

Abstract
Retinal degeneration is a leading cause of vision impairment and blindness worldwide and medical care for advanced disease does not exist. Stem cell-derived retinal organoids (RtOgs) became an emerging tool for tissue replacement therapy. However, existing RtOg production methods are highly heterogeneous. Controlled and predictable methodology and tools are needed to standardize RtOg production and maintenance. In this study, we designed a shear stress-free micro-millifluidic bioreactor for nearly labor-free retinal organoid maintenance. We used a stereolithography (SLA) 3D printer to fabricate a mold from which Polydimethylsiloxane (PDMS) was cast. We optimized the chip design using in silico simulations and in vitro evaluation to optimize mass transfer efficiency and concentration uniformity in each culture chamber. We successfully cultured RtOgs at three different differentiation stages (day 41, 88, and 128) on an optimized bioreactor chip for more than 1 month. We used different quantitative and qualitative techniques to fully characterize the RtOgs produced by static dish culture and bioreactor culture methods. By analyzing the results from phase contrast microscopy, single-cell RNA sequencing (scRNA seq), quantitative polymerase chain reaction (qPCR), immunohistology, and electron microscopy, we found that bioreactor-cultured RtOgs developed cell types and morphology comparable to static cultured ones and exhibited similar retinal genes expression levels. We also evaluated the metabolic activity of RtOgs in both groups using fluorescence lifetime imaging (FLIM), and found that the outer surface region of bioreactor cultured RtOgs had a comparable free/bound NADH ratio and overall lower long lifetime species (LLS) ratio than static cultured RtOgs during imaging. To summarize, we validated an automated micro-millifluidic device with significantly reduced shear stress to produce RtOgs of comparable quality to those maintained in conventional static culture.
Keywords: Retinal organoids, Microfluidics, Millifluidics, 3D printing, Fluorescence lifetime imaging, Phasor approach, Single-Cell RNA Sequencing
INTRODUCTION:
Retinal degeneration (RD) is a leading cause of vision impairment and blindness worldwide. Visual degeneration can originate in any of the cell types in the retina. Some of the more common visual degenerations arise from death and/or dysfunction of the photoreceptors (PR) and retinal pigmented epithelial (RPE) cells. Irreversible cell damage is the root to vision loss in diseases like age-related macular degeneration (AMD) and retinitis pigmentosa (RP)1, 2. Retinal sheets and dissociated retinal cells are candidates for retinal tissue replacement therapy. However, both tissue sources have inherent limitations. Historically, retinal sheets derived from fetal neurosensory retina and RPE transplanted into the subretinal space demonstrated utility to restore vision and neurosensory functions3–9 in animals10–12 and humans13. However, the use of fetal tissue carried complex social, ethical, and political implications. Transplantation of dissociated photoreceptor precursors overcame the ethical issues and demonstrated some visual function improvements14, 15 but dissociated cell transplantation16–19 suffered from insufficient cell type differentiation, lack of cellular polarization and eventual cell death.
With the advent of new techniques to manipulate human embryonic (hESCs)20 and induced pluripotent stem cells (iPSCs),21 stem cell-derived retinal organoids (RtOgs) have emerged as tools that exhibit the combined advantages of retinal sheets and differentiated retinal cells. RtOgs are 3D spheroid tissues that arise from stem cells and self-organize into layered retinal tissues containing retinal ganglion cells, rods and cones22–24. Transplantation of RtOgs have been shown to restore vision in retina degenerated rats25, mouse26 and primate27 models with RD. Even so, current state-of-the-art RtOg production methods are highly heterogeneous due to their use of different cell lines, tissue maintenance methods, high manual labor and imprecise tissue selection for use in multiple applications28. A comparative study revealed that RtOgs differentiated from iPSCs showed stage specific, cell line and methodological differences29. This heterogeneity and imprecision limit human RtOg procurement for preclinical trials28 and in vitro investigations. Many approaches, including bioreactors30–36 and optimized production protocols28, 37 are investigated to standardize RtOg production and maintenance over months. Controlled and predictable RtOg production is important to ensure a quality-controlled tissue product that is suitable for transplantation.
In recent years, many in vitro cell culture platforms have emerged for organoid differentiation and maintenance at the macro-38, milli-39, and microscales40. Macro-scaled platforms are typically utilized for their ease and effectiveness in producing organoids, while milli-scaled systems (≥ 1 mm) are employed for relatively high flow rates, cell-cell interaction, and less frequent media changes and thus less organoid perturbation and lower probability for damage31. Considering the costs associated with the relatively high media volumes required by the macro-scaled bioreactors, microscale devices (< 100 μm) are steadily growing in popularity41. Microfluidic devices share the advantages of millifluidic devices, with the advantage of lower media consumption. However, the dimensional limits of traditionally fabricated microfluidics devices hinder their application to organoids research since organoids are 3D spherical tissues that can grow up to several millimeters in size. Fig. 1 summarizes published organoid bioreactors and their advantages and disadvantages. The integration of micro- and millifluidic device is a promising solution for organoids differentiation and maintenance.
Figure 1: Review of Organoid Bioreactors.
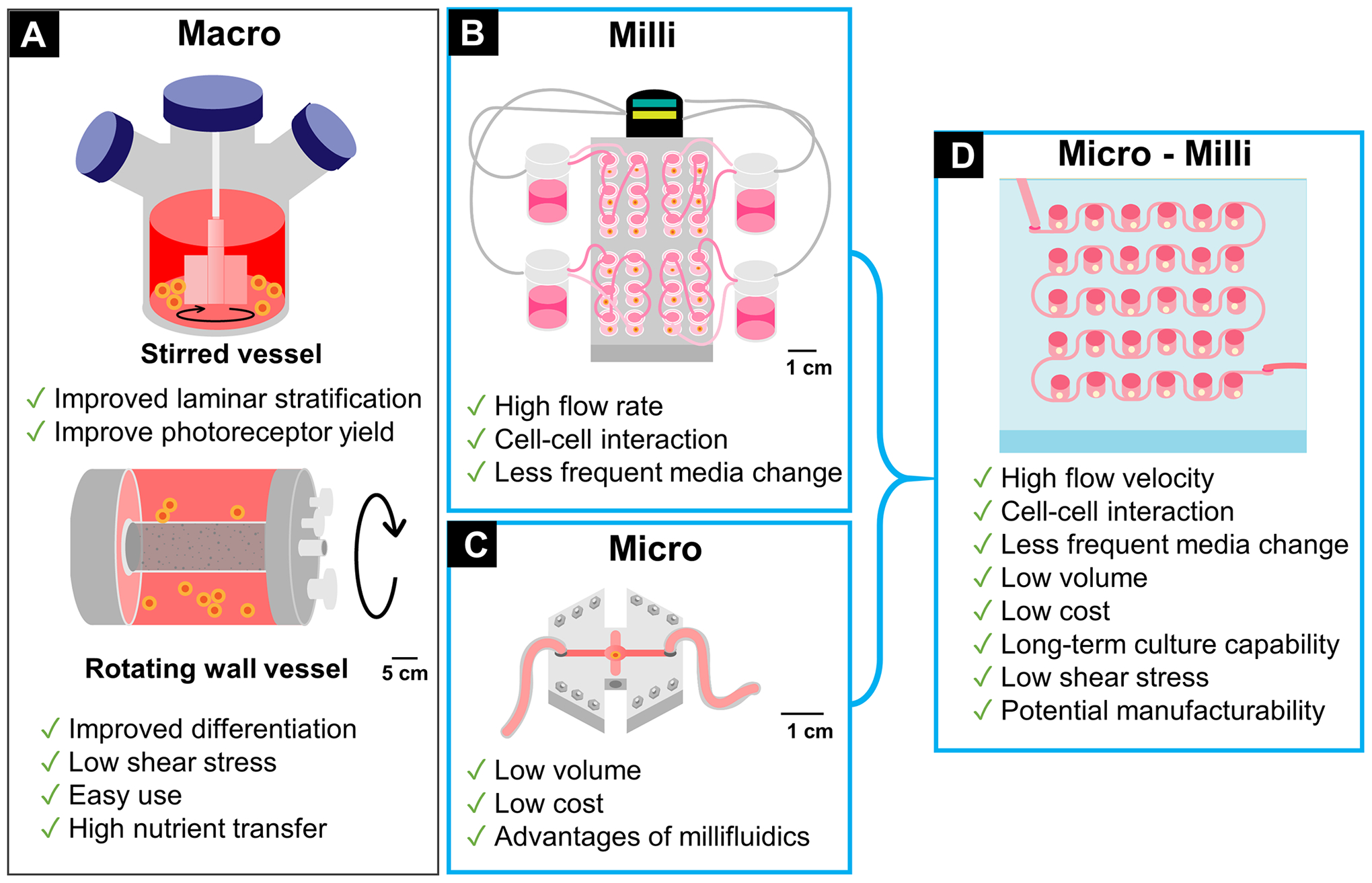
(A) Macro scale bioreactors: stirred/spinning and rotating wall vessels38; (B) Millifluidic bioreactor39; (C) Microfluidic bioreactor40; (D) Micro-millifluidic bioreactor in this article.
In this study, we designed and fabricated a shear stress-free micro-millifluidic bioreactor for use in RtOg culture and maintenance. We used a high resolution (25 μm) stereolithography (SLA) 3D printer to fabricate the mold for Polydimethylsiloxane (PDMS) molding, which easily combined micro and millimeter features in one design with very low cost and short manufacturing time. First, we simulated the fluidic design parameters in COMSOL to optimize the fluidic transports in the chip design. We evaluated 3 different factors that could affect mass transfer efficiency and uniformity. We then successfully cultured RtOgs in 3 different differentiation stages on the designed chip platform for more than one month (31~37 days). Finally, we did a comparative study to characterize the RtOgs produced by dish culture (denoted as “static” because there was no constant media flow through the dish) and chip culture. We compared live and fixative organoids qualitatively and quantitatively through various characterization methods.
METHODS
COMSOL simulation
The simulation was performed using finite element analysis software, COMSOL Multiphysics (COMSOL, Inc, Palo Alto, CA, USA). COMSOL was used to evaluate different chip designs and flow channel configurations to optimize mass-transport dynamics in culture chambers with different heights. Three major factors that affected the mass transfer rate were taken into consideration: 1) channel width (1000 or 500 μm wide), 2) channel configuration relative to culture chambers (linear single-sided chambers, serpentine alternating side chambers, serpentine with integrated mixer) and 3) the culture chamber height (2 or 4 mm tall).
The simulation parameters are listed in Table 1. The initial concentration of the whole system was set to zero, which was considered the most extreme condition. The left end of the channel was set as the inlet with concentration of 1 mol/m3 as the boundary condition.
Table 1:
Simulation parameters
| Physics | Laminar flow & Transport of diluted species |
| Study type | Time dependent |
| Material | Water |
| Diffusion coefficient (m 2 /s) | 6.00E–10 |
| Boundary conditions (mol/m 3 ) | cinitial = 0, cinlet = 1 |
Chip design and fabrication
The mold was designed using SolidWorks (SolidWorks Corp., Waltham, MA, USA) and the final design used for RtOg culture had the dimensions shown in Fig. 2A with channel height of 200 μm and chamber height of 2 mm. The chambers were arranged in a 6 × 5 array with the distance between each chamber at 9 mm, which was the same as that of a 96-well plate for compatibility with subsequent imaging steps. The mold was produced with 25 μm resolution with the Formlabs Form 3B printer (Formlabs, Somerville, MA, USA) using standard clear resin (Formlabs) (Fig. 2B). After printing, the mold was cleaned with 90% isopropanol to remove any resin residue. The mold was then air dried for 24 hours and cured with ultraviolet light for 30 minutes.
Figure 2: Fabrication methods.
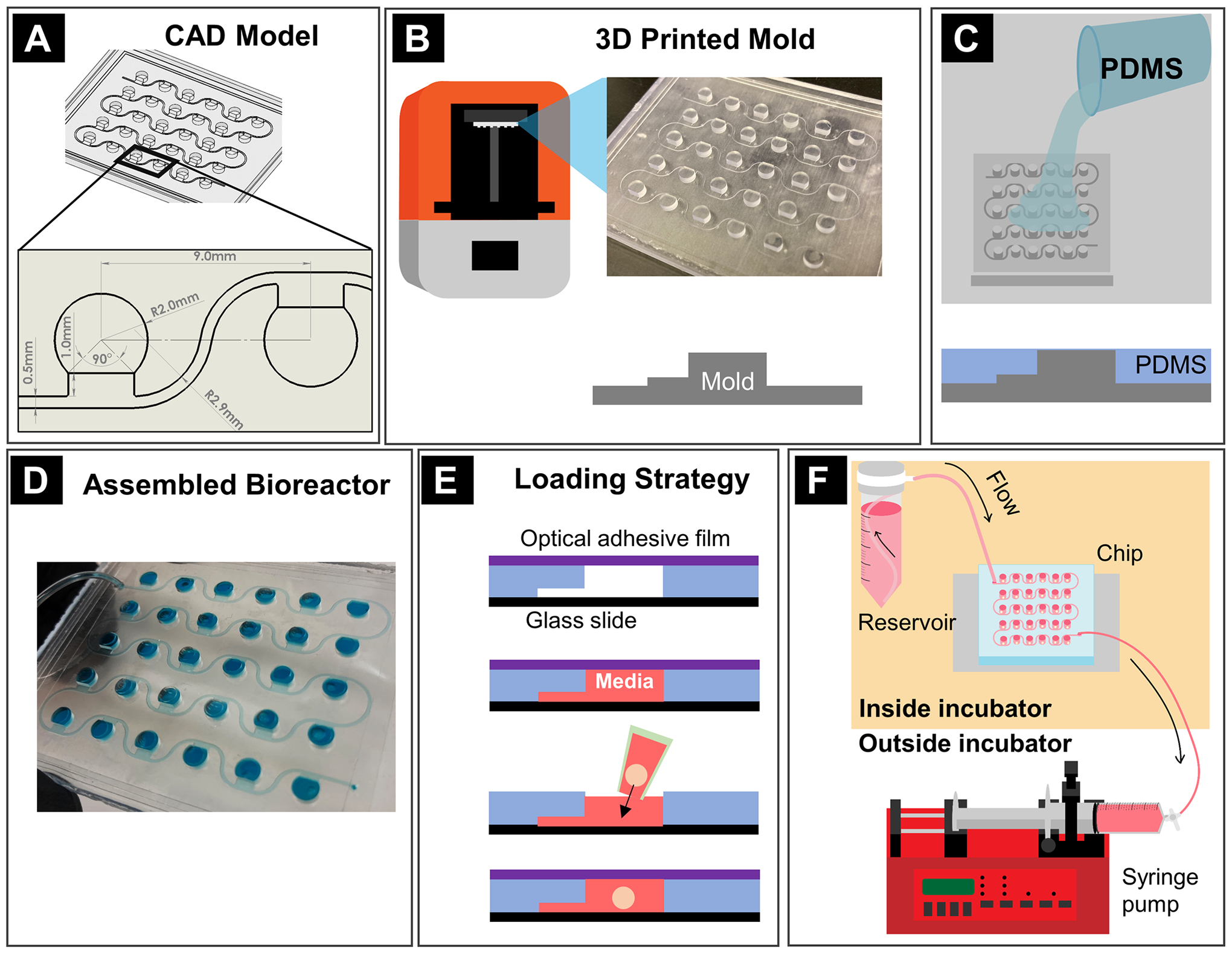
(A) Mold design with CAD software; (B) Mold printing; (C) PDMS casting on the mold; (D) Assembled bioreactor; (E) Cross-section view of organoid loading procedure whereby microchannels were filled with media first, then an organoid was placed in the open well, and the wells were sealed using adhesive optical film; (F) On-chip culturing system assembly.
The bioreactor was fabricated from the printed mold similar to the molding steps in soft lithography42. Polydimethylsiloxane (PDMS) Sylgard 184 (Dow Corning, Midland, MI, USA) was mixed manually for 10 minutes at a 10:1 ratio (base elastomer/curing agent). After degassing in a vacuum chamber, the PDMS was poured over the 3D-printed mold until the level reached the top of the culture chamber features and degassed again in a vacuum desiccator to remove bubbles (Fig. 2C). After 48 hours of curing under room temperature, the molded PDMS piece was carefully peeled off from the mold. The fluidic inlet and outlet were created with a biopsy punch. Finally, the PDMS piece was treated with air plasma (Harrick) (Harrick Plasma, Ithaca, NY, USA) for 1 min. to promote adhesion and then pressure-bonded to a cover slip (#1.5, 64*50 mm, ClariTex) (Ted Pella, Inc., Redding, CA, USA) (Fig. 2D).
Stem cell culture and retinal organoids initiation
Stem cell line 1
Retinal organoids were differentiated from genetically modified NIH-registered cell line H9 human embryonic stem cells (hESCs) with green fluorescent protein (GFP) tagged to CRX gene which encodes cone-rod homeobox protein and is specifically expressed in photoreceptor cells43–45. Stem cells were maintained by feeding mTeSR 1 media (STEMCELL Technologies, Vancouver, BC, Canada) daily and passaged every 4–7 days by ReLeSR (STEMCELL Technologies) when cells reached ~80% confluency. Cells were expanded on Vitronectin XF™ (STEMCELL Technologies) coated plates at 37°C in a humidified 5% CO2 incubator (Nuaire, Plymouth, MN, USA).
To initiate organoid formation, Accutase (Nacalai Inc, Kyoto, Japan) was added to the stem cells into a single cell suspension when 2-dimensional culture reached ~80% confluency. The cells were then placed in an 800-μm micro-well EZSPHERE 12-well plate (Nacalai USA, Inc., San Diego, CA, USA) and centrifuged at 100g for 3 min. to evenly distribute the stem cells throughout the bottom of each well. From day 1 to 7, the stem cells self-aggregated into embryonic bodies (EBs) in the EZSPHERE microwells. From day 8, the EBs were seeded onto a 1% growth factor reduced Matrigel (Corning, Corning, NY, USA) coated culture dish. The EBs spread onto the Matrigel and began 2D differentiation. Retinal eye fields were cut from the Matrigel between day 38 and 50 and transferred to ultra-low attachment 24-well plates (Corning Costar) (Corning, Corning, NY, USA) for 3D culture to be loaded into the bioreactor chip. Media used for retinal organoid differentiation was modified from Zhong et al.46 From day 0 to 18, the organoids were gradually transitioned from mTeSR1 medium into neural induction media (NIM) containing Dulbecco’s modified eagle medium (DMEM)/F12 (1:1) (Gibco, Waltham, MA, USA), 1% N2 supplement (Gibco), 1x minimum essential media non-essential amino acids (NEAA) (Gibco), 1x L-glutamine (Gibco), and 2 μg/ml heparin (Sigma-Aldrich, St. Louis, MO, USA), with daily media changes. From day 19 to 41, the media was switched to NIM containing DMEM/F12 (1:1) supplemented with 2% B27 supplement (50X) (minus vitamin A, Gibco), 1x NEAA, 1x L-glutamine, and 2 mg/ml heparin. From day 42 and beyond, the organoids were cultured with media containing DMEM/F12 (1:1) supplemented with 2% B27 Plus Supplement (50X) (Gibco), 1x NEAA, 1x L-glutamine, 2 ug/ml heparin, 100 μM taurine (Sigma), and 10% fetal bovine serum (FBS; Gibco). The media was changed 3 times a week and the organoids were maintained at 37°C in a humidified 5% CO2 incubator.
On day 41, 87, and 128 of differentiation, 12~15 RtOgs were randomly selected to load one each into every other chamber in the bioreactor chip. After about one month (30~37 days) of on-chip culture, RtOgs were used for histology, single-cell RNA sequencing. Same tests for the same age RtOgs in static culture group were performed. The detailed information of experimental groups was summarized in Table S1.
Stem cell line 2
We applied a second stem cell line in this study. The hESCs (cell line CSC14, NIH registration no. 0284; AIVITA Biomedical, Inc) were maintained on Matrigel coated flasks and cultured in a xeno-free custom formulated media supplemented with low levels of bFGF and Activin-A (Peprotech, Rocky Hill, NJ, USA). Media was replaced daily, and flasks kept in a 37°C, 5% CO2 tissue culture incubator. Every 4–5 days, colonies were passaged by enzymatic dissociation using collagenase IV (Gibco, 2 mg/ml) and transferred to a fresh Matrigel coated TC flask.
To initiate differentiation, growth factors are omitted, and media is replaced with a serum-free composition containing a GMP manufactured basal media, and Vitamin-A free B27 supplement (Gibco). Stem colonies are enzymatically released with collagenase IV (2 mg/mL) and aggregates allowed to form embryoid bodies (EB) for seven days in ultra-low adherence flasks. After seven days, EBs were transferred to Matrigel coated dishes and allowed to attach. Culture continues for 21 to 36 days with media replacement every 2–3 days. When refringent annular structures showing visible laminated morphology appear in the culture, these are the retina organoids to dissect and place in suspension culture. At day 55 of 3D culture, media is changed to B27 with Vitamin A (Gibco) and 10% (v/v) fetal bovine serum (Gibco) for long term culture. Retina organoids are fed every 2–3 days until needed.
Ten RtOgs on day 70 of differentiation were randomly selected and cultured on the chip for a month (35 days) until day 105. The organoids were divided into two groups for gene expression qPCR analysis afterwards. The same tests were performed for RtOgs in the static culture group.
Bioreactor system assembly and organoid loading
The chip and the associated tubing were disinfected with 70% ethanol and 30 min. in a UV and ozone cool clave (CoolCLAVE Plus) (Genlantis, San Diego, CA, USA). Each chamber was treated with anti-cell adherence solution twice (STEMCELL Technology) and washed by Dulbecco’s phosphate-buffered saline (DPBS) without calcium and magnesium (STEMCELL Technology). The on-chip culturing system was assembled as shown in Fig. 2F. The media reservoir was comprised of a 50 mL Steriflip-GP sterile centrifuge tube (MilliporeSigma, Burlington, MA, USA) and a filter cap with a pore size of 0.22 μm.
Before loading the organoids, the chip chambers were sealed by pasting a slice of MicroAmp™ optical adhesive film (Thermo Fisher Scientific, Waltham, MA, USA) on the top surface. Then the syringe was slowly withdrawn to apply negative pressure to fill the channel with fresh media drawn from the media reservoir. Tubing clamps were then applied to block both the inlet and outlet tubing, so that the adhesive film could be removed without disturbing the fresh media level in the channel. One organoid was loaded into each chamber by 20 μL pipette tips with tips cut off. Lastly, the top of the chambers was resealed with sterile optical adhesive film (Fig. 2E). The flow rate used for long-term culture was 250 μL/h. Under this flow rate, 50 mL media was sufficient for about 8 days of culture. When changing the media, the inlet and outlet tubing were clamped, and fresh media was refilled in the centrifuge tube. All these steps were performed in an ESCO Class II Type A2 biosafety cabinet (Labculture, ESCO) (ESCO Micro Pte. Ltd., Singapore) to avoid contamination.
In vitro dye test
The dye test experiment was performed to compare the uniformity of the concentration in chambers between the four different channel designs. Four chips with 3×3 chamber array were fabricated with 2-mm chamber height. The channels were first filled with blue food dye solution following similar steps as the organoid loading procedure (Fig. 2E). The flow was then blocked by clamping both the inlet and outlet tubing, and the inlet was switched to a yellow dye solution. Lastly, a syringe pump was used to draw the yellow dye solution into the chip at a rate of 600 μL/h. The whole flow process was recorded with a camera. The grayscale value of each chamber was obtained by ImageJ to quantify concentration changes of each chamber from the images.
Fluorescence life-time imaging
Fluorescence lifetime imaging (FLIM) was used to study the intrinsic fluorophore Nicotinamide adenine dinucleotide (NADH) in the RtOg. The fluorophore’s emission decay curve was obtained by photon counting to calculate the fluorescent lifetime (SFig. 1A). FLIM data was displayed on a phasor plot after Fourier transform, with the intensity decay curve of fluorescence for each pixel represented by the g and s coordinates. Using this method, the decay and spectrum for each pixel could be depicted on the phasor plot (SFig. 1B).
The metabolic trajectory was visualized using the phasor approach47. The phasor plot has a universal circle, with boundaries of each point representing a single exponential lifetime of one type of molecule. Different components on the phasor plot followed a linear relationship, thus, the ratio of the linear combination could be used to determine the fraction of each component. The lifetime of free and lactate dehydrogenase-bound NADH was about 0.37 ns and 3.4 ns, respectively48. Free NADH was linked to more glycolysis and a more proliferative state, while bound NADH was correlated with more oxidative phosphorylation and a more differentiated state49. The lifetime of lipid was 7.89 ns associated with long lifetime species (LLS) (SFig. 1B), the presence of which indicated oxidative stress50. The fraction of each component was calculated as SFig. 1B suggested, F1/F2 was the free/bound NADH ratio, and F3 was the ratio of LLS. Based on the above mechanism, we evaluated the metabolic state of RtOgs quantitatively by calculating the free/bound NADH ratio and LLS ratio in representative image cross-sections. Qualitatively, the metabolic differences were visualized by applying a pseudo color gradient to the phasor plot (SFig. 1C).
Images were taken by a Zeiss LSM 780 microscope using a Plan-Apochromat 20x/0.8 M27 objective (Carl Zeiss, Jena, Germany). The excitation wavelength was 740 nm, produced by Mai Tai multi-photon laser source (Spectra-Physics Mai Tai, Mountain View, CA). Imaging settings used were as follow: 256 × 256 frame size, 1.66 μm pixel size, 25.21 μs pixel dwell time and 8-bit pixel depth. Emission laser passed through an MBS 690+ and an SBS SP 610 filters and the lifetime data was collected by the photomultiplier tube (H7422p-40, Hamamatsu, Japan) and a320 FastFLIM FLIMbox (ISS, Champaign, IL). Before imaging, the system was calibrated on frequency factor and lifetime by coumarin 6 solution with the known lifetime of 2.5 ns. FLIM data were collected after 100 counts in the brightest pixel of the image were acquired. During imaging, fresh medium flowed into the bioreactor continuously, while RtOgs in static groups were moved into Nunc® Lab-Tek® II Chambered Coverglass (Thermo Fisher) for imaging.
Phase contrast imaging
The phase contrast microscopy images were acquired using an Olympus IX71 (Olympus, Tokyo, Japan) and a QICAM FAST1394 CCD camera (Teledyne QImaging, Surrey, BC, Canada) under two magnifications by UPlanFL N 4x/0.13 PhL and UPlanFI 10x/0.30 PhL objectives.
Green fluorescent protein imaging
Green fluorescent protein images were acquired using a Zeiss LSM 780 microscope using Plan-Apochromat 20x/0.8 M27 objective (Carl Zeiss, Jena, Germany). The excitation wavelength was 488nm with a pixel dwell time of 1.58 μs. We used the frame size of 512 × 512 pixels and each pixel is 0.42 μm.
Quantitative polymerase chain reaction analysis
The primers for qPCR test are listed in Table S2 (Qiagen, Germantown, MD, USA). We used 12 retinal progenitor and photoreceptor genes and 1 housekeeping gene to identify and quantify the gene expression profile in retinal organoids. Human adult retinal tissue was used as a positive control (n = 3). For CRX-GFP hESCs derived RtOgs, each sample was analyzed on days 122 to 124 of differentiation (n = 3 for both static and chip groups); for CSC14 hESCs differentiated RtOgs, each sample was analyzed on day 105 (n = 2 for both static and chip groups). Each sample consisted of 4 RtOgs. Trizol reagent (Qiagen), DNase I digestion (Invitrogen, TURBO, Waltham, MA, USA), and phenol-chloroform extraction (Fisher) were used to isolate RNA, and an RT2 cDNA synthesis kit (Qiagen) was used to synthesize cDNA. RT2 SYBR Green with ROX qPCR master mix (Qiagen) was used for amplification, which was performed under the following conditions: 95°C (15 minutes), 40 cycles at 95°C (15 seconds each), 55°C (30 seconds each) and 72°C (30 seconds each). The annealing temperature was 60°C. The double delta cycle threshold (Ct) method was used to calculate the fold expression, and day 0 undifferentiated hESC (line CSC14) was used as a control. For analysis and heatmap generation, non-detected amplification in the control tissue and organoids were assigned cycle threshold values of 40. Heat maps were generated using Graphpad Prism software (Graphpad Software LLC, La Jolla, CA, USA), the heat map has the value of log2(Fold Expression).
Single cell dissociation
Eight to twelve RtOgs on day 72 and 159 (chip vs. static, four experimental groups in total) were dissociated using papain-based enzymatic digestion by Worthington papain dissociation system (Worthington, Lakewood, NJ, USA), followed the standard dissociation protocol provided by Worthington. Briefly, the papain vial was dissolved in 5 mL of EBSS buffer in 37°C water bath for 10 minutes to yield a solution at 20 units of papain per ml in 1 mM L-cysteine with 0.5 mM EDTA. After adding 250 μl DNase (2000 units/ml deoxyribonuclease in EBSS) into the papain solution. The RtOgs were added in the papain solution and incubated at 37°C incubator on a rocker platform for 1 hour. Post incubation, the tissue was further triturated using 18G needle and syringe. The dissociated tissue mixture was centrifuged at 300g for 5 minutes at room temperature. After removing the supernatant, the cells were resuspended in the albumin-inhibitor solution with 2.7 ml EBSS, 150 μl DNase and 300 μl ovomucoid solution (10 mg/ml Egg White/BSA in EBSS). The single-cell solution was then carefully layered on top of 5 ml albumin-inhibitor solution and centrifuged at 70g for 6 minutes at room temperature. The supernatant of dead cells was discarded, and the pelleted cells were immediately resuspended in 1% BSA/PBS solution. The cell viability was tested by 0.4% trypan blue using a hemocytometer (>90%) and the concentration was adjusted to ~870 live cells/ul. The samples were sent for scRNA-seq library preparation within 5 minutes.
Single-cell RNA-seq library preparation
Sequencing libraries were prepared using the protocol from 10X Genomics Chromium Single Cell 3’ Reagent Kit v3.1 (10X Genomics, Pleasanton CA). Briefly, the 10X workflow was followed using 10,000 cells as the capture target. The resulting Gel-in-Emulsions (GEMs) were transferred to PCR tubes and incubated in a Bio-Rad C1000 Thermocycler (Bio-Rad Laboratories, Hercules, CA) for the reverse transcription protocol. The GEMs were cleaned up using Dynabeads MyOne SILANE (Life Technologies, Carlsbad CA) and then amplified using 11 cycles according the 10X workflow. The cDNA was cleaned using 0.6X SPRIselect (Beckman Coulter, Indianapolis, IN) size selection and then quality control assays using Qubit DNA HS assay (Life Technologies, Carlsbad CA) and Agilent 2100 Bioanalyzer DNA HS (Agilent, Santa Clara, CA) were performed. The endogenous cDNA fraction was then processed according to the 10X workflow for library construction. The cDNA was fragmented, end repaired and then A-tailed. After a SPRIselect cleanup the adapters were ligated on the cDNA. Sample indexes were added by PCR and a double-sided size selection using SPRIselect was performed. The libraries were assayed for quality using Qubit DNA HS assay, Agilent 2100 Bioanalyzer and quantified by Kapa qPCR Library (Roche, Basel, Switzerland) quantification for Illumina platform. The libraries were sequenced in the Illumina NovaSeq 6000 (Illumina, San Diego, CA) using 28 cycles for read 1, 8 cycles for the index read and 100 cycles for read 2.
Single-cell RNA-seq data analysis
Raw reads were first subjected to quality control QC analysis with FASTQC software and aligned to the reference transcriptome Grch38 using a short-read aligner STAR68 through the 10X pipeline software cellRanger v.3.1.0. Gene level expression for each valid cell was then quantified using UMI (Unique Molecular Identifier) and normalization was performed. Dimension reduction was then used to visualize and explore major features in single cell RNA-seq data. PCA, t-distributed Stochastic Neighbor Embedding (t-NSE) and UMAP was performed using cellRanger followed by unsupervised clustering methods such as K mean clustering to identify sub populations and cell types in the sample. Loupe browser v.5.0.1 was then used to visualize the further explore marker gene expression.
Immunohistology
RtOgs were fixed with cold 4% paraformaldehyde in 0.1M Na-phosphate buffer for 1 hour, cryoprotected (30% sucrose) and frozen in optimum cutting temperature (OCT) compound (PolarStat Plus, StatLab, McKinney, TX, USA). Organoids were then cryo-sectioned into 10 μm serial sections and stored at −20°C. Histo-VT One (Nacalai) was used for antigen retrieval at 70°C. Primary and secondary antibodies used are listed in Table S3. Organoid sections were incubated in primary antibody dilutions at the concentrations listed overnight at 4°C. The following day, sections were left incubating in primaries at room temperature for an hour before washing. Sections were then incubated at room temperature for at least 30 minutes in fluorescent secondary antibodies. Following 30 minutes of incubation in 4,6-diamidino-2-phenylindole (DAPI) at a concentration of 50 μg/ml, slides were coverslipped using Vectashield Vibrance Antifade Mounting Medium (Vector Labs, Burlingame, CA, USA).
Fluorescent sections were imaged using a Zeiss LSM700 confocal microscope (Zeiss, Oberkochen, Germany). Tiled stacks of 5–8 μm thickness were taken at 20X and 40X magnifications. Images were extracted using the Zen 3.3 Software (Zeiss). Regions of interest for cell counting were outlined in Adobe Photoshop software (San Jose, CA, USA). Cell counting was performed using ImageJ Software (U.S. NIH).
SEM & TEM sample preparation and imaging
Samples were fixed in Karnovsky’s fixative (2% Paraformaldehyde/2.5% Glutaraldehyde in 0.2 M sodium cacodylate buffer) and stored at 4°C overnight. The tissue was then washed by 0.1 M cacodylate buffer and post fixed in the solution (1:1 mixture of 0.1 M cacodylate buffer: 0.2 M cacodylate buffered 2% osmium tetroxide) for 2 hours on ice. The tissue was dehydrated in 35%, 50%, 70%, and 95% ETOH for 15 minutes each.
The organoids were cut into halves in 100% ETOH and washed again with 100% ETOH. Starting from this step, half of each organoid was prepared for SEM and the other half was used for TEM. For TEM samples, after two changes of propylene oxide (15 minutes each), the tissue was then infiltrated in a 1:1 mixture of propylene oxide:Epon Araldite resin overnight. The next morning, this mixture was changed out to fresh Epon Araldite for 2 hours. The sample was then placed into flat embedding molds and polymerized at 60°C for 48 hours. The resin blocks were then cut by Leica EM UC7/FC7 cryo-ultramicrotome (Leica, Wetzlar, Germany). The TEM used in this study was JEOL 2100 (JEOL USA Inc, Pleasanton, CA, USA). The montages were processed by the program Etomo (University of Colorado, Boulder).
For SEM samples, the organoids samples were processed by a Leica critical point dryer. The surface of the sample was sputter coated with platinum using a Leica ACE200 sputter coater before imaging. The SEM used in this study was FEI Magellan 400 XHR (FEI Company, Fremont, CA, USA) with an Everhart-Thornley detector (ETD) and a Through-the-Lens detector (TLD).
Statistical analysis
Data in the plot were presented as means with standard deviations. Graphpad Prism software was used for all statistical analyses. In the GFP MFI, immunohistology cell count, free/bound NADH and LLS ratio figures, one-way ANOVA tests were performed. In the qPCR heatmap, two-way ANOVA tests were performed. The significance was determined by a p value less than 0.05.
RESULTS
Microfluidics design and testing
The bioreactor chip was designed with the distance between chambers matching a 96-well plate to retain microscope compatibility. Preliminary designs in which chambers were located on one side of a 1000 μm wide perfusion channel revealed two problems: 1) heterogeneous media concentration changes between chambers and 2) low mass transfer efficiency (SFig. 2). Therefore, not all wells in the preliminary design received comparable fresh media exchange. To optimize the design and improve mass transfer rates, three different variables were evaluated with COMSOL simulation: channel width, channel alignment and chamber height.
The channel width determined the cross-section area and thus affected the flow velocity (v) as indicated in Equation (1). Holding volume flow rate (Q) constant, the larger the cross-sectional area (A), the slower the flow velocity (v) would be.
| (1) |
According to the definition of Péclet number (PeL, the ratio of advective transport rate to diffusive transport rate, Equation (2)), a larger flow rate would lead to a higher advective transport rate, accelerating mass transport.
| (2) |
where L is the characteristic length, v the local flow velocity, and D the mass diffusion coefficient. Therefore, narrowing the channel width would facilitate an increase in flow velocity. Based on the simulation results, under both flow rates, the narrow channel (500 μm) designs showed faster mass transfer (Figs. 3A–B) and therefore, a theoretically faster delivery of media to each culture chamber.
Figure 3: COMSOL simulation and dye test of 4 different channel designs.
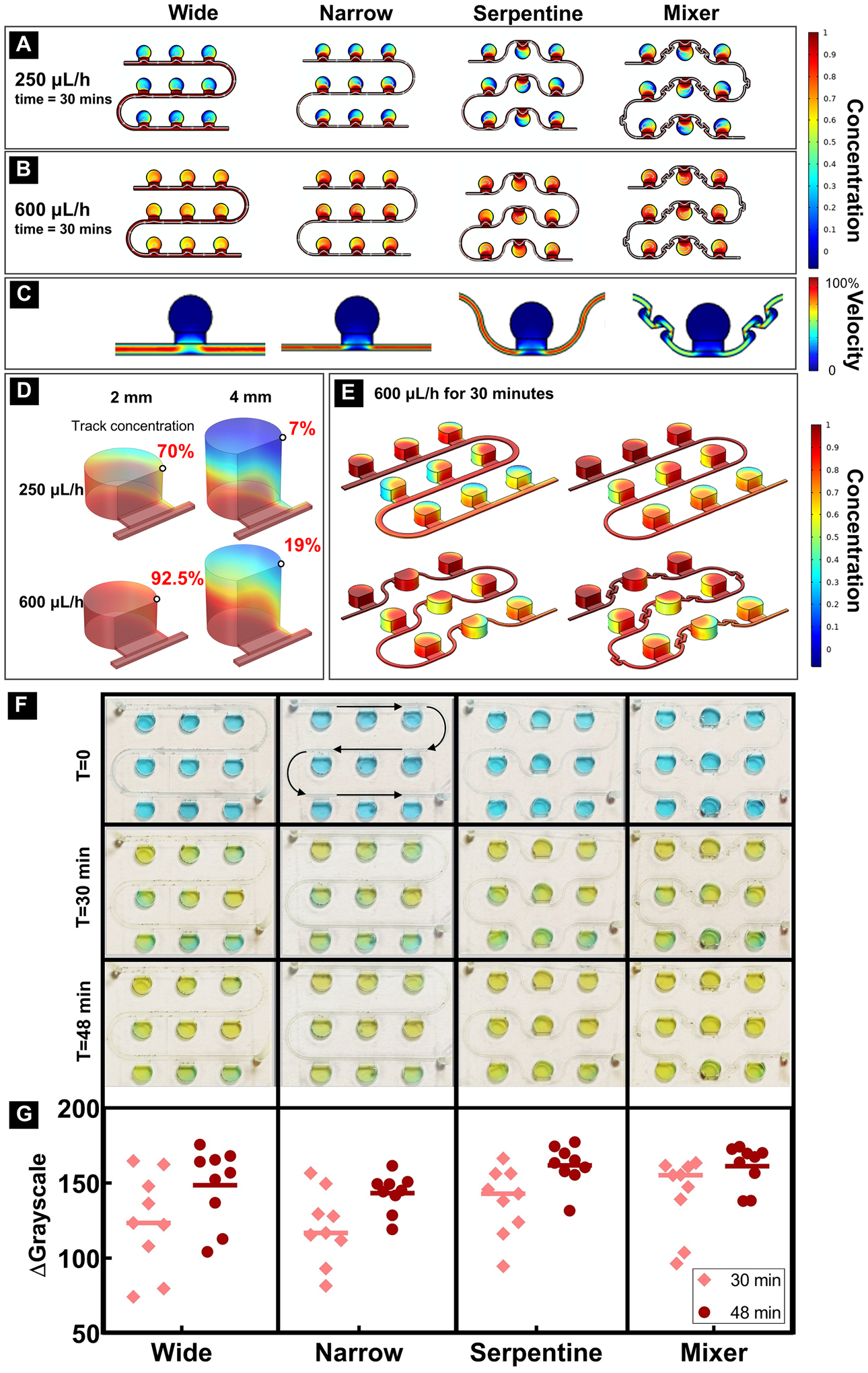
(A) Concentration distribution after 30 minutes of slow flow (250 μL/h); (B) Concentration distribution after 30 minutes of fast flow (600 μL/h); (C) Velocity distribution – zero velocity in all chambers demonstrated shear stress-free culture environment. A single culture chamber and adjacent flow channels is shown because focal flow velocity was identical for every culture chamber and interconnecting microfluidic channels in the linear series; (D) Mass transfer efficiency comparison between different height chambers under two different flow rates after 30 minutes. Black circles represent the location of concentration determination at 30 minutes; (E) 3D concentration pattern of four different designs. (F) Diffusion pattern of four different designs (flow rate was 600 μL/h); (G) Grayscale change of each well after 30 minutes and 48 minutes.
Incompressible fluid flow within the microfluidic device, due to its small size, should be laminar with a parabolic velocity profile when fully developed. As a result, the velocity next to the channel walls should be close to zero. Thus, the designs with all chambers on one side of each channel should show a higher velocity in the fluid close to the channel wall connected to a chamber than the flow velocity on the opposite wall without a connected chamber. Figs. 3A–B demonstrate that single-sided channel had a concentration gradient from the first chamber to the last chamber in each row and then entire series of chambers. This difference was even more pronounced in a larger series of 5 × 6 chambers (SFig. 2). To minimize this effect, a serpentine channel was designed to promote comparable media diffusion from both sides of the channel. To further improve concentration distribution, a mixer unit was added between each chamber51. Simulation demonstrated that narrow channels with or without mixer showed comparable qualitative performance as indicated with the color map representation of the concentration variations between the first and last culture chambers in each row and those between rows.
The third variable evaluated with simulation was the chamber height. By tracking the point concentration on the same top corner of each chamber, the 3D COMSOL simulation results showed that doubling the height of the chamber to 4 mm caused a dramatic change (4~10 folds difference) in mass transport efficiency (Fig. 3D). To maximize the transport efficiency, we chose 2 mm as our final chamber height for bioreactor fabrication. Fig. 3E shows the 3D concentration patterns in four different bioreactor designs.
To confirm simulation results and examine the functionalities of the four designs, a dye test was performed to confirm the optimum design for culturing RtOgs. A 3 × 3 chamber array was fabricated for each channel design with a 2 mm tall culture chamber. Blue dye was used to fill each channel followed by 30 and 48 minutes of 600 μL/h flow of yellow dye (Fig. 3F). The grayscale photogrammetry from pictures taken on each chamber were quantified (Fig. 3G). The serpentine channel with mixer design showed the smallest standard deviation, indicating that this design had the most uniform concentration among the four. The serpentine channel without mixer exhibited the next best performance based on variability after 48 minutes of flow. The simple serpentine channel without an integrated mixer showed higher fabrication success with 3D printing and lower probability of trapped air bubbles in the microfluidic channels than the serpentine channels with mixer.
Retinal organoid culture methods comparison
Phase contrast imaging
Representative phase contrast images in SFigs. 3A–D showed the key stages of RtOg differentiation from human embryonic stem cells. The EZSPHERE microwell aggregated stem cells into uniformly sized embryonic bodies which were then plated on Matrigel coated dishes. Eye fields cut from Matrigel were maintained in ultra-low attachment 24-well plate as they assembled into RtOgs. In this study, RtOgs were put on the bioreactor on days 41, 87 and 128 of differentiation, respectively.
At an early differentiation stage from day 41 to day 71, RtOgs in both static and bioreactor groups showed a significant size change (Figs. 4A–B) and developed hollow center and transparent edge. Figs. 4C–D showed a representative RtOg in both groups on day 88 and day 124 of differentiation, respectively. The transparent and laminar outer surface, which was observed in both groups, indicated the development of photoreceptor layer. In later differentiation stages from day 128 to day 158, the RtOg’s edge became more mature and developed outer segment-like structures on their surface (Figs. 4E–F). Overall, there was no observable morphological difference between static and bioreactor cultured organoids.
Figure 4: Representative phase contrast images of organoid differentiation in bioreactors and static culture during different stages of development.
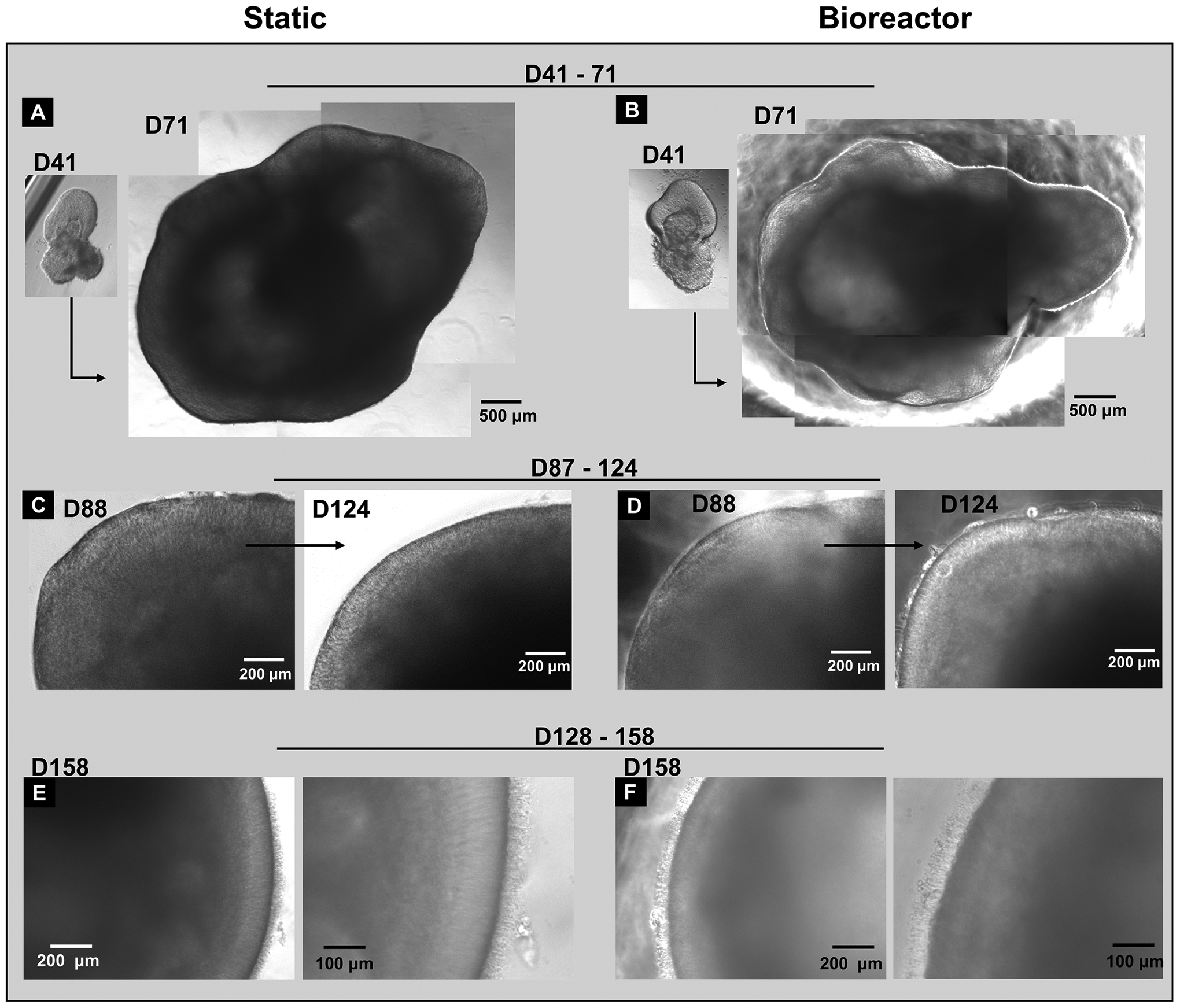
The same RtOg in static culture (A) and bioreactor culture (B) from day 41 to 71 demonstrating the magnitude of RtOg growth (Day 41 insets share the same 500 μm scale bar as Day 71 larger insets); (C) The same RtOg in static culture from day 88 to 124; (D) The same RtOg in bioreactor culture from day 88 to 124; (E-F) RtOgs on day 158 of differentiation showed outer segment structures in both static and bioreactor groups. (C-F) Higher magnification figures were shown on the right.
Fluorescence lifetime imaging
The bioreactor chip platform was continuously supplied with nutrients while the RtOgs in conventional dish culture received nutrient exchanges every 3 days. FLIM was used to measure the metabolic activity in a non-invasive and non-destructive way as described in the method section.
Four imaging modalities were used to visualize the same cross-section in RtOgs. Conventional fluorescence microscopy demonstrated green fluorescent protein (GFP) in photoreceptors and their progenitors in the CRX-GFP organoids (SFigs. 3A–D). Multiphoton infrared stimulation was used to acquire total autofluorescent images showing the total NADH (Figs. 5A–D), which delineated cellular structures and viability of RtOgs. Multiphoton lifetime imaging revealed metabolic changes in NADH from its free to bound form and their associated free:bound ratio (f/b NADH) (Figs. 5E–H). Long lifetime species analysis highlighted oxidative stresses in the tissues (Figs. 5I–L). The above two values were calculated based on the location of the datapoints on the phasor plot (Fig.5M).
Figure 5: Qualitative and quantitative comparison of RtOgs in two culture methods.
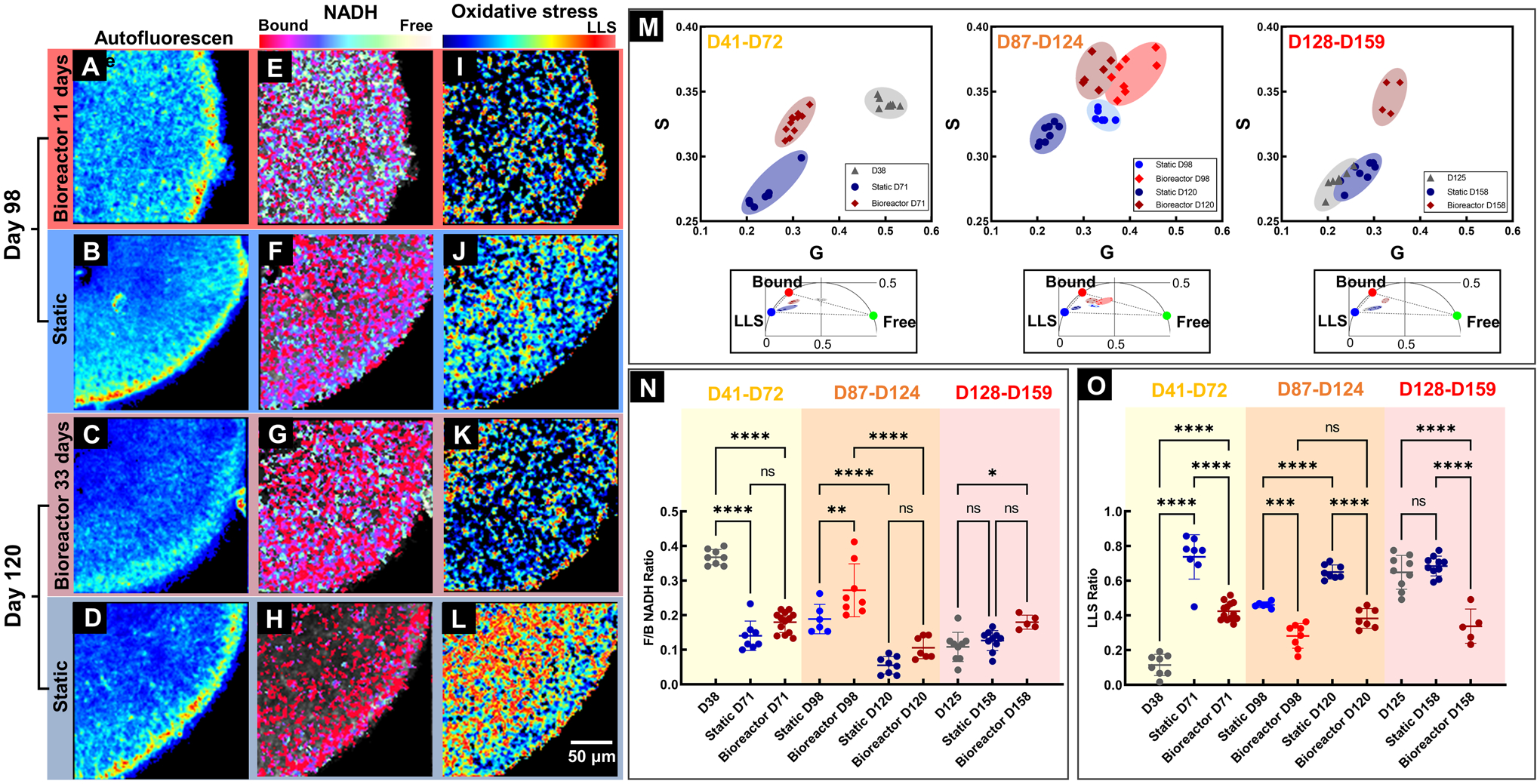
(A-D) Total NADH autofluorescence images demonstrated the cellular structures within RtOg cross sections; Pseudo color-coded free/bound NADH distribution (E-H) and LLS distribution (I-L) images were generated based on two photon lifetime within the 2-dimensional phasor space; (M) Scatter plots of and the clustering of different groups of RtOgs on the FLIM phasor diagram; (N) Plot of free/bound NADH ratio to evaluate metabolism (higher f/b value represented glycolysis, and lower f/b indicated greater oxidative phosphorylation.) Metabolism is not significantly different between static and bioreactor RtOgs after 1 month in culture for RtOgs of different ages; (O) Plot of LLS ratio to evaluate oxidative stress. LLS is significantly different between static and bioreactor maintained RtOgs of different ages after 1 month in culture. The values of f/b NADH ratio and LLS ratio reflect the average lifetimes of the organoids cross-section imaging frame. (One-way ANOVA test was performed: D38, n = 8; Static D71, n = 8; Bioreactor D71, n = 13; Static D98, n = 6; Bioreactor D98, n = 8; Static D120 n = 8; Bioreactor D120, n = 7; D125, n = 9; Static D158, n = 10; Bioreactor D158, n = 4; The RtOgs placed into the bioreactor D41–72 were imaged on D38 at the outset of the experiment. The RtOgs placed into the bioreactor D128–159 were imaged on D125 at the outset of the experiment.)
RtOgs at different differentiation stages were imaged. For the D41–72 and D128–158 groups, RtOgs were imaged 3 days before the bioreactor and static comparison experimen started (D38 and D125). After approximately one month of culture under two conditions, RtOgs were imaged again at the endpoint (D71 and D158). For the D87–124 group, RtOgs were imaged at two time points (D98 and D120) during the culture period.
RtOg differentiation in both static and bioreactor groups demonstrated a shift from more glycolytic to more oxidative phosphorylated metabolism according to the f/b NADH ratio (Fig.5N). On day 38 of differentiation the f/b ratio was the highest (Fig.5N). The pseudo color-coded f/b NADH distribution from day 98 to day 120 of differentiation visualized the developmental trend from more glycolytic (yellow-green) to more oxidative phosphorylation (red) (Figs. 5E–H). A higher total fluoscence NADH metabolic signature was present in bioreactor cultured organoids (Comparing Figs. 5G and 5H). When comparing the f/b ratio of bioreactor and static culture RtOgs, no significant difference was identified in the f/b NADH ratio on day 71, 120 and 158 (Fig. 5N).
LLS is a marker for oxidative stress and RtOgs in both groups showed a significant increase of LLS ratio over time (Fig.5O). RtOgs on the bioreactor experienced significantly lower LLS signatures on FLIM imaging than RtOgs in static culture at all imaged timepoints (Fig.5O). False color LLS images showed a distinct color difference between two groups (Figs. 5K–L). Fig.5L highlights that the innermost layer (where progenitor cells, ganglion cells and Müller glia are located) of the static cultured organoid experienced a higher LLS signal (more red) than the bioreactor cutlured RtOgs. The outer layer (where photoreceptors are located) of static cultured RtOgs experienced lower LLS signal (more blue) than bioreactor cultured RtOgs. The time-dependent metabolic shifts and the metabolic difference between two groups were visualized on G-S phasor plot, which highlights the metbolic fingerprint of RtOgs before and after culture in static or bioreactor conditions. The G-S plot demosntrates differential clustering of RtOgs cultured under static or bioreactors conditions at 3 stages of differentiation (Fig. 6M).
Figure 6: Gene profiles of RtOgs at different ages.
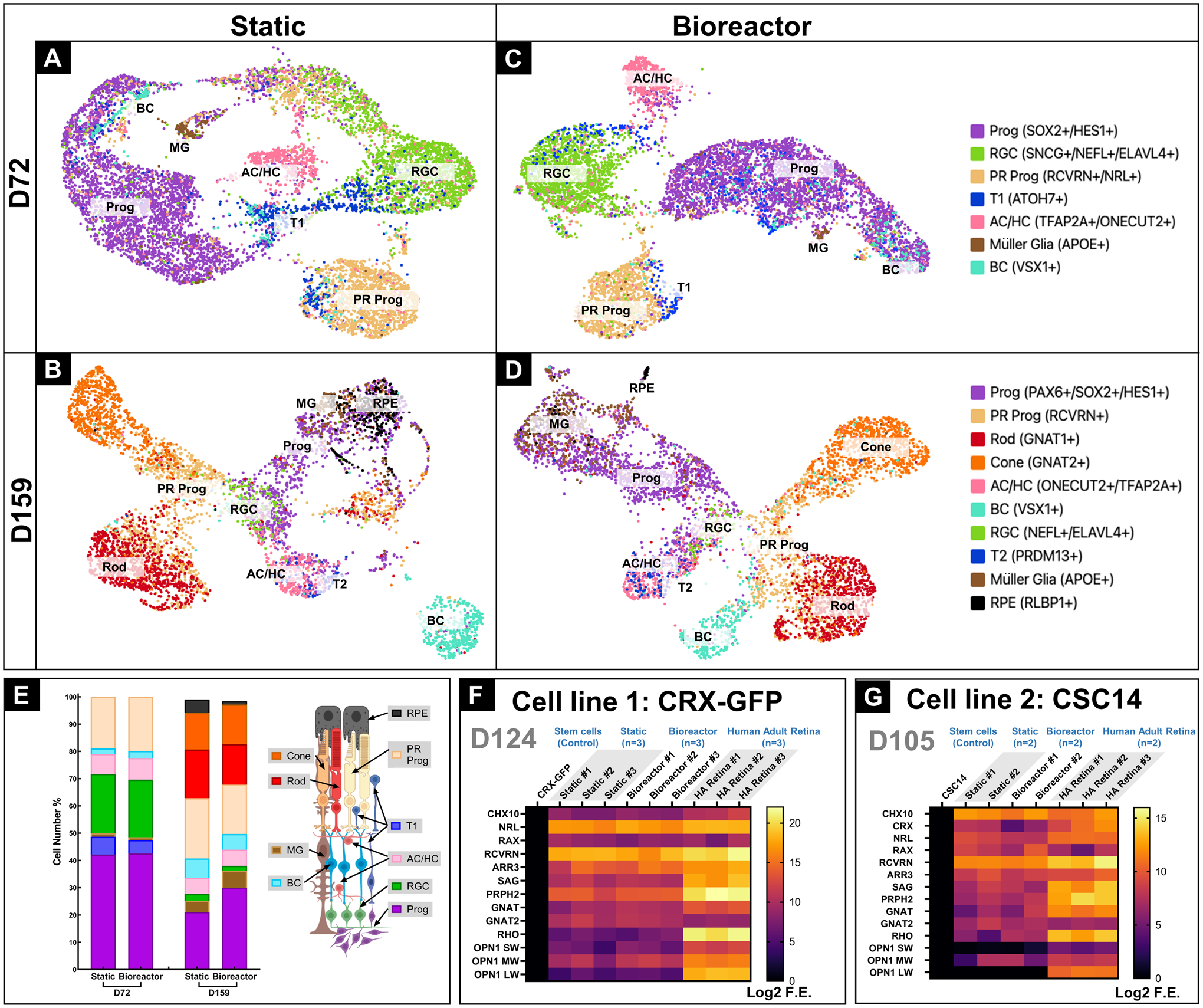
Single-cell RNA seq generated UMAP identified cell types of RtOgs cultured under static on day 72 (A) and day 159 (B),under bioreactor culture on day 72 (C) and day 159 (D); (E) Cell number quantification: Cell number percentage of different type of cells, organized by the order of photoreceptor layers and the schematic image was shown on the right side; (F) qPCR gene analysis of CRX-GFP hESCs (negative control) generated RtOgs on day 124 of differentiation; (G) qPCR gene analysis of CSC-14 hESCs (negative control) generated RtOgs on day 105 of differentiation; Log2 F.E – Log2 (Fold Expression); Cell identities in (A-E): Prog – retinal progenitor cell; RGC – retinal ganglion cell; PR prog – photoreceptor progenitor cell; T1 – transition phase 1; AC/HC – amacrine cells and horizontal cells; BC – bipolar cells; T2 – transition phase 2; RPE – retinal pigment epithelium cell.
Gene expression profile by single-cell RNA sequencing and qPCR
We focused on the gene expression profile of RtOgs at several stages of differentiation and compared their cellular profiles maintained in bioreactors and static culture condition. We used scRNA seq to study static- and bioreactor-cultured RtOgs on day 72 and day 159 of differentiation. The genes to distinguish and identify specific cell populations were previously described52, 53 (Figs. 6A–D). We also performed qPCR analysis for two different stem cell lines – CRX-GFP (day 124) and CSC-14 (day 105) (Figs. 6F–G).
Single-cell RNA seq provided a comprehensive overview of cell types within RtOgs. RtOgs that had been maintained in either static culture or bioreactor culture for approximately 1 month were studied at two different time points: D72 and D159. For both static and bioreactor cultured RtOgs, the three predominant cell types on day 72 were retinal progenitor cells (Prog), retinal ganglion cells (RGC) and photoreceptor progenitor cells (PR Prog). Many cells were also in the transition phase 1 (T1) as identified by ATO7 (a marker cells differentiating from retinal progenitor cells to other cells types)52, 53(Figs. 6A, C). The population difference of each cell type between static and bioreactor group on day 72 was very small (Fig. 6E).
Within mature RtOgs after 1 month of static or bioreactor culture on day 158, more advanced cell types emerged and formed more distinct clusters on the scRNA seq UMAP (Figs. 6B, D). The percentage of RGC decreased, while the proportion of bipolar cells (BC) and Müller glia (MG) increased. PR progenitor cells further differentiated into rods and cones. Compared to static culture RtOgs, those in the bioreactor group contained a higher percentage of retinal progenitor cells. The bioreactor group showed a similar population of rods and cones, while static group RtOgs contained more rods. Both groups have differentiated cell types that corresponded to cell types found in vivo mature human retina.
Analysis using qPCR included a short list of retinal genes (detailed information in Table S2). RtOgs derived from CRX-GFP hESCs expressed retinal progenitor genes (CHX10, NRL and RAX) that were comparable to those of human adult retina in both static and bioreactor groups (Fig. 6F). Both groups also expressed rod and cone genes including RCVRN, ARR3, SAG, PRPH2, GNAT and GNAT2. However, both static and bioreactor cultured RtOgs showed low mature photoreceptor gene expression. Gene expression levels by qPCR were not significantly difference between the static and the bioreactor groups (two-way ANOVA test, p>0.05). Similar results were obtained from the CSC-14 hESCs derived RtOgs at 105 days of differentiation (Fig. 6G); there was no significant gene expression difference between static and bioreactor culture conditions (two-way ANOVA test, p>0.05).
Immunohistology and electronic microscopy
RtOgs maintained in both conventional static culture or the bioreactor for approximately 1 month were fixed on day 72 and 159 of differentiation. Cryostat sectioning was performed to acquire immunohistology images to visualize cell types and structures. On day 72 of differentiation, RtOgs in both groups demonstrated layered cellular structures (Fig.7). The apical aspect was composed of photoreceptor progenitor cells, marked by orthodenticle homeobox 2 (OTX2), and retinal progenitor cells, which were immunoreactive for visual system homeobox 2 (CHX10/VSX2) (Figs.7A, C). The basal aspect contained amacrine cells which were immunoreactive for calretinin (CAL2) (Figs.7A, C). There were also some retinal ganglion cells marked by brain-specific homeobox/POU domain protein 3A (BRN3A, also known as POU4F1) (Figs.7B, D). The expression of synaptophysin (SYP) indicated synaptogenesis (Figs.7B, D).
Figure 7: Immunohistology images of RtOgs on day 72 of differentiation after 1 month of tissue culture in static or bioreactor conditions.
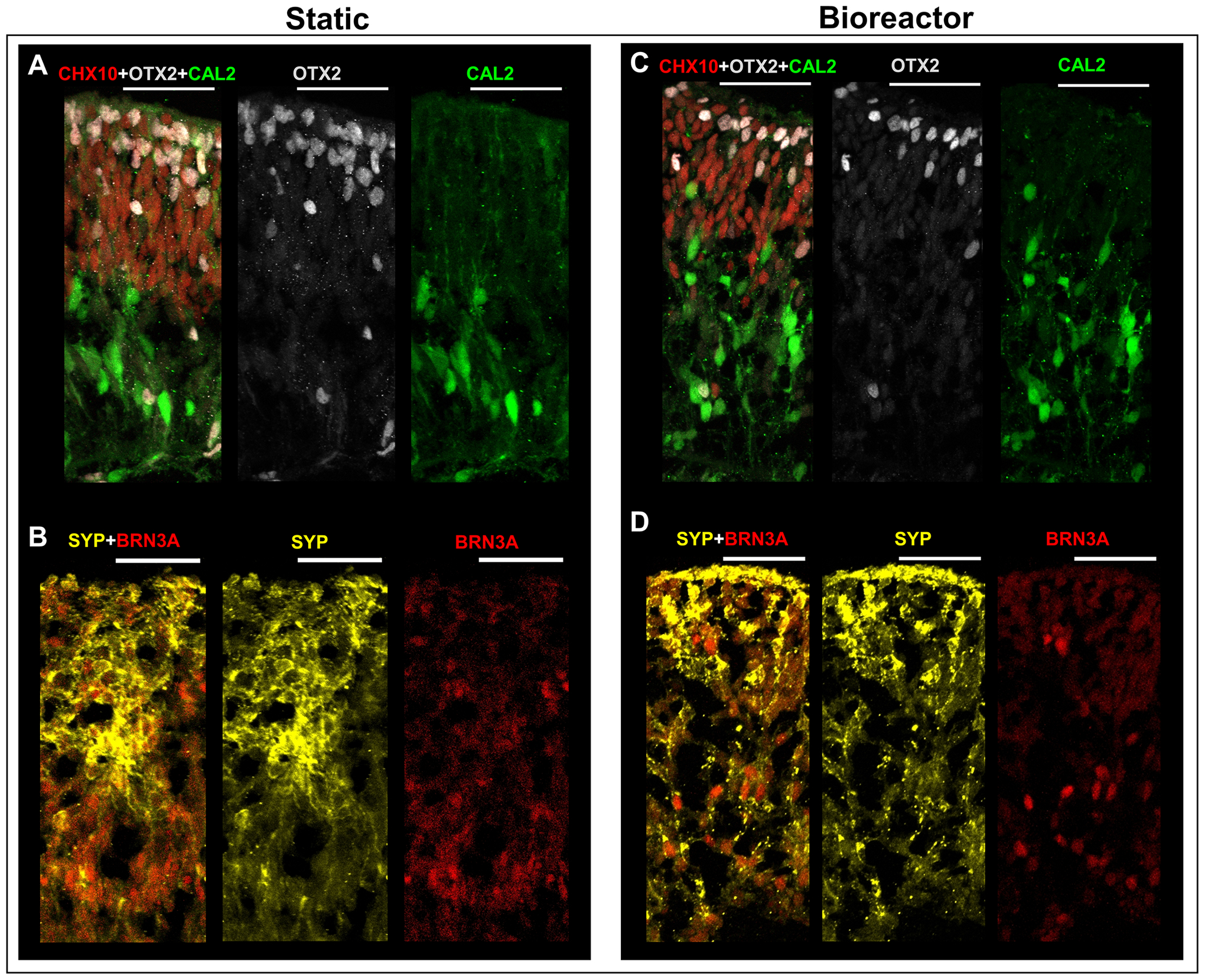
(A-B) Static cultured RtOgs; (C-D) Bioreactor cultured RtOgs. Antibody marked cells: CHX10 – retinal progenitor cells; OTX2 – photoreceptor progenitor cells; CAL2– amacrine cells; SYP – evidence of synaptogenesis; BRN3A – retinal ganglion cells. (scale bar: 50 μm)
On day 159 of differentiation, RtOgs in both groups (Fig.8) demonstrated a distinct and compact photoreceptor outer nuclear layer (ONL), marked by the immunoreactivity for neural retina-specific leucine zipper protein (NRL) (Fig.8A, D) and OTX2 (Fig.8C, F). When comparing Fig. 8A and Fig. 8D, the bioreactor group had a thicker ONL. However, this difference is not significant, as shown in the NRL+ cell counting result in Fig. 8H. Photoreceptor outer segment structures were shown on the apical aspect, marked by rhodopsin (RHO). The basal aspects were composed of retinal progenitor cells (CHX10) and amacrine cells (CAL2) (Figs. 8C, F). Rod bipolar cells immunoreactive for protein kinase (PKC)-α formed the inner nuclear layer (INL) (Figs. 8B, E). The expression of synaptophysin (SYP) indicated synaptogenesis through the inner plexiform layer (IPL) to ONL (Figs. 8B, D). High-resolution SEM images showed that RtOgs in both static and bioreactor groups differentiated matured photoreceptor cells with inner segment (IS), connecting cilium (CC) and outer segment (OS) (Fig. 8G, I). More electron microscopic images are shown in SFig. 4. Cell counting from the immunohistology staining sections showed no significant difference between static and bioreactor groups (Fig. 8H).
Figure 8: Immunohistology and SEM images of RtOgs on day 159 of differentiation.
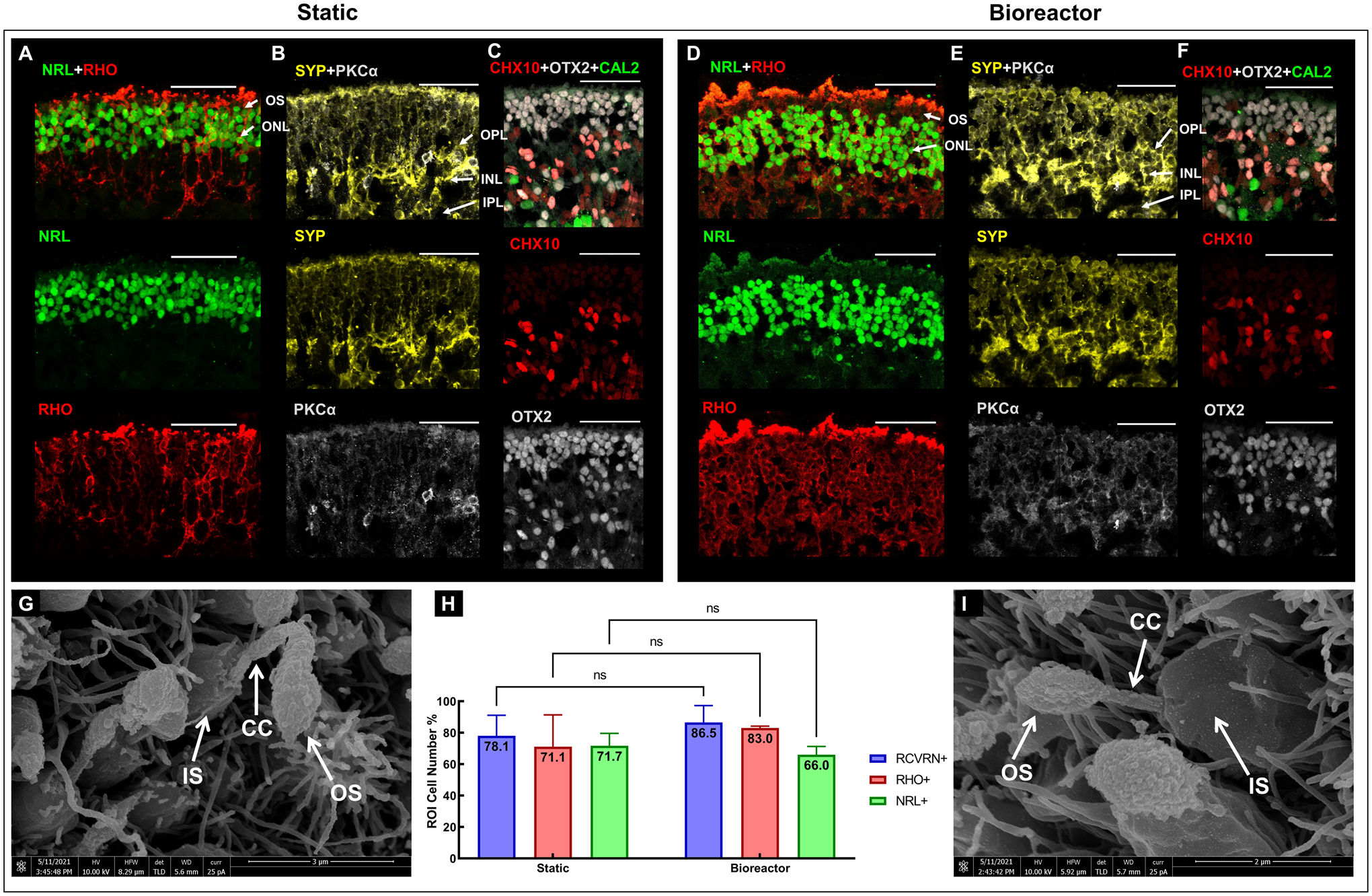
Immunostaining images of static (A-C) and bioreactor (D-F) cultured RtOgs. SEM images of static (G) and bioreactor (I) cultured RtOgs; (H) Cell counting from selected immunohistology slides (RCVRN+: nstatic = 2, nbioreactor = 3; RHO+: nstatic = 3, nbioreactor = 3; NRL+: nstatic = 3, nbioreactor = 3). Antibody marked cells: RHO – rod photoreceptors; NRL – photoreceptors; CHX10 – retinal progenitor cells; OTX2 – photoreceptor progenitor cells; CAL2 – amacrine cells; SYP – synaptophysin; PKCα – rod bipolar cells. Arrow markers: OS – outer segment; IS – inner segment; CC – connection cilium. ONL – outer nuclear layer; OPL – outer plexiform layer; INL – inner nuclear layer; IPL – inner plexiform layer. (scale bar: 50 μm)
DISCUSSION
The overall goal of this study was to improve current RtOgs culture techniques by reducing manual labor required for organoid culture and improve the RtOgs reproducibility and quality. Two main differences between in vivo retina and conventional in vitro RtOg culture were targeted: consistency of nutrition supply and fluid mechanical stability. In the human body, the visual system is the highest energy-consuming system in the brain54 and photoreceptor cells are identified as the most metabolically active cells. Retinas in the human body are continuously nourished by the dual blood supply from the choriocapillaris and the retinal vasculature. Additionally, the retina in vivo is isolated from mechanical forces, by virtue of the non-compressible fluid contents for the globe and the outermost structural support provided by the sclera. In its natural configuration, the photoreceptors are isolated from dynamic fluid forces, whereas organoids in tissue culture develop photoreceptors surrounded by turbulence in multi-well culture plates. Shear stress can impact the stem cell differentiation phenotypes55, destroy circulating tumor cells56, induce cells’ metabolite production57, and cause RtOgs to lose the outer segment like structures58. Loss of photoreceptor outer segments is also seen clinically in patients with retinal detachments who develop glaucoma as a consequence of outer segment shedding59.
Bioreactors for organoid culture to overcome the drawbacks of conventional tissue culture have been described in recent years. Existing challenges for bioreactor designs include minimizing the volume of media used, minimizing shear stresses on tissues and reducing their incompatibility with longitudinal non-invasive imaging. At the macro level, stirred and rotating wall vessel (RWV)38 platforms have been used for retinal organoid differentiation. The former has been shown to produce retinal organoids with improved laminar stratification and increased yield of photoreceptor cells with outer segment structure, with drawbacks of damage to these fragile structures from sheer stress58. The latter has the advantages of improved differentiation, easy use, and high nutrient transfer, and has also been used to culture bladder, lung, intestinal, and vaginal epithelial cell types into three-dimensional cell aggregates33,60–63. However, these larger systems share the disadvantage of high cost due to the high volume of media required to maintain the organoids36. At an intermediate scale is the millifluidic system used to manipulate fluids for organoid maintenance41. These bioreactors have been used for development of kidney organoids and long-term maintenance of human midbrain and liver organoids31, 64, 65. Millifluidic systems have the advantages of supporting relatively high flow rates, cell-cell interaction, and less frequent media changes and thus less organoid perturbation but has the disadvantage of intermediately high volume and cost, and low throughput. At the microscale, microfluidic devices have the added advantages of lower volume and lower cost compare to millifluidics41. Microfluidic devices have been used to culture human intestinal, lung, hepatocyte, and cardiac organoids,66–68. For both the milli- and microfluidic devices, shear stress can be minimized by placing organoids in wells or chambers at a set distance from the flow channels. Therefore, we sought to reduce shear stress while creating a perfused environment to house and isolate individual organoids for long term non-invasive imaging.
First, we developed a hybrid bioreactor design that incorporated both micro- and millifluidic components. This design was made possible with the novel fabrication method based on SLA 3D printers to create a mold incorporating micro-, milli- and even macroscopic features (Figs. 2A–D). 3D printing also enabled rapid prototyping bioreactor designs to iteratively optimize the design. This additive manufacturing offers cost savings and reduced facility requirement compared with traditional microfabrication methods and serves as an attractive alternative to manufacturing bioreactors69.
We used computer simulation to first demonstrate that each millifluidic culture chamber could be supplied with media from a microfluidic channel. We evaluated flow velocity inside culture chambers and found no active flow (Fig. 4C), which satisfied the design goals to minimize turbulence and shear stresses on retinal organoids by eliminating fluidic movement in the culture chamber. We further optimized channel geometry relative to the culture chambers and flow rate of media through the bioreactor. The endpoint for determining success in each design iteration was comparing uniformity of media composition in each culture chamber. We performed both COMSOL simulations in silico (Fig. 4) as well as dye tests in vitro (Fig. 5). In silico simulations demonstrated that narrow microfluidic channels (500 μm wide × 200 μm tall) allowed greater mass transfer than wider microfluidic channels (1 mm wide × 200 μm tall). We also observed in silico that high flow rate (250 vs 600 μL/hr) also improved mass transfer into culture chambers (Fig. 4B). In vitro dye tests to confirm in silico modeling predictably revealed that bioreactor designs with all culture chambers arranged on the same side of the microfluidic channel suffered from diffusion from a single side of the channels laminar flow. This resulted in the first chamber in each row of the microfluidic series to have the highest mass transfer of fresh media, while the last chamber had the lowest (Fig. 5A). To overcome this limitation, we designed a bioreactor with serpentine microfluidic flow line and culture chambers on alternating sides of the microfluidic flow line. These designs were simulated in silico to reveal improved concentration uniformity in each culture chamber compared with straight channel designs. In vitro dye testing confirmed that media concentration variability between all wells was improved by the serpentine design (Fig. 5B). Finally, we introduced mixers in the flow channel to determine if mixing would improve culture chamber concentration uniformity. In silico simulation demonstrated improved chamber concentration uniformity over the serpentine channel design (Fig. 5A). In vitro dye testing demonstrated a marginal improvement when the mixer was included than when it was not. A decision based on practical implementation was made to exclude the mixer because of the higher probability of trapping bubbles in the mixer elements as well as the mixer having tapered features that exceeded the resolution of the 3D printers employed.
A second major requirement for our design was to enable imaging of retinal organoids maintained in perfused culture. The bioreactor chip design included glass cover slips to seal the microfluidic circuit. Glass cover slips are thinner than microscope slides and, therefore, suitable for both multiphoton imaging and conventional fluorescence microscopy. Multiphoton imaging relies upon optimally efficient photon capture, and thicker glass slide reduces captured photons below threshold of practical imaging.
A third major requirement for our design is to facilitate RtOgs’ long-term maintenance in automated culture. Archberger et al. demonstrated a chip platform containing tissue chambers seeded with RPE and fed with media via a porous membrane to mimic vasculature in the retina70. This platform emphasized human physiological fidelity and minimized shear stress. However, a limitation was the relatively short 7-day maintenance of the chip platform for organoid culture. After optimizing our bioreactor design, we evaluated its performance in sustaining retinal organoids. The protocol for loading organoids into the bioreactor was determined as described in Figs. 2E–F. In this body of work, we sought to evaluate the bioreactor’s ability to maintain RtOgs for 1 month. We compared organoids in three different differentiation stages (41, 88 and 128 days) that were either placed in the bioreactor for 31 to 37 days or remained in conventional plate culture. Non-invasive functional imaging of metabolism and oxidative stress, sustained development of photoreceptors on the organoids outer layer, and terminal gene and immunohistology analysis of RtOg tissue were endpoints for comparing culture conditions. Phase contrast microscopy revealed that RtOgs maintained in conventional culture and bioreactors developed a comparable semi-translucent outer layer on day 128 and outer segment-like structures on day 158 of differentiation (Fig. 4C–F).
We previously used FLIM for live RtOg characterization71. The hypothesis in this study was that chip cultured RtOgs would experience less oxidative stress caused by reactive oxygen species (ROS), and the sufficient nutrients supply would benefit RtOgs survival and maturation. On day 38 of differentiation the f/b ratio was the highest (Fig. 5N) since the RtOgs were just cut from the Matrigel. The value decreased over time, which suggested that RtOgs were more differentiated from a stem cell state (glycolytic)72, 73. Bioreactor cultured RtOgs at all timepoints presented similar f/b NADH ratio as those in static culture, indicating similar differentiated state74 (Fig. 5N). Furthermore, organoids in the bioreactor demonstrated significant lower LLS levels suggesting that they experienced less oxidative stress than organoids maintained in conventional tissue culture while imaging (Fig. 5O). A significant increase of LLS ratio was shown over time (Fig.5O), which suggested a higher demand for oxygen and a trend to a hypoxic environment as RtOgs became more mature.
One potential problem for long-term bioreactor culture that needs to be solved is tissue adherence. We observed that some RtOgs in both day 124 and 158 groups tended to grow beside the chamber wall after 3 weeks of culture on the bioreactor. While this phenomenon was not observed in day 41 to day 72 group. Thus, adding auxiliary steps to prevent adhesion (e.g. a slow-motion rotating device) should be pursued in future refinements. Understanding of cell migration, adhesion and mechanics may be further clarified using scRNA seq.
Differences between different stem cell lines were further confirmed by qPCR. For the selected retinal genes, there was no significant difference between RtOgs maintained in conventional culture or the bioreactor in both CRX-GFP and CSC-14 hESC lines (Figs. 6F–G). However, both static and bioreactor cultured RtOgs on day 105 and 124 showed low mature photoreceptor gene expression, which was expected, as RtOgs typically do not reach full maturity until day 150 of differentiation. Immunohistology and scRNA seq analysis of organoids maintained in the bioreactor or in conventional culture showed cellular and structural similarities. Finally, we observed outer segment-like structures through high resolution SEM imaging in day 159 organoids in both culture conditions (Figs. 8G, I).
CONCLUSION
In this study, we designed and optimized a bioreactor for long term RtOg culture in a low shear stress environment that was also compatible with multimodal imaging. We found that higher flow rate through narrower channel with culture chambers on alternating sides of the perfusion channel enabled optimal and practical concentration uniformity between culture chambers. We subsequently achieved RtOgs culture on a shear stress-free micro-millifluidic bioreactor for 1 month and identified key similarities and differences between RtOgs maintained in either static culture or the bioreactor. We found that: 1) bioreactor cultured RtOgs developed cell types and morphology comparable to static cultured ones and exhibited similar retinal genes expression levels; 2) the outer surface region of bioreactor cultured RtOgs had comparable free/bound NADH ratio and overall lower long lifetime species (LLS) ratio than static culture RtOgs during imaging. Therefore, the micro-millifluidic bioreactor in this study has demonstrated its potential to sustain RtOgs of comparable quality to those maintained in static culture, while achieving this goal with reduced labor and a sheer stress-free system. Additional investigation is warranted to understand the differences in oxidative stress between RtOgs maintained in static and bioreactor tissue culture.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by CIRM (#TRAN1-10995), RPB unrestricted grant to UCI Department of Ophthalmology and ICTS KL2 (#KL2 TR001416). The authors thank Dr. Majlinda Lako (Newcastle University, UK.) for donating the CRX GFP hESC cell line, Drs. Michelle Digman (Dept. of BME, UCI) and Enrico Gratton (Dept. of BME, UCI) for their help and advice on fluorescence lifetime imaging. The authors thank the Stem Cell Research Center Core for qPCR and histology service. The authors thank Optical Biology Core Facility of the Developmental Biology Center (OBC) for imaging assistance (supported by Cancer Center Support Grant (CA-62203) and Center for Complex Biological Systems Support Grant (GM-076516) at UCI). The authors acknowledge the use of facilities and instrumentation at the UC Irvine Materials Research Institute (IMRI), which is supported in part by the National Science Foundation through the UC Irvine Materials Research Science and Engineering Center (DMR-2011967). This work was made possible, in part, through access to the Genomics High Throughput Facility (GHTF) Shared Resource of the Cancer Center Support Grant (P30CA-062203) at the University of California, Irvine and NIH shared instrumentation grants 1S10RR025496-01, 1S10OD010794-01, and 1S10OD021718-01. Figure 6E was created with BioRender.com.
Finally, we thank all the undergraduate students that participated in this project: Johnny Garcia (now B.S.), Yuming Ye, Xiyue Yao, Lelin Zhong, Jacqueline Chen, Kaylee Chew, Nicolas Lee, Vanna Giang and Mojan Rouhizadeh.
Grant support:
California Institute of Regenerative Medicine (CIRM) grant number TRAN1-10995
RPB unrestricted grant to UCI Department of Ophthalmology
ICTS KL2 Grant number is KL2 TR001416
Footnotes
CONFLICTS OF INTEREST
There are no conflicts to declare.
REFERENCES
- 1.Trapani I, Puppo A and Auricchio A, Progress in retinal and eye research, 2014, 43, 108–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dias MF, Joo K, Kemp JA, Fialho SL, da Silva Cunha A Jr, Woo SJ and Kwon YJ, Progress in retinal and eye research, 2018, 63, 107–131.29097191 [Google Scholar]
- 3.Seiler MJ, Aramant RB and Ball SL, Vision Res, 1999, 39, 2589–2596. [DOI] [PubMed] [Google Scholar]
- 4.Woch G, Aramant RB, Seiler MJ, Sagdullaev BT and McCall MA, Invest Ophthalmol Vis Sci, 2001, 42, 1669–1676. [PubMed] [Google Scholar]
- 5.Sagdullaev BT, Aramant RB, Seiler MJ, Woch G and McCall MA, Invest Ophthalmol Vis Sci, 2003, 44, 1686–1695. [DOI] [PubMed] [Google Scholar]
- 6.Thomas BB, Seiler MJ, Sadda SR and Aramant RB, Exp Eye Res, 2004, 79, 29–39. [DOI] [PubMed] [Google Scholar]
- 7.Yang PB, Seiler MJ, Aramant RB, Yan F, Mahoney MJ, Kitzes LM and Keirstead HS, Exp Eye Res, 2010, 91, 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seiler MJ, Lin RE, McLelland BT, Mathur A, Lin B, Sigman J, De Guzman AT, Kitzes LM, Aramant RB and Thomas BB, Invest Ophthalmol Vis Sci, 2017, 58, 614–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas BB, Aramant RB, Sadda SR and Seiler MJ, in Retinal Degenerative Diseases, eds. Hollyfield JG, Anderson RE and LaVail MM, Springer, New York, NY, 2006, pp. 367–376. [PubMed] [Google Scholar]
- 10.Aramant RB and Seiler MJ, Prog Retin Eye Res, 2004, 23, 475–494. [DOI] [PubMed] [Google Scholar]
- 11.Seiler MJ, Aramant RB and Keirstead HS, Optics and Photonics News, 2008, 19, 37–47. [Google Scholar]
- 12.Seiler MJ and Aramant RB, Prog Retin Eye Res, 2012, 31, 661–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radtke ND, Aramant RB, Petry HM, Green PT, Pidwell DJ and Seiler MJ, Am J Ophthalmol, 2008, 146, 172–182. [DOI] [PubMed] [Google Scholar]
- 14.Tucker BA, Park IH, Qi SD, Klassen HJ, Jiang C, Yao J, Redenti S, Daley GQ and Young MJ, PLoS ONE, 2011, 6, e18992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearson RA, Barber AC, Rizzi M, Hippert C, Xue T, West EL, Duran Y, Smith AJ, Chuang JZ, Azam SA, Luhmann UF, Benucci A, Sung CH, Bainbridge JW, Carandini M, Yau KW, Sowden JC and Ali RR, Nature, 2012, 485, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh MS, Charbel Issa P, Butler R, Martin C, Lipinski DM, Sekaran S, Barnard AR and MacLaren RE, Proc Natl Acad Sci U S A, 2013, 110, 1101–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamba DA, Gust J and Reh TA, Cell Stem Cell, 2009, 4, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacLaren RE, Pearson RA, MacNeil A, Douglas RH, Salt TE, Akimoto M, Swaroop A, Sowden JC and Ali RR, Nature, 2006, 444, 203–207. [DOI] [PubMed] [Google Scholar]
- 19.Mansergh FC, Vawda R, Millington-Ward S, Kenna PF, Haas J, Gallagher C, Wilson JH, Humphries P, Ader M and Farrar GJ, Exp Eye Res, 2010, 91, 500–512. [DOI] [PubMed] [Google Scholar]
- 20.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS and Jones JM, science, 1998, 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K and Yamanaka S, Cell, 2007, 131, 861–872. [DOI] [PubMed] [Google Scholar]
- 22.Fligor CM, Langer KB, Sridhar A, Ren Y, Shields PK, Edler MC, Ohlemacher SK, Sluch VM, Zack DJ and Zhang C, Scientific reports, 2018, 8, 14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wahlin K, Maruotti J, Sripathi S, Ball J, Angueyra J, Kim C, Grebe R, Li W, Jones B and Zack D, Journal, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakano T, Ando S, Takata N, Kawada M, Muguruma K, Sekiguchi K, Saito K, Yonemura S, Eiraku M and Sasai Y, Cell Stem Cell, 2012, 10, 771–785. [DOI] [PubMed] [Google Scholar]
- 25.McLelland BT, Lin B, Mathur A, Aramant RB, Thomas BB, Nistor G, Keirstead HS and Seiler MJ, Investigative ophthalmology & visual science, 2018, 59, 2586–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Assawachananont J, Mandai M, Okamoto S, Yamada C, Eiraku M, Yonemura S, Sasai Y and Takahashi M, Stem cell reports, 2014, 2, 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirai H, Mandai M, Matsushita K, Kuwahara A, Yonemura S, Nakano T, Assawachananont J, Kimura T, Saito K and Terasaki H, Proceedings of the National Academy of Sciences, 2016, 113, E81–E90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Llonch S, Carido M and Ader M, Dev Biol, 2018, 433, 132–143. [DOI] [PubMed] [Google Scholar]
- 29.Mellough CB, Collin J, Queen R, Hilgen G, Dorgau B, Zerti D, Felemban M, White K, Sernagor E and Lako M, Stem cells translational medicine, 2019, 8, 694–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ao Z, Cai H, Havert DJ, Wu Z, Gong Z, Beggs JM, Mackie K and Guo F, Anal Chem, 2020, 92, 4630–4638. [DOI] [PubMed] [Google Scholar]
- 31.Berger E, Magliaro C, Paczia N, Monzel AS, Antony P, Linster CL, Bolognin S, Ahluwalia A and Schwamborn JC, Lab Chip, 2018, 18, 3172–3183. [DOI] [PubMed] [Google Scholar]
- 32.Boutin ME, Hampton C, Quinn R, Ferrer M and Song MJ, Adv Exp Med Biol, 2019, 1186, 171–193. [DOI] [PubMed] [Google Scholar]
- 33.DiStefano T, Chen HY, Panebianco C, Kaya KD, Brooks MJ, Gieser L, Morgan NY, Pohida T and Swaroop A, Stem cell reports, 2018, 10, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goto-Silva L, Ayad NME, Herzog IL, Silva NP, Lamien B, Orlande HRB, da Costa Souza A, Ribeiro S, Martins M, Domont GB, Junqueira M, Tovar-Moll F and Rehen SK, BMC Dev Biol, 2019, 19, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ovando-Roche P, West EL, Branch MJ, Sampson RD, Fernando M, Munro P, Georgiadis A, Rizzi M, Kloc M, Naeem A, Ribeiro J, Smith AJ, Gonzalez-Cordero A and Ali RR, Stem Cell Res Ther, 2018, 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phelan MA, Lelkes PI and Swaroop A, Stem Cell Investig, 2018, 5, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Artero Castro A, Rodriguez Jimenez FJ, Jendelova P and Erceg S, Stem Cells, 2019, 37, 1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith D, et al. , 2014.
- 39.Berger E, Magliaro C, Paczia N, Monzel AS, Antony P, Linster CL, Bolognin S, Ahluwalia A and Schwamborn JC, Lab on a Chip, 2018, 18, 3172–3183. [DOI] [PubMed] [Google Scholar]
- 40.Sidar B, Jenkins BR, Huang S, Spence JR, Walk ST and Wilking JN, Lab Chip, 2019, 19, 3552–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beauchamp MJ, Nordin GP and Woolley AT, Anal Bioanal Chem, 2017, 409, 4311–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin D, Xia Y and Whitesides GM, Nature protocols, 2010, 5, 491–502. [DOI] [PubMed] [Google Scholar]
- 43.Collin J, Mellough CB, Dorgau B, Przyborski S, Moreno‐Gimeno I and Lako M, Stem Cells, 2016, 34, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collin J, Queen R, Zerti D, Dorgau B, Hussain R, Coxhead J, Cockell S and Lako M, Stem Cells, 2019, 37, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collin J, Zerti D, Queen R, Santos‐Ferreira T, Bauer R, Coxhead J, Hussain R, Steel D, Mellough C and Ader M, Stem Cells, 2019, 37, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong X, Gutierrez C, Xue T, Hampton C, Vergara MN, Cao L-H, Peters A, Park TS, Zambidis ET and Meyer JS, Nature communications, 2014, 5, 4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Digman MA, Caiolfa VR, Zamai M and Gratton E, Biophys J, 2008, 94, L14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ranjitǂ S, Malacridaǂ L, Stakic M and Gratton E. [DOI] [PMC free article] [PubMed]
- 49.Stringari C, Nourse JL, Flanagan LA and Gratton E, PloS one, 2012, 7, e48014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Datta R, Alfonso-García A, Cinco R and Gratton E, Sci Rep, 2015, 5, 9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong C-C, Choi J-W and Ahn CH, Lab on a Chip, 2004, 4, 109–113. [DOI] [PubMed] [Google Scholar]
- 52.Sridhar A, Hoshino A, Finkbeiner CR, Chitsazan A, Dai L, Haugan AK, Eschenbacher KM, Jackson DL, Trapnell C and Bermingham-McDonogh O, Cell reports, 2020, 30, 1644–1659. e1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J and Goldman M, Cell, 2016, 166, 1308–1323. e1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niven JE and Laughlin SB, The Journal of experimental biology, 2008, 211, 1792–1804. [DOI] [PubMed] [Google Scholar]
- 55.Wolfe RP and Ahsan T, Biotechnology and bioengineering, 2013, 110, 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Regmi S, Fu A and Luo KQ, Scientific Reports, 2017, 7, 39975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frangos JA, McIntire LV and Eskin SG, Biotechnology and bioengineering, 1988, 32, 1053–1060. [DOI] [PubMed] [Google Scholar]
- 58.Ovando-Roche P, West EL, Branch MJ, Sampson RD, Fernando M, Munro P, Georgiadis A, Rizzi M, Kloc M, Naeem A, Ribeiro J, Smith AJ, Gonzalez-Cordero A and Ali RR, Stem Cell Research & Therapy, 2018, 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwartz A, Transactions of the American Ophthalmological Society, 1972, 70, 178. [PMC free article] [PubMed] [Google Scholar]
- 60.Smith YC, Grande KK, Rasmussen SB, & O’Brien AD, 2006.
- 61.Carterson AJ, Höner zu Bentrup K, Ott CM, Clarke MS, Pierson DL, Vanderburg CR, Buchanan KL, Nickerson CA, Schurr MJ, Infection and Immunity, 2005, 73, 1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salerno-Goncalves R, Fasano A and Sztein MB, J Vis Exp, 2016, DOI: 10.3791/54148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hjelm BE, Berta AN, Nickerson CA, Arntzen CJ and Herbst-Kralovetz MM, Biol Reprod, 2010, 82, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Homan KA, Gupta N, Kroll KT, Kolesky DB, Skylar-Scott M, Miyoshi T, Mau D, Valerius MT, Ferrante T, Bonventre JV, Lewis JA and Morizane R, Nat Methods, 2019, 16, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramachandran SD, Schirmer K, Munst B, Heinz S, Ghafoory S, Wolfl S, Simon-Keller K, Marx A, Oie CI, Ebert MP, Walles H, Braspenning J and Breitkopf-Heinlein K, PLoS One, 2015, 10, e0139345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kasendra M, Tovaglieri A, Sontheimer-Phelps A, Jalili-Firoozinezhad S, Bein A, Chalkiadaki A, Scholl W, Zhang C, Rickner H, Richmond CA, Li H, Breault DT and Ingber DE, Sci Rep, 2018, 8, 2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang YS, Aleman J, Shin SR, Kilic T, Kim D, Mousavi Shaegh SA, Massa S, Riahi R, Chae S, Hu N, Avci H, Zhang W, Silvestri A, Sanati Nezhad A, Manbohi A, De Ferrari F, Polini A, Calzone G, Shaikh N, Alerasool P, Budina E, Kang J, Bhise N, Ribas J, Pourmand A, Skardal A, Shupe T, Bishop CE, Dokmeci MR, Atala A and Khademhosseini A, Proc Natl Acad Sci U S A, 2017, 114, E2293–E2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mattei G, Giusti, Serena, Ahluwalia, Arti, 2014, 2, 548–569.
- 69.Macdonald NP, Cabot JM, Smejkal P, Guijt RM, Paull B and Breadmore MC, Analytical Chemistry, 2017, 89, 3858–3866. [DOI] [PubMed] [Google Scholar]
- 70.Archberger K P. C; Haderspeck J; Bolz S; Rogal J; Chuchuy J; Nikolova M; Cora V; Antowiak L; Haq W; Shen N; Schenke-Layland K; Ueffing M; Liebau S; Loskill P , eLife 2019, DOI: 10.7554/eLife.46188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Browne AW, Arnesano C, Harutyunyan N, Khuu T, Martinez JC, Pollack HA, Koos DS, Lee TC, Fraser SE, Moats RA, Aparicio JG and Cobrinik D, Invest Ophthalmol Vis Sci, 2017, 58, 3311–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stringari C, Edwards RA, Pate KT, Waterman ML, Donovan PJ and Gratton E, Scientific reports, 2012, 2, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wright BK, Andrews LM, Markham J, Jones MR, Stringari C, Digman MA and Gratton E, Biophysical journal, 2012, 103, L7–L9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Datta R, Heylman C, George SC and Gratton E, Biomedical optics express, 2016, 7, 1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.