

Summary
Fluorescence-based sensors are powerful molecular tools for studying the spatiotemporal regulation of cell signaling, which is often organized into discrete microdomains. Here, we present a protocol for using fluorescent sensors targeted to endogenous proteins (FluoSTEPs), a new class of fluorescent sensors in which the functional probe is exclusively reconstituted at an endogenously expressed protein of interest associated with a specific microdomain. FluoSTEPs allow microdomain-specific signaling activities to be measured with high selectivity without perturbing the native stoichiometry of signaling components.
For complete details on the use and execution of this protocol, please refer to Zhang et al. (2020) and Tenner et al. (2021).
Subject areas: Cell-based Assays, Microscopy, Molecular Biology, Signal Transduction, Molecular/Chemical Probes
Graphical abstract
Highlights
-
•
Protocol for fluorescent sensors targeted to endogenous proteins
-
•
Steps for creating gene-edited cells with short fluorescent protein fragment
-
•
Details for imaging gene-edited cells and image analysis
-
•
Allows measurement of cellular signaling events at specific microdomains
Fluorescence-based sensors are powerful molecular tools for studying the spatiotemporal regulation of cell signaling, which is often organized into discrete microdomains. Here, we present a protocol for using fluorescent sensors targeted to endogenous proteins (FluoSTEPs), a new class of fluorescent sensors in which the functional probe is exclusively reconstituted at an endogenously expressed protein of interest associated with a specific microdomain. FluoSTEPs allow microdomain-specific signaling activities to be measured with high selectivity without perturbing the native stoichiometry of signaling components.
Before you begin
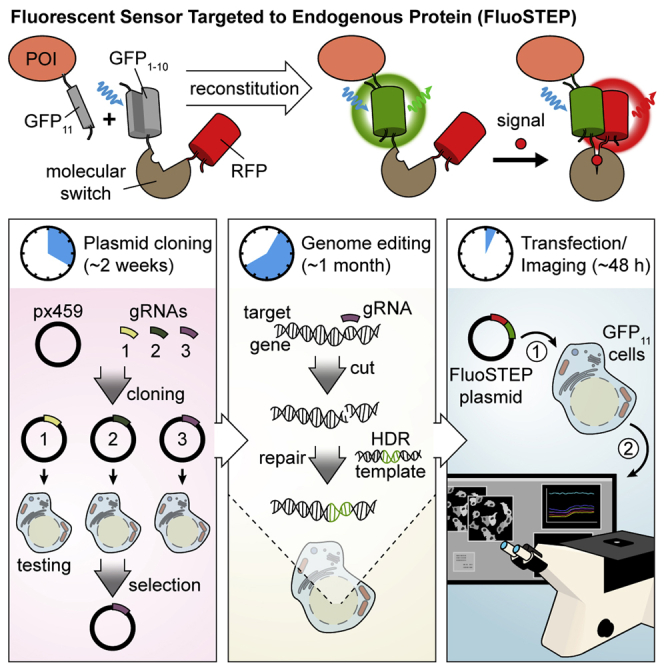
Design of FluoSTEPs
To achieve specificity, intracellular signaling pathways are organized into numerous compartments, or microdomains, with distinct signaling dynamics. These compartments are often formed through the assembly of various signaling proteins, such as a scaffolding proteins, signaling enzymes, and receptors. Genetically encoded fluorescence-based biosensors are instrumental in measuring the unique spatiotemporal dynamics associated with these signaling microdomains (Zhang et al., 2021). Fluorescent sensors typically contain one or more fluorescent protein(s) coupled to a molecular switch that is sensitive to a molecule of interest such as a signaling molecule. The conformation of the molecular switch modulates the fluorescent properties of the sensor based on the real-time signaling state. For the interrogation of specific signaling microdomains, a genetically encoded fluorescent probe can be directly fused to a protein of interest (POI) that localizes to the microdomain. However, this strategy necessarily involves overexpressing the POI, which can perturb the native signaling environment.
The purpose of FluoSTEPs is to allow monitoring of native signaling activities around a POI without overexpressing the POI. To achieve this, the fluorescent sensor is split into two components (Figure 1): 1) the 11th strand of superfolder GFP (GFP11) tagged to the POI and 2) the remaining strands of superfolder GFP (GFP1-10) tethered to RFP and a molecular switch sensitive to a molecule of interest. Although CFP and YFP are a more optimal FRET pair than GFP and RFP, reconstitution of split GFP yields brighter fluorescence compared with split CFP and is thus more robust for protein tagging (Leonetti et al., 2016). When both components are present, GFP spontaneously reconstitutes and thus can act as a Förster Resonance Energy Transfer (FRET) donor, allowing the sensor to function. With the FRET donor now available, FRET between GFP and RFP can occur, and the FRET efficiency will be regulated by the molecular switch, which changes its conformation in response to a specific signaling event. To utilize FluoSTEPs, the GFP11 fragment is attached to the POI at its endogenous genomic locus via CRISPR/Cas9. The first step is achieved through GFP11 knock-in at the gene encoding the POI via CRISPR/Cas9. Following the generation of this gene-edited cell line, component 2 is transiently transfected to permit real-time fluorescence imaging of the sensor. This way, the signaling activities surrounding the POI at the native expression level can be monitored to interrogate the dynamic regulation and functional behaviors of the POI.
Figure 1.

Schematic of FluoSTEP
To measure the signaling activities around a protein of interest (POI) expressed at endogenous levels, the POI is tagged with GFP11 via CRISPR/Cas9 gene editing (component 1). In the presence of a GFP1-10-containing FluoSTEP (component 2), GFP will then reconstitute and become fluorescent. The reconstituted GFP functions as a FRET donor, and FRET efficiency will be modulated by signaling based on the conformation of the signal-specific sensing domain contained within the probe. In this example, a FluoSTEP Indicator of cAMP Using Epac (FluoSTEP-ICUE) contains a fragment of the cAMP-binding guanine-nucleotide exchange factor Epac1 sandwiched between GFP1-10 and RFP. The binding of cAMP to the Epac1 domain induces a conformation change that alters the distance and orientation between the fluorescent proteins, leading to a FRET change. Monitoring cAMP dynamics next to the POI allows interrogation of dynamic regulation and functional behaviors of the POI.
FluoSTEPs follow the design of existing FRET-based sensors (Greenwald et al., 2018; Zhang et al., 2021). For instance, FluoSTEP indicator of cAMP using Epac (FluoSTEP-ICUE; Figure 1) contains the same Epac1-based molecular switch found in several previous FRET-based cAMP indicators (DiPilato and Zhang, 2009; DiPilato et al., 2004). cAMP binding induces a conformational change that increases the distance between GFP and RFP, resulting in decreased FRET efficiency. Several additional FluoSTEPs have been generated by swapping out the molecular switch domain to measure protein kinase A (PKA), protein kinase B (PKB)/Akt, extacellular response kinase (Erk), and c-Jun N-terminal kinase (Jnk) activity, as well as RhoA GTPase activation (Tenner et al., 2021). Although the modularity and generalizability of FluoSTEPs should allow for their application to many biological systems, this system may not be suitable for all POIs, as the endogenous levels of protein expression may limit the fluorescence signal.
Cell line and protein of interest selection
CRITICAL: One of the major bottlenecks for using FluoSTEPs is the expression of the POI around which localized signaling activity will be monitored. The RNA expression of a target, which can be approximated by reads per kilobase of transcript per million mapped reads (RPKM), generally correlates with protein level. The RPKM value for a certain POI may depend on the cell type of choice; thus, it is important to estimate the RPKM value for a given POI in your specific experimental context. Luckily, genome-wide RPKM values from RNAseq data are available for some cell lines (Li et al., 2014; Williams et al., 2014). Proteins with higher RPKM values tend to be amenable to split GFP tagging, which is also the basis for FluoSTEPs. Leonetti et al. found that when targeting proteins with RPKM values greater than 27 for split GFP tagging, GFP fluorescence intensities were approximately 2 standard deviations above background; however, this is not a hard cutoff (Leonetti et al., 2016). The average RPKM value for proteins to be detectable through this split GFP endogenous labeling tool is 180 (Leonetti et al., 2016). In addition, protein localization can also contribute to the detectability of a POI. Proteins that are concentrated within certain areas such as membrane-bound (Feng et al., 2017; Kamiyama et al., 2016; Leonetti et al., 2016; Tenner et al., 2021) or membraneless organelles (Zhang et al., 2020) are easier to detect compared to diffuse proteins.
Design, generation, and testing of gRNAs
Timing: 2 weeks
-
1.
Choose whether to target the N- or C-terminus of the POI.
Note: A complete, reconstituted FluoSTEP is a bulky multi-domain protein; thus, the selection of N- or C-terminal tethering to the POI may be critical. Several factors should be considered: Are there important binding or catalytic sites near the chosen terminus? Is this terminus within the correct region where the signaling activity occurs (e.g., intracellular versus extracellular)? How flexible is this terminus?
-
2.
Design at least 3 gRNAs for the target region of the gene of interest (GOI), which encodes the POI. We use the CRISPR tool from Benchling, but other gRNA design resources are available as listed on https://zlab.bio/guide-design-resources. PAM sequences should be as close as possible to the target region and can be outside the coding region.
-
3.
Clone each gRNA individually into the px459 plasmid (Ran et al., 2013), which contains Cas9, the gRNA scaffold, and the puromycin resistance gene. The px459 plasmid is optimized for using Golden Gate assembly (Engler and Marillonnet, 2014), but alternative cloning methods can be used in combination with other sgRNA and Cas9-containing plasmids. A detailed protocol for Golden Gate cloning for CRISPR/Cas9 plasmids can be found here: http://sam.genome-engineering.org/static/SAM%20sgRNA%20spacer%20cloning%20protocol.pdf.
-
4.Quantify the gRNA efficiency in editing the genomic target.
-
a.Transfect the cloned gRNA plasmid into HEK293T cells using PolyJet.
-
b.After 1 day of transfection, select cells using 1 μg mL−1 puromycin.
-
c.3 days after selection, harvest the cells and extract the genomic DNA.
-
d.Amplify the target genomic region using PCR.Note: Primers should be at least 100 bp away from the target site for the GOI. Our amplified region was 400–800 bp in total due to Sanger sequencing limitations.
-
e.PCR amplification of genomic DNA tends to yield non-specific products even with well-designed primers. If this occurs, gel-purify PCR products and send them for Sanger sequencing. For issues with PCR using genomic DNA, we refer the reader to Problem 1 in the troubleshooting portion of this protocol.
-
f.If gel-purified PCR products give rise to messy chromatogram reads, clone the PCR products into the TOPO vector (Taylor et al., 2007) and then send them for Sanger sequencing.
-
g.To estimate the gRNA efficiency, upload the Sanger sequencing chromatograms and the target region into the Tracking of Indels by Decomposition (TIDE) tool (https://tide.nki.nl) (Brinkman et al., 2014). The TIDE program will estimate the percentage of sequences with indels and break down what kind of indels were induced.
-
a.
Design and generation of HDR templates
-
5.Once a gRNA has been shown to induce indels at a high rate, an HDR template can be designed. An example HDR template is shown in Figure 2. Things to include and consider when designing the HDR template:
-
a.Include: The GFP11 sequence (green) and a linker region (blue) in between GFP11 and the protein-coding sequence. Linkers of 3–6 amino acids in length have been tested successfully, with GGTGGS serving as a 6 amino-acid linker. Linkers should be flexible (through glycines) but also short for practical reasons due to the 200-nucleotide length limit of IDT ultramers (for HDR template). The amino acid sequences for GFP11 and the linker region are as follows:Targeting N-terminus: RDHMVLHEYVNAAGITGGGTargeting C-terminus: GGGRDHMVLHEYVNAAGIT
-
b.Include: Silent mutation of the PAM sequence for the gRNAs to reduce additional rounds of genome editing. If a silent mutation is not possible, change the next possible nucleotide within the gRNA targeting region without altering the amino acid sequence. If multiple gRNAs are shown to efficiently edit the genome, silent mutations for every usable gRNA can be implemented into the HDR template.
-
c.Include: N-terminal methionine at the 5′ end if targeting the N-terminus and a stop codon at the 3′ end if targeting the C-terminus.
-
d.Include: HDR arms that are of equal length.
-
e.Consider: Both double- and single-stranded HDR templates work for CRISPR/Cas9-mediated genome editing, but the efficiency and cellular context should be considered (Bai et al., 2020).
-
f.Consider: Feasibility and price of the HDR template. Although longer HDR arms enable higher specificity and efficiency, long HDR templates can be more expensive.
-
a.
-
6.
Once designed, the HDR templates can be synthesized in-house or obtained commercially. We purchased customized HDR templates as IDT ultramers.
Figure 2.

Schematic of CRISPR/Cas9 experiment design
In this example, GFP11 with a short linker is fused to the PRKA1RA gene. The gRNA should be designed to cut near the 3′ end of the PRKAR1A coding sequence (CDS). After gRNA-directed cutting (denoted as the line through the PRKAR1A CDS), the provided homology-directed repair (HDR) template enables cells to undergo HDR of the PRKAR1A gene. The HDR template also includes a mutation in the PAM sequence (NGG) to reduce re-cutting by the gRNA. For genotyping, design forward and reverse primers that span the entire insertion area.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Q5 High-Fidelity Polymerase | NEB | Cat# M0491S |
| PolyJet | SignaGen | Cat# SL100688 |
| DNase I | Thermo Fisher | Cat# EN0525 |
| Poly-D-Lysine | Sigma-Aldrich | Cat# P6407 |
| Lipofectamine LTX | Invitrogen | Cat# 15338500 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| BSA | Roche | Cat# 10738328103 |
| HEPES | Sigma-Aldrich | Cat# H3375 |
| EDTA | Sigma-Aldrich | Cat# E6758 |
| DAPI | Thermo Fisher | Cat# D21490 |
| Forskolin | Calbiochem | Cat# 344281 |
| IBMX | Sigma-Aldrich | Cat# I7018 |
| Critical commercial assays | ||
| TOPO PCR Vector | Invitrogen | Cat# K450002 |
| DNeasy Blood and Tissue Kit | QIAGEN | Cat# 69504 |
| Q5 High-Fidelity PCR Kit | NEB | Cat #E0555S |
| PureLink Quick Gel Extraction Kit | Thermo Fisher | Cat# K220001 |
| 35-mm Glass bottom imaging dish | Cellvis | Cat# D35-20-1.5-N |
| Deposited data | ||
| FluoSTEP-ICUE sequence | GenBank | MT800777.1 |
| FluoSTEP-AKAR sequence | GenBank | MT800778.1 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | ATTC Cat# CRL-11268 |
| HEK293A | Thermo Fisher | Cat# R70507 |
| 293-RIα | (Zhang et al., 2020) | N/A |
| Oligonucleotides | ||
| 293-RIα cell line validation F | 5′-TTTGTTGAAGTGGGAAGATTGG-3′ | N/A |
| 293-RIα cell line validation R | 5′-TCAATAGGTGCTGGGATCTGC-3′ | N/A |
| IDT Ultramer for generation of 293-RIα cell line | 5′-GTTCTTGGCCCATGCTCAGACATCCTC AAACGAAACATACAGCAGTACAACAGTT TTGTGTCACTGTCTGTCGGTGGCGGCCG TGACCACATGGTCCTTCATGAGTATGTA AATGCTGCTGGGATTACATGAAATCTGC CTCCTGTGCCTCCCTTTTCTCCTCTCCCC AATCCATGCTTCACTCATGCAAACTGCTTTAT-3′ |
N/A |
| Recombinant DNA | ||
| px459 | (Ran et al., 2013) | Addgene plasmid #62988 |
| px459 human RIα gRNA | (Zhang et al., 2020) | N/A |
| pcDNA3.1 GFP1-10 | (Kamiyama et al., 2016) | Addgene plasmid #70219 |
| pcDNA3.1 FluoSTEP-AKAR | (Zhang et al., 2020) | N/A |
| pcDNA3.1 FluoSTEP-ICUE | (Zhang et al., 2020) | N/A |
| pcDNA3.1 FluoSTEP-AKAR(T/A) | (Zhang et al., 2020) | N/A |
| pcDNA3.1 FluoSTEP-ICUE(R279E) | (Zhang et al., 2020) | N/A |
| Software and algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov |
| Other | ||
| Zeiss Axiovert 200M Microscope | Carl Zeiss | N/A |
| ORCA-flash4.0LT digital CMOS camera | Hamamatsu | C11440-42U30 |
| Lambda 10-2 filter changer | Sutter Instruments | LB10-B/IQ |
Materials and equipment
For Fluorescence Activated Cell Sorting (FACS): Sort Buffer (pH 7)
| Reagent | Final concentration |
|---|---|
| BSA | 0.5% w/v |
| HEPES | 25 mM |
| EDTA | 1 mM |
| DNase I | 2.5 μg mL−1 |
| DAPI | 0.1 μg mL−1 |
| DPBS | 1× |
Note: FACS sorting is optional.
Hank’s Balanced Salt Solution (HBSS) for Imaging: Imaging Buffer (pH 7.4)
| Reagent | Final concentration |
|---|---|
| HEPES | 20 mM |
| NaCl | 137 mM |
| KCl | 5.4 mM |
| Na2HPO4 | 0.25 mM |
| KH2PO4 | 0.44 mM |
| CaCl2 | 1.3 mM |
| MgSO4 | 1 mM |
| NaHCO3 | 4.2 mM |
| Glucose | 5.5 mM |
Note: Other buffers may be used for imaging.
Note: We used the following excitation/emission filter combinations in performing this protocol (center/bandwidth in nm). GFP - EX480/30, EM535/45; RFP - EX568/55, EM653/95; GFP/RFPFRET - EX480/30, EM653/95.
Step-by-step method details
Split GFP tagging of a POI
The purpose of this step is to generate a cell line where GFP11 is attached to a POI via CRISPR/Cas9. The details of this protocol apply to HEK293 cells (both T and A versions), thus some details such as confluency and transfection method may differ depending on your cells.
Note: HEK293A cells are more adherent and flatter, and these properties are beneficial for live-cell imaging (Hurt et al., 2014).
(A) Plating and transfection of cells
-
1.
Plate HEK cells in 6-well plates at approximately 50% confluency.
Note: Expression of FluoSTEP component 2 can alternatively be driven by an inducible promoter (e.g., TetOn) to achieve more precise control. To avoid overexpression of both the POI and the sensor, a stable cell line allowing for dose-dependent FluoSTEP expression can thus be generated, which can also be used as the target cell line for GFP11 tagging. In this case, GFP11-edited cells can be FACS selected using gain of GFP fluorescence in the presence of the inducer (see step 7 below).
-
2.
After 24 h, transfect each well with 1 μg of px459 plasmid encoding the customized gRNA and 20 pmol of ssDNA ultramer HDR template using Polyjet, following the manufacturer’s protocol. Transfection of multiple gRNAs into the same cell is possible and may increase the chances of successful knock-in. Leave one well untransfected as a negative control.
-
3.
The next day, passage cells from each well into separate 60-mm dishes.
(B) Selection of cells
-
4.
1 day after passaging, add 1 μg mL−1 puromycin to each 60-mm dish (this concentration is specific to our HEK293 cells).
-
5.
When no viable cells remain in the non-transfected dish (around 2–3 days), exchange the media for puromycin-free media to ease the stress on the cells.
-
6.
1 day after relieving puromycin, passage and resuspend cells in FACS sorting buffer, which includes DAPI.
-
7.
Using FACS, sort for DAPI-negative cells with the expected size (Figure 3), and plate single cells into a 96-well plate with media containing 15 mg/L phenol red and 20% FBS.
Note: The absence of DAPI in cells is due to the cell membrane being intact, thus indicating that these cells survived puromycin selection.
Alternatives: Transfection of a GFP1-10 expression plasmid into both gene-edited and wild-type cells (negative control) 1 day prior to FACS may allow for sorting of cells based on GFP fluorescence (Figure 3). However, due to the expected low signal from a tagged protein expressed at endogenous levels, this strategy may be challenging. After sorting for GFP-positive cells, a subsequent negative FACS sort to select cells without GFP signal is necessary to prevent genomic integration of GFP1-10.
-
8.
3 weeks after incubation (may be longer depending on the doubling time of different cell lines), wells containing single-cell colonies should be nearly confluent.
Note: Using media that contains phenol red allows for easy identification of wells containing single-cell colonies, as the color of the media will change from pink to yellow.
Figure 3.

Example FACS workflow
To identify cells with GFP11 successfully incorporated into the target genomic locus, puromycin (Puro)-selected cells are stained with DAPI and subjected to FACS to identify the DAPI− cell population. DAPI− cells can immediately be single-cell sorted into 96-well plates to obtain clones for expansion and sequencing. Alternatively, DAPI− cells can be transfected with a GFP1-10 expression plasmid, followed by FACS to select GFP+ cells. These cells should then be cultured and subjected to another round of FACS to select GFP− cells and avoid stable integration of GFP1-10, followed by single-cell sorting.
(C) Genotyping of cells
-
9.
Passage single-cell colonies, keeping half for further expansion in 6- or 12-well plates in normal FBS concentrations and the other half for genomic DNA extraction using kits such as the DNeasy Blood & Tissue Kit (Qiagen).
-
10.
Perform PCR on the extracted genomic DNA using kits such as the Q5 High-Fidelity Kit (New England Biolabs) and with primers designed to amplify the target region of the GOI. Genomic PCR may be tricky and typically leads to multiple bands; for these issues, we refer the reader to Problem 1 in the troubleshooting portion of this protocol.
-
11.
To evaluate the copy number of correct gene edits, PCR products should be gel extracted using kits such as the PureLink Quick Gel Extraction kit (Invitrogen), cloned into TOPO PCR vectors, and subjected to Sanger sequencing.
Note: Some cell lines such as HEK293 cells are aneuploid; thus, it may be difficult to discern if cells are homozygous or heterozygous. Sequencing multiple TOPO PCR vectors allows for a rough estimation of the percentage of gene loci with successful knock-in of GFP11.
(D) Imaging of cells to determine feasibility for FluoSTEP imaging
-
12.
Passage cells that are identified to have successful knock-in, as well as wild-type cells, into 35-mm glass-bottom dishes at approximately 30% confluency.
-
13.
1 day after passaging, transfect 1 μg of GFP1-10 plasmid into both knock-in and wild-type cells via PolyJet.
Alternatives: Other transfection reagents, such as Lipofectamine, may suffice as well depending on the specific cell type.
-
14.Image cells 1 day after transfection.
-
a.Aspirate the cell media without touching the bottom of the dish, and wash twice with 1 mL of HBSS. Add 2 mL of HBSS for imaging.
-
b.Mount the imaging dish on the microscope stage and image in the GFP channel. Use the GFP1-10-transfected wild-type cells as a guide for determining the autofluorescence background signal.
-
a.
Live-cell imaging of FluoSTEP sensors to measure native, microdomain-specific signaling
Note: To increase cell adhesion especially for cell lines that easily detach (e.g., HEK), we recommend coating the imaging dish with poly-D-lysine (PDL). Reconstitute PDL in sterile water at a concentration of 0.1 mg/mL. Coat the 35-mm glass-bottom dish with 400 μL Poly-D-Lysine (PDL) (just enough to cover the imaging area) in a biosafety cabinet. Incubate the dishes with PDL for 30 min in a sterile, 37°C incubator and then wash 3 times with sterile PBS in a biosafety cabinet. Let the coated dishes dry for 30 min in a biosafety cabinet, and then wrap the dishes in parafilm and store at 4°C. As an alternative, 35-mm glass-bottom dishes pre-coated with poly-D-lysine can be purchased commercially.
Note: To determine the influence of the microdomain on the measured activity/signaling, control experiments using untargeted biosensors containing the full donor fluorophore should be performed, similar to what was done previously (Tenner et al., 2021).
(E) Real-time imaging of FluoSTEP-expressing cells
-
15.
Passage FP11-cells and transfect 1 day later with 1 μg of the FluoSTEP probe of your choice.
-
16.
Image cells 1 day after transfection (wash and place cells in HBSS as above). Prepare drugs at 1000× stock concentration. Aliquot 2 μL of 1000× drug stocks into separate 1.5 mL tubes.
Note: Store drug aliquots properly during imaging. Most drugs are best kept on ice, but this will depend on the specific compound.
-
17.Real-time imaging of cells with acute stimulation.CRITICAL: There are multiple factors to consider when choosing the parameters for real-time imaging of cells, especially FP11-cells, as there is a balance between obtaining enough fluorescence signal and minimizing photobleaching. Select appropriate excitation and emission filters for monitoring GFP and RFP fluorescence intensities, gain, ND filters, exposure times, and image acquisition interval. For example, our imaging used a 0.3 ND filter (50% transmittance), 500 ms exposure times for the FRET and GFP channel, and 30 s acquisition interval. In addition, select an appropriate objective lens to record imaging with sufficient detail. For example, higher-magnification objectives (e.g., ≥40×) are preferable for visualizing fluorescence signals localized to subcellular structures (e.g., plasma membrane, organelles, cytoskeleton, etc.). Details of our microscope system are provided in the Key Resources Table and the Materials and Equipment section.
-
a.Image cells in 3 channels: green direct (GFP excitation and emission), red direct (RFP excitation and emission), and green-red FRET (GFP excitation and RFP emission).
-
b.Image the cells for at least 5 min to acquire a baseline signal prior to the first drug addition.
-
c.Pause the imaging cycles to add the first drug to the dish that is fixed to the microscope stage: Use a P1000 micropipette to transfer ∼0.5 mL of HBSS from the imaging dish into a 1.5 mL tube containing an aliquot of 1000× drug stock; mix briefly, and carefully add the HBSS + drug mixture back to the imaging dish, mixing again (at least 3 times) to ensure homogeneous distribution of the drug. When adding a drug to the dish, pipet gently and away from the center of the dish to avoid displacing cells from the surface. Also, avoid directly touching the imaging dish when performing drug addition, as you may shift the microscope field of view.CRITICAL: The cells may be subjected to significant shear stress from pipetting during acute drug addition. HEK cells are not as adherent as other cells and can easily detach from the imaging dish surface; therefore, using poly-D-lysine-coated imaging dishes is important to limit this effect. Alternatively, performing drug addition using a smaller volume of HBSS (e.g., 0.2 mL and a P200) may reduce shear stress.Note: The exact stimulation conditions will depend on the probe/activity being monitored. FluoSTEPs have been tested with adenylyl cyclase activators, phosphodiesterase inhibitors, and β-adrenergic agonists to induce cAMP and PKA signaling (FluoSTEP-ICUE and FluoSTEP-AKAR); growth factor stimulation to induce Akt or Erk signaling (FluoSTEP-AktAR or FluoSTEP-EKAR); stress induction to induce Jnk signaling (FluoSTEP-JNKAR); and G-protein coupled receptor stimulation to induce RhoA activity (FluoSTEP-RhoA) (Tenner et al., 2021).
-
d.Restart the imaging cycles and image the cells until the fluorescence signal reaches a stable plateau. Repeat for subsequent drug additions.Note: Imaging for at least 5 min before the first drug addition is required, as FluoSTEPs in our experience can exhibit significant baseline drift, mostly due to photobleaching of the donor fluorophore. Having a stable baseline emission ratio before any drug addition is critical to see the effect of the added drug.
-
a.
Expected outcomes
Due to the small size of GFP11, knock-in efficiencies should be on par with CRISPR/Cas9-mediated knock-in of other proteins. POIs that have relatively high RPKM values (more than 50), especially POIs that localize to particular subcellular compartments, should be amenable to imaging when tagged with GFP11.
Successful tagging and reconstitution of the POI with GFP1-10 will allow for monitoring of signaling dynamics using FluoSTEPs. Figure 4 shows example data from FluoSTEP sensing around a POI expressed at endogenous levels. Briefly, PRKAR1A was tagged at its N-terminus with GFP11 in HEK293T cells. These cells were transfected with FluoSTEP-ICUE, which monitors cAMP levels, and imaged after stimulation with the adenylyl cyclase activator forskolin. The change in the FluoSTEP-ICUE GFP/RFP emission ratio reflects changes in intracellular cAMP accumulation.
Figure 4.

Example FluoSTEP response data
PRKAR1A-FP11 cells expressing FluoSTEP-ICUE were stimulated with 50 μM forskolin, an adenylyl cyclase activator. A time course shows the FluoSTEP-ICUE green/red emission ratio recorded from the imaged cells, which corresponds to cAMP levels. Data represent the mean±SEM. Inset: GFP fluorescence image of PRKAR1A-FP11 cells, which are a HEK293T cells gene edited to tag GFP11 at the N-terminus of endogenously expressed RIα, expressing FluoSTEP-ICUE. The fluorescence localizes to discrete puncta within cells, which correspond to RIα phase-separated droplets. Scale bar, 10 μm.
Quantification and statistical analysis
FluoSTEP imaging analysis
The analysis is typically performed by selecting regions of interest (ROIs) corresponding to whole-cell fluorescence, but this will depend on your application and can be extended to subcellular regions (Tenner et al., 2021; Zhang et al., 2020). Raw fluorescence intensities are corrected by subtracting the background fluorescence intensity of a cell-free ROI from the emission intensities of FluoSTEP-expressing FP11-cells in each channel. Green/red (e.g., FluoSTEP-ICUE) or red/green (e.g., FluoSTEP-AKAR) emission ratios (R) should then be calculated at each time point. The specific formulas are outlined here:
Normalize the resulting emission ratio time courses by dividing the emission ratio at each time point by the basal ratio value at time zero (R/R0), which is the emission ratio at the time point immediately before the first drug addition (R0).
We recommend repeating experiments at least 3 times (3 separate dishes), pooling the time-course emission ratios together, and calculating the average emission ratio and standard error of the mean.
Limitations
As this protocol involves imaging a POI expressed at endogenous levels, as described above in “Cell Line and Protein of Interest Selection”, the fluorescence signal may be undetectable for proteins with low expression; thus, not all POIs can are compatible with FluoSTEPs. In addition, peptide tagging to protein termini is a potential issue, which has been seen for HA tags (Saiz-Baggetto et al., 2017). Although the sequence for GFP11 plus linker is only 19 amino acids long, tethering of GFP11 to the N- or C-terminus of a POI may alter protein dynamics.
Troubleshooting
Problem 1
No bands or many non-specific bands after PCR of genomic DNA. Related to “Genotyping of cells”.
Potential solution
If no bands are seen, molecules from the source DNA such as lipids may be inhibiting DNA polymerase activity (Schrader et al., 2012). Therefore, we suggest using 50–100 ng of DNA for PCRs to dilute inhibitory molecules. If many non-specific bands are seen, we suggest designing multiple primers and comparing the specificity after PCR of wild-type genomic DNA via gel electrophoresis and subsequent Sanger sequencing.
Problem 2
No detectable fluorescence signal but successful GFP11 knock-in. Related to “Imaging of cells to determine feasibility for FluoSTEP imaging”.
Potential solution
There are several options to consider, which are listed from easiest to most burdensome:
Image at higher illumination, longer exposure, or higher EM gain.
Knock-in several tandem copies of GFP11 separated by flexible linkers (Leonetti et al., 2016; Tenner et al., 2021).
Change the POI target to one that localizes to the same compartment and has higher RPKM values.
Problem 3
Significant drifting baseline before first drug addition (Figure 5). Related to “Real-time imaging of FluoSTEP-expressing cells”.
Figure 5.

Performing baseline correction of emission-ratio time courses
Due to the low fluorescence intensities associated with tagged proteins expressed at endogenous levels, photobleaching of the reconstituted GFP may significantly contribute to FluoSTEP emission ratio measurements in the form of a drifting baseline. One method to correct for baseline drift is to perform a linear fit of the baseline (i.e., time points preceding drug addition), after which corrected emission ratios (Rcorrected,t) can be calculated by subtracting the fitted slope (slopefit) from the original emission ratio (Rraw,t) value at each time point t. Data shown represent the mean±SEM.
Potential solution
There are several options to consider:
Acquire more images before adding the first drug to allow the baseline to be stabilized.
Reduce the exposure time to decrease photobleaching.
Use higher ND filters to decrease photobleaching.
Increase the time interval between image acquisitions to reduce light damage.
If the drifting baseline is consistent throughout the image, calculate the slope before any drug addition and correct the baseline for all the values in the time course (Figure 5).
Problem 4
Does intermolecular FRET contribute to the signal? Related to “Real-time imaging of FluoSTEP-expressing cells”.
Potential solution
According to the biosensor design, FRET should occur between GFP and RFP within each biosensor molecule (i.e., intramolecular FRET). However, if the concentration of the biosensor molecules is high, it is possible that one domain of a biosensor molecule binds to another domain of a second biosensor molecule, especially with the bipartite molecular switches used in kinase activity reporters (i.e., intermolecular FRET). Plot the single cell response amplitude versus their fluorescence intensities in the red direct (RFP excitation and emission) channel for a cell population. A dependence on the fluorescence intensity may suggest a contribution from intermolecular FRET.
Problem 5
FRET signal is low and unsure if this signal is above background. Related to “Real-time imaging of FluoSTEP-expressing cells”.
Potential solution
To ensure that the signal in the FRET channel is truly from FRET, perform acceptor photobleaching by illuminating RFP without any ND filters for 10 min. After confirming that most of the RFP is photobleached, take another image with an ND filter. FRET efficiency is calculated as 1 − (GFPbefore/GFPafter), where GFPbefore and GFPafter are the GFP intensities before and after acceptor photobleaching, respectively. If FRET efficiency is statistically significantly greater than 0, then the detected FRET signal has some contribution from FRET.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jin Zhang (jzhang32@ucsd.edu).
Materials availability
FluoSTEP-AKAR and FluoSTEP-ICUE expression constructs are available upon request by contacting the lead contact.
Data and code availability
Not applicable
Acknowledgments
This work was supported by a National Science Foundation predoctoral fellowship DGE-1650112 to J.Z.Z. and NIH R35 CA197622 and R01 DK073368 to J.Z.
Author contributions
S.M. conceived of and B.T. and J.Z.Z. developed FluoSTEPs. J.Z.Z. wrote the initial draft of the manuscript. B.T., S.M., and J.Z. edited the manuscript. J.Z. supervised the project. All authors read and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- Bai H., Liu L., An K., Lu X., Harrison M., Zhao Y., Yan R., Lu Z., Li S., Lin S. CRISPR/Cas9-mediated precise genome modification by a long ssDNA template in zebrafish. BMC Genomics. 2020;21:67. doi: 10.1186/s12864-020-6493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman E.K., Chen T., Amendola M., van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPilato L.M., Zhang J. The role of membrane microdomains in shaping beta2-adrenergic receptor-mediated cAMP dynamics. Mol. Biosyst. 2009;5:832–837. doi: 10.1039/b823243a. [DOI] [PubMed] [Google Scholar]
- DiPilato L.M., Cheng X., Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C., Marillonnet S. Golden Gate cloning. Methods Mol. Biol. 2014;1116:119–131. doi: 10.1007/978-1-62703-764-8_9. [DOI] [PubMed] [Google Scholar]
- Feng S., Sekine S., Pessino V., Li H., Leonetti M.D., Huang B. Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-00494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald E.C., Mehta S., Zhang J. Genetically encoded fluorescent biosensors illuminate the spatiotemporal regulation of signaling networks. Chem. Rev. 2018;118:11707–11794. doi: 10.1021/acs.chemrev.8b00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt C.M., Björk S., Ho V.K., Gilsbach R., Hein L., Angelotti T. REEP1 and REEP2 proteins are preferentially expressed in neuronal and neuronal-like exocytotic tissues. Brain Res. 2014;1545:12–22. doi: 10.1016/j.brainres.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama D., Sekine S., Barsi-Rhyne B., Hu J., Chen B., Gilbert L.A., Ishikawa H., Leonetti M.D., Marshall W.F., Weissman J.S. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 2016;7:11046. doi: 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti M.D., Sekine S., Kamiyama D., Weissman J.S., Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. U S A. 2016;113:E3501–E3508. doi: 10.1073/pnas.1606731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.-W., Burkhardt D., Gross C., Weissman J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz-Baggetto S., Méndez E., Quilis I., Igual J.C., Bañó M.C. Chimeric proteins tagged with specific 3xHA cassettes may present instability and functional problems. PLoS One. 2017;12:e0183067. doi: 10.1371/journal.pone.0183067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader C., Schielke A., Ellerbroek L., Johne R. PCR inhibitors – occurrence, properties and removal. J. Appl. Microbiol. 2012;113:1014–1026. doi: 10.1111/j.1365-2672.2012.05384.x. [DOI] [PubMed] [Google Scholar]
- Taylor D.L., Herriott I.C., Long J., O’Neill K. TOPO TA is A-OK: a test of phylogenetic bias in fungal environmental clone library construction. Environ. Microbiol. 2007;9:1329–1334. doi: 10.1111/j.1462-2920.2007.01253.x. [DOI] [PubMed] [Google Scholar]
- Tenner B., Zhang J.Z., Huang B., Mehta S., Zhang J. FluoSTEPs: Fluorescent biosensors for monitoring compartmentalized signaling within endogenous microdomains. Sci. Adv. 2021;7:eabe4091. doi: 10.1126/sciadv.abe4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.C., Jan C.H., Weissman J.S. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science. 2014;346:748–751. doi: 10.1126/science.1257522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.-F., Mehta S., Zhang J. Signaling microdomains in the spotlight: visualizing compartmentalized signaling using genetically encoded fluorescent biosensors. Annu. Rev. Pharmacol. Toxicol. 2021;61:587–608. doi: 10.1146/annurev-pharmtox-010617-053137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.Z., Lu T.-W., Stolerman L.M., Tenner B., Yang J., Zhang J.-F., Falcke M., Rangamani P., Taylor S.S., Mehta S. Phase separation of a PKA regulatory subunit controls cAMP compartmentation and oncogenic signaling. Cell. 2020;182:1531–1544.e15. doi: 10.1016/j.cell.2020.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable