

Abstract
We describe a 46,XX girl with Denys-Drash syndrome, showing both kidney disease and genital abnormalities, in whom a misdiagnosis of hyperandrogenism was made. A 15 year-old girl was affected by neonatal nephrotic syndrome, progressing to end stage kidney failure. Hair loss and voice deepening were noted during puberty. Pelvic ultrasound and magnetic resonance imaging showed utero-tubaric agenesis, vaginal atresia and urogenital sinus, with inguinal gonads. Gonadotrophin and estradiol levels were normal, but testosterone was increased up to 285 ng/dL at Tanner stage 3. She underwent prophylactic gonadectomy. Histopathology reported fibrotic ovarian cortex containing numerous follicles in different maturation stages and rudimental remnants of Fallopian tubes. No features of gonadoblastoma were detected. Unexpectedly, testosterone levels were elevated four months after gonadectomy (157 ng/dL). Recent medical history revealed chronic daily comsumption of high dose biotin, as a therapeutic support for hair loss. Laboratory immunoassay instruments used streptavidin-biotin interaction to detect hormones and, in competitive immunoassays, high concentrations of biotin can result in false high results. Total testosterone, measured using liquid chromatography tandem mass spectrometry, was within reference intervals. Similar testosterone levels were detected on repeat immunoassay two weeks after biotin uptake interruption. Discordance between clinical presentation and biochemical results in patients taking biotin, should raise the suspicion of erroneous results. Improved communication among patients, health care providers, and laboratory professionals is required concerning the likelihood of biotin interference with immunoassays
Keywords: Denys-Drash syndrome, testosterone, biotin, disorder of sex development
What is already known on this topic?
WT1 gene mutations are associated with Denys-Drash syndrome (DDS), characterized by steroid-resistant nephrotic syndrome, Wilms tumor, disorder of sex development with dysgenetic gonads and gonadoblastoma risk in males. The renal manifestations are generally the only pathological condition in females. Hormone assays support endocrinological assessment and the suspicion of gonadal dysgenesis.
What this study adds?
We describe a girl with an unusual presentation of DDS, with end stage renal failure, severe genital abnormalities, signs of hyperandrogenism, and suspected dysgenetic gonads. Recent clinical history revealed that the patient consumed biotin, and the high levels of testosterone were due to analytical interference of the laboratory immunoassay. Enquiring about biotin supplementation should be conducted, since patients may not consider biotin as a medication and therefore may not mention it in their medication list.
Introduction
Wilms’ tumor suppressor gene 1 (WT1, OMIM *607102) is essential for kidney and gonadal development (1,2). Mutations in the WT1 gene are associated with Denys-Drash syndrome (DDS). In 46,XY subjects, WT1 mutations are associated with steroid-resistant nephrotic syndrome, Wilms tumor, disorder of sex development (DSD) with dysgenetic gonads and gonadoblastoma risk. In contrast, the impact of WT1 gene on the genital development of 46,XX subjects is not clear and most affected subjects only show the renal manifestations of the condition (1,2).
We describe a girl with end stage renal failure, severe genital abnormalities, signs of hyperandrogenism, and suspected dysgenetic gonads. Recent clinical history revealed that the patient consumed biotin, and the erroneous high levels of testosterone were due to an analytical interference of laboratory immunoassay.
Case Report
A 15 year-old Caucasian Italian girl had exhibited steroid-resistant nephrotic syndrome in the first month of life. Kidney biopsy at onset showed mesangial proliferative glomerulonephritis with focal segmental glomerulosclerosis. End-stage renal failure was reached by two years of age. Cytogenetic analysis showed a normal 46,XX female karyotype. Sanger sequencing of the WT1 gene, performed at five years of age at another center, showed the missense mutation c.1097G>A in exon 8, causing the amino acid change Arg366His affecting the zinc finger 2 region. The mutation was de novo and present in the heterozygous state.
She underwent left nephrectomy at one year of age. Right nephrectomy and a first renal transplantation were performed at 3.3 years of age. Chronic primary Epstein-Barr virus infection was diagnosed early after transplantation and did not respond to reduction in immunosuppression therapy and rituximab. In the following years progressive chronic allograft nephropathy developed and renal function worsened. At the age of 12 years hemodialysis was restarted and 10 months later the transplanted kidney was removed.
Puberty started at 13 years. A few months later, hair loss and voice deepening were observed. Repeated hormone assays, measured by chemiluminescence on an ADVIA Centaur XPT Immunoassay System (Siemens Healtineers Diagnostics, Erlangen, Germany) showed normally increasing pubertal female levels of gonadotrophins and estradiol, but testosterone level progressively increased up to abnormally high concentrations (285 ng/dL) when the girl reached Tanner stage 3 of breast and pubic hair development. The levels of adrenal androgens and precursors including delta4 androstendione, dehydroepiandrosterone sulfate and 17-hydroxyprogesterone, were in the normal range for a pubertal female, excluding the adrenal origin of hyperandrogenism. These data suggested the presence of dysgenetic gonads. Pelvic ultrasound and magnetic resonance imaging showed absence of uterus and Fallopian tubes, vaginal atresia and urogenital sinus. Both gonads were located at the internal inguinal ring. The right gonad appeared small, with a relatively homogeneous, streak-like, structure and rare anechoic areolas. The left gonad was larger and showed an anechoic area consistent with a dominant follicle. As a second step, targeted next generation sequencing was performed using a customized panel for DSD, including all coding exons and flanking introns of the following genes: AR, FOXL2, FST, HSD3B2, NR5A1, RSPO1, SOX3, SOX9, SRY, WNT4, WT1. Sequence enrichment was performed using the NimbleGen SeqCap Target Enrichment kit (Nimblegen Roche, Basel, Switzerland) and sequenced on the Illumina NextSeq550 platform (Illumina, San Diego, California). The WT1 Arg366His mutation was confirmed, while no others mutations were found (Figure 1).
Figure 1.
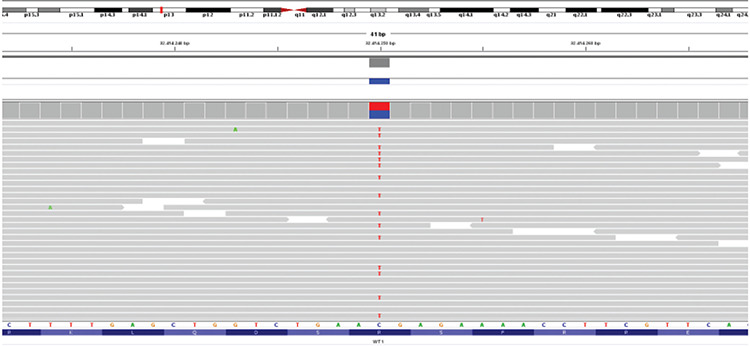
Next generation sequencing analysis WT1: variant visualization on integrative genome viewer (IGV). Patient DNA was sequenced using a custom panel including genes involved in 46,XX disorder of sex development. Sequence enrichment was performed using the NimbleGen SeqCap Target Enrichment kit (Roche) and sequenced on the Illumina NextSeq550 platform (Illumina, San Diego, California). VariantStudio software (Illumina, http://variantstudio.software.illumina.com/) was used for variants annotation. Each single variant has been evaluated for the coverage and the Qscore, and visualized via IGV software. The variant was analyzed in silico using prediction pathogenicity software (Scale-Invariant Feature Transform-SIFT and Polymorphism Phenotyping v2 -PolyPhen2) and database of variants frequency
The neoplastic risk associated with WT1 mutations, the need to plan a second renal transplantation with the related long-term immunosuppressive therapy, the absence of Mullerian structures with potentially dysgenetic gonads producing testosterone led to consideration of prophylactic gonadectomy. She underwent gonadectomy at the age of 14 years. Gross examination revealed small multicystic ovaries. Microscopy showed fibrotic ovarian cortex containing numerous follicles in different maturation stages, from primordial to secondary follicles (Figure 2); some follicles were cystic and scattered corpora lutea were observed. No features of gonadoblastoma were detected. Rudimental remnants of Fallopian tubes were present.
Figure 2.
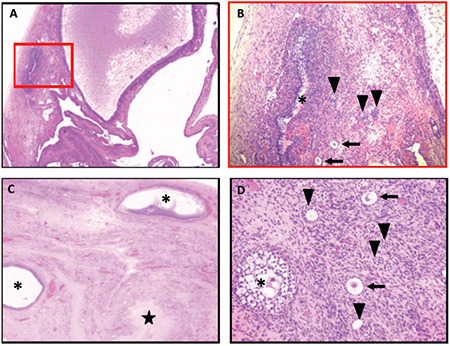
Gonadal histology. (A) Right ovary: multiple cystic follicles in the fibrotic cortex (hematoxylin and eosin x2.5). (B) Higher magnification of the red insert in A) Follicles in different maturation stages: primordial (arrows), primary (arrowheads) and late stage secondary (asterisk) follicles (hematoxylin and eosin x10). (C) Left ovary: fibrotic cortex containing some dilated follicles (asterisks) and a small corpus luteum (star) (hematoxylin and eosin x2.5). (D) Left ovary: Follicles in different maturation stages: primordial (arrows), primary (arrowheads) and early stage secondary (asterisk) follicles (hematoxylin and eosin x20)
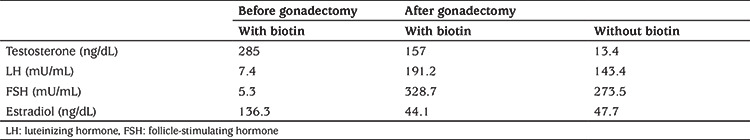
Four months after gonadectomy, hair loss appeared improved but hormonal tests unexpectedly showed elevated testosterone levels (157 ng/dL). In-depth interview on recent medical history revealed daily consumption of high dose biotin, started eight months before, as a therapeutic support for hair loss. Plasma level of biotin was higher than 800 mg/L.
The patient’s total testosterone levels, collected during biotin intake and after its cessation, were measured using liquid chromatography tandem mass spectrometry and were both found within reference intervals, at 4 and 5 ng/dL, respectively. Similar testosterone levels were confirmed using immunoassay testing two weeks after biotin uptake interruption (Table 1).
Table 1. Laboratory measurements before and after gonadectomy and with or without assumption of biotin.
Discussion
WT1 encodes a DNA-binding protein, containing four zinc finger structures, which is essential for normal mammalian urogenital development (3). WT1 knockout mice lack gonads in both sexes, suggesting a role of this gene during the formation of the genital ridge, an early stage of genital development when the gonad is still undifferentiated (4). Classically, its pathogenic variants are associated with abnormal testis development, leading to 46,XY DSD, while 46,XX subjects generally show normal female genitalia (5,6,7).
WT1 mutations have been described in two 46,XX patients with premature ovarian insufficiency (8). Minor genital abnormalities, such as streak ovaries or bicornuate uterus have been reported sporadically (2). Steroid-resistant nephrotic syndrome, associated with absence of both ovaries, has been described in a single case (9). A 46,XX woman showing adult onset of both focal segmental glomerulosclerosis and hypergonadotropic hypogonadism has been reported recently (10). Laparoscopy showed myomas of uterus and cervix, and streak gonads. Both tubes were lying face up with absent fimbrian funnel. None of the reported 46,XX patients showed abnormalities of the external genitalia.
Recently, a novel frameshift WT1 variant (c.1453_1456del; p.Arg485Glyfs*14) has been reported in a SRY-negative 46,XX girl with clitoridomegaly, single perineal opening, and short blind-ending vagina. At 10 years of age, basal gonadotrophins were low, but gonadotrophin releasing hormone analog stimulation test showed a significant elevation of testosterone levels, without an increase in estradiol levels. She underwent bilateral gonadectomy, confirming bilateral testes with seminiferous tubules containing predominantly Sertoli cells and rare germ cells. An immature right uterine tube was also identified (11).
The Arg366His mutation found in our patient, was first described in a 46,XY subject with early onset renal disease, female external genitalia with right dysgenetic testis, left streak-gonad and absence of both Mullerian and Wolffian structures. Histopathology of the removed gonads showed a gonadoblastoma (2). The Arg366His mutation has been subsequently described in several 46,XY subjects with DDS, while 46,XX patients with this mutation generally show normal female genitalia and normal pubertal development (5,6,7). An exception are the two identical twins described by Dharnidharka et al (12). Both were phenotypically females and died a few weeks after birth due to multiorgan failure. At autopsy, the gonads were normal sized ovaries in both twins. Twin A had a complete duplication of uterus and vagina. Mesonephric remnants were prominent in the mesovarium of both twins. Twin B had a microscopic cluster of tubules within the mesovarium consisting of germ cells and supporting cells, reminiscent of testicular architecture. Fluorescent in situ hybridization analysis for detection of the Y chromosome was negative in both twins.
Biotin (also known as vitamin H, vitamin B7, and coenzyme R) is a water-soluble vitamin, naturally present in some foods, with plasma levels between 100-250 ng/L and undergoes urinary excretion. In Western populations, dietary biotin intake is estimated to be 35 to 70 µg daily, a level in line with the recommended dietary allowance. In recent years, high-dose supplementation (doses greater than 1 mg/d) has played a role in the treatment of several diseases, including biotinidase deficiency, mitochondrial metabolic disorders, and multiple sclerosis. Furthermore, advised doses up to 10 mg/day are frequently encountered in nutritional supplements taken to improve hair, skin, and nail health. Many common blood tests employ a biotin-streptavidin reaction as part of the test procedure. While the expected amount of dietary biotin intake is not expected to be high enough to affect these tests, biotin supplementation at doses greater than 1 mg per day can cause either falsely low or falsely high test results, depending on the analyte and platform used for testing (13). Briefly, excess biotin in blood competes with biotinylated antibody of the assay, which produces falsely decreased hormone concentrations in sandwich or non-competitive immunoassays and falsely increased hormone concentrations in competitive immunoassays. Several reports have shown analytical biotin interference, especially in thyroid function tests, but only one included total testosterone measurement (14). Our analytical platform measured testosterone by a competitive immunoassay: elevated concentrations of biotin compete with biotin-antibody-(labeled) analyte complexes for binding to the streptavidin-coated well, leading to the detection of a diminished signal causing a falsely high analyte result.
Our case confirms that the clinical phenotype of subjects with WT1 mutation and 46,XX karyotype may include a severe DSD. The clinical signs of hyperandrogenism, partially improved after gonadectomy, suggest that the girl had hypersecretion of ovarian androgens during puberty, but the “male” levels of testosterone mostly resulted from the analytical interference. Unfortunately, the “true” testosterone levels before gonadectomy are unknown, because there were no testosterone measurements made without in periods when there was no biotin consumption. Several different factors primarily impacted the decision-making process leading to prophylactic gonadectomy: the potential risk of gonadoblastoma associated with WT1 mutations; the need for a second renal transplantion with long-term immunosuppressive therapy; and the absence of Mullerian structures associated with abnormally located and potentially dysgenetic gonads. However, the spuriously elevated testosterone levels apparently confirmed clinical hyperandrogenism, and supported the suspicion of gonadal dysgenesis. Surprisingly, histology showed normal appearance of ovarian tissue, but we cannot exclude the presence of abnormal clusters of androgen secreting cells. Experimental studies on mouse models demonstrated that WT1 gene expression controls the differentiation of genital ridge somatic cells into granulosa or Sertoli cells in genetically female and male gonads, respectively. When WT1 is deleted, these somatic cells turn into steroidogenic cells, hyper-expressing enzymes involved in androgen synthesis, without any sex dimorphism (15).
During patient history taking, enquiring about biotin supplementation should be conducted, since patients may not consider biotin as a medication and therefore may not mention it in their medication list. In the presence of discordance between clinical presentation and biochemical results in patients taking biotin-containing medications, considering biotin half-life of 15 hours (16), we recommend to repeat specimen collection after at least 48 hours of interruption.
Acknowledgments
We thank Simona Pancotti for technical support.
Footnotes
Ethics
Informed Consent: Informed consent was obtained from all individual participants included in the study
Peer-review: Externally and internally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Carla Bizzarri, Luca Dello Strologo, Isabella Guzzo, Francesco Emma, Marco Cappa, Concept: Carla Bizzarri, Marco Cappa, Ottavia Porzio, Luca Dello Strologo, Isabella Guzzo, Francesco Emma, Design: Carla Bizzarri, Marco Cappa, Ottavia Porzio, Data Collection or Processing: Carla Bizzarri, Germana Antonella Giannone, Jacopo Gervasoni, Sabina Benedetti, Federica Albanese, Francesca Diomedi Camassei, Analysis or Interpretation: Carla Bizzarri, Germana Antonella Giannone, Jacopo Gervasoni, Sabina Benedetti, Federica Albanese, Luca Dello Strologo, Isabella Guzzo, Mafalda Mucciolo, Ottavia Porzio, Literature Search: Carla Bizzarri, Ottavia Porzio, Writing: Carla Bizzarri, Ottavia Porzio.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Pelletier J, Bruening W, Li FP, Haber DA, Glaser T, Housman DE. WT1 mutations contribute to abnormal genital system development and hereditary Wilms’ tumor. Nature. 1991;353:431–434. doi: 10.1038/353431a0. [DOI] [PubMed] [Google Scholar]
- 2.Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, Houghton DC, Junien C, Habib R, Fouser L, Fine RN, Silverman BL, Haber DA, Housman D. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell. 1991;67:437–447. doi: 10.1016/0092-8674(91)90194-4. [DOI] [PubMed] [Google Scholar]
- 3.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, Jones C, Housman DE. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 4.Hastie ND. Wilms’ tumour 1 (WT1) in development, homeostasis and disease. Development. 2017;144:2862–2872. doi: 10.1242/dev.153163. [DOI] [PubMed] [Google Scholar]
- 5.Hillen LM, Kamsteeg EJ, Schoots J, Tiebosch AT, Speel EJ, Roemen GM, Peutz-Koostra CJ, Stumpel CT. Refining the diagnosis of congenital nephrotic syndrome on long-term stored tissue: c.1097G>A (p.(Arg366His) WT1 mutation causing denys drash Syndrome. Fetal Pediatr Pathol. 2016:112–119. doi: 10.3109/15513815.2016.1139018. [DOI] [PubMed] [Google Scholar]
- 6.Antonius T, van Bon B, Eggink A, van der Burgt I, Noordam K, van Heijst A. Denys-Drash syndrome and congenital diaphragmatic hernia: another case with the 1097G > A(Arg366His) mutation. Am J Med Genet. 2008;146A:496–499. doi: 10.1002/ajmg.a.32168. [DOI] [PubMed] [Google Scholar]
- 7.Cho HY, Lee BS, Kang CH, Kim WH, Ha IS, Cheong HI, Choi Y. Hydrothorax in a patient with Denys-Drash syndrome associated with a diaphragmatic defect. Pediatr Nephrol. 2006:1909–1912. doi: 10.1007/s00467-006-0273-5. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Li G, Zhang J, Gao F, Li W, Qin Y, Chen ZJ. Novel WT1 missense mutations in han Chinese women with premature ovarian failure. Sci Rep. 2015;5:13983. doi: 10.1038/srep13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JH, Han KH, Lee H, Kang HG, Moon KC, Shin JI, Hahn H, Park YS, Pai KS, Cho BS, Kim SY, Lee SJ, Ha IS, Choi Y, Cheong HI. Genetic basis of congenital and infantile nephrotic syndromes. Am J Kidney Dis. 2011;58:1042–1043. doi: 10.1053/j.ajkd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Hoefele J, Kemper MJ, Schoenermarck U, Mueller S, Klein HG, Lemke A. Truncating Wilms Tumor suppressor gene 1 mutation in an xx female with adult-onset focal segmental glomerulosclerosis and streak ovaries: a case report. Nephron. 2017;135:72–76. doi: 10.1159/000450709. [DOI] [PubMed] [Google Scholar]
- 11.Gomes NL, de Paula LCP, Silva JM, Silva TE, Lerário AM, Nishi MY, Batista RL, Faria Júnior JAD, Moraes D, Costa EMF, Hemesath TP, Guaragna-Filho G, Leite JCL, Carvalho CG, Domenice S, Costa EC, Mendonca BB. A 46,XX testicular disorder of sex development caused by a Wilms’ tumour Factor-1 (WT1) pathogenic variant. Clin Genet. 2019;95:172–176. doi: 10.1111/cge.13459. [DOI] [PubMed] [Google Scholar]
- 12.Dharnidharka VR, Ruteshouser EC, Rosen S, Kozakewich H, Harris HW Jr, Herrin JT, Huff V. Pulmonary dysplasia, Denys-Drash syndrome and Wilms tumor 1 gene mutation in twins. Pediatr Nephrol. 2001;16:227–231. doi: 10.1007/s004670000537. [DOI] [PubMed] [Google Scholar]
- 13.Health C for D and R. Safety Communications - The FDA Warns that Biotin May Interfere with Lab Tests: FDA Safety Communication. Center for Devices and Radiological Health. AVAİLABLE FROM: [Internet] https://www.fda.gov/medical-devices/safety-communications/update-fda-warns- biotin-may-interfere-lab-tests-fda-safety-communication .
- 14.Stieglitz HM, Korpi-Steiner N, Katzman B, Mersereau JE, Styner M. Suspected Testosterone-Producing Tumor in a Patient Taking Biotin Supplements. J Endocr Soc. 2018;2:563–569. doi: 10.1210/js.2018-00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen M, Zhang L, Cui X, Lin X, Li Y, Wang Y, Wang Y, Qin Y, Chen D, Han C, Zhou B, Huff V, Gao F. Wt1 directs the lineage specification of sertoli and granulosa cells by repressing Sf1 expression. Development. 2017;144:44–53. doi: 10.1242/dev.144105. [DOI] [PubMed] [Google Scholar]
- 16.Clevidence BA, Marshall MW and Canary JJ. Biotin levels in plasma and urine of healthy adults consuming physiological doses of biotin. Nutr Res. 1988;8:1109–1118. [Google Scholar]