

Abstract

The classical Butler equation used to describe surface tension and the surface composition of liquid mixtures is revisited. A straightforward derivation is presented, separating basic chemical thermodynamics and assumptions proper to Butler’s model. This model is shown to conceal an approximation not recognized by other researchers. The shortcoming identified consists of not allowing surface standard values to vary with surface tension by virtue of the changing composition. A more rigorous equation is derived and shown to yield the Butler equation in case of incompressible surface phases. It is concluded that the Butler equation slightly overestimates ideal surface tensions. Butler’s surface-phase concentrations of the surface-active component are also slightly overestimated in the surface-active component dilute range, being just underestimated at higher concentrations. Despite this, Butler’s model stands as a very good standard due to its versatility.
Introduction
Predicting the surface tension and the composition of layers overlying liquid mixtures has been the goal of much research over more than a century.1,2 The renowned Gibbs adsorption isotherm2−5 (GAI) is a fundamental equilibrium equation that is the foundation stone of this field of inquiry. However, in the case of a binary mixture, GAI only links the surface tension to the relative adsorption, which is the difference between adsorption of one component and that of the other.5 Among the milestones in this pursuit, the Butler equation6 published in 1932 is of the foremost influence. Indeed, it was not only the first workable proposal for estimating both the surface composition and surface tension but also the starting point for the advancements due to Hoar and Melford,7 Sprow and Prausnitz,8 and Kaptay.9 This notwithstanding, it has been claimed by Rusanov10 and by Santos and Reis11,12 that Butler’s model is not so rigorous as generally regarded. Furthermore, it has been emphasized by different authors3,13 that molecular-thermodynamic models must be validated against the GAI, a condition that Radke3 suspects that is not met by the Butler equation. Present examination of the Butler equation was prompted by Kaptay’s9 recently improved derivation that removed some, not all, previous reservations to its acceptance. A straightforward derivation of the Butler equation is advanced in terms of formal chemical thermodynamics. Properties of the ideal surface region proposed by Butler are carefully examined since the ideal model for liquid mixtures has proven very useful to describe the essential features of mixtures.14 It will be shown that indeed Butler’s original and subsequently improved derivations conceal an approximation, namely, that the chosen standard state for describing surface energy changes with varying compositions is not a constant.
Based on a previous thermodynamic development,11,12 an equation for the surface tension of ideal liquid mixtures is concisely derived. This alternative approach accounts for the variation of standard chemical potentials with changing surface tension from its value for a pure component to its current value at the composition of interest. Remarkably, the alternative equation reduces to the classical Butler equation for incompressible surface phases, as claimed by Rusanov10 on different grounds. A quantitative evaluation of the effect of this hidden approximation is presented for the molecular mixture, water–ethanol, at 298.15 K and for the metallic liquid alloy, cooper–tin, at 1400 K and is shown to be insignificant.
Butler Equation
Formal Thermodynamics Derivation
Considering a physical planar surface phase15 of molecular components A and B at fixed temperature T and pressure p that is in thermodynamic equilibrium with an underlying liquid phase, the differential of the surface-phase Gibbs energy is
| 1 |
where G stands for Gibbs energy, S for entropy, V for volume, γ for surface tension, A for surface area, and μi and ni for chemical potential and the amount of the substance, respectively, of component i = A, B. The superscript “s” denoting the surface phase is only attached to properties whose equilibrium values are different in both phases.
| 2 |
| 3 |
The variation of surface Gibbs energy with the composition at constant T and p can be established from eq 3 by applying the rule of partial differentiation to introduce an extra restriction. This gives
| 4 |
It has been shown2,16−18 that the derivative (∂A/∂nBs)T,p,nA is the partial molar surface area of B designed
for conditions of constant T and p, and denoted18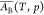
| 5 |
Inserting eqs 2, 3, and 5 into eq 4 leads to
| 6 |
Equation 6 is not new, but now it has been concisely derived using classical thermodynamics only. This is an important step because the Butler equation is based on it. Furthermore, it is noted that eq 6 is identical to eq 21e of ref (2), eq 13 of ref (9), and eq 2d of ref (19) although a misprint certainly occurred in the last two equations, where A appears as an extra constraint in the partial derivative of Gs. For simplicity, the following shorthand is introduced
| 7 |
In the bulk phase, μB is described by
| 8 |
where R is the gas constant and fB is the rational activity coefficient of B at mole fraction xB; an asterisk denotes a pure-component property. Hence, the standard state is the pure liquid B.
The equilibrium condition between the surface and bulk phases requires the uniformity of μB so that from eqs 6 and 8, and considering eq 7, one obtains
| 9 |
Equation 9 is valid over the full composition range. Since fB = 1 for xB = 1, then
| 10 |
By noting that gB = (∂G/∂nB)T,p,nA = μB, it follows from eq 10 that gB*,s ≠ gB = μB*.
Subtracting eq 10 from eq 9 yields
| 11 |
| 12 |
It is remarked that proceeding from eqs 6 to 12 required only well-established chemical thermodynamics (see eq 8) and the phase-equilibria thermodynamics expressed in eq 9. Hence, eq 12 is a rigorous expression for the dependence of surface tension on the mixture composition at constant T and p that does not involve any approximation or extra thermodynamic assumption.
Butler’s original contribution was to put forward an equation for the difference (gBs – gB). Assuming that this difference can be expressed by an equation similar to eq 8, for changes with the composition at fixed T and p, Butler6 proposed the following expression
| 13 |
where gB0,s is the corresponding standard-state value.
From eqs 6, 7, and 13, one obtains
| 14 |
Using the artifact gB0,s = gB + gB0,s – gB and recalling eq 10, eq 14 leads to
| 15 |
Hence, this approach implicitly assumes a surface standard chemical potential, μB0,s, having the form
| 16 |
However, Butler considered gB0,s to be a constant, independent of the surface-phase composition and thus equal to gB. Therefore, from eqs 12 and 13 one obtains the commonly accepted form of the Butler equation
| 17 |
In accordance with this treatment, Butler’s standard chemical potential, μB0,s(Butler), becomes
| 18 |
It is interesting to note that Butler’s approach still makes some allowance for a change in the value of the surface standard chemical potential with varying surface compositions.
It is emphasized that in the abovementioned derivation of the Butler equation, no assumption was made regarding the surface-phase thickness, in agreement with the improved derivation of the Butler equation recently achieved by Kaptay9 without assuming a surface monolayer. This notwithstanding, why some authors3,10−12 have argued that the Butler equation still contains an approximation? If so, it must be found in a hidden assumption within eq 13. To clarify this matter, the focus is directed to Butler’s ideal surface phase.
The ideal model14 acquires special relevance in surface thermodynamics because experimentally, the surface-region composition is not easily accessible.20−22 Since for an ideal liquid mixture according to Raoult’s law, xB(ideal) = xB, then the corresponding ideal surface phase should be defined so that xBs(ideal) = xB. These equalities hold for equilibrium attained from the joint conditions μB(ideal) = μB and μB(real) = μB.23
According to Butler, the ideal model for the surface region is obtained from eq 13 by setting fBs = 1
| 19 |
| 20 |
To progress, pure molar and ideal partial
molar surface areas need to be modeled.24 Usually, they are calculated from well-known relationships as proportional
to some power of the pure molar volume2,8,9,11,19 or the partial molar volume in the ideal bulk phase.2,8,9,19 Since
the latter does not vary with the composition, then 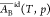 = AB* and eq 20 simplifies
to
= AB* and eq 20 simplifies
to
| 21 |
Noting that an analogous expression holds for component A
| 22 |
equating eqs 21 to 22 allows the estimate Butler’s surface-phase composition by solving the nonexplicit expression25,26
| 23 |
where rA/B = AA*/AB.
Butler’s ideal surface tensions are then obtained by inserting these xBs values in eq 21.
Alternatively, solving eq 21 for xBs and eq 22 for xA and considering that xAs + xB = 1, a nonexplicit
equation is obtained for the calculation of γid. Using these values in eq 21 gives xBs. In fact, this alternative way
is the most frequently employed. However, it is not certain that Butler’s
ideal surface-phase compositions thus calculated, and which should
be used in eq 19, match
those of the corresponding real surface phase. In other words, it
is legitimate to look for hidden assumptions in the modern derivation
of ideal Butler equations leading to eqs 21 and 22. In addition
to equating 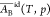 in eq 20 to AB, there is no guarantee that the standard value gBid,0,s in eq 19 is rigorously
composition independent. Indeed, at fixed T and p, standard states in the ideal surface phase with composition xB should be the pure liquid components under the surface tension of
the ideal mixture at that composition because, as demonstrated by
Rudisill and LeVan,13 only these pure-component
standard states satisfy the GAI.
in eq 20 to AB, there is no guarantee that the standard value gBid,0,s in eq 19 is rigorously
composition independent. Indeed, at fixed T and p, standard states in the ideal surface phase with composition xB should be the pure liquid components under the surface tension of
the ideal mixture at that composition because, as demonstrated by
Rudisill and LeVan,13 only these pure-component
standard states satisfy the GAI.
Finally, it is noticed that Gs in eq 1 is defined as the Legendre transform of the surface-phase internal energy, Us, with respect to the variables T and p, Gs = Us – TSs + pVs. Guggenheim27 made a different choice by introducing the surface-phase transformed Gibbs energy, herein denoted G’s, defined as G’s = Us – TSs + pVs – γA = Gs – γA. However, it is paradoxical that using the differential of G’s does not lead to the useful Butler equation.
Hidden Approximation
The suspected variation of gBid,0,s with the composition can be estimated using the Taylor formula for a single variable around gB, which provides a sound mathematical answer to this question. Its application gives
| 24 |
Differentiation of eq 10 in order of γB* gives
| 25 |
The Gibbs–Duhem equation for the surface phase of pure liquid B is27
| 26 |
Division by dγB* at fixed T and p gives
| 27 |
| 28 |
Singling out the first summation term in eq 24 and considering eq 28 leads to
| 29 |
By noting that (∂AB*/∂γB)T,p > 0 is a thermodynamic requirement for a stable surface phase,28 even disregarding the higher-order terms of Taylor expansion, one concludes that gBid,0,s ≠ gB for xBs < 1. In other words, Butler’s assumption of constant standard states gi while the composition is changed at fixed T and p conceals a presumably small approximation.
On the other hand, Butler’s approach leading to eq 20 for γid does not provide a clue to calculate 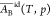 , which has been considered constant and
equal to AB*. However, using an advanced thermodynamic
treatment,18
, which has been considered constant and
equal to AB*. However, using an advanced thermodynamic
treatment,18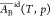 and
and 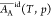 have been calculated for the system water
(A)–ethanol (B) at 298 K and shown to vary slightly with the
composition. Over the full range from pure water to pure ethanol,
have been calculated for the system water
(A)–ethanol (B) at 298 K and shown to vary slightly with the
composition. Over the full range from pure water to pure ethanol, 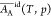 increases 6% and
increases 6% and 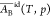 decreases 2% (see Table S2 of ref (18)).
decreases 2% (see Table S2 of ref (18)).
Alternative Equation
Ideal Surface Phase
According to the Lewis–Randall formulation of chemical thermodynamics, the chemical potential of B in a bulk ideal mixture is
| 30 |
It is reasonable to base the formulation of surface ideality on the following expression11−13
| 31 |
Then, at equilibrium
| 32 |
Turning to the Taylor formula, standard chemical potentials become related as follows
| 33 |
Separating the first term and considering eq 27 give
| 34 |
Assuming constant surface thickness of a pure liquid, it is shown in the Supporting Information that4,11
| 35 |
and
 |
36 |
Inserting the latter result into eq 34 leads to
| 37 |
Recalling the series expansion
| 38 |
and setting the variable x as
| 39 |
one finds that
| 40 |
Finally, combining eqs 32 and 40 gives
| 41 |
Bearing in mind that e(c In z) = zc, eq 41 is transformed into
 |
42 |
where (∂AB*/∂p) can be estimated using the model-based eq S7.
Since an analogous equation holds for xAs and recalling xA + xBs = 1, γid can be computed from the resulting nonexplicit expression and thereby the surface-phase composition calculated. Although eqs 40 and 42 are not new,11 the foregoing proof is shorter. Note that this alternative approach is not amenable to yield an explicit equation for γid as eq 21 nor an equation for xB similar to eq 23, which are both proper to Butler’s approach.
Results and Discussion
Considering a given binary system at fixed T, p, and xB, it is clear that calculated values for xBs and γid depend on the chosen model, the Butler equation, or the alternative equation. However, these models are not too dissimilar. Indeed, inasmuch as ln(1 – x) ≈ −x for |x| < 1, x being given by eq 39, eq 41 reduces to eq 21, a fact appreciated by other authors,29,30 and consequently xB (Butler’s approach) ≈ xBs (alternative approach). Considering eqs 39, S9, and S10, this equivalence is best for binary systems of components with not significantly different surface tensions, large molar volumes, and low isothermal compressibilities. This same outcome arises from retaining only the first term of Taylor expansion in eq 33, which amounts to considering (∂AB/∂p)T null (see eq 36). Equivalently, in view of eqs 28 and 35, (∂gB*,s/γB)T,p = 0 for an incompressible surface phase so that from eq 29, gBid,0,s = gB, which is Butler’s nonstated assumption. This is an important result on the account of Rusanov’s harsh criticism of Butler’s model “because it involves compressible bulk phases and incompressible monolayer”.10 It is underlined that the alternative model makes provision for compressible surface phases. Present arguments have been based on ideal surface properties that provide reference values for estimating excess quantities. Excess surface tensions γE = γ – γid are linked to surface-phase activity coefficients.31 Hence, any approximation made while computing Butler’s ideal surface tension values will be included in Butler’s activity coefficients to describe real surfaces.
Comparison of Models
Two binary systems were used to compare ideal surface tension and the surface composition predicted by Butler’s and alternative equations. Labeling B as a surface-active constituent, the examined systems are water (A)–ethanol (B) at 298.15 K, which is a mixture of molecular liquids, and the liquid alloy cooper (A)–tin (B) at 1400 K. Molar surface areas of the pure components were estimated on the basis of hexagonally close-packed disks in a monolayer of close-packed equal spheres (packing factor fi(hcp) = 1.091);7,32 additional calculations were made using the packing factor for cubic cells fi(cc) = 1.000, as recommended by Kaptay19,33 for simple liquid metals, and which is also employed for molecular liquids.8 The required physical data for water and ethanol (refs (34−36)) and for copper and tin (refs (37−39)) are presented in Table S1. For Butler’s model values, xBs was calculated using eq 23, and then eq 21 was employed to obtain γid. For the alternative model values, an equivalent procedure was used, namely, resorting to eq 42 and its analogue for xA and solving xAs + xB = 1 for γid. Plots of ideal surface tension (fi(hcp) = 1.091) and the difference between surface compositions, expressed as % deviation, calculated by both models against the bulk concentration are shown in Figures 1 and 2, respectively, for both binary systems.
Figure 1.

Ideal surface tension, γid, versus the bulk mole fraction of component B, xB. Solid lines for the alternative model and broken lines for Butler’s model. Surface packing factor fi(hcp) = 1.091.
Figure 2.

Difference between estimated surface mole fractions by the alternative and Butler’s equation expressed as % deviation from the alternative model. Surface packing factor fi(hcp) = 1.091.
Figure 1 shows that Butler’s ideal surface tensions are marginally larger than those calculated with the alternative equation. Consequently, the corresponding excess surface tensions are slightly different.
The unexpected behavior observed in Figure 2 deserves some clarification. Indeed, solving eq 21 for Butler’s xBs values, dividing by eq 42 for the alternative xB values, and taking logarithms
and resorting to the series expansion 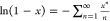 (|x| ≤ 1 and x given by eq 39), yield
(|x| ≤ 1 and x given by eq 39), yield
| 43 |
Analysis of eq 43 shows that the first term on the r.h.s. is positive and the remainder negative; the magnitude of the remainder decreases with increasing xB since (γid – γB*) varies between (γA – γB*) and zero. Therefore, xB(Butler) < xBs(alternative) for low xB values and opposite for large xB values. Moreover, since these differences are negligible, particularly for xB ≥ 0.2, one may conclude that Butler’s model introduces unimportant errors in the calculated surface-phase compositions.
The impact of the adopted packing factor on theoretical calculations of ideal surface tensions was evaluated for both representative binary systems. The outcome of this analysis is condensed in Figure 3, which shows that a smaller fi entails smaller γid values as well as smaller differences between alternative and Butler’s models. The generally small differences depend on the composition, being imperceptible at high concentrations of the surface-active component. The chemical nature of the mixture components having a noteworthy effect on γid estimates which depend on the computed molar surface areas, that are calculated using fi values. In sum, for both binary systems, γid (Butler, fi = 1) > γid (alternative, fi = 1) > γid (Butler, fi = 1.091) > γid (alternative, fi = 1.091), despite an overall modest impact of the packing factor value on the reliability of Butler’s model.
Figure 3.

Effect of the packing factor fi-value on the difference between calculated ideal surface tensions by the alternative and Butler’s models, expressed as % deviation from the alternative model for (⧫), fi(hcp) = 1.091 and (●), fi(cc) = 1.000.
Finally, one may speculate about the major factor contributing to the deviation of Butler’s model from the alternative approach herein presented. Clearly, it is the isothermal compressibility of the pure compounds, which is of the order of 1 GPa–1 for molecular liquids,40 while not amounting to more than a few 0.01 GPa–1 for liquid metals.41 The magnitude difference between isothermal compressibilities offsets the pure-component surface tension differences γH2O* – γEtOH = 49.72 mN m–1 and γCu* – γSn = 801 mN m–1, resulting in minor deviations of γid and xBs for both systems.
Conclusions
The Butler equation merits a good score in this examination. As widely acknowledged, it has provided a good basis to estimate the surface tension and the surface-phase composition of planar liquid mixtures. The Butler equation is, herein, deduced using formal thermodynamics allowing the disclosure of an underlying hidden approximation. This consists of implicitly assuming that in the thermodynamic description of surface phases at fixed T and p, the standard-state values of mixture constituents are not affected by the surface tension variation that accompanies composition changes. An alternative equation allowing for the effect of surface tension on surface standard chemical potentials is worked out. It is shown that it reduces to the conventional Butler equation when molar surface areas of the pure components are not affected by surface tension or pressure variations. Interestingly, this conclusion corroborates Rusanov’s10 inference that the Butler equation is only exact for incompressible surface phases.
Summing up, even though imperfect, the Butler equation has the advantage of providing an explicit and mathematically simple expression for the surface tension of liquid mixtures. This feature has allowed various surface phenomena to be successfully modeled2,19,26,42−46 while employing the Butler equation. It has been, herein, demonstrated that it can continue to be confidently applied because of the small, largely inconsequent numerical errors involved.
Acknowledgments
Centro de Química Estrutural acknowledges the financial support of Fundação para a Ciência e a Tecnologia (UIDB/00100/2020).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c02606.
Author Contributions
† M.S.C.S.S. and J.C.R.R. contributed equally. This manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Adamson A. W.; Gast A. P.. Physical Chemistry of Surfaces, 6th ed.; John Wiley & Sons: New York, 1997; pp 48–81. [Google Scholar]
- Kaptay G. The chemical (not mechanical) paradigm of thermodynamics of colloid and interface science. Adv. Colloid Interface Sci. 2018, 256, 163–192. 10.1016/j.cis.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Radke C. J. Gibbs adsorption equation for planar fluid–fluid interfaces: Invariant formalism. Adv. Colloid Interface Sci. 2015, 222, 600–614. 10.1016/j.cis.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Marmur A. Surface tension and adsorption without a dividing surface. Langmuir 2015, 31, 12653–12657. 10.1021/acs.langmuir.5b03647. [DOI] [PubMed] [Google Scholar]
- Marmur A. Interfaces at equilibrium: A guide to fundamentals. Adv. Colloid Interface Sci. 2017, 244, 164–173. 10.1016/j.cis.2016.05.007. [DOI] [PubMed] [Google Scholar]
- Butler J. A. V. The thermodynamics of the surfaces of solutions. Proc. R. Soc. London, Ser. A 1932, 135, 348–375. 10.1098/rspa.1932.0040. [DOI] [Google Scholar]
- Hoar T. P.; Melford D. A. The surface tension of binary liquid mixtures: lead + tin and lead + indium alloys. Trans. Faraday Soc. 1957, 53, 315–326. 10.1039/tf9575300315. [DOI] [Google Scholar]
- Sprow F. B.; Prausnitz J. M. Surface tensions of simple liquid mixtures. Trans. Faraday Soc. 1966, 62, 1105–1111. 10.1039/tf9666201105. [DOI] [Google Scholar]
- Kaptay G. Improved derivation of the Butler equations for surface tension of solutions. Langmuir 2019, 35, 10987–10992. 10.1021/acs.langmuir.9b01892. [DOI] [PubMed] [Google Scholar]
- Rusanov A. I. The essence of the new approach to the equation of the monolayer state. Colloid J. 2007, 69, 131–143. 10.1134/S1061933X07020019. [DOI] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. New thermodynamics for evaluating the surface-phase enrichment in the lower surface tension component. ChemPhysChem 2014, 15, 2834–2843. 10.1002/cphc.201402100. [DOI] [PubMed] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. Thermodynamic evaluation of molar surface area and thickness of water + ethanol mixtures. J. Mol. Liq. 2018, 255, 419–428. 10.1016/j.molliq.2018.01.136. [DOI] [Google Scholar]
- Rudisill E. N.; LeVan M. D. Standard states for the adsorbed-solution theory. Chem. Eng. Sci. 1992, 47, 1239–1245. 10.1016/0009-2509(92)80245-8. [DOI] [Google Scholar]
- Prausnitz J. M.; Lichtenthaler R. N.; Gomes de Azevedo E.. Molecular Thermodynamics of Fluid-Phase Equilibria, 3rd ed.; Prentice Hall: Upper Saddle River, NJ, 1999; p 214. [Google Scholar]
- Eriksson J. C. On the thermodynamics of surface systems. Adv. Chem. Phys. 1964, 6, 145–174. [Google Scholar]
- Lupis C. H. P.Chemical Thermodynamics of Materials; North-Holland: New York, 1983; p 141. [Google Scholar]
- Lucassen-Reynders E. H.; Cagna A.; Lucassen J. Gibbs elasticity, surface dilational modulus and diffusional relaxation in non-ionic surfactants monolayers. Colloids Surf., A 2001, 186, 63–72. 10.1016/S0927-7757(01)00483-6. [DOI] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. Partial molar surface areas in liquid mixtures. Theory and evaluation in aqueous ethanol. J. Mol. Liq. 2019, 273, 525–535. 10.1016/j.molliq.2018.10.057. [DOI] [Google Scholar]
- Kaptay G. A coherent set of model equations for various surface and interface energies in systems with liquid and solid metals and alloys. Adv. Colloid Interface Sci. 2020, 283, 102212 10.1016/j.cis.2020.102212. [DOI] [PubMed] [Google Scholar]
- Bermúdez-Salguero C.; Gracia-Fadrique J. Gibbs excess and the calculation of the absolute surface composition of liquid binary mixtures. J. Phys. Chem. B 2015, 119, 5598–5608. 10.1021/acs.jpcb.5b01436. [DOI] [PubMed] [Google Scholar]
- Hyde A. E.; Ohshio M.; Nguyen C. V.; Yusa S.-i.; Yamada N. L.; Phan C. M. Surface properties of ethanol/water mixtures: Thickness and composition. J. Mol. Liq. 2019, 290, 111005 10.1016/j.molliq.2019.111005. [DOI] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. Comment on “Surface properties of ethanol/water mixtures: Thickness and composition”. J. Mol. Liq. 2019, 296, 112071 10.1016/j.molliq.2019.112071. [DOI] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. A semi-empirical equation for describing the surface tension of aqueous organic liquid mixtures. Fluid Phase Equilib. 2016, 423, 172–180. 10.1016/j.fluid.2016.04.025. [DOI] [Google Scholar]
- Kaptay G. Partial surface tension of components of a solution. Langmuir 2015, 31, 5796–5804. 10.1021/acs.langmuir.5b00217. [DOI] [PubMed] [Google Scholar]
- Goldsack D. E.; White B. R. An iterative technique for calculating surface tensions of non-electrolyte solutions. Can. J. Chem. 1983, 61, 1725–1729. 10.1139/v83-295. [DOI] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. Surface tension of liquid mixtures and metal alloys. Thermodynamic conditions for the occurrence of a positive temperature coefficient. J. Alloys Compd. 2021, 864, 158839 10.1016/j.jallcom.2021.158839. [DOI] [Google Scholar]
- Guggenheim E. A. The thermodynamics of interfaces in systems of several components. Trans. Faraday Soc. 1940, 35, 397–412. 10.1039/tf9403500397. [DOI] [Google Scholar]
- Bennes R.; Douillard J.-M.; Privat M.; Tronel-Peyroz E. The composition and tension fluctuations in a surface layer: the stability criteria. J. Colloid Interface Sci. 1987, 117, 574–575. 10.1016/0021-9797(87)90419-X. [DOI] [Google Scholar]
- Lu G.; He M.; Kang Z. Study on surface tension and surface phase of molten MCl-CeCl3 (M = Li, Na, K, Cs): A comparison of Butler’s equation and a newly-proposed model. Fluid Phase Equilib. 2016, 427, 345–352. 10.1016/j.fluid.2016.07.017. [DOI] [Google Scholar]
- Liao C.; Cai B.; Wang X.; Jiao Y.; Chen S.; Li L. Study on surface tension of Dy-Cu intermediate alloy prepared using LiF-DyF3 molten salt system. J. Mol. Liq. 2020, 301, 112453 10.1016/j.molliq.2020.112453. [DOI] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. Activity coefficients in the surface phase of liquid mixtures. ChemPhysChem 2015, 16, 470–475. 10.1002/cphc.201402624. [DOI] [PubMed] [Google Scholar]
- Santos M. S. C. S.; Barbosa E. F. G. Adsorption of Et3N and Bu3N in toluene on AgI, in the very dilute region. Surface models and cross-sectional areas. J. Phys. Chem. B 1998, 102, 6040–6048. 10.1021/jp980891l. [DOI] [Google Scholar]
- Kaptay G. A unified model for the cohesive enthalpy, critical temperature, surface tension and volume thermal expansion coefficient of liquid metals of bcc, fcc and hcp crystals. Mater. Sci. Eng., A 2008, 495, 19–26. 10.1016/j.msea.2007.10.112. [DOI] [Google Scholar]
- Strey R.; Viisanen Y.; Aratono M.; Kratohvil J. P.; Yin Q.; Friberg S. E. On the necessity of using activities in the Gibbs equation. J. Phys. Chem. B 1999, 103, 9112–9116. 10.1021/jp990306w. [DOI] [Google Scholar]
- Lampreia I. M. S.; Santos A. F. S.; Moita M.-L. C. J.; Figueiras A. O.; Reis J. C. R. Ultrasound speeds and molar isentropic compressions of aqueous 1-propoxypropan-2-ol mixtures from T = (283.15 to 303.15) K. Influence of solute structure. J. Chem. Thermodyn. 2012, 45, 75–82. 10.1016/j.jct.2011.09.008. [DOI] [Google Scholar]
- Dávila M. J.; Alcade R.; Atilhan M.; Aparicio S. PρT measurements and derived properties of liquid 1-alkanols. J. Chem. Thermodyn. 2012, 47, 241–259. 10.1016/j.jct.2011.10.023. [DOI] [Google Scholar]
- Lee J.; Shimoda W.; Tanaka T. Surface tension and its temperature coefficient of liquid Sn-X (X=Ag, Cu) alloys. Mater. Trans. 2004, 45, 2864–2870. 10.2320/matertrans.45.2864. [DOI] [Google Scholar]
- Assael M. J.; Kalyva A. E.; Antoniadis K. D.; Banish R. M.; Egry I.; Wu J.; Kaschnitz E.; Wakeham W. A. Reference data for the density and viscosity of liquid copper and liquid tin. J. Phys. Chem. Ref. Data 2010, 39, 033105 10.1063/1.3467496. [DOI] [Google Scholar]
- Singh R. N.; Arafin S.; George A. K. Temperature-dependent thermo-elastic properties of s-, p- and d-block liquid metals. Phys. B 2007, 387, 344–351. 10.1016/j.physb.2006.04.029. [DOI] [Google Scholar]
- Marcus Y.The Properties of Solvents; John Wiley & Sons: Chichester, 1998; pp 70–77. [Google Scholar]
- Marcus Y. On the compressibility of liquid metals. J. Chem. Thermodyn. 2017, 109, 11–15. 10.1016/j.jct.2016.07.027. [DOI] [Google Scholar]
- a Kaptay G. A method to calculate equilibrium surface transition lines in monotectic systems. Calphad 2005, 29, 56–67. 10.1016/j.calphad.2005.04.004. [DOI] [Google Scholar]; b Erratum. Calphad 2005, 29, 262.
- Mekler C.; Kaptay G. Calculation of surface tension and surface phase transition line in Ga–Tl system. Mater. Sci. Eng., A 2008, 495, 65–69. 10.1016/j.msea.2007.10.111. [DOI] [Google Scholar]
- Kaptay G. On the negative surface tension of solutions and on spontaneous emulsification. Langmuir 2017, 33, 10550–10560. 10.1021/acs.langmuir.7b01968. [DOI] [PubMed] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. Shape and curvature of surface tension isotherms for liquid mixtures. Colloids Surf., A 2017, 518, 208–217. 10.1016/j.colsurfa.2017.01.013. [DOI] [Google Scholar]
- Santos M. S. C. S.; Reis J. C. R. What happens when the surface and bulk phases of liquid metal alloys attain the same density at equilibrium?. Mater. Today Commun. 2020, 24, 100932 10.1016/j.mtcomm.2020.100932. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.