

Abstract
Summary
The expansion of targeted panel sequencing efforts has created opportunities for large-scale genomic analysis, but tools for copy-number quantification on panel data are lacking. We introduce ASCETS, a method for the efficient quantitation of arm and chromosome-level copy-number changes from targeted sequencing data.
Availability and implementation
ASCETS is implemented in R and is freely available to non-commercial users on GitHub: https://github.com/beroukhim-lab/ascets, along with detailed documentation.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Aneuploidy, or copy-number alterations of chromosomes and chromosome arms, is the most frequent somatic alteration in cancer (Taylor et al., 2018). Arm-level somatic copy-number alterations (aSCNAs) can drive cancer progression (Liu et al., 2016), provide information about cancer type (The Cancer Genome Atlas Research Network, 2015) and serve as predictive and prognostic biomarkers (Cairncross et al., 2013; Lamberti et al., 2020). Thus, quantitation of aSCNAs is useful in research and clinical decision-making.
Targeted next-generation sequencing (targeted NGS) provides clinicians with rapidly available information about genomic events including copy-number alterations (Garcia et al., 2017). However, there are several challenges in detecting aSCNAs from targeted NGS data. The lower breadth of coverage (BOC; fraction of chromosome arm encompassed by copy-number segments) and significant noise in these data, often obtained from fresh-frozen paraffin embedded (FFPE) samples, often contradict assumptions underlying existing algorithms designed for research-quality whole-exome or genome sequencing data. Perhaps as a result, existing methods for calling copy-number in targeted NGS data do not provide aSCNA calls (Markham et al., 2019; Shen and Seshan, 2016; Talevich et al., 2016) and aSCNAs are not commonly reported or systematically determined. Therefore, novel algorithms are needed.
2 Implementation
We introduce ASCETS (Arm-level Somatic Copy-number Events in Targeted Sequencing), a method for efficient and robust aSCNA detection in targeted sequencing data (Fig. 1). ASCETS utilizes segmentation files representing de-noised copy-number calls and (optionally) raw log2 copy-ratios (LCRs) for each locus (Supplementary Methods). When provided, LCRs are used to determine the noise per segment by separating alternating LCRs within a given segment into two groups and calculating their difference in means. These differences are compiled across all segments and samples to determine a noise threshold representing one standard deviation above the mean. This threshold and its additive inverse are taken as amplification and deletion thresholds, respectively. Optionally, segments of ≥5 markers with above-threshold noise levels can excluded from further analysis. If LCRs are not supplied, the user may specify the threshold (default: 0.2).
Fig. 1.
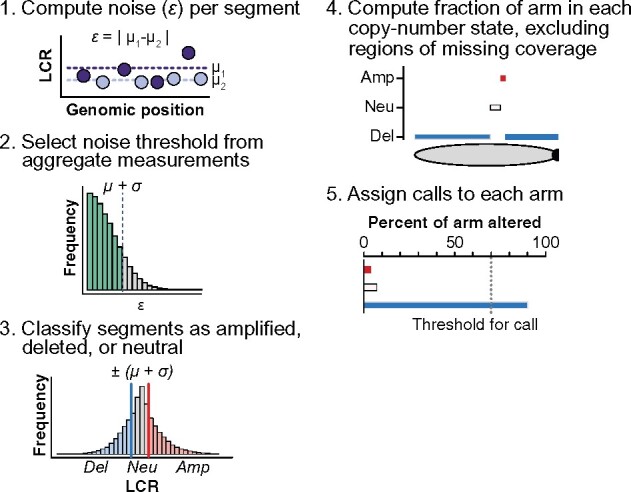
The ASCETS algorithm workflow
After stratifying amplified and deleted segments, ASCETS computes the BOC of each arm corresponding to each segment class (amplified, deleted or neutral). Arms for which more than a user-specified fraction of territory (default 70%; Supplementary Table S3) are encompassed by one class are assigned the respective call (AMP, DEL or NEUTRAL). Arms that do not meet these criteria are not called (NC). Optionally, arms with BOC less than a specified threshold (default: 50%) can be left uncalled (LOWCOV). The supplied arm coordinates can also be replaced with any set of genomic coordinates to call any region of interest, such as cytobands or genes.
3 Results
We evaluated the accuracy of ASCETS calls on two datasets: (i) 9,945 TCGA samples and (ii) 407 glioma samples from the Dana-Farber OncoPanel assay (Garcia et al., 2017). To assess its applicability to publicly available data, we also applied ASCETS to all 34,798 MSK-IMPACT samples in GENIE v7.0 (The AACR Project GENIE Consortium, 2017; Supplementary Table S1). This task completed in 104 min on a consumer-grade laptop with a 3.1 GHz i7 processor and 16 GB of RAM.
We used TCGA data to compare ASCETS calls to ‘gold-standard’ aSCNA calls from SNP array copy-number data (Taylor et al., 2018). Because TCGA data have a greater BOC, resulting in a lower level of noise and more accurate segmentation, we modified the TCGA data to simulate the lower BOC found in targeted sequencing data (Supplementary Methods and Fig. S1). After excluding 50,071 arms with low BOC (<50%) or no call from ASCETS or TCGA, 337,594 chromosome arms were retained (Supplementary Table S2). Of the excluded arms, ASCETS alone called 18,857 arms; TCGA alone called 6,160 and 25,244 were no-called in either dataset.
ASCETS and TCGA calls were 90.3% concordant (Kappa statistic 0.768, P-value [accuracy > no information rate (NIR)] < 2.2e−16). The positive predictive value (PPV) and negative predictive value (NPV), respectively, were 83.5% and 97.0% for amplifications and 95.8% and 94.6% for deletions (Supplementary Table S4). Discordant aSCNA calls tended to occur in samples with higher subclonal genome fractions (med. 0.19 versus 0.06, Wilcoxon P < 2.2e−16, Supplementary Fig. S2a), lower tumor purity (med. 0.50 versus 0.66, Wilcoxon P < 2.2e−16, Supplementary Fig. S2b; Carter et al., 2012) and lower magnitude average LCRs (med. 0.17 versus 0.37, Wilcoxon P < 2.2e−16, Supplementary Fig. S2c), but exhibited no differences in median alteration fraction (med. 0.96 versus 0.96, Wilcoxon P < 2.2e−16, Supplementary Fig. S2d). Similar concordance was observed across each chromosome arm and no relationship was seen with arm length (Supplementary Fig. S2e and f).
We also compared 1p/19q arm-level calls from ASCETS applied to OncoPanel data with results from OncoCopy, a 2 million-probe array comparative genomic hybridization (aCGH) assay (Ramkissoon et al., 2017), across 407 gliomas (Touat et al., 2020). We called arms with average aCGH LCRs of −0.15 in both 1p and 19q as codeleted. ASCETS exhibited PPV and NPV of 81.6% and 99.7%, respectively (Kappa statistic 0.875, P-value [accuracy > NIR] = 2.1e−07; Supplementary Fig. S3 and Table S5). However, 6/7 cases called codeleted by ASCETS but not aCGH were determined to be true positives after expert review of clinical reports (Supplementary Table S6), corresponding to a PPV of 97.4% and NPV of 99.7%. In addition, 37/38 (97.4%) of cases called codeleted by ASCETS, compared to 80/369 (21.7%) non-codeleted samples, had pathogenic IDH1/2 mutations (Touat et al., 2020), known to co-occur with 1p/19q codeletion (The Cancer Genome Atlas Research Network, 2015).
To assess the effects of the fraction of genomic territory interrogated (FGI) by sequencing and BOC on the performance of ASCETS, we performed a titration analysis (Supplementary Methods). As expected, the performance of ASCETS in calling 1p/19q codeletion increases with FGI and, as a result, BOC. Predictive values were consistently above 90% when at least 0.05% of the bases on each arm were interrogated (Supplementary Fig. S4a–d).
4 Discussion
aSCNAs cover more of the genome than any other somatic alteration (Taylor et al., 2018), but thus far have received insufficient focus in clinical decision-making as unifying events in cancer. ASCETS allows for the leveraging of large publicly available targeted sequencing datasets like GENIE to identify novel relationships between aSCNAs and clinical or tumoral features. In addition, ASCETS can potentially be employed to call clinically relevant aSCNAs such as 1p/19q codeletion in a research or diagnostic setting. Indeed, ASCETS has already been utilized in recent publications to subclassify brain tumors (Touat et al., 2020) and call aSCNAs affecting 9p (Lamberti et al., 2020). We conclude that ASCETS calls are highly concordant with calls from exome and array-based methods, and that ASCETS can be applied broadly to panel NGS datasets. While ASCETS is designed to account for challenges inherent to targeted sequencing data, it can in principle be applied broadly to copy-number data from whole-exome or whole-genome sequencing data. However, like all algorithms for copy-number analysis, ASCETS performs least well in samples and regions with low signal-to-noise ratios, including areas of subclonal or low-level copy-number change, samples with low tumor purity, and regions with low BOC. Users should consider the length of the sequenced regions when applying ASCETS to their own data to ensure they have sufficient BOC to generate accurate calls. Overall, ASCETS is an efficient, accurate method for quantifying aneuploidy and helps address the need for copy-number analysis tools for targeted NGS data.
Funding
This work has been supported by the National Cancer Institute [U24CA210978 to A.D.C. and R.B.], the Fund for Innovation in Cancer Informatics, the Gray Matters Brain Cancer Foundation, Pediatric Brain Tumor Foundation and a generous gift from Alison Poorvu Jaffe.
Conflict of Interest: M.T. reports consulting/advisory roles from Agios Pharmaceuticals, Integragen, and Taiho Oncology; travel, accommodations, expenses from Merck Sharp & Dome. A.M.T. reports research funding from Ono Pharmaceuticals. M.L.M. reports consultant/advisory board/equity in OrigiMed; research funding from Ono and Bayer Pharmaceuticals; patent for EGFR mutation diagnosis in lung cancer licensed to LabCorp. A.D.C. reports research funding from Bayer. Y.L. reports equity in g. Root Biomedical Services. R.B. reports equity in/advisory role to Scorpion Therapeutics and grant funding from Novartis. All COI are outside the submitted work and all other authors report no COI.
Supplementary Material
Contributor Information
Liam F Spurr, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
Mehdi Touat, Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA; Department of Oncologic Pathology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Sorbonne Université, Inserm, CNRS, UMR S 1127, Institut du Cerveau et de la Moelle épinière, ICM, AP-HP, Hôpitaux Universitaires La Pitié Salpêtrière—Charles Foix, Service de Neurologie, Paris, France.
Alison M Taylor, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA; Department of Pathology and Cell Biology and Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, 10032 NY, USA.
Adrian M Dubuc, Department of Pathology, Brigham and Women’s Hospital, Boston, 02215 MA, USA.
Juliann Shih, Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
David M Meredith, Department of Oncologic Pathology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Department of Pathology, Brigham and Women’s Hospital, Boston, 02215 MA, USA.
William V Pisano, Department of Oncologic Pathology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA.
Matthew L Meyerson, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
Keith L Ligon, Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA; Department of Oncologic Pathology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Department of Pathology, Brigham and Women’s Hospital, Boston, 02215 MA, USA; Center for Patient Derived Models, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Department of Pathology, Boston Children’s Hospital, Boston, 02215 MA, USA.
Andrew D Cherniack, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
Yvonne Y Li, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA.
Rameen Beroukhim, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Cancer Program, Broad Institute of MIT and Harvard, Cambridge, 02142 MA, USA; Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, 02215 MA, USA; Department of Medicine, Brigham and Women’s Hospital, Boston, 02215 MA, USA.
References
- Cairncross G. et al. (2013) Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J. Clin. Oncol., 31, 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S.L. et al. (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol., 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia E.P. et al. (2017) Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med., 141, 751–758. [DOI] [PubMed] [Google Scholar]
- Lamberti G. et al. (2020) Clinicopathological and genomic correlates of Programmed Cell Death Ligand 1 (PD-L1) expression in nonsquamous non-small cell lung cancer. Ann. Oncol., 31, 807–814. [DOI] [PubMed] [Google Scholar]
- Liu Y. et al. (2016) Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature, 531, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham J.F. et al. (2019) CNspector: a web-based tool for visualisation and clinical diagnosis of copy number variation from next generation sequencing. Sci. Rep., 9, 6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkissoon S.H. et al. (2017) Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: precision medicine analysis of 203 pediatric brain tumors. Neuro Oncol., 19, 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R., Seshan V.E. (2016) FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res., 44, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E. et al. (2016) CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol., 12, e1004873. 10.1371/journal.pcbi.1004873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.M. et al. (2018) Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell, 33, 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The AACR Project GENIE Consortium (2017) AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov., 7, 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2015) Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med., 372, 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touat M. et al. (2020) Mechanisms and therapeutic implications of hypermutation in gliomas. Nature, 580, 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.