

Abstract
Cervical cancer is one of the leading causes of cancer-associated mortality in gynecological diseases and ranks third among female cancers worldwide. Although early detection and vaccination have reduced incidence rates, cancer recurrence and metastasis lead to high mortality due to the lack of effective medicines. The present study aimed to identify novel drug candidates to treat cervical cancer. In the present study, lanatoside C, an FDA-approved cardiac glycoside used for the treatment of heart failure, was demonstrated to have anti-proliferative and cytotoxic effects on cervical cancer cells, with abrogation of cell migration in a dose-dependent manner. Lanatoside C also triggered cell apoptosis by enhancing reactive oxygen species production and reducing the mitochondrial membrane potential, which induced cell cycle arrest at the S and G2/M phases. Furthermore, lanatoside C inhibited the phosphorylation of Janus kinase 2 (JAK2) and signal transducer and activator of transcription 6 (STAT6), while inducing the expression of suppressor of cytokine signaling 2, a negative regulator of JAK2-STAT6 signaling. Taken together, the results of the present study suggest that lanatoside C suppresses cell proliferation and induces cell apoptosis by inhibiting JAK2-STAT6 signaling, indicating that lanatoside C is a promising agent for the treatment of cervical cancer.
Keywords: lanatoside C, cervical cancer, apoptosis, JAK2, STAT6
Introduction
Cervical cancer is the most frequently occurring malignancy worldwide, ranking third in terms of cancer mortality in gynecological tumors (1). It has an estimated incidence of >560,000 in women and >311,000 mortalities are reported annually worldwide (2,3). Although screening and prevention with human papilloma virus (HPV) vaccines have significantly contributed to lower incidence rates of cervical cancer, clinical prognosis remains diverse and unpredictable (4). In developing countries, >70% of cervical cancer cases are metastatic or invasive, resulting in high mortality rates (5,6). Systemic first-line treatment with chemotherapy significantly prolongs the 5-year overall survival rate; however, lack of effective and alternative therapy is still a major cause of cervical cancer-associated mortality (7,8). Thus, it is important to develop novel drug candidates to treat cervical cancer.
Resistance to cell death and enhancement of cell proliferation are hallmarks of cancer (9). Thus, induction of apoptosis by chemotherapy or radiotherapy is widely used to treat cervical cancer (10–12), which is usually dependent on intrinsic apoptotic signaling pathways that are mitochondrial-associated and predominantly take place in the mitochondria (13). At the molecular level, pro-apoptotic and anti-apoptotic signals in the mitochondria maintain a balance for homeostasis (14). Once the cell is exposed to stimuli, such as oxidative stress or UV damage, disruption of this balance occurs, with cytochrome C released from mitochondria to the cytoplasm, leading to the activation of caspase cascades, which cleave proteins, such as caspase-9, caspase-3 and poly-ADP ribose polymerase (PARP) (15). Similarly, cell cycle arrest limits the sustained proliferation of cancer cells: Cell cycle regulators, such as p21 and cyclin B1, which play diverse roles in mediating cycle progression (16). The expression of p21 is associated with the obstruction of G1/S progression via cyclin-dependent kinase (17). Conversely, cyclin B1 is an essential molecule for controlling the cell cycle by initiating mitosis (18). However, the role of p21 and cyclin B1 in G2/M cycle progression remains controversial, given that different findings have reported opposite results of these two proteins in G2/M cycle inhibition (19).
Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling is involved in various cellular functions, including cell proliferation, differentiation and inflammation, and has been implicated in several diseases, such as immune dysregulation and cancer (20–23). Specific combinations of JAKs (JAK1, 2, 3, and Tyk2) and STAT (STAT1, 2, 3, 4, 5A, 5B and 6) isoforms possess different abilities to transduce cell signaling (24). For example, activation of JAK3/STAT6 signaling contributes to the progression of renal fibrosis, while higher expression and activity of JAK2/STAT6 are associated with Hodgkin lymphoma (25,26). The suppressor of cytokine signaling (SOCS) family (SOCS1-7 and CIS) serves as a negative regulator in controlling the activity and degradation of JAKs (25). SOCS expression is downregulated in human breast cancer (27). Thus, the potential role of JAK/STAT signaling and SOCS family in cancer progression has provoked considerable interest in identifying novel inhibitors targeting JAK/STAT signaling in cancer treatment.
Lanatoside C is an FDA-approved cardiac glycoside that is widely used for treating arrhythmias and heart failure due to its ability to arrest Na+ and K+ interchange across cell membranes (28). Lanatoside C has recently been reported to possess several other advantages in treating dengue virus infection (29), pulmonary fibrosis (30) and cancer (28). Lanatoside C exerts its anti-proliferative effect in gastric cancer in vitro (26) and liver cancer in vivo (31); however, the effect and molecular mechanism of lanatoside C in cervical cancer remain unknown.
In the present study, the anti-proliferative effect of lanatoside C on human cervical cancer HeLa cells was evaluated. The results demonstrated that Lanatoside C promoted cell apoptosis and induced cell cycle arrest at the G2/M phase, which was closely associated with downregulation of JAK2/STAT6 signaling, and increased SOCS2 expression. The present study provides novel insight on the molecular mechanism of lanatoside C in cervical cancer, which suggests that lanatoside C is a potential chemotherapy candidate for cervical treatment.
Materials and methods
Cell lines and culture
HeLa and BEAS-2B cells were purchased from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences. Cells were maintained in RPMI-1640 medium (Thermo Fisher Scientific, Inc.) supplemented with 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin (both purchased from Thermo Fisher Scientific, Inc.), at 37°C with 5% CO2.
Reagents and antibodies
Lanatoside C was purchased from Sigma-Aldrich; Merck KGaA, dissolved in DMSO and stored as stock, at a final concentration of 100 mM at −20°C. A crystal violet stain kit was purchased from Nanjing Jiancheng Bioengineering Institute. The MMP assay kit with JC-1, the reactive oxygen species (ROS) assay kit, Cell Counting Kit-8 (CCK-8) assay, PBS and loading buffer (1X) were purchased from Beyotime Institute of Biotechnology. DMSO, crystal violet, propidium iodide (PI), JAK inhibitor I and RNase were purchased from Sigma-Aldrich; Merck KGaA. The Annexin V-FITC/PI apoptosis detection kit was purchased from MultiSciences Biotech Co. Ltd (https://www.multisciences.net/). Primary antibodies against cleaved caspase-9 (9505), cleaved caspase-3 (9664), cleaved PARP (9548), cyclin B1 (12231), p21(2947), phosphorylation-JAK2 (3771), total-JAK2 (3230), phosphorylation-STAT6 (56554), total-STAT6 (5397), SOCS2 (2779) and β-actin (3700) were purchased from Cell Signaling Technology, Inc. The dilution concentration of primary antibody was 1:1,000. The secondary antibodies (Goat anti-Mouse IgG; cat. no. 115-005-003 and Goat anti-Rabbit IgG; cat. no. 111-005-003) were purchased from Jackson ImmunoResearch Laboratories, Inc. The dilution concentration of secondary antibody was 1:5,000. Methanol and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd. The BCA protein Assay kit was purchased from Beyotime Institute of Biotechnology.
Colony formation assay
The colony formation assay was performed to assess the effect of lanatoside C on cell proliferation. Briefly, HeLa cells were seeded into 6-well plates at a density of 500 cells/well. Due to the high sensitivity of the colony formation assay for unicellular proliferation (32), the doses of lan atoside C in this experiment were 0, 50, 100 and 500 nM. Cells were treated with different concentrations of lanatoside C. After 10 days, the culture medium was discarded and cells were fixed in methanol for 20 min, followed by staining with 0.5% crystal violet solution at roomtemprature. Cells were rinsed with water and dried at room temperature. The number of cell colonies were manually counted.
Cell viability assay
The cytotoxic effect of lanatoside C on HeLa cells was assessed via the CCK-8 assay. HeLa cells were seeded into 96-well culture plates at a density of 5,000 cells/well and incubated overnight at 37°C. Cells were subsequently incubated with different concentrations of lanatoside C (0, 0.1, 0.5, 1, 2.5, 5 and 10 µM or lanatoside C (2.5 µM) plus IL-4 (10 ng/ml) for 24 h. Following incubation, 10 µl CCK-8 solution was added to each well for 2 h at 37°C and the absorbance was measured at a wavelength of 450 nm, using a microplate reader (Thermo Fisher Scientific, Inc.).
Cell migration assay
The effect of lanatoside C on HeLa cell migration was assessed via the wound healing and Transwell assays. For the wound healing assay, HeLa cells were seeded into 12-well plates at a density of 5×105 cells/well, cultured overnight and replaced with serum-free medium to minimize cell proliferation. Sterilized 200 µl pipette tips were subsequently used to scratch the wounds and cells were washed with PBS. Fresh culture medium was subsequently added, along with different concentrations of lanatoside C (0, 0.25, 0.5 and 1 µM) for 24 h. A light microscope was used to observe the changes in the scratch area (×200).
For the Transwell assay, Transwell chambers with 8 µm pores (Corning, Inc.) were used. Briefly, HeLa cells (2×105 cells/well) were pretreated with different concentrations of lanatoside C and plated into upper chambers with 8-µm pores without FBS, while the lower chambers were filled with cell culture medium supplemented with 10% FBS. Following incubation for 24 h at 37°C, cells in the upper chambers were removed using a cotton swab, while the migratory cells were fixed with methanol for 20 min at room temperature and stained with 0.5% crystal violet solution at room temperature for 20 min. Cells were washed twice with PBS, and the stained cells were observed under a light microscope (×200).
Apoptosis assay
The effect of lanatoside C on apoptosis was detected using the Annexin V-FITC/PI apoptosis detection kit. Cells (5×105 cells/well) were seeded into 12-well plates and cultured overnight at 37°C. Cells were incubated with different concentations of lanatoside C (0, 0.5, 1 and 2.5 µM) for 24 h. Cells were subsequently harvested and incubated with 10 µl Annexin V-FITC and 5 µl of PI solution for 5 min at room temperature in the dark. Apoptotic cells were subsequently analyzed using a flow cytometer (FACS Calibur; BD Biosciences), and Flowjo 7.6 software (BD Biosciences).
Detection of MMP
To investigate the effect of lanatoside C on MMP, a JC-1 stain kit was used to detect changes in MMP. HeLa cells were seeded into 12-well plates at a density of 5×105 cells/well and cultured overnight at 37°C. Cells were treated with different concentrations (0, 0.5, 1 and 2.5 µM) of lanatoside C for 24 h, collected following centrifugation (500 × g, at 4°C for 3 min) and stained with a JC-1 dye working solution for 20 min at 37°C. After washing twice with pre-cooled JC-1 dye buffer, the cells were analyzed via flow cytometry (FACS Calibur; BD Biosciences).
Intracellular ROS determination
Changes in intracellular ROS levels were detected using the fluorescence probe DCFH-DA, according to the instructions of the ROS assay kit. Briefly, cells were treated with 2.5 µM lanatoside C for 0, 4 and 6 h, and stained with DCFH-DA solution at 37°C for 20 min. Cells were washed three times with serum-free cell culture solution to remove the free DCFH-DA, and DCF fluorescence was detected via fluorescence microscopy (Leica, Germany) in four randomly selected fields. Results were analyzed using ImageJ software 1.8.0_172 (National Intitutes of Health) to determine the mean fluorescence intensity (MFI), which reflects ROS levels.
Cell cycle analysis
Cell cycle distribution was assessed via PI staining. HeLa cells (5×105 cells/well) were seeded into 12-well plates for 12 h and subsequently treated with different concentrations of lanatoside C for 24 h. Cells were digested using trypsin and fixed in pre-cooled 70% ethanol overnight at 4°C. Cells were washed three times with PBS and centrifuged (500 × g, at 4°C for 3 min) for collection. Cells were treated with 10 µg/ml RNase solution for 20 min at room temperature and stained with 40 µg/ml PI solution for 20 min at room temperature. Cell cycle distribution was subsequently measured using a flow cytometer (FACS Calibur).
Western blotting
HeLa cells were seeded into 6-well plates at a density of 1×106 cells/well. Following incubation overnight at room temperature, cells were treated with different concentrations of lanatoside C (0, 0.5, 1 and 2.5 µM) for 24 h. Cells were washed twice with PBS, harvested using RIPA lysis buffer (Beyotime Institute of Biotechnology) and protein concentration was determined using the BCA method. The samples were subsequently heated at 100°C for 5 min. Proteins were separated by 10% SDS-PAGE, with 20 µg protein loaded per lane. And then protein were transferred onto nitrocellulose membranes and blocked with 5% milk at room temperature for 2 h. After washing with washing buffer, the membranes were incubated with the corresponding primary antibodies overnight at 4°C, and subsequently incubated with secondary HRP-linked antibody at room temperature for 2 h. Membranes were re-washed three times with washing buffer for 15 min. Protein bands were visualized using ECL western blotting substrate (Thermo Fisher Scientific, Inc.) and ImageJ software 1.8.0_172 (National Intitutes of Health) was used to measure the band density.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software, Inc.). All experiments were performed in triplicate and data are presented as the mean ± SEM. Data were analyzed using one-way ANOVA followed by Bonferroni correction. P<0.05 was considered to indicate a statistically significant difference.
Results
Lanatoside C inhibits HeLa cell proliferation
To determine whether lanatoside C has an anti-proliferative effect on cervical cancer cells, a colony formation assay was performed using HeLa cells. Following treatment with 50, 100 and 500 nM lanatoside C for 10 days, the number of HeLa cell colonies significantly decreased in a dose-dependent manner (Fig. 1A and B). The CCK-8 assay was performed to assess the cellular toxicity of lanatoside C. As presented in Fig. 1C, treatment with different concentrations of lanatoside C produced an inhibitory curve and the half maximal inhibitory concentration for HeLa was 378.5 nM. The cell toxicity of lanatoside C on normal human epithelial BEAS-2B cells was also assessed. As presented in Fig. 1D, lanatoside C did not have a toxic effect on BEAS-2B cells at 5 mM, indicating that lanatoside C has no anti-proliferative effect on normal epithelial cells. Taken together, these results suggest that lanatoside C inhibits the proliferation of HeLa cells.
Figure 1.

Lanatoside C inhibits HeLa cell proliferation. (A) HeLa cells were treated with different concentrations of lanatoside C (0, 50, 100 and 500 nM) for 10 days, and the colony formation assay was performed. (B) Quantification analysis of colony formation assay of HeLa cells was performed. (C) Cellular cytotoxicity of lanatoside C on HeLa cells detected via the CCK-8 assay and the value of IC50 was calculated. (D) The viability of BEAS-2B cells was assessed via the CCK-8 assay following treatment with lanatoside C. Data are presented as the mean ± SEM (n=3). *P<0.05; ***P<0.001. IC50, half maximal inhibitory concentration; CCK-8, Cell Counting Kit-8.
Lanatoside C reduces HeLa cell migration
Cancer cell migration leads to cancer recurrence and metastasis (33). To determine whether lanatoside C affects cell migration, the wound healing assay was performed to detect HeLa cell migration. Lanatoside C significantly decreased the formation of the wound healing area in HeLa cells (Fig. 2A and B). To further confirm this result, the Transwell assay was performed to determine the inhibitory effect of lanatoside C on cell migration. Following treatment with lanatoside C, fewer HeLa cells migrated from the upper chamber without serum to the lower chamber with 10% serum (Fig. 2C and D), indicating that lanatoside C significantly impeded cell migration. Collectively, these results suggest that lanatoside C inhibits HeLa cell migration.
Figure 2.

Lanatoside C decreases HeLa cell migration. (A) HeLa cells were treated with different concentrations of lanatoside C (0, 0.25, 0.5 and 1 µM) for 24 h, and the wound healing assay was performed. (B) Quantification analysis of HeLa cell migration was performed. (C) The Transwell assay was performed to assess HeLa cell migration. HeLa cells were allowed to migrate from the upper chamber without serum to the lower chamber which contained 10% serum. (D) Quantification analysis of HeLa cell migration was performed. Data are presented as the mean ± SEM (n=3). *P<0.05; **P<0.01; ***P<0.001.
Lanatoside C promotes cell apoptotic response in HeLa cells
Given that lanatoside C has an inhibitory effect on HeLa cell proliferation, the pro-apoptotic activity of lanatoside C in HeLa cells was assessed via Annexin V/PI staining. Following treatment with lanatoside C, the number of apoptotic HeLa cells increased in a dose-dependent manner (Fig. 3A and B). Apoptosis-related proteins in lanatoside C-treated HeLa cells were detected via western blotting. The expression levels of cleaved caspase-9 (Fig. 3C and D), cleaved caspase-3 (Fig. 3C and E) and cleaved PARP (Fig. 3C and F) were significantly upregulated following treatment with lanatoside C, in a dose-dependent manner. Taken together, these results suggest that lanatoside C induces apoptosis in HeLa cells.
Figure 3.

Lanatoside C induces HeLa cell apoptosis. (A) HeLa cells were treated with lanatoside C for 24 h and Annexin-V/PI staining with flow cytometry detection was performed. (B) Quantification analysis of apoptotic rates in lanatoside C-treated HeLa cells was performed. (C) Expression of apoptosis-related proteins, cleaved caspase-9, cleaved caspase-3 and cleaved PARP was detected via western blotting. β-actin served as the loading control. The expression levels of (D) cleaved caspase-9, (E) cleaved caspase-3 and (F) cleaved PARP were quantified using ImageJ software. Data are presented as the mean ± SEM (n=3). *P<0.05; **P<0.01; ***P<0.001. LC, lanatoside C.
Lanatoside C decreases MMP and enhances intracellular ROS production
Given that cleaved caspase-9, cleaved caspase-3 and cleaved PARP are involved in mitochondria-related cell death (34), the effect of lanatoside C on mitochondrial function was determined. JC-1 staining was performed to assess MMP. The JC-1 polymer decreased substantially following treatment with lanatoside C (Fig. 4A and B), suggesting that lanatoside C decreases MMP in HeLa cells. In addition, intracellular ROS are accompanied by mitochondrial dysfunction (35); thus, ROS production was detected using a DCFH-DA probe. Treatment with lanatoside C (2.5 µM) significantly increased ROS generation in a time-dependent manner (Fig. 4C and D). Collectively, these results suggest that lanatoside C decreases MMP in HeLa cells and increases intracellular ROS generation.
Figure 4.

Lanatoside C decreases MMP and increases cellular ROS production in HeLa cells. (A) Following treatment with lanatoside C for 24 h, MMP was detected via JC-1 staining and flow cytometry in HeLa cells. (B) JC-1 polymer in HeLa cells was analyzed to determine quantitative changes in the loss of MMP. (C) Cells were treated with lanatoside C at 2.5 µM for 4 or 6 h, and cellular ROS production in HeLa cells was monitored via DCFH-DA probe with fluoresce microscopy detection. (D) Statistical analysis of ROS production (MFI, mean fluorescence intensity) in HeLa cells using ImageJ software. Data are presented as the mean ± SEM (n=3). *P<0.05; **P<0.01; ***P<0.001. MMP, mitochondria membrane potential; ROS, reactive oxygen species; LC, lanatoside C.
Lanatoside C inhibits G2/M cell cycle arrest in HeLa cells
Given that cancer cells maintain cell proliferation and resist cell death by accentuating the cell cycle (36), the effect of lanatoside C on the cell cycle of HeLa cells was determined. PI staining and flow cytometry demonstrated that lanatoside C significantly induced cell cycle arrest in HeLa cells (Fig. 5A). Following treatment with lanatoside C, the percentage of cells in the S phase and G2/M phase increased in a dose-dependent manner, while the number of HeLa cells in the G1 phase decreased (Fig. 5B). Furthermore, the expression of cell cycle-related proteins was detected, and the expression levels of p21 and cyclin B1 significantly decreased and increased, respectively, in a dose-dependent manner (Fig. 5C-E). Taken together, these results suggest that lanatoside C induces cell cycle S and G2/M arrest, in which the expression of cyclin B1 is induced and p21 is attenuated.
Figure 5.
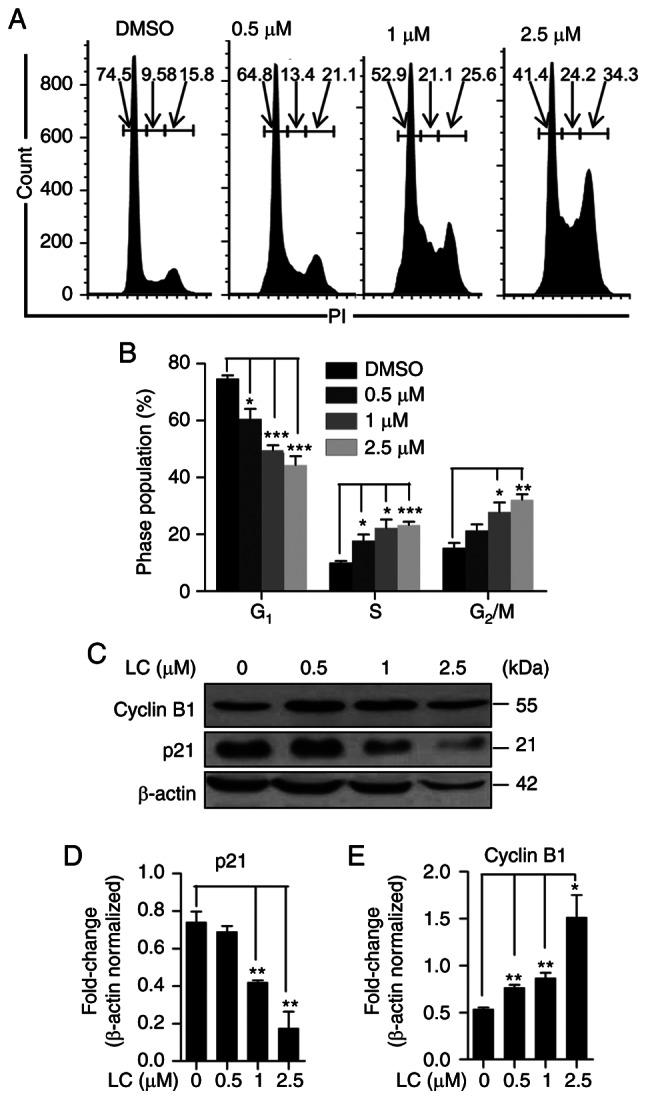
Lanatoside C suppresses HeLa cell cycle. (A) HeLa cells were treated with different concentrations of lanatoside C (0, 0.5, 1 and 2.5 µM) for 24 h and cell cycle was assessed via PI staining with flow cytometry detection. (B) Quantification of cell cycle phase distribution in HeLa cells was performed. (C) Western blot analysis was performed to detect the expression of cell cycle related proteins, cyclin B1 and p21, in HeLa cells treated with different concentrations of lanatoside C for 24 h. The alternations of (D) p21 and (E) cyclin B1 in HeLa cells were quantified. Data are presented as the mean ± SEM (n=3). *P<0.05; **P<0.01; ***P<0.001. LC, lanatoside C.
Lanatoside C inhibits the JAK2/STAT6/SOCS2 signaling pathway
Activation of JAK2/STAT6 signaling is associated with cancer progression, and SOCS family proteins are negative modulators of JAK/STAT signaling (37). Thus, the effect of lanatoside C on the JAK2/STAT6 signaling pathway was determined. As presented in Fig. 6A, lanatoside C suppressed JAK2 and STAT6 phosphorylation in a dose-dependent manner (Fig. 6B and C). Following treatment with lanatoside C, the negative regulator, SOCS2, increased in a dose-dependent manner (Fig. 6D). To further confirm the effect of S on the JAK2/STAT6 signaling pathway, the effect of IL-4 on the inhibitory effect of lanatoside C was examined. As presented in Fig. 6E, the inhibition of lanatoside C on p-STAT6 was counteracted by IL-4 (Fig. 6E). Furthermore, the expression of SOCS2 induced by lanatoside C decreased in the presence of IL-4 (Fig. 6E). Furthermore, the inhibition of lanatoside C proliferation in HeLa cells was recovered by IL-4 (Fig. 6F). To further determine the inhibition of lanatoside C on HeLa cell proliferation via JAK2/STAT6/SOCS2 signaling, JAK inhibitor was used to decrease JAK2 activation. The JAK inhibitor was used to further clarify the inhibition of lanatoside C on JAK2. (Fig. 6G). The results demonstrated that the interference of JAK inhibited the proliferation of HeLa cells. However, combined treatment with lanatoside C and JAK inhibitor I did not increase the anti-proliferation effect of lanatoside C on HeLa cells, which indicates that lanatoside C inhibits the downstream signal of JAK2. Taken together, these results suggest that lanatoside C inhibits HeLa cell proliferation via JAK2/STAT6/SOCS2 signaling.
Figure 6.

Lanatoside C inhibits the JAK2-STAT6 signaling pathway. (A) Alternations of p-JAK2, total-JAK2, p-STAT6, total-STAT6 and SOCS2 in HeLa cells treated with lanatoside C for 24 h were detected via western blotting. β-actin served as the loading control. Statistical analysis of alternated level of (B) JAK2 phosphorylation, (C) STAT6 phosphorylation and (D) SOCS2 was measured using ImageJ software. (E) Western blot analysis was performed to detect the expression levels of p-STAT6, total-STAT6 and SOCS2 in HeLa cells treated with lanatoside C (2.5 µM) or lanatoside C (2.5 µM) plus IL-4 (10 ng/ml) for 24 h. (F) The CCK-8 assay was performed to assess the viability of HeLa cells treated with lanatoside C (2.5 µM) or lanatoside C (2.5 µM) plus IL-4 (10 ng/ml) for 24 h. (G) The CCK-8 assay was performed to assess the viability of HeLa cells treated with lanatoside C (2.5 µM), JAK inhibitor I (0.1 µM) or lanatoside C plus JAK inhibitor I for 24 h. Data are presented as the mean ± SEM (n=3). *P<0.05; **P<0.01; ***P<0.001. p, phospho; CCK-8, Cell Counting Kit-8; JAK2, Janus kinase 2; STAT6, signal transducer and activator of transcription 6; SOCS, suppressor of cytokine signaling; LC, lanatoside C.
Discussion
The results of the present study demonstrated that lanatoside C had an antitumor effect on cervical cancer cells, with a strong inhibition of cell proliferation and migration, and the induction of cell cycle arrest and apoptosis via a mitochondria-related pathway. Inhibition of JAK2-STAT6 signaling was observed in lanatoside C-treated cervical cancer cells, which was associated with the induction of SOCS2. These results are similar to those of a previous study, in which chemical JAK2 inhibition resulted in decreased tumor growth of Hodgkin lymphoma both in vitro and in vivo (38). Lanatoside C was approved for treating heart failure by the FDA (28), and its potential application in the treatment of cervical cancer is demonstrated in the present study.
Although the recovery rate for early stage of cervical cancer is up to 80% due to effective treatments, such as concurrent chemotherapy and surgery, prognosis remains poor in patients with recurrent or metastatic cervical cancer (39,40). Metastatic cancer cells require migratory and invasive abilities to invade and migrate through the stoma toward the vasculature and lymphatics (41). Given that lanatoside C significantly inhibits cell migration (28), its application in preventing cervical cancer metastasis or treating patients with metastasis has been proposed. Furthermore, the cervical cancer cell cycle was blocked by lanatoside C. Several clinical drugs, such as Taxol, also target cell cycle progression (42). Taxol can induce G2/M arrest, whereas lanatoside C initiates both S and G2/M arrest (43). The pattern of molecular regulation of cyclins may be diverse, including the expression level of cyclin B, which is either upregulated or downregulated in the G2/M cell cycle-inhibited condition (44). This suggests that the excessive accumulation and degradation of cyclin B may contribute to G2/M cycle arrest, and cyclin B is a potential target for maintaining cell cycle progression (45).
Aberrant activation and mutation of JAK2 has been implicated in cancer progression, including cell motility, invasion and proliferation (46). Preclinical studies have developed JAK2 inhibitors, such as ruxolitinib for myelofibrosis (46,47), suggesting that the clinical application of JAK2 blockade is warranted. However, inhibiting JAK/STAT signaling remains a challenge in controlling cancer due to the pleiotropic roles of JAKs in immune regulation (48). JAK2 serves as a kinase following cytokine binding to the receptor during oligomerization (49). Phosphorylated JAK2 phosphorylates Src homology 2 (SH2) domain-containing proteins, such as STATs, which dock on cytokine receptors (46,50). In the JAK2/STAT6 canonical signaling pathway, STAT6 is phosphorylated by JAK2, which promotes STAT6 dimerization and nuclear translocation to regulate the transcription of genes affecting cell proliferation and apoptosis (51). The present study investigated the activated profiles of JAK2 and STAT6, and lanatoside C inhibited their activation. Constitutively active STAT6 is also associated with increased cell proliferation in leukemia and lymphomas (52). Although a previous study revealed the association of single nucleotide polymorphisms in JAK2 and STAT6 in cervical cancer populations, direct evidence of STAT6 activation in cervical cancer is still insufficient (53). Notably, the JAK/STAT signaling pathway is one of many control pathways that promote cell motility by regulating actin dynamics and activating key metastasis-promoting genes (54). Activation of STAT6 may contribute to inducing the epithelial-to-mesenchymal transition (EMT) process and aggressiveness (55). STAT6 activation is associated with the expression of the EMT core regulator, ZEB1 (56).
In the present study, the presence of IL-4 recovered the inhibition of lanatoside C on cancer cell proliferation, indicating that the mechanism of lanatoside C on cancer cell proliferation is associated with inhibition of the JAK2/STAT6/SOCS2 signaling pathway. It has been reported that Na+, K+-ATPase is the target of lanatoside C, which inhibits Na+, K+-ATPase pump to induce cancer cell apoptosis (57). Based on the results of the present study, lanatoside C may have several potential targets on antitumor cell proliferation. The results of the present study demonstrated that the SOCS2/JAK2/STAT6 signaling pathway is a newly identified signal for lanatoside C.
In non-cancerous cells, STAT6 activation by IL-4 induction leads to the upregulation of growth factor independence-1 and downregulation of the cell cycle inhibitor p27Kip1, causing cell proliferation (52). From an immunological perspective, the immune system plays a critical role in controlling cervical cancer progression in HPV-induced cases (58). Given that JAK/STAT signaling contributes to the activation and cytokine production of multiple immune cells, such as macrophages and NK cells, inhibition of STAT6 activation may abrogate cancer cell immune evasion (59,60). For example, supernatants from HeLa cells render macrophages from M1 to M2 with STAT6 activation, which upregulates the expression pattern of cytokines, such as IL-10 and GM-CSF, providing a favorable environment for tumor growth (61). Thus, it is presumed that drugs targeting JAK2/STAT6 signaling in both cancer cells and tumor-associated immune cells can enhance their efficacy. Furthermore, the inhibitory effect of lanatoside C on other signaling pathways is also worthy of attention. Lanatoside C has been reported to suppress cancer cell proliferation by attenuating the MAPK, Wnt, and PI3K/AKT/mTOR pathways (57). The MAPK pathway is critical for the proliferation and migration of cancer cells and cancer progression (62). Activation of the MAPK signaling pathway can keep cancer cells in the G2/M phase (57). The PI3K/AKT/mTOR pathway is associated with autophagy; the inhibitor of PI3K/AKT/mTOR is regarded as a novel and efficient drug for cancer therapy (63). Although the inhibitory effect of lanatoside C on JAK2/STAT6 signaling in cervical cancer cells has been reported, whether lanatoside C inhibits JAK/STAT signaling in immune cells requires further investigation.
In addition to the positive regulators of JAK/STAT signaling, inhibitory factors, such as protein inhibitors of activated STATs and suppressors of cytokine signaling are involved in the negative feedback loop of JAK/STAT activation (20). Directly transcribed by STATs, the SOCS family can exert its inhibitory effect via spatial competition for cytokine receptor phosphotyrosines in an SH2-dependent manner, or via direct inhibition of JAK proteins with kinase inhibitory regions, while promoting JAK protein degradation in a ubiquitin-mediated signaling pathway (64). In the present study, the JAK2 protein expression was not affected, while the phosphorylation of STAT6 was significantly inhibited, suggesting that SOCS2 may exert its regulatory effect mainly by decreasing the activity of JAK to inhibit JAK/STAT signal transduction. Similarly, trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling by upregulating SOCS1 and SOCS3 by histone modifications in human colorectal cancer cells (65). Although these results suggest the regulatory role of SOCS2 in the JAK2/STAT6 signaling pathway, the molecular mechanism by which SOCS2 mediates JAK2/STAT6 signaling requires further investigation.
A cervical cancer mouse model and clinical samples treated with lanatoside C should be investigated to confirm that lanatoside C can reduce this signal and to further clarify the mechanism of lanatoside C in cancer therapy.
In conclusion, the results of the present study demonstrated that lanatoside C is as a promising anticancer agent for treating cervical cancer, in terms of inhibiting cell proliferation, migration and inducing apoptosis. Furthermore, lanatoside C inhibits JAK2/STAT6 signal transduction by upregulating SOCS2 expression. Taken together, these findings suggest that lanatoside C is a potential chemotherapy candidate for cervical cancer treatment. However, the present study is not without limitations. A cervical cancer mouse model and clinical samples treated with lanatoside C should be investigated to confirm that lanatoside C can reduce this signal and to further clarify the mechanism of lanatoside C in cancer therapy.
Acknowledgements
Not applicable.
Funding Statement
The present study was supported by the Shanghai Municipal Health Commission (grant no. 201840354).
Funding
The present study was supported by the Shanghai Municipal Health Commission (grant no. 201840354).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Authors' contributions
MY and JZ conceived the present study. YD and LC made substantial contributions to conception and design. JS, CJ, YZ, RZ and MY designed, performed and interpreted the experimental data. YL and HK analyzed the data. YD and MY drafted the initial manuscript. YD and MY confirm the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chrysostomou AC, Stylianou DC, Constantinidou A, Kostrikis LG. Cervical cancer screening programs in Europe: The transition towards HPV vaccination and population-based HPV testing. Viruses. 2018;10:729. doi: 10.3390/v10120729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends-an update. Cancer Epidemiol Biomarkers Prev. 2016;25:16–27. doi: 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 3.Shrestha AD, Neupane D, Vedsted P, Kallestrup P. Cervical cancer prevalence, incidence and mortality in low and middle income countries: A systematic review. asian Pac J Cancer Prev. 2018;19:319–324. doi: 10.22034/APJCP.2018.19.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radu MC, Boeru C, Pop-Tudose ME, Necsulescu A, Dumitrescu A, Iancu CF, Nita I, Limbau AM, Zaharia C. Human papillomavirus infection at the time of delivery. Cureus. 2021;13:e15364. doi: 10.7759/cureus.15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman A. HPV testing as a screen for cervical cancer. BMJ. 2015;350:h2372. doi: 10.1136/bmj.h2372. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Wu L, Tong R, Yang F, Yin L, Li M, You L, Xue J, Lu Y. PD-1/PD-L1 inhibitors in cervical cancer. Front Pharmacol. 2019;10:65. doi: 10.3389/fphar.2019.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang S, Wu X, Tan M, Gong J, Tan W, Bian B, Chen M, Wang Y. Fighting fire with fire: Poisonous Chinese herbal medicine for cancer therapy. J Ethnopharmacol. 2012;140:33–45. doi: 10.1016/j.jep.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 8.Minion LE, Tewari KS. Cervical cancer-State of the science: From angiogenesis blockade to checkpoint inhibition. Gynecol Oncol. 2018;148:609–621. doi: 10.1016/j.ygyno.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heeran AB, Berrigan HP, O'Sullivan J. The radiation-induced bystander effect (RIBE) and its connections with the hallmarks of cancer. Radiat Res. 2019;192:668–679. doi: 10.1667/RR15489.1. [DOI] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Qin AQ, Liang ZG, Ye JX, Li J, Wang JL, Chen CX, Song HL. Significant efficacy of additional concurrent chemotherapy with radiotherapy for postoperative cervical cancer with risk factors: A systematic review and meta-analysis. Asian Pac J Cancer Prev. 2016;17:3945–3951. [PubMed] [Google Scholar]
- 12.Cao YP, Sun JY, Li MQ, Dong Y, Zhang YH, Yan J, Huang RM, Yan X. Inhibition of G9a by a small molecule inhibitor, UNC0642, induces apoptosis of human bladder cancer cells. Acta Pharmacol Sin. 2019;40:1076–1084. doi: 10.1038/s41401-018-0205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu GJ, Chen JT, Tsai HC, Chen TL, Liu SH, Chen RM. Protection of dexmedetomidine against ischemia/reperfusion-induced apoptotic insults to neuronal cells occurs via an intrinsic mitochondria-dependent pathway. J Cell Biochem. 2017;118:2635–2644. doi: 10.1002/jcb.25847. [DOI] [PubMed] [Google Scholar]
- 14.Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol. 2012;942:157–183. doi: 10.1007/978-94-007-2869-1_7. [DOI] [PubMed] [Google Scholar]
- 15.Bhola PD, Letai A. Mitochondria-judges and executioners of cell death sentences. Mol Cell. 2016;61:695–704. doi: 10.1016/j.molcel.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan J, Li X, Zhang G, Cheng W, Wang W, Lei Y, Ma Q, Song G. USP39 mediates p21-dependent proliferation and neoplasia of colon cancer cells by regulating the p53/p21/CDC2/cyclin B1 axis. Mol Carcinog. 2021;60:265–278. doi: 10.1002/mc.23290. [DOI] [PubMed] [Google Scholar]
- 17.Pestell RG, Albanese C, Reutens AT, Segall JE, Lee RJ, Arnold A. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr Rev. 1999;20:501–534. doi: 10.1210/edrv.20.4.0373. [DOI] [PubMed] [Google Scholar]
- 18.Strauss B, Harrison A, Coelho PA, Yata K, Zernicka-Goetz M, Pines J. Cyclin B1 is essential for mitosis in mouse embryos, and its nuclear export sets the time for mitosis. J Cell Biol. 2018;217:179–193. doi: 10.1083/jcb.201612147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang SH, Kim AR, Park NH, Park JW, Han IS. DRG2 regulates G2/M progression via the cyclin B1-Cdk1 complex. Mol Cells. 2016;39:699–704. doi: 10.14348/molcells.2016.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong S, Laimins LA. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013;9:e1003295. doi: 10.1371/journal.ppat.1003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M, Decker T. Canonical and non-canonical aspects of JAK-STAT signaling: Lessons from interferons for cytokine responses. Front Immunol. 2017;8:29. doi: 10.3389/fimmu.2017.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welsch K, Holstein J, Laurence A, Ghoreschi K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur J Immunol. 2017;47:1096–1107. doi: 10.1002/eji.201646680. [DOI] [PubMed] [Google Scholar]
- 23.Shan H, Yao S, Ye Y, Yu Q. 3-Deoxy-2β,16-dihydroxynagilactone E, a natural compound from Podocarpus nagi, preferentially inhibits JAK2/STAT3 signaling by allosterically interacting with the regulatory domain of JAK2 and induces apoptosis of cancer cells. Acta Pharmacol Sin. 2019;40:1578–1586. doi: 10.1038/s41401-019-0254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pattison MJ, Mackenzie KF, Arthur JS. Inhibition of JAKs in macrophages increases lipopolysaccharide-induced cytokine production by blocking IL-10-mediated feedback. J Immunol. 2012;189:2784–2792. doi: 10.4049/jimmunol.1200310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mottok A, Renné C, Willenbrock K, Hansmann ML, Bräuninger A. Somatic hypermutation of SOCS1 in lymphocyte-predominant Hodgkin lymphoma is accompanied by high JAK2 expression and activation of STAT6. Blood. 2007;110:3387–3390. doi: 10.1182/blood-2007-03-082511. [DOI] [PubMed] [Google Scholar]
- 26.von Hoff L, Kärgel E, Franke V, McShane E, Schulz-Beiss KW, Patone G, Schleussner N, Kolesnichenko M, Hübner N, Daumke O, et al. Autocrine LTA signaling drives NF-κB and JAK-STAT activity and myeloid gene expression in Hodgkin lymphoma. Blood. 2019;133:1489–1494. doi: 10.1182/blood-2018-08-871293. [DOI] [PubMed] [Google Scholar]
- 27.Ghafouri-Fard S, Oskooei VK, Azari I, Taheri M. Suppressor of cytokine signaling (SOCS) genes are downregulated in breast cancer. World J Surg Oncol. 2018;16:226. doi: 10.1186/s12957-018-1529-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Y, Yu K, Wang G, Zhang D, Shi C, Ding Y, Hong D, Zhang D, He H, Sun L, et al. Lanatoside C inhibits cell proliferation and induces apoptosis through attenuating Wnt/β-catenin/c-Myc signaling pathway in human gastric cancer cell. Biochem Pharmacol. 2018;150:280–292. doi: 10.1016/j.bcp.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 29.Cheung YY, Chen KC, Chen H, Seng EK, Chu JJ. Antiviral activity of lanatoside C against dengue virus infection. Antiviral Res. 2014;111:93–99. doi: 10.1016/j.antiviral.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 30.Nie Y, Zhang D, Jin Z, Li B, Wang X, Che H, You Y, Qian X, Zhang Y, Zhao P, Chai G. Lanatoside C protects mice against bleomycin-induced pulmonary fibrosis through suppression of fibroblast proliferation and differentiation. Clin Exp Pharmacol Physiol. 2019;46:575–586. doi: 10.1111/1440-1681.13081. [DOI] [PubMed] [Google Scholar]
- 31.Durmaz I, Guven EB, Ersahin T, Ozturk M, Calis I, Cetin-Atalay R. Liver cancer cells are sensitive to Lanatoside C induced cell death independent of their PTEN status. Phytomedicine. 2016;23:42–51. doi: 10.1016/j.phymed.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 32.Kheraldine H, Gupta I, Alhussain H, Jabeen A, Cyprian FS, Akhtar S, Al Moustafa AE, Rachid O. Substantial cell apoptosis provoked by naked PAMAM dendrimers in HER2-positive human breast cancer via JNK and ERK1/ERK2 signalling pathways. Comput Struct Biotechnol J. 2021;19:2881–2890. doi: 10.1016/j.csbj.2021.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mierke CT. The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep Prog Phys. 2019;82:064602. doi: 10.1088/1361-6633/ab1628. [DOI] [PubMed] [Google Scholar]
- 34.Sun BB, Fu LN, Wang YQ, Gao QY, Xu J, Cao ZJ, Chen YX, Fang JY. Silencing of JMJD2B induces cell apoptosis via mitochondria-mediated and death receptor-mediated pathway activation in colorectal cancer. J Dig Dis. 2014;15:491–500. doi: 10.1111/1751-2980.12166. [DOI] [PubMed] [Google Scholar]
- 35.Singh A, Kukreti R, Saso L, Kukreti S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules. 2019;24:1583. doi: 10.3390/molecules24081583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calaf GM, Ponce-Cusi R, Carrión F. Curcumin and paclitaxel induce cell death in breast cancer cell lines. Oncol Rep. 2018;40:2381–2388. doi: 10.3892/or.2018.6603. [DOI] [PubMed] [Google Scholar]
- 37.Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS proteins to control JAK-STAT signalling in disease. Trends Pharmacol Sci. 2019;40:298–308. doi: 10.1016/j.tips.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res. 2014;20:2674–2683. doi: 10.1158/1078-0432.CCR-13-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Wu X, Cheng X. Advances in diagnosis and treatment of metastatic cervical cancer. J Gynecol Oncol. 2016;27:e43. doi: 10.3802/jgo.2016.27.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim MC, Lee M, Shim SH, Nam EJ, Lee JY, Kim HJ, Lee YY, Lee KB, Park JY, Kim YH, et al. Practice guidelines for management of cervical cancer in Korea: A Korean society of gynecologic oncology consensus statement. J Gynecol Oncol. 2017;28:e22. doi: 10.3802/jgo.2017.28.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oudin MJ, Weaver VM. Physical and chemical gradients in the tumor microenvironment regulate tumor cell invasion, migration, and metastasis. Cold Spring Harb Symp Quant Biol. 2016;81:189–205. doi: 10.1101/sqb.2016.81.030817. [DOI] [PubMed] [Google Scholar]
- 42.Hua S, Kong X, Chen B, Zhuang W, Sun Q, Yang W, Liu W, Zhang Y. Anticancer mechanism of lobaplatin as monotherapy and in combination with paclitaxel in human gastric cancer. Curr Mol Pharmacol. 2018;11:316–325. doi: 10.2174/1874467211666180813095050. [DOI] [PubMed] [Google Scholar]
- 43.Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event, which is independent of taxol-mediated G2/M arrest. J Biol Chem. 2004;279:15196–15203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 44.Pan B, Liu C, Zhan X, Li J. Protegrin-1 regulates porcine granulosa cell proliferation via the EGFR-ERK1/2/p38 signaling pathway in vitro. Front Physiol. 2021;12:673777. doi: 10.3389/fphys.2021.673777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Striz A, DePina A, Jones R, Jr, Gao X, Yourick J. Cytotoxic, genotoxic, and toxicogenomic effects of dihydroxyacetone in human primary keratinocytes. Cutan Ocul Toxicol. 2021 Jul 11; doi: 10.1080/15569527.2021.1931877. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 46.Harry BL, Eckhardt SG, Jimeno A. JAK2 inhibition for the treatment of hematologic and solid malignancies. Expert Opin Investig Drugs. 2012;21:637–655. doi: 10.1517/13543784.2012.677432. [DOI] [PubMed] [Google Scholar]
- 47.Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 48.Pencik J, Pham HT, Schmoellerl J, Javaheri T, Schlederer M, Culig Z, Merkel O, Moriggl R, Grebien F, Kenner L. JAK-STAT signaling in cancer: From cytokines to non-coding genome. Cytokine. 2016;87:26–36. doi: 10.1016/j.cyto.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hedl M, Proctor DD, Abraham C. JAK2 disease-risk variants are gain of function and JAK signaling threshold determines innate receptor-induced proinflammatory cytokine secretion in macrophages. J Immunol. 2016;197:3695–3704. doi: 10.4049/jimmunol.1600845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quintás-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10:127–140. doi: 10.1038/nrd3264. [DOI] [PubMed] [Google Scholar]
- 51.Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 2006;17:173–188. doi: 10.1016/j.cytogfr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 52.Bruns HA, Kaplan MH. The role of constitutively active Stat6 in leukemia and lymphoma. Crit Rev Oncol Hematol. 2006;57:245–253. doi: 10.1016/j.critrevonc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Z, Fye S, Borecki IB, Rader JS. Polymorphisms in immune mediators associate with risk of cervical cancer. Gynecol Oncol. 2014;135:69–73. doi: 10.1016/j.ygyno.2014.07.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trivedi S, Starz-Gaiano M. Drosophila Jak/STAT signaling: Regulation and relevance in human cancer and metastasis. Int J Mol Sci. 2018;19:4056. doi: 10.3390/ijms19124056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McGaha TL, Le M, Kodera T, Stoica C, Zhu J, Paul WE, Bona CA. Molecular mechanisms of interleukin-4-induced up-regulation of type I collagen gene expression in murine fibroblasts. Arthritis Rheum. 2003;48:2275–2284. doi: 10.1002/art.11089. [DOI] [PubMed] [Google Scholar]
- 56.Cao H, Zhang J, Liu H, Wan L, Zhang H, Huang Q, Xu E, Lai M. IL-13/STAT6 signaling plays a critical role in the epithelial-mesenchymal transition of colorectal cancer cells. Oncotarget. 2016;7:61183–61198. doi: 10.18632/oncotarget.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reddy D, Kumavath R, Ghosh P, Barh D. Lanatoside C induces G2/M cell cycle arrest and suppresses cancer cell growth by attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR signaling pathways. Biomolecules. 2019;9:792. doi: 10.3390/biom9120792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berman TA, Schiller JT. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer. 2017;123:2219–2229. doi: 10.1002/cncr.30588. [DOI] [PubMed] [Google Scholar]
- 59.Bottos A, Gotthardt D, Gill JW, Gattelli A, Frei A, Tzankov A, Sexl V, Wodnar-Filipowicz A, Hynes NE. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun. 2016;7:12258. doi: 10.1038/ncomms12258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carlsson C, Johansson T. The psychological effects of propranolol in the abstinence phase of chronic alcoholics. Br J Psychiatry. 1971;119:605–606. doi: 10.1192/bjp.119.553.605. [DOI] [PubMed] [Google Scholar]
- 61.Sánchez-Reyes K, Pedraza-Brindis EJ, Hernández-Flores G, Bravo-Cuellar A, López-López BA, Rosas-González VC, Ortiz-Lazareno PC. The supernatant of cervical carcinoma cells lines induces a decrease in phosphorylation of STAT-1 and NF-κB transcription factors associated with changes in profiles of cytokines and growth factors in macrophages derived from U937 cells. Innate Immun. 2019;25:344–355. doi: 10.1177/1753425919848841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fan J, Ren D, Wang J, Liu X, Zhang H, Wu M, Yang G. Bruceine D induces lung cancer cell apoptosis and autophagy via the ROS/MAPK signaling pathway in vitro and in vivo. Cell Death Dis. 2020;11:126. doi: 10.1038/s41419-020-2317-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol. 2019;59:125–132. doi: 10.1016/j.semcancer.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 64.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. 2008;19:414–422. doi: 10.1016/j.semcdb.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiong H, Du W, Zhang YJ, Hong J, Su WY, Tang JT, Wang YC, Lu R, Fang JY. Trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing the promoter-associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells. Mol Carcinog. 2012;51:174–184. doi: 10.1002/mc.20777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.