

Abstract
Motivation
Genome-wide association studies have successfully identified multiple independent genetic loci that harbour variants associated with human traits and diseases, but the exact causal genes are largely unknown. Common genetic risk variants are enriched in non-protein-coding regions of the genome and often affect gene expression (expression quantitative trait loci, eQTL) in a tissue-specific manner. To address this challenge, we developed a methodological framework, E-MAGMA, which converts genome-wide association summary statistics into gene-level statistics by assigning risk variants to their putative genes based on tissue-specific eQTL information.
Results
We compared E-MAGMA to three eQTL informed gene-based approaches using simulated phenotype data. Phenotypes were simulated based on eQTL reference data using GCTA for all genes with at least one eQTL at chromosome 1. We performed 10 simulations per gene. The eQTL-h2 (i.e. the proportion of variation explained by the eQTLs) was set at 1%, 2% and 5%. We found E-MAGMA outperforms other gene-based approaches across a range of simulated parameters (e.g. the number of identified causal genes). When applied to genome-wide association summary statistics for five neuropsychiatric disorders, E-MAGMA identified more putative candidate causal genes compared to other eQTL-based approaches. By integrating tissue-specific eQTL information, these results show E-MAGMA will help to identify novel candidate causal genes from genome-wide association summary statistics and thereby improve the understanding of the biological basis of complex disorders.
Availability and implementation
A tutorial and input files are made available in a github repository: https://github.com/eskederks/eMAGMA-tutorial.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Genome-wide association studies (GWAS) have identified thousands of single nucleotide polymorphisms (SNPs) associated with disease risk (Visscher et al., 2017). However, the functional relevance of most SNPs remains unknown, due in part to their position in non-protein coding regions of the genome (Gamazon et al., 2018). Mapping trait-associated SNPs to their nearest gene often fails to identify the functional gene, since regulatory effects on gene expression, known as expression quantitative trait loci (eQTLs), can be long-range (Smemo et al., 2014). Gene-based mapping methods that rely on arbitrary genomic windows to assign SNPs to genes, such as MAGMA (de Leeuw et al., 2015), do not allow inferences on causal genes. Furthermore, assignment of SNPs to the nearest gene for gene-level association testing does not eliminate the necessity of functionally connecting SNPs to genes (e.g. via genetic regulation) for improved understanding of possible underlying mechanisms.
In recent years, several methods have been developed to integrate GWAS and gene expression information to improve our understanding of the functional mechanisms that underlie statistical genetic associations, known as a transcriptome-wide association study (TWAS). These methods are now widely used as GWAS secondary analyses using software packages such as FUSION (Gusev et al., 2016) and S-PrediXcan (Barbeira et al., 2018), and have identified novel genes and mechanisms underlying a range of diseases (Gamazon et al., 2018, 2019). Another related method, summary-based Mendelian Randomization (SMR) (Zhu et al., 2016), integrates GWAS summary statistics with eQTL data to identify pleiotropic effects (i.e. a single causal variant affecting with gene expression and the manifestation of a phenotype). Both TWAS FUSION and S-PrediXcan rely on a two-stage regression procedure. In the first stage, they train multi-variant prediction models in a sample with both genotype and gene expression data. In the second stage, these weights are then combined with summary-level data from GWAS to perform association analysis of imputed gene expression with a phenotype. SMR and its extension, the HEIDI test, aims to test for pleiotropic association between the expression level of a gene and a complex trait of interest using summary-level data from GWAS and expression quantitative trait loci (eQTL) studies within a Mendelian Randomization framework.
TWAS methods test the association between the genetically determined component of gene expression and disease risk, ideally removing unwanted influences of environmental and technical factors on gene expression. However, this means only those genes whose expression can be reliably imputed from genotype data (i.e. moderately highly heritable genes) can be tested for an association with a trait. Indeed, only 6759 genes in GTEx (v8) whole blood—a relatively highly powered tissue—can be tested using S-PrediXcan, and 6006 genes using FUSION. While the number of significant cis-heritable genes detected by each approach is a function of sample size, the relatively small number of testable genes reduces the search space for prioritizing candidate causal genes. We therefore created an alternative method, called E-MAGMA, which modifies the MAGMA pipeline by mapping variants to genes based on tissue-specific eQTL information. We have used eQTL information from 48 tissues of the GTEx reference panel version 8 (The GTEx Consortium atlas of genetic regulatory effects across human tissues, 2020), although the method can be easily extended to other eQTL reference datasets. This approach was developed to identify functional gene associations that may be missed using proximity-based SNP assignment in MAGMA and may therefore identify alternative causal pathways from SNPs to trait.
In this article, we introduce the E-MAGMA gene-based annotation approach and perform a systematic comparison of four different methods using data simulations and a real-life example using summary statistics from GWAS of attention-deficit hyperactivity disorder (Demontis et al., 2019), autism spectrum disorder (Grove et al., 2019), bipolar disorder (Stahl et al., 2019), depression (Howard et al., 2019) and schizophrenia (Pardiñas et al., 2018). Our aims are to: (i) compare the statistical power of E-MAGMA and other gene-based methods to detect a true association; (ii) compare type-I error rates; (iii) test the influence of the number of eQTLs on statistical power (i.e. weak instrument bias); and (iv) compare the strength of association across methods. We plan to extend our simulations by modelling the performance of each method across different estimates of trait heritability and prevalence, and the proportion of overlap between causal GWAS variants and eQTL variants. A tutorial and input files are made available in a github repository: https://github.com/eskederks/E-MAGMA-tutorial.
2 Materials and methods
2.1 Gene-based methods
We used five gene-based methods: S-PrediXcan (Barbeira et al., 2018), TWAS FUSION (Gusev et al., 2016), SMR (version 1.0) (Zhu et al., 2016), conventional MAGMA (de Leeuw et al., 2015) and our newly developed E-MAGMA (Gerring et al., 2019). S-PrediXcan and TWAS FUSION are prediction-based approaches that impute the genetically regulated component of gene expression from SNP genotype data and regress the imputed expression on a given phenotype. SMR uses a Mendelian randomization approach to estimate the effect of gene expression on a phenotype due to a single genetic marker (i.e. SNP), and tests whether a SNP’s association with gene expression is due to linkage or pleiotropy (HEterogeneity In Dependent Instruments [HEIDI] test). Conventional MAGMA simply links SNPs to genes based on physical proximity, before combining the SNP-level P values using a modified version of Brown’s method that adjusts for linkage disequilibrium. Our E-MAGMA approach, a modification of MAGMA, leverages significant (FDR < 0.05) tissue-specific cis-eQTL information from GTEx (v8) to assign SNPs to putative genes.
2.2 SNP genotype data for simulation analyses
The original genotype file from the QIMR Adult Twin Study (Duffy et al., 2018; Medland et al., 2009) included 3 738 240 SNPs from 28 110 individuals. We excluded non-founders (N = 20 825), SNPs with >1% missingness (N = 1 023 785) and SNPs with minor allele frequency (MAF) < 0.05 (N = 2 653 824). We subsequently excluded individuals with >1% missing data (N = 147). SNP identifiers were transformed to chr_chrposition to enable matching with GTEx eQTL reference data. This resulted in 43 duplicate SNPs, which were excluded from further analysis. Finally, we selected only SNPs from chromosome 1. The cleaned dataset included 7138 subjects and 60 585 SNPs. eQTL information was obtained from whole blood samples of the GTEx eQTL database. (Whole_Blood.v8.signif_variant_gene_pairs.txt.gz). Significant eQTLs (FDR < 0.05) were included in subsequent analyses. This eQTL reference database included 655 939 eQTL-gene combinations for 8235 unique genes.
2.3 Phenotype simulation
Phenotypes were simulated using GCTA (Yang et al., 2011) using genotype and eQTL reference data from chromosome 1 (N = 811 genes). For each gene, a phenotype was simulated using all significant (FDR < 0.05) cis-eQTLs as predictors, based on the eQTL regression coefficients from the GTEx reference dataset. We performed 10 simulations per gene. Only those genes with at least one significant eQTL are included in the analysis (N = 651). The eQTL-h2 (i.e. the proportion of variation explained by the eQTLs was set at 1%, 2% or 5%). We evaluated the type-I error rate across methods by calculating the proportion of genes that are significant in the absence of true association between eQTLs and phenotypic values. For this purpose, phenotypes were simulated using GCTA with the proportion of variation explained by the eQTLs set at 0%.
2.4 GWAS analysis
GWAS analyses of the 6510 generated phenotypes were performed using the linear regression option in Plink (Purcell et al., 2007). SNP identifiers were replaced with rs identifiers using a lookup table to enable alignment with the annotation files in subsequent statistical analyses. We used the same significance level (P = 6.25 × 10−5) for all analyses and corrected for the total number of genes in the GTEx whole blood reference dataset located at chromosome 1 (i.e. 0.05/811 = 1.2 × 10−3).
2.5 E-MAGMA gene-level analysis
Since we are primarily interested in identifying variants with prior functional support associated with complex disorders, we leveraged eQTL data from 48 tissues in GTEx (version 8). Using the tissue-specific GTEx datasets, we generated SNP-gene pairs (FDR < 0.05) that reflect functional relationships between SNPs and genes (cis-eQTLs), which serves as an input annotation file for the MAGMA software. We use the statistical framework from MAGMA to calculate gene-based P values using the updated (eQTL) annotation files. Gene-level analysis was done using default parameters and snp-wise=mean gene analysis model. We share comprehensive instructions on how to run E-MAGMA in a github repository: https://github.com/eskederks/eMAGMA-tutorial
2.6 Real-life example
We compared each gene-based method using real GWAS summary statistics for five neuropsychiatric disorders: attention hyperactivity disorder (ADHD) (Demontis et al., 2019), autism spectrum disorder (ASD) (Grove et al., 2019), bipolar disorder (BIP) (Stahl et al., 2019), depression (DEP) (Howard et al., 2019) and schizophrenia (SCZ) (Pardiñas et al., 2018). eQTL data from GTEx (v8) brain tissues were used to calculate the expression-phenotype association for all gene-based methods, with the exception of conventional MAGMA which does not use eQTL data. For conventional MAGMA, the gene-based analysis was done using default parameters and snp-wise=mean gene analysis model. We compared the number of unique Bonferroni-corrected genes for each method, using all gene-based results and after restricting each method to genes with genetically regulated gene expression. We calculated the correlations between the test statistics of each method using Pearson’s correlation coefficients.
2.7 Comparative gene-level analyses
For the comparative analysis with S-PrediXcan, FUSION and SMR, we applied prediction models trained in whole blood (GTEx v8) to analyse the generated simulated phenotype files, using the gene expression weight files provided by each package. We used software-specific default options for our analyses and used 1000 Genomes (Delaneau et al., 2014) data as the reference panel. For the MAGMA annotation, we specified an annotation window 5 kb upstream and 1.5 kb downstream of each gene. For E-MAGMA, we assigned SNPs to genes using significant (FDR < 0.05) eQTL data from GTEx (v8). Gene-level analyses for E-MAGMA were done using default parameters and snp-wise=mean gene analysis model.
3 Results
We first counted the number of genes included in each post-GWAS method, after running the expression-phenotype association analysis in GTEx (v8) whole blood (Table 1). There was a wide range in the number of genes output from each approach, ranging from 401 in SMR to 628 in S-PrediXcan. A total of 6510 (651 genes with 10 simulations each) causal genes were used as input for phenotypic simulations. We first assessed the false positive rate (type-I error) of each method (i.e. under simulated conditions with no significant eQTLs/non-eQTLs) (Supplementary Fig. S1), and found all methods showed good control of the type-I error rate. We subsequently evaluated statistical power to detect association at a gene-based level, for varying levels of eQTL-h2. We assessed the proportion of significant associations relative to both the total number of causal genes (Fig. 1A) and when accounting for the total number of causal genes included in each method (Fig. 1B). E-MAGMA outperformed all methods across different proportions of variance explained by gene expression. After correcting for the number of genes included in each gene-based method, E-MAGMA still outperformed other methods (Fig. 1B).
Table 1.
Number of genes and causal genes in each gene-based method
| Method | Number of genes in model | Causal genesa | Proportion of causal genes |
|---|---|---|---|
| E-MAGMA | 565 | 530 | 0.81 |
| S-PrediXcan | 628 | 490 | 0.75 |
| SMR | 401 | 387 | 0.59 |
| FUSION | 588 | 186 | 0.29 |
From simulations.
Fig. 1.
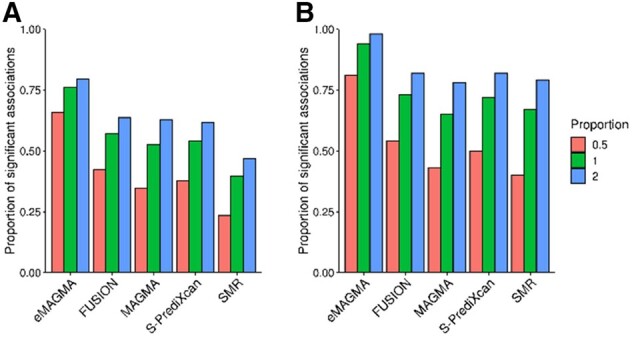
Proportion of significant associations (A) relative to the total number of causal genes and (B) relative to the total number of causal genes per method. Legend: 0.5%, 1% and 2% denote the percentage of phenotypic variance explained by eQTLs
All of the gene-based methods, with the exception of SMR, combine statistical evidence across multiple SNPs to derive a gene-based association. We therefore estimated statistical power as a function of the number of eQTLs per gene, with 1% of phenotypic variance explained by eQTLs. Power significantly increased with the number of eQTLs per gene (Fig. 2; Supplementary Table S1). There was a significant association between the number of eQTLs per gene and statistical power for all methods (P < 0.01), however E-MAGMA was less sensitive to the number of eQTLs than the other methods.
Fig. 2.
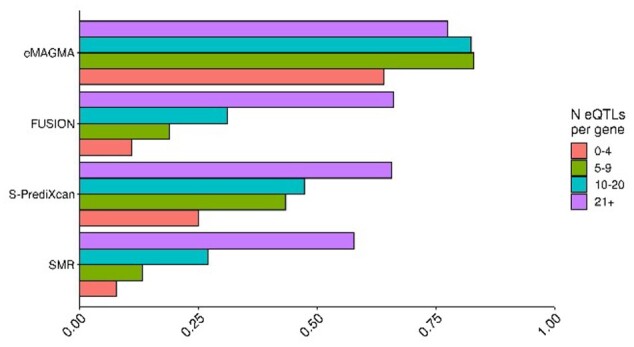
Statistical power as a function of the number of eQTLs per gene
We assessed the overlap in genes between eQTL-based methods at 1% of phenotypic variance explained (Supplementary Fig. S2). The number of genes unique to each method far outweighed the overlap between any two methods, however there was good overlap across all four methods (n=851 from a total of 6511 tests). We calculated the pairwise correlation of the Z-scores between gene-based methods. Effect sizes of transcriptome-imputation methods were strongly correlated, particularly S-PrediXcan and FUSION (r = 0.97, P < 2.2 × 10-16, df = 402), but only low-moderate correlation of the absolute z-scores was observed with E-MAGMA (e.g. S-PrediXcan versus E-MAGMA; r = 0.47, P < 2.22 × 10-16, df = 429) (Supplementary Table S2).
Finally, we compared the number of significant (Bonferroni-corrected for the number of tests performed) risk genes detected by each gene-based approach using GWAS summary statistics for five neuropsychiatric disorders and (if applicable) expression weights from 13 brain tissues in GTEx (version 8). E-MAGMA results for each of the five neuropsychiatric disorders is displayed in Supplementary Table S3; MAGMA results are displayed in Supplementary Table S4; S-PrediXcan results are displayed in Supplementary Table S5; FUSION results are displayed in Supplementary Table S6; and SMR results are displayed in Supplementary Table S7. Compared to eQTL-based methods, E-MAGMA identified more significant associations for bipolar disorder (N = 32), depression (N = 119) and schizophrenia (n = 254), and a comparable number of gene associations for attention deficit hyperactivity disorder (N = 5) (Table 2), thereby improving gene discovery for hypothesis generation. It should be noted, however, that E-MAGMA gene discovery is driven by genes with significant eQTLs in GTEx, rather than significant genetically regulated gene expression in TWAS approaches. After restricting to genes with genetically regulated gene expression, the number of unique significant genes found by E-MAGMA was less than the TWAS approaches (Supplementary Tables S8 and S9), highlighting the value of running multiple gene-based tests with different underlying models and assumptions.
Table 2.
Number of unique significant (Bonferroni corrected) associations for 5 neuropsychiatric disorders across different gene-based methods
| E-MAGMA | MAGMA | S-PrediXcan | FUSION | SMR | |
|---|---|---|---|---|---|
| ADHD | 5 | 17 | 7 | 10 | – |
| AUT | 1 | 5 | 2 | 4 | – |
| BIP | 32 | 47 | 20 | 31 | 0 |
| DEP | 119 | 190 | 89 | 45 | 7 |
| SCZ | 254 | 460 | 210 | 236 | 37 |
Note: Association statistics using SMR for ADHD and AUT could not be calculated because publicly available summary statistics for these disorders do not include SNP minor allele frequency. Supplementary Table S6 shows the number of associations for each method after restricting to genes with genetically regulated levels of gene expression, as defined by S-PrediXcan and TWAS FUSION.
4 Discussion
We developed a gene-based method called E-MAGMA which uses functional tissue-specific eQTL information from GTEx to assign SNPs to genes with the aim of improved annotation and interpretation of GWAS association signals. Our approach uses the statistical framework from MAGMA, but rather than assigning SNPs to genes based on physical proximity during gene annotation (i.e. mapping SNPs to genes using a pre-defined and arbitrary genomic window), we use significant (FDR < 0.05) SNP-gene expression associations (eQTL) in GTEx. Our extension therefore provides more biologically meaningful and interpretable results compared to conventional MAGMA. We compared E-MAGMA to three other eQTL-informed gene-based approaches (S-PrediXcan, FUSION and SMR) using both simulated and observed GWAS data. We show that E-MAGMA maintains appropriate control of the type-I error rate while outperforming other methods in detecting causal associations.
We used the methodological framework of MAGMA, rather than similar gene-based methods such as EUGENE (Ferreira et al., 2017), because it is one of the most widely used secondary analyses for the interpretation of GWAS results. Furthermore, the framework can be modified to include any type of annotation that maps SNPs to genes. For example, recent work to integrate chromatin interaction data from relevant tissues using the MAGMA framework increased power to identify putative risk genes and biological pathways for a range of neuropsychiatric traits (Sey et al., 2020). With the availability of tissue-specific multi-omic (transcriptome, chromatin, Hi-C, DNA methylation) datasets through projects such as GTEx (The GTEx Consortium atlas of genetic regulatory effects across human tissues, 2020) and psychENCODE (Wang et al., 2018), it will be possible to link SNPs to target genes using the most functionally relevant data and improve the biological interpretation of GWAS results.
Recent gene-based methods integrate genetic and transcriptomic information to estimate the effect of genetically determined gene expression on phenotypic variation. No systematic comparison of the three most commonly used methods—S-PrediXcan, FUSION and SMR—has been done. However, a head-to-head comparison of S-PrediXcan and FUSION found both methods recapitulate known associations between genotype and expression, and produced accurate and reliable results when compared to observed eQTL data (Fryett et al., 2020). Furthermore, both methods produced highly correlated results when applied to the same eQTL reference data. These data suggest the models underlying TWAS FUSION and S-PrediXcan perform similarly, and produce negligible differences in gene-based results.
We found all of the tested methods maintained control of the false positive rate under simulated conditions, where no single variant contributes to phenotypic variation. Under simulated conditions where 0.5%, 1% or 2% of the phenotypic variation was explained by eQTLs (or non-eQTLs), S-PrediXcan captured more causal genes compared to SMR and FUSION. The performance of each method improved when measured against the actual total number of causal genes tested, correcting for the fact that some methods test fewer genes than others. E-MAGMA was least influenced by the number of eQTLs of a gene, while all other methods tended to show a monotonic relationship with the number of eQTLs.
Our framework provides a more functionally valid gene-based test of association for GWAS compared to conventional MAGMA. However, it is prone to many of the same limitations of existing eQTL gene-based approaches. First, E-MAGMA is not immune for the influence of linkage—where two or more variants in linkage disequilibrium independently affect gene expression and phenotypic variation—and pleiotropic SNP effects—where a single causal variant affects both gene expression and phenotypic variation. Our method may therefore yield non-causal SNP-gene associations in the disease-associated region. Second, the power of E-MAGMA is limited by the sample size of the annotation eQTL dataset and including genes with weaker eQTL signals (based on less stringent 5% false discovery rate) may increase false positive associations. This is especially problematic with brain tissue eQTL datasets, which tend to be underpowered given the inaccessibility of brain tissue. The meta-analysis of multiple independent brain eQTL datasets, performed by the psychENCODE consortium (Wang et al., 2018), will improve the power and interpretation of E-MAGMA. Third, unlike TWAS methods, E-MAGMA does not provide information on the gene expression effect direction (that is, whether a gene is predicted to be up-regulated or down-regulated in cases). This limits the translation of results to higher order molecular mechanisms. Furthermore, the lack of effect direction may increase false positive associations, as opposite effects (for the same gene across different tissues) may be counted as valid associations. Fourth, our simulations might favour E-MAGMA over the other TWAS approaches because the eQTLs used in the annotation files were derived from the same reference eQTL dataset (GTEx) used to simulate gene expression. Future simulations using independent reference eQTL datasets will be required to confirm the better performance of E-MAGMA. Finally, gene expression is highly cell-type specific (Mathys et al., 2019). The use of bulk tissue eQTL datasets may therefore reduce power to identify cell-type specific disease signals. The use of existing (Mathys et al., 2019) and impending (Wang et al., 2018) single cell expression datasets may therefore improve the resolution of eQTL-based gene-mapping.
Future work will refine both the methodological framework of E-MAGMA and the simulated data comparisons. First, our simulations were developed to compare statistical power of transcriptome imputation methods with MAGMA and E-MAGMA. The simulations might be improved upon by modelling the impact of the proportion of causal eQTLs that contribute to phenotypic variation; that is, how do the methods perform under scenarios where only a subset of cis-eQTLs contribute of gene expression variation. Furthermore, we will assess the performance of each method across different estimates of trait heritability and prevalence. These additional analyses will provide a biologically valid and comprehensive assessment of model performance. Second, the tissue-specificity of E-MAGMA may provide novel insights into biological mechanisms of disease, but at the cost of limited sample size—and statistical power—of tissue-specific eQTL datasets. Future work will annotate genes with eQTL from larger datasets blood-based eQTL datasets to improve gene discovery, before prioritising genes using tissue-specific results. Furthermore, when sample sizes become sufficient powered, trans-eQTL effects may be integrated into the E-MAGMA annotation files. Finally, the use of a co-localisation method such as ENLOC (Wen et al., 2017) to calculate the probability that the top eQTL from the eMAGMA association and GWAS signals share the same causal variant would further refine a credible set of causal genes.
In conclusion, we present a modified MAGMA framework, E-MAGMA that aggregates eQTL summary statistics into gene level association statistics for gene-level analyses. Using simulated data, we showed E-MAGMA has greater power to detect causal associations compared to other popular gene-based approaches, while maintaining appropriate control of the type I error rate. Therefore, E-MAGMA can provide a functionally relevant alternative to existing methods to identify genes and pathways from GWAS. A tutorial and input files can be found in the github repository: https://github.com/eskederks/E-MAGMA-tutorial.
Supplementary Material
Acknowledgements
The authors thank Nick Martin (QIMR Berghofer) for providing access to the QIMR Adult Twin Study cohort. E.R.G. is grateful to the President and Fellows of Clare Hall, University of Cambridge for the stimulating intellectual environment during his fellowship in the college. E.R.G. is supported by the National Human Genome Research Institute of the National Institutes of Health under Award Numbers R35HG010718 and R01HG011138. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
E.R.G. was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Numbers R35HG010718 and R01HG011138.
Conflict of Interest: none declared.
Contributor Information
Zachary F Gerring, Mental Health, Translational Neurogenomics Laboratory, QIMR Berghofer Medical Research Institute, Brisbane, QLD 4006, Australia.
Angela Mina-Vargas, Mental Health, Translational Neurogenomics Laboratory, QIMR Berghofer Medical Research Institute, Brisbane, QLD 4006, Australia.
Eric R Gamazon, Division of Genetic Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Department of Medicine, Vanderbilt Genetics Institute, Vanderbilt University Medical Center, Nashville, TN 37232, USA; Clare Hall, University of Cambridge, Cambridge CB3 9AL, UK.
Eske M Derks, Mental Health, Translational Neurogenomics Laboratory, QIMR Berghofer Medical Research Institute, Brisbane, QLD 4006, Australia.
References
- Barbeira A.N., GTEx Consortium. et al. (2018) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun., 9, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O. et al. ; 1000 Genomes Project Consortium. (2014) Integrating sequence and array data to create an improved 1000 Genomes Project haplotype reference panel. Nat. Commun., 5, 3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis D. et al. ; ADHD Working Group of the Psychiatric Genomics Consortium (PGC). (2019) Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet., 51, 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy D.L. et al. ; Melanoma GWAS Consortium. (2018) Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat. Commun., 9, 4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M.A.R. et al. (2017) Gene-based analysis of regulatory variants identifies 4 putative novel asthma risk genes related to nucleotide synthesis and signaling. J. Allergy Clin. Immunol., 139, 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryett J.J. et al. (2020) Correction: comparison of methods for transcriptome imputation through application to two common complex diseases. Eur. J. Hum. Genet., 28, 1135–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon E.R. et al. (2019) Multi-tissue transcriptome analyses identify genetic mechanisms underlying neuropsychiatric traits. Nat. Genet., 51, 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon E.R. et al. ; GTEx Consortium. (2018) Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet., 50, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerring Z.F. et al. ; for the Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. (2019) A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet., 15, e1008245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove J. et al. ; Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium. (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet., 51, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A. et al. (2016) Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet., 48, 245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard D.M. et al. ; 23andMe Research Team. (2019) Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci., 22, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw C.A. et al. (2015) MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol., 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H. et al. (2019) Single-cell transcriptomic analysis of Alzheimer’s disease. Nature, 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medland S.E. et al. (2009) Common variants in the Trichohyalin gene are associated with straight hair in Europeans. Am. J. Hum. Genet., 85, 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardiñas A.F. et al. ; GERAD1 Consortium. (2018) Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet., 50, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S. et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sey N.Y.A. et al. (2020) A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci., 24, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smemo S. et al. (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature, 507, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl E.A. et al. (2019) Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet., 51, 793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science, 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher P.M. et al. (2017) 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet., 101, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., PsychENCODE Consortium. et al. (2018) Comprehensive functional genomic resource and integrative model for the human brain. Science, 362, eaat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X. et al. (2017) Integrating molecular QTL data into genome-wide genetic association analysis: probabilistic assessment of enrichment and colocalization. PLoS Genet., 13, e1006646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. et al. (2011) GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet., 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z. et al. (2016) Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet., 48, 481–487. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.