

Abstract

PB1 is a bromodomain-containing protein hypothesized to act as the nucleosome-recognition subunit of the PBAF complex. Although PB1 is a key component of the PBAF chromatin remodeling complex, its exact role has not been elucidated due to the lack of potent and selective inhibitors. Chemical probes that target specific bromodomains within the complex would constitute highly valuable tools to characterize the function and therapeutic pertinence of PB1 and of each of its bromodomains. Here, we report the design and synthesis of lead compound LM146, which displays strong stabilization of the second and fifth bromodomains of PB1 as shown by DSF. LM146 does not interact with bromodomains outside of sub-family VIII and binds to PB1(2), PB1(5), and SMARCA2B with KD values of 110, 61, and 2100 nM, respectively, providing a ∼34-fold selectivity profile for PB1(5) over SMARCA2.
Introduction
ε-N-Acetylation of lysine residues on histone tails is one of the most fundamental and dynamic epigenetic transformations that controls changes in chromatin accessibility.1 This post-translational modification plays a key role in regulating gene expression and is often associated with transcriptional activation.2 Bromodomains are selective protein motifs that act as epigenetic readers by mediating the interaction with acetylated lysine of histones and transcriptional regulators.3−5 They are defined by four α helices linked by two flexible loops forming a hydrophobic pocket and are usually composed of about 110 amino acids.6 The human proteome expresses 61 bromodomains distributed across 46 proteins, which can be clustered into eight sub-families based on both their structure similarities and their sequence homology.7
Bromodomain-containing proteins (BCPs) are often part of multivalent complexes, and many possess other chromatin recognition domains such as PHD fingers, PWWP domains, or multiple bromodomains.8 Polybromo-1 protein (PB1, also referred to as PBRM1 or BAF180) is a component of the BRG1/BRM-associated factor (PBAF) complex, a human analog of the yeast switch/sucrose non-fermenting (SWI/SNF) complex. PB1 is a unique epigenetic reader that contains six distinct bromodomains, while other known bromodomain-containing proteins possess at most two bromodomains. The multi-subunit PBAF complex contains three BCPs: BRD7, a member of the sub-family IV, and SMARCA4 (also known as BRG1) and PB1, two members of the sub-family VIII. While it is known that PBAF acts as a chromatin remodeling complex by repositioning nucleosomes through ATP hydrolysis,9 the exact role of each component remains ambiguous. However, it has been established that the BCPs are necessary components for the complex’s cellular function.10
Intensive research has been dedicated to the SWI/SNF family complexes as their components are recurrently mutated in many cancers.11−15 In light of the biological importance of PBAF, chemical probes that target different bromodomains within the complex would constitute highly valuable tools to elucidate the exact function and therapeutic pertinence of each member.
Molecular probes targeting sub-family IV have been reported, including I-BRD9, a BRD9 selective inhibitor with a pIC50 of 7.3, and more than 70-fold selectivity over a panel of other bromodomains,16 as well as numerous BRD9/BRD7 dual inhibitors such as LP99,17 BI-9564,18 TP-472,19 or GSK6776.20 However, chemical probes targeting sub-family VIII are fewer and lack selectivity within the group. PFI-3 potently targets the bromodomains of SMARCA2A/B (two isoforms derived from alternative splicing: a long transcript, named SMARCA2A, and a short transcript, SMARCA2B), SMARCA4, as well as the fifth bromodomain of PB1 (PB1(5)) with KD values of 81, 86, 97, and 54 nM, respectively (Figure 1).21 Compound 1, an analog of PFI-3, was also described as targeting simultaneously SMARCA2, SMARCA4, and PB1(5) as well as the second bromodomain of PB1 (PB1(2)) with KD values of 37, 53, 30, and 190 nM, respectively.22 In 2016, Sutherell et al. reported compound 2 that binds to PB1(5), SMARCA2B, and SMARCA4 with KD values of 124, 262, and 417 nM, respectively, and analog 3 that showed stronger interaction with PB1 than SMARCAs.23 The same year, pyridazine derivatives of generic structure 4 were reported by Constellation Pharmaceuticals and Genentech as pan-inhibitors of SMARCA2/4 and PB1(5).24 In 2020, some of these patented molecules were fully characterized by the SGC, revealing KD values of 35, 36, and 13 nM against SMARCA2, SMARCA4, and PB1(5), respectively, for compound 5.25 The only inhibitor showing some degree of selectivity within the sub-family VIII for PB1(5) is the tricyclic compound 6. However, with a KD value of 3.3 μM, 6 lacks potency for PB1(5).26 To the best of our knowledge, there are currently no potent inhibitors of PB1 with selectivity toward specific bromodomains.
Figure 1.

Reported sub-family VIII bromodomain inhibitors.
Given that PB1 is predicted to act as the nucleosome-recognition subunit of the PBAF complex and that somatic PB1 mutations occur in many cancers, including up to 50% of clear cell renal cell carcinomas (ccRCC),27,28 we aimed to develop potent and selective PB1 inhibitors as tool compounds for in vitro functional characterization. Because it has been recently demonstrated that bromodomains 2, 4, and 5 are critical for PB1 activity by mediating the binding to acetylated histone peptides as well as to modified recombinant and cellular nucleosomes29 and since their collaboration is key for PB1 tumor suppressor functions,30 we elected to focus on the development of chemical probes targeting PB1(2), PB1(4), and PB1(5). Herein, we report the synthesis and biological evaluation of a new potent derivative of compound 3 that binds to PB1 with significant selectivity over SMARCA2 and with preference for distinct bromodomains of PB1.
Results and Discussion
We began by resynthesizing compound 3 for use as a reference in our assays. Since the binding pocket around the aromatic ring of 3 is slightly larger in PB1(5) than in SMARCA2A, we then prepared a small ensemble of derivatives where substituents are introduced on the aromatic ring, as shown in Table 1. These compounds were easily accessed using the synthetic route reported by Sutherell et al. (see Experimental Section for details).23 The stereochemistry of the double bond was previously demonstrated to be E for these types of compounds.23 The proton NMR spectrum of resynthesized known compound 3 was identical to the data from the literature.23 Other new compounds had similar proton NMR spectra and were assumed to have the E stereochemistry. Differential scanning fluorimetry (DSF) was used to quickly evaluate the binding of our compounds against PB1(2), PB1(4), PB1(5), and SMARCA2A. This thermal shift assay was previously reported to be suitable for rapid determination of apparent potency and selectivity for bromodomains.22,23 However, since proteins can behave differently, the extent of thermal stabilization should not be interpreted as an absolute scale of compound affinity.31 In agreement with the literature results, resynthesized 3 showed strong binding to PB1(2) and PB1(5), thus validating our thermal shift biophysical assay. The presence of the chloride ortho to the carbonyl moiety in 3 was previously shown to be beneficial for PB1(5) protein stabilization due to a halogen bond interaction with the carbonyl of Met731.23 However, this chloride also led to good affinity with SMARCA2A, probably as a result of a similar halogen bond interaction with the carbonyl of Leu1456. We therefore studied the impact of moving or replacing the chloride in 3 on the stabilization of PB1 and SMARCA2A. Compounds 3, 7, and 8 confirmed that the optimal position for the chlorine is in R1, that is, adjacent to the carbonyl. Indeed, complete loss of affinity to all domains of PB1 as well as to SMARCA2A was observed when the chloride was moved to the R2 position, as shown by compound 7. Interestingly, marginal but specific stabilization of PB1(2) was retained when the chloride was moved to R3, as indicated by compound 8. Conversely, dichloride 9 showed a reversal of apparent selectivity by weakly binding to PB1(5) and SMARCA2A, suggesting that the negative effect of the chloride at R2 is compensated by the chloride at R1. Sutherell et al. reported that a methoxy in R1 leads to complete loss of stabilization of PB1(5) in the related enamine series.23 Similarly, we observed that compounds 10 and 11 that contain a methoxy at R2 or R3 do not bind to PB1 nor to SMARCA2A. However, polar hydroxy groups were tolerated by PB1(2), as indicated by compounds 12 and 13, leading to inhibitors with first known apparent selectivity over the other sub-family VIII bromodomains.
Table 1. Structure Activity Relationship (SAR) of Left-Hand Side (LHS) Substituted Derivatives of 3 against Sub-Family VIII Bromodomains by the DSF Assay.
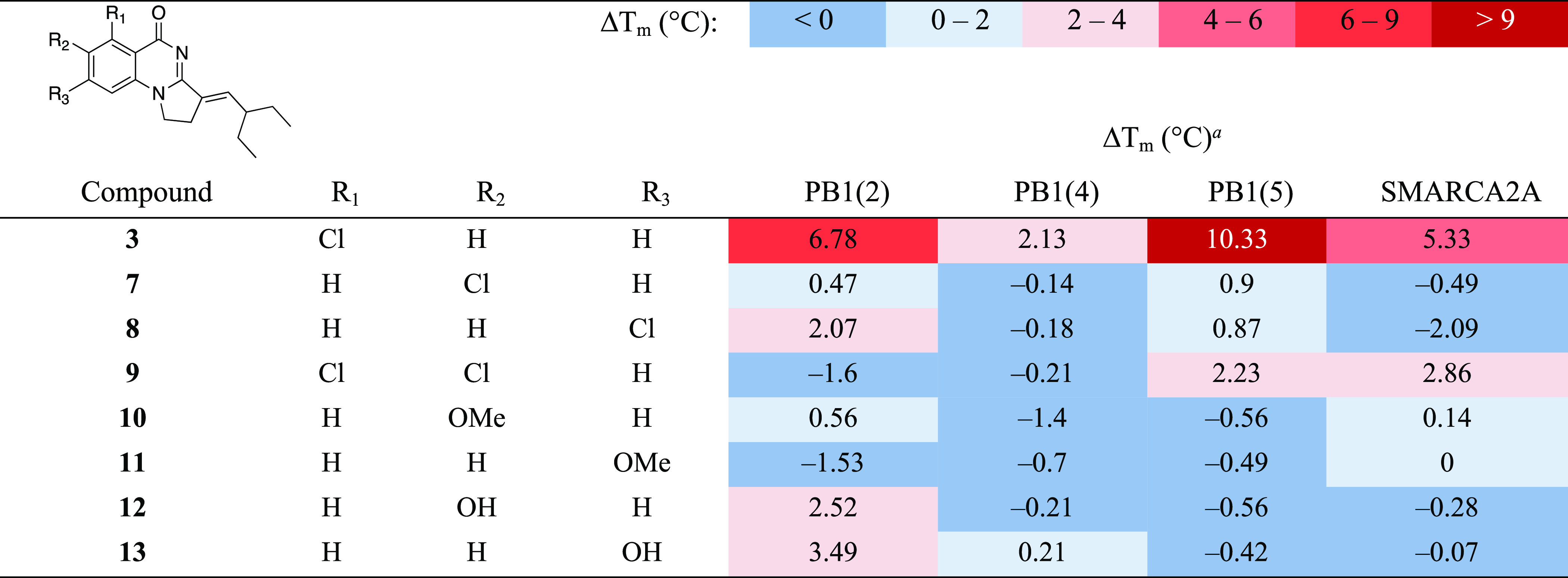
Values shown are the average of at least two replicates with a compound concentration of 100 μM.
As gaining selectivity over SMARCA2A while keeping high binding affinity with PB1 proved to be challenging when the aromatic ring was not substituted with a chloride in R1, we decided to move our efforts to the right-hand side R′ alkylidene chain (Table 2). Although it is solvent-exposed, the olefinic R′ chain was previously shown to play a major role in orienting the core inside the pocket.23 Replacing the 3-pentyl group in compound 3 by a sec-butyl (compound 14) resulted in erosion of apparent affinity for all targets. Compound 15 that possesses a linear n-propyl chain showed reduced stabilization of PB1(2) compared to 3 but retained the stabilization of PB1(5). Shortening the chain length provided compound 16, which showed exclusive binding to PB1(5). The stabilization of PB1(2) and PB1(5) could be increased by building on steric hindrance with the introduction of a tert-butyl group in compound 17. We also found that the iso-butyl and neopentyl derivatives 18 and 19 have high apparent affinity for PB1(5), with 19 also showing strong stabilization of the second domain of PB1.
Table 2. Structure Activity Relationship (SAR) of Right-Hand Side (RHS) Substituted Derivatives of 3 against Sub-Family VIII Bromodomains by the DSF Assay.
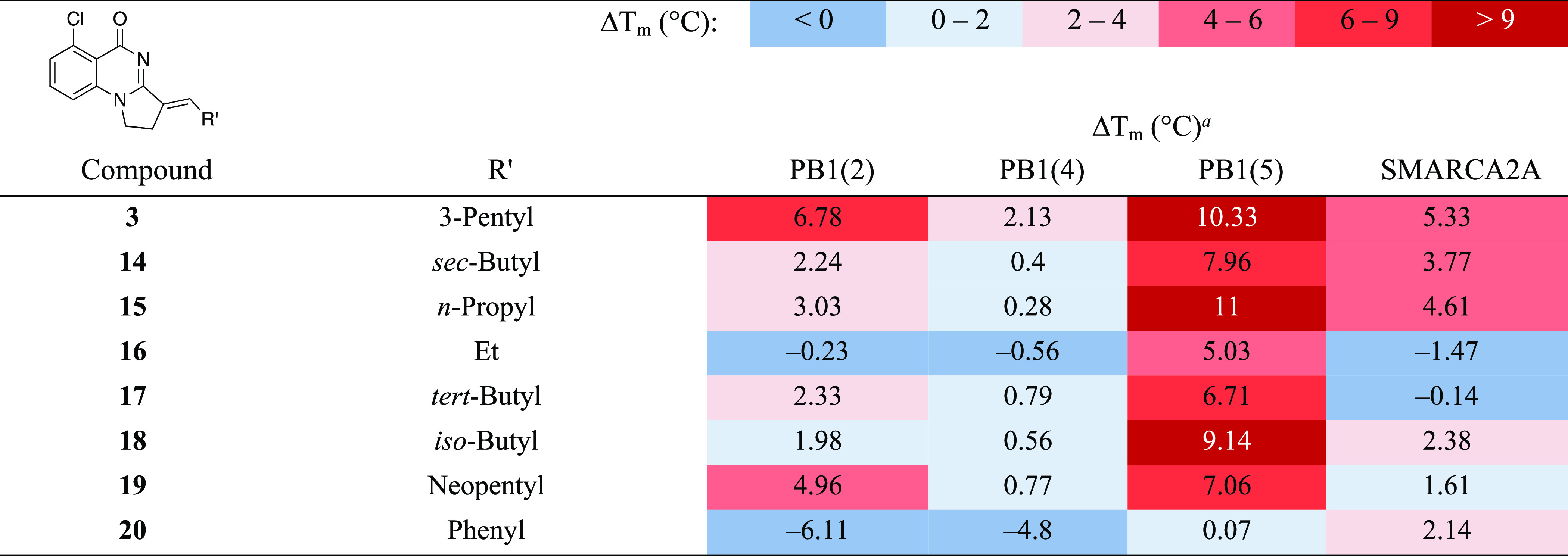
Values shown are the average of at least two replicates with a compound concentration of 100 μM.
Sutherell et al. reported that replacement of the alkyl chain by an aromatic ring in the des-chloro version of 3 leads to retention of affinity against PB1(5).23 Surprisingly, in the chloro series, we observed no stabilization of PB1(5), weak binding to SMARCA2A, and even a drastic destabilization of PB1(2) and PB1(4) for the phenyl derivative 20. Compounds 14 to 20 provide valuable information on the structural elements that are required for optimal stabilization of PB1 domains and SMARCA while also offering diversified binding profiles.
As the entrance of the pocket appears to be a key region to selectively inhibit PB1 over SMARCA2A, we decided to expand the lower right-hand side cycle size as illustrated in Table 3 with the hypothesis that these compounds would not fit in the narrower entrance of SMARCA2. This modification could also help deviate from planarity by introducing extra tetrahedral carbons while also increasing the overall solubility of the compounds. To evaluate the impact of this ring expansion, we prepared the tricyclic compound 21 in the olefinic series and compound 22 in the enamine series using routes from the literature (see Experimental Section for details).32 As anticipated, the expansion of the lower ring completely canceled the affinity for SMARCA2A. In addition, this modification also abrogated the binding to PB1(4), considerably reduced the binding to PB1(2), but retained good affinity to PB1(5).
Table 3. Structure Activity Relationship (SAR) of Ring Expanded, Saturated, and Cyclopropanated Substituted Derivatives of 3 against Sub-Family VIII Bromodomains by the DSF Assay.
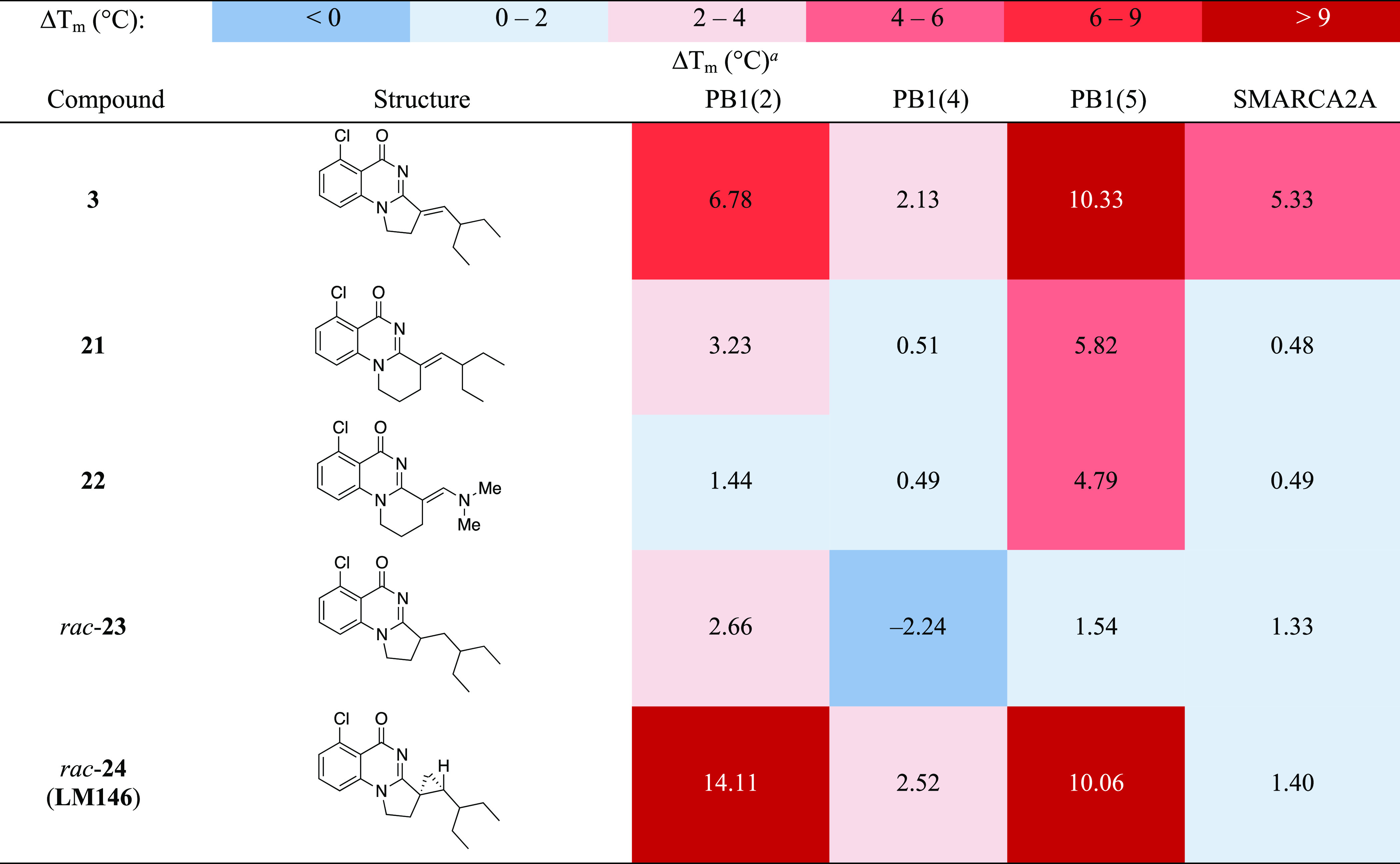
Values shown are the average of at least two replicates with a compound concentration of 100 μM.
Motivated by the impact of geometrical features on the apparent selectivity profile, we then synthesized the saturated analog 23 using the sequence illustrated in Scheme 1. For initial assessment and for ease of synthesis, the racemic version was targeted. Intermediate 28 was first prepared according to Sutherell et al. by hydrolysis of the cyano group in 25 followed by amide formation between 26 and 4-chlorobutyryl chloride and cyclization of 27.23 Condensation of 28 with 2-ethylbutanal afforded derivative 3. At this stage, we were faced with the challenge of chemoselectively saturating the olefin in the presence of the aryl chloride. After testing various conditions, we found that reacting 3 with Raney nickel under a hydrogen atmosphere provided the desired saturated product 23 in low yield, albeit with the chlorine still in place.
Scheme 1. Synthesis Route for 23.

DSF results indicate that 23 binds weakly to PB1(2) but none of the other bromodomains nor to SMARCA2A. Since the alkene seemed to be the key for maintaining binding, cyclopropyl derivative 24 (LM146) was designed in order to keep the orientation and conformational rigidity of the olefin. Considered a common alkene replacement in medicinal chemistry, the cyclopropane would also add some steric hindrance at the entrance of the cavity, which was previously hypothesized to be essential for gaining selectivity over SMARCA2. Therefore, we synthesized the cyclopropyl derivative LM146 as a racemic mixture by engaging compound 3 in a Johnson–Corey–Chaykovsky cyclopropanation reaction (Scheme 2). The relative stereochemistry of LM146 was confirmed by X-ray analysis, with the obtention of a structure in a centrosymmetric space group (P21/c) typical of racemic mixtures. DSF results indicate that LM146 provides a clear increase in ΔTm for PB1(2) compared to 3 while keeping the stabilization of PB1(4) and PB1(5). Moreover, LM146 showed very minor apparent interaction with SMARCA2A.
Scheme 2. Synthesis and X-ray Structure of Cyclopropyl Derivative LM146 (rac-24) CCDC No.: 2070470.

In order to confirm its utility as a potential tool compound for biological studies of PB1, LM146 was tested against a panel of 32 bromodomains, including BET bromodomains, in a bromoMAX assay performed at Eurofins (see the Supporting Information). Results show that at 10 μM, LM146 only binds to bromodomains within the sub-family VIII, with less affinity for SMARCA4, as illustrated by the interaction map in Figure 2. These results are in agreement with the binding mode of the parent compounds 2 and 3 (and presumably our derivatives), which displace conserved water molecules within the acetylated lysine pocket. These water molecules were previously reported to have stronger binding in other bromodomain sub-families, partly explaining the selectivity observed for sub-family VIII.33
Figure 2.
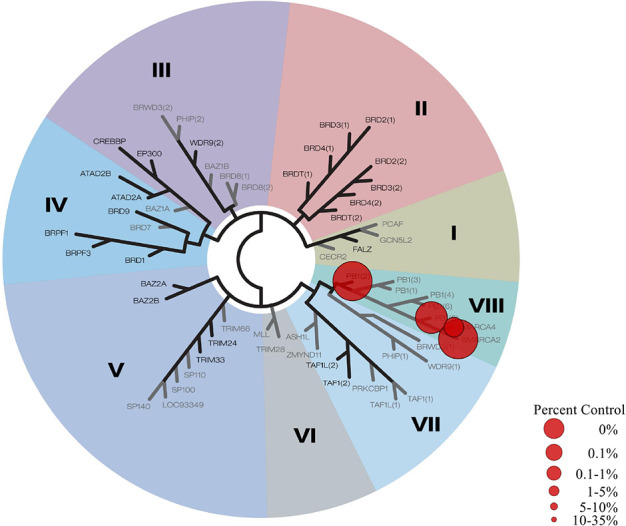
Selectivity profile for LM146 screened at 10 μM against 32 selected bromodomains in a bromoMAX assay.
Key compounds were then selected for dissociation constant determination using a bromoSCAN assay performed at Eurofins (see the Supporting Information) in order to accurately evaluate the binding affinity of our compounds against PB1(2), PB1(5) and SMARCA2B. Compound 18 was selected for its apparent selectivity profile, which favors binding to PB1(5), while compounds 21 and 24 were chosen for their structural features that diverge from existing literature compounds combined with the absence of apparent interaction with SMARCA2A. As shown in Table 4, compound 18 is a moderate inhibitor of PB1(5) with a KD value of 490 nM, a ∼3-fold selectivity over PB1(2), and ∼10-fold selectivity over SMARCA2B. Analog 21, even if less potent, shows high selectivity for the PB1 bromodomains with an unprecedented ∼24-fold selectivity for PB1(5) over SMARCA2B. Finally, cyclopropyl derivative LM146 is as potent as PFI-3 and its analog 1 but shows increased selectivity toward PB1 over SMARCA2B with a ∼34-fold selectivity profile in favor of PB1(5) and a ∼19-fold selectivity profile in favor of PB1(2). To our knowledge, LM146 constitutes the first potent modulator of PB1 bromodomains with a high selectivity profile over SMARCA2. Representative full inhibition curves for LM146 against PB1(5) and SMARCA2B show a gradual dose–response with increasing compound concentration (Figure 3).
Table 4. KD Values for Key Compounds.
|
KD (nM) |
|||
|---|---|---|---|
| cmpd | PB1(2) | PB1(5) | SMARCA2B |
| PFI-3a | N/Ad | 48 | 81 |
| 1a | 190 | 30 | 37 |
| 2b | N/Ad | 124 | 262 |
| 3c | 190 | 47 | 290 |
| 18c | 1600 | 490 | 4800 |
| 21c | 5400 | 1100 | 26,000 |
| LM146c | 110 | 61 | 2100 |
Values were assessed by ITC and reported in ref (21).
Values were assessed by ITC and reported in ref (23).
Values were assessed by bromoSCAN (see the Supporting Information).
N/A = not applicable.
Figure 3.

Representative bromoSCAN traces for KD determination of LM146 against (a) PB1(5) and (b) SMARCA2B.
Conclusions
In summary, we described the optimization of reported compound 3 with the aim of developing potent and selective PB1 inhibitors. DSF biophysical characterization allowed the assessment of SAR and led to the discovery of compounds with various apparent selectivity profiles that should allow further in vitro functional characterization of PB1 subdomains. Lead compound LM146 showed no binding to any bromodomains outside the sub-family VIII at 10 μM and displayed KD values of 110, 61, and 2100 nM against PB1(2), PB1(5), and SMARCA2B, respectively. LM146 is a PB1-pan inhibitor with a ∼34-fold selectivity profile for PB1(5) over SMARCA2B and a ∼19-fold selectivity profile for PB1(2) over SMARCA2B. The inhibitors reported herein should contribute to expanding the toolbox for studying the role of PB1 in chromatin remodeling and disease development.
Experimental Section
General Information
Unless otherwise stated, reactions were performed in non-flame dried glassware, and commercial reagents were used without further purification. Anhydrous solvents were obtained using an encapsulated solvent purification system and were further dried over 4 Å molecular sieves. The evolution of reactions was monitored by analytical thin-layer chromatography (TLC) using silica gel 60 F254 precoated plates visualized by ultraviolet radiation (254 nm). Flash chromatography was performed employing 230–400 mesh silica using the indicated solvent system according to standard techniques. Nuclear magnetic resonance 1H spectra were recorded on a Bruker Avance-III 300 or 600 MHz, and 13C spectra were recorded on a Bruker Avance-III 75 or 151 MHz spectrometer. Chemical shifts for 1H NMR spectra were recorded in parts per million from tetramethylsilane with the solvent resonance as the internal standard (chloroform-d, δ 7.26 ppm; methanol-d4, δ 3.31 ppm; and dimethyl sulfoxide-d6, δ 2.50 ppm). Data is reported as follows: chemical shift, multiplicity (s = singlet, s(br) = broad singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, m = multiplet, dd = doublet of doublet, dt = doublet of triplet, ddd = doublet of doublet of doublet, dtd = doublet of triplet of doublet), coupling constant J in Hz, and integration. Chemical shifts for 13C NMR spectra are recorded in parts per million from tetramethylsilane using the solvent resonance as the internal standard (chloroform-d, δ 77.16 ppm; methanol-d4, δ 49.00 ppm; and dimethysulfoxide-d6, δ 39.52 ppm). Purity was assessed on an Agilent 1260 infinity HPLC system equipped with an Agilent Eclipse Plus C18 (3.5 μM and 4.6 × 100 mm) column using a 20 min gradient method (0 to 100% MeCN + 0.06% TFA in water + 0.06% TFA; the absorbance was measured at 254 nm). HRMS was performed on a TOF LCMS analyzer using the electrospray (ESI) mode.
General Procedure A
The appropriate 2-aminobenzonitrile (1.0 equiv) and K2CO3 (0.2 equiv) were added to a microwave tube in nanopure water. After irradiation under microwave at 150 °C for 1 h 30 min, the reaction mixture was cooled and extracted with EtOAc. The combined organic phases were dried over Na2SO4, filtered, and evaporated under vacuum. When indicated, the crude residue was purified by flash column chromatography to give the title compound.
General Procedure B
A solution of the appropriate substituted 2-aminobenzamide (1.0 equiv) in THF (2.5 mL per mmol of substrate) was cooled to 0 °C. Triethylamine (2.0 equiv) followed by the appropriate acid chloride (1.2 equiv) in THF (2 mL per mmol substrate) was added to the stirred solution. The reaction was stirred at room temperature overnight. The mixture was then diluted with EtOAc and washed with a saturated aqueous solution of NaHCO3. The aqueous phase was back extracted with EtOAc, and the combined organic phases were dried over Na2SO4, filtered, and evaporated under vacuum. The crude residue was purified by flash column chromatography to give the desired compound.
General Procedure C
To a solution of the appropriate benzamide (1.0 equiv) in THF (10 mL per mmol of substrate) was added t-BuOK (2.0 equiv). The reaction was stirred at room temperature overnight, and then the solvent was removed in vacuo. The resulting residue was dissolved in CH2Cl2, and the resulting solution was washed with a saturated aqueous solution of NaHCO3. The aqueous layer was back extracted with CH2Cl2, and the combined organic phases were dried over Na2SO4, filtered, and evaporated under vacuum. The crude residue was purified by flash column chromatography to give the title compound.
General Procedure D
t-BuOK (1.2 equiv) was added to a solution of the appropriate cyclized substrate (1.0 equiv) in CH2Cl2 (0.4 M). The mixture was vigorously stirred for 5 min, and then the appropriate aldehyde (1.1 equiv) was added. After 1 h, a saturated aqueous solution of NaHCO3 and CH2Cl2 was added. The phases were separated, and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over Na2SO4, filtered, and evaporated under vacuum. The crude residue was purified by flash column chromatography to give the title compound.
2-Amino-6-chlorobenzamide (26)
2-Amino-6-chlorobenzonitrile (2.00 g and 13.1 mmol) was hydrolyzed in 18 mL of nanopure water according to general procedure A to afford 2-amino-6-chlorobenzamide 26 (2.22 g, 13.0 mmol, and 99%) as a white powder. The crude compound was taken directly in the next step. Spectral data are consistent with literature values.231H NMR (300 MHz, CDCl3) δ 7.06 (t, J = 8.0 Hz, 1H), 6.73 (dd, J = 7.9, 1.0 Hz, 1H), 6.60 (dd, J = 8.2, 1.0 Hz, 1H), 6.19 (s(br), 1H), 5.97 (s(br), 1H), 4.84 (s(br), 2H).
2-Chloro-6-(4-chlorobutanamido)benzamide (27)
2-Amino-6-chlorobenzamide 26 (2.22 g and 13.0 mmol) and 4-chlorobutanoyl chloride (1.75 mL and 15.6 mmol) were reacted according to general procedure B. Purification by flash column chromatography (SiO2, hexanes/EtOAc 60/40 to 30/70) afforded 2-chloro-6-(4-chlorobutanamido)benzamide 27 (3.11 g, 11.3 mmol, and 87%) as a white solid. Spectral data are consistent with literature values.231H NMR (300 MHz, CDCl3) δ 9.26 (s(br), 1H), 8.11 (d, J = 8.3 Hz, 1H), 7.31 (t, J = 8.2 Hz, 1H), 7.15 (dd, J = 8.1, 1.1 Hz, 1H), 6.44 (s(br), 2H), 3.62 (t, J = 6.3 Hz, 2H), 2.55 (t, J = 7.2 Hz, 2H), 2.15 (quint, J = 6.6 Hz, 2H).
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (28)
2-Chloro-6-(4-chlorobutanamido)benzamide 27 (220 mg and 0.800 mmol) was cyclized according to general procedure C. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) provided 6-chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (175 mg, 0.793 mmol, and 99%) as a white solid. Spectral data are consistent with literature values.231H NMR (300 MHz, CDCl3) δ 7.52 (t, J = 8.1 Hz, 1H), 7.39 (dd, J = 8.0, 1.1 Hz, 1H), 7.07 (dd, J = 8.2, 1.1 Hz, 1H), 4.19 (t, J = 7.4 Hz, 2H), 3.15 (t, J = 8.1 Hz, 2H), 2.40 (quint, J = 7.6 Hz, 2H).
(E)-6-Chloro-3-(2-ethylbutylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (3)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (156 mg and 0.707 mmol) and 2-ethylbutanal (96 μL and 0.78 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 3 (146 mg, 0.482 mmol, and 68%) as a white solid. Spectral data are consistent with literature values.231H NMR (300 MHz, CDCl3) δ 7.45 (t, J = 8.1 Hz, 1H), 7.30 (dd, J = 7.9, 1.1 Hz, 1H), 7.04 (dd, J = 8.3, 1.1 Hz, 1H), 6.85 (dt, J = 10.8, 2.7 Hz, 1H), 4.20–4.09 (m, 2H), 2.99 (ddd, J = 7.9, 6.6, 2.7 Hz, 2H), 2.22–2.07 (m, 1H), 1.63–1.47 (m, 2H), 1.45–1.28 (m, 2H), 0.85 (t, J = 7.4 Hz, 6H); HRMS (ESI) calcd for [C17H19ClN2O + H]+: 303.12587, found 303.12554, calcd for [C17H19ClN2O + Na]+: 325.10781, found 325.10795; HPLC purity: >99%.
2-Amino-5-chlorobenzamide (29)
2-Amino-5-chlorobenzonitrile (500 mg and 3.28 mmol) was hydrolyzed in 6 mL of nanopure water according to general procedure A. Purification by flash column chromatography (SiO2, hexanes/EtOAc 50:50 to 0:100) provided 2-amino-5-chlorobenzamide 29 (421 g, 2.47 mmol, and 75%) as a white solid. Spectral data are consistent with literature values.321H NMR (300 MHz, methanol-d4) δ 7.53 (d, J = 2.4 Hz, 1H), 7.14 (dd, J = 8.8, 2.5 Hz, 1H), 6.72 (d, J = 8.8 Hz, 1H).
5-Chloro-2-(4-chlorobutanamido)benzamide (30)
2-Amino-5-chlorobenzamide 29 (421 mg and 2.47 mmol) and 4-chlorobutanoyl chloride (0.331 mL and 2.96 mmol) were reacted according to general procedure B. Purification by flash column chromatography (SiO2, hexanes/EtOAc 60/40 to 30/70) provided 5-chloro-2-(4-chlorobutanamido)benzamide 30 (463 mg, 1.68 mmol, and 68%) as a yellow solid. Spectral data are consistent with literature values.321H NMR (300 MHz, CDCl3) δ 11.08 (s(br), 1H), 8.61 (d, J = 8.9 Hz, 1H), 7.51–7.43 (m, 2H), 6.14 (s, 1H), 5.75 (s, 1H), 3.65 (t, J = 6.3 Hz, 2H), 2.60 (t, J = 7.2 Hz, 2H), 2.19 (quint, J = 6.6 Hz, 2H).
7-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (31)
5-Chloro-2-(4-chlorobutanamido)benzamide 30 (463 mg and 1.68 mmol) was cyclized according to general procedure C. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) provided 7-chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 31 (73 mg, 0.33 mmol, and 20%) as a beige solid. Spectral data are consistent with literature values.321H NMR (300 MHz, CDCl3) δ 8.19 (d, J = 2.4 Hz, 1H), 7.63 (dd, J = 8.7, 2.4 Hz, 1H), 7.15 (d, J = 8.8 Hz, 1H), 4.24 (t, J = 7.4 Hz, 2H), 3.20 (t, J = 8.1 Hz, 2H), 2.42 (quint, J = 7.8 Hz, 2H).
(E)-7-Chloro-3-(2-ethylbutylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (7)
7-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 31 (73 mg and 0.33 mmol) and 2-ethylbutanal (45 μL and 0.36 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 7 (61 mg, 0.20 mmol, and 61%) as a beige solid. Spectral data are consistent with literature values.321H NMR (300 MHz, CDCl3) δ 8.19 (d, J = 2.4 Hz, 1H), 7.56 (dd, J = 8.7, 2.4 Hz, 1H), 7.13 (d, J = 8.7 Hz, 1H), 6.89 (dt, J = 10.8, 2.7 Hz, 1H), 4.19 (dd, J = 7.9, 6.7 Hz, 2H), 3.02 (ddd, J = 8.0, 6.6, 2.8 Hz, 2H), 2.25–2.08 (m, 1H), 1.63–1.47 (m, 2H), 1.45–1.28 (m, 2H), 0.85 (t, J = 7.4 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 169.32, 160.40, 143.08, 137.32, 133.86, 131.82, 131.03, 128.25, 120.73, 116.17, 45.99, 44.27, 27.58, 23.06, 12.05; HRMS (ESI) calcd for [C17H19ClN2O + H]+: 303.12587, found 303.12598; HPLC purity: >99%.
2-Amino-4-chlorobenzamide (32)
2-Amino-4-chlorobenzonitrile (500 mg and 3.28 mmol) was hydrolyzed in 6 mL of nanopure water according to general procedure A. Purification by flash column chromatography (SiO2, hexanes/EtOAc 50:50 to 0:100) provided 2-amino-4-chlorobenzamide 32 (400 mg, 2.34 mmol, and 71%) as a beige solid. 1H NMR (300 MHz, methanol-d4) δ 7.47 (d, J = 8.5 Hz, 1H), 6.74 (d, J = 2.1 Hz, 1H), 6.54 (dd, J = 8.5, 2.1 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 169.91, 140.83, 136.62, 130.30, 122.11, 119.43, 117.96.
4-Chloro-2-(4-chlorobutanamido)benzamide (33)
2-Amino-4-chlorobenzamide 32 (400 mg and 2.35 mmol) and 4-chlorobutanoyl chloride (0.316 mL and 2.82 mmol) were reacted according to general procedure B. Purification by flash column chromatography (SiO2, hexanes/EtOAc 60/40 to 30/70) provided 4-chloro-2-(4-chlorobutanamido)benzamide 33 (357 mg, 1.30 mmol, and 55%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 11.36 (s(br), 1H), 8.76 (d, J = 2.1 Hz, 1H), 7.44 (d, J = 8.5 Hz, 1H), 7.06 (dd, J = 8.4, 2.1 Hz, 1H), 6.06 (s(br), 1H), 5.77 (s(br), 1H), 3.65 (t, J = 6.3 Hz, 2H), 2.61 (t, J = 7.2 Hz, 2H), 2.20 (quint, J = 6.7 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 171.01, 170.58, 141.41, 139.83, 128.38, 122.93, 121.54, 116.43, 44.35, 35.08, 27.98.
8-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (34)
4-Chloro-2-(4-chlorobutanamido)benzamide 33 (346 mg and 1.26 mmol) was cyclized according to general procedure C. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) provided 8-chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 34 (80 mg, 0.36 mmol, and 29%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 8.06 (d, J = 8.5 Hz, 1H), 7.28 (dd, J = 8.5, 1.9 Hz, 1H), 7.11 (d, J = 1.9 Hz, 1H), 4.17 (t, J = 7.4 Hz, 2H), 3.14 (t, J = 8.1 Hz, 2H), 2.39 (quint, J = 7.8 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 169.36, 167.26, 140.02, 139.43, 130.35, 126.38, 116.94, 114.70, 48.95, 32.87, 18.70.
(E)-8-Chloro-3-(2-ethylbutylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (8)
8-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 34 (80 mg and 0.36 mmol) and 2-ethylbutanal (49 μL and 0.40 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 8 (107 mg, 0.353 mmol, and 98%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 8.29 (d, J = 8.5 Hz, 1H), 7.41 (dd, J = 8.5, 1.9 Hz, 1H), 7.22 (d, J = 1.9 Hz, 1H), 7.00 (dt, J = 10.8, 2.7 Hz, 1H), 4.19 (dd, J = 8.0, 6.8 Hz, 2H), 3.06 (ddd, J = 9.6, 6.8, 2.8 Hz, 2H), 2.27–2.12 (m, 1H), 1.65–1.51 (m, 2H), 1.49–1.32 (m, 2H), 0.89 (t, J = 7.4 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 169.76, 160.79, 143.46, 140.03, 139.84, 130.96, 130.69, 126.64, 118.20, 114.37, 45.96, 44.41, 27.65, 23.15, 12.11; HRMS (ESI) calcd for [C17H19ClN2O + H]+: 303.12587, found 303.12559, calcd for [C17H19ClN2O + Na]+: 325.10781, found 325.10818; HPLC purity: >99%.
6-Amino-2,3-dichlorobenzamide (35)
6-Amino-2,3-dichlorobenzonitrile (500 mg and 2.67 mmol) was hydrolyzed in 4.5 mL of nanopure water according to general procedure A to afford 6-amino-2,3-dichlorobenzamide 35 (547 mg, 2.67 mmol, quant.) as a beige powder. Compound was used in the next step without further purification. 1H NMR (300 MHz, DMSO-d6) δ 7.92 (s(br), 1H), 7.66 (s(br), 1H), 7.23 (d, J = 8.8 Hz, 1H), 6.66 (d, J = 8.8 Hz, 1H), 5.30 (s, 2H); 13C NMR (75 MHz, DMSO-d6) δ 166.83, 145.51, 129.96, 127.74, 123.50, 117.25, 115.00.
2,3-Dichloro-6-(4-chlorobutanamido)benzamide (36)
6-Amino-2,3-dichlorobenzamide 35 (499 mg and 2.43 mmol) and 4-chlorobutanoyl chloride (0.327 mL and 2.92 mmol) were reacted according to general procedure B. Purification by flash column chromatography (SiO2, hexanes/EtOAc 60/40 to 30/70) provided 2,3-dichloro-6-(4-chlorobutanamido)benzamide 36 (269 mg, 0.869 mmol, and 36%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.88 (s(br), 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 9.0 Hz, 1H), 6.21 (s(br), 1H), 6.14 (s(br), 1H), 3.64 (t, J = 6.2 Hz, 2H), 2.56 (t, J = 7.2 Hz, 2H), 2.17 (quint, J = 6.6 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 171.34, 165.66, 134.51, 134.03, 129.92, 128.12, 127.98, 125.84, 44.94, 33.08, 28.25.
6,7-Dichloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (37)
2,3-Dichloro-6-(4-chlorobutanamido)benzamide 36 (94 mg and 0.30 mmol) was cyclized according to general procedure C. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) provided 6,7-dichloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 37 (21 mg, 0.082 mmol, and 27%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 7.96 (d, J = 9.0 Hz, 1H), 7.45 (d, J = 9.0 Hz, 1H), 4.19 (t, J = 7.3 Hz, 2H), 3.00 (t, J = 8.0 Hz, 2H), 2.24 (quint, J = 7.7 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 166.69, 166.60, 140.26, 134.50, 132.00, 130.10, 116.86, 116.73, 49.92, 32.47, 18.51.
(E)-6,7-Dichloro-3-(2-ethylbutylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (9)
6,7-Dichloro-2,3-dihydropyrrolo[1,2-a]qui]]nazolin-5(1H)-one 37 (21 mg and 0.082 mmol) and 2-ethylbutanal (11 μL and 0.089 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 9 (14 mg, 0.042 mmol, and 51%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 8.9 Hz, 1H), 7.04 (d, J = 8.9 Hz, 1H), 6.90 (dt, J = 10.8, 2.7 Hz, 1H), 4.18 (dd, J = 8.0, 6.8 Hz, 2H), 3.03 (ddd, J = 8.0, 6.7, 2.8 Hz, 2H), 2.26–2.11 (m, 1H), 1.66–1.50 (m, 2H), 1.48–1.31 (m, 2H), 0.88 (t, J = 7.4 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 159.35, 146.35, 143.42, 139.53, 134.20, 134.05, 131.88, 130.62, 117.89, 113.87, 46.51, 44.47, 27.66, 23.01, 12.16; HRMS (ESI) calcd for [C17H18Cl2N2O + H]+: 337.08690, found 337.08631, calcd for [C17H18Cl2N2O + Na]+: 359.06884, found 359.06781; HPLC purity: >99%.
2-Amino-5-methoxybenzamide (38)
2-Amino-5-methoxybenzonitrile (500 mg and 3.37 mmol) was hydrolyzed in 6 mL of nanopure water according to general procedure A. Purification by flash column chromatography (SiO2, hexanes/EtOAc 50:50 to 0:100) provided 2-amino-5-methoxybenzamide 38 (385 mg, 2.32 mmol, and 69%) as a beige solid. Spectral data are consistent with literature values.321H NMR (300 MHz, methanol-d4) δ 7.09 (d, J = 2.9 Hz, 1H), 6.89 (dd, J = 8.9, 2.9 Hz, 1H), 6.73 (d, J = 8.9 Hz, 1H), 3.74 (s, 3H).
2-(4-Chlorobutanamido)-5-methoxybenzamide (39)
2-Amino-5-methoxybenzamide 38 (245 mg and 1.47 mmol) and 4-chlorobutanoyl chloride (0.197 mL and 1.76 mmol) were reacted according to general procedure B. Purification by flash column chromatography (SiO2, hexanes/EtOAc 60/40 to 30/70) provided 2-(4-chlorobutanamido)-5-methoxybenzamide 39 (396 mg, 1.46 mmol, and 99%) as a beige solid. Spectral data are consistent with literature values.321H NMR (300 MHz, CDCl3) δ 10.76 (s(br), 1H), 8.50 (d, J = 9.0 Hz, 1H), 7.08–7.01 (m, 2H), 6.17 (s(br), 1H), 5.72 (s(br), 1H), 3.82 (s, 3H), 3.65 (t, J = 6.3 Hz, 2H), 2.57 (t, J = 7.2 Hz, 2H), 2.19 (quint, J = 6.7 Hz, 2H).
7-Methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (40)
2-(4-Chlorobutanamido)-5-methoxybenzamide 39 (396 mg and 1.46 mmol) was cyclized according to general procedure C. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) provided 7-methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 40 (217 mg, 1.00 mmol, and 68%) as a beige solid. Spectral data are consistent with literature values.321H NMR (300 MHz, CDCl3) δ 7.57 (d, J = 2.9 Hz, 1H), 7.21 (dd, J = 9.0, 2.9 Hz, 1H), 7.09 (d, J = 9.0 Hz, 1H), 4.20 (t, J = 7.3 Hz, 2H), 3.83 (s, 3H), 3.12 (t, J = 8.0 Hz, 2H), 2.36 (quint, J = 8.1 Hz, 2H).
(E)-3-(2-Ethylbutylidene)-7-methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (10)
7-Methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 40 (111 mg and 0.513 mmol) and 2-ethylbutanal (69 μL and 0.56 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 10 (123 mg, 0.412 mmol, and 80%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 2.8 Hz, 1H), 7.13 (dd, J = 8.9, 2.8 Hz, 1H), 7.06 (d, J = 8.9 Hz, 1H), 6.78 (dt, J = 10.8, 2.7 Hz, 1H), 4.12 (dd, J = 7.8, 6.6 Hz, 2H), 3.77 (s, 3H), 2.93 (ddd, J = 8.0, 6.6, 2.7 Hz, 2H), 2.17–2.02 (m, 1H), 1.57–1.37 (m, 2H), 1.37–1.19 (m, 2H), 0.78 (t, J = 7.4 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 170.32, 158.90, 157.75, 141.14, 132.94, 131.27, 123.16, 120.82, 115.96, 108.65, 55.74, 45.84, 43.93, 27.46, 22.94, 11.85; HRMS (ESI) calcd for [C18H22N2O2 + H]+: 299.17540, found 299.17538; HPLC purity: >99%.
2-Amino-4-methoxybenzamide (41)
2-Amino-4-methoxybenzonitrile (500 mg and 3.37 mmol) was hydrolyzed in 6 mL of nanopure water according to general procedure A. Purification by flash column chromatography (SiO2, hexanes/EtOAc 50:50 to 0:100) provided 2-amino-4-methoxybenzamide 41 (133 mg, 0.800 mmol, and 24%) as a beige solid. 1H NMR (300 MHz, DMSO-d6) δ 7.48 (d, J = 8.8 Hz, 1H), 6.73 (s, 2H), 6.19 (d, J = 2.6 Hz, 1H), 6.06 (dd, J = 8.8, 2.6 Hz, 1H), 3.69 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 170.99, 162.18, 152.42, 130.47, 106.85, 102.11, 99.28, 54.77.
2-(4-Chlorobutanamido)-4-methoxybenzamide (42)
2-Amino-4-methoxybenzamide 41 (133 mg and 0.800 mmol) and 4-chlorobutanoyl chloride (0.11 mL and 0.98 mmol) were reacted according to general procedure B. Purification by flash column chromatography (SiO2, hexanes/EtOAc 60/40 to 30/70) provided 2-(4-chlorobutanamido)-4-methoxybenzamide 42 (180 mg, 0.665 mmol, and 83%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 11.71 (s(br), 1H), 8.35 (d, J = 2.6 Hz, 1H), 7.44 (d, J = 8.8 Hz, 1H), 6.59 (dd, J = 8.8, 2.6 Hz, 1H), 6.02 (s(br), 2H), 3.85 (s, 3H), 3.65 (t, J = 6.3 Hz, 2H), 2.61 (t, J = 7.2 Hz, 2H), 2.20 (quint, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.50, 171.18, 163.70, 142.85, 128.96, 110.21, 109.98, 105.10, 55.67, 44.39, 35.29, 28.08.
8-Methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (43)
2-(4-Chlorobutanamido)-4-methoxybenzamide 42 (180 mg and 0.665 mmol) was cyclized according to general procedure C. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) provided 8-methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 43 (64 mg, 0.30 mmol, and 45%) as a beige solid. 1H NMR (600 MHz, CDCl3) δ 8.04 (d, J = 8.8 Hz, 1H), 6.85 (dd, J = 8.9, 2.3 Hz, 1H), 6.40 (d, J = 2.3 Hz, 1H), 4.09 (t, J = 7.4 Hz, 2H), 3.85 (s, 3H), 3.10–3.04 (m, 2H), 2.33 (quint, J = 7.8 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 169.86, 166.44, 163.82, 140.35, 130.40, 113.67, 112.21, 97.85, 55.83, 48.79, 32.74, 18.57.
(E)-3-(2-Ethylbutylidene)-8-methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (11)
8-Methoxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 43 (64 mg and 0.30 mmol) and 2-ethylbutanal (39 μL and 0.32 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 11 (43 mg, 0.14 mmol, and 47%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 8.14 (d, J = 8.9 Hz, 1H), 6.91–6.81 (m, 2H), 6.45 (d, J = 2.3 Hz, 1H), 4.08 (dd, J = 8.0, 6.7 Hz, 2H), 3.86 (s, 3H), 2.97 (ddd, J = 8.0, 6.6, 2.7 Hz, 2H), 2.22–2.07 (m, 1H), 1.60–1.45 (m, 2H), 1.43–1.26 (m, 2H), 0.84 (t, J = 7.4 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 170.23, 163.82, 160.15, 141.94, 140.59, 131.29, 130.49, 113.66, 113.32, 97.60, 55.85, 45.76, 44.11, 27.56, 23.02, 11.99; HRMS (ESI) calcd for [C18H22N2O2 + H]+: 299.17540, found 299.17565; HPLC purity: >99%.
(E)-3-(2-Ethylbutylidene)-7-hydroxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (12)
A solution of the methoxy substrate 10 (40 mg, 0.13 mmol, and 1 equiv) in dry CH2Cl2 (2.7 mL) was cooled to 0 °C. Boron tribromide 1 M in CH2Cl2 (1.3 mL, 1.3 mmol, and 10 equiv) was added dropwise to the stirred reaction mixture. The reaction was stirred at room temperature for 24 h. The mixture was then diluted with CH2Cl2 and a saturated aqueous solution of NaHCO3. The aqueous phase was extracted with CH2Cl2, and the combined organic phases were dried over Na2SO4, filtered, and evaporated under vacuum. The crude residue was purified by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) to give the title compound 12 (36 mg, 0.13 mmol, quant.) as a yellow solid. 1H NMR (300 MHz, methanol-d4) δ 7.57 (d, J = 1.5 Hz, 1H), 7.55 (d, J = 4.5 Hz, 1H), 7.38 (dd, J = 9.0, 2.7 Hz, 1H), 6.84 (dt, J = 10.6, 2.6 Hz, 1H), 4.53–4.43 (m, 2H), 3.15 (td, J = 7.5, 7.0, 2.7 Hz, 2H), 2.41–2.23 (m, 1H), 1.72–1.58 (m, 2H), 1.54–1.37 (m, 2H), 0.95 (t, J = 7.4 Hz, 6H); 13C NMR (75 MHz, methanol-d4) δ 165.95, 159.20, 157.80, 145.40, 132.41, 132.04, 125.48, 122.23, 119.85, 112.72, 45.57, 28.54, 24.37, 12.24; HRMS (ESI) calcd for [C17H20N2O2 + H]+: 285.15975, found 285.16012, calcd for [C17H20N2O2 + Na]+: 307.1417, found 307.1417; HPLC purity: >99%.
(E)-3-(2-Ethylbutylidene)-8-hydroxy-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (13)
A solution of the methoxy substrate 11 (38 mg, 0.13 mmol, and 1 equiv) in dry CH2Cl2 (2.7 mL) was cooled to 0 °C. Boron tribromide 1 M in CH2Cl2 (1.3 mL, 1.3 mmol, and 10 equiv) was added dropwise to the stirred reaction mixture. The reaction was stirred at room temperature for 24 h. The mixture was then diluted with CH2Cl2 and a saturated aqueous solution of NaHCO3. The aqueous phase was extracted with CH2Cl2, and the combined organic phases were dried over Na2SO4, filtered, and evaporated under vacuum. The crude residue was purified by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) to give the title compound 13 (7.8 mg, 0.027 mmol, and 21%) as a yellow solid. 1H NMR (300 MHz, methanol-d4) δ 8.06 (d, J = 8.8 Hz, 1H), 6.99 (dd, J = 8.8, 2.2 Hz, 1H), 6.80–6.73 (m, 2H), 4.33–4.25 (m, 2H), 3.09 (td, J = 7.5, 2.8 Hz, 2H), 2.39–2.20 (m, 1H), 1.70–1.56 (m, 2H), 1.51–1.35 (m, 2H), 0.94 (t, J = 7.4 Hz, 6H); 13C NMR (151 MHz, methanol-d4) δ 172.95, 164.81, 161.92, 142.63, 142.56, 133.95, 131.04, 116.97, 112.99, 101.04, 47.53, 45.21, 28.71, 24.17, 12.29; HRMS (ESI) calcd for [C17H20N2O2 + H]+: 285.15975, found 285.15902, calcd for [C17H20N2O2 + Na]+: 307.1417, found 307.14081; HPLC purity: 98%.
(E)-6-Chloro-3-(2-methylbutylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (14)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (66 mg and 0.30 mmol) and 2-methylbutanal (35 μL and 0.33 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 14 (50 mg, 0.17 mmol, 57%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.42 (t, J = 8.1 Hz, 1H), 7.27–7.23 (dd, J = 7.9, 0.7 HZ, 1H), 7.02 (dd, J = 8.2 Hz, 0.7 Hz, 1H), 6.84 (dt, J = 10.3, 2.7 Hz, 1H), 4.12 (t, J = 7.3 Hz, 2H), 3.02–2.92 (m, 2H), 2.39–2.26 (m, 1H), 1.50–1.33 (m, 2H), 1.04 (d, J = 6.7 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 168.01, 159.43, 143.42, 141.00, 135.77, 132.92, 129.66, 128.59, 116.35, 113.45, 46.46, 36.68, 29.52, 22.56, 19.48, 12.04; HRMS (ESI) calcd for [C16H17ClN2O + H]+: 289.11022, found 289.1108; HPLC purity: >99%.
(E)-3-Butylidene-6-chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (15)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (75 mg and 0.34 mmol) and butyraldehyde (33 μL and 0.37 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 15 (11 mg, 0.040 mmol, and 12%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.46 (t, J = 8.1 Hz, 1H), 7.29 (dd, J = 7.9, 0.7 Hz, 1H), 7.06 (dd, J = 8.2, 0.7 Hz, 1H), 7.03–6.96 (m, 1H), 4.20–4.13 (m, 2H), 2.99 (t, J = 6.1 Hz, 2H), 2.21 (q, J = 7.4 Hz, 2H), 1.53 (sext, J = 7.3 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 168.19, 159.42, 141.06, 138.39, 136.08, 133.10, 130.95, 128.87, 116.41, 113.56, 46.65, 32.09, 22.72, 21.78, 14.07; HRMS (ESI) calcd for [C15H15ClN2O + H]+: 275.09457, found 275.09405, calcd for [C15H15ClN2O + Na]+: 297.07651, found 297.07538; HPLC purity: >99%.
(E)-6-Chloro-3-propylidene-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (16)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (75 mg and 0.34 mmol) and propionaldehyde (28 μL and 0.37 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 16 (0.6 mg, 0.002 mmol, and 1%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.55 (t, J = 8.1 Hz, 1H), 7.44 (d, J = 7.5 Hz, 1H), 7.22–7.15 (m, 1H), 7.11 (d, J = 7.3 Hz, 1H), 4.20 (t, J = 6.8 Hz, 2H), 3.09–2.96 (m, 2H), 2.37–2.21 (m, 2H), 1.13 (t, J = 7.5 Hz, 3H); HRMS (ESI) calcd for [C14H13ClN2O + H]+: 261.07892, found 261.07797; HPLC purity: 98%.
(E)-6-Chloro-3-(2,2-dimethylpropylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (17)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (75 mg and 0.34 mmol) and pivalaldehyde (40 μL and 0.37 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 17 (17 mg, 0.059 mmol, and 17%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 7.46 (t, J = 8.1 Hz, 1H), 7.33 (dd, J = 7.9, 1.0 Hz, 1H), 7.12 (t, J = 2.7 Hz, 1H), 7.05 (dd, J = 8.2, 1.0 Hz, 1H), 4.17–4.07 (m, 2H), 3.16 (td, J = 7.4, 7.0, 2.8 Hz, 2H), 1.21 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 168.05, 160.72, 147.36, 141.06, 136.09, 132.93, 128.79, 127.18, 116.64, 113.40, 46.70, 34.30, 29.65, 23.19; HRMS (ESI) calcd for [C16H17ClN2O + H]+: 289.11022, found 289.10894, calcd for [C16H17ClN2O + Na]+: 311.09216, found 311.09099; HPLC purity: >99%.
(E)-6-Chloro-3-(2,2-dimethylpropylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (18)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (75 mg and 0.34 mmol) and 3-methylbutanal (40 μL and 0.37 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 18 (5 mg, 0.02 mmol, and 5%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 7.50 (t, J = 8.1 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 7.12 (dt, J = 8.3, 2.5 Hz, 2H), 7.07 (d, J = 8.1 Hz, 1H), 4.21–4.11 (m, 2H), 2.99 (t, J = 6.2 Hz, 2H), 2.14 (t, J = 7.2 Hz, 2H), 1.94 – 1.75 (m, 1H), 0.97 (d, J = 6.6 Hz, 6H); HRMS (ESI) calcd for [C16H17ClN2O + H]+: 289.11022, found 289.10879, calcd for [C16H17ClN2O + Na]+: 311.09216, found 311.09085; HPLC purity: 97%.
(E)-6-Chloro-3-(3,3-dimethylbutylidene)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (19)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (70 mg and 0.32 mmol) and 3,3-dimethylbutanal (44 μL and 0.35 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 19 (10 mg, 0.033 mmol, and 10%) as a beige solid. 1H NMR (300 MHz, CDCl3) δ 7.51 (t, J = 8.1 Hz, 1H), 7.39 (dd, J = 7.9, 1.0 Hz, 1H), 7.23–7.14 (m, 1H), 7.08 (dd, J = 8.2, 1.0 Hz, 1H), 4.21–4.11 (m, 2H), 3.00 (t, J = 6.0 Hz, 2H), 2.15 (d, J = 7.9 Hz, 2H), 0.99 (s, 9H); 13C NMR (75 MHz, CDCl3) δ 168.16, 159.14, 141.02, 135.84, 132.96, 132.22, 128.64, 116.34, 113.49, 113.42, 46.44, 44.30, 32.31, 29.57, 22.86; HRMS (ESI) calcd for [C17H19ClN2O + H]+: 303.12587, found 303.12722, calcd for [C17H19ClN2O + Na]+: 325.10781, found 325.10619; HPLC purity: >99%.
(E)-3-Benzylidene-6-chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (20)
6-Chloro-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one 28 (75 mg and 0.34 mmol) and benzaldehyde (38 μL and 0.37 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 20 (2 mg, 0.006 mmol, and 2%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.02 (t, J = 2.6 Hz, 1H), 7.58–7.51 (m, 3H), 7.48–7.37 (m, 4H), 7.14 (dd, J = 8.2, 0.9 Hz, 1H), 4.34–4.26 (m, 2H), 3.42 (td, J = 7.7, 2.7 Hz, 2H); HRMS (ESI) calcd for [C18H13ClN2O + H]+: 309.07892, found 309.07753, calcd for [C18H13ClN2O + Na]+: 331.06086, found 331.05951; HPLC purity: 97%.
2-Chloro-6-(5-chloropentanamido)benzamide (44)
2-Amino-6-chlorobenzamide 26 (2.18 g and 12.8 mmol) and 5-chloropentanoyl chloride (1.97 mL and 15.3 mmol) were reacted according to general procedure B. Purification by flash column chromatography (SiO2, hexanes/EtOAc 60/40 to 30/70) provided 2-chloro-6-(5-chloropentanamido)benzamide 44 (2.70 g, 9.3 mmol, and 73%) as a beige solid. Spectral data are consistent with literature values.321H NMR (300 MHz, CDCl3) δ 9.32 (s(br), 1H), 8.23 (dd, J = 8.4, 1.1 Hz, 1H), 7.34 (t, J = 8.2 Hz, 1H), 7.16 (dd, J = 8.1, 1.1 Hz, 1H), 6.31 (s(br), 1H), 6.12 (s(br), 1H), 3.57 (td, J = 5.6, 4.9, 2.8 Hz, 2H), 2.41 (td, J = 6.5, 5.4, 3.1 Hz, 2H), 1.90–1.83 (m, 4H).
7-Chloro-1,2,3,4-tetrahydro-6H-pyrido[1,2-a]quinazolin-6-one (45)
2-Chloro-6-(5-chloropentanamido)benzamide 44 (655 mg and 2.27 mmol) was cyclized according to general procedure C. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 95:5) provided 7-chloro-1,2,3,4-tetrahydro-6H-pyrido[1,2-a]quinazolin-6-one 45 (237 mg, 1.01 mmol, and 44%) as a beige solid. Spectral data are consistent with literature values.321H NMR (300 MHz, CDCl3) δ 7.25 (dd, J = 8.6, 7.8 Hz, 1H), 7.12 (dd, J = 8.7, 1.1 Hz, 1H), 7.04 (dd, J = 7.8, 1.0 Hz, 1H), 3.80 (t, J = 6.2 Hz, 2H), 2.74 (t, J = 6.6 Hz, 2H), 2.03–1.93 (m, 2H), 1.82–1.72 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 165.59, 160.33, 142.81, 134.39, 132.25, 128.12, 116.50, 112.96, 47.06, 32.38, 22.17, 18.55.
(E)-7-Chloro-4-(2-ethylbutylidene)-1,2,3,4-tetrahydro-6H-pyrido[1,2-a]quinazolin-6-one (21)
7-Chloro-1,2,3,4-tetrahydro-6H-pyrido[1,2-a]quinazolin-6-one 45 (70 mg and 0.30 mmol) and 2-ethylbutanal (41 μL and 0.33 mmol) were reacted according to general procedure D. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 96:4) provided 21 (43 mg, 0.14 mmol, and 45%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 7.53–7.44 (m, 1H), 7.38–7.29 (m, 3H), 4.04–3.96 (m, 2H), 2.61 (ddd, J = 7.9, 4.3, 1.6 Hz, 2H), 2.32–2.18 (m, 1H), 2.13 (quint, J = 6.2 Hz, 2H), 1.62–1.44 (m, 2H), 1.44–1.26 (m, 2H), 0.82 (t, J = 7.4 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 166.56, 155.71, 147.53, 143.62, 135.35, 132.51, 128.36, 127.54, 117.89, 113.00, 47.64, 42.85, 27.74, 23.55, 22.04, 12.20; HRMS (ESI) calcd for [C18H21ClN2O + H]+: 317.14152, found 317.14249; HPLC purity: >99%.
(E)-7-Chloro-4-((dimethylamino)methylene)-1,2,3,4-tetrahydro-6H-pyrido[1,2-a]quinazolin-6-one (22)
To a solution of 7-chloro-1,2,3,4-tetrahydro-6H-pyrido[1,2-a]quinazolin-6-one 45 (70 mg, 0.30 mmol, and 1.0 equiv) in DMF was added phosphoryl trichloride (56 μL, 0.60 mmol, and 2.0 equiv). The reaction was heated to 70 °C for 16 h, and then the solution was cooled to room temperature, diluted with CH2Cl2, and slowly quenched using a saturated aqueous solution of NaHCO3. The aqueous layer was extracted twice with CH2Cl2, and the combined organic phases were dried over Na2SO4, filtered, and evaporated under vacuum. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 98:2 to 94:6) provided 22 (46 mg, 0.16 mmol, and 53%) as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 8.17 (s, 1H), 7.30 (t, J = 8.2 Hz, 1H), 7.17 (d, J = 7.7 Hz, 1H), 7.07 (d, J = 8.4 Hz, 1H), 3.82–3.73 (m, 2H), 3.06 (s, 6H), 2.69–2.60 (m, 2H), 1.99 (quint, J = 6.1 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 165.61, 158.40, 150.89, 144.18, 134.62, 131.58, 126.64, 117.36, 111.84, 94.06, 46.81, 43.67, 23.10, 22.28; HRMS (ESI) calcd for [C15H16ClN3O + H]+: 290.10547, found 290.10590, calcd for [C15H16ClN3O + Na]+: 312.08741, found 312.08816; HPLC purity: 97%.
6-Chloro-3-(2-ethylbutyl)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (23)
A solution of substrate 3 (59 mg, 0.19 mmol, and 1.0 equiv) in MeOH was purged with argon for 30 min at room temperature before treatment with Raney nickel (41 mg, 0.70 mmol, 3.5 equiv, and pre-washed 3 times with MeOH). The reaction mixture was then purged with hydrogen before being stirred under a hydrogen atmosphere for 1 h at room temperature. The mixture was then filtered through a pad of celite, washed with MeOH, and concentrated under pressure. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 98:2 to 96:4) provided 23 (8 mg, 0.03 mmol, 13%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.52 (t, J = 8.1 Hz, 1H), 7.39 (dd, J = 7.9, 0.8 Hz, 1H), 7.07 (dd, J = 8.2, 0.7 Hz, 1H), 4.17 (td, J = 9.7, 9.2, 3.9 Hz, 1H), 4.04 (dt, J = 10.3, 8.0 Hz, 1H), 3.25 (dtd, J = 12.0, 8.4, 3.8 Hz, 1H), 2.55 (dtd, J = 12.5, 8.3, 3.9 Hz, 1H), 2.18–2.07 (m, 1H), 2.06–1.92 (m, 1H), 1.53–1.39 (m, 3H), 1.37–1.27 (m, 4H), 0.88 (td, J = 7.2, 5.4 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 168.24, 141.12, 136.64, 132.99, 128.72, 116.30, 113.29, 47.73, 41.95, 38.27, 35.97, 26.05, 26.00, 24.35, 11.10, 10.52; HRMS (ESI) calcd for [C17H21ClN2O + H]+: 305.14152, found 305.14167; HPLC purity: >99%.
6′-Chloro-2-(pentan-3-yl)-1′,2′-dihydro-5′H-spiro[cyclopropane-1,3′-pyrrolo[1,2-a]quinazolin]-5′-one (24 = LM146)
A solution of trimethylsulfoxonium iodide (40 mg, 0.18 mmol, and 1.5 equiv) in dry DMSO (0.7 mL) was cooled to 0 °C. Sodium hydride 95% (5.0 mg, 0.18 mmol, and 1.5 equiv) was added to the stirred solution. The mixture was allowed to warm-up to room temperature over 1 h. A solution of substrate 3 (37 mg, 0.12 mmol, and 1.0 equiv) in dry DMSO (0.8 mL) was added dropwise to the reaction mixture, which was then stirred at 50 °C for 1 h 30 min under an argon atmosphere. The mixture was diluted with CH2Cl2 and quenched with a saturated aqueous solution of NH4Cl. The aqueous phase was extracted with CH2Cl2 (three times). Combined organic phases were washed with brine, dried over Na2SO4, filtered, and evaporated under vacuum. Purification by flash column chromatography (SiO2, CH2Cl2/MeOH 97:3) provided racemic 24 (LM146) (31 mg, 0.098 mmol, and 82%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.49 (t, J = 8.1 Hz, 1H), 7.34 (dd, J = 7.9, 0.9 Hz, 1H), 7.04 (dd, J = 8.0, 0.9 Hz, 1H), 4.19 (dtd, J = 19.6, 10.2, 5.7 Hz, 2H), 2.49–2.25 (m, 2H), 1.74–1.63 (m, 2H), 1.62–1.47 (m, 2H), 1.46–1.33 (m, 2H), 0.90 (dt, J = 23.4, 7.5 Hz, 7H), 0.79 (dd, J = 6.7, 3.7 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 168.79, 168.36, 141.29, 136.41, 132.84, 128.32, 116.40, 112.89, 47.05, 41.48, 32.80, 28.89, 26.58, 26.10, 23.50, 22.75, 11.16, 10.96; HRMS (ESI) calcd for [C18H21ClN2O + H]+: 317.14152, found 317.14255, calcd for [C18H21ClN2O + Na]+: 339.12346, found 339.1219; HPLC purity: 93%. 1H NMR analysis indicates that the material purified by flash chromatography contains traces of the starting material 3. LM-MS analysis confirmed the presence of the starting material 3 (m/z = 303.3) and also showed traces of a compound with a m/z ratio of 317.0 that is most likely the diasteromeric product of 24. These impurities could only be removed by reversed phase preparative HPLC using a Sunfire C18 column (19 × 100 mm, 5 um) and 25 to 55% MeCN in water (+0.01% formic acid) as the eluent (21.6 mL/min) to afford small amounts sufficient for 1H NMR. LM146 was crystallized by the solvent diffusion technique using chloroform. The X-ray structure has been deposited into the Cambridge Crystallographic Data Centre CCDC (no. 2070470).
Acknowledgments
We thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for a Collaborative Research and Development Grant (CRD).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01555.
Experimental procedures, NMR spectra of all final analogs, X-ray analysis report of 24 (LM146), additional bromoSCAN traces, and bromoMAX data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Shahbazian M. D.; Grunstein M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation. Nat. Rev. Drug Disc. 2014, 13, 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Zeng L.; Zhou M.-M. Bromodomain: An Acetyl-Lysine Binding Domain. FEBS Lett. 2002, 513, 124–128. 10.1016/S0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- Schiedel M.; Moroglu M.; Ascough D. M. H.; Chamberlain A. E. R.; Kamps J. J. A. G.; Sekirnik A. R.; Conway S. J. Chemical Epigenetics: The Impact of Chemical and Chemical Biology Techniques on Bromodomain Target Validation. Angew. Chem., Int. Ed. 2019, 58, 17930–17952. 10.1002/anie.201812164. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Smith S. G.; Zhou M.-M. Discovery of Chemical Inhibitors of Human Bromodomains. Chem. Rev. 2015, 115, 11625–11668. 10.1021/acs.chemrev.5b00205. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A.-C.; Arrowsmith C. H.; Knapp S. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Li H.; Patel D. J.; Allis C. D. Multivalent Engagement of Chromatin Modifications by Linked Binding Modules. Nat. Rev. Mol. Cell Biol. 2007, 8, 983–994. 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. G.; Roberts C. W. M. SWI/SNF Nucleosome Re-Modellers and Cancer. Nat. Rev. Cancer 2011, 11, 481–492. 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- Thompson M. Polybromo-1: the Chromatin Targeting Subunit of the PBAF Complex. Biochimie 2009, 91, 309–319. 10.1016/j.biochi.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shain A. H.; Pollack J. R. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLoS One 2013, 8, e55119 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah-Planchon J.; Bièche I.; Guinebretière J.-M.; Bourdeaut F.; Delattre O. SWI/SNF Chromatin Remodeling and Human Malignancies. Annu. Rev. Pathol. 2015, 10, 145–171. 10.1146/annurev-pathol-012414-040445. [DOI] [PubMed] [Google Scholar]
- Hodges C.; Kirkland J. G.; Crabtree G. R. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026930. 10.1101/cshperspect.a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C.; Crabtree G. R. Mammalian SWI/SNF Chromatin Remodeling Complexes and Cancer: Mechanistic Insights Gained from Human Genomics. Sci. Adv. 2015, 1, e1500447 10.1126/sciadv.1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Pierre R.; Kadoch C. Mammalian SWI/SNF Complexes in Cancer: Emerging Therapeutic Opportunities. Curr. Opin. Genet. Dev. 2017, 42, 56–67. 10.1016/j.gde.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou N. H.; Bamborough P.; Bannister A. J.; Becher I.; Bit R. A.; Che K. H.; Chung C.-w.; Dittmann A.; Drewes G.; Drewry D. H.; Gordon L.; Grandi P.; Leveridge M.; Lindon M.; Michon A.-M.; Molnar J.; Robson S. C.; Tomkinson N. C. O.; Kouzarides T.; Prinjha R. K.; Humphreys P. G. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark P. G. K.; Vieira L. C. C.; Tallant C.; Fedorov O.; Singleton D. C.; Rogers C. M.; Monteiro O. P.; Bennett J. M.; Baronio R.; Müller S.; Daniels D. L.; Méndez J.; Knapp S.; Brennan P. E.; Dixon D. J. LP99: Discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew. Chem., Int. Ed. 2015, 54, 6217–6221. 10.1002/anie.201501394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L. J.; Koegl M.; Bader G.; Cockcroft X.-L.; Fedorov O.; Fiegen D.; Gerstberger T.; Hofmann M. H.; Hohmann A. F.; Kessler D.; Knapp S.; Knesl P.; Kornigg S.; Müller S.; Nar H.; Rogers C.; Rumpel K.; Schaaf O.; Steurer S.; Tallant C.; Vakoc C. R.; Zeeb M.; Zoephel A.; Pearson M.; Boehmelt G.; McConnell D. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475. 10.1021/acs.jmedchem.5b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakim M.; Clark P. G. K.; Hay D. A.; Dixon D. J.; Brennan P. E. Chemical Probes and Inhibitors of Bromodomains Outside the BET Family. Med. Chem. Commun. 2016, 7, 2246–2264. 10.1039/C6MD00373G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg M. A.; Bamborough P.; Chung C.-w.; Craggs P. D.; Gordon L.; Grandi P.; Leveridge M.; Lindon M.; Liwicki G. M.; Michon A.-M.; Molnar J.; Rioja I.; Soden P. E.; Theodoulou N. H.; Werner T.; Tomkinson N. C. O.; Prinjha R. K.; Humphreys P. G. Application of Atypical Acetyl-Lysine Methyl Mimetics in the Development of Selective Inhibitors of the Bromodomain-Containing Protein 7 (BRD7)/Bromodomain-Containing Protein 9 (BRD9) Bromodomains. J. Med. Chem. 2020, 63, 5816–5840. 10.1021/acs.jmedchem.0c00075. [DOI] [PubMed] [Google Scholar]
- Fedorov O.; Castex J.; Tallant C.; Owen D. R.; Martin S.; Aldeghi M.; Monteiro O.; Filippakopoulos P.; Picaud S.; Trzupek J. D.; Gerstenberger B. S.; Bountra C.; Willmann D.; Wells C.; Philpott M.; Rogers C.; Biggin P. C.; Brennan P. E.; Bunnage M. E.; Schüle R.; Günther T.; Knapp S.; Müller S. Selective Targeting of the BRG/PB1 Bromodomains Impairs Embryonic and Trophoblast Stem Cell Maintenance. Sci. Adv. 2015, 1, e1500723 10.1126/sciadv.1500723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstenberger B. S.; Trzupek J. D.; Tallant C.; Fedorov O.; Filippakopoulos P.; Brennan P. E.; Fedele V.; Martin S.; Picaud S.; Rogers C.; Parikh M.; Taylor A.; Samas B.; O’Mahony A.; Berg E.; Pallares G.; Torrey A. D.; Treiber D. K.; Samardjiev I. J.; Nasipak B. T.; Padilla-Benavides T.; Wu Q.; Imbalzano A. N.; Nickerson J. A.; Bunnage M. E.; Müller S.; Knapp S.; Owen D. R. Identification of a Chemical Probe for Family VIII Bromodomains through Optimization of a Fragment Hit. J. Med. Chem. 2016, 59, 4800–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherell C. L.; Tallant C.; Monteiro O. P.; Yapp C.; Fuchs J. E.; Fedorov O.; Siejka P.; Müller S.; Knapp S.; Brenton J. D.; Brennan P. E.; Ley S. V. Identification and Development of 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one Inhibitors Targeting Bromodomains within the Switch/Sucrose Non-Fermenting Complex. J. Med. Chem. 2016, 59, 5095–5101. 10.1021/acs.jmedchem.5b01997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht B. K.; Côté A.; Crawford T.; Duplessis M.; Good A. C.; Leblanc Y.; Magnuson S. R.; Nasveschuk C. G.; Romero F. A.; Tang Y.; Taylor A. M.. Therapeutic Pyridazine Compounds and Uses Thereof. International Patent WO 2016/138114 A1, 2016.
- Wanior M.; Preuss F.; Ni X.; Krämer A.; Mathea S.; Göbel T.; Heidenreich D.; Simonyi S.; Kahnt A. S.; Joerger A. C.; Knapp S. Pan-SMARCA/PB1 Bromodomain Inhibitors and Their Role in Regulating Adipogenesis. J. Med. Chem. 2020, 63, 14680–14699. 10.1021/acs.jmedchem.0c01242. [DOI] [PubMed] [Google Scholar]
- Myrianthopoulos V.; Gaboriaud-Kolar N.; Tallant C.; Hall M.-L.; Grigoriou S.; Brownlee P. M.; Fedorov O.; Rogers C.; Heidenreich D.; Wanior M.; Drosos N.; Mexia N.; Savitsky P.; Bagratuni T.; Kastritis E.; Terpos E.; Filippakopoulos P.; Müller S.; Skaltsounis A.-L.; Downs J. A.; Knapp S.; Mikros E. Discovery and Optimization of a Selective Ligand for the Switch/Sucrose Nonfermenting-Related Bromodomains of Polybromo Protein-1 by the Use of Virtual Screening and Hydration Analysis. J. Med. Chem. 2016, 59, 8787–8803. 10.1021/acs.jmedchem.6b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I.; Tarpey P.; Raine K.; Huang D.; Ong C. K.; Stephens P.; Davies H.; Jones D.; Lin M.-L.; Teague J.; Bignell G.; Butler A.; Cho J.; Dalgliesh G. L.; Galappaththige D.; Greenman C.; Hardy C.; Jia M.; Latimer C.; Lau K. W.; Marshall J.; McLaren S.; Menzies A.; Mudie L.; Stebbings L.; Largaespada D. A.; Wessels L. F.; Richard S.; Kahnoski R. J.; Anema J.; Tuveson D. A.; Perez-Mancera P. A.; Mustonen V.; Fischer A.; Adams D. J.; Rust A.; Chan-on W.; Subimerb C.; Dykema K.; Furge K.; Campbell P. J.; Teh B. T.; Stratton M. R.; Futreal P. A. Exome Sequencing Identifies Frequent Mutation of the SWI/SNF Complex Gene PBRM1 in Renal Carcinoma. Nature 2011, 469, 539–542. 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comprehensive Molecular Characterization of Clear Cell Renal Cell Carcinoma. Nature 2013, 499, 43–49. 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter M. J.; Shanle E. K.; McFadden A. W.; Hollis E. S.; Suttle L. E.; Strahl B. D.; Davis I. J. PBRM1 Bromodomains Variably Influence Nucleosome Interactions and Cellular Function. J. Biol. Chem. 2018, 293, 13592–13603. 10.1074/jbc.RA118.003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L.; Alicea-Velázquez N. L.; Langbein L.; Niu X.; Cai W.; Cho E.-A.; Zhang M.; Greer C. B.; Yan Q.; Cosgrove M. S.; Yang H. High Affinity Binding of H3K14ac through Collaboration of Bromodomains 2, 4 and 5 is Critical for the Molecular and Tumor Suppressor Functions of PBRM1. Mol. Oncol. 2019, 13, 811–828. 10.1002/1878-0261.12434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp M. M.; Weïwer M.; Koehler A. N. Unbiased Binding Assays for Discovering Small-Molecule Probes and Drugs. Bioorg. Med. Chem. 2012, 20, 1979–1989. 10.1016/j.bmc.2011.11.071. [DOI] [PubMed] [Google Scholar]
- Sutherell C. L.; Ley S. V. On the Synthesis and Reactivity of 2,3-Dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones. Synthesis 2017, 49, 135–144. [Google Scholar]
- Aldeghi M.; Ross G. A.; Bodkin M. J.; Essex J. W.; Knapp S.; Biggin P. C. Large-Scale Analysis of Water Stability in Bromodomain Binding Pockets with Grand Canonical Monte Carlo. Commun. Chem. 2018, 1, 10.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.