety of model systems with broad applications for cellular engineering.
Keywords: Compartmentalization, membraneless organelles, disordered protein, RGG domain, synthetic condense, programmable behavior, mesoscale protein switch
INTRODUCTION
Cells can enhance the rate and fidelity of biochemical reactions through subcellular compartmentalization1. For example, membrane bound organelles such as the nucleus and lysosome display highly selective partitioning of biological cargo. Their restricted permeability increases reactivity via enforced proximity and ensures specificity by insulating components from competing reactions2–6. Cells also contain membraneless organelle subcompartments such as the nucleolus and P granules, that form through the self-assembly and coacervation of disordered proteins and RNA into mesoscale biomolecular condensates7. By harnessing principles of protein self-assembly, it is possible to construct nano- or micro-compartments inside a cell that encapsulate enzymes and substrates to control or augment their functions in living systems8–10. One such strategy has been to assemble designer compartments that co-localize components to enhance reaction rates of exogenous pathways11, 12. A second important strategy, the use of synthetic organelles to sequester user-defined, native proteins for control of cellular decision-making, has yet to be demonstrated.
Synthetic condensates or membraneless organelles can be assembled in a cell from expression of disordered protein sequences above their saturation concentration. Low-complexity sequences from Fus and other FET family members, Reslin-like sequences, and RGG domains from Laf-1 have been used to generate synthetic condensates in bacterial, yeast, and mammalian systems13–18. We previously showed the utility of a disordered protein platform for generating condensates in vitro, in synthetic-cell like comparments18. The 168 aa disordered Arg/Gly rich (RGG) domain of C. elegans P granule protein, LAF-1, is necessary and sufficient for phase separation and does not require RNA for self-assembly18–20. Importantly, the valency of RGG domain tunes the critical concentration for liquid-liquid phase separation (LLPS) and real-time reduction of valency promotes condensate disassembly. Further, enzymatic and optical release of a solubilization domain from RGG initiates condensate assembly21, 22. In addition, transient expression in cells leads to formation of liquid-like micron size condensates18.
In living cells, biomolecular condensates and membraneless organelles sequester client enzymes or RNAs to either increase enzymatic flux or to insulate them from other cellular machinery. For example, in response to various stresses, mammalian cells form stress granules to sequester proteins, RNA, and elongation factors, a response that prevents stress-induced cellular senescence23. Guided by this insulation mechanism we sought to develop our own synthetic membraneless organelle platform that functions to sequester and insulate native enzymes for modular control over cellular functions. For these designer organelles to have broad utility in cell biology and engineering applications, they should exhibit restricted permeability, highly selective and efficient enrichment of specific cargos, and be capable of controllable client release. Throughout this article, we refer to our platform as synthetic organelles or condensates interchangeably.
Enforced localization of exogenously expressed clients in cells has been demonstrated using synthetic condensate systems11, 13, 15, 18, 24. A common strategy tags the exogenous client with the same disordered protein sequence domain present on the IDP scaffold to direct partitioning to synthetic condensates12, 15, 16, 18. However, concerns arise about integrating large, disordered domains into endogenous gene loci, particularly whether they are orthogonal or may alter endogenous protein functionality. Further, it is not clear whether this IDP-tagging approach is generalizable and capable of sequestering a majority of the endogenously expressed target protein in the cell. Therefore, a substantial advance would be the development of a synthetic condensate platform in which a majority of the scaffold protein partitions to the condensate to achieve high fractional client recruitment. Combined with a modular strategy for localizing clients without disrupting their native function, for example using coiled coil interaction motifs, a key capability would be functional insulation of native enzymes. An additional engineering demand is reversibility of client recruitment, enabling controlled release from a designer organelle to restore pathway function and switch cells between functional states.
In this study, we developed such a synthetic membraneless organelle system to insulate and functionally knockdown essential native enzymes via compartmentalization and achieve modular control of cellular behavior. We demonstrate successful engineering of a number of platform functions: we achieve nearly full partitioning of scaffold and native clients to the synthetic organelle by screening through IDP valencies and recruitment tags. By genomic tagging of native gene loci, we show functional insulation of enzymes that regulate the cell cycle control system and actin cytoskeleton, thereby switching cells from proliferation to arrested states, and from polarized to isotropic cytoskeletal organization. We demonstrate the feasibility of rapid induced client recruitment and switching of cell behavior. Further, we demonstrate thermal and optical strategies for controlled release of clients localized to the synthetic organelle, for reversible control of the cell activity state. Finally, we demonstrate the feasibility of implementing this platform in mammalian cells by CRISPR-tagging endogenous gene loci to efficiently partition and relocalize native enzymes. We propose that this designer membraneless organelle system, embedded with interaction tags, offers a powerful and generalizable chemical biology tool for controlling cellular activities. The applications of our approach range from real-time probing of pathways in cell biology to mesoscale protein switches for cellular engineering and synthetic biology.
RESULTS
Targeting clients to synthetic membraneless organelles
Our first goal was to augment living cells with synthetic compartments, screening them for temperature stability and critical concentration to achieve a high fraction of IDP scaffold in condensates. Constructs containing a single RGG domain have poor LLPS activity in vivo, consistent with previous in vitro findings (Extended Data Fig. 1a, b, c)18. Addition of a second RGG domain allowed condensate formation at 25°C, but was not stable at higher temperatures (Extended Data Fig. 1a, c). A scaffold encoding three RGG domains, however, allowed for robust condensate formation and stability over a wide range of temperatures (Extended Data Fig. 1a, b, c). Importantly, these in vivo structures maintained liquid-like features (Extended Data Fig. 1d, e).
Our second goal was to test various protein interaction motifs for tagging clients to stably or reversibly enforce their proximity to our synthetic organelle (Fig. 1a). We encoded cognate interaction motifs on the N-terminus of the IDP scaffold protein and C-terminus of client proteins. Our testing set included (i) short coiled-coil SYNZIP pairs (SZ1, SZ2), (ii) thermally reversible coiled coil domains, TsCC(A) and (B), which are shortened forms of a bacterial thermometer (TlpA) engineered to form heterodimers25 whose DNA binding domain has been removed, and (iii) small molecule inducible dimerization domains, FRB and FKBP (Fig. 1b).
Figure 1. Robust cargo recruitment to synthetic condensates via protein-protein interaction domains.
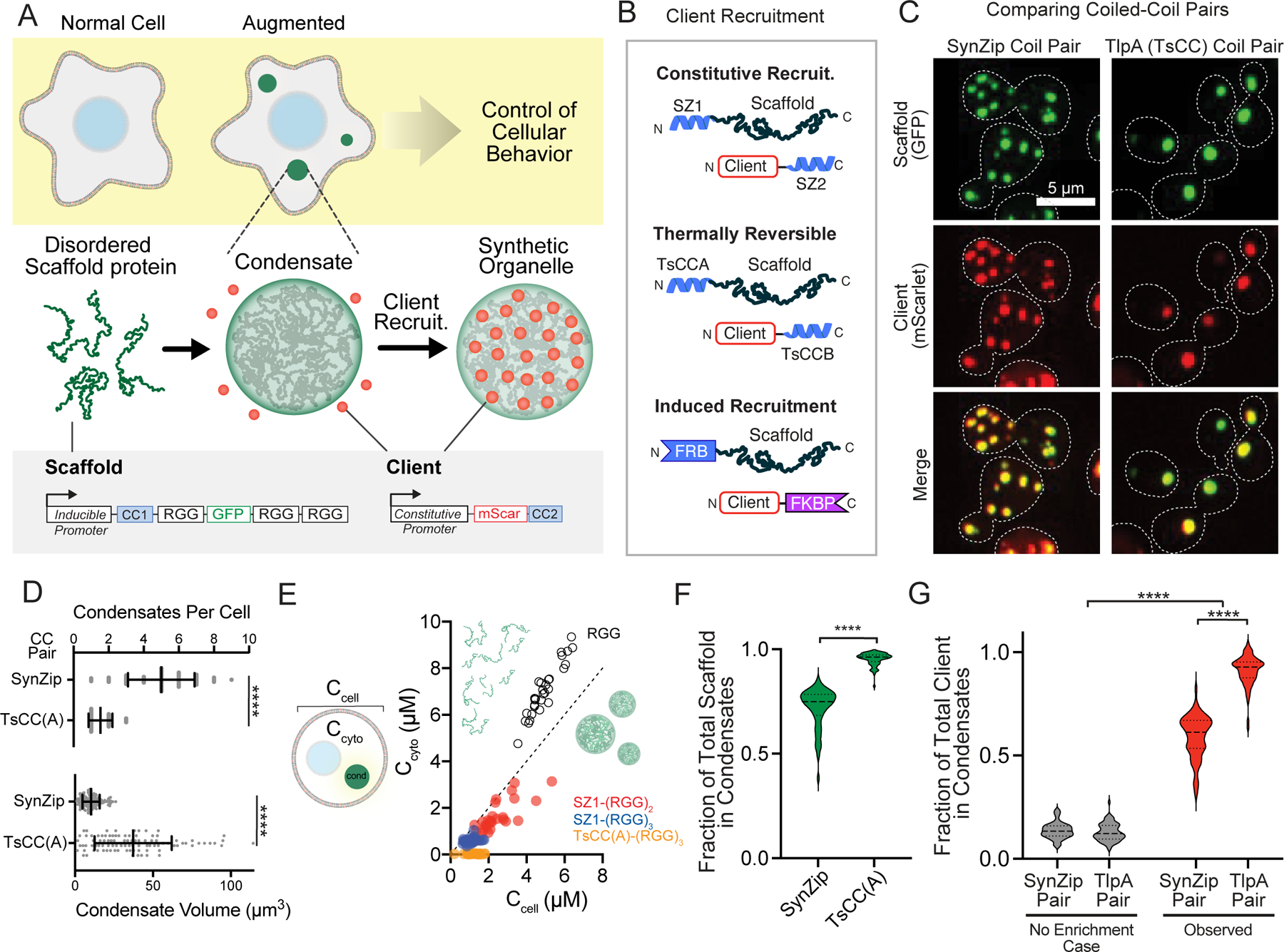
a, Schematic overview assembling synthetic organelles from disordered scaffold proteins to target clients and predictably modulate cellular functions. Scaffold: triple RGG, GFP tag, and a high affinity coiled-coil (CC) tag under the control of an inducible promoter. Client: fluorophore with a cognate CC under the control of a constitutive promoter. b, Client recruitment strategies: Synzips, thermally responsive coiled-coils, and rapamycin-induced dimerization domains. c, Representative images of yeast cell expressing a scaffold and client with cognate Synzip (left) or thermally responsive (right) CC pairs. Merged images show strong recruitment of client to condensates. d, Comparison of mean condensate number (top) and volume (bottom) for each scaffold type n = 60 cells for SynZip and n = 75 cells for TsCC(A) scaffolds. Error bars, s.d. Significance calculated by unpaired, two-tailed t-test (****, p ≤ 0.0001). e, Steady-state cytoplasmic concentration of scaffold outside of condensates (Ccyto) as a function of total cellular concentration (Ccell) for 30 cells per scaffold type. Dashed line, slope of 1. f, Violin plots of fraction of total scaffold protein present in condensates for cells as in d. Significance calculated by unpaired, two-tailed t-test (****, p ≤ 0.0001). g, Violin plots of fraction of total client recruitment to condensates with each CC pair compared to expected percentage for condensates of same size without recruitment in cells as in d. Significance calculated by one-way ANOVA (****, p ≤ 0.0001).
Next, we determined the in vivo phase boundaries for the various RGG scaffolds and characterized the number and size of condensates per cell. When fused to N-terminal SZ1 coiled, the (RGG)3-GFP scaffold formed an average of 5 condensates per cell (Fig. 1c, d). Addition of the TsCC(A) domain to (RGG)3-GFP scaffolds led to fusion and formation of 1–2 large condensates per cell (Fig. 1c, d). To better evaluate the phase behavior of these condensates in vivo, we measured the intracellular phase boundaries for scaffolds containing various RGG domains and tags (Fig. 1e). A SZ1-(RGG)2-GFP scaffold protein has a saturation concentration (Csat) of approximately 1610 nM. Addition of a third RGG domain lowered the Csat to ~ 600 nM, in agreement with previous in vitro findings18. And the TsCC(A)-(RGG)3-GFP scaffold has an even further reduction of Csat to ~ 29 nM, likely due to some coiled coil homodimerization activity (Fig. 1e, Extended Data Fig. 1f).
The steady-state fraction of scaffold protein that will partition to the condensate versus remain in the cytosol is determined by the Csat and protein expression levels. This parameter is essential because it may impact the fraction of client recruited via cognate interaction motifs. We measured the fraction of total scaffold and client integrated intensity present in cells after inducing scaffold expression. We found that over 95% of total TsCC(A)-(RGG)3-GFP scaffold protein and approximately 72% of total SZ1-(RGG)3-GFP scaffold protein localized to condensates (Fig. 1f, Extended Data Fig. 1g). Importantly, while an exogenously expressed mScarlet client fused to an interaction motif is diffusely localized through the cell (Extended Data Fig. 1h), expression of a scaffold with the cognate protein interaction motif results in robust localization of mScarlet to our synthetic condensates. Over 91% of the client tagged with TsCC(B) was recruited to TsCC(A)-(RGG)3-GFP condensates (Fig. 1g), demonstrating sequestration of a vast majority of a client protein in cells at room temperature, under normal growth conditions.
We also tested the feasibility of induced cargo recruitment. Our rationale was to allow a tagged client to localize and function normally in the presence of synthetic condensates under basal conditions and then rapidly induce dimerization and sequestration to the synthetic condensates. We fused FRB to the scaffold and FKBP to a client. In the absence of dimerizer, the tagged client diffused freely throughout the cytosol (Extended Data Fig. 1h), and upon the addition of rapamycin (Rap), the client was quickly relocalized to the condensates (Extended Data Fig. 1i, Supplementary Video 1). We found that this strategy could partition approximately 50% of cargo with a time for half maximal recruitment of ~ 12 minutes (Extended Data Fig. 1j).
Collectively, we achieved both stable and inducible client recruitment to our synthetic condensates and found that the TsCC(A)-(RGG)3-GFP scaffold was capable of recruiting over 90% of a client tagged with the cognate interaction motif. Based on these results we proceeded with the TsCC(A)-(RGG)3-GFP scaffold for sequestering native enzymes to control cell behaviors.
Control of cell behavior by sequestering native enzymes
We tested the utility of our synthetic membraneless organelle platform as a protein-based switch to regulate cell decision-making. To modulate both sides of the cell proliferation control system, we chose as targets for sequestration, the GEF Cdc24 and the kinase Cdc5 (Fig. 2a). Knockout or depletion of Cdc24 prevents polarized growth and proliferation and loss of Cdc5 prevents cells from undergoing cell division26–29. Our hypothesis was that by tagging these proteins with coiled coils at their genomic loci, we would sequester a sufficient fraction of the endogenous enzyme to our designer condensates, functionally insulating them and preventing pathway activity (Fig. 2b).
Figure 2. Control of cellular behavior through targeted insulation of native enzymes in synthetic organelles.
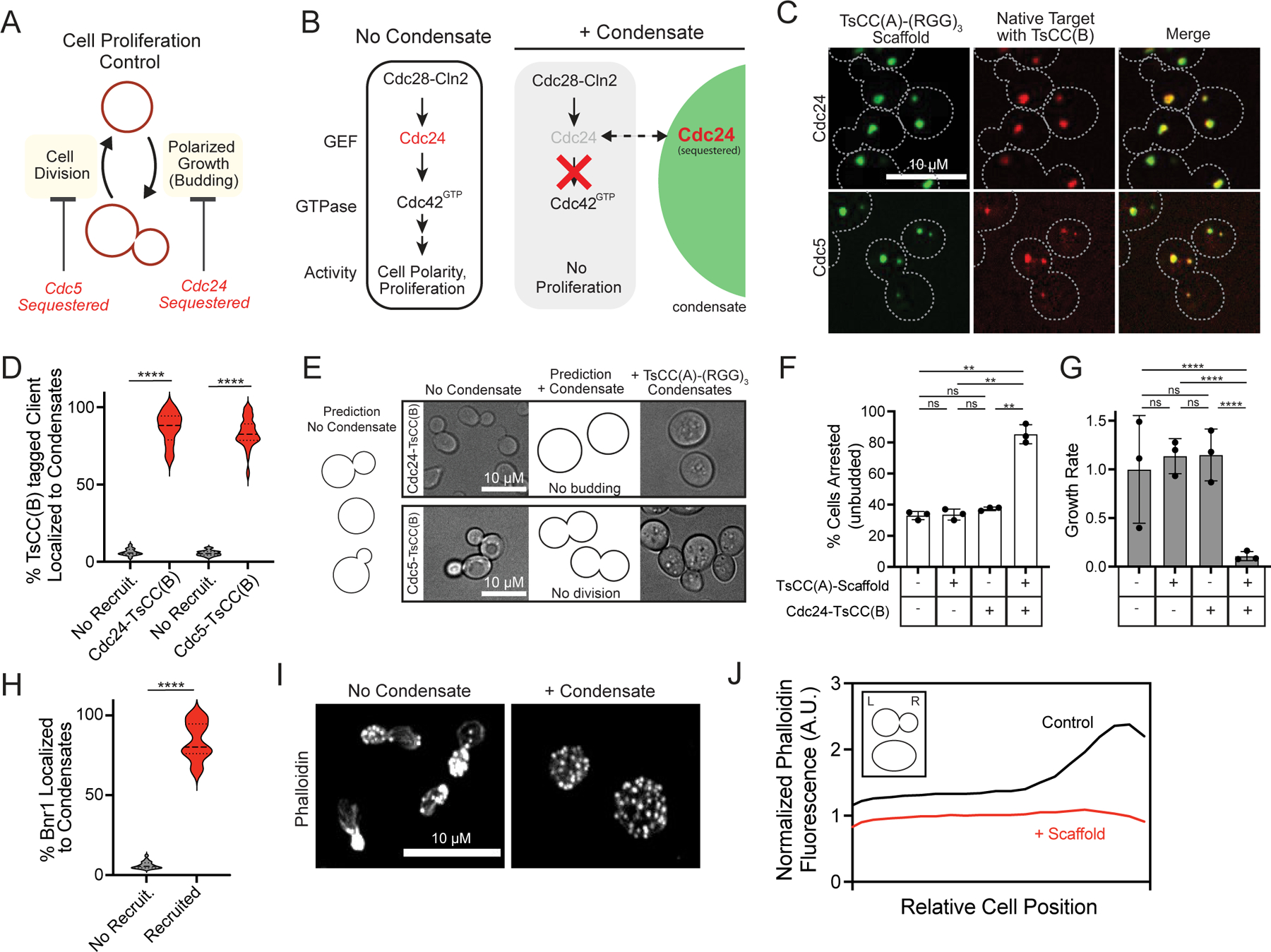
a, Scheme for regulation of cell cycle control system. b, Hypothesis: insulation of Cdc24 within synthetic organelle blocks signaling and cell polarization; no proliferation. c, Representative images of cells expressing TsCC(A)-(RGG)3 scaffolds and tagged clients, showing native Cdc24-mScarlet-TsCC(B) and Cdc5-mScarlet-TsCC(B) enriched in synthetic condensates. d, Violin plots of fraction of total client protein in synthetic condensates compared to expected for condensates with no recruitment for 50 cells per strain from 3–5 fields of view (FOV). Significance calculated by unpaired, two-tailed t-test (****, p ≤ 0.001). e, Predictions and representative brightfield images of cell morphologies for tagged Cdc24 (top) and Cdc5 (bottom), before (left) and after (right) inducing expression of scaffold to form condensates. f, Percentage of cells arrested (unbudded) in the indicated strains, n=150 cells per strain pooled from three independent trials. Error bars, s.d. Significance calculated by one-way ANOVA (ns, no significance; **, p ≤ 0.01). g, Growth rate in liquid culture for indicated strains as in f, averaged from three independent trials. Error bars, s.d. Significance calculated by linear regression (ns, no significance; ****, p ≤ 0.0001). h, Violin plots of fraction of total Bnr1-mScarlet-TsCC(B) in condensates compared to expected for condensates with no recruitment; 50 cells from 305 FOVs. i, Representative images of phalloidin stained Bnr1ΔDAD-mScarlet-TsCC(B) bni1Δ cells with or without condensates shows altered spatial organization of actin cytoskeleton upon Brn1 sequestration. j, Distribution of average phalloidin fluorescence across cell body for 50 cells/condition from 5 FOVs. Cells lose polarization. Inset: diagram of cell orientation.
Tagging with a fluorophore and the TsCC(B) coiled coil does not affect the normal localization of Cdc24 to polarity sites like the yeast bud neck and tip or that of Cdc5 at spindle pole bodies30, 31 (Extended Data Fig. 2a, b). Natively expressed Cdc24-mScarlet-TsCC(B) is strongly recruited to condensates formed from TsCC(A)-(RGG)3-GFP expression (Fig. 2c, Extended Data Fig. 2c), but not recruited to control scaffolds that lack the cognate coiled coil tag (Extended Data Fig. 2d). Enforced localization of Cdc24-mScarlet-TsCC(B) to the synthetic condensates competes it away from its native localization sites (Extended Data Fig. 2e) and this relocalization can be observed in real-time upon induced expression of the scaffold (Extended Data Fig. 2f, Supplementary Video 2). Importantly, both tagged Cdc24 and tagged Cdc5 are efficiently recruited (Fig. 2c, d), demonstrating greater than 86% and 83% of the native enzymes, respectively, can be sequestered within condensates.
The behavior of cells containing endogenously tagged clients are dramatically altered by the expression of synthetic condensates functionalized with cognate recruitment tags. Cells containing tagged Cdc24 or Cdc5 grow and proliferate normally in the absence of TsCC(A)-(RGG)3-GFP condensates. However, their cell cycle control systems are blocked upon formation of condensates. Cdc24-mScarlet-TsCC(B) cells can no longer polarize or bud (Fig. 2e). Thus, localization of Cdc24 to condensates arrests cells (Fig. 2f, Extended Data Fig. 2g, h), leading to nearly 12-fold drop in the rate of cell proliferation in liquid culture (Fig. 2g, Extended Data Fig. 2g) and causes cells to significantly expand in area (Extended Data Fig. 2i). Importantly, only cells expressing both a tagged Cdc24 and TsCC(A)-(RGG)3-GFP scaffold show growth arrest; other cells behave similar to wildtype. Sequestration of Cdc5-mScarlet-TsCC(B) disrupts cell division, and cells remain dumbbell shaped (Fig. 2e). As a result, sequestration and functional insulation of Cdc5 also stalls cell proliferation (Extended Data Fig. 2j).
In addition to switching cell growth control, we wanted to test regulation of the spatial organization of the actin cytoskeleton by our synthetic condensates. To do this we targeted a yeast formin, Bnr1, which generates linear actin cables for intracellular trafficking and polarized cell growth32, 33. By tagging a native, constitutively active form of Bnr1 in a cell that otherwise lacks formins, we efficiently sequestered 83% of it to TsCC(A)-(RGG)3-GFP condensates34 (Fig. 2h). This functional insulation of the formin prevented normal formation of actin cables and spatial polarization of the cytoskeleton (Fig. 2i, j).
Finally, to demonstrate rapid, inducible recruitment of a native enzyme, we tagged the endogenous locus of Cdc24 with an FKBP tag in cells containing FRB-(RGG)3-GFP condensates (Fig. 3a). In the absence of dimerizer, the native enzyme localizes normally. Upon addition of Rap, Cdc24-mScarlet-FRB protein is relocalized to our synthetic condensates (Fig. 3b). Nearly 54% of the total cellular pool of tagged Cdc24 protein is sequestered to the synthetic organelle within approximately 10 minutes (Fig. 3c). Further, cell proliferation is effectively stalled in the presence of Rap and expressed condensates (Fig. 3d), whereas no phenotype is observed when the scaffold is expressed in the absence of dimerizer or when Rap is added to cells that lack condensates.
Figure 3. Control of cell proliferation by induced target sequestration.
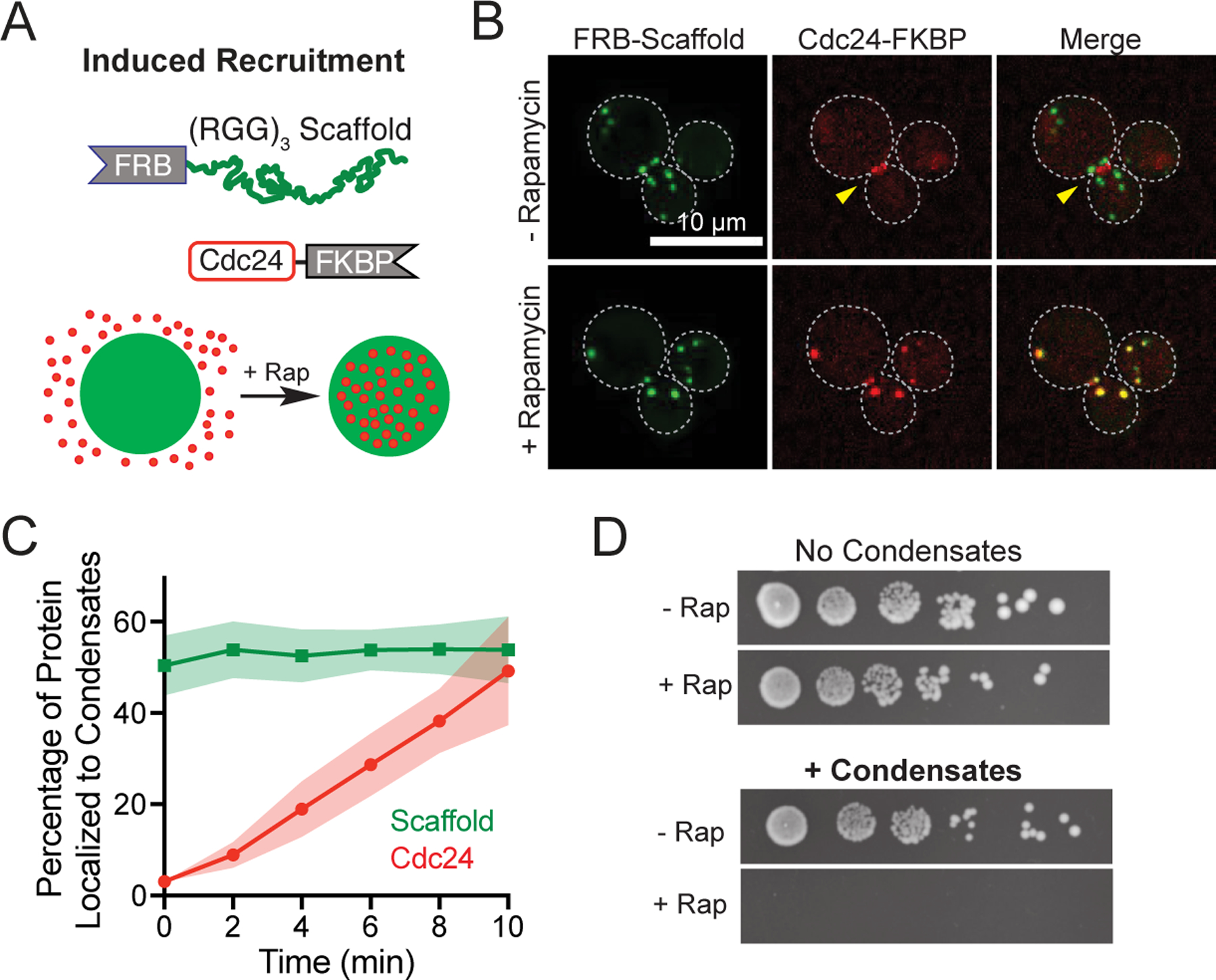
a, Scheme for rapamycin inducible client recruitment: Cdc24-mScarlet-FKBP to FRB-(RGG)3 scaffold condensates. b, Representative images of cells expressing Cdc24-mScarlet-FKBP and FRB-Scaffold before (top) and after (bottom) rapamycin addition. c, Quantification of fraction of scaffold and client (Cdc24) in synthetic condensates over time as in b, for 16 cells. Shaded area, 95% CI. d, Spot dilution assays of the same yeast strain grown, containing Cdc24-mScarlet-FKBP, either lacking or expressing condensates, without or with 100 nM rapamycin in the media.
These results demonstrate the utility of our disordered domain-based scaffold to generate orthogonal membraneless organelles in vivo. With the addition of high-affinity coiled coil interaction domains or inducible recruitment tags, endogenous clients are effectively sequestered and insulated in membraneless organelles. As a result, we demonstrate modular control over cell decision making via designer compartments.
Controlled release of clients from synthetic condensates
Having demonstrated efficient functional insulation of endogenous enzymes to synthetic organelles, we next sought to develop handles for controlled intracellular release. By utilizing the thermally response TsCC(A)-TsCC(B) coiled coil interaction pair, we hypothesized that client recruitment would be reversed above a critical temperature (Fig. 4a, b). We used Cdc24-mScarlet-TsCC(B) cells and expressed the cognate scaffold for 6 hours at room temperature, during which the client was sequestered and cells arrested while control cells that do not express the scaffold were unresponsive (Fig. 4c). We then reversed client recruitment by raising the temperature to 37°C or 42°C (Fig. 4c), temperatures known to dissociate the heterodimer pair in in vitro and in vivo25. Strikingly, we found that thermal induction successfully reversed the arrest phenotype of Cdc24-mScarlet-TsCC(B) cells expressing TsCCA-(RGG)3-GFP condensates (Fig. 4c). This reversal was dose-dependent and concomitant with a reduction of Cdc24 sequestered to the organelle (Fig. 4d); higher temperature restored nearly wild-type levels of polarized cells. Additionally, temperature reversal of the phenotype was maintained overnight (Fig. 4c, Extended Data Fig. 3a), indicating that cells could polarize at these higher temperatures, sustaining client release.
Figure 4. Optical and thermal client release for reversible cell cycle control.
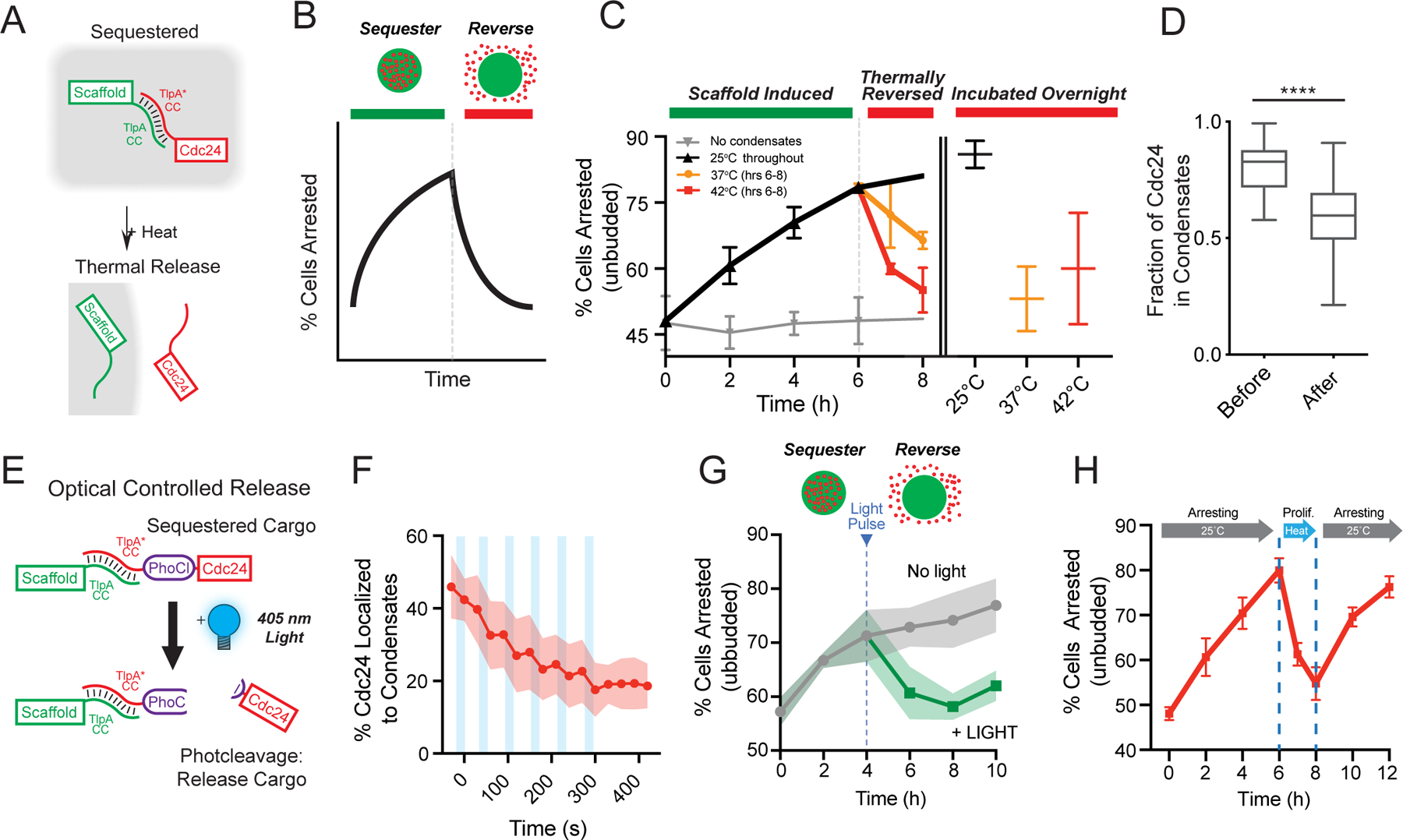
a, Schematic of cargo release using thermally responsive coiled coils. b, Schematic prediction of switch from cell arrest to proliferation upon release of Cdc24 from synthetic condensates. c, Average percentage of cells arrested over time. Scaffold expressed at 25°C to form synthetic condensates and induce arrest. After 6 hrs, cell heated to indicated temperatures to induce client release. Cells with no condensate expression represent baseline (light gray). Orange and red lines represent temperature increase to 37°C and 42°C, respectively for one and two hours (left), and overnight (right). Cells with scaffold expression maintained at 25°C (black). Data from three trials, n = 4369 cells in total. Error bars, s.d. d, Quantification of the fraction of Cdc24 in condensates before and after heating; 10 cells. e, Schematic: light-induced release of Cdc24 from condensates via PhoCl photocleavage on scaffold. f, Quantification of loss of Cdc24-mScarlet-TsCC(B) signal from condensates after short pulses of illumination (light blue bars, 10 sec each). n = 11 cells. g, Percentage of cells arrested over time after inducing synthetic condensate expression, comparing no illumination to 10 min UV light exposure of at 4 hr timepoint. n = 4928 cells pooled from three trials Shaded area, s.d. h, Cycling cells between arrest proliferation-arrest. 25°C from 0–6 hrs: scaffold expressed; 42°C for 6–8 hrs: client is released and arrest reversed; 25°C from 8–12 hrs: client re-recruited and arrest reimposed. Data are averaged from three trials and total of 4988 cells. Error bars, s.d.
We also sought to develop a light-based client release strategy from our synthetic organelles. Optogenetic dimerization domains has been leveraged to reverse condensate clustering or to release exogenous cargos11, 15. However, these strategies require sustained illumination and have not been demonstrated as effective in sequestering a large fraction of endogenous clients. We therefore decided to test an optogenetic strategy that would require short durations of illumination to achieve cargo release and reverse the programmed cell phenotype. In one strategy, we encoded a photocleavable domain, PhoCl35, 36, between the interaction tag and disordered domains of the scaffold, and fluorescently tagged Cdc24 to monitor light-induced client release (Fig. 4e). We found that upon short pulses of illumination, Cdc24 quickly accumulated in the cytosol (Extended Data Fig. 3b), achieving half client release from condensates in approximately 100 seconds (Fig. 4f). In a second strategy, we encoded a photocleavable domain, PhoCl, between the endogenous Cdc24 client and TsCC(B) interaction tag (Extended Data Fig. 3c) and tested the functional effect of client release on switching cells between an arrested and proliferative state. We initiated condensate formation to arrest the cell cycle by sequestering Cdc24 and then tested whether a pulse of illumination was sufficient to reverse the effect. We found that illumination was sufficient to stably reverse the arrest phenotype, returning cells to near normal levels of arrest, and that this was maintained for up to 6 hours after light exposure (Fig. 4g). Importantly, cells containing condensates and tagged client, but lacking a photocleavable domain did not respond to illumination (Extended Data Fig. 3d).
Finally, to determine whether it was possible to achieve cyclical control of client sequestration-release, we devised a multi-step proof-of-concept experiment to cycle through cell proliferation and arrest. Scaffold expression was induced at 25°C to first sequester client in condensates and arrest the cell cycle. In the next step, cargo was thermally released to reverse to the imposed arrest, and finally the arrest would be re-induced by returning the temperature to 25°C. We tested this strategy using Cdc24-mScarlet-TsCC(B) as the client and quantified cell arrest throughout the induction and release cycles. Indeed, we achieved robust arrest upon organelle induction at room temperature, followed by thermal reversal of the phenotype via heating, and finally restoration of the arrest by returning the system to 25°C (Fig. 4h, Extended Data Fig. 3e, f).
Taken together, we demonstrate two distinct approaches for release of native clients from synthetic organelles. The use of thermally responsive coiled-coils enables cyclical modulation of cellular control systems through client sequestration-release-sequestration and an optogenetic approach for irreversible client release from condensates which requires only a short period of illumination to stably reverse the imposed cell phenotype.
Sequestrating CRISPR-tagged targets in mammalian cells
In addition to single-cell organisms with industrial applications, we wanted to determine whether our platform to sequester native enzymes within synthetic membraneless organelles is generalizable in mammalian cells. We used a CRISPR knock-in approach to tag the 3’ end of genomic loci in U2OS cells (Extended Data Fig. 4a, b), so that clients are expressed from their endogenous promoters. We selected enzymes, GTPase Rac1 and the kinase ERK1, that have central roles in control of cell signaling pathways controlling cell motility and proliferation37. In migrating cells, Rac1 activity is required at leading edge, and in proliferating cells Erk1 is required to transmit mitogen signals from surface receptors to downstream transcriptional effectors. Cells harboring mCherry-TsCC(B) tagged Rac1 and ERK1 showed diffuse red localization throughout the cytosol and were not recruited to condensates that lacked the correct interaction motif (Fig. 5a, Extended Data Fig. 4c). In contrast, when scaffold containing the cognate TsCC(A) tagged was expressed, clients robustly localized to condensates and showed substantial enrichment in the organelles relative to the cytosol (Fig. 5a, b). Quantitation of the fraction of enzyme protein partitioned to the condensates revealed that on average over 70% of the scaffold protein and nearly 50% of each endogenous tagged client were localized to our synthetic organelle (Fig. 5c). To determine whether client sequestration impacts native pathway organization, we tagged the polarity protein Par6, which is normally enriched on the plasma membrane. After expressing synthetic condensates, Par6 is de-enriched from its native sites of localization and sequestered within the condensates (Fig. 5d, e, Extended Data Fig. 4d, e).
Figure 5. CRISPR-tagged endogenous clients enrich within synthetic condensates expressed in mammalian cells.
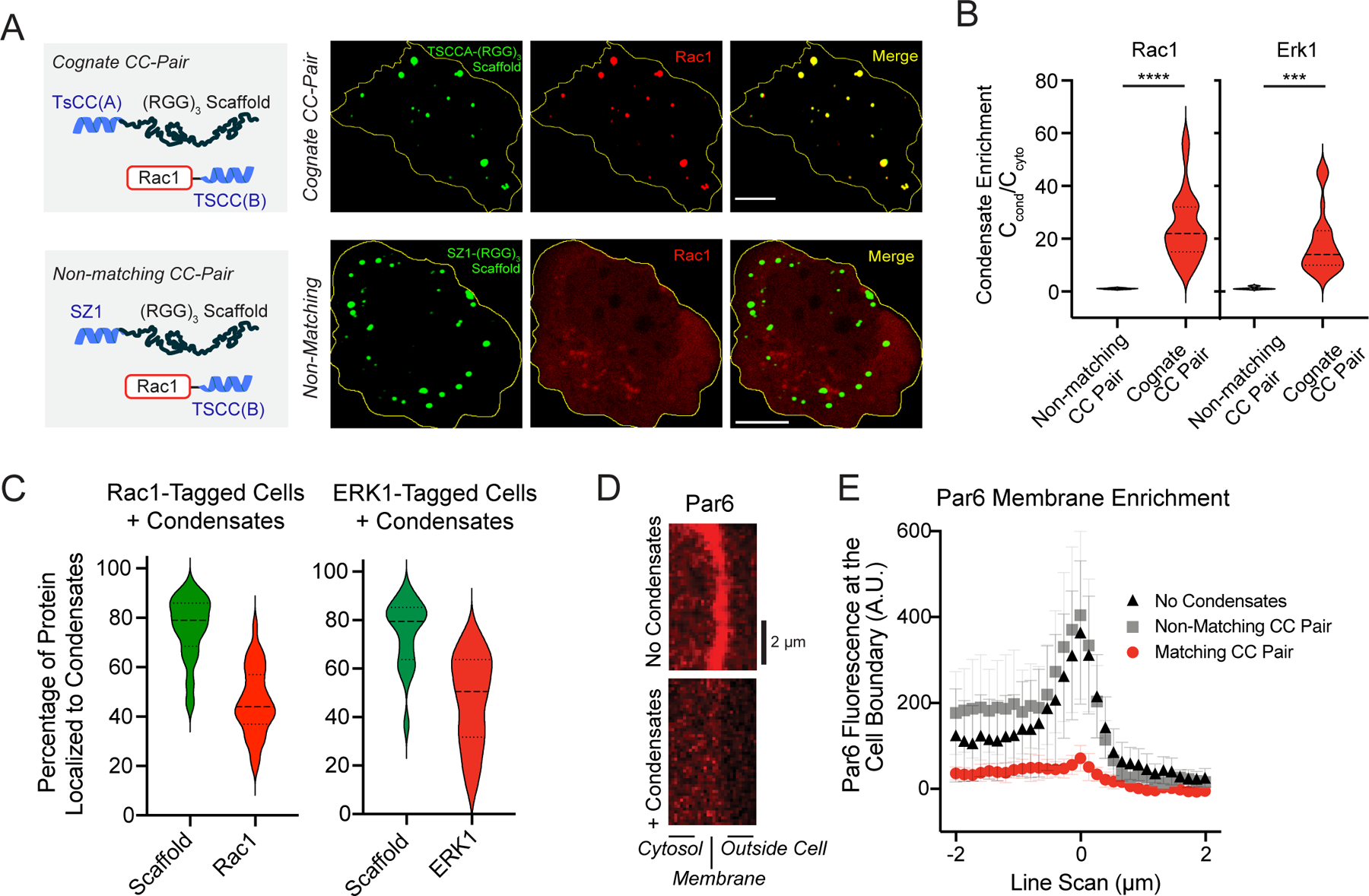
a, Representative images of Rac1-mCherry-TsCC(B) localization in U2OS cells expressing (RGG)3 scaffold with the cognate (TsCCA) or non-matching (SZ1) coiled coil. Scale bars, 10 μm. b, Violin plots of client enrichment for Rac1-mCherry-TsCC(B) and ERK1-mCherry-TsCC(B) in the presence of synthetic condensates with matching and non-matching coiled coils. n = 65 and 20 cells from four and three independent experiments, respectively. c, Percentage of scaffold and indicated endogenously tagged protein in condensates. For b and c, n = 65 and 20 cells pooled from four and three independent experiments for Rac1 and ERK1, respectively. d, Representative images of Par6-mCherry-TsCC(B) localization at the plasma membrane with no scaffold expression (top) and with scaffold expression and condensate formation. e, Average of line scans of Par6 at the cell boundary for cells without scaffold expression, non-matching scaffolds, and scaffolds with cognate coiled coil. n = 10 cells for each line scan. Error bars, s.d.
Taken together, these data demonstrate the utility of our synthetic membraneless organelle system for modular control of essential proteins and activities in multiple cell types. By combining expression of a designed scaffold and tagging of an endogenous genomic locus with high-affinity coiled-coil interaction motifs, it is feasible to impose cell behavioral states in real-time through functional sequestration of enzymes to designer membraneless compartments in cells.
DISCUSSION
Protein engineers have only recently begun to target the self-assembly of polypeptides into mesoscale membraneless compartments expressed in living cells13, 17, 38. Concurrently, metabolic engineers have leveraged these and other compartments to cluster exogenous enzymes to produce novel products11, 12. Cellular engineers interested in programming cellular behaviors and decision-making embed new molecules into cells that function as receptors or switches, to augment or redirect native behaviors39–44. Here, we expand the toolkit for cellular engineering by constructing a designer membraneless organelle system from disordered proteins that is capable of efficiently client sequestration and release. When recruited to synthetic condensates a targeted client is insulated from its native pathway, thereby generating predictable switching of cell behavior. Additionally, we demonstrate controlled release of sequestered clients from synthetic organelles using optical and thermal induction, which complement existing strategies, such as light-regulated condensate disassembly. The platform is generalizable to control of a variety of native components and pathways, and we demonstrate its application in multiple cell types, including cells used for bioproduction, and for mammalian tissue culture.
Cells enhance pathway enhance flux and selectivity by enforcing the proximity of pathway components. This can be achieved by binding the components to platforms such as macromolecular scaffold proteins or anchoring them to the plasma membrane1, 45. Co-localization increases the effective concentration of proteins and reduces interactions with other competing factors in the cells. Additionally, cells achieve even higher levels of specificity through physical compartmentalization, trafficking components into membrane bound organelles, such as the nucleus or lysosome. Although these are attractive strategies for reengineering subcellular reaction schemes, they have a number of drawbacks. It is currently not feasible to rewire native lipid metabolism to create a new orthogonal compartment bounded by a lipid bilayer membrane. Also, although assembly of enzymes and substrates onto a single nanometer size macromolecular scaffold protein can enhance flux, this reaction scheme is quite sensitive to scaffold protein concentrations and titration effects, and thus fluctuations in protein levels may lower reaction efficiency. Protein-based compartments offer a number of potential solutions to these engineering challenges.
Construction of synthetic subcompartments inside a cell from protein-based materials relies on polypeptides that that assemble into nanocapsules or mesoscale condensates. At the nanoscale, exogenous assemblies of encapsulins or designer protein cages provide one avenue for targeting components46–48. However, these compartments are tens of nanometers in diameter, limiting their cargo capacity. Further, their highly restrictive permeability often prevents the diffusion in and out of macromolecules. At the microscale, multivalent binding proteins can undergo complex coacervation10, 38, 49 or disordered polypeptide polymers can self-assemble to form mesoscale condensates7. Native membraneless organelles, such as P granules and the nucleolus, contain disordered protein components that condense and contribute to proteinaceous compartment self-assembly19, 50. These dynamic liquid-like compartments demonstrate selective composition and restricted permeability but are also highly porous to molecular and macromolecular clients18. An important feature of designer protein condensates is that they can achieve large sizes and therefore offer high payload capacities. Additionally, the dimensions and permeability of protein condensates are tunable, for example by increasing protein polypeptide length or expression levels above the saturation concentration. Therefore, membraneless organelles provide a means to scale the output of reactions localized to the compartment, something that is harder to achieve via endogenous membrane bound organelles.
Disordered protein sequences have been leveraged to generate synthetic liquid-like condensates in living systems. Examples in model eukaryotic culture systems include Corelets, OptoDroplets, REPS, and SPLIT among others12–14, 18, 21. More recently, resilin-like polypeptide sequences have been redesigned to assemble designer condensates in prokaryotic systems17, 51. In this work, we leveraged a disordered RGG domain from Laf-1, whose sequence displays UCST behavior and phase separation can be tuned by sequence mutation or controlling domain valency18, 20 and is amenable to engineering cytosolic condensates. We optimized the Csat by changing RGG polymer valency and through interaction motifs to generate a robust condensate system that partitions more than ninety percent of the cellular pool of scaffold to the synthetic organelle in budding yeast. Many phase separating proteins, including those of the FET family, include RNA binding RGG domains, which have been shown to enhance LLPS alone and in the presence of RNA52. Although we cannot exclude the idea that our RGG platform may still interact with RNAs, it does not require RNA to phase separate in biochemical reconstitution experiments18, 19, and the temperature dependent phase behavior in cells match those from in vitro experiments (Extended Data Fig. 1b, c).
There are a variety of strategies to enrich clients in synthetic compartments, although there are strength and limitations of each approach. Similar to localization motifs used in cells, short coiled coil sequence pairs can be used to target a client protein to a disordered scaffold53. Alternatively, a disordered sequence can be appended directly to a protein of interest to target its partitioning to the scaffold only in the condensed state11. A challenge of fusing low complexity polypeptide sequence to a native protein is that it may alter stability or endogenous interactions and functions. Because we wanted to target essential proteins at their native encoding genomic loci, we chose to use coiled-coil interaction pairs. These high affinity tags have been shown to be functional and orthogonal in vivo in other cellular engineering studies25, 53 and we demonstrate here that tagging of the GEF, Cdc24, or the kinase, Cdc5, with coiled coil interaction domains does not disrupt localization and essential activities.
Additional challenges, to which our system is also subject, are design considerations including IDR/folded protein ratio of the scaffold and the limitations to protein expression inherent to in vivo studies. Because we rely on coacervation to form condensates capable of sequestering high levels of native clients, the scaffold must necessarily be expressed at levels well above its Csat. In yeast, GAL1 promoters lead to high expression levels, allowing us to achieve up to 90% client partitioning and control over cell behavior. However, in our transient transfections of mammalian cells, we do not achieve as high a level of scaffold expression and only approximately 80% scaffold partitioning to condensates. This reduced partitioning relative to expression in yeast helps explain by we achieve lower client partitioning in mammalian cells. Future work that enhances scaffold expression for example via multicopy viral integration would ensure higher fractional client partitioning. Nevertheless, using the current iteration of our platform transiently expressed in mammalian cells, we were able to recruit substantial amounts of native enzymes, Rac1 and Erk1, and sequestered Par6, insulating it from its normal localization along the cell cortex.
Further, one must also consider that client size, subcellular localization, and stoichiometry relative to the disordered sequences of the scaffold may affect the levels of client partitioning. We demonstrate efficient functional insulation of GTPase Cdc24 and kinase Cdc5. Efficacy is likely high because the substrates of these enzymes are dozens of kilodaltons and therefore do not easily diffuse inside the condensates. Additionally, the normal subcellular positioning of Cdc24 to the plasma membrane and Cdc5 to spindle pole bodies, likely enhances the functional effect of sequestration at shutting down pathway activity. It may be more challenging to insulate metabolic enzymes whose reactants and products are small molecules that more readily diffuse in and out of synthetic condensates. One additional unknown is whether client sequestration to synthetic condensates will exhibit an inverse size dependence at some critical size. In our current study we effectively sequestered clients whose molecular weight is greater 100 kDa of folded domains, when including recruitment tags and fluorophores.
Our system is ostensibly similar to other inducible sequestration or inducible knockdown systems. The anchors-away approach leverages small-molecule dependent protein dimerization to anchor targets, such as transcription, outside of the nucleus54. However, these systems are often less effective for achieving functional knockdown of highly expressed cytoplasmic proteins and by anchoring targets to native structures such as plasma membrane, ER or golgi membranes, may interfere with function. Additionally, achieving reversibility of these systems by small molecular washout is challenging. RNA interference strategies, although useful can be incomplete and quite slow, taking days to sufficiently clear pre-existing transcripts. Auxin Induced Degradation (AID) systems overcome the time limitations of RNAi, enabling knockdown of protein levels within tens of minutes to hours55. However, these systems are difficult to reverse, often requiring extensive washing out of the small molecule and multiple rounds of cell division to restore protein levels. We propose that the synthetic membraneless organelle system we developed has a number of advantages: It is orthogonal, offers a high payload capacity, is capable of ultrahigh sequestration of targeted clients, and demonstrates controlled client release, readily reversing cell activity state both by thermal and optical means.
Unique features of our condensate platform including regulatory handles for thermal and optical control of client release. Using thermally responsive coiled-coils as interaction motifs, reversal of client recruitment to synthetic condensates can be achieved by transient shifts to elevated temperatures of 37–42°C. Although yeast grow normally at 37°C, maintaining temperatures as high as 42°C for long periods of time is not advisable and will produce a heat shock stress response. Additionally, although temperature transients are possible through ultrasound heating of mammalian cells, we would largely recommend thermal client release only for yeast. On the other hand, light-based client release is highly effective in both yeast and mammalian cells, and has a number of clear applications for cell biology and cellular engineering. A simple experimental setup would be to express our disordered scaffold along with an exogenous client that one would like to release for regulation of cellular behavior or cell fate, and to illuminate the system to achieve sustained client release on the timescale of minutes. For example, sequestered signaling enzymes or transcription factors could be rapidly released to modulate a cellular decision. In effect, this system can be considered an intracellular drug delivery or controlled release platform, one in which the kinetics of client accumulation in cytoplasm would be substantially faster than inducible transcription and translation
In conclusion, we offer a new strategy for programmed control of cellular decision-making by modular targeting of cellular machinery to synthetic membraneless compartments. As a proof-of-concept we demonstrate near complete targeting and insulation of endogenously expressed enzymes upon organelle induction. Sequestration to our designer organelles blocks its native localization and function, thereby switching off cell polarity and proliferation control systems in a single cell system with industrial applications. Using thermosensitive interaction motifs or photocleavable domains, we show effective and cyclical reversal of client recruitment and subsequent reversal of cellular phenotypes. Further, we extend this platform to mammalian cells and show efficient client recruitment and insulation from native targeting sites, demonstrating our membraneless organelle system as generalizable across cell types and applications. Our study reveals that de novo compartmentalization of native enzymes can be used engineer cellular systems capable of responding to specific stimuli with predictable outcomes. This synthetic organelle approach can be leveraged a hub to insulate and rewire native biochemical pathways to reveal principles of pathway organization, or as a protein switch based for cellular engineering.
METHODS
Molecular Biology
All plasmids were constructed using InFusion cloning (Takara Bio; Mountain View, CA) and verified by DNA sequencing. Yeast plasmids expressing RGG domain scaffolds are encoded in the integrating Yiplac211 (URA3, AmpR) plasmid backbone downstream of the inducible GAL1 promoter. GAL1, interaction tag, RGG and GFP sequences were generated by PCR were cloned into the plasmid backbone between the XbaI and AgeI restriction sites. Plasmids expressing exogenous client (mScarlet) fused to a C-terminal interaction domain are encoded in the integrating Yiplac128 (LEU2, AmpR) plasmid backbone downstream of a constitutive MET17 promoter. PCR products encoding the MET17 promoter sequence, mScarlet and an interaction tag were cloned into Yiplac128 between the XbaI and AgeI cut sites. To generate PCR products for yeast knock-ins, fluorophore and interaction domain sequences were cloned into pfa6a::KANMX6 or pfa6a::HIS3 plasmid backbones. PCR products of TsCC(B), mScarlet-TsCC(B) and mScarlet-FKBP, were generated from the previously cloned Yiplac128 plasmids above and cloned into the pfa6a vectors using the PacI and AscI restriction sites. To generate a plasmid to knock-in PhoCl-TsCC(B), PCR products of the PhoCl and TsCC(B) sequences were cloned into pfa6a::KANMX6 between PacI and AscI restriction sites via InFusion ligation. For mammalian cell work, plasmids encoding scaffolds with interaction domains were cloned into pcDNA vectors, downstream of a CMV promoter. GFP and RGG domains were cloned from gene fragments codon optimized for human expression (Integrated DNA Technologies). Sequences were cloned into the pcDNA backbone sequentially between the BamHI and XbaI restriction sites. For mammalian CRISPR knock-ins, Cas9 plasmids with the appropriate guide RNA (gRNA) and donor plasmids encoding a fluorophore and coiled coil interaction domains were generated. To construct Cas9 plasmids, the pCas9-Guide (OriGene Technologies; Rockville, MD) was used as a backbone and a 20 nt sequencing encoding the gRNA targeting the C-terminal end of the gene of interest was assembled using duplexed oligos and cloned between BamHI and BsmBI restriction sites according to the manufacturer instructions. Donor plasmids were constructed using the pUC19 donor backbone (Takara) and encoded 600–1000 bp homology arms, along with mCherry-TsCC(B) and a NAT resistance cassette in between the homology arms. The mCherry-TsCC(B) sequence was first cloned into a pcDNA backbone using the BamHI and XbaI cut sites. A 1000 bp 5’ homology arm was generated by PCR from synthesized gene fragments (Integrated DNA Technologies; Coralville, IA) and cloned upstream of the mCherry sequence using the NheI and BamHI restriction sites. The NATR cassette and a 600 – 800 bp 3’ homology arm were then amplified and fused by a 2-step PCR. These 5’ and 3’ sequences were then PCR amplified and cloned into pUC19 between the HindIII and SacI restriction sites. In each case, the PAM site, located in one of the homology arms was changed to prevent persistent cleavage by Cas9.
Yeast Procedures
Standard methodologies were followed for all experiments involving S. cerevisiae56,57. In all cases, the scaffold was under the control of the galactose inducible GAL1 promotor and was incorporated into the yeast URA3 locus using an integrating vector (Yiplac211) cut with EcoRV and standard lithium acetate transformation. Exogenously expressed clients under the control of the MET17 promotor was similarly integrated into the LEU2 locus with an integrating vector (Yiplac128) after EcoRV digestion. To tag native genomic loci, PCR products of tags and drug resistance cassettes containing 40 to 50 bp of homology on either end to the C-terminus of the target gene were transformed into yeast cells by lithium acetate/PEG transformation as previously described58. BNI1 was deleted by replacing the ORF with a TRP1 marker. The DAD domain of Bnr1 was internally deleted by Cas9 mediated gene editing by co-transforming yeast strains with a plasmid expressing Cas9 and a gRNA targeting the DAD domain of BNR1 and an 80 nt oligo with homology to sequences upstream and downstream of the DAD domain59. All yeast strains used in this study are listed in Supplementary Table 1 and plasmids are listed in Supplementary Table 2.
For scaffold induction, yeast cells were first grown to saturation overnight in liquid YPD media in a 25°C shaking incubator. Cells were then washed three times in sterile water and diluted in YP + 2% Raffinose and incubated in a 25°shaking incubator for 6 to 8 hours. Finally, yeast cells were diluted to an OD600 of 0.3 in YP + 2% Galactose induction proceeded overnight in the same shaking incubator or for hours on a microscope slide to track scaffold induction and cargo recruitment in the same cells. Final OD600 of cultures used for experiments were between 0.4–0.8 except for the strain harboring TsCC(A)-Scaffold and endogenous Cdc24 or Cdc5 tagged with mScarlet-TsCC(B) as their growth arrests with scaffold induction. For client partitioning studies in the presence of scaffold, cells were incubated overnight in galactose, ~ 14 hrs.
For thermal reversal experiments, yeast cells harboring TsCC(A)-Scaffold and Cdc24-TsCC(B) were grown in YPD media as above and then washed and transferred to YP + 2% Raffinose overnight. Cells were then transferred into YP + 2% Galactose to induce scaffold induction. Thermal reversal was performed after 6 hours of scaffold expression by transferring 1 mL of each cell culture to a heated water bath for 1 and 2 hours. For overnight thermal reversion, cells were maintained at 37°C or 42°C. Samples were taken at the indicated timepoints and cells were fixed by 4% paraformaldehyde (PFA) for 10 min (Ricca Chemical Company; Arlington, TX), centrifuged and washed three times with 1 mL PBS, and stored at 4°C until imaging. For light-induced client release, yeast cells harboring a combination of TsCC(A)-PhoCl2f-Scaffold and Cdc24-mScarlet-TsCC(B) or TsCC(A)-Scaffold and Cdc24-PhoCl-TsCC(B) were grown, and scaffolds were expressed by switching to galactose, as above. After four hours of scaffold expression, cells expressing TsCC(A)-PhoCl2f-Scaffold and Cdc24-mScarlet-TsCC(B) were imaged and exposed to 10 second pulses of 405 nm light on an Olympus IX81 inverted confocal microscope (Olympus Life Science; Tokyo, Japan) with a Yokogawa CSU-X1 spinning disk, Mercury lamp, 488 and 561 nm lasers, and an iXon3 EMCCD camera (Andor; Belfast, UK) controlled by Metamorph software (Molecular Devices; Downingtown, PA). Cells expressing TsCC(A)-Scaffold and Cdc24-PhoCl-TsCC(B) were induced as above, heated to 37°C, exposed to 10 min pulse of UV light, and then allowed to continue growth in YP + 2% Galactose media. Samples were taken at the indicated timepoints and fixed/stored as above until imaging.
For phalloidin staining, 1mL of cell cultures were fixed with 4% PFA for one hour and centrifuged and washed three times in 1 mL PBS. Fixed cells were resuspended in 49 μl PBS plus 1 μl of AlexaFluor568-Phalloidin (Invitrogen; Carlsbad, CA, USA) and rotated in the dark at room temperature overnight. Before imaging, cells were centrifuged and washed twice with PBS.
Mammalian Cell Procedures
U2OS human osteosarcoma cells were cultured in Eagle’s minimal essential medium [EMEM, Quality Biological] supplemented with 10% fetal bovine serum (Gibco; Gaithersburg, MD), 2 mM L-glutamine (Gibco), and 10 U/mL penicillin-streptomycin (Gibco) and maintained at 37°C in a humidified atmosphere with 5% CO2. Cells were split in 1:3 ratio every 3 days and had been passaged for less than 2 months, were negative for known infection, and experiments were done with confirmed viability >95% by Trypan Blue staining (Gibco). For drug selection, cells were cultured in EMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 0.75 mg/mL G418 sulfate (MediaTech, Inc; Manassas, VA).
A CRISPR knock-in strategy was implemented to tag Rac1 and ERK1 at their native genomic loci. pUC19 donor plasmids (Takara Bio) were cloned harboring mCherry-TsCC(B) and a Neomycin resistant cassette along with 600 to 1000 bp homology arms as described above in Molecular Biology (Supplementary Table 2). Donor plasmids were co-transfected with Cas9 plasmids (OriGene Technologies) cloned with the 2–3 distinct guide RNAs to target the gene of interest. Co-transfection of donor plasmids and pCas-gRNA plasmids were performed using Lipofectamine 2000 (Invitrogen) according to the manufacture protocol. Briefly, Cells were seeded at 70% confluency in 6-well flat-bottom tissue culture plates (CELLTREAT; Pepperell, MA) 24 hours prior to the transfection. On the day of transfection, 1500 ng of donor plasmids and 500ng of each pCas-gRNA plasmid were mixed in Opti-MEM Reduced Serum Media (Gibco). Lipofectamine 2000 were added in 1:5 DNA to reagent ratio and incubated for 15 minutes before adding to the cells dropwise. 24 hours post transfection, cells were trypsinzed and moved to 100mm cell culture dishes (ThermoFisher Scientific; Waltham, MA). Cells were selected with drug for 7 days. After selection, cells were rested in media without drug for 24 hours, and sorted based on mCherry expression using BD FACSAria™ III Cell Sorter (BD Bioscience; San Jose, CA) with the help from flowcytometry core at University of Pennsylvania. Briefly, cells were resuspended at 10*106 cells/ml in media supplied with 25mM HEPES (Gibco). Before the sort, 1ul of 1ug/ml DAPI (Invitrogen) was added to the sample for live/dead staining. Cells were sorted into medium and high-expression bins and maintained for 2 weeks in complete media until confluency. mCherry-positive cells were confirmed by fluorescence microcopy. CRISPR knock-in of tags to endogenous loci was confirmed via PCR.
For scaffold expression, post-selection cells were seeded on 24-well glass bottom plate (Greiner Bio-one) at 70% confluency. 24 hours later, cells were transfected with 1000 ng of GFP-tagged scaffold cloned into a pcDNA vector (Supplementary Table 2) using X-tremeGENE 9 DNA Transfection Reagent (Sigma-Aldrich; St. Louis, MO) at 1:3 DNA to reagent ratio according to the manufacture protocol. In all cases, cells imaged were first tested for mycoplasma using MycoAlertTM Mycoplasma Detection Kit (Lonza; Basel, Switzerland) according to the manufacture protocols. All of the cells reported in this study were determined to be mycoplasma free.
Microscopy
Fluorescence microscopy imaging of yeast and mammalian cells was performed on an Olympus IX81 inverted confocal microscope (Olympus Life Science) equipped with a Yokogawa CSU-X1 spinning disk, Mercury lamp, 488 and 561 nm laser launches, and an iXon3 EMCCD camera (Andor). Multidimensional acquisition was controlled by MetaMorph software (Molecular Devices). Samples were illuminated using a 488 nm laser and/or 561 nm laser and imaged through a 100x/1.4 NA oil-immersion objective. Z-stacks were collected at sampling depth appropriate for three-dimensional reconstitution. Brightfield transmitted light images used to assess yeast cell morphologies were also captured on a Nikon Eclipse Ti-U confocal microscope (Nikon, Tokyo, Japan) equipped with Yokogawa CSU-X1 spinning-disk and Photometrics Evolve Delta EMCCD Camera (Teledyne Photometrics; Tucson, AZ).
To image mesoscale condensates, budding yeast in YP media containing 2% galactose were immobilized to glass coverslips treated with Concanavalin A (ConA). For chemogenic induction of client recruitment, yeast cells in the same media were first allowed to adhere to glass coverslips coated with ConA, and subsequently Rapamycin was added to a final concentration of 20 μM
For yeast photobleaching experiments (FRAP and FLIP), a Roper iLas2 photoactivation system controlling a 405 nm laser was used. For FRAP, individual condensates were selected, photobleached, and fluorescence recovery in the bleached region was analyzed in ImageJ. For FLIP, half of a cell body was photobleached and fluorescence loss from the condensate on the opposite half of the cell was analyzed in ImageJ. Mammalian U2OS cells were imaged 40 hours after transient transfection with the after adhering to a 24-well glass bottom plate (Greiner Bio-one). In all cases, Z-stacks were collected to visualize the scaffold at the 488 nm wavelength and client at 561 nm wavelength using a 100x/1.4 NA oil-immersion objective.
Image Analysis
Analysis of condensates and clients in cells was performed in ImageJ. To quantify in vivo phase plots and determine Csat, cells expressing scaffold were imaged alongside wells containing purified GFP fusion proteins to generate a standard curve for fluorescence. Ccyto was calculated from average background subtracted fluorescence intensity of cytoplasmic signal, coverted to concentration using the calibration curve. To quantify scaffold and client recruitment to synthetic condensates in yeast and U2OS cells, we segmented cells and condensates using in ImageJ. Objects were masked in the z-plane of the image stack containing the largest portion of cells. Because U2OS cells are adherent and spread, masks were generated in 488 nm channel by automatic thresholding using the MaxEntropy algorithm in ImageJ, and lower boundary were manually set to be 3-fold higher than the average cytosolic signal. The particle analysis function in ImageJ was used to segment condensates larger than a 5-pixel area. Background subtracted measurements of 488 nm and 561 nm pixel intensity for masks for of the condensates and cells were used to calculate an enrichment index (background corrected fluorescence Intensitycondensates / Intensitycytosol). To estimate the fraction of scaffold or client partitioned to the organelles, we divided the background subtracted integrated pixel intensity for condensate mask areas by the background subtracted integrated pixel intensity in of the cell mask (∑ Intensitycondensates / ∑ Intensitycell). Par6 signal at the cell cortex was analyzed by line scans in ImageJ using a line 30 pixels in length and thickened by 10 pixels. Line scans were then averaged, and background intensity was subtracted.
Quantification of cellular phenotypes (e.g. budding indices and cell size) were performed in ImageJ using brightfield images of live cells or of fixed cells from timecourse experiments. Multiple fields of view (FOVs) were captured per experiment and budding indices were generated by counting the fraction of cells that had a daughter cell (bud). Cell size measurements were performed by manual tracing of the outline of the mother cell to determine cell area. Distribution of AlexaFluor561-Phalloidin staining was quantified drawing a box, that encompasses the entire cell body, along the long axis of the cell and plotting summed intensity as a function of position. Box position was determined by position of bud or location of polarized signal to the end of the mother cell. The longest cell axis was used in cases, such as in cells with sequestered Bnr1, where a polarity axis could not be determined. Datasets were normalized to average mother cell fluorescence in cells that lacked condensates. The fluorescence profile for at least individual 50 cells from each strain were rescaled by defining the back of the mother cell as 0 and the tip of polarized signal in G1 cells, or tip of the bud in other cell cycle stages, as 1.
Statistics and Reproducibility
Experiments were reproducible. All statistical analyses were performed in GraphPad Prism 9. To test the significance of two categories, an unpaired two tailed t-test was used. To test significance of more than two categories, a one-way ANOVA was used. To compare difference in growth curves, significance was determined by linear regression analysis. In all cases, ns indicates not significant; *, p ≤ 0.05; **, p ≤ 0.01, ***, p ≤ 0.001; and ****, p ≤ 0.0001.
DATA AVAILABILITY.
All data supporting the findings of this study are included in the published article and its supplementary information files. Original data are available from the corresponding authors upon reasonable request.
Extended Data
Extended Data Fig. 1. Properties of in vivo synthetic condensates.
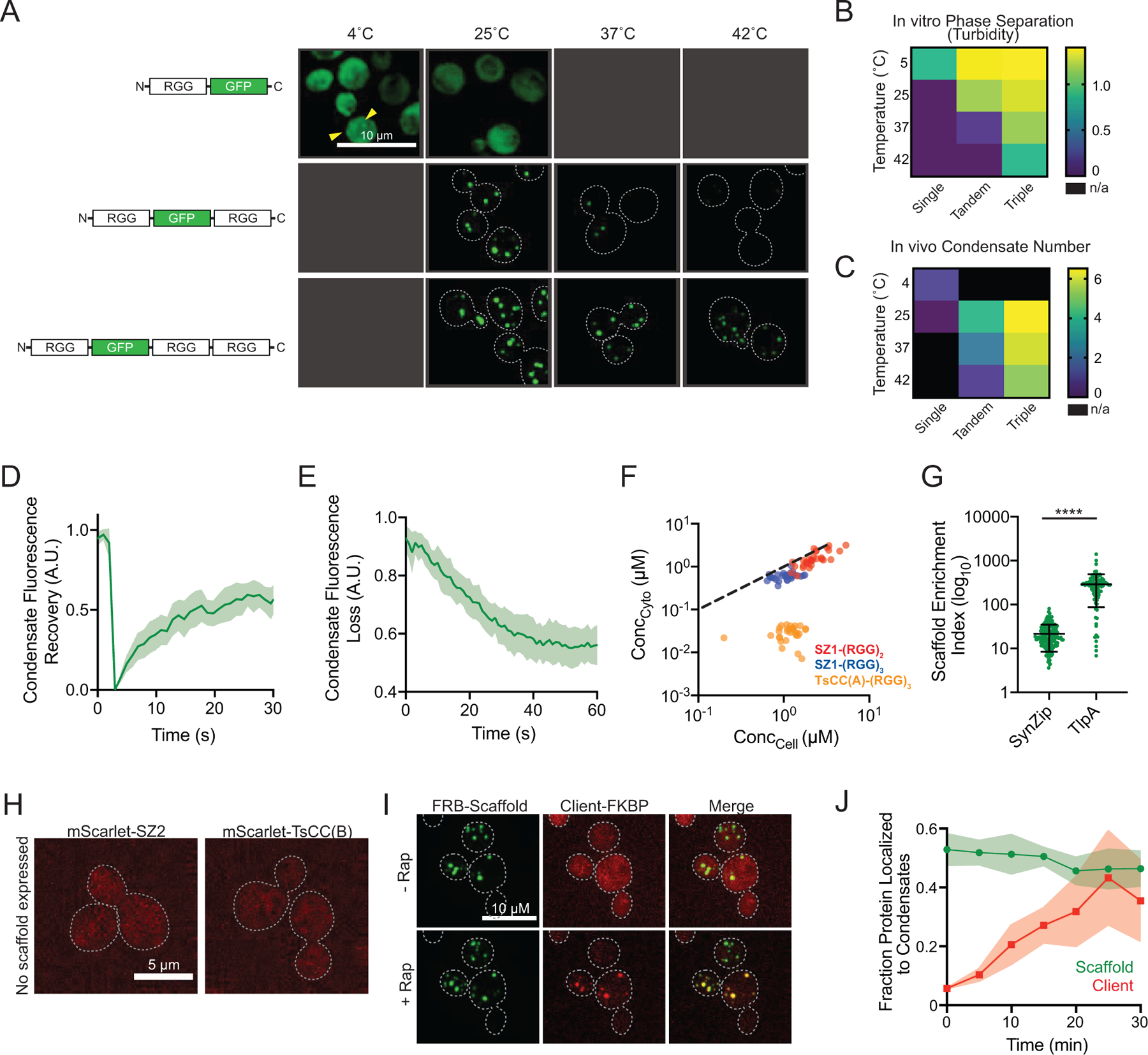
a, Temperature dependence of condensate assembly as a function of scaffold RGG domain valency. Representative images of yeast cells expressing galactose induced GFP tagged scaffold with 1, 2, or 3 RGG domains at different temperatures. b, Heat mao: quantitation of turbidity data of purified proteins from Schuster et al., 2018. c, Heat map: number of condensates per cell as a function of temperature and RGG domain valency. d, Fluorescence recovery after photobleaching (FRAP) of condensates formed by (RGG)3 scaffold; n = 10 condensates. Shaded area, 95% CI. e, Fluorescence loss in photobleaching (FLIP) of condensates formed by (RGG)3 scaffold. n = 13 cells. f, Steady state cytoplasmic scaffold concentration outside of condensates (Ccyto) as a function of cellular concentration (Ccell) for 30 cells per scaffold type. Dashed line, slope of 1. g, Average enrichment of scaffold protein in condensates for SZ1-(RGG)3 or TsCC(A)-(RGG)3. n = 164 and 97 condensates respectively. Error bars, s.d. h, Representative images of exogenously expressed mScarlet-SZ2 and mScarlet-TsCC(B) diffusely distributed in cytosol in the absence of condensates. i, Representative images of cells expressing FRB-(RGG)3 scaffold and mScarlet-FKBP as a client. The client is diffuse in the cytosol before the addition of Rap and concentrated in condensates after Rap addition. j, Quantitation of the fraction of client protein as in i localized to condensates over time after Rap addition. n = 15 cells. Shaded area 95% CI.
Extended Data Fig. 2. Condensate expression relocalizes tagged native clients and regulates cell growth.
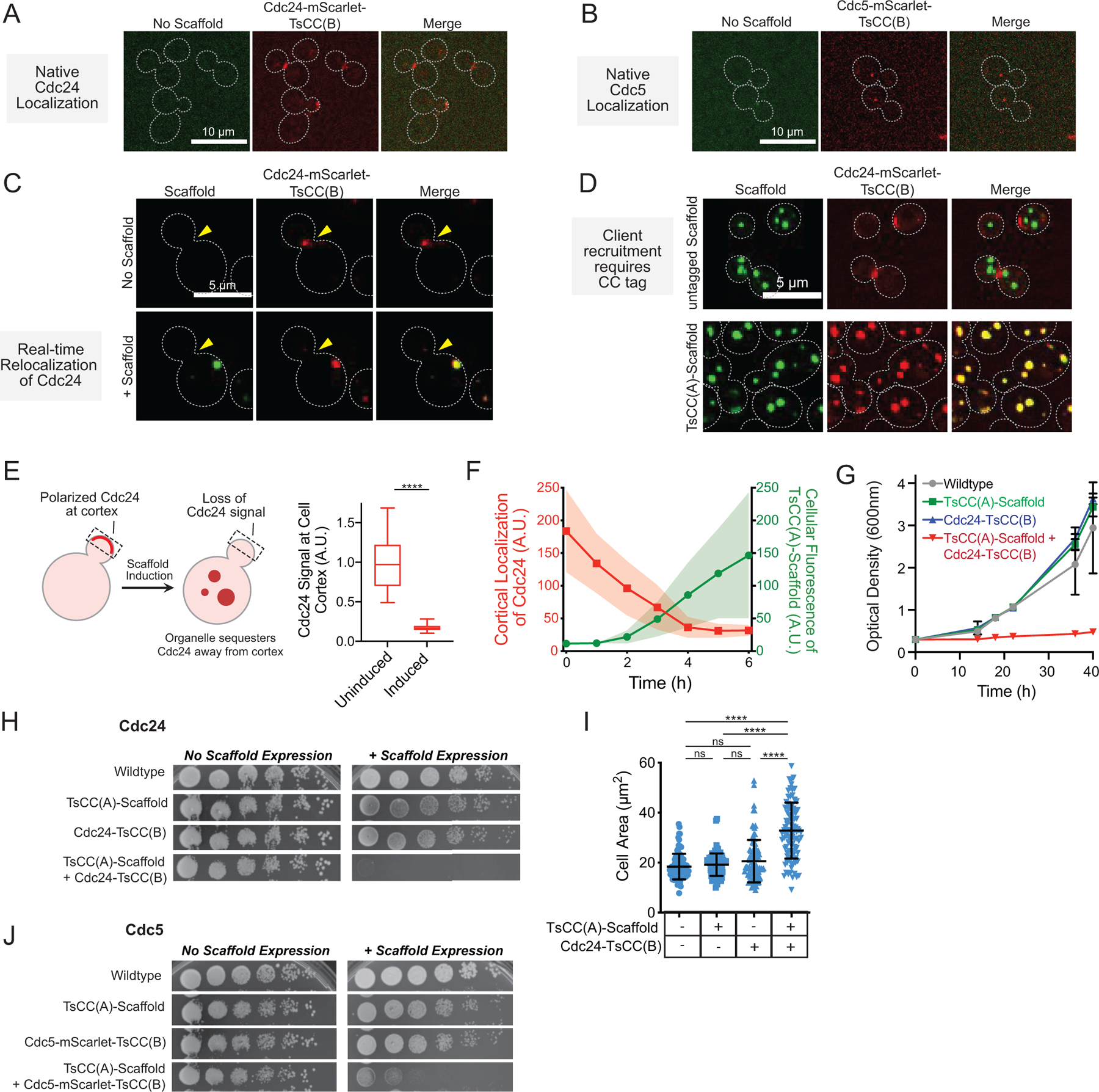
a, Representative images of tagged natively expressed Cdc24 show its cortical localization. b, Representative images of tagged natively expressed Cdc5 show its punctate localization to spindle pole body. c, Images of the same Cdc24-mScarlet-TsCC(B) cell before and after induced expression of TsCC(A)-(RGG)3 scaffold for 6 hr, show loss of cortical Cdc24 signal and partitioning to synthetic condensate. d, Client recruitment to condensates specifically depends on CC tag interaction; Cdc24 does not interact with (RGG)3 condensates that lack the interaction tag. e, Left, scheme: cortical Cdc24 is relocalized from cortex to synthetic condensates after induction of scaffold. Right, Average cortical Cdc24-mScarlet-TsCC(B) signal before and after TsCC(A)- (RGG)3 scaffold expression (6h). n = 20 cells before and after hours of galactose induction. Significance calculated by unpaired, two-tailed, t-test (****, p ≤ 0.0001). f, Kinetics of loss of cortical Cdc24-mCherry-TsCC(B) signal (red) concomitant with cellular accumulation of expressed TsCC(A)-(RGG)3 scaffold (green) upon induction with galactose for 20 cells over 6 hours. Shaded area, s.d. g, Cell proliferation: measurements of cell density (OD600) over time for indicated Cdc24 strains in liquid media containing galactose. h, Growth assay for Cdc24 strains: five-fold serial dilution of indicated strains grown on solid- media containing glucose or galactose. i, Average cell area of mother cells only increases upon TsCC(A)-(RGG)3 expression in Cdc24-mScarlet-TsCC(B) cells, consistent with cell cycle arrest in G1. j, Growth assay for Cdc5 strains: five-fold serial dilution of indicated strains grown on solid-media containing glucose or galactose. In all cases, growth defect depends on presence of tagged client and expression of scaffold to form condensates. Phenotype is not observed with only native client tagging or only scaffold expression.
Extended Data Fig. 3. Reversible control of cell proliferation-arrest state.
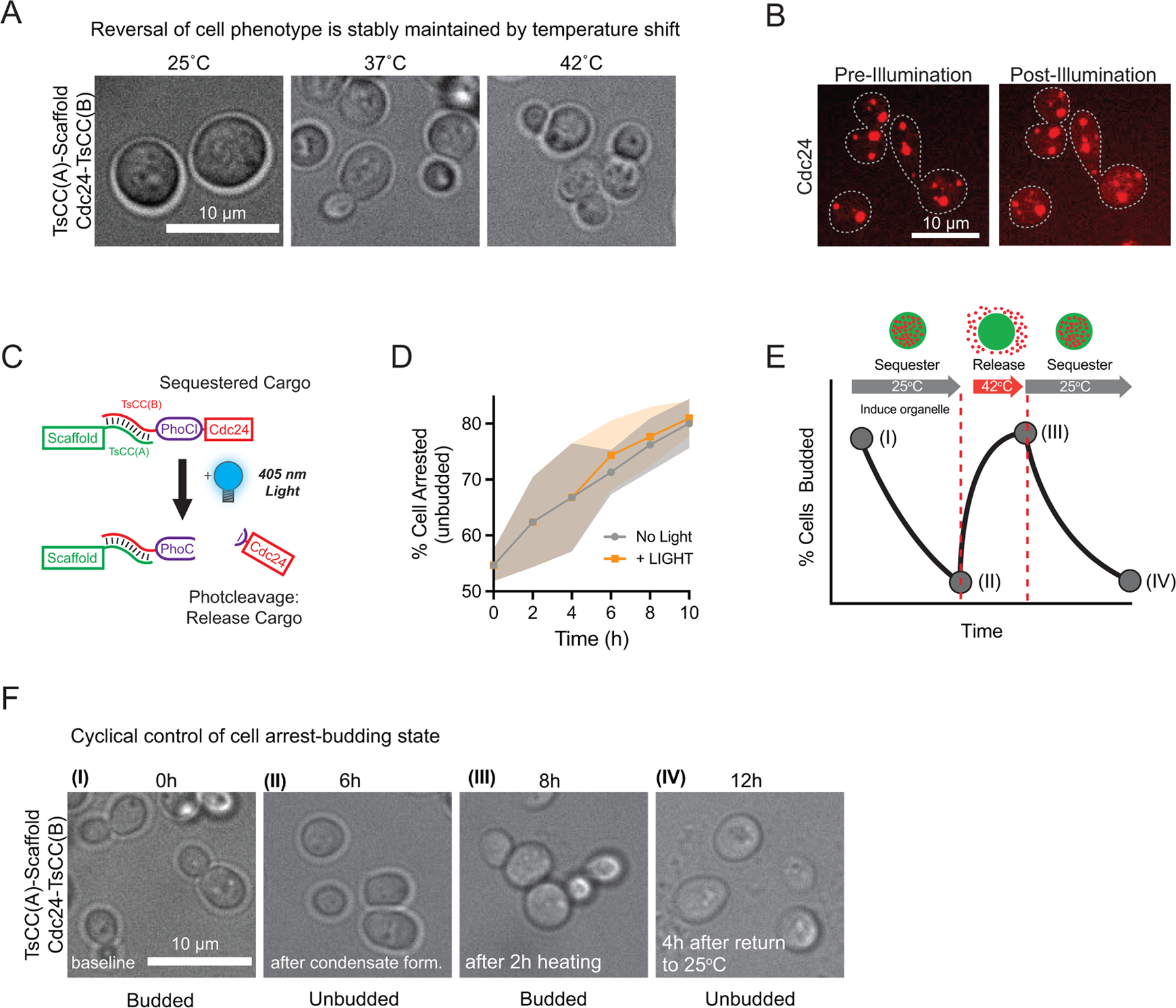
a, Representative images of Cdc24-mScarlet-TsCC(B) cells expressing TsCC(A)-(RGG)3 scaffold at the indicated temperatures for 14 hours. Thermally responsive coiled-coil pair dissociate upon heating to 42°C, releasing client to promote cell polarity and reversing the cell cycle arrest associated with Cdc24 sequestration to condensates. b, Representative images of Cdc24-mScarlet-TsCC(B) in the presence of TsCC(A)-PhoCl2f-(RGG)3 before and after illumination with 405 nm light. c, Schematic of client release strategy: Cdc24 is tagged with PhoCl-TsCC(B). 405 nm light results in PhoCl cleavage and client release. d, Percentage of cells expressing Cdc24-mScarlet-TsCC(B) arrested (unbudded cells) over time after scaffold induction +/− illumination. n = 4048 cells in total pooled from three trials. e, Prediction: cycling of cell state between budded-arrested-budded-arrested. f, Representative images of cells at the indicated time points. Wildtype levels of budding at time 0 h. Cells incubated in galactose at 25°C from 0–6h timepoints to induce condensate formation, blocking budding, then heated from 6h to 8h timepoints, promoting polarization and budding and cooled back to 25°C and arrested by 12h.
Extended Data Fig. 4. Sequestration to synthetic condensates in mammalian cells.
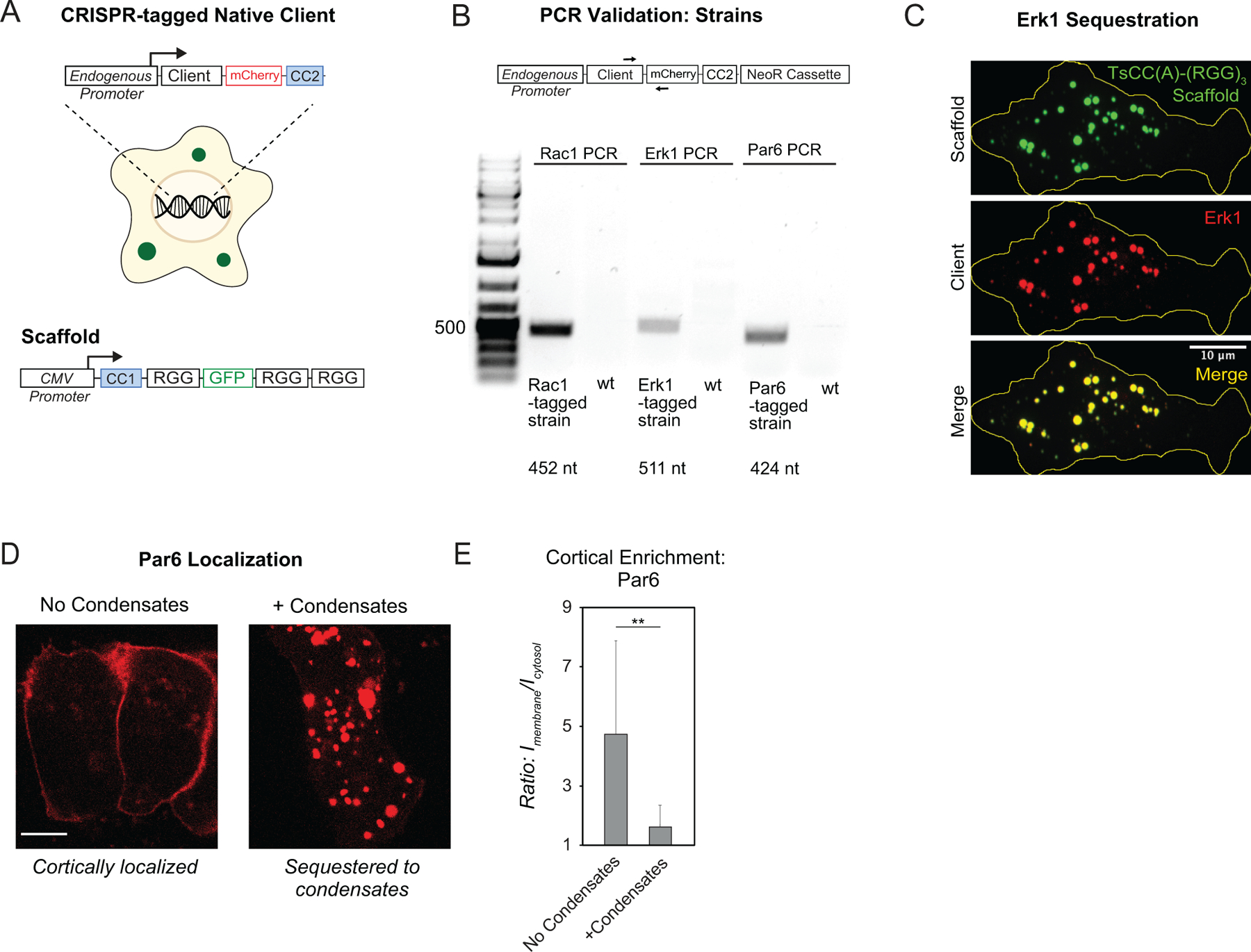
a, Schematic of CRISPR tagging approach to endogenous loci in mammalian cells and expression of the scaffold by a CMV promoter. b, PCR validation of CRISPR tagging in mammalian cell lines. Only tagged strains show a PCR product of the expected size as indicated. c, Representative images of tagged ERK1 robustly partitions to synthetic condensates formed by expression of TsCC(A)-(RGG)3 scaffold. d, Representative images of tagged Par6 localized to the cell cortex in the absence of scaffold expression (left) and to condensate structures when scaffold is expressed (right). e, Quantitation of cortical Par6-mCherry-TsCC(B) in the absence and presence of condensates with cognate coiled coil. n = 10 cells for each condition. Error bars, s.d. Significance calculated by unpaired, two-tailed t-test. (**, p ≤ 0.01).
Supplementary Material
Supplementary Video 1 Rapamaycin-induced client recruitment to preformed condensates in cells. Green: FRB-(RGG)3 GFP-tagged scaffold.
Supplementary Video 2 Inducible expression of scaffold disrupts native cortical localization of Cdc24, sequestering it to newly formed synthetic condensates. Green: TsCC(A)-(RGG)3 GFP-tagged scaffold. Red: native client, Cdc24-mScarlet-TsCC(B)
Supplementary Video 3 Induced condensate sequestration of endogenous client, Cdc24, upon rapamycin addition. Green: FRB-(RGG)3 GFP-tagged scaffold. Red: native client, Cdc24-mScarlet-FKBP
ACKNOWLEDGEMENTS
We thank Anuj Kumar for sharing yeast strains, Erfei Bi lab for yeast plasmids and technical support, Boao Xia and Haidar Ahmed for cloning, Holly Ramage for U2OS cell lines, Mikhail Shapiro lab for TlpA plasmids, Daniel Hammer lab for critical reading of manuscript, and Andrea Stout and the Penn CDB Microscopy Core for imaging and support. This cellular engineering study was supported by a National Institute of Biomedical Imaging and Bioengineering R01 grant, EB028320 (M.C.G). Biochemical characterization of disordered proteins was partly funded by National Science Foundation (NSF) iSuperseed grant, DMR1720530 (M.C.G). Synthesis of optochemical dimerizers was supported by NSF grant CHE-1404836 (A.D.). Conceptual development of condensates as decision hubs and investigator salary support was supported in part by Department of Energy BES Biomolecular Materials grant DE-SC0007063 (M.C.G).
Footnotes
COMPETING INTERESTS
Authors declare no competing interests.
REFERENCES
- 1.Good MC, Zalatan JG & Lim WA Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.al-Mohanna FA, Caddy KW & Bolsover SR The nucleus is insulated from large cytosolic calcium ion changes. Nature 367, 745–750 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Burack WR & Shaw AS Signal transduction: hanging on a scaffold. Curr Opin Cell Biol 12, 211–216 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Burack WR, Cheng AM & Shaw AS Scaffolds, adaptors and linkers of TCR signaling: theory and practice. Curr Opin Immunol 14, 312–316 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharyya RP, Reményi A, Yeh BJ & Lim WA Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu Rev Biochem 75, 655–680 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Scott JD & Pawson T Cell signaling in space and time: where proteins come together and when they’re apart. Science 326, 1220–1224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banani SF, Lee HO, Hyman AA & Rosen MK Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choudhary S, Quin MB, Sanders MA, Johnson ET & Schmidt-Dannert C Engineered protein nano-compartments for targeted enzyme localization. PLoS One 7, e33342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dueber JE et al. Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol 27, 753–759 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Aumiller WM Jr., Pir Cakmak F, Davis BW & Keating CD RNA-Based Coacervates as a Model for Membraneless Organelles: Formation, Properties, and Interfacial Liposome Assembly. Langmuir 32, 10042–10053 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Zhao EM et al. Light-based control of metabolic flux through assembly of synthetic organelles. Nat Chem Biol 15, 589–597 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reinkemeier CD, Girona GE & Lemke EA Designer membraneless organelles enable codon reassignment of selected mRNAs in eukaryotes. Science 363 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin Y et al. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168, 159–171.e114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bracha D et al. Mapping Local and Global Liquid Phase Behavior in Living Cells Using Photo-Oligomerizable Seeds. Cell 175, 1467–1480.e1413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dine E, Gil AA, Uribe G, Brangwynne CP & Toettcher JE Protein Phase Separation Provides Long-Term Memory of Transient Spatial Stimuli. Cell Syst 6, 655–663.e655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song D, Jo Y, Choi JM & Jung Y Client proximity enhancement inside cellular membrane-less compartments governed by client-compartment interactions. Nat Commun 11, 5642 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei SP et al. Formation and functionalization of membraneless compartments in Escherichia coli. Nat Chem Biol 16, 1143–1148 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Schuster BS et al. Controllable protein phase separation and modular recruitment to form responsive membraneless organelles. Nat Commun 9, 2985 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elbaum-Garfinkle S et al. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc Natl Acad Sci U S A 112, 7189–7194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuster BS et al. Identifying sequence perturbations to an intrinsically disordered protein that determine its phase-separation behavior. Proc Natl Acad Sci U S A 117, 11421–11431 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed EH, Schuster BS, Good MC & Hammer DA SPLIT: Stable Protein Coacervation Using a Light Induced Transition. ACS Synth Biol 9, 500–507 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caldwell RM et al. Optochemical Control of Protein Localization and Activity within Cell-like Compartments. Biochemistry 57, 2590–2596 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao X, Jin X & Liu B The involvement of stress granules in aging and aging-associated diseases. Aging Cell 19, e13136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banani SF et al. Compositional Control of Phase-Separated Cellular Bodies. Cell 166, 651–663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piraner DI, Wu Y & Shapiro MG Modular Thermal Control of Protein Dimerization. ACS Synth Biol 8, 2256–2262 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Iida H & Yahara I Specific early-G1 blocks accompanied with stringent response in Saccharomyces cerevisiae lead to growth arrest in resting state similar to the G0 of higher eucaryotes. J Cell Biol 98, 1185–1193 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams AE, Johnson DI, Longnecker RM, Sloat BF & Pringle JR CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J Cell Biol 111, 131–142 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woods B, Kuo CC, Wu CF, Zyla TR & Lew DJ Polarity establishment requires localized activation of Cdc42. J Cell Biol 211, 19–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida S et al. Polo-like kinase Cdc5 controls the local activation of Rho1 to promote cytokinesis. Science 313, 108–111 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Toenjes KA, Simpson D & Johnson DI Separate membrane targeting and anchoring domains function in the localization of the S. cerevisiae Cdc24p guanine nucleotide exchange factor. Curr Genet 45, 257–264 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Botchkarev VV Jr., Rossio V & Yoshida S The budding yeast Polo-like kinase Cdc5 is released from the nucleus during anaphase for timely mitotic exit. Cell Cycle 13, 3260–3270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagot I, Klee SK & Pellman D Yeast formins regulate cell polarity by controlling the assembly of actin cables. Nat Cell Biol 4, 42–50 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Moseley JB & Goode BL The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol Mol Biol Rev 70, 605–645 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chesarone M, Gould CJ, Moseley JB & Goode BL Displacement of formins from growing barbed ends by bud14 is critical for actin cable architecture and function. Dev Cell 16, 292–302 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W et al. Optogenetic control with a photocleavable protein, PhoCl. Nat Methods 14, 391–394 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Lu X et al. Improved Photocleavable Proteins with Faster and More Efficient Dissociation. bioRxiv, 2020.2012.2010.419556 (2020). [Google Scholar]
- 37.Tanimura S & Takeda K ERK signalling as a regulator of cell motility. J Biochem 162, 145–154 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Nakamura H et al. Intracellular production of hydrogels and synthetic RNA granules by multivalent molecular interactions. Nat Mater 17, 79–89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gordley RM, Bugaj LJ & Lim WA Modular engineering of cellular signaling proteins and networks. Curr Opin Struct Biol 39, 106–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordley RM et al. Engineering dynamical control of cell fate switching using synthetic phospho-regulons. Proc Natl Acad Sci U S A 113, 13528–13533 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu CY, Roybal KT, Puchner EM, Onuffer J & Lim WA Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toda S, Blauch LR, Tang SKY, Morsut L & Lim WA Programming self-organizing multicellular structures with synthetic cell-cell signaling. Science 361, 156–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y et al. Modular construction of mammalian gene circuits using TALE transcriptional repressors. Nat Chem Biol 11, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Najem JS et al. Assembly and Characterization of Biomolecular Memristors Consisting of Ion Channel-doped Lipid Membranes. J Vis Exp (2019). [DOI] [PubMed] [Google Scholar]
- 45.Bashor CJ, Helman NC, Yan S & Lim WA Using engineered scaffold interactions to reshape MAP kinase pathway signaling dynamics. Science 319, 1539–1543 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Lau YH, Giessen TW, Altenburg WJ & Silver PA Prokaryotic nanocompartments form synthetic organelles in a eukaryote. Nat Commun 9, 1311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sigmund F et al. Bacterial encapsulins as orthogonal compartments for mammalian cell engineering. Nat Commun 9, 1990 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giessen TW et al. Large protein organelles form a new iron sequestration system with high storage capacity. Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li P et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feric M et al. Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 165, 1686–1697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dzuricky M, Rogers BA, Shahid A, Cremer PS & Chilkoti A De novo engineering of intracellular condensates using artificial disordered proteins. Nat Chem 12, 814–825 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chong PA, Vernon RM & Forman-Kay JD RGG/RG Motif Regions in RNA Binding and Phase Separation. J Mol Biol 430, 4650–4665 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Thompson KE, Bashor CJ, Lim WA & Keating AE SYNZIP protein interaction toolbox: in vitro and in vivo specifications of heterospecific coiled-coil interaction domains. ACS Synth Biol 1, 118–129 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haruki H, Nishikawa J & Laemmli UK The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31, 925–932 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T & Kanemaki M An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Sambrook J, Fritsch EF, Maniatis T Molecular Cloning: A Laboratory Manual. (1989).
- 57.Guthrie GC, Fink G Guide to yeast genetics and molecular biology. Methods Enzymol 194, 1–863 (1991). [PubMed] [Google Scholar]
- 58.Longtine MS, Fares H & Pringle JR Role of the yeast Gin4p protein kinase in septin assembly and the relationship between septin assembly and septin function. J Cell Biol 143, 719–736 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anand R, Memisoglu G & Haber J Cas9-mediated gene editing in Saccharomyces cerevisiae. (2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1 Rapamaycin-induced client recruitment to preformed condensates in cells. Green: FRB-(RGG)3 GFP-tagged scaffold.
Supplementary Video 2 Inducible expression of scaffold disrupts native cortical localization of Cdc24, sequestering it to newly formed synthetic condensates. Green: TsCC(A)-(RGG)3 GFP-tagged scaffold. Red: native client, Cdc24-mScarlet-TsCC(B)
Supplementary Video 3 Induced condensate sequestration of endogenous client, Cdc24, upon rapamycin addition. Green: FRB-(RGG)3 GFP-tagged scaffold. Red: native client, Cdc24-mScarlet-FKBP
Data Availability Statement
All data supporting the findings of this study are included in the published article and its supplementary information files. Original data are available from the corresponding authors upon reasonable request.