

Abstract

Molecular traffic across lipid membranes is a vital process in cell biology that involves specialized biological pores with a great variety of pore diameters, from fractions of a nanometer to >30 nm. Creating artificial membrane pores covering similar size and complexity will aid the understanding of transmembrane molecular transport in cells, while artificial pores are also a necessary ingredient for synthetic cells. Here, we report the construction of DNA origami nanopores that have an inner diameter as large as 30 nm. We developed methods to successfully insert these ultrawide pores into the lipid membrane of giant unilamellar vesicles (GUVs) by administering the pores concomitantly with vesicle formation in an inverted-emulsion cDICE technique. The reconstituted pores permit the transmembrane diffusion of large macromolecules, such as folded proteins, which demonstrates the formation of large membrane-spanning open pores. The pores are size selective, as dextran molecules with a diameter up to 28 nm can traverse the pores, whereas larger dextran molecules are blocked. By FRAP measurements and modeling of the GFP influx rate, we find that up to hundreds of pores can be functionally reconstituted into a single GUV. Our technique bears great potential for applications across different fields from biomimetics, to synthetic biology, to drug delivery.
Keywords: DNA origami, nanopores, liposomes, transmembrane transport, cDICE
Introduction
In the past decade, advancements in the field of DNA nanotechnology have enabled the fabrication of a great variety of “DNA origami” nanostructures,1,2 including transmembrane structures that resemble the biological pores found in cells.3 Drawing inspiration from protein pores such as α-hemolysin,4 artificial pores have been engineered with DNA origami1,2 that can insert into lipid bilayers and allow for transmembrane diffusion of ions5,6 and small molecules such as fluorophores,7,8 DNA oligomers,5,7 short PEG,9 and dextran.8,10 To favor partitioning into the lipid bilayer, a popular strategy relies on the chemical modification of the outer nanopore surface with hydrophobic groups, such as cholesterols, that anchor and stabilize the strongly hydrophilic DNA-based nanopores into the lipid membrane.3 As DNA origami structures can be designed with virtually any shape or size11 up to the dimensions comparable to those of viruses,12 the approach bears great potential for various applications, from recapitulating the function of complex enzymes like flippase,13 to mimicking large protein transporters such as the nuclear pore complex.14,15
However, so far, only small pores with an inner diameter of a few nanometers have been realized,8,10 which is because wider pores are increasingly difficult to insert into a membrane. A commonly used strategy to reconstitute pores into a bilayer relies on spontaneous insertion into a preformed lipid membrane.3 This method relies on transient membrane instabilities16 and becomes quickly ineffective for larger size objects. This is because the work required to open up a hole in a membrane grows quadratically with the diameter of the hole,6,8,17 and spontaneous creation of such a hole occurs in a Boltzmann weighted fashion, which exponentially decreases the chances of success for inserting large pores into spontaneously occurring membrane defects. Scaling up the size of artificial pores thus requires an alternative insertion process that can circumvent the barriers associated with spontaneous insertion.
Here, we overcome these challenges by employing a continuous droplet interface crossing encapsulation (cDICE) technique18 to incorporate ultrawide (∼30 nm inner diameter, ∼55 nm outer diameter) DNA origami pores into the membrane of giant unilamellar vesicles (GUVs). As we show below, by administering the pores concomitantly with vesicle formation, the pores can be localized efficiently at the membrane. To demonstrate the transmembrane transport through the pores, we measured the influx of multiple fluorescent macromolecules with different sizes using confocal microscopy. We find that the origami pores indeed support the transit of large (up to 28 nm) molecular structures, consistent with the 30 nm inner diameter of the pores. Using fluorescence recovery after photobleaching (FRAP), we estimate that up to hundreds of pores can be functionally reconstituted in a single liposome. These ultrawide pores represent an exciting tool for various applications, from the mimicking of large biological pores such as the nuclear pore complex to applications in synthetic biology and drug delivery.
Results and Discussion
Design and Assembly of Ultrawide DNA Origami Pores
Using DNA origami folding of a template strand,1,2 we designed and assembled a rigid octagonal ring-like structure (see Figure 1). A multilayer DNA origami object was designed in square-lattice helical packing and folded from a 7560 bases long scaffold single strand with 240 individual oligonucleotide single strands, arranged in a 4 × 4 double-helical pattern. It was designed to form a closed-loop octagonal shape, formed by 8 corner design motifs19 with single-stranded poly-T strands at the corner sites. Deletions were introduced every 32 bases to correct for global twist.20 The origami pore was designed with a nominal outer diameter of 55 nm, an inner diameter of 35 nm, and a height of 10 nm (Figure 1a), which represents the channel length. The design includes 48 single-stranded handles that were distributed evenly on the interior surface facing the central cavity, as sites for future functionalization purposes (Figure S15). The octagon furthermore included 32 cholesterol modifications on the outer surface that were arranged evenly with a ∼5.5 nm horizontal spacing and ∼3 nm vertical distance between neighboring cholesterols (Figure 1a). An asymmetric feature was added (Figure 1b) consisting of four additional double helices on top of the octagon, arranged in a 2 × 2 double helical pattern on the top side of the octagon, spanning over 3 corners. The octagon further features 12 Atto647N dyes for fluorescence imaging, one at each corner on the bottom side of the octagon and four more on both ends of the asymmetric feature (Figure 1a). We iteratively refined the octagon design using gel electrophoretic mobility analysis as the read-out in order to minimize the occurrence of higher-order aggregates and improve the folding quality of the octagon object (Figures S3–S10).
Figure 1.

DNA origami pore design and incorporation approach. (a) DNA origami pore in front (left) and side view (right). Blue cylinders represent DNA double helices. The outer diameter is 55 nm, and the inner diameter is 35 nm. Green objects along the outer side of the DNA origami pore indicate the 32 cholesterol modifications. They vertically span over a width of ∼3 nm (purposely slightly less than the hydrophobic thickness of ∼3.7 nm of a DOPC lipid bilayer37). Black cylinders represent an asymmetric feature that was introduced for class averaging of single particles from TEM. Violet spots along the outer surface of the DNA origami pore indicate the positions of 12 Atto647N dye modifications. The pore is designed with a height of 10 nm. (b) Schematic representation of K10-PEG5K coating of the origami pore. (c) Schematic representation of the DNA origami pore incorporated into the lipid bilayer of a GUV. Left: GUV with pores incorporated in the lipid bilayer. Middle: zoom-in on one of the incorporated origami pores resulting in a 30 nm diameter hole. Right: further zoom-in showing individual lipid molecules of the bilayer; white spheres represent lipid heads, white strokes represent lipid tails, and the green object represents a cholesterol.
The origami pore was coated with K10-PEG5K molecules, which consist of 10 positively charged lysine amino acids (K) linked to a short polyethylene glycol chain (PEG, 5 kDa) (Figure 1b) as described in Ponnuswamy et al.(21) This coating serves two purposes: (i) it stabilizes the octagon in low-ionic-strength environments, where uncoated DNA origami structures would otherwise disassemble, and (ii) it prevents the cholesterol-modified origami pores from aggregating in bulk. Figure 1c shows a schematic illustration of the incorporation of origami pores into the lipid membrane of a liposome (left). The zoom-ins highlight the interaction of the cholesterol-modified origami pores with the lipid molecules.
Characterization of the proper folding and verification of the intended dimensions of the structure were performed using negative-stain transmission electron microscopy (TEM) imaging and subsequent class averaging of single particles (Figure 2). Figure 2a shows a high-pass-filtered image of a typical field of view with several octagon pores. The overall shape of the majority of the particles matches the intended design, without obvious defects, in which 8 straight edges and 8 corners form an octagonal ring-like structure with a ∼35 nm inner diameter and an ∼11 nm thickness. Most of the structures are oriented in a flat orientation on the surface, while a few can be seen in their side view.
Figure 2.

Negative-staining TEM and class averaging of the DNA origami pores. (a) Typical field of view of a negative-stained DNA origami sample. The pores in this image were prepared as described in the Methods section. The image is high-pass filtered with a radius of 20 nm and autoleveled in Photoshop. Scale bar: 50 nm. (b) Top: Front view class average of N = 682 single particles (Figure S12, left). Bottom: Side view class average of N = 80 single particles (Figure S12, right). Scale bar: 25 nm. (c) Two exemplary slices of negative-stained TEM tomograms calculated from EM tilt series, showing octagons (indicated by white arrows) embedded in the membrane of liposomes at a z-height away from the TEM support surface. See Figure S23 for a z-stack of the TEM tomography data. Scale bar: 50 nm.
We computed 2D class averages from single-particle micrographs (Figure S12), which resulted in two distinct images (Figure 2b), corresponding to front and side view transmission projections of the particles. The front view displays four distinct layers of double-stranded DNA. The asymmetric feature is visible on the outer two helices on the top side (Figure 2b, top). The class-averaged image of the side view shows less detail of the expected 4 layers of DNA and the asymmetric feature. This is likely due to the staining with uranyl formate and the tendency of the structures to adhere to the grid surface predominantly with their front side up, resulting in less side-view particles. From these images, we measured an inner diameter of ∼35 nm and a thickness of ∼11 nm (Figure S13). Accounting for the K10-PEG5K coating by including a layer with a thickness of 2.4 ± 1.3 nm, as measured by Ponnuswamy et al.,21 would result in an effective inner diameter of ∼30 nm and an effective height of ∼16 nm including the unstructured PEG brushes. Class-averaged images of K10-PEG5K-coated and bare octagons match well (Figure S14), which shows that the coating preserves the global shape of the octagon pore.
Liposomes with octagons were imaged using negative-stained EM tomography, showing that the octagons can be well incorporated within the lipid membrane (Movie 4 and Movie 5). The tomograms were calculated from the EM tilt series. Slices through the tomograms show octagons located at different heights on the surface of the liposomes (Figure S23). The top slices of the tomograms show octagons embedded in the lipid membrane on top of liposomes (Figure 2c). The global shape and the inner diameter of these octagons remained intact.
DNA Origami Pore Reconstitution in Liposomes by cDICE
We first tested spontaneous insertions observed for our 55 nm (outer diameter) large DNA origami pores into free-standing planar lipid bilayers or into preformed GUVs, under various conditions and with different nanopore versions with either 32 or 64 cholesterols. As expected, these attempts were unsuccessful. While membrane interactions drove the DNA origami objects to adhere on the GUVs (Figure S16), there was a total lack of influx of molecules (Figure S17), indicating that no actual pores were created with a functional transmembrane opening.
We therefore used an alternative approach where we incorporate the DNA origami pores during the process of the formation of the GUVs. We employed an inverted emulsion technique called cDICE,18 which yields unilamellar liposomes with good encapsulation of a large variety of macromolecules.22 Briefly, in cDICE, layers of buffer solution and lipid-in-oil suspension are deposited subsequently in a rotating chamber. As illustrated in Figure 3a, an inner buffer solution containing the origami pores is then injected through a capillary into the rotating lipid-in-oil suspension at a constant flow rate. Shear forces due to the rotation result in detachment of droplets from the capillary orifice. While traveling through the oil phase, these droplets acquire a first monolayer of lipids where the origami pores can insert. A second monolayer is subsequently formed when the droplets cross the oil–water interface, resulting in GUVs that are eventually collected from the outer buffer solution. In this cDICE process, we thus aimed to reconstitute the pore within the lipid monolayer that is formed in the water-in-oil droplets during the first step of cDICE. Whereas the origami rings with 64 cholesterols mainly resulted in formation of large aggregates at the droplet interface (Figure S18), the rings with 32 cholesterols appeared to interact with the lipid monolayer without inducing significant aggregation, suggesting a more promising route. Hereafter, we report the results for the 32 cholesterol version of the DNA origami rings.
Figure 3.

Reconstitution of wide DNA origami pores in GUVs by cDICE. (a) Schematics of the cDICE workflow. A section of the rotating chamber is depicted, including the oil and outer buffer phases. Origami pores are indicated in yellow. (b) Comparison of the efficiency of DNA origami pore localization at the membrane by addition of 2 MDa dextran in the inner buffer. Without dextran, origami pores were observed to remain dispersed in bulk, whereas inclusion of dextran drove the origami pores to the lipid surface. (c) FRAP analysis of origami pores reconstituted in GUV that shows that the pores are mobile within the lipid bilayer. The dashed rectangle indicates the bleached area, which recovers within a few minutes. (d) Example of a large field of GUVs with reconstituted origami pores. (e) Example of a GUV retaining the 2 MDa dextran in the lumen after 24 h incubation in buffer. Scale bars: 10 μm.
In a standard cDICE protocol,22 the inner buffer solution that is injected contains either sucrose or Optiprep22 to obtain an optimal density of the inner GUV solution. We found, however, that in these conditions the reconstitution of 32 cholesterol origami rings was inefficient, with most of the origami pores remaining in solution after vesicle formation (Figure 3b). When we included high molecular weight dextran (∼2 MDa) in the inner buffer, we found that 32 cholesterol origami rings could be fully localized at the membrane (Figure 3b). Note that the choice of such a large dextran molecule (2 MDa) was also motivated by the high diameter of gyration (∼76 nm; ref (23)), which makes it large enough to stay in the vesicle despite the presence of the 30 nm wide DNA origami pores.
The origami pores appeared to be evenly distributed on the GUV surface, only occasionally forming clusters, which then were also enriched in lipids (Movie 1 and Movie 2: z-stack and 3D rendering). Moreover, fluorescence recovery after photobleaching (FRAP) analysis revealed that DNA origami pores could freely diffuse within lipid bilayer of the vesicle membrane (Figure 3c). Hundreds of GUVs could be obtained from a single preparation (Figure 3d). The resulting GUVs retained the 2 MDa dextran over prolonged periods of time (>24 h, Figure 3e) and were remarkably stable, remaining spherical in spite of osmotic changes, suggesting that the origami pores were very well able to equilibrate osmotic differences by allowing osmolytes to rapidly cross the membrane. All in all, the data indicate the successful insertion of wide DNA origami pores in the membrane of GUVs.
Protein Influx through the Pores Demonstrates Their Transmembrane Transport Capabilities
To assess whether the DNA origami pores were functional, that is, inserted in the intended orientation that allows for transmembrane transport through the central cavity of the octagon, we tested the influx of IBB-mEGFP-Cys, a 34.6 kDa variant of the soluble green fluorescent protein that includes an importin β-binding domain24 (see Supporting Information S22), henceforth referred to as “GFP” for simplicity. We measured the normalized fluorescence intensity difference In,diff = (Iout – Iin)/Iout of the vesicles, that is, the difference between in-vesicle intensity (Iin) and the outer bulk (Iout) divided by Iout (Figure 4a) after overnight incubation of the GUVs in an outer solution with 2.3 μM of GFP. While poreless vesicles (control, Figure 4b) remained empty, yielding a finite value In,diff = 0.13 ± 0.05 (errors are standard deviation, N = 34), the pore-containing liposomes showed an influx of GFP for about 50% of the vesicles (Figure 4c): half of the vesicles exhibited an almost complete saturation to In1,diff = 0.01 ± 0.03 (N = 45), while the other half yielded In2,diff = 0.09 ± 0.03 (N = 45), that is, no significant change compared to the control. We note that heterogeneity in GUV samples is a widely reported phenomenon25 that apparently is associated with variations in the efficiency of pore insertion during vesicle formation in cDICE. Most importantly, a clear fraction of the liposomes unambiguously showed transmembrane transport of the folded proteins, indicating transport through the ultrawide DNA origami pores.
Figure 4.

Test of GFP influx through origami pores. (a) Schematic of the influx experiment. Left: Sketch of a vesicle (red) containing pores (blue) that is incubated with GFP (green). Right: Fluorescence intensities Iin and Iout that are expected to be measured in a time-lapse experiment on the dashed purple and yellow areas, respectively. The right part of the graph illustrates the end-point intensities after ∼24 h, after an initial transient. (b) Influx experiment with vesicles that were not subjected to DNA pore insertion. Top right: sketch of a poreless vesicle that excludes GFP. Top left: example of fluorescence image for a poreless vesicle (cyan) that prevents GFP (green) from entering the vesicle, showing up as a darker area signifying the absence of GFP. Bottom: Histogram of the normalized end-point intensity difference (Iout – Iin)/Iout for the poreless vesicles, verifying that no vesicles showed GFP influx. (c) Influx experiment with vesicles that were functionalized with origami pores. Top right: sketch of a pore-containing vesicle filled with GFP. Top left: example of fluorescence image for a pore-containing vesicle (cyan) that allows GFP to diffuse (green) into the vescicle, resulting in Iout – Iin ≈ 0. Bottom: Histogram showing normalized end-point intensity difference (Iout – Iin)/Iout for the poreless vesicles. We find that ∼50% of the vesicle population manifested GFP influx. Scale bars: 10 μm.
Next, we characterized the GFP influx rate quantitatively using a FRAP assay (Figure 5a). In this experiment, we selected pore-containing vesicles that showed that influx occurred during the overnight incubation (i.e., those with In,diff ≈ 0), then photobleached the inner part, and subsequently measured the recovery of the fluorescence signal. An example of the FRAP experiment is shown in Figure 5b (top) and Movie 3. Starting from an equilibrium state where Iin ≈ Iout (Figure 5b, left), the vesicle fluorescence drops upon photobleaching to a level comparable to the one of an empty vesicle, to subsequently fully recover within a few minutes (Figure 5b, right). Notably, this time scale is much faster compared to those of previously reported fluorescence recovery with DNA origami pores.7,8 A control with vesicles lacking pores (Figure 5b, bottom) resulted in no appreciable recovery upon photobleaching. Figure 5c displays Iin and Iout for a pore-containing GUV as a function of time where, after initial transient behavior (gray area), Iout is constant, whereas Iin continues to slowly increase over time, which represents the influx of GFP through the ultrawide DNA origami pores.
Figure 5.

FRAP assay, modeling, and extraction of number of functional pores. (a) Schematic of the FRAP assay: (1) the vesicle is left to equilibrate with bulk solution containing GFP; (2) GFP molecules inside the vesicle are photobleached; (3) new GFP molecules are driven through the ultrawide origami pores from the outer solution, causing a recovery of the fluorescence signal. (b) Example of a FRAP experiment. Top left: image showing the pore-containing vesicle (origami pores in cyan) at equilibrium with the outer GFP (green) solution. Top right: series of frames showing recovery (within minutes) of the GFP signal after photobleaching. Bottom left: image showing a pore-less vesicle (cyan) that excludes GFP (green) present in the outer solution. Bottom right: series of frames showing the absence of a signal increase after photobleaching. (c) Fluorescence intensities Iin (red) and Iout (purple) that reduce and recover after photobleaching for the vesicle shown in (b). Gray area denotes the initial transient, after which Iout can be considered constant. Inset: Highlight of the regions where Iin (red) and Iout (purple) were measured. (d) Normalized intensity difference In,diff over time for the vesicle illustrated in (b). Data are fitted by eq 4. Gray area was excluded from the fit as the model assumes a constant Iout. For such a 13.3 μm diameter vesicle, we estimated a number of functional pores Np = 250. (e) Number of origami pores extracted from fits for different vesicles, as a function of vesicle diameter (gray circles). Averaged data over 2.5 μm bins (red squares, error bars are standard deviation) show an increase of the number of pores as a function of vesicle diameter. Black line represents a cubic fit to the data. Scale bars: 10 μm.
To obtain the number of functional pores, we model the influx rate using a diffusion model derived from Fick’s first law:26
| 1 |
which describes the flux ϕ
of GFP molecules
with diffusion constant D, as a result of the concentration
gradient dc/dx between the outer
(cout) and inner (cin) environment of the GUV. The flux ϕ is defined as
number of molecules dN crossing a membrane area dA in a time dt. Since the vesicle volume V is constant over time, the in-vesicle concentration cin(t) is a time-dependent variable
that reflects the fact that the vesicle is filling up over time: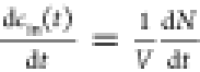 . The outside concentration cout instead is assumed to be constant over time, which
is reasonable given the large volume (∼200 μL) of the
bulk solution as compared to the ∼picoliter volume of the vesicle.
Assuming that the diffusion occurs through a number Np of DNA origami pores that each have a length Lp and an area Ap, yielding a total integrated area A = ApNp, we can rewrite eq 1 as the first-order differential
equation
. The outside concentration cout instead is assumed to be constant over time, which
is reasonable given the large volume (∼200 μL) of the
bulk solution as compared to the ∼picoliter volume of the vesicle.
Assuming that the diffusion occurs through a number Np of DNA origami pores that each have a length Lp and an area Ap, yielding a total integrated area A = ApNp, we can rewrite eq 1 as the first-order differential
equation
| 2 |
which, with cin(t = 0) = cin, start, has a solution
| 3 |
Note, however, that for the transport through the pores, we need to employ an effective diffusion constant Deff which is reduced compared to the bulk diffusion constant D to account for the confined transport through the pores, as reported by Dechadilok and Deen.27 For our pore dimensions, this results in almost a factor of 2 decrease of the GFP diffusivity, that is, Deff = 0.54Dbulk. Upon rearranging eq 3, we then finally obtain
| 4 |
As the measured fluorescence intensities are a good measure for the protein concentrations of the fluorescent GFP, we can use eq 4 to fit the In,diff(t) data, with cin,start/cout and Np as fit parameters. Figure 5d provides an example, showing an excellent fit to the FRAP data. From fitting FRAP curves acquired from different vesicles, we find that the estimated number of pores Np varies between a few to a few hundred per GUV. Despite the substantial scatter in the extracted Np (gray circles in Figure 5e), which we attribute to the stochasticity inherent to the vesicle production and origami insertion during cDICE, we do observe an overall increase of Np with the vesicle diameter. Assuming that all origami pores would move from the volume into the membrane during GUV formation, Np should scale with the cube of the vesicle diameter, which is consistent with the behavior observed (black line in Figure 5e).
Ultrawide DNA Origami Pores Act As a Molecular Sieve with ∼30 nm Cutoff
To provide evidence that our DNA origami pores indeed form open channels with an effective size of the order of the inner pore diameter of 30 nm, we tested the influx of dextran-FITC molecules with a variety of sizes, viz., Dg ≈ 15 nm (70 kDa), 22 nm (150 kDa), 28 nm (250 kDa), 39 nm (500 kDa), and 76 nm (2 MDa) (see Figure 6a), where Dg is the diameter of gyration as measured by Hanselmann and Burchard.23 As in the GFP influx experiment, we incubated the pore-containing vesicles with 2 μM of dextran-FITC and measured the normalized intensity difference In,diff after ∼24 h. This was done independently for each of the 5 molecules, keeping the same molar concentration of dextran-FITC molecules in bulk solution. Similar to the GFP experiments (Figure 4c), In,diff of pore-containing vesicles yielded two populations when incubated with 70 kDa dextran-FITC (Figure 6b, left), where 58% manifested influx (In1,diff,70k = 0.02 ± 0.03, errors are standard deviation, N = 47), while 42% remained empty (In2,diff,70k = 0.13 ± 0.02, N = 34). Incubation with 150 kDa dextran-FITC (Figure 6b, center-left) led to a similar result, with 47% GUVs that were filled (In1,diff,150k = 0.03 ± 0.03, N = 24) against 53% of empty vesicles (In2,diff,150k = 0.11 ± 0.03, N = 27). Influx of the larger 250 kDa dextran-FITC (Figure 6b, center) resulted in a reduced (19%), yet present, population of filled vesicles (In1,diff,250k = 0.01 ± 0.02, N = 8), while 79% (In2,diff,250k = 0.10 ± 0.02, N = 34) showed no sign of influx. Importantly, this demonstrates that very large (250 kDa; 28 nm) macromolecules can be transported across the ultrawide DNA origami nanopores. By contrast, incubation with the even larger 500 kDa and 2 MDa dextran-FITC (Figure 6b, center-right and right) resulted in only one population of empty vesicles, with In,diff,500k = 0.13 ± 0.03 (N = 49) and In,diff,2M = 0.09 ± 0.04 (N = 98), respectively, indicating a lack of any transmembrane transport during 24 h. This is fully consistent with expectations as their size clearly exceeds the 30 nm size of the origami pore. It is also consistent with the use of 2 MDa dextran as a macromolecular crowder (cf. Figure 3e).
Figure 6.

Size selectivity of DNA origami nanopores. (a) Diameter of gyration of dextran Dgvs molecular weight Mw. Black line is a fit of Dg = 0.072 × Mw0.48 (ref (23)). Dextran sizes of 70 kDa, 150 kDa, 250 kDa, 500 kDa, and 2 MDa employed in our study are highlighted in blue, yellow, magenta, and black, respectively. Gray area underlies dextran sizes larger than the origami pore (red) which are not expected to translocate. (b) Histograms showing normalized intensity difference of pore-containing vesicles after overnight incubation the dextran molecules. Blue indicate vesicles that showed influx of 70 kDa, yellow bars for 150 kDa, magenta bars for 250 kDa, whereas gray bars represent empty vesicles. Dashed lines represent hypothetical populations of vesicles that would have been present if influx of 500 kDa or 2 MDa through the origami pores had occurred.
Conclusions
In this work, we achieved functional reconstitution of ultrawide DNA origami pores in giant liposomes, obtaining stable transmembrane pores of 30 nm inner diameter within the lipid bilayer. We successfully achieved the pore reconstitution by inserting the origami pore into a lipid monolayer followed by bilayer formation. This methodology lowers the energy barrier for pore insertion, allowing functional reconstitution of pores with much larger diameter than previously possible.
Our results show great potential for a number of biomimetic applications. For example, future efforts can be directed toward the functionalization of the origami pore with FG-nucleoporins from the nuclear pore complex, the macromolecular complex mediating transport across the nuclear envelope in eukaryotes,28 as reported in previous works.14,15 This will enable the creation of selective biomimetic pores that permit the exclusive transport of specific transport receptor proteins. While such biomimetic pores constitute an exciting platform for investigating nucleocytoplasmic transport in vitro, they may also be employed as a building block for synthetic cells, thus contributing to the in vitro reconstitution of an artificial model of a nucleus. This will, for example, aid the study of emergent properties of liposome-confined genomes29 by allowing the controlled injection of genome-processing proteins. Indeed, our DNA origami pores may be a great asset for building a synthetic cell as they provide a stable portal that is large enough for biological macromolecules like proteins and RNAs to enter a liposome. The long-term stability and functionality of these liposomes is a particularly useful feature in such synthetic biology applications. Finally, responsive drug delivery systems that release clinically relevant macromolecules such as antibodies upon stimulation by specific biomolecules could be envisioned, as well. We expect that the ultrawide DNA origami nanopores may thus find a broad variety of different applications.
Methods
Design of Scaffolded DNA Origami Objects
The objects were designed using caDNAno v0.2.30
Optimal Folding Conditions of the Octagon
The folding reaction mixture for the octagon contained scaffold DNA31 at a final concentration of 10 nM and oligonucleotide strands (IDT Integrated DNA Technologies) at 300 nM each. The folding reaction buffer contained 5 mM Tris, 1 mM EDTA, 5 mM NaCl (pH 8), and 24 mM MgCl2. The folding reaction mixtures were subjected to the following thermal annealing ramp using TETRAD (MJ Research, now Biorad) thermal cycling devices: 15 min at 65 °C, followed by 16 h at 50 °C, followed by incubation at 20 °C before further sample preparation steps.
Purification of DNA Origami Nanostructures
Excess oligonucleotides were removed via ultrafiltration (Amicon Ultra 0.5 mL Ultracel filters, 50K) with FoB5 (5 mM Tris, 1 mM EDTA, 5 mM NaCl, and 5 mM MgCl2) buffer.19 All centrifugation steps were performed at 10 kG for 3 min at 25 °C. Step 1: 0.5 mL of FoB5 buffer was added to the filters and centrifuged. Step 2: 0.1 mL of folded object sample and 0.4 mL of FoB5 buffer were added to the filters and centrifuged. Step 3: 0.45 mL of FoB5 buffer was added to the filters and centrifuged. Step 3 was repeated 3 times before a final retrieving step: the filter insets were turned upside down, placed into a new tube, and centrifuged. This concludes one round of ultrafiltration.
Coating of the Octagon
Different volumes of purified octagons were mixed with PEG-oligolysine (K10-PEG5k). The optimal amount of K10-PEG5K added was determined based on the ratio of nitrogen in amines to the phosphates in DNA (Ponnuswamy et al.(21)) to a final ratio of 0.75:1 (N/P).
Negative-Stain TEM
A carbon Formvar grid (Electron Microscopy Sciences) was plasma-treated (45 s, 35 mA) right before use. Five microliters of uncoated octagon sample (10–50 nM DNA object concentrations, 5–20 mM MgCl2 concentration) was pipetted onto the grid. Coated octagon samples were pipetted onto the grids without plasma treatment in order to reduce positive staining effects. The sample droplets were incubated for 30–120 s on the grids and then blotted away with filter paper. A 5 μL droplet of 2% aqueous uranyl formate (UFO) solution containing 25 mM sodium hydroxide was pipetted onto the grid and immediately blotted away. A 20 μL UFO droplet was then pipetted onto the grid, incubated for 30 s, and blotted away. The grids were then air-dried for 20 min before imaging on a Philips CM100 and an FEI Tecnai 120 microscope. Particles were picked automatically using crYOLO32 and subsequently class averaged using Relion3.0.33
Negative-stain EM tomography was used to visualize octagons at the surface of liposomes. The samples (5 μL) containing liposomes with incorporated octagons were incubated on the grid surface until the liquid evaporated (∼15 min). The sample was not blotted away using filter paper in an effort not to damage the liposomes through shear forces. All other grid preparation steps were the same as described before.
The tilt series were acquired at a magnification of 30000× using an FEI Tecnai 120. The stage was tilted from −50° to +50°, and micrographs were acquired every 2°. The tilt series were then processed using Etomo (IMOD)34 to acquire tomograms. The micrographs were first aligned by calculating a cross-correlation of the consecutive images. The tomograms were then generated using a filtered back projection. The Gaussian filter used a cutoff of 0.35 and a falloff of 0.035.
Gel Electrophoresis of Octagon Samples
Octagon samples were electrophoresed on 1.5 and 2.0% agarose gels containing 0.5× Tris-borate-EDTA and 5 mM MgCl2 (unless noted otherwise) for around 1–2 h at 70 or 90 V bias voltage in a gel box, cooled in a water or ice water bath. The loaded samples contained final monomer concentrations of 10 nM (unless otherwise noted). The gels were scanned with a Typhoon FLA 9500 laser scanner (GE Healthcare) at a resolution of 25 or 50 μm/pixel. Further enhancement of the resulting images or parts of images was performed using Photoshop CS5.
DNA Origami Sample Preparation Protocol
-
1.
Folding of DNA origami samples at previously described optimal folding conditions.
-
2.
Three rounds of ultrafiltration to purify the samples of excess oligonucleotides.
-
3.
Addition of Atto647N modified oligonucleotide at a final oligonucleotide/DNA origami ratio of 48:1 (12 handles per DNA origami; effective 4:1 ratio).
-
4.
One round of ultrafiltration to purify the samples of excess Atto647N oligonucleotides.
-
5.
Addition of PEG-oligolysine at N/P ratio of 0.75:1, incubation at room temperature overnight.
-
6.
Addition of cholesterol modified oligonucleotide at a final oligonucleotide/DNA origami ratio of 128:1 (32 handles per DNA origami; effective 4:1 ratio).
-
7.
One round of ultrafiltration to purify the samples of excess cholesterol oligonucleotides.
Reagents
The 2 MDa dextran (D5376), MgCl2 (M8266), glucose (G7021), Optiprep (60% (w/v) iodixanol in water, D1556), silicone oil (317667), and mineral oil (M3516-1L) were purchased from Sigma-Aldrich. Tris-HCl (10812846001) was purchased from Roche. DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) (850375) and DOPE-Rhodamine (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt)) (810150C) were purchased from Avanti Lipids. Lipids were stored and resuspended in anhydrous chloroform (288306, Sigma Alrich). Sodium dodecyl sulfate employed for cleaning steps was purchased from Sigma-Aldrich. UltraPure bovine serum albumin used for passivation of the glass coverslips was purchased by ThermoFisher. For the influx experiments, we employed 70 kDa dextran-FITC (FITC/glucose (mol/mol) = 0.004) (Sigma-Aldrich), 150 kDa dextran-FITC (FITC/glucose (mol/mol) = 0.004) (Sigma-Aldrich), 250 kDa dextran-FITC (FITC/glucose (mol/mol) = 0.003–0.020) (Sigma-Aldrich), 500 kDa dextran-FITC (FITC/glucose (mol/mol) = 0.003–0.020) (Sigma-Aldrich), and 2 MDa dextran-FITC (FITC/glucose (mol/mol) = 0.001–0.004) (Sigma-Aldrich).
Expression and Purification of IBB-eGFP
For the expression of His14-TEV-IBB-mEGFP-Cys (“IBB-GFP”), plasmid pSF807 (Eisele et al.(35)) was transformed into Escherichia coli BLR cells. Cultures were grown in TB medium supplemented with 50 μg/mL kanamycin, and expression was induced with 0.5 mM IPTG during overnight incubation at 18 °C, with shaking at 150 rpm in baffled flasks. Cells were harvested by centrifugation (10 min at 4000 rpm, JLA8.1000 rotor), washed in 1× phosphate-buffered saline, and lysed using a French Press (Constant Systems) at 20 kpsi at 4 °C in buffer A (2 M NaCl, 50 mM Tris/HCl pH 8.0, 5 mM MgCl2). Unbroken cells, debris, and aggregates were pelleted in a Ti45 rotor (30 min, 40000 rpm, 4 °C), and the lysate resulting from 1 L cell culture was applied to 2 mL (bed volume) of Ni-NTA matrix prewashed with buffer A. After 60 min incubation while slowly inverting the tube in a cold room (4 °C), the matrix was washed extensively with buffer A supplemented with 60 mM imidazole and buffer B (50 mM Tris/HCl pH 7.5, 300 mM NaCl, 5 mM MgCl2, 1 mM βME, 10% glycerol). Following overnight elution by homemade TEV protease in buffer B, the eluate was concentrated by ultracentrifugation and further purified by size exclusion chromatography on a Superdex 200 column (GE Healthcare) pre-equilibrated with buffer B supplemented with 0.5 mM EDTA. The final IBB-GFP sample with a concentration of 138 μM was aliquoted, snap frozen in liquid nitrogen, and stored at −80 °C.
cDICE Buffers and Solutions
The encapsulated inner solution contains 50 mM Tris-HCl pH 7.4, 5 mM MgCl2, 15 μM 2 MDa dextran, 2 nM DNA origami pores. The outer solution contains glucose in Milli-Q water and is osmotically matched to the inner solution. The oil phase is a mixture of silicone and mineral oil (4:1 ratio) and contains lipids dissolved at 0.2 mg/mL. The lipid-in-oil suspension was prepared as in Van de Cauter et al.(22) and used immediately.
Preparation of Lipid-in-Oil Suspension
DOPC and DOPE-Rhodamine lipids solubilized in chloroform were mixed at 99.9:0.1 ratio and blow-dried with pure nitrogen. Inside a glovebox filled with pure nitrogen, lipids were subsequently resolubilized with anhydrous chloroform. The freshly prepared mixture of silicone and mineral oil was added to the lipids dropwise while vortexing at 1400 rpm. The lipid-in-oil solution was finally vortexed at 2800 rpm for 2 min and further sonicated in an ice bath for 15 min.
cDICE Equipment and GUV Production
The rotating component of the cDICE device is built from a magnetic stirrer L-71 (LAbinco). A Teflon adaptor connects the rotating shaft with the cDICE chamber, assembled from a standard 3.5 cm cell culture dish (corning) modified to decrease the height. The chamber has an inner height of 8 mm and a circular opening of 10 mm on the top to allow insertion of the capillary and collection of the GUVs. The detailed protocol for GUV production is reported in Van de Cauter et al.,22 while here we describe it briefly. First, the cDICE chamber is set to rotate at 300 rpm. Next, 350 μL of outer aqueous solution is injected into the rotating the chamber, followed by 1 mL of lipid-in-oil solution, resulting in the two phases being stacked, as depicted in Figure 3a. The tip of a 100 μm PEEK capillary tube (211633-3, BGB) is then inserted into the oil phase of the rotating chamber, allowing for a continuous supply of inner aqueous solution into the oil phase by a pressure pump (MFCS-EZ, Fluigent) set at 900 mbar. After 15 min, the rotating speed is set to zero, and GUVs are collected from the outer aqueous solution and moved to a prepassivated imaging well to carry out the fluorescence experiments.
Data Collection and Analysis
Fluorescence images were acquired using spinning disk confocal laser microscopy (Olympus IXB1/BX61 microscope, 60× objective, iXon camera) with Andor iQ3 software. To induce GFP photobleaching, we employed raster scanning with a 491 nm laser (at 9.8 mW) over the region of interest. To measure the recovery signal, frames were collected every 15 s, starting right after the photobleaching event. Fluorescence images were analyzed and processed using ImageJ (v2.1.0). The extracted fluorescence data were plotted and fitted using MATLAB.
Diffusion Model
Fluorescence recovery data were fitted with a diffusion model, described by eq 4, derived from Fick’s law of diffusion. Whereas cin,start/cout and Np were free parameters of the fit, all the other parameters were known or could be calculated. More particularly, the area of the pore Ap = 745.6 nm2 was calculated from assuming an octagonal pore with an effective radius of 15 nm (as discussed in the text). The effective diffusion constant Deff = 47 μm2/s of GFP was calculated using the formula provided by Dechadilok and Deen:27
 |
where Rg is the molecule radius of gyration (for GFP, Rg = 1.77 nm), Rp is the pore radius (here 15 nm), and Dbulk is the diffusion constant of the molecule in free solution (for GFP, Dbulk = 87 μm2/s, ref (36)). The channel length L = 16 nm was assumed to equal the effective height of the origami, which was measured by TEM imaging (Figure S13b). Finally, V was the volume of the vesicle, which was estimated from the fluorescence images.
Acknowledgments
We thank Kristina Ganzinger and Gijsje Koenderink for helpful discussions and technical advice in setting up cDICE; Federico Fanalista, Nils Klughammer, and Jaco van der Torre for discussions; Jeremie Capoulade for technical support on confocal microscopy; and Dimitri de Roos for constructing the cDICE steup. This work was supported by a European Research Council Consolidator Grant to H.D. (GA No. 724261), the Deutsche Forschungsgemeinschaft through grants provided within the Gottfried-Wilhelm-Leibniz Program and the SFB863 Project ID 111166240 TPA9 (to H.D.) C.D. acknowledges funding support from the ERC Advanced Grant 883684, NWO grants 16PR3242-1, BBOL.737.016.016, and OCENW.GROOT.2019.068, and the NanoFront and BaSyC programs.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c01669.
Design, layout, sequence of the DNA origami pore; agarose gels for the screening protocol, TEM single particles used for class averaging, additional images on the EM tomography of the liposomes, fluorescence images showing results of control experiments and raw data used for fitting, captions describing Movies 1–5 (PDF)
Movies 1–5 (ZIP)
Author Contributions
A.F., H.D., and C.D. conceived the research. N.D.F. and C.D. set up cDICE and devised the reconstitution protocol. P.S. and H.D. devised the DNA origami nanopores. P.S. carried out assembly, quality control, and TEM imaging of the DNA origami nanopores. A.F. and N.D.F. carried out influx experiments. A.F. performed analysis and modeling of influx data. E.O.v.d.S. expressed and purified the GFP. A.F., N.D.F., P.S., H.D., and C.D. wrote the manuscript. A.F., N.D.F., and P.S. contributed equally to this work.
The authors declare no competing financial interest.
Notes
The manuscript has been previously deposited on a preprint server as Alessio Fragasso, Nicola De Franceschi, Pierre Stömmer, Eli O. van der Sluis, Hendrik Dietz, Cees Dekker. Reconstitution of Ultrawide DNA Origami Pores in Liposomes for Transmembrane Transport of Macromolecules. bioRxiv 2021, 432733; 10.1101/2021.02.24.432733 (accessed February 24, 2021).
Supplementary Material
References
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440 (7082), 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Dietz H.; Liedl T.; Högberg B.; Graf F.; Shih W. M. Self-Assembly of DNA into Nanoscale Three-Dimensional Shapes. Nature 2009, 459 (7245), 414–418. 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Ainsa S.; Keyser U. F. DNA Origami Nanopores: Developments, Challenges and Perspectives. Nanoscale 2014, 6 (23), 14121–14132. 10.1039/C4NR04094E. [DOI] [PubMed] [Google Scholar]
- Sugawara T.; Yamashita D.; Kato K.; Peng Z.; Ueda J.; Kaneko J.; Kamio Y.; Tanaka Y.; Yao M. Structural Basis for Pore-Forming Mechanism of Staphylococcal α-Hemolysin. Toxicon 2015, 108, 226–231. 10.1016/j.toxicon.2015.09.033. [DOI] [PubMed] [Google Scholar]
- Martin T. G.; Simmel F. C.; Arnaut V.; Mayer M.; Dietz H.; Langecker M.; Renner S.; List J. Synthetic Lipid Membrane Channels Formed by Designed DNA Nanostructures. Science (Washington, DC, U. S.) 2012, 338 (6109), 932–936. 10.1126/science.1225624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyenes B.; Göpfrich K.; Winterhalter M.; Li C.-Y.; Ohmann A.; Yoo J.; Aksimentiev A.; Ricci M.; Bhamidimarri S. P.; Keyser U. F. Large-Conductance Transmembrane Porin Made from DNA Origami. ACS Nano 2016, 10 (9), 8207–8214. 10.1021/acsnano.6b03759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S.; Ziegler D.; Arnaut V.; Martin T. G.; Kapsner K.; Henneberg K.; Bausch A. R.; Dietz H.; Simmel F. C. Molecular Transport through Large-Diameter DNA Nanopores. Nat. Commun. 2016, 7, 1–7. 10.1038/ncomms12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen R. P.; Malle M. G.; Okholm A. H.; Krishnan S.; Bohr S. S.; Sørensen R. S.; Ries O.; Vogel S.; Simmel F. C.; Hatzakis N. S.; Kjems J. A Large Size-Selective DNA Nanopore with Sensing Applications. Nat. Commun. 2019, 10, 1–10. 10.1038/s41467-019-13284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns J. R.; Seifert A.; Fertig N.; Howorka S. A Biomimetic DNA-Based Channel for the Ligand-Controlled Transport of Charged Molecular Cargo across a Biological Membrane. Nat. Nanotechnol. 2016, 11 (2), 152–156. 10.1038/nnano.2015.279. [DOI] [PubMed] [Google Scholar]
- Iwabuchi S.; Kawamata I.; Murata S.; Nomura S. M. Large, Square-Shaped, DNA Origami Nanopore with Sealing Function on Giant Vesicle Membrane. Chem. Commun. 2021, 57, 2990–2993. 10.1039/D0CC07412H. [DOI] [PubMed] [Google Scholar]
- Ramezani H.; Dietz H. Building Machines with DNA Molecules. Nat. Rev. Genet. 2020, 21 (1), 5–26. 10.1038/s41576-019-0175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenbauer K. F.; Sigl C.; Dietz H. Gigadalton-Scale Shape-Programmable DNA Assemblies. Nature 2017, 552 (7683), 78–83. 10.1038/nature24651. [DOI] [PubMed] [Google Scholar]
- Ohmann A.; Li C. Y.; Maffeo C.; Al Nahas K.; Baumann K. N.; Göpfrich K.; Yoo J.; Keyser U. F.; Aksimentiev A. A Synthetic Enzyme Built from DNA Flips 107 Lipids per Second in Biological Membranes. Nat. Commun. 2018, 9 (1), 1–9. 10.1038/s41467-018-04821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketterer P.; Ananth A. N.; Laman Trip D. S.; Mishra A.; Bertosin E.; Ganji M.; Van Der Torre J.; Onck P.; Dietz H.; Dekker C. DNA Origami Scaffold for Studying Intrinsically Disordered Proteins of the Nuclear Pore Complex. Nat. Commun. 2018, 9 (1), 1–8. 10.1038/s41467-018-03313-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher P. D. E.; Shen Q.; Akpinar B.; Davis L. K.; Chung K. K. H.; Baddeley D.; Šarić A.; Melia T. J.; Hoogenboom B. W.; Lin C.; Lusk C. P. A Programmable DNA Origami Platform for Organizing Intrinsically Disordered Nucleoporins within Nanopore Confinement. ACS Nano 2018, 12 (2), 1508–1518. 10.1021/acsnano.7b08044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exerowa D.; Kashchiev D.; Platikanov D. Stability and Permeability of Amphiphile Bilayers. Adv. Colloid Interface Sci. 1992, 40 (C), 201–256. 10.1016/0001-8686(92)80077-B. [DOI] [PubMed] [Google Scholar]
- Akimov S. A.; Volynsky P. E.; Galimzyanov T. R.; Kuzmin P. I.; Pavlov K. V.; Batishchev O. V. Pore Formation in Lipid Membrane I: Continuous Reversible Trajectory from Intact Bilayer through Hydrophobic Defect to Transversal Pore. Sci. Rep. 2017, 7 (1), 1–20. 10.1038/s41598-017-12127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abkarian M.; Loiseau E.; Massiera G. Continuous Droplet Interface Crossing Encapsulation (CDICE) for High Throughput Monodisperse Vesicle Design. Soft Matter 2011, 7 (10), 4610–4614. 10.1039/c1sm05239j. [DOI] [Google Scholar]
- Wagenbauer K. F.; Engelhardt F. A. S.; Stahl E.; Hechtl V. K.; Stömmer P.; Seebacher F.; Meregalli L.; Ketterer P.; Gerling T.; Dietz H. How We Make DNA Origami. ChemBioChem 2017, 18 (19), 1873–1885. 10.1002/cbic.201700377. [DOI] [PubMed] [Google Scholar]
- Dietz H.; Douglas S. M.; Shih W. M. Folding DNA into Twisted and Curved Nanoscale Shapes. Science (Washington, DC, U. S.) 2009, 325 (5941), 725–730. 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnuswamy N.; Bastings M. M. C.; Nathwani B.; Ryu J. H.; Chou L. Y. T.; Vinther M.; Li W. A.; Anastassacos F. M.; Mooney D. J.; Shih W. M. Oligolysine-Based Coating Protects DNA Nanostructures from Low-Salt Denaturation and Nuclease Degradation. Nat. Commun. 2017, 8 (May), 1–9. 10.1038/ncomms15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Cauter L.; Fanalista F.; van Buren L.; De Franceschi N.; Godino E.; Bouw S.; Danelon C.; Dekker C.; Koenderink G. H.; Ganzinger K. A.. Optimized CDICE for Efficient Reconstitution of Biological Systems in Giant Unilamellar Vesicles. bioRxiv 2021, 10.1101/2021.02.24.432456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanselmann R.; Burchard W.; Lemmes R.; Schwengers D. Characterization of DEAE-Dextran by Means of Light Scattering and Combined Size-Exclusion Chromatography/Low-Angle Laser Light Scattering/Viscometry. Macromol. Chem. Phys. 1995, 196 (7), 2259–2275. 10.1002/macp.1995.021960715. [DOI] [Google Scholar]
- Görlich D.; Henklein P.; Laskey R. A.; Hartmann E. A 41 Amino Acid Motif in Importin-α Confers Binding to Importin-β and Hence Transit into the Nucleus. EMBO J. 1996, 15 (8), 1810–1817. 10.1002/j.1460-2075.1996.tb00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baykal-Caglar E.; Hassan-Zadeh E.; Saremi B.; Huang J. Preparation of Giant Unilamellar Vesicles from Damp Lipid Film for Better Lipid Compositional Uniformity. Biochim. Biophys. Acta, Biomembr. 2012, 1818 (11), 2598–2604. 10.1016/j.bbamem.2012.05.023. [DOI] [PubMed] [Google Scholar]
- Fick A. Über Diffusion [Translated: (1995) “On Liquid Diffusion”. Journal of Membrane Science]. Ann. Phys. 1855, 170 (1), 59–86. 10.1002/andp.18551700105. [DOI] [Google Scholar]
- Dechadilok P.; Deen W. M. Hindrance Factors for Diffusion and Convection in Pores. Ind. Eng. Chem. Res. 2006, 45 (21), 6953–6959. 10.1021/ie051387n. [DOI] [Google Scholar]
- Wente S. R.; Rout M. P. The Nuclear Pore Complex and Nuclear Transport. Cold Spring Harbor Perspect. Biol. 2010, 2 (10), a000562. 10.1101/cshperspect.a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnie A.; Dekker C. Genome-in-a-Box: Building a Chromosome from the Bottom Up. ACS Nano 2021, 15 (1), 111–124. 10.1021/acsnano.0c07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S. M.; Marblestone A. H.; Teerapittayanon S.; Vazquez A.; Church G. M.; Shih W. M. Rapid Prototyping of 3D DNA-Origami Shapes with CaDNAno. Nucleic Acids Res. 2009, 37 (15), 5001–5006. 10.1093/nar/gkp436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kick B.; Praetorius F.; Dietz H.; Weuster-Botz D. Efficient Production of Single-Stranded Phage DNA as Scaffolds for DNA Origami. Nano Lett. 2015, 15 (7), 4672–4676. 10.1021/acs.nanolett.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T.; Merino F.; Stabrin M.; Moriya T.; Antoni C.; Apelbaum A.; Hagel P.; Sitsel O.; Raisch T.; Prumbaum D.; Quentin D.; Roderer D.; Tacke S.; Siebolds B.; Schubert E.; Shaikh T. R.; Lill P.; Gatsogiannis C.; Raunser S. SPHIRE-CrYOLO Is a Fast and Accurate Fully Automated Particle Picker for Cryo-EM. Commun. Biol. 2019, 2 (1), 1–13. 10.1038/s42003-019-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J.; Nakane T.; Forsberg B.; Kimanius D.; Hagen W. J. H.; Lindahl E.; Scheres S. H. W. New Tools for Automated High-Resolution Cryo-EM Structure Determination in RELION-3. eLife 2018, 7, e42166. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J. R.; Mastronarde D. N.; McIntosh J. R. Computer Visualization of Three-Dimensional Image Data Using IMOD. J. Struct. Biol. 1996, 116 (1), 71–76. 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Eisele N. B.; Frey S.; Piehler J.; Görlich D.; Richter R. P. Ultrathin Nucleoporin Phenylalanine-Glycine Repeat Films and Their Interaction with Nuclear Transport Receptors. EMBO Rep. 2010, 11 (5), 366–372. 10.1038/embor.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potma E. O.; De Boeij W. P.; Bosgraaf L.; Roelofs J.; Van Haastert P. J. M.; Wiersma D. A. Reduced Protein Diffusion Rate by Cytoskeleton in Vegetative and Polarized Dictyostelium Cells. Biophys. J. 2001, 81 (4), 2010–2019. 10.1016/S0006-3495(01)75851-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt D.; Heberle F. A.; Greathouse D. V.; Koeppe R. E.; Standaert R. F.; Van Oosten B. J.; Harroun T. A.; Kinnun J. J.; Williams J. A.; Wassall S. R.; Katsaras J. Lipid Bilayer Thickness Determines Cholesterols Location in Model Membranes. Soft Matter 2016, 12 (47), 9417–9428. 10.1039/C6SM01777K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.